
ABSTRACT
Background: Hereditary hemochromatosis gene (HFE) mutations have a role in iron overload in pediatric acute lymphoblastic leukemia (ALL) survivors. We aimed to evaluate the genotype frequency and allelic distribution of the two HFE gene mutations (C282Y and H63D) in a sample of Egyptian pediatric ALL survivors and to detect the impact of these two mutations on their iron profile.
Patients and methods: This study was performed on 35 ALL survivors during their follow-up visits to the Hematology and Oncology Unit, Pediatric Department, Menoufia University Hospitals. Thirty-five healthy children of matched age and sex were chosen as controls. After completing treatment course, ALL survivors were screened for the prevalence of these two mutations by polymerase chain reaction-restriction fragment length polymorphism. Serum ferritin levels were measured by an enzyme-linked immunosorbent assay technique (ELISA).
Results: C282Y mutation cannot be detected in any of the 35 survivors or the 35 controls. The H63D heterozygous state (CG) was detected in 28.6% of the survivors group and in 20% of controls, while the H63D homozygous (GG) state was detected in 17.1% of survivors. No compound heterozygosity (C282Y/H63D) was detected at both groups with high G allele frequency (31.4%) in survivors more than controls (10%). There were significant higher levels of iron parameters in homozygote survivors than heterozygotes and the controls.
Conclusion: H63D mutation aggravates the iron overload status in pediatric ALL survivors.
Introduction
Hereditary hemochromatosis (HH) is a genetic disorder affecting iron metabolism resulting in iron overload-associated tissue injury. It is an autosomal recessive disorder caused by mutations in the HFE gene (on 6p21.3) [Citation1]. It is characterized by increased iron absorption and storage, resulting in progressive and multisystem oxidative organ damage [Citation2]. HFE gene variants correlate with body iron levels and associated with cancer risk such as childhood acute lymphoblastic leukemia (ALL) [Citation3]. The HFE protein plays a key role in the regulation of body iron uptake through interaction with the transferrin receptor (TfR) on the plasma membrane in which it modulates the interaction of transferrin (Tf) with the TfR, thereby limiting the amount of iron that is internalized [Citation4]. HFE is a protein of 343 amino acids that includes a signal peptide, and its extracellular domain consists of three loops with intramolecular disulfide bonds within the second and third loops which are an extracellular TfR-binding region (α1 and α2), an immunoglobulin-like domain (α3), a trans-membrane region and a short cytoplasmic tail [Citation5]. Two mutations of the HFE gene were included as potential confounders based on their association with high iron absorption (C282Y and H63D) [Citation6]. C282Y means substitution of tyrosine for cysteine at the 282nd amino acid position in the protein sequence. H63D is a point mutation that changes histidine to aspartic acid at HFE residue number 63. C282Y is the most common gene mutation associated with HFE-HH. Approximately 82–90% of individuals diagnosed with HFE-HH are homozygous for this gene, and this genotype has the highest risk for iron overload when inherited in this state [Citation7]. Heterozygote mutations in C282Y/H63D account for nearly 3–8% of individuals with HFE-HH, and this genotype can result in iron overload but at lesser risk than those homozygous for C282Y [Citation8]. Genetic mutations of H63D/H63D (homozygotes) account for approximately 1% of those with gene mutation [Citation8]. The mechanism, by which mutations increase cancer risk, is that excess iron promotes oxidative DNA damage and free radical activity [Citation9]. The iron-catalyzed free radical reactions cause cellular injury by lipid peroxidation, stimulation of collagen formation by activation of hepatic stellate cells, and interaction of reactive oxygen species and iron directly with DNA [Citation10]. Altered iron metabolism affects carcinogenesis through a number of signaling pathways [Citation11].
Objectives and aim
The aim of this work is to study the genotype frequency and allelic distribution of the two HFE gene point mutations (C282Y and H63D) in a sample of Egyptian pediatric ALL survivors and to detect the impact of these two mutations on their iron profile.
Patients and methods
This study was performed on 35 ALL survivors during their follow-up visits to the Hematology and Oncology Unit, Department of Pediatrics, Menoufia University Hospitals after completing the course of treatment. The study was held in the period from March 2015 to February 2016. Thirty-five age- and gender-matched healthy children were taken as the control group. They were volunteers recruited from schools. ALL survivors were treated by chemotherapy according to St Jude Total XV Chemotherapy Protocol [Citation12]. None of them received radiotherapy. This study was approved by the ethical committee of Faculty of Medicine, Menoufia University. A written informed consent was obtained from the parents of all children, and oral assent was obtained from all children.
Inclusion criteria included survivors of pediatric ALL, children aged from 1 to 18 years, and survivors treated with chemotherapy only. Exclusion criteria included other types of childhood cancer, children less than 1 year or more than 18 years old, survivors treated with radiotherapy or bone marrow transplantation and patients with any disease affecting the iron metabolism.
Sample collection and preparation
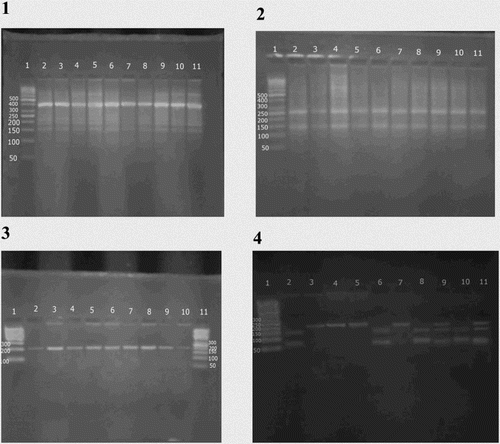
Four milliliters of venous blood was withdrawn from every subject, then 2 ml was transferred into a plain tube, centrifuged for 10 min at 4000 r.p.m. The serum obtained was kept frozen at −20°C till analysis (determination of serum total iron, total iron-binding capacity (TIBC), and ferritin). Two milliliters was transferred into an EDTA tube for DNA extraction and polymerase chain reaction (PCR) at the research laboratory of Medical Biochemistry Department, Faculty of Medicine, Menoufia University. Serum ferritin levels were measured to assess the iron status of our patients using an enzyme-linked immunosorbent kit (Ramco Laboratories, Inc., Stafford, Texas, USA) [Citation13]. Total iron and total iron-binding capacity (TIBC) were determined in serum samples by using the colorimetric method [Citation14, Citation15]. Transferrin saturation index (TSI) was calculated by the following formula: serum iron concentration divided by serum TIBC and multiplied by 100. The TSI > 16% values were regarded as correct ones [Citation16]. Analysis of HFE gene mutations: First, DNA was extracted from whole blood using Thermo Scientific Gene JET Genomic DNA purification kit, Lithuania. DNA was eluted and stored at −20°C for further PCR procedure. The mutation of HFE gene was determined using a PCR-restriction fragment length polymorphism (PCR-RFLP) technique. PCR for HFE gene was carried out with a total volume of 25 μl, containing 10 μl of genomic DNA; 1 μl of each primer; 12.5 μl of Master Mix using Thermo Scientific Dream Taq Green PCR Master Mix, USA, and 0.5 μl of distilled water. C282Y mutation was analyzed using the following designed primers (Midland, Texas): forward: 5′-TGGCAAGGGTAAACAGATCC-3′; reverse: 5′-CTCAGGCACTCCTCTCAACC-3′. Primer sequences of H63D mutation were as follows: forward: 5′-ACATGGTTAAGGCCTGTTGC-3′; reverse: 5′-GCCACATCTGGCTTGAAATT-3′. PCR amplification was performed using Applied Bio systems 2720 thermal cycler (Singapore). The PCR conditions were as follows: 94°C for 3 min, and then 35 cycles of 94°C for 60 s, 55°C for 45 s and 72°C for 1.5 min, and finally 72°C for 2 min. The amplified products were visualized and photographed using a UV transilluminator. Upon digestion with two restriction enzymes RsaI and BclI (provided by Fermentas), 10 μl of the PCR products for the HFE was mixed with 1 μl (1 unit) of Fast Digest® restriction enzyme with 6.5 μl nuclease-free water and 2.5 of 10× Fast Digest® buffer. After good mix, incubation was carried out at 37°C for 2 h, then 10 μl of the products were loaded into a 3% agarose gel containing ethidium bromide for electrophoresis. The digestion products were two fragments of 247 and 140 bp in all patients and in healthy controls. The digestion products of BclI restriction enzyme were two fragments of (138 bp) and (70 bp) in all healthy controls. The mutant alleles resist digestion giving band with (208 bp) size ().
Figure 1. Agarose gel electrophoresis of (1) amplification product of HFE gene at 387 bp. Lane 1 shows 50 bp ladder; lanes (2, 3, 4, 5, 6, 7, 8, 9, 10, and 11) show the amplification product of HFE gene at 387 bp. (2) Digestion products of HFE gene by RsaI at 247 and 140 bp. Lane 1 shows 50 bp ladder; lanes (2, 3, 4, 5, 6, 7, 8, 9, and 10) show GG homozygote (normal alleles) at (247–140) bp, lane 11 shows 100 bp ladder. (3) Amplification product of HFE gene at 208 bp. Lane (1) shows 100 bp ladder; lanes (2, 3, 4, 5, 6, 7, 8, 9, and 10) show the amplification product of HFE gene at 208 bp; lane (11) shows 50 bp ladder. (4) Digestion products of HFE gene by BclI at 70, 138, and 208 bp. Lane 1 shows 50 bp ladder; lanes (2, 6, 8, and 10) show CC homozygote at (70–138) bp, lanes (3, 4, 5, and 7) show GG homozygote at (208) bp and lanes (9 and 11) show CG heterozygote at (70–138 and 208) bp.
Statistical analysis
Results were collected, tabulated and statistically analyzed by an IBM personal computer and Statistical Package for the Social Sciences (SPSS), version 20. Data were expressed in two phases: (1) descriptive phase: including the mean and standard deviation (SD) for quantitative data, frequency and percentage for qualitative date; (2) analytic phase: including U-test (Mann–Whitney test): comparison of two independent quantitative variables not normally distributed, X2 (Chi-square): comparison between two or more independent qualitative variables normally distributed, T test: for comparison of two independent quantitative variables normally distributed, F-test = ANOVA test (analysis of variance) modified t-test for comparison between more than two groups and Fischer’s exact test: for comparison between two or independent qualitative variables not normally distributed.
Results
The age of children survivors ranged between 4 and 18 years. Fourteen of them (40%) were females, and 21 (60%) were males. The control group was age- and gender-matched (). The weight and BMI were significantly higher in the ALL survivors group than the control group, but no significant difference was detected between them regarding height (). Eight of our survivors (22.85%) were overweight (>85th percentile) and 10 (28.51%) were obese (>95th percentile) (). By comparing iron parameters of both survivors and controls, we found that the serum ferritin levels and TSI are significantly higher in the survivors group than controls (p value = 0.006 and 0.03, respectively), while no significant difference was detected between them regarding serum iron and TIBC (). The C282Y mutation was not present in any of the 35 survivors group or controls. The H63D heterozygous state (CG) was detected in 10 (28.6%) of survivors group and in 3 (20%) of controls, while H63D homozygous mutant allele (GG) was detected in six survivors (17.1%) and none of the controls. There is a high allele frequency of mutant genotype of H63D in survivors than controls (p value <0.05) with high incidence of mutant G allele in the survivors (31.4%) more than controls (10%), while C allele frequency was 68.6% in the survivors and 90% in the controls (). Regarding iron profile of different H63D genotypes of the studied groups, our results illustrated that the survivors of the mutant genotypes (GG and CG) were associated with the increase in the iron parameters more than non-mutant genotype (CC) and controls. There was a significant increase in serum ferritin, iron, and TIBC and highly significant increase in transferrin saturation in homozygous mutant (GG) than heterozygote (CG) and wild (CC) genotypes. In addition, a highly significant increase was found between homozygous mutant genotype (GG) and controls regarding serum ferritin and transferrin saturation and significant increase regarding serum iron, while no significant differences were detected regarding TIBC. Furthermore, no significant differences were detected between wild (CC) and heterozygote (CG) genotypes; and between controls and each of wild (CC) and heterozygote (CG) genotypes regarding all iron parameters except for serum ferritin where there was a significant increase in heterozygote genotype (CG) as compared with the controls (). No significant statistical differences were detected between male and female ALL survivors with different H63D genotypes (). There was a significant statistical increase of serum ferritin levels in male and female ALL survivors, while no significant statistical differences were detected between them regarding other iron parameters such as serum iron, TIBC, and TSI ().
Table 1. Demographic data of the studied groups.
Table 2. Anthropometric measures of the studied groups.
Table 3. Iron profile of the studied groups.
Table 4. Genotype frequencies and allelic distribution of C282Y and H63D mutations among studied groups.
Table 5. Iron profile with different H63D genotypes in studied groups.
Table 6. H63D genotype frequency among male and female ALL survivors.
Table 7. Iron profile among male and female ALL survivors.
Discussion
ALL is the most common cancer in children in the USA, accounting for 26% of new cancer diagnoses at the birth to 14-year-age group. ALL is more common in boys than in girls [Citation17]. Genetic susceptibility increases the risk of childhood leukemia or ALL. Genetic associations’ studies have found gender-specific associations (increased male-to-female ratio) including the HFE association [Citation18]. HFE is one of the molecules that participate in iron homeostasis [Citation16]. Most studies of iron overload in cancer survivors have been among those diagnosed as adults, and few studies have been conducted among survivors of childhood cancer [Citation19]. We aimed to evaluate the genotype frequency and allelic distribution of the two HFE gene mutations (C282Y and H63D) in pediatric ALL survivors and to detect the impact of them on iron profile. In the current study, the weight and BMI were significantly higher in ALL survivors than in controls. This is in agreement with Zhang et al., [Citation20] who reported that the obesity is prevalent in pediatric ALL survivors, the mean BMI z score was 0.83 which corresponds to the 80th BMI percentile, indicating a significantly higher BMI in pediatric ALL survivors than the reference population. In our study, there were no carriers for the C282Y gene mutation in the cancer survivors group or in controls. On the other hand, we detect that 45.7% of our cancer survivors had H63D gene mutation, while 54.3% did not. The allelic frequencies of C282Y and H63D are widely variable between different populations. C282Y frequency ranged from 0 to 9.9% and seen to be nearly 0% in North African population. On the other hand, the allelic frequency of H63D in different populations ranged from 0 to 20.4% [Citation21]. These results are in line with Settin et al. [Citation22] who studied HFE gene mutations in Egyptians with HCV infection with liver cirrhosis. They stated that C282Y was not detected among patients or controls. In this study, the H63D mutation analysis revealed that CG (heterozygous mutant) genotype had higher frequency among ALL survivors when compared with controls, while CC (wild) genotype had higher frequency among controls when compared with ALL survivors. This is in contrast to Settin et al. [Citation22] who found that the prevalence of heterozygosity for H63D was similar in both patients and controls. Also, El-Rashidi et al. [Citation23] reported that H63D heterozygous state was detected in 25.4% of thalassemic patients with no significant difference between them and controls. Assunta et al. [Citation24] reported that the H63D mutation was significantly more frequent in adult ALL patients than controls. In the current study, the mean values of biochemical iron parameters were analyzed in survivors according to their H63D genotypes and compared to controls. Survivors who carry GG mutant genotype showed a significant increase in serum ferritin, iron, and TIBC, and a highly significant increase in TSI when compared to CG and CC genotypes. This is in line with Bruno et al. [Citation25] who stated that an iron overload phenotype is observed in individuals presenting only the HFE-H63D. In addition, a highly significant increase was found between GG genotype and the controls regarding serum ferritin, transferrin saturation, and iron. This is in accordance with Kennedy et al. [Citation3] who reported that HFE gene variants H63D correlating with body iron levels are associated with cancer risk, including childhood ALL. Another study from Egypt by Abdel Rahman et al. [Citation26] on sickle cell anemia patients showed that patients carrying HFE-H63D heterozygote CG did not show statistically significant difference in iron parameters in relation to patients with CC, and multivariate regression analysis revealed that the number of mutations harbored by the patients tends to affect the serum ferritin level. Furthermore, the present study revealed that there were no significant differences in all iron parameters found between wild CC and CG genotypes; or between controls and each of CC and CG genotypes except for serum ferritin where there was a significant increase in CG genotype when compared to the controls. A study on Chinese patients with myelodysplastic syndrome was carried out by Ling et al. [Citation27] and compared serum ferritin, iron, and TSI values between HFE CG and CC genotypes and found no significant difference. Andreani et al. [Citation28] reported that the HFE-H63D gene mutation is a common variant and associated with iron overload, usually in the homozygous state or in compound heterozygote individuals with C282Y mutation of HFE gene.
Study limitations
Sample size is small. Further prospective studies are thus needed to confirm our results.
Conclusion
Our study concluded that H63D mutation has a positive association with iron overload and seems to aggravate the individuals’ iron status in ALL survivors. Carriers of the mutant G allele have genetic risk factor for iron overload and carcinogenesis.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Farida H. El-Rashedi is professor and head of Pediatric Hematology and Oncology unit, Pediatrics Department, Faculty of Medicine, Menoufia University. She had a wide experience, many researches in the field of Pediatric Hematology and Oncology. She supervised and discussed many theses in this field in most of Egypt universities. She is the mother of all researchers in this field in Menoufia University.
Mahmoud A. El-Hawy is Lecturer of Pediatric Hematology and Oncology, Pediatrics Department, Faculty of Medicine, Menoufia University, Menoufia, Egypt.
Sally M. El-Hefnawy is Lecturer of Medical Biochemistry, Faculty of Medicine, Menoufia University, Menoufia, Egypt.
Mona M. Mohammed is pediatric resident, Pediatrics Department, Benha Educational Hospital, Benha, Egypt. She is Msc student at Menoufia University.
ORCID
Mahmoud A. El-Hawy http://orcid.org/0000-0002-3420-922X
References
- Neghina A, Anghel A. Hemochromatosis genotypes and risk of iron overload – a meta-analysis. Ann Epidemiol. 2011;21:1–14. doi: 10.1016/j.annepidem.2010.05.013
- Griffiths W, Cox T. Haemochromatosis: novel gene discovery and the molecular pathophysiology of iron metabolism. Hum Mol Genet. 2005;9(16):2377–82. doi: 10.1093/hmg/9.16.2377
- Kennedy A, Kamdar K, Lupo P, et al. Examination of HFE association with childhood leukemia risk and extension to other iron regulatory genes. Leuk Res. 2014;38:1055–60. doi: 10.1016/j.leukres.2014.06.016
- Lebron J, Bennett M, Vaughn D, et al. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell. 1998;93:113–23. doi: 10.1016/S0092-8674(00)81151-4
- Feder J, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399
- Bacon B, Adams P, Kowdley K. Diagnosis and management of hemochromatosis: 2011 practice guideline by the American Association for the Study of Liver Diseases. Hepatology. 2011;54:328–43. doi: 10.1002/hep.24330
- Seckington R, Powell L. HFE-associated hereditary hemochromatosis. 2000 Apr 3 [Updated 2015 Sep 17]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. Reviews [Internet]. Seattle (WA): University of Washington; 1993–2016 (www.ncbi.nlm.nih.gov/books/NBK1440/).
- Pedersen P, Milman N. Genetic screening for HFE hemochromatosis in 6,020 Danish men: penetrance of C282Y, H63D and S65C variants. Ann Hematol. 2009;88(8):775–84. doi: 10.1007/s00277-008-0679-1
- Diwan B, Kasprzak K, Anderson L. Promotion of dimethyl benz[a]anthracene-initiated mammary carcinogenesis by iron in female Sprague-Dawley rats. Carcinogenesis. 1997;18(9):1757–62. doi: 10.1093/carcin/18.9.1757
- Puliyel M, Mainous A, Berdoukas V, Coates TD. Iron toxicity and its possible association with treatment of Cancer: lessons from hemoglobinopathies and rare, transfusion-dependent anemias. Free Radic Biol Med. 2015;79:343–51. doi: 10.1016/j.freeradbiomed.2014.10.861
- Zhang C, Zhang F. Iron homeostasis and tumor genesis: molecular mechanisms and therapeutic opportunities. Protein Cell. 2015;6:88–100. doi: 10.1007/s13238-014-0119-z
- Pui C, Sandlund J, Pei D. Improved outcome for children with acute lymphoblastic leukemia: results of Total Therapy Study XIIIB at St Jude Children’s Research Hospital. Blood. 2004;104:2690–6. doi: 10.1182/blood-2004-04-1616
- Jacobs A, Miller F, Worwood M, et al. Ferritin in the serum of normal subjects and patients with iron deficiency and iron overload. Brit Med J. 1972;4:206–208. doi: 10.1136/bmj.4.5834.206
- Stookey L. Ferrozine – a new spectrophotometric reagent for iron. Anal Chem. 1970;42:779–781. doi: 10.1021/ac60289a016
- Fairbanks V, Klee G. Biochemical aspects of hematology. In: Tietz NW, editor. Fundamentals of clinical chemistry. 3rd ed. Philadelphia (PA): WB Saunders. 1987. p. 789–824.
- Kamer B, Dolka E, Pasowska R, Swiatkowska E. The usefulness of Soluble Transferrin Receptor (sTfR) in differentiating anemia occurring in young children. Folia Histochem Cytobiol. 2012;50(3):473–9. doi: 10.5603/FHC.2012.0066
- Ward E, Desantis C, Robbins A, et al. Childhood and adolescent cancer statistics. CA Cancer J Clin. 2014;64(2):83–103. doi: 10.3322/caac.21219
- Linet M, Wacholder S, Zahm S. Interpreting epidemiologic research: lessons from studies of childhood cancer. Pediatrics. 2003;112:218–232.
- Ruccione K, Mudambi K, Sposto R, et al. Association of projected transfusional iron burden with treatment intensity in childhood cancer survivors. Pediatr Blood Cancer. 2012;59:697–702. doi: 10.1002/pbc.24046
- Zhang F, Kelly M, Saltzman E, et al. Obesity in pediatric ALL survivors – a meta-analysis. Pediatrics. 2014;133:e704–e715. doi: 10.1542/peds.2013-3332
- El Attar E, Hassan N, Mounir M, et al. Childhood cancer NCI (2002–2005). Department of Biostatistics and Cancer Epidemiology, NCI Egypt; 2006. http://www.nci.cu.edu.eg/lecturesregisteration/nci200205.ppt.2006
- Settin A, El Bendary M, Abo Al Kassem R, et al. Molecular analysis of A1AT (S and Z) and HFE (C282Y and H63D) gene mutations in Egyptian cases with HCV liver cirrhosis. J Gastrointestin Liver Dis. 2006;15(2):131–5.
- El-Rashidi F, Elshafey A, Ragab S, et al. Haemochromatosis gene mutation H63D is a risk factor for iron overload in Egyptian beta-thalassemic children Egypt. J Med Hum Genet. 2008;9(2):149–59.
- Assunta V, Leonilde P, Daniela L, et al. HFE gene mutations in patients with acute leukemia. Leuk Lymphoma. 2006;47(11):2331–2334. doi: 10.1080/10428190600821898
- Bruno F, Bonalumi S, Camaschella C, et al. The c-582A>G variant of the HAMP promoter is not associated with high serum ferritin levels in normal subjects. Haematologica. 2010;95:849–50. doi: 10.3324/haematol.2009.018986
- Abdel Rahman HA, Abou-Elew HH, El-Shorbagy RM, et al. Influence of iron regulating genes mutations on iron status in Egyptian patients with sickle cell disease. Ann Clin Lab Sci. 2014;44:304–9.
- Ling N, Lin L, Lin Y, et al. HFE genotype and iron metabolism in Chinese patients with myelodysplastic syndromes and aplastic anemia. Ann Hematol. 2010;89:1249–1253. doi:10.1007/s00277-010-1016-z
- Andreani M, Radio F, Testi M, et al. Association of hepcidin promoter c.-582 A>G variant and iron overload in thalassemia major. Haematologica. 2009;94(9):1293–6. doi: 10.3324/haematol.2009.006270