
ABSTRACT
Objectives: Redox imbalance and genotoxic damage are commonly observed in β thalassaemic patients. The aim of this study was to assess the role of anaemia in oxidative and genotoxic damage in regularly transfused thalassaemic patients, undergoing iron chelation therapy.
Methods: We studied the relationships of haematological, biochemical and clinical parameters with oxidative (reactive oxygen species and 8-oxo-7,8-dihydro-2′-deoxyguanosine) and genotoxic biomarkers (Comet assay and cytokinesis-block micronucleus test) in blood samples from 105 patients. To reduce the early effect of redox-active iron, samples were collected when pharmacokinetics of the iron chelators ensured their maximum effectiveness. The transfusion regimen, cardiac and hepatic magnetic resonance imaging T2* were evaluated to characterize the patient cohort. Labile plasma iron (LPI) was also assayed.
Results: Haemoglobin level had a significant effect on ROS, %DNA in the tail and micronuclei-micronucleated cell frequency (p < 0.05). Higher Hb values reduced redox imbalance. LPI, detectable in 50.5% of patients, was related to the number of apoptotic and necrotic lymphocytes (p = 0.03), demonstrating the cytotoxic effect of iron.
Discussion: The results highlight that an adequate transfusion regimen is essential to limit oxidative and genotoxic damage in β-thalassemic patients undergoing chelation therapy.
Conclusion: Owing to the higher risk of cancer in the thalassaemic cohorts, specific genotoxicity/oxidative biomarkers should be monitored in order to ameliorate and formulate more personalized disease management.
Introduction
The pathophysiology of β-thalassaemia is characterized by anaemia, due to a reduction or absence of β-globin, and excessive free α-globin that leads to the premature death of erythrocytes [Citation1] and induces ineffective erythropoiesis. β-Thalassaemia major patients must receive regular blood transfusions to improve haemoglobin (Hb) levels, suppress bone marrow activity and reduce gastrointestinal iron absorption. This treatment is designed to maintain pre-transfusion Hb levels between 9.5 and 10 g/dl, in accordance with the guidelines for the clinical management of thalassaemia and the Thalassaemia International Federation [Citation1].
Blood transfusion therapy increases non-transferrin-bound iron (NTBI) and can lead to multi-organ complications due to haemosiderosis [Citation2]. Owing to the lack of active iron excretion, life-long iron chelation therapy and good compliance are necessary to prevent morbidity and mortality caused by iron overload [Citation1,Citation3]. In addition to parenterally administered deferoxamine (DFO), for which poor compliance is common [Citation4], orally active drugs such as deferiprone (DFP) and deferasirox (DFX) and combined therapy, such as DFO + DFP, are currently available [Citation5–7]. In severe haemosiderosis, combination therapy is more effective than monotherapy due to the ‘shuttle effect’, i.e. the ability of DFP to increase the NTBI fraction that is chelatable by DFO [Citation8,Citation9].
Redox-active free iron and weakly bound iron catalyse the generation of hydroxyl radicals, triggering radical chain reactions [Citation10–12]. Furthermore, haemolysis contributes to a greater redox imbalance due to the pro-oxidant action of iron–binding haemoproteins [Citation13]. The genotoxic properties of pro-oxidant iron, which can cause oxidative DNA damage, are thought to explain the relationship between carcinogenesis and iron overload [Citation14]. Hydroxyl radicals, produced by the redox-active iron in Fenton chemistry, form 8-hydroxy-guanine adducts, causing DNA point-mutations and genomic instability. An increased risk of hepatocellular carcinoma has been observed in hereditary haemochromatosis [Citation15] and in thalassaemia [Citation16].
To date, redox imbalance in thalassaemic patients has mainly been attributed to iron. Anaemia-induced hypoxia is an additional source of oxidative damage that has been underestimated in thalassaemia. We observed, for the first time, that redox imbalance was inversely related to Hb levels in these patients [Citation17]. Overall, we observed that untransfused thalassaemic patients with more severe anaemia and lower iron overload, had higher levels of reactive oxygen species (ROS) and lipid hydroperoxides than transfused patients. The measurement of mitochondrial transmembrane potential, which was lower in untransfused patients, revealed that mitochondrial impairment could be the cause of ROS overproduction [Citation17]. In addition to anaemia-induced hypoxia [Citation18], several studies have highlighted that chronic oxidative stress and DNA damage, as observed in iron deficient anaemia, are due to decreased anti-oxidant defences [Citation19–21].
Examining a large group of transfusion-dependent patients, we assessed the role of anaemia in oxidative and genotoxic damage in thalassaemic patients. The study aimed to improve the effectiveness of therapeutic management of thalassaemic patients and to identify new biomarkers for monitoring oxidative/genotoxic damage.
Oxidative biomarkers included ROS and 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxo-dG), whereas genotoxicity was evaluated by alkaline single cell gel electrophoresis (Comet assay) and the cytokinesis-block micronucleus (CBMN) test.
To better evaluate the pro-oxidant role of anaemia, minimizing the effect of redox-active iron, the endpoints were assayed on samples collected when pharmacokinetics of the iron chelators ensured maximum effectiveness. However, our protocol did not affect chronic iron-induced cito/genotoxicity that was ‘stored’ in lymphocytes and it could be expressed after in vitro activation increasing lymphocytic micronuclei (MNi) frequency [Citation22].
Materials and methods
Patients
One hundred and five β-thalassaemic transfusion-dependent patients were recruited from Messina University Hospital, Riuniti Hospital in Reggio Calabria and Sant'Agata Militello Hospital, Italy. Patients (58 women and 47 men) under the age of 55 years (range 26–53) were selected a priori. They gave informed consent to participate in the study and approval was obtained from the Ethics Committees of the hospitals. All patients underwent iron chelation therapy which was not suspended before sampling. Drugs were administered as follows: 33 patients were treated with DFX (30 mg/kg once daily in the morning per os), 27 with DFO (40 mg/kg per day subcutaneously overnight from 8:00 pm until 8:00 am), 17 with DFP (75 mg/kg per os in three daily doses) and 28 with combined DFO + DFP (40 mg/kg of DFO for three days of the week and 75 mg/kg of DFP in three daily doses on the remaining four days). A control cohort was formed by enrolling twenty healthy individuals matched with patients for age and gender.
Five millilitres of blood were collected from each subject in the morning and, in the patients, immediately before packed red blood cell (RBC) transfusion, in order to better assess the effects of anaemia. In the patient group, samples were collected about 2 hours after taking DFX or DFP and to about 3 hours after the end of DFO treatment. The transfusion regimen for each subjects was based on haematological parameters; the average interval between transfusions was 15–20 days. The number of total transfused RBCs (ml/kg/year) was calculated using the equation:
Iron intake was calculated by multiplying the total transfused RBCs by 1.08 [Citation23]. Pre-transfusion Hb, % reticulocytes, serum ferritin, iron, transferrin and percentage of transferrin saturation (%TSAT) were used to assess the relationships with the assayed endpoints. Although nucleated red blood cells (NRBC) would be a more appropriate marker of ineffective erythropoiesis, they did not routinely monitored at the time of sampling. Cardiac and hepatic MRI T2* were considered to evaluate organ siderosis.
Endpoints
The parameters were analysed in lymphocyte suspensions prepared using previously reported protocols [Citation17]. Briefly, after separation of blood mononuclear cells by Lymphoprep Separation MediumTM (Axis-Shield Oslo, Norway), the buffy layer was collected and resuspended in 0.8% NH4Cl in 0.1 mM EDTA (pH 7.2) in order to lyse NRBC that could affect results, contaminating lymphocytes. After incubation at 37°C for 15 minutes the suspensions were centrifuged and the purified pellets were aliquoted for later analysis.
Intracellular ROS and oxidative DNA damage were detected by cytofluorimetric measurement. We used membrane-permeable lipophilic 2′,7′-dichloro-fluorescein-diacetate as a probe for ROS, and fluorescein isothiocyanate (FITC)-labelled avidin as a probe for 8-oxo-dG [Citation17]. The weighted average of emission values per 100 cells was calculated for both probes and the results of FACS analyses were expressed in arbitrary fluorescence units (AFU). To detect DNA strand breaks, the Comet assay was performed in duplicate according to the method proposed by Tice et al. [Citation24]. For each sample DNA unwinding and lysis were performed for 20 minutes and 1 hour, respectively. Electrophoresis was carried out for 30 minutes at 300 mA and 25 V (0.86 V/cm). Images of at least 100 randomly selected nuclei (ethidium bromide stained) were analysed.
The CBMN assay allows the evaluation of chromosomal damage or the instability status of lymphocytes by detecting MNi. MNi, originated from chromosome breakage and/or aneuploidy, can be the result of cumulative effects, due to recent or previous exposures and they are strongly linked to cancer in humans [Citation25]. Apoptotic and necrotic cells were also scored and the nuclear division index (NDI) was calculated as a marker of mitotic status. To perform the CBMN assay lymphocytes were previously stimulated with phytohemagglutinin (2%) then treated with cytochalasin B (4.5 mg/ml) to block cytokinesis [Citation17]. The cells were fixed, dropped onto microscopic slides and Giemsa stained. According to CBMN criteria [Citation25], micronucleated cells (MNed), binucleated cells with MNi, and the MNi frequency were scored in 1000 binucleated cells for each sample while NDI and necrotic and apoptotic cells were scored in 500 cells.
Labile plasma iron (LPI) which, unlike the Fe(III)-transferrin-bound (TBI), catalyses ROS formation, was analysed following the method devised by Esposito et al. [Citation26]. This fluorimetric assay measures iron-specific redox cycling capacity. Redox reactions were detected by the oxidation of the non-fluorescent probe dihydrorhodamine (DHR) to its fluorescent form rhodamine. The assessment of labile forms of iron, independently of other DHR oxidation mechanisms, samples were analysed in parallel by the addition of the iron chelator DFO which quenches only iron-induced fluorescence. Briefly, serum samples were assayed in quadruplicate in 96-well plates by adding the DHR (50 µM) in reagent solution (pH 7.3) containing 40 µM of ascorbate to regenerate Fe(II) after its oxidation to Fe(III). DFO (50 µM) was added to this solution in two of the wells. A Fe:NTA (1:7 mM) complex, starting from freshly prepared ferrous ammonium sulphate and nitrilotriacetic acid (NTA) (pH 7.0), was used to build a calibration curve (0.2–4 µM). Emitted fluorescence was recorded every 2 minutes starting from 15 up to 40 minutes using excitation/emission filters 485/538 nm (Tecan, Brescia, Italia). The differences between samples with and without DFO, due to redox-active iron, were used to calculate fluorescence units per minute (FU/minute). LPI values (µM) were extrapolated from the calibration curves.
Statistical analyses
Analyses were performed using the software Statistica (StatSoft®, version 10). Lilliefors and Shapiro–Wilk normality tests were used to assess data distribution patterns. Continuous variables were expressed either as mean ± standard deviation (SD) or as median and percentiles; the non-parametric Mann–Whitney or Kruskal–Wallis tests were used in addition to the Spearman test. Multivariate regression analysis was performed using a priori models.
Results
Haematological parameters and iron status
reports the biochemical and haematological parameters, blood and iron intake per year, and cardiac and hepatic MRI T2* in the patient cohort, formed by 105 regularly transfused thalassaemic patients (age 35.4 ± 8.72 years). The values highlight the very high intra-group variability observed for many of the parameters considered (in particular ferritin and transferrin values), as confirmed by SD, skewness and kurtosis values. In addition, the Shapiro–Wilk test showed that several data sets were not normally distributed.
Table 1. Biochemical and haematological parameters, blood and iron intake per year, cardiac and hepatic MRIT2* in the patient cohort.
Although the transfusion treatment was adequate for the majority of patients, 18.9% had pre-transfusion Hb values below the recommended range (9.5–10 g/dl). In this group erythropoiesis enhancement was not effectively blocked, as confirmed by the higher number of reticulocytes in comparison to those with Hb values >10 g/dl (5.91 ± 7.28 vs. 1.31 ± 2.26, Z = 7.91, p < 0.0001).
As shown by MRI T2*, 21.0% of the patients had myocardial siderosis and 36.8% had hepatic siderosis (<20 and <6.3 ms, respectively). Heart and hepatic MRI T2* were inversely related to serum ferritin (r = −0.33, p = 0.02 and r = −0.48, p < 0.001, respectively); the latter was also inversely related to %TSAT (r = −0.27, p = 0.03).
There were no differences between iron chelation treatments for biochemical and haematological parameters, blood and iron intake, cardiac or hepatic MRI T2* (data not shown).
LPI values are not reported in the table since the assay gave results below the detection limit (0.2 µM) in a high percentage of patients. As expected considering the short interval between chelating treatment and blood withdrawal, only 50.5% of patients had detectable LPI (median value in this subgroup: 0.43, interquartile range: 0.29–1.21).
The LPI was >2 µM in 5 patients, probably due to poor compliance with chelation therapy. Moreover, 30 sera samples from both patients and controls gave negative values of LPI. This did not allow statistical analysis to be performed for the entire patient group by correlating the LPI values to the other endpoints investigated. In the patients with detectable LPI, were no differences on the basis iron chelation treatments for (data not shown). Control group sera with detectable LPI (45%) were consistently below 0.3 µM.
Oxidative and genotoxic damage
shows the results of oxidative and genotoxicity biomarkers. In comparison to controls, higher values of biomarkers were obtained in thalassaemic patients. The graphs highlight the high intra-group variability in patients, suggesting that, due to the complexity of the disease, several variables influenced the endpoints assayed. Moreover, in the patient group, ROS and 8-oxo-dG values were directly related to MNi, MNed and TDNA% (p < 0.01), underlining the inter-relations between oxidative damage and genotoxicity.
Figure 1. Results of bivariate analysis by Mann–Whitney test to compare lymphocytic oxidative damage and genotoxic effect in patient cohort and health controls. (a) ROS as assessed in function of DCF emission values (p = 0.0001). (b) 8-oxo-dG as assessed in function of emission values of Avidin-FITC emission values (p < 0.0001). (c) Oxidative DNA damage to Comet assay evaluated by %TDNA (p = 0.01). (d) MNi frequency (p = 0.027). (e) NDI used as markers of proliferative activity (p = 0.001).
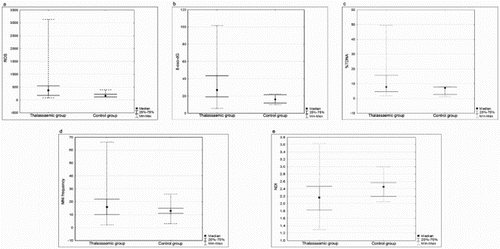
To evaluate the roles of anaemia in the assayed endpoints, patients were grouped on the basis of Hb (<10 vs. ≥10 g/dl). As shown in , highly significant differences were observed in the two subgroups since a small decrease (10.9%) in Hb (9.29 ± 0.44 g/dl vs. 10.43 ± 0.3 g/dl) remarkably increased oxidative and genotoxic damage. NDI values (a marker of proliferative activity) and cytotoxicity (measured by the number of necrotic and apoptotic cells) were also affected by the severity of anaemia.
Table 2. Oxidative and genotoxic damage in patients grouped according the levels of Hb (g/dl).
A decrease in lymphocytic ROS was observed in patients with more severe hyperferritinemia and ROS were inversely related to ferritin levels (r = −0.39; p < 0.001). However, this correlation was non-causal but attributable to better anaemia control, as shown by Hb values that were, on average, 3% lower in the patients with more severe hyperferritinemia (values >500 ng/ml). Although a more intensive blood transfusion therapy increased iron load, it also reduced anaemia-induced oxidative and genotoxic damage. Despite the predictive role for siderosis, as reported above, ferritin was inversely related to percentage of DNA in the tail (%TDNA), MNi and MNed (r = −0.26, p = 0.049; r = −0.32, p = .008; r = −0.29, p = 0.039) while it was directly related to NDI (r = 0.31, p = 0.022). These non-causal relationships strengthened the role of Hb levels in oxidative and genotoxic damage in transfused thalassaemic patients.
The short interval between chelating treatment and blood withdrawal did not allow us to assess the pro-oxidant effect of free and/or labile-bound iron which might occur at points during the day. In the subgroup with detectable LPI values, no significant correlations were observed between LPI and oxidative and genotoxicity endpoints. Instead, LPI was related to apoptotic and necrotic lymphocytes (r = 0.28; p = 0.03), revealing the cytotoxicity of labile iron. Moreover, as demonstrated by the significant LPI association with reticulocytes (%) (r = 0.3; p = 0.04), the degree of anaemia was more severe in patients with higher LPI levels. Similar results were obtained for transferrin level, %TSAT and serum iron; the latter seemingly was more predictive of iron genotoxicity, showing direct relations with %TDNA and TM (p < 0.05 for all percentiles; data not shown). There were no differences between iron chelation treatments oxidative and genotoxicity biomarkers (data not shown).
Hypersplenism and biological variables
Overall, the observed variability revealed a large number of confounding factors, due to the complexity of the disease.
Hypersplenism, a common condition in β-thalassaemia patients, was partially responsible for the variability in the assayed endpoints, causing premature and rapid removal of damaged blood cells. This increases the need for blood transfusion to counteract the worsening of anaemia. After stratification of patients into splenectomized (S) and non-splenectomized (NS), it emerged that the hypersplenism prevalence in the US group led to higher blood consumption (S: 118.1 ± 23.7 vs. NUS: 149.6 ± 25 ml/kg/year; Z = 4.45, p < 0.001). The increase in iron intake in NS patients led to higher ferritin levels (S: 1026.9 ± 1311.2 vs. NS: 1494.4 ± 1381.9 ng/ml; Z = 2.32, p = 0.02) and, although not statistically significant, to lower hepatic MRI T2* levels (S: 12.0 vs. US: 9.91 ms). In addition, due to the premature removal of damaged lymphocytes caused by hypersplenism, cyto- and genotoxicity biomarkers were underestimated. Bivariate analysis showed a significant difference between S and NS patients regarding 8-oxo-dG (S: 13.6 ± 6.0 vs. NS: 7.2 ± 3.9 AFU; Z = −2.2; p < 0.05). Other differences, although not significant, were also found for intracellular ROS, MNi frequency, apoptotic and necrotic cells.
Further factors that influenced the assayed parameters in the patient cohort were biological variables such as gender and age. A greater number of necrotic cells were observed in males than in females (median value 8.1 vs. 4.4; p < 0.05) whereas, as expected, MNi rose with increasing age (>30 vs. ≤30 median value: 20.9 vs. 15.5; p = 0.02).
Assessment of oxidative and genotoxic damage by multivariate analysis
Multivariate analysis using the a priori models of multiple regression was carried out to better evaluate the independent variables most responsible for oxidative/genotoxic damage. The model included six continuous covariates () and the results indicated a pivotal protective role of Hb level and consequently, of blood transfusion. Owing to the confounding effect of hypersplenism, the amount of RBCs transfused was not considered in the regression model. %TDNA was inversely related to Hb values and the effect was observed indiscriminately throughout the lymphocyte population (all percentiles) of each patient. Moreover, higher Hb values improved lymphocyte proliferation, as assessed by NDI. None of the covariates included in the model to evaluate iron overload contributed significantly to genotoxicity endpoints. However, serum iron and hepatic and cardiac siderosis were significantly related to cytotoxicity, increasing the number of apoptotic and necrotic lymphocytes. Moreover, hepatic siderosis significantly depressed lymphocyte proliferation.
Table 3. Multiple regression analysis for oxidative and genotoxic effects.
Discussion
The results, obtained reducing the effect of redox-active iron, underline the impact of anaemia on oxidative and genotoxic damage in transfused thalassaemic patients. The significant inverse relationship between Hb and oxidative damage highlights the protective role of blood transfusions which maintain sufficiently high Hb levels to counteract oxidative damage. As assessed previously [Citation17], free radical overproduction can be due to mitochondrial impairment, i.e. to the imbalance between O2 and electron flow caused by anaemia-induced tissue hypoxia [Citation27,Citation28]. In addition, anaemia may cause redox imbalance due to a decrease in anti-oxidant enzymes such as catalase, superoxide dismutase and glutathione peroxidase [Citation19,Citation20].
Thanks to the effect of iron chelation therapy, the LPI values observed in this study were generally low (0–0.4 µM), and overlapped with the values reported in well-chelated thalassaemic patients [Citation29]. As observed in other studies [Citation30–32], some sera gave negative LPI values. This could be attributed to the removal of contaminant Fe by the Fe-binding activity in the sera [Citation32]. Although the aim of our study was to assess the anaemia-induced cito/genotoxicity, it is also noteworthy that chelating treatment could not ensure a complete removal of the redox-active iron. Depending on the pharmacokinetics of the chelator and the patient's iron status, daily LPI recrudescence may occur during the washout period of the drugs [Citation33]. This can occur in particular when monotherapy with DFO or DFP is used, causing daily and nightly LPI increases, respectively [Citation33].
On this basis, chelation therapy does not entirely prevent organ failure due to the cytotoxic effect of iron overload. The significant increase in apoptosis and necrosis, observed in our study in lymphocytes, could be responsible for the higher incidence of hepatocellular carcinoma in thalassaemic patients [Citation16]. By increasing cell turnover, iron cytotoxicity could promote the multistep process of carcinogenesis in the liver [Citation10,Citation11,Citation34], as observed in hereditary haemochromatosis [Citation14,Citation15,Citation35]. This disease is broadly adopted as a model to evaluate the impairment of iron homeostasis caused by secondary siderosis, that characterizes thalassaemia.
The analogy, although only partial, between hereditary haemochromatosis and thalassaemia led to the undervaluation of the effects of chronic anaemia in the latter. Excluding hepatocellular carcinoma, malignancies were once considered rare in β-thalassaemia, but the frequency of leukaemia and other cancers is now rising [Citation36,Citation37]. The greater incidence of tumours is not necessarily linked to iron overload; it could also be attributable to anaemia-induced ROS overproduction which, until recently, has been underestimated in thalassaemia.
Contrary to the widely accepted thesis that iron overload is the main contributory factor in oxidative impairment [Citation38], this is, to the best of our knowledge, the first study highlighting the role of anaemia in oxidative/genotoxic damage of poly-transfused thalassaemic patients subjected to iron chelation. Owing to the short half-life of ROS and of DNA oxidative damage, which is quickly eliminated by efficient enzymatic repair systems [Citation39], our results of the ROS, 8-oxo-dG and Comet assays may be attributed almost entirely to anaemia.
Despite the strong associations observed in our patient group between MNi frequency, ROS, 8-oxo-dG and TDNA%, the high percentage of MNi could also be caused by LPI recrudescence. This is attributable to an MNi feature that, unlike the other assayed endpoints, underlines previous genotoxic damage which has gone unrepaired and then been transmitted to daughter cells [Citation22].
Our data highlight how even small Hb increases (i.e. ∼11%) are sufficient to significantly reduce oxidative and genotoxic damage. They indicate the importance of a regular transfusion regimen coupled to iron chelation therapy that complies strictly with the guidelines for thalassaemia management [Citation40]. In this regard it is useful to underline that 18.9% of the assayed patients had pre-transfusion Hb values below the recommended range.
In addition to the oxidative/genotoxic effect of chronic anaemia, we assessed the contribution of hypersplenism, a complication common in β-thalassaemia, to anaemia of these patients [Citation1]. Our results, consistent with a recent study [Citation41], demonstrated the protective effect of splenectomy that allows anaemia and siderosis containment in thalassaemia. The eradication of splenic haemocatheresis ensures higher Hb levels, reducing the need for RBC transfusion and, consequently, decreasing ferritin and iron load. This effect was further supported by the higher values of hepatic MRI T2* found in splenectomized versus non-splenectomized patients.
Conclusion
Despite the complexity of β-thalassaemia and the many confounding factors, our study indicates that appropriate blood transfusion therapy is essential to restrain oxidative and genotoxic damage. The increased life expectancy from improved disease management might make the onset of cancer more likely [Citation37,Citation42]. The significantly higher risk of haematological malignancies, recently observed in the thalassaemic cohorts [Citation43], corroborate our results that showed a strong link between severe anaemia and genotoxicity. Considering the relationship between genotoxicity and carcinogenesis [Citation44], a transfusion regimen designed to maintain pre-transfusion Hb levels as close as possible to 10 g/dl is essential for the correct management of transfused β-thalassaemia patients. The increase in iron load, due to an appropriate transfusion treatment, is partially offset by reduced intestinal iron absorption, which is hypoxia-activated [Citation18,Citation45].
The prevention of oxidative/genotoxic damage should not overshadow the role of iron overload that has to be efficiently contained by life-long chelation therapy. The effectiveness of chelation therapy should be periodically evaluated to minimize LPI recrudescences that may occur during the washout period of the drugs.
Finally, specific genotoxicity/oxidative biomarkers should be monitored in order to ameliorate and formulate more personalized disease management for thalassaemic patients.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Giuseppa Visalli http://orcid.org/0000-0002-9072-1148
Angela Di Pietro http://orcid.org/0000-0002-7273-6493
Notes on contributors
Dr Elisa Ferro is a biologist specialized in Medical Genetics and Ph.D. in ‘Applied Biology and Experimental Medicine’ of Messina University. She has experience in the field of cytogenetics, cytogenetic tests for genotoxicity, diagnosis of carrier status and prevention of haemoglobinopathies by using HPLC and molecular genetic tests. She has collaborated and still actively participates in research projects in the field of clinical genetics and particularly of haemoglobinopathies.
Dr Giuseppa Visalli is a biologist specialized in Clinical Biochemistry and Ph.D. She is currently Researcher at the Department of Biomedical and Dental Sciences and Morphofunctional Imaging, University of Messina. She teaches ‘General and Applied Hygiene’ (SSD Med 42) at the University of Messina. She is a member of Italian Society of Hygiene (SITI) and of Italian Society of Environmental Mutagenesis (SIMA). She is the author of several publications and proceedings regarding both the interactions between environment and health and the infectious disease. In particular she studied the role of oxidative stress in several diseases both inherited and mediated by environmental pollutants, as transition metals adsorbed to fine and ultrafine particulate matter. She has also studied sexually transmitted diseases, with particular regard to HIV and HPV.
Dr Maria Angela La Rosa is a biologist manager of Thalassemia Laboratory at U.O.C. of Genetics and Pediatric Immunology of Messina University Hospital. She is a member of the Italian Society of Biochemistry (SIBioC) and of the Italian Society Thalassemia and Hemoglobinopathies (SITE). She is a tutor in both the School of Specialization in Medical Genetics and in the doctoral school ‘Applied Biology and Experimental Medicine’ of Messina University. She has collaborated and still actively participates in research projects in the field of haemoglobinopathies.
Dr Rosa Civa is a biologist manager of Cytogenetics Laboratory at U.O.C. of Genetics and Pediatric Immunology of Messina University Hospital. She is a tutor in the School of Specialization in Medical Genetics and she has had teaching assignments in degree course of Medicine and Surgery of Messina University. She has collaborated in research projects in the field of medical genetic and in monitoring studies of subjects exposed to physical, chemical and environmental genotoxic agents by genotoxicity tests.
Dr Gaetano Randazzo Papa, specialist in clinical pathology, is a biologist manager in Clinical Pathology Service of A.S.P. N.5, Messina. His primarily interest is on haematological diseases and he collaborates in research projects in haematology and cell cultures of bone marrow. He is a lecturer at the Training Department of the Company ECM courses.
Dr Domenico Giuseppe D’Ascola is the director of the Thalassemia Center in U.O.C. Riuniti Hospital of Reggio Calabria, Italy. He participates to prospective multicentre randomised clinical trials promoted by the Italian Society for Thalassemia and Hemoglobinopathies and he is the author of several study published in international and national journals.
Dr Gaetano Roccamo is the director of the Thalassemia Center in Sant’ Agata Militello Hospital, Messina, Italy. He participates to prospective multicentre randomised clinical trials promoted by the Italian Society for Thalassemia and Hemoglobinopathies.
Dr Basilia Piraino, specialist in Medical Genetics, is a geneticist/manager in clinical thalassemia at the UOC of Genetics and Paediatric Immunology – Center Microcythemia of Messina University Hospital. She is an investigator of the clinical study (Protocol Novartis Farma – CICL670A2203) to assess the long-term effectiveness and to provide access to treatment with ICL670 (Deferasirox) in patients with congenital chronic anemias and transfusional haemosiderosis. She collaborated to the multicentre study MIOT (Myocardial Iron Overload in Thalassemia) aimed at the validation of the MRI sequences Imaging (MRI) multi-slice multi-echo T2 * level and cardiac T2 * gradient-echo in the liver. She is the principal investigator of the multicentre observational study ‘Quality of life, patient satisfaction and cost of the disease in patients with β-thalassemia major undergoing iron chelation treatment.’ SPONSOR – Consortium for Biological Evaluation and pharmacological – Pavia.
Dr Carmelo Salpietro is a full professor of the Department of Human Pathology of Adult and Developmental Age ‘Gaetano Barresi’, University Hospital of Messina. He is also the director of the ‘Genetics and Pediatric Immunology Society’ and of the School of Specialization of Medical Genetics and teaches Paediatrics, Immunology, Immunogenetics, Medical Genetics, and Pediatric Therapy in Messina University. He is author of about 300 publications published in international and national journals and book chapters.
Dr Angela Di Pietro is an associate professor of Hygiene and public health (SSD Med 42) in the Department of Biomedical and Dental Sciences and Morphofunctional Imaging of Messina University, Italy. She is a component of the teaching staff of the specialization school ‘Hygiene and preventive medicine’ and of the doctoral school ‘Applied Biology and Experimental Medicine.’ She is a member of Italian Society of Hygiene (SITI) and of Italian Society of Environmental Mutagenesis (SIMA). She is the author of several publications and proceedings regarding the interactions environment-health. More recent researches concern the pathogenic mechanisms of endogenous transition metals and metals adsorbed to fine and ultra-fine particulate matter. By means of in vitro studies she studied the metal role in the pathogenesis of chronic obstructive pulmonary disease (COPD). By ex vivo studies she highlighted the role of oxidative stress in several diseases both inherited and mediated by environmental pollutants.
Additional information
Funding
References
- Cao A, Galanello R. Beta-thalassaemia. Genet Med. 2010;12:61–76. doi: 10.1097/GIM.0b013e3181cd68ed
- Delea TE, Edelsberg J, Sofrygin O, et al. Consequences and costs of noncompliance with iron chelation therapy in patients with transfusion-dependent thalassaemia: a literature review. Transfusion. 2007;47:1919–1929. doi: 10.1111/j.1537-2995.2007.01416.x
- Taher A, Musallam KM, El Rassi F, et al. Levels of non-transferrin-bound iron as an index of iron overload in patients with thalassaemia intermedia. Br J Haematol. 2009;146:569–572. doi: 10.1111/j.1365-2141.2009.07810.x
- Cianciulli P. Treatment of iron overload in thalassaemia. Pediatr Endocrinol Rev. 2008;6:208–213.
- Borgna-Pignatti C, Cappellini MD, De Stefano P, et al. Cardiac morbidity and mortality in deferoxamine- or deferiprone-treated patients with thalassaemia major. Blood. 2006;107:3733–3737. doi: 10.1182/blood-2005-07-2933
- Nick H, Acklin P, Lattmann R, et al. Development of tridentate iron chelators: from desferrithiocin to ICL67. Curr Med Chem. 2003;10:1065–1076. doi: 10.2174/0929867033457610
- Crisponi G, Remelli M. Iron chelating agents for the treatment of iron overload. Coord Chem Rev. 2008;252:1225–1240. doi: 10.1016/j.ccr.2007.12.014
- Evans P, Kayyali R, Hider RC, et al. Mechanism for the shuttling of plasma non transferrin-bound iron (NTBI) onto Deferoxamine by Deferiprone. Transl Res. 2010;156:55–67. doi: 10.1016/j.trsl.2010.05.002
- Alpendurada F, Smith GC, Carpenter JP, et al. Effects of combined deferiprone with deferoxamine on right ventricular function in thalassaemia major. J Cardiovasc Magn Reson. 2012;14:8. doi: 10.1186/1532-429X-14-8
- Gutteridge JM, Rowley DA, Halliwell B. Superoxide-dependent formation of hydroxyl radicals and lipid peroxidation in the presence of iron salts. Detection of ‘catalytic’ iron and anti-oxidant activity in extracellular fluids. Biochem J. 1982;206:605–609. doi: 10.1042/bj2060605
- Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol. 2005;202:199–211. doi: 10.1016/j.taap.2004.06.021
- Fibach E, Rachmilewitz EA. The role of oxidative stress in haemolytic anaemia. Curr Mol Med. 2008;8:609–619. doi: 10.2174/156652408786241384
- Ryter SW, Tyrrell RM. The heme synthesis and degradation pathways: role in oxidant sensitivity. Hemeoxygenase has both pro- and antioxidant properties. Free Radic Biol Med. 2000;28:289–309. doi: 10.1016/S0891-5849(99)00223-3
- Toyokuni S. Iron-induced carcinogenesis: the role of redox regulation. Free Radic Biol Med. 1996;20:553–566. doi: 10.1016/0891-5849(95)02111-6
- Huang X. Iron overload and its association with cancer risk in humans: evidence for iron as a carcinogenic metal. Mutat. Res. 2003;533:153–171. doi: 10.1016/j.mrfmmm.2003.08.023
- Borgna-Pignatti C, Vergine G, Lombardo T, et al. Hepatocellular carcinoma in the thalassaemia syndromes. Br J Haematol. 2004;124:114–117. doi: 10.1046/j.1365-2141.2003.04732.x
- Ferro E, Visalli G, Civa R, et al. Oxidative damage and genotoxicity biomarkers in transfused and untransfused thalassaemic subjects. Free Radic Biol Med. 2012;53:1829–1837. doi: 10.1016/j.freeradbiomed.2012.08.592
- Ferro E, Visalli G, Currò M, et al. HIF1α and Glut1 receptor in transfused and untransfused thalassemic patients. Br J Haematol. 2016;174:824–6. doi: 10.1111/bjh.13815
- Isler M, Delibas N, Guclu M, et al. Superoxide dismutase and glutathione peroxidase in erythrocytes of patients with iron deficiency anaemia: effects of different treatment modalities. Croat Med J. 2002;43:16–19.
- Aslan M, Horoz M, Kocyigit A, et al. Lymphocyte DNA damage and oxidative stress in patients with iron deficiency anaemia. Mutat Res/Fund Mol Mech Mutagen. 2006;601:144–149. doi: 10.1016/j.mrfmmm.2006.06.013
- Coghetto Baccin A, Lauerman Lazzaretti L, Duarte Martins Brandao V, et al. Oxidative stress in older patients with iron deficiency anaemia. J Nutr Health Aging. 2009;13:666–670. doi: 10.1007/s12603-009-0195-6
- Vral A, Fenech M, Thierens H. The micronucleus assay as a biological dosimeter of in vivo ionising radiation exposure. Mutagenesis. 2011;26:11–17. doi: 10.1093/mutage/geq078
- Cohen AR, Glimm E, Porter JB. Effect of transfusional iron intake on response to chelation therapy in β-thalassaemia major. Blood. 2008;111:583–587. doi: 10.1182/blood-2007-08-109306
- Tice RR, Augurell E, Andersen D, et al. Single cell gel/comet assay: guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen. 2000;35:206–221. doi: 10.1002/(SICI)1098-2280(2000)35:3<206::AID-EM8>3.0.CO;2-J
- Fenech M. Cytokinesis-block micronucleus cytome assay. Nat Protoc. 2007;2:1084–1104. doi: 10.1038/nprot.2007.77
- Esposito BP, Breuer W, Sirankapracha P, et al. Labile plasma iron in iron overload: redox activity and susceptibility to chelation. Blood. 2003;102:2670–2677. doi: 10.1182/blood-2003-03-0807
- Semenza GL. Hypoxia-inducible factor 1: regulator of mitochondrial metabolism and mediator of ischemic preconditionin. Biochim Biophys Acta (BBA) – Mol Cell Res. 2011;1813:1263–1268. doi: 10.1016/j.bbamcr.2010.08.006
- Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology. 2009;24:97–106. doi: 10.1152/physiol.00045.2008
- Daar S, Pathare A, Nick H, et al. Reduction in labile plasma iron during treatment with deferasirox, a once-daily oral iron chelator, in heavily iron-overloaded patients with beta-thalassaemia. Eur J Haematol. 2009;82:454–457. doi: 10.1111/j.1600-0609.2008.01204.x
- Gosriwatana I, Loreal O, Lu S, et al. Quantification of non-transferrin-bound iron in the presence of unsaturated transferrin. Anal Biochem. 1999;273:212–220. doi: 10.1006/abio.1999.4216
- Breuer W, Ronson A, Abramov A, et al. The assessment of serum non-transferrin bound iron (NTBI) in chelation therapy and iron supplementation. Blood. 2000;95:2975–2982.
- Breuer W, Ermers MJ, Pootrakul P, et al. Desferrioxamine-chelatable iron, a component of serum non-transferrin-bound iron, used for assessing chelation therapy. Blood. 2001;97:792–798. doi: 10.1182/blood.V97.3.792
- Zanninelli G, Breuer W, Cabantchik ZI. Daily labile plasma iron as an indicator of chelator activity in Thalassaemia major patients. Br J Haematol. 2009;147:744–751. doi: 10.1111/j.1365-2141.2009.07907.x
- Prá D, Franke SI, Henriques JA, et al. Iron and genome stability: an update. Mutat Res. 2012;733:92–99. doi: 10.1016/j.mrfmmm.2012.02.001
- Chen J, Chloupková M. Abnormal iron uptake and liver cancer. Cancer Biol Ther. 2009;8:1699–1708. doi: 10.4161/cbt.8.18.9146
- Alavi S, Sadeghi E, Amiri S. Hematological malignancies complicating β-thalassemia syndromes: a single center experience. Blood Res. 2013;48:149–151. doi: 10.5045/br.2013.48.2.149
- Baldini M, Serafino S, Zanaboni L, et al. Thyroid cancer in β-thalassaemia. Haemoglobin. 2012;36:407–408. doi: 10.3109/03630269.2012.695308
- Fibach E, Rachmilewitz EA. The role of antioxidants and iron chelators in the treatment of oxidative stress in thalassaemia. Ann N Y Acad Sci. 2010;1202:10–16. doi: 10.1111/j.1749-6632.2010.05577.x
- Spivak G. Nucleotide excision repair in humans. DNA Repair (Amst). 2015;36:13–18. doi: 10.1016/j.dnarep.2015.09.003
- Cappellini MD, Cohen A, Porter J, et al. Guidelines for the management of transfusion dependent thalassaemia (TDT). 3rd ed. Nicosia (CY): Thalassaemia International Federation; 2014.
- Casale M, Cinque P, Ricchi P, et al. Effect of splenectomy on iron balance in patients with β-thalassaemia major: a long-term follow-up. Eur J Haematol. 2013;91:69–73. doi: 10.1111/ejh.12121
- Otrock ZK, Shamseddine AI, Taher AT. Non-Hodgkin disease in beta-thalassaemia major. Am J Hematol. 2006;81:62–64. doi: 10.1002/ajh.20463
- Zanella S, Garani MC, Borgna-Pignatti C. Malignancies and thalassaemia: a review of the literature. Ann N Y Acad Sci. 2016;1368:140–148. doi: 10.1111/nyas.13005
- Iarmarcovai G, Ceppi M, Botta A, et al. Micronuclei frequency in peripheral blood lymphocytes of cancer patients: a meta-analysis. Mutat Res/Rev Mutat Res. 2008;659:274–283. doi: 10.1016/j.mrrev.2008.05.006
- Ganz T. Hepcidin and iron regulation, 10 years later. Blood. 2011;117:4425–4433. doi: 10.1182/blood-2011-01-258467