
ABSTRACT
Objective: To investigate the cause(s) of a Thai male proband presenting low oxygen saturation by pulse oximetry (SpO2) and severe anemia.
Methods: As Hb variant was suspected, Hb typing was determined by high-performance liquid chromatography and capillary electrophoresis, and subsequently Hb variant was identified by DNA sequencing. Complete blood counts were performed using automated blood cell counter and oxygen saturation was measured by pulse oximetry.
Results: Proband was compound heterozygous for Hb Louisville [β42(CD1)Phe→Leu] and Hb La Desirade [β129(H7)Ala→Val]. Of the proband’s two sons, one was compound heterozygous for Hb Louisville and Hb E and the other for Hb La Desirade and Hb E. The former son had similar clinical features and laboratory findings with those of the proband while the latter showed had no abnormal clinical manifestations.
Conclusion: This the first report of compound heterozygosity of Hb Louisville and Hb La Desirade in an individual of Southeast Asian ethnicity. Hb variant identification is crucial for genetic counseling and appropriate treatment in regions where hemoglobinopathies are common.
Introduction
Hemoglobinopathies are the most common inherited disorder worldwide, with high prevalence in many malaria endemic sub-tropical and tropical countries, such as Thailand [Citation1]. More than 1150 hemoglobin (Hb) variants have been described [Citation2]. The majority of heterozygous Hb variants are not of clinical significance but on-going screening for their identification is still important because certain variants, viz. homozygous Hb S, can be the cause of pathology or when combined with thalassemia, viz. β-thalassemia/Hb E [Citation3].
Differences between low measured oxygen saturation by pulse oximetry (SpO2) and normal calculated oxygen saturation (SaO2) and partial pressure of oxygen (PaO2) in arterial blood gas (‘saturation gap’) may arise from methemoglobinemia or sulfhemoglobinemia [Citation4]. However, the presence of Hb variant with abnormal oxygen binding property should also be suspected.
Here we report the first case of compound heterozygosity of two rare β-Hb variants, Hb Louisville and Hb La Desirade, in a Thai individual presenting severe anemia during febrile illness and low oxygen saturation by SpO2.
Materials and methods
Subjects
The proband was a 30-year-old Thai married man presenting at the Emergency Room, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, Thailand with feeling of dizziness and near-syncope for previous several days. He had a history of fever for the past few days and had no history of taking herbal medicine or prescription drug, had previous history of fever with anemia and of receiving blood transfusion at the age of 10 years. He had a marked pallor but vital signs and physical examination were normal except for tachycardia and low SpO2 (82%). As the proband’s 3-year-old son also had a previous history of low SpO2 (75%), the family was requested to undergo a physical and laboratory examinations. The parents (father and mother of 50 and 49 years of age, respectively) of the proband were reportedly healthy, but were not available for a hospital visit.
The study was approved by the Ethical Committee on Human Rights Related to Research Involving Human Subjects of Ramathibodi Hospital, Mahidol University (approval no. MURA2016/145).
Hematological analysis
Hematological indices were analyzed using Sysmex S-1000i automated blood cell counter (Sysmex, Kobe, Japan). Hemoglobin typing was performed with a Variant-II high-performance liquid chromatography (HPLC) (Bio-Rad, France) and Capillarys-2 capillary electrophoresis (CE) (Sebia, France).
DNA analysis
Common deletional (--SEA, --THAI, --FILL, -α3.7, and -α4.2) and non-deletional (Hb Constant Spring [HBA2:c.427T>C; α142Term→Gln(TAA→CAA at α2)] and Hb Pakse [HBA2:c.429A>T; α142Term→Tyr(TAA→TAT at α2)] were identified by multiplex PCR as described previously [Citation5,Citation6]. Beta-Hb variant sequences were identified by DNA sequencing (1st BASE Pte Ltd., Singapore) using forward and reverse primers 5′ ACGATCTTCAATATGCTTACCAAGC 3′ and 5′ AGACCACCAGCAGCCTAAGG 3′ for exon 1, 5′ TGCCTATTGGTCTATTTTCCCA 3′ and 5′ CAAATAGTAATGTACTAGGCAGACTGTG 3′ for exon 2, and 5′ CAGTGATAATTTCTGGGTTAAGGC 3′ and 5′ GCTGTTGCCAATGTGCATTAG 3′ for exon 3 of the β-globin gene.
Results
In the first visit, the proband had in room air a SpO2 value of 76–80% that increased to 84–86% after oxygen cannula therapy at 5 L/min. The proband had pancytopenia with low Hct and RBC counts, anisocytosis, low WBC counts (69% neutrophils), and low platelet counts (). Blood chemistry showed mildly elevated transaminase activity (aspartate aminotransferase = 70 U/L and alanine aminotransferase = 72 U/L) and indirect bilirubin level (total bilirubin = 1.7 mg/dL and direct bilirubin = 0.4 mg/dL). Arterial blood under room air had a pH of 7.45, PaO2 of 92 mmHg, PaCO2 of 29.9 mmHg, and SaO2 of 97%. Chest X-ray was normal and methemoglobinemia was ruled out (0.3% methemoglobin detected). Measurement by CO-oximetry revealed low oxyHb (28.9%), normal carboxyHb (0.8%) and high deoxyHb (70%) levels. The proband was diagnosed as having acute hemolysis, pancytopenia and low oxygen saturation (but with normal arterial oxygen saturation). The proband was treated with two units of pack red cells transfusion and discharged. A follow-up examination after 2 weeks showed normal vital signs with mild pale conjunctiva and low SpO2 (76–82%) with unexplained oxygen saturation gap. CBC returned to nearly normal values (WBC counts of 7.17 × 109/L, PLT counts of 192 × 109/L, but low RBC number (2.99 × 1012/L) and hematocrit (27.5%)). Reticulocyte production index (RPI) was calculated to yield 0.02 and 1.19 in first and second visits indicating ineffective erythropoiesis cause of anemia (). Arterial blood gas revealed normal oxygenation. Resolving from infection was suspected owing to the spontaneous improvement in CBC parameters.
Table 1. Hematological parameters and globin genotypes of proband and family.
In order to understand the underlying cause of the proband’s anemia, Hb typing was performed. HPLC profile demonstrated abnormal peak shape in A- and A2-windows representing 63.6% and 26.9% respectively of total Hb peaks ((a)), but normal peaks representing 92.6% and 4.3% of total Hb fractions were obtained by CE analysis ((b)). PCR assays rules out the presence of common α-thalassemia genotypes (data not shown). As a result of a combination of the proband’s medical history and abnormal Hb typing results, of the hemoglobinopathy was suspected. DNA sequencing identified the proband as compound heterozygote of Hb Louisville (LV) [β42(CD1)Phe→Leu; HBB:c.127T>C] and Hb La Desirade (LD) [β129(H7)Ala→Val; HBB:c.389C>T] ().
Figure 1. Hb typing by HPLC (A, C, and D) and CE (B). (A) Proband (I-1) showing two overlapping peaks at A- and A2-window. (B) Proband (I-1) showing peaks of Hb A and Hb A2. (C) Proband’s wife (I-2) showing single peak of Hb A2/Hb E. (D) Proband’s son (II-1) showing overlapping peak of variant Hb, Hb E, and Hb A2 and peak of Hb F.
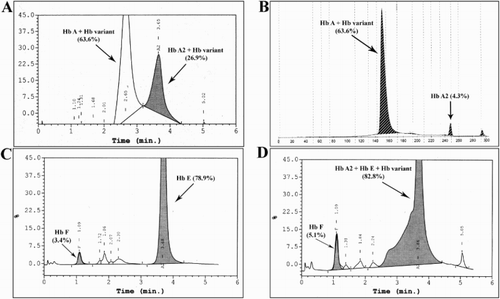
Figure 2. DNA sequencing chromatogram of proband’s β-globin gene. (A) Heterozygous Hb Louisville [β42(CD1)Phe→Leu] as shown by presence of both T and C at codon 42. (B) Heterozygous Hb La Desirade [β129(H7)Ala→Val] as shown by presence of both C and T at codon 129.
![Figure 2. DNA sequencing chromatogram of proband’s β-globin gene. (A) Heterozygous Hb Louisville [β42(CD1)Phe→Leu] as shown by presence of both T and C at codon 42. (B) Heterozygous Hb La Desirade [β129(H7)Ala→Val] as shown by presence of both C and T at codon 129.](/cms/asset/2372fa54-ab89-43bc-80fc-41deecabb887/yhem_a_1231989_f0002_c.jpg)
Examinations of the proband’s wife and the two infant sons showed that the older child (II-1) had low SpO2 (74–80%) while the other two family members had normal values (). Son (II-1) was born healthy and had normal development, but with a history of acute hemolysis whenever there was fever. Low SpO2 occurred after 7 months of age but the subject had normal oxygenation (PaO2 of 97.1% and SaO2 of 95.1%). CBC of II-1 revealed anemia (). HPLC Hb typing of the proband’s wife indicated homozygosity of Hb E ((c)) and II-1 HPLC profile had an abnormal shape Hb peak in A2-window ((d)). DNA sequencing of II-1 β-globin gene identified compound heterozygosity of Hb Louisville and Hb E (data not shown). Medical history of the younger son (II-2) indicated normal growth and normal SaO2 (96–100%) in room air, and DNA sequencing showed compound heterozygosity of Hb La Desirade and Hb E (data not shown). The family’s pedigree is summarized in .
Figure 3. Pedigree of the proband and family. Hb E, [β26(B8)Glu→Lys]; Hb LD, La Desirade [β129(H7)Ala→Val]; Hb LV, Louisville [β42(CD1)Phe→Leu].
![Figure 3. Pedigree of the proband and family. Hb E, [β26(B8)Glu→Lys]; Hb LD, La Desirade [β129(H7)Ala→Val]; Hb LV, Louisville [β42(CD1)Phe→Leu].](/cms/asset/e52c661d-c2f6-417b-8f35-6ff2f91fb851/yhem_a_1231989_f0003_b.gif)
Discussion
We report the first Thai compound heterozygous Hb Louisville and Hb La Desirade resulting in severe anemia with low SpO2. Though premature destruction due to these unstable hemoglobins precipitation in circulating RBC was suspected, ineffective erythropoiesis was seemed to be the cause of anemia. However, of the proband’s two sons, one with compound heterozygous Hb Louisville and Hb E and the other compound heterozygous La Desirade, the former presented clinical and laboratory parameters similar to those of the proband. Hb E is also a mildly unstable, so the compound heterozygosity with Hb Louisville and Hb La Desirade is also very important. It seems like that the substitution of the beta-42Phe in the heme pocket is more crucial for the stability of hemoglobin molecule compared to the alpha1beta1 interface beta-129Ala substitution, resulting in pronounced anemia.
Hb Louisville, also known as Hb Bucuresti [Citation7], has been reported in Canadian [Citation8], Cuban and Rumanian individuals [Citation9]. The mutation [β42(CD1)Phe→Leu] alters the heme contact at F42 of α1β2 dimer resulting in decreased oxygen affinity but normal Bohr effect [Citation10,Citation11]. Carriers of Hb Louisville risk hemolytic crisis when exposed to fever or oxidizing agents or drugs [Citation7,Citation10]. A previous report of a family with Hb Louisville also showed presentation of anemia and low SpO2 reading, but the index has low arterial blood PaO2 owing to other underlying complications [Citation12]. On the other hand, the proband and elder son had normal PaO2 indicating that their clinical profiles and laboratory findings can be attributed to the presence of Hb Louisville. Other similar Hb variants, viz. Hb Hamersmith [β42(CD1)Phe→Ser], Hb Sendagi [β42(CD1)Phe→Val] are associated with similar clinical picture [Citation13,Citation14]. Hb La Desirade also exhibits low oxygen affinity results in at least 1.5 fold increase of P50 values [Citation15]. The synergistic-additive effect of Hb La Desirade on Hb Louisville could be expected and explained the low Hb levels of the proband in the non-febrile state. Similarly, Hb E/β-thalassemia patients are able to adapt to anemia by reducing the oxygen affinity of their RBC [Citation15]. It is worth noting that the proband’s stained blood smear demonstrated 10–20% inclusion bodies. We surmise that this might be due to the existence (of small amounts) of unstable hybrid α2βLouisvilleβLa Desirade when incubation at 37°C for 30 minutes though Hb Louisville was precipitated at 65°C incubation [Citation10], but further investigations are needed.
In conclusion, presence of low SpO2 in the absence of methemoglobinemia or sulfhemoglobinemia but accompanied with fever-related anemia led to the identification of a proband with compound heterozygosity of (mildly unstable) Hb Louisville and Hb La Desirade, the first such case in Thailand. Identification of Hb Louisville in the proband’s two offsprings helped to account for their similar clinical picture. Awareness and subsequent identification of the causative molecule(s) of low SpO2 accompanied with anemia presenting at an emergency room setting will allow appropriate care and treatment for the index case and family.
Acknowledgements
The authors thank Professor Prapon Wilairat for critical reading of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Parin Kamseng is a post graduate student in Doctoral Degree Program in Clinical Pathology at Faculty of Medicine, Ramathibodi hospital, Mahidol University, Thailand. He has studied on various hemoglobin variants detection and identification.
Satariya Trakulsrichai is an Assistant Professor at Ramathibodi Poison Center, Department of Medicine, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Thailand. She is also a clinician in Emergency Medicine at Ramathibodi hospital, Thailand.
Objoon Trachoo is an Assistant Professor in Internal Medicine and Consultant Medical Geneticist, Department of Medicine and Center for Medical Genomics, Faculty of Medicine, Ramathibodi hospital, Mahidol University, Thailand.
Walaiporn Yimniam is a post graduate student in Master Degree Program in Clinical Pathology at Faculty of Medicine, Ramathibodi hospital, Mahidol University, Thailand. She just graduated with the thesis work on hemoglobin variants detection by high resolution melting (HRM) analysis.
Bhakbhoom Panthan is a research assistant at Department of Pathology, Faculty of Medicine, Ramathibodi hospital, Mahidol University, Thailand.
Paisan Jittorntam is a research assistant at Research Center, Faculty of Medicine, Ramathibodi hospital, Mahidol University, Thailand.
Pimjai Niparuck is an Assistant Professor in Internal Medicine. She is also a hematologist at Division of Hematology, Department of Medicine at Faculty of Medicine, Ramathibodi hospital, Mahidol University, Thailand. She has worked in field of hematologic malignancy and stem cell transplantation.
Pitsucha Sanguanwit is a physician and lecturer at Department of Emergency Medicine, Faculty of Medicine, Ramathibodi Hospital, Mahidol University, Thailand.
Winai Wananukul is a Professor of Medicine. He is also a Head of deputy director of Ramathibodi hospital, Head of Ramathibodi Poison Center, Division of Clinical Pharmacology and Toxicology, Department of Medicine at Faculty of Medicine, Ramathibodi hospital, Mahidol University, Thailand.
Sumalee Jindadamrongwech is an Associated Professor in Clinical Pathology teaching and coaching research for post graduate students of Clinical Pathology Programs at Department of Pathology, Faculty of Medicine, Ramathibodi hospital, Mahidol University, Thailand. She has special expertise on hematology and molecular genetics. She works in the field of red blood cell disorders such as thalassemia and hemoglobinopathies.
ORCiD
Objoon Trachoo http://orcid.org/0000-0001-7867-3238
References
- Fucharoen S, Winichagoon P. Haemoglobinopathies in Southeast Asia. Indian J Med Res. 2011;134(4):498–506.
- Giardine B, Borg J, Viennas E, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014;42( Database issue):D1063–1069. doi: 10.1093/nar/gkt911
- Clarke GM, Higgins TN. Laboratory investigation of hemoglobinopathies and thalassemias: review and update. Clin Chem. 2000;46(8 Pt 2):1284–1290.
- Hamirani YS, Franklin W, Grifka RG, et al. Methemoglobinemia in a Young Man. Texas Heart Inst J. 2008;35(1):76–77.
- Chong SS, Boehm CD, Higgs DR, et al. Single-tube multiplex-PCR screen for common deletional determinants of alpha-thalassemia. Blood 2000;95(1):360–362.
- Fucharoen S, Sanchaisuriya K, Fucharoen G, et al. Interaction of hemoglobin E and several forms of alpha-thalassemia in Cambodian families. Haematologica 2003;88(10):1092–1098.
- von Planta M, Humbert J, Wacker P, et al. Hypothesis for generation of the unstable Hb Bucuresti (β 42 Phe→Leu) mutation. Hematol J. 2001;2(1):61–66. doi: 10.1038/sj.thj.6200085
- Smiley RK, Gravely ME, Wilson JB, et al. Hemoglobin louisville (B42 (CDI) PHE→LEU) occurring as a fresh mutation in a Canadian woman. Hemoglobin 1978;2(1):89–90. doi: 10.3109/03630267808999195
- Colombe B, Benitez MP, Bernini L, et al. A new case of haemoglobin Bucuresti in a Cuban family: further functional studies. J Med Genet. 1975;12(3):297–298. doi: 10.1136/jmg.12.3.297
- Keeling MM, Ogden LL, Wrightstone RN, et al. Hemoglobin Louisville (β42 (CD1) Phe→Leu): an unstable variant causing mild hemolytic anemia. J Clin Invest. 1971;50(11):2395–2402. doi: 10.1172/JCI106738
- Villegas A, Malcorra JJ, Balda I, et al. A new Spanish family with Hb Louisville. Am J Med Genet. 1989;32(1):9–14. doi: 10.1002/ajmg.1320320103
- Wu Y, Ramani GV, Gai Q, et al. Rare hemoglobinopathy presenting as progressive dyspnea. Am J Hematol. 2010;85(5):355–357.
- Akiyama M, Murayama S, Yokoi K, et al. Hemoglobin Hammersmith [β 42(CD1) Phe→Ser] causing severe hemolytic anemia in a Japanese girl. Pediatric Blood Cancer. 2006;47(6):839–841. doi: 10.1002/pbc.20533
- Honig GR, Vida LN, Rosenblum BB, et al. Hemoglobin Warsaw (Phe beta 42(CD1)----Val), an unstable variant with decreased oxygen affinity. Characterization of its synthesis, functional properties, and structure. J Biol Chem. 1990;265(1):126–132.
- Merault G, Keclard L, Garin J, et al. Hemoglobin La Desiradb αA2β2 129 (H7) Ala→Val: a new unstable hemoglobin. Hemoglobin 1986;10(6):593–605. doi: 10.3109/03630268609036564