
ABSTRACT
Introduction: GATA2 mutations are associated with several conditions, including Emberger syndrome which is the association of primary lymphedema with hematological anomalies and an increased risk for myelodysplasia and leukemia.
Objective: To describe a family with Emberger syndrome with incomplete penetrance.
Methods: A DNA sequencing of GATA2 gene was performed in the parents and offspring (five individuals in total).
Results: The family consisted of 5 individuals with a GATA2 null mutation (c.130G>T, p.Glu44*); three of them were affected (two of which were deceased) while two remained unaffected at the age of 40 and 13 years old. The three affected siblings (two boys and one girl) presented with lymphedema of the lower limbs, recurrent warts, epistaxis and recurrent infections. Two died due to hematological abnormalities (AML and pancytopenia). In contrast, the two other family members who carry the same mutation (the mother and one brother) have not presented any symptoms and their blood tests remain normal.
Discussion: Incomplete penetrance may indicate that GATA2 haploinsufficiency is not enough to produce the phenotype of Emberger syndrome. It could be useful to perform whole exome or genome sequencing, in cases where incomplete penetrance or high variable expressivity is described, in order to probably identify specific gene interactions that drastically modify the phenotype. In addition, skewed gene expression by an epigenetic mechanism of gene regulation should also be considered.
Introduction
GATA binding protein 2 (GATA2) is a zinc finger transcription factor associated with the development of different tissues including hematological [Citation1,Citation2], lymphatic [Citation3,Citation4], urogenital [Citation5] and neural [Citation6,Citation7]. GATA2 germline mutations in the heterozygous state have been found in different pathologies including the Emberger syndrome [Citation8], familial myelodysplastic syndrome (MDS), acute myeloid leukemia (AML) [Citation9], dendritic cell, mono and lymphocytopenia syndrome (DCM) [Citation10], and monocytopenia and Mycobacterium avium complex infection (MonoMac syndrome) [Citation11]. However, other clinical manifestations have also been associated with GATA2 mutations. These include pulmonary (including diffusion and ventilatory defects, pulmonary arterial hypertension and pulmonary alveolar proteinosis), dermatologic (warts and panniculitis), neoplastic (associated with human papilloma virus or Epstein-Barr virus), and vascular (venous thrombosis) alterations, lymphedema, sensorineural hearing loss, miscarriage, and hypothyroidism [Citation12]. To date, a vague genotype-phenotype correlation has been established, where null mutations have been observed more frequently in severe viral infections and have been the only mutations reported (together with regulatory mutations) in lymphedema [Citation12]; nevertheless, the phenotype within the same family has varied widely, with hematological manifestations being the most prevalent [Citation12,Citation13]. In this report we present a family with a null mutation in GATA2 in which Emberger syndrome was diagnosed in three members and two carriers of the mutation remain unaffected.
Case report
Three siblings of the family were referred to the genetic service with hereditary lymphedema (). The mother was 33 years old and the father 34 years old (at the first consultation), both with high school education. They were not consanguineous and apparently healthy. The mother had six pregnancies, all delivered normally (without complications) and there was no relevant family history of similar problems.
Figure 1. The family tree showing the affected members (with filled quadrants), the carriers (black circle inside) and the death members (with a transversal line).
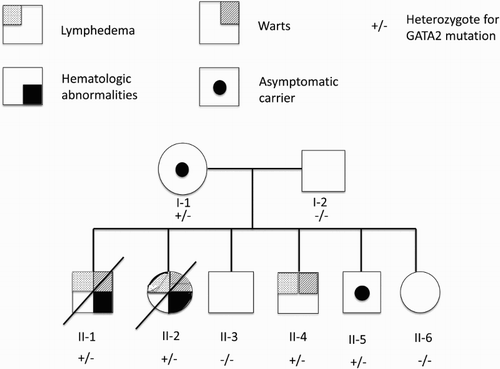
At the time of the first genetic consultation three of the six children presented with lymphedema of the lower limbs, aged 13 (boy, II-1), 12 (girl, II-2) and 8 (boy, II-4) years old. The unaffected siblings were aged 11 (boy, II-3), 6 (boy, II-5) and 5 (girl, II-6) years old (). The oldest boy (II-1) had developed lymphedema of the right lower limb and genitalia at the age of 7 years old after a fall. He also exhibited recurrent warts, cellulitis in the right leg, epistaxis and pancytopenia episodes. He had 4 hospitalizations, one at age 18 months for a gastrointestinal infection and the second at 12 years for cellulitis of the right limb, where pancytopenia was detected. Two months later, he was hospitalized for a phimosis that did not require surgery. He was admitted a fourth time at 13 years old with cellulitis and septic shock. On that occasion, pancytopenia was accompanied by hypogammaglobulinemia (with normal IgA levels). Three days into his last hospitalization he was transferred to intensive care but, unfortunately, died of the septicemia.
His younger sister (II-2), presented with lymphedema of both lower limbs (mainly in the left leg) and genitalia since the age of 3 months. She presented with an unspecified hepatitis at 4 years old, chickenpox at 5 years old and was hospitalized at 8 years old for cellulitis in the legs. At 13 years old, she developed hyperkeratotic lesions in both legs; at 14 years old, she had an episode of chronic cough secondary to flu symptoms; and at 15 years old, she died of acute myelocytic leukemia (AML). The blood tests at the time of the AML diagnosis showed anemia, thrombocytopenia and leukocytosis. The family reported that she had chronic cellulitis of legs (four in a year), epistaxis and recurrent warts.
The youngest affected boy (II-4), who is currently 16 years old, developed lymphedema during the first year of life. Both feet (the left more prominently) and his left lower limb were affected. Later, in the adolescence, it extended to his genitalia. He has had recurrent episodes of cellulitis of his left limb requiring hospitalization, recurrent tinea pedis with erythema and peeling of the skin, and occasional epistaxis and warts. The hematological biometric tests in the rest of the family were normal, although all the siblings presented occasional epistaxis.
Mutational analysis
DNA was extracted and screened for GATA2 mutations by Sanger sequencing for all the family members (). A heterozygous GATA2 mutation (c.130G>T, p.Glu44*) leading to a premature stop codon in amino acid 44 was detected by two independent laboratories in two affected siblings (the oldest had already died), the unaffected mother and one unaffected son (I-1, II-5), who now is 13 years old. The resulting messenger RNA would encode a truncated GATA protein, but the mRNA may be degraded via the non-sense mediated mRNA decay (NMD) pathway. Mendola et al. [Citation14] previously reported the mutation of the two affected siblings (II-2 and II-4) in a mutational screening report, without the clinical data.
Discussion
Emberger et al. [Citation15] first described the association of lymphedema of the lower limbs, sensorineural deafness and hematological anomalies (MIM#614038) as an autosomal dominant trait. Later, Mansour et al. [Citation16] further delineated the phenotype in seven unrelated cases. Finally, Ostergaard et al. [Citation8] described the genetic alterations in GATA2.
In this report, we describe a family with a null mutation in GATA2 associated with Emberger syndrome. Three siblings were symptomatic (two are deceased) but two additional family members are unaffected carriers. Although the presence of lymphedema has been reported as a low-penetrant phenotype in families with the same genetic mutation in GATA2 [Citation3,Citation12,Citation13], this is one of the few cases that exemplify a highly variable expressivity, including the possibility of incomplete penetrance in a family with Emberger syndrome. In this report, although the youngest affected brother (II-4) has not had documented hematological anomalies yet, the presence of recurrent infections and epistaxis episodes are suggestive. Considering the ages of the unaffected family members, it is likely that some clinical manifestations associated with GATA2 mutations may arise in the future. This has, indeed, been observed in other mutation carriers who presented with hematologic symptoms only after 70 years of age [Citation12,Citation13], in agreement with the age-dependent penetrance in GATA2 mutation carriers. Lymphedema can also have a late-onset appearance.
Previous work has shown a wide clinical variability in individuals with GATA2 mutations [Citation3,Citation12,Citation13]. Non-penetrance for any condition associated with GATA2 mutations had a frequency of 7% (n = 4) in a series of 57 individuals with these mutations [Citation12]. Lymphedema is one of the least frequent manifestations (found in 11%) and hematological abnormalities the most frequent (>80%) [Citation12]. This high variability, even within the same family, accompanied with cases of incomplete penetrance [Citation3,Citation10] supports the notion that GATA2 haploinsufficiency is not enough to produce any of the clinical manifestations. Thus, many genes with their respective variants, as well as some environmental factors, may interact to produce the phenotype. Considering the high number of phenotypes associated with hematological abnormalities, and its high penetrance in GATA2 mutation carriers, GATA2 seems to play a more preponderant role in the hematological development and function than in the development of the lymphatic, urogenital or neural tissues. A poorly explored mechanism involved in variable expression and incomplete penetrance is the epigenetic regulation of gene expression, as reported in homozygous twins discordant for multifactorial diseases [Citation17,Citation18]. Interestingly, epigenetic regulation of GATA2 leading to skewed allele-specific expression (ASE) has been reported in patients with acute myeloid leukemia and normal karyotype [Citation19]. Although this ASE was not observed in normal tissues, we cannot exclude its potential role in this family and other similar cases.
Considering the high inter-individual variability within the same family in earlier reports and the current family, it appears that a precise genotype-phenotype correlation cannot be established [Citation12]. Because all the syndromes previously described as produced by GATA2 mutations can present clinical overlap, these should be considered as part of the phenotypic spectrum of the same genetic deficiency. Nevertheless, as previously observed [Citation10,Citation12], the type of mutation (null mutation) in this family was associated with the presence of lymphedema in the three affected members.
In this particular genetic condition, familial screening is essential for a correct genetic counseling of risk and for the early detection of suitable hematopoietic stem cell donors in order to perform bone marrow transplantation, which has been reported as an effective treatment for patients with GATA2 mutations [Citation20]. However, the precise moment in which this should be done remains to be determined particularly in cases with intermittent immunodeficiency and pancytopenia. Therefore, a close surveillance of hematological alterations is needed in GATA2 mutation carriers.
In conclusion, we report a family with a null mutation in GATA2 and Emberger syndrome with variable expressivity and incomplete penetrance. This case emphasizes the need of genetic interactions to produce specific phenotypes in the clinical spectrum of GATA2 deficiency, where no precise genotype-phenotype correlation has been found. This and other cases, where incomplete penetrance or a highly variable expressivity is described, are good candidates for whole genome sequencing in order to identify specific gene interactions that drastically modify the phenotype, these genomes could be submitted in an electronic database that allow worldwide users to access it.
Acknowledgements
We are grateful to all the family members for their invaluable contributions. We want to thank Dr Sahar Mansour and Dr Pia Ostergaard for their valuable help in the mutational analysis of the family as well as in the critical review of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Aniel Jessica Leticia Brambila-Tapia obtained her MD and PhD in human genetics at the ‘University of Guadalajara’, the last grade in 2012. Since then, she has obtained 2 postdoctoral fellowships in bioinformatics at the ‘Universidad Nacional Autónoma de México (UNAM)’ and has published research work since 2009 in different scientific areas including human genetics, microbiology, rheumatology, and alternative therapies. She has been a member of the National System of Researchers in Mexico (SNI) since 2012 and has received scientific scholarships from the governmental institutions such as CONACyT, IMSS, and UNAM.
José Elías García-Ortiz is a senior researcher in Division of Genetics, Centro de Investigación Biomédica de Occidente, IMSS. He obtained his MD in Autonomous University of Coahuila and MSc and PhD in University of Guadalajara, Guadalajara, Mexico; He undertook Postdoc position in Laboratory of Genetics, National Institute on Aging, National Institutes of Health, Baltimore, Maryland, USA. His research interests include female infertility and recurrent pregnancy loss, genetic counseling of infertile couples; prenatal diagnosis and preimplantation diagnosis of genetic diseases; biochemical and molecular diagnosis of lysosomal disorders.
Pascal Brouillard graduated in Biochemistry at the University of Liège, Belgium, in 1997. The same year, he started pursuing PhD in the Laboratory of Human Molecular Genetics (Prof. Vikkula), de Duve Institute, Université catholique de Louvain (UCL), Brussels, Belgium, where he studied vascular anomalies. More precisely, he discovered the glomulin gene, mutations in which cause glomuvenous malformations. Moreover, he found that the lesions need a second-hit mutation for development. It was the first time such a mechanism was reported in vascular anomalies. He obtained his PhD. in 2003. Since then, he has pursued his postdoctoral studies in the same laboratory, mainly focusing on the generation of various glomulin mouse models. Between 2003 and 2007, he was a research associate of the Fonds National de la Recherche Scientifique (FNRS, Belgium), and is now a research associate and senior platform manager of UCL. Pascal was a laureate of the FBBF Belgian Biotechnology Fund Research Award in 2005 and is a member of the Belgian Society of Human Genetics and of NAVBO.
Ha-Long Nguyen obtained her PhD in biomedical sciences at the University of Florida, under the supervision of Dr. S. Paul Oh, in 2011. She joined the laboratory of Prof. Miikka Vikkula as a postdoctoral researcher in 2011.
Prof Miikka Vikkula obtained his MD at the University of Helsinki in 1992 and his PhD in molecular genetics, in 1993. He was a research associate at Harvard Medical School 1993–1997, during which time he became interested in vascular and lymphatic anomalies. He and his wife, Prof Laurence Boon, Plastic Surgeon, coordinator of the Vascular Anomaly Center, Brussels, discovered the gene for familial venous malformation in 1996, and since then were involved in many discoveries. They settled in Brussels in 1997, where Dr Vikkula developed his own laboratory. He obtained a ‘docentship PhD’ in 2000, and became an assistant professor at the Faculty of Medicine in UCL. He has been a member of the Directorate of the de Duve Institute since 2004, and a full professor of Human Genetics since 2013. He has received numerous honours and awards; most recently, the Inbev-Baillet Latour Clinical Prize in 2013. He served as the president of the Belgian Society of Human Genetics during 2004–2008, and as a member of the Scientific Program Committee of the European Society of Human Genetics during 2008–2012. He has been a member of the Royal Belgian Academy of Medecine since 2012. Prof Vikkula is well known internationally as a major contributor to the understanding of molecular basis of vascular anomalies and lymphedema with >150 peer-reviewed publications and numerous chapters in major bio-medical text books.
Blanca Estela Ríos-González is a medical doctor (University of Guadalajara). She obtained her MSc and PhD (Human Genetics) in University of Guadalajara/Division of Genetics, at the ‘Centro de Investigación Biomédica de Occidente, IMSS’. Guadalajara, México. Family Medicine Specialty, University of Guadalajara/IMSS, Guadalajara, Jalisco, Mex. Certified in Clinical Genetics and Family Medicine Specialty. She is currently working for Instituto Mexicano del Seguro Social.
Roberto de Jesús Sandoval-Muñiz is pursuing his PhD in Human Genetics in the Laboratory of chronic and degenerative diseases. He obtained his MD and MSc at the University of Guadalajara, Jalisco, Mexico. His clinical activities are in the ‘Nuevo Hospital Civil de Guadalajara, Dr. Juan I. Menchaca’, División of genetics. He has research interests in diabetes mellitus, chronic diseases, glucose metabolism, phytopharmaceutical compounds, and gene expression.
Ana Karen Sandoval-Talamantes is a graduate of the Faculty of Medicine of the University of Guadalajara. She specialized in Medical Genetics at ‘Nuevo Hospital Civil de Guadalajara, Dr. Juan I. Menchaca’. She was the editor of the magazine ‘Hippocrates Médica’, and is a reviewer of research articles for some magazines. Her research interests include genetics, immunology, psoriasis, and autism. She has published 4 original articles in journals indexed.
Lucina Bobadilla-Morales obtained MD at the University of Guadalajara in 1990. She obtained her Master’s degree in 1993 postulating Seckel syndrome as a chromosomal instability syndrome, later on she obtained her PhD in 2000, with a research in people occupationally exposed to radiation. She is a professor in Human Genetics in the Universidad de Guadalajara and researcher at the Genomic and Molecular Biology department from the same institution. She has been a member of the National System of Researchers in Mexico (SNI) since 2000, and PRODEP certified since 2006. She is a member of the academic board of the Human Genetics Doctorate program and thus director of thesis of multiple undergraduate and graduate students. She works as a clinical MD in Nuevo Hospital Civil de Guadalajara Dr. Juan I. Menchaca (HCJIM), giving genetic consultations and genetic counseling. In 2007, Dr. Bobadilla, in collaboration with Alfredo Corona-Rivera, PhD, founded the Cytogenetic Unit from the Hemato-Oncology Service in the HCJIM, targeting mainly childhood cancer state-of-the-art services and research. Her research interests include dysmorphology, instability syndromes, cytogenetic, and cancer genetics.
Jorge Román Corona-Rivera is the Head of Genetics Service, Division of Pediatrics, ‘Nuevo Hospital Civil de Guadalajara, Dr. Juan I. Menchaca, Guadalajara, Jalisco, Mexico. He is a senior researcher in ‘Dr. Enrique Corona-Rivera’ Institute of Human Genetics, Department of Molecular Biology and Genomics, Health Sciences University Center, University of Guadalajara. He obtained his MD in University of Guadalajara; specialty in Pediatrics, University of Guadalajara and MSc and PhD (Genetics) in University of Guadalajara. His research interests include clinical genetics and dysmorphology, genetic markers associated with congenital anomalies, syndromology, and syndrome definitions, and into the study of risk factors and prevalence of birth defects.
Lisette Arnaud-Lopez is working in the department of Clinical Genetics, Division of Pediatrics, Nuevo Hospital Civil de Guadalajara ‘Dr. Juan I. Menchaca’, Guadalajara, Jalisco, Mexico. She obtained her MD in Autonomous University Benito Juárez of Oaxaca, Oaxaca, Mexico and PhD in University of Guadalajara, Jalisco, Mexico. She pursued her postdoctoral studies in Laboratory of Genetics, National Institute on Aging, National Institute of Health, Baltimore, Maryland, USA.
ORCID
Aniel Jessica Leticia Brambila-Tapia http://orcid.org/0000-0002-6455-3662
José Elías García-Ortiz http://orcid.org/0000-0003-1504-1457
Pascal Brouillard http://orcid.org/0000-0001-9548-8229
Ha-Long Nguyen http://orcid.org/0000-0002-4976-1615
Miikka Vikkula http://orcid.org/0000-0002-6236-338X
Blanca Estela Ríos-González http://orcid.org/0000-0001-8197-455X
Roberto de Jesús Sandoval-Muñiz http://orcid.org/0000-0001-7111-1793
Ana Karen Sandoval-Talamantes http://orcid.org/0000-0002-2702-5625
Lucina Bobadilla-Morales http://orcid.org/0000-0001-7324-2287
Jorge Román Corona-Rivera http://orcid.org/0000-0001-7968-2267
Lisette Arnaud-Lopez http://orcid.org/0000-0002-3681-6365
Additional information
Funding
References
- Tsai FY, Keller G, Kuo FC, et al. An early haematopoietic defect in mice lacking the transcription factor GATA-2. Nature. 1994;371:221–226. doi: 10.1038/371221a0
- Rodrigues NP, Janzen V, Forkert R, et al. Haploinsufficiency of GATA-2 perturbs adult hematopoietic stem-cell homeostasis. Blood. 2005;106:477–484. doi: 10.1182/blood-2004-08-2989
- Kazenwadel J, Secker GA, Liu YJ, et al. Loss-of-function germline GATA2 mutations in patients with MDS/AML or MonoMAC syndrome and primary lymphedema reveal a key role for GATA2 in the lymphatic vasculature. Blood. 2012;119:1283–1291. doi: 10.1182/blood-2011-08-374363
- Kazenwadel J, Betterman KL, Chong CE, et al. GATA2 is required for lymphatic vessel valve development and maintenance. J Clin Invest. 2015;125:2979–2994. doi: 10.1172/JCI78888
- Zhou Y, Lim KC, Onodera K, et al. Rescue of the embryonic lethal hematopoietic defect reveals a critical role for GATA-2 in urogenital development. EMBO J. 1998;17:6689–6700. doi: 10.1093/emboj/17.22.6689
- Charles MA, Saunders TL, Wood WM, et al. Pituitary-specific Gata2 knockout: effects on gonadotrope and thyrotrope function. Mol Endocrinol. 2006;20:1366–1377. doi: 10.1210/me.2005-0378
- Kala K, Haugas M, Lilleväli K, et al. Gata2 is a tissue-specific post-mitotic selector gene for midbrain GABAergic neurons. Development. 2009;136:253–262. doi: 10.1242/dev.029900
- Ostergaard P, Simpson MA, Connell FC, et al. Mutations in GATA2 cause primary lymphedema associated with a predisposition to acute myeloid leukemia (Emberger syndrome). Nat Genet. 2011;43:929–931. doi: 10.1038/ng.923
- Hahn CN, Chong CE, Carmichael CL, et al. Heritable GATA2 mutations associated with familial myelodysplastic syndrome and acute myeloid leukemia. Nat Genet. 2011;43:1012–1017. doi: 10.1038/ng.913
- Dickinson RE, Griffin H, Bigley V, et al. Exome sequencing identifies GATA-2 mutation as the cause of dendritic cell, monocyte, B and NK lymphoid deficiency. Blood. 2011;118:2656–2658. doi: 10.1182/blood-2011-06-360313
- Hsu AP, Sampaio EP, Khan J, et al. Mutations in GATA2 are associated with the autosomal dominant and sporadic monocytopenia and mycobacterial infection (MonoMAC) syndrome. Blood. 2011;118:2653–2655. doi: 10.1182/blood-2011-05-356352
- Spinner MA, Sanchez LA, Hsu AP, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014;123:809–821. doi: 10.1182/blood-2013-07-515528
- Mutsaers PGNJ, van de Loosdrecht AA, Tawana K, et al. Highly variable clinical manifestations in a large family with a novel GATA2 mutation. Leukemia. 2013;27:2247–2248. doi: 10.1038/leu.2013.105
- Mendola A, Schlögel MJ, Ghalamkarpour A, et al. Mutations in the VEGFR3 signaling pathway explain 36% of familial lymphedema. Mol Syndromol. 2013;4:257–266. doi: 10.1159/000354097
- Emberger JM, Navarro M, Dejean M, et al. Deaf-mutism, lymphedema of the lower limbs and hematological abnormalities (acute leukemia, cytopenia) with autosomal dominant transmission. J Genet Hum. 1979;27:237–245.
- Mansour S, Connell F, Steward C, et al. Emberger syndrome-primary lymphedema with myelodysplasia: report of seven new cases. Am J Med Genet A. 2010;152A:2287–2296. doi: 10.1002/ajmg.a.33445
- Galetzka D, Hansmann T, El Hajj N, et al. Monozygotic twins discordant for constitutive BRCA1 promoter methylation, childhood cancer and secondary cancer. Epigenetics. 2012;7:47–54. doi: 10.4161/epi.7.1.18814
- Sugawara H, Iwamoto K, Bundo M, et al. Hypermethylation of serotonin transporter gene in bipolar disorder detected by epigenome analysis of discordant monozygotic twins. Transl Psychiatry. 2011;1:e24. doi: 10.1038/tp.2011.26
- Celton M, Forest A, Gosse G, et al. Epigenetic regulation of GATA2 and its impact on normal karyotype acute myeloid leukemia. Leukemia. 2014;28:1617–1626. doi: 10.1038/leu.2014.67
- Cuellar-Rodriguez J, Gea-Banacloche J, Freeman AF, et al. Successful allogeneic hematopoietic stem cell transplantation for GATA2 deficiency. Blood. 2011;118:3715–3720. doi: 10.1182/blood-2011-06-365049