
ABSTRACT
Background: The spectrum of thalassemias is wide ranging from thalassemia minor, which consists of mild hypochromic microcytic anemia without obvious clinical manifestations, to thalassemia major (TM), which is characterized by severe anemia since the first years of life and is transfusion dependent. Thalassemia intermedia (TI) describes those patients with mild or moderate anemia.
Objective: To describe the genetic features and major clinical complications of TI, and the therapeutic approaches available in the management of this disease.
Methods: Publications from potentially relevant journals were searched on Medline.
Results and discussion: Over the past decade, the understanding of TI has increased with regard to pathophysiology and molecular studies. It is now clear that clinical presentation and specific complications make TI different from TM. It is associated with greater morbidity, a wider spectrum of organ dysfunction and more complications than previously thought.
Conclusion: TI is not a mild disease. The interplay of three hallmark pathophysiologic factors (ineffective erythropoiesis, chronic anemia, and iron overload) leads to the clinical presentations seen in TI. New treatment modalities are currently being investigated to broaden the options available for TI management.
1. Introduction
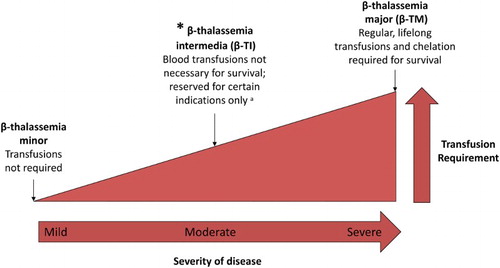
Thalassemia syndromes constitute the most common genetic diseases worldwide [Citation1]. Beta(β)-thalassemia comprises a heterogeneous group of hemoglobin (Hb) disorders characterized by a reduction or complete absence of β-globin gene expression. They are inherited in an autosomal recessive fashion [Citation2] and are characterized by an extreme diversity in phenotype, making diagnosis a real challenge. The spectrum of β-thalassemias is wide ranging from thalassemia minor, which consists of mild hypochromic microcytic anemia without obvious clinical manifestations, to β-thalassemia major (β-TM), which is characterized by severe anemia since the first years of life and is transfusion dependent [Citation3]. In the middle lies β-TI, a term developed to describe patients with manifestations too mild to be considered β-TM and too severe to be called thalassemia minor (). TI belongs to the group of non-transfusion-dependent thalassemias (NTDT), which also includes mild/moderate HbE/β thalassemia, and α-TI (HbH disease). The aim of this review is to describe the genetic features and major clinical complications in β-TI, and the therapeutic approaches available in the management of this disease.
Figure 1. Spectrum of β-thalassemias according to disease severity and transfusion requirement. aRefer to for indications for transfusion in β-TI.
2. Genetic features
β-Thalassemia is caused by an imbalance in globin chain synthesis. Disease severity depends on the extent of this imbalance. The β-thalassemias, including TI, arise from a defective gene function leading to partial suppression of β-globin protein production [Citation4]. The phenotypic diversity of β-TI results from its underlying genetic diversity, which can be explained by primary, secondary or tertiary modifiers [Citation5,Citation6]. The primary modifier is the severity of the β-globin gene mutation itself. They are the numerous different alleles at the beta-chain locus that can cause either complete or marked reduction in β-chain synthesis. Secondary modifiers include co-inheritance of α-thalassemia, and increased synthesis of the γ-chains and fetal Hb production after infancy [Citation7,Citation8]. Anything that modifies the magnitude of the surfeit of α-chains should have an important impact on the phenotype [Citation9]. The co-inheritance of α thalassemia leads to a reduction in the excess of α-chain pool and inclusion-body formation in erythroid precursors [Citation9]. As the spectrum of molecular forms of α-thalassemia is broad, this interaction leads to a wide range of different β-thalassemia phenotypes. The α-hemoglobin stabilizing protein is another described modifier. It is an abundant, erythroid-specific protein that forms a stable complex with free α-Hb. It specifically protects free α-Hb from precipitation, reducing the deleterious effects of free α-Hb precipitation and modulates pathological states of α-Hb excess, in patients with β thalassemia [Citation6,Citation9,Citation10]. Similarly, the variation of fetal hemoglobin (HbF) production is one of these secondary modifiers. The degree of globin-chain imbalance can be reduced by the more effective synthesis of the gamma chains of HbF after birth. Different genes are involved in modifying the gamma chain response, some that are encoded in the β-globin gene cluster, others that are on different chromosomes. Tertiary modifiers include genetic and environmental factors that alter specific complication rates [Citation8,Citation11,Citation12].
β-TI may also result from the increased production of α-globin chains by a triplicated or quadruplicated α-genotype associated with β-heterozygosity, or when a single β-globin locus is affected while the other is completely normal (dominant inclusion body β-thalassemia) [Citation2,Citation8,Citation13].
3. Clinical features and complications
Three main factors are responsible for the clinical sequelae of β-TI: ineffective erythropoiesis, chronic anemia, and iron overload () [Citation4]. summarizes the clinical complications of β-TI, along with their management.
Figure 2. Mechanism of iron overload development due to ineffective erythropoiesis in β-TI.
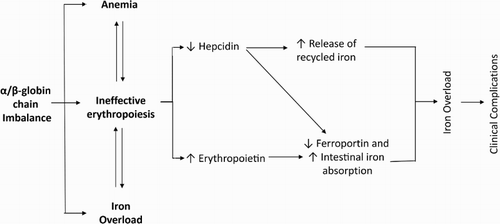
Table 1. Complications of β-TI, and their management options.
3.1. Extramedullary hematopoietic pseudotumors
Transfusion independence comes at the cost of an important hypertrophy of erythroid marrow at the medullary and extramedullary sites. It results in characteristic deformities of the skull and the face. Masses of erythropoietic tissue primarily affect the spleen, liver, lymph nodes, broad ligaments, kidneys, adrenal glands, pleura, retroperitoneal tissue, skin, peripheral and cranial nerves, and spinal canal [Citation14–18]. More frequently than in β-TM, extramedullary hematopoiesis occurs in 15–20% of β-TI cases by the age of 20–30 years old and in more than one-third of cases after the age of 30 [Citation4,Citation19]. While usually causing mild compression symptoms, some extramedullary hematopoietic lesions are present as pseudotumors that can cause various neurological symptoms due to spinal compression [Citation4].
3.2. Thrombosis and pulmonary hypertension
Thromboembolic events in general are more common in β-TI than in β-TM [Citation20]. There are several factors that contribute to this hypercoagulable state in patients β-TI. These include formation and precipitation of hemichromes, formation of reactive oxygen species, expression of negatively charged phospholipids, increased platelet aggregation, increased expression of activation markers, presence of platelet morphologic abnormalities, expression of endothelial adhesion molecules and tissue factor on endothelial cells, formation of microparticles, monocyte and granulocyte activation [Citation21]. These factors have been observed at a higher rate in splenectomized β-TI patients [Citation22].
Pulmonary hypertension (PHTN) is another hematological complication in patients with β-TI. It is five times more prevalent in β-TI than in β-TM as-revealed by right heart catheterization [Citation23]. Although the exact mechanism underlying the association between PHTN and thalassemia remains unknown [Citation24,Citation25], it is believed to be due to vasculopathy, resulting from excessive hemolysis combined with nitric oxide depletion and enhanced platelet activation. Transfusions have been shown to significantly reduce PHTN in patients with thalassemia [Citation8,Citation26,Citation27]. This, however, needs to be reproduced in clinical trials before any conclusions can be drawn.
3.3. Silent brain infarcts
Although strokes are uncommon in β-TI, silent brain infarctions have been detected on magnetic resonance imaging (MRI) in patients with β-TI [Citation28,Citation29]. Transfusion independence and age showed a significant correlation with the presence of either single or multiple white matter lesions in MRI, which remains a gold standard in the detection of ischemic lesions in the brain [Citation29,Citation30]. There currently exists no data in order to determine whether these silent abnormalities are really clinically asymptomatic. We believe that further studies are needed to evaluate the long-term implications and eventual complications of silent brain infarcts in β-TI.
3.4. Hepatocellular carcinoma
The accumulation of the majority of iron in the liver leads to an increased risk of developing fibrosis, cirrhosis, and hepatocellular carcinoma (HCC) mainly in non-chelated patients. The incidence of HCC in patients with thalassemia has been increasing with time [Citation31]. HCC seems to be more commonly seen in patients with β-TI than βTM [Citation32–34]. This is because β-TI patients usually have improved survival compared with those who have β-TM, which enables them to live long enough to develop HCC [Citation32]. The first well recognized risk factor for HCC in β-TI is iron overload [Citation35,Citation36]. Another risk factor for HCC in patients with β-TI is chronic viral infection, namely, with HCV or HBV, related to blood transfusion exposure. Current management options for HCC in patients with thalassemia include screening and prevention plans in addition to treatment strategies that target the abovementioned HCC risk factors.
3.5. Leg ulcers
TI patients are generally at a higher risk of developing leg ulcers when compared with regularly transfused β-TM patients [Citation4,Citation37]. The risk of developing these leg ulcers further increases with age [Citation37,Citation38]. Their pathogenesis has been attributed to reduced tissue oxygenation which is believed to be due to the combination anemia, hypercoagulability, and ineffective erythropoiesis [Citation2,Citation26,Citation39]. Once it develops, the management of leg ulcers is often painful and difficult. Chelation therapy, hydroxyurea, oxygen chambers, and blood transfusions could be beneficial without any sufficient evidence to be clearly recommended [Citation8,Citation40,Citation41].
3.6. Endocrine disease
Although less frequent than in β-TM, endocrine complications are among the most common complications in β-TI. They are attributed to iron overload and to suboptimal iron chelation [Citation42,Citation43]. Delayed puberty and hypogonadism are the most common endocrine complications in β-TI and are attributed to iron-mediated damage leading to dysregulation of the hypothalamic-pituitary axis. Hypothyroidism can be observed late in life [Citation43]. It is a late consequence of iron deposition in the thyroid gland ultimately leading to parenchymal fibrosis [Citation43]. Splenectomy is a specific risk factor for hypothyroidism in β-TI. Hypoparathyroidism, which is seen in up to 6.7% of β-TM patients, is not well studied in β-TI [Citation43].
3.7. Diabetes mellitus and glucose intolerance
Diabetes mellitus (DM) and glucose intolerance are common complications in thalassemia patients. While glucose intolerance occurs at an earlier stage during adolescence in TI patients, DM frequently occurs at later stages and is usually secondary to iron overload and subsequent chronic liver disease [Citation43]. The prevalence of DM and glucose intolerance is 2 and 24%, respectively, in β-TI [Citation42,Citation44]. The development of DM in thalassemia is attributed to impaired insulin excretory function secondary to chronic iron overload in the pancreas, selective immune system activation against pancreatic β-cells leading to cell damage, and/or pancreatic cell death due to fat transformation [Citation45–47]. Iron-mediated diabetes can be reversed if treated early.
3.8. Bone disease
Osteoporosis is commonly seen in β-TI patients and can be related to increased bone resorption or decreased bone formation [Citation43]. Iron overload, in addition to nutritional imbalance and increased erythron (due to ineffective erythropoiesis), also explains the occurrence of osteoporosis, osteopenia, and other low bone mineral density states in β-TI patients [Citation14,Citation39,Citation48].
3.9. Pregnancy and fertility
In most β-TI patients, fertility is not affected and most pregnancies can be achieved spontaneously [Citation49,Citation50]. However, pregnancy is considered a high risk one, as it is associated with intra-uterine growth retardation, spontaneous abortions, and thrombotic events [Citation51,Citation52]. This is due to many factors including anemia, hypoxia, acute hypersplenism, splenomegaly, and hypercoagulability state [Citation51]. Increased risk of thromboembolism may necessitate short-term anticoagulation with low-molecular weight heparin and platelet anticoagulants followed by a long-term oral anticoagulant [Citation41,Citation51]. Introduction of transfusion therapy should depend on Hb level, fetal growth status and maternal general and cardiac status. Such transfusions should be considered at high risk of alloimmunization [Citation53]. Splenectomy may be required before conception or in postpartum or in cases of hypersplenism or splenomegaly [Citation54]. Folic acid deficiency is commonly seen and can cause neural tube defects so folic acid supplementation is recommended [Citation41,Citation55]. Optimal management of pregnant women with β-TI requires a multidisciplinary approach with close maternal and fetal surveillance [Citation54].
4. Conventional management
There are a number of options currently available for managing patients with β-TI including transfusion therapy, iron chelation therapy, splenectomy, and hemoglobin F inducers.
4.1. Transfusion therapy
Whether to initiate transfusion and when are challenging decisions in β-TI. Although β-TI patients are transfusion independent, they might require blood transfusions in certain clinical scenarios (). Transfusion therapy in patients with β-TI is guided by clinical necessity. The decision to transfuse a patient with any thalassemia should be based not only on Hb level but also on other individual factors such as activity level, feeding, growth, and development. Occasional blood transfusions in β-TI are indicated in pregnancy, surgical settings in anticipation of acute blood loss, or in cases of serious infections [Citation8,Citation53]. Frequent blood transfusions should be considered for children with growth failure and poor performance at school, and for adults being treated for specific complications [Citation8,Citation53]. Preventive transfusions should be initiated for patients at high risk of developing thrombotic disease, PHTN, extramedullary hematopoiesis or leg ulcers [Citation8,Citation53].
Table 2. Indications for transfusion and splenectomy in β-TI.
4.2. Iron chelation therapy
Iron overload in β-TI develops from increased intestinal absorption. Results from the OPTIMAL CARE study have suggested that β-TI is associated with high morbidity rates. Moreover, there exists an age-related increase in the severity of the TI-associated complications [Citation26]. Several modalities are currently available for the diagnosis and monitoring of iron overload in β-TI, each carrying their own advantages and disadvantages. Serum ferritin assay is widely available and remains to be heavily relied on in resource-poor countries where MRI technology is not available [Citation56]. Current available guidelines recommend measurement of serum ferritin every 3 months [Citation8]. In patients with β-TI, serum ferritin values of >800 and <300 μg/l are used to indicate the need for iron chelation initiation or interruption [Citation8,Citation57]. However, studies have showed that serum ferritin underestimates the iron burden in NTDT [Citation58,Citation59].
Measurement of liver iron concentration (LIC) is another modality that is commonly used to diagnose iron overload in β-TI. Since LIC and total body iron stores are linearly related [Citation60], LIC quantification with R2 or R2* MRI is currently the test of choice for estimation of total body iron in all thalassemia patients. In patients with β-TI, LIC values ≥5 mg have been associated with more complications and thus increased morbidity [Citation61]. As per the available guidelines, assessment of LIC should be performed every 6, 12, or 24 months. This will depend on the severity of the iron overload [Citation8]. Other available markers for iron overload that have been investigated in β-TI include non-transferrin bound iron and transferrin saturation [Citation62].
Iron chelation therapy is currently the cornerstone of managing β-TI patients and minimizing disease-related complications. In β-TI, it is indicated in patients ≥10 years of age (or 15 years in hemoglobin H disease) if their LIC ≥5 mg Fe/g DW or when their serum ferritin level is ≥800 μg/l (when LIC is not available) [Citation63]. Iron-chelating drugs currently available for the treatment of iron overload in thalassemia patients include deferoxamine in subcutaneous or intravenous injection, oral deferiprone in tablet or solution form, and oral deferasirox in dispersible tablet (DT) () [Citation8,Citation64–66], whereas these three agents are currently available in TDT, deferasirox remains to be the only chelator to have received Food and Drug Administration (FDA) and European Medicines Agency (EMA) approval in NTDT based on results from the THALASSA trial [Citation2,Citation67]. In this study, 1-year deferasirox treatment in β-TI patients older >10 years was found to decrease LIC by a mean of 2.33 ± 0.70 and 4.18 ± 0.69 mg Fe/g dw at a daily dose of 5 and 10 mg/kg, respectively, compared to placebo [Citation67]. Doses were doubled after 6 months for patients with LIC >7 mg Fe/g dry weight and <15% reduction from baseline. Sub-analyses from the study showed that reduction in LIC with deferasirox 5 and 10 mg/kg/day starting dose groups is consistent irrespective of baseline LIC/serum ferritin, age, gender, race, splenectomy status, and underlying NTDT form. The analyses also showed that greater reductions in LIC were achieved in patients dose-escalated at 6 months from deferasirox 10 mg/kg/day starting dose to 20 mg/kg/day. The overall incidence of adverse effects was comparable between the deferasirox and placebo arms, and the main side effects were nausea, gastrointestinal discomfort, and headache. These side effects, which were mild to moderate in severity, resolved spontaneously without discontinuation of the drug.
Table 3. Characteristics of the currently available iron chelators in thalassemia management [Citation8,Citation65].
The THETIS study, a phase IV, multicenter efficacy and safety study of deferasirox targeting a larger population of patients with NTDT, showed at 1-year analysis of the results that deferasirox is effective in reducing iron overload in NTDT at a starting dose of 10 mg/kg/day, with dose escalations starting at week 4 up to 30 mg/kg/day according to the LIC response [Citation68]. In addition, the THESIS study provided more evidence about the satisfactory safety profile of the drug and reported on cases of pancreatitis and ocular toxicity possibly related to treatment with deferasirox [Citation68].
Successful management of iron overload extends beyond chelator efficacy and safety. Ensuring adherence is essential, and applicable to most chelators irrespective of administration form. Currently, some of the barriers to optimal adherence to deferasirox DTs include palatability, preparation time, and requirements for a fasting state at the time of dosing. A new film-coated tablet (FCT) formulation has been developed. This formulation can be swallowed once-daily, whole or crushed, with or without a light meal. The open-label, phase II ECLIPSE study [Citation69] evaluated the patient-reported outcomes in TDT or lower-risk myelodysplastic syndromes patients randomized to receive deferasirox DT or FCT over a 24-week period. FCT recipients consistently reported better adherence, greater satisfaction, and fewer concerns, with a safety profile consistent with the known DT formulation [Citation69]. These findings suggest that there exists a preference in favor of the new formulation, with better patient satisfaction and adherence reported which translates into reduced iron overload-related complications [Citation69]. The utility of this new formulation in patients with NTDT remains to be investigated.
4.3. Splenectomy
Splenectomy is common practice in NTDT patients, and serves to increase total Hb level by 1–2 g/dl [Citation2,Citation37]. However, cumulative evidence confirms an association with a variety of adverse outcomes. The spleen functions to scavenge procoagulant platelets and RBCs, which together are the key factors underlying the hypercoagulable state observed in NTDT. Splenectomized NTDT patients are at increased risk for venous thromboembolism, PHTN, leg ulcers and silent cerebral infarction than their non-splenectomized counterparts [Citation70]. Nonetheless, splenectomy may be indicated in certain clinical settings such as hypersplenism (resulting in worsening anemia, leucopenia, or thrombocytopenia or their clinical manifestations), worsening anemia leading to poor growth (when transfusion therapy is not possible) or splenomegaly (accompanied by left upper quadrant pain, early satiety or concern of splenic rupture) () [Citation8].
4.4. Hemoglobin F inducers
HbF inducers work by increasing γ-globin production, a β-like globin molecule which can bind excess α-chains, thus decreasing the α/β-chain imbalance inherent to thalassemia, and improving effective erythropoiesis [Citation71]. Hydroxyurea (or hydroxycarbamide) has been the most studied HbF inducer in β-TI. Early case reports documented hematological improvements in β-thalassemia patients treated with hydroxyurea, and since then several studies have evaluated this drug in β-TI and have considered it to be clinically effective with a satisfactory long-term safety [Citation71]. However, the data is conflicting and mostly comes from single-arm trials or retrospective cohort studies. Although HbF inducers are a potentially promising aspect in NTDT treatment, large, randomized, controlled trials are needed before these agents or their derivatives are widely used in management.
5. New therapeutic approaches
Recently, the discovery of previously unknown mechanisms leading to anemia has enabled the development of novel therapies. The aim is to improve the treatment, and possibly to cure the disease. Newly emerging therapies include minihepcidins, transmembrane protease serine 6 (TMPRSS6), Janus Kinase 2 (JAK2) inhibitors, apo-transferin therapy, activin receptor fusion proteins, stem cell transplantation, and gene therapy and gene editing.
5.1. Minihepcidins
It is known that hepcidin plays a key role in the limitation of both iron absorption and utilization. Minihepcidins, or long-acting hepcidin analogs, have been shown to restrict iron absorption and utilization in the setting of iron overload, with beneficial effects on ineffective erythropoiesis [Citation72–74]. They are known to increase the levels of hepcidin therefore decreasing iron absorption from the gastrointestinal tract, increasing the redistribution of iron to macrophages, and limiting end-organ toxicity [Citation75]. Studies on mice have also shown that minihepcidin therapy not only increases Hb concentrations but also decreases reticulocyte counts, and reduces spleen size [Citation75,Citation76].
5.2. TMPRSS6
TMPRSS6 has been found to play a key role in hepcidin expression from the liver. Its inactivation leads to increased hepcidin levels and subsequent amelioration of iron overload and improved ineffective erythropoiesis [Citation77,Citation78]. Anti-sense oligonucleotides and small interfering RNA (siRNAs) targeting TMPRSS6 have been effectively used in β-TI murine models to stimulate hepcidin, leading to a reduction in iron burden [Citation79–81]. Genetic ablation of TMPRSS6 also improved ineffective erythropoiesis and decreased splenomegaly in β-TI, without a concomitant decrease in erythropoietin production [Citation78]. Normalization of RBC survival is a significant component of the effects of TMPRSS6 inhibition on both hemoglobin and spleen size. The abovementioned data on TMPRSS6 provide proof of principle that pharmacologic manipulation of hepcidin may be an effective treatment for human diseases of iron dysregulation.
5.3. Janus Kinase 2 inhibitors
Thalassemia patients tend to demonstrate an increased expression of phosphorylated JAK2. This is turn leads to excessive proliferation and decreased differentiation of erythroid progenitors [Citation82,Citation83]. JAK2 inhibitors might therefore be effective in thalassemia [Citation14]. Ruxolitinib is a JAK2 kinase inhibitor that is already being used in the treatment of myeloproliferative diseases. Studies in thalassemic mice indicated that a short treatment with a JAK2 inhibitor can ameliorate ineffective erythropoiesis and decrease spleen size [Citation82,Citation84,Citation85]. For example, the TRUTH study, a phase IIa study, revealed up to a 26.8% decrease of spleen volume during the 30-week study period, in addition to slightly improved pre-transfusion Hb levels, with a benign safety profile overall [Citation84]. Although all subjects of the study had TM, the results of this clinical trial might be promising for TI patients with enlarged spleens.
5.4. Apo-transferrin therapy
Transferrin can circulate in the blood in three major forms: monoferric transferrin, dimeric transferrin, and apo-transferrin [Citation86]. Daily apo-transferrin injections on thalassemic mice increased Hb levels, decreased apoptosis of erythroid precursors and improved their maturation, and decreased the size of the spleen [Citation86]. These findings have promising clinical implications in TI patients.
5.5. Activin receptor fusion proteins
Sotatercept (ACE-011), an activin type IIA receptor (ActRIIA) fusion protein, acts mainly on late-stage erythropoiesis leading to increased hemoglobin production. Results from a study by Ruckle et al. [Citation87] have confirmed that sotatercept therapy increases RBC counts and Hb concentrations. In NTDT murine models, sotatercept therapy caused a decrease in ineffective erythropoiesis and bilirubin levels and markedly improved anemia [Citation88]. Another study by Porter and co-workers [Citation88] concluded that the subcutaneous administration of sotatercept every 3 weeks might improve anemia in NTDT patients with a good safety profile. Other fusion proteins of the same family include luspatercept (ACE-536), which is currently undergoing extensive research, especially in TDT [Citation89].
5.6. Hematopoietic stem cell transplantation
Hematopoietic stem cell transplantation (HSCT), which involves the replacement of mutant hematopoetic cells, is the only existing curative therapy that is available for both β-TI and β-TM patients [Citation90]. It is now an established approach to correct the defective erythropoiesis, particularly, when matched sibling donors are available. It is now widely applied with a disease free survival exceeding 80% with HLA-matched sibling donor transplants [Citation91–94]. For example, a retrospective study by Baronciani et al. [Citation90] on the natural history of thalassemia patients who received HSCT showed a 2-year overall survival rate of 88 ± 1% and a 2-year event-free survival incidence of 81 ± 1%. Patients who received a transplant from an HLA-identical sibling had the best results, with 2-year overall and event-free survival rates of 91 ± 1 and 83 ± 1%, respectively.
5.7. Gene therapy and genome editing
Gene therapy is another promising treatment modality in the management of thalassemia. Some recent studies have described the long-term correction of murine models of human β-thalassemia and sickle cell anemia by lentivirus-mediated gene transfer [Citation95,Citation96]. Furthermore, evidence of high gene transfer and expression in transduced hematopoietic cells in humans has also been noted.
The emergence of gene editing technology, whether by direct correction of genetic mutations in the endogenous DNA of the cell or by disruption of specific DNA sequences in the genome, offers a new approach for treating β-thalassemias. This is facilitated by site specific double strand breaks which can be induced with zinc finger nucleases, transcription activator-like effector nucleases, meganucleases and more recently with Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system [Citation97]. Other alternatives that could allow more efficient and immediate treatment of β-thalassemia with genome editing include the disruption of factors that silence the γ-globin genes, such as BCL11A, or γ-globin repressive elements within the β-globin gene locus [Citation77,Citation98].
6. Conclusion
Β-TI is a subset of thalassemia patients that do not require regular transfusion therapy for survival. It encompasses several genotypes and is mainly a clinical diagnosis. The interplay of three hallmark pathophysiologic factors (ineffective erythropoiesis, chronic anemia, and iron overload) leads to the clinical presentations seen in β-TI. Therapy involves a tailored combination of transfusions, HbF inducers, splenectomy, and iron chelation. New treatment modalities are currently being investigated to broaden options available for β-TI management, with ultimate goals of prolonging longevity, promoting greater compliance and better adherence and improving quality of life. Cappellini et al. [Citation99] have recently developed a new scoring system for NTDT patients in order to assess disease severity and thus tailor therapy. Though promising, this scoring system has yet to be validated.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Naouel Ben Salah http://orcid.org/0000-0003-0166-7715
References
- Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassaemia. Lancet. 2012;379(9813):373–383.
- Musallam KM, Rivella S, Vichinsky E, et al. Non-transfusion-dependent thalassemias. Haematologica. 2013;98(6):833–844.
- Rund D, Rachmilewitz E. Beta-thalassemia. N Engl J Med. 2005;353(11):1135–1146.
- Haddad A, Tyan P, Radwan A, et al. beta-thalassemia intermedia: a bird’s-eye view. Turk J Haematol. 2014;31(1):5–16.
- Weatherall DJ. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl. 1):S3–S6.
- Galanello R, Cao A. Relationship between genotype and phenotype – thalassemia intermedia. Cooleys Anemia. 1998;850:325–333.
- Danjou F, Anni F, Galanello R. Beta-thalassemia: from genotype to phenotype. Haematol-Hematol J. 2011;96(11):1573–1575.
- Taher A, Vichinsky E, Musallam K, et al. Guidelines for the management of non transfusion dependent thalassaemia (NTDT). Nicosia, Cyprus: Thalassaemia International Federation; 2013.
- Weatherall DJ. Phenotype-genotype relationships in monogenic disease: lessons from the thalassaemias. Nat Rev Genet. 2001;2(4):245–255.
- Camaschella C, Mazza U, Roetto A, et al. Genetic interactions in thalassemia-intermedia – analysis of beta-mutations, alpha-genotype, gamma-promoters, and beta-Lcr hypersensitive site-2 and site-4 in Italian patients. Am J Hematol. 1995;48(2):82–87.
- Weatherall D. 2003 William Allan Award address – the thalassemias: the role of molecular genetics in an evolving global health problem. Am J Hum Genet. 2004;74(3):385–392.
- Thein SL. Genetic insights into the clinical diversity of beta thalassaemia. Br J Haematol. 2004;124(3):264–274.
- Musallam KM, Taher AT, Rachmilewitz EA. β-thalassemia intermedia: a clinical perspective. Csh Perspect Med. 2012;2(7): a013482.
- Rivella S. The role of ineffective erythropoiesis in non-transfusion-dependent thalassemia. Blood Rev. 2012;26(Suppl. 1):S12–S15.
- Ross P, Logan W. Roentgen findings in extramedullary hematopoiesis. Am J Roentgenol Radium Ther Nucl Med. 1969;106(3):604–613.
- Aessopos A, Tassiopoulos S, Farmakis D, et al. Extramedullary hematopoiesis-related pleural effusion: the case of beta-thalassemia. Ann Thorac Surg. 2006;81(6):2037–2043.
- Chuang CK, Chu SH, Fang JT, et al. Adrenal extramedullary hematopoietic tumor in a patient with beta-thalassemia. J Formos Med Assoc. 1998;97(6):431–433.
- Kumar A, Aggarwal S, de Tilly LN. Case of the season. Thalassemia major with extramedullary hematopoiesis in the liver. Semin Roentgenol. 1995;30(2):99–101.
- Thuret I. Current management of thalassemia intermedia. Transfus Clin Biol. 2014;21(4-5):143–149.
- Taher A, Isma’eel H, Mehio G, et al. Prevalence of thromboembolic events among 8,860 patients with thalassaemia major and intermedia in the Mediterranean area and Iran. Thromb Haemost. 2006;96(4):488–491.
- Taher AT, Otrock ZK, Uthman I, et al. Thalassemia and hypercoagulability. Blood Rev. 2008;22(5):283–292.
- Taher AT, Musallam KM, Karimi M, et al. Splenectomy and thrombosis: the case of thalassemia intermedia. J Thromb Haemost. 2010;8(10):2152–2158.
- Derchi G, Galanello R, Bina P, et al. Prevalence and risk factors for pulmonary arterial hypertension in a large group of beta-thalassemia patients using right heart catheterization: a Webthal study. Circulation. 2014;129(3):338–345.
- Gladwin MT, Vichinsky E. Pulmonary complications of sickle cell disease. N Engl J Med. 2008;359(21):2254–2265.
- Gladwin MT, Lancaster JR, Jr., Freeman BA, et al. Nitric oxide’s reactions with hemoglobin: a view through the SNO-storm. Nat Med. 2003;9(5):496–500.
- Taher AT, Musallam KM, Karimi M, et al. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: the OPTIMAL CARE study. Blood. 2010;115(10):1886–1892.
- Karimi M, Musallam KM, Cappellini MD, et al. Risk factors for pulmonary hypertension in patients with beta thalassemia intermedia. Eur J Intern Med. 2011;22(6):607–610.
- Manfre L, Giarratano E, Maggio A, et al. MR imaging of the brain: findings in asymptomatic patients with thalassemia intermedia and sickle cell-thalassemia disease. AJR Am J Roentgenol. 1999;173(6):1477–1480.
- Musallam KM, Taher AT, Karimi M, et al. Cerebral infarction in beta-thalassemia intermedia: breaking the silence. Thromb Res. 2012;130(5):695–702.
- Karimi M, Haghpanah S, Bagheri MH, et al. Frequency and distribution of asymptomatic brain lesions in patients with beta-thalassemia intermedia. Ann Hematol. 2012;91(12):1833–1838.
- Moukhadder HM, Halawi R, Cappellini MD, et al. Hepatocellular carcinoma as an emerging morbidity in the thalassemia syndromes: a comprehensive review. Cancer. 2017;123(5):751–758.
- Borgna-Pignatti C, Vergine G, Lombardo T, et al. Hepatocellular carcinoma in the thalassaemia syndromes. Br J Haematol. 2004;124(1):114–117.
- Restivo Pantalone G, Renda D, Valenza F, et al. Hepatocellular carcinoma in patients with thalassaemia syndromes: clinical characteristics and outcome in a long term single centre experience. Br J Haematol. 2010;150(2):245–247.
- Fragatou S, Tsourveloudis I, Manesis G. Incidence of hepatocellular carcinoma in a thalassemia unit. Hemoglobin. 2010;34(3):221–226.
- Kowdley KV. Iron, hemochromatosis, and hepatocellular carcinoma. Gastroenterology. 2004;127(5 Suppl. 1):S79–S86.
- Bruix J, Sherman M, Llovet JM, et al. Clinical management of hepatocellular carcinoma. Conclusions of the Barcelona-2000 EASL conference. European Association for the Study of the Liver. J Hepatol. 2001;35(3):421–430.
- Olivieri NF, Muraca GM, O’Donnell A, et al. Studies in haemoglobin E beta-thalassaemia. Br J Haematol. 2008;141(3):388–397.
- Musallam KM, Cappellini MD, Daar S, et al. Serum ferritin levels and morbidity in beta-thalassemia intermedia: a 10-year Cohort study. Blood. 2012;120(21):1021.
- Musallam KM, Taher AT, Duca L, et al. Levels of growth differentiation factor-15 are high and correlate with clinical severity in transfusion-independent patients with beta thalassemia intermedia. Blood Cells Mol Dis. 2011;47(4):232–234.
- Matta BN, Abbas O, Maakaron JE, et al. Leg ulcers in patients with beta-thalassaemia intermedia: a single centre’s experience. J Eur Acad Dermatol Venereol. 2014;28(9):1245–1250.
- Borgna-Pignatti C. Modern treatment of thalassaemia intermedia. Br J Haematol. 2007;138(3):291–304.
- Sanctis VD, Tangerini A, Testa MR, et al. Final height and endocrine function in thalassaemia intermedia. J Pediatr Endocrinol Metab. 1998;11(Suppl. 3):965–971.
- Inati A, Noureldine MA, Mansour A, et al. Endocrine and bone complications in beta-thalassemia intermedia: current understanding and treatment. Biomed Res Int. 2015;2015:813098.
- Karamifar H, Shahriari M, Sadjadian N. Prevalence of endocrine complications in beta-thalassaemia major in the Islamic Republic of Iran. East Mediterr Health J. 2003;9(1–2):55–60.
- Monge L, Pinach S, Caramellino L, et al. The possible role of autoimmunity in the pathogenesis of diabetes in B-thalassemia major. Diabetes Metab. 2001;27(2 Pt 1):149–154.
- Argyropoulou MI, Kiortsis DN, Astrakas L, et al. Liver, bone marrow, pancreas and pituitary gland iron overload in young and adult thalassemic patients: a T2 relaxometry study. Eur Radiol. 2007;17(12):3025–3030.
- Chern JP, Lin KH, Lu MY, et al. Abnormal glucose tolerance in transfusion-dependent beta-thalassemic patients. Diabetes Care. 2001;24(5):850–854.
- Haidar R, Musallam KM, Taher AT. Bone disease and skeletal complications in patients with beta thalassemia major. Bone. 2011;48(3):425–432.
- Skordis N, Christou S, Koliou M, et al. Fertility in female patients with thalassemia. J Pediatr Endocrinol Metab. 1998;11(Suppl. 3):935–943.
- Nassar AH, Usta IM, Rechdan JB, et al. Pregnancy in patients with beta-thalassemia intermedia: outcome of mothers and newborns. Am J Hematol. 2006;81(7):499–502.
- Nassar AH, Usta IM, Taher AM. Beta-thalassemia intermedia and pregnancy: should we anticoagulate? J Thromb Haemost. 2006;4(6):1413–1414.
- Nassar AH, Naja M, Cesaretti C, et al. Pregnancy outcome in patients with beta-thalassemia intermedia at two tertiary care centers, in Beirut and Milan. Haematologica. 2008;93(10):1586–1587.
- Taher AT, Musallam KM, Cappellini MD, et al. Optimal management of beta thalassaemia intermedia. Br J Haematol. 2011;152(5):512–523.
- Tuck SM, Jensen CE, Wonke B, et al. Pregnancy management and outcomes in women with thalassaemia major. J Pediatr Endocrinol Metab. 1998;11(Suppl. 3):923–928.
- Mojtahedzadeh F, Kosaryan M, Mahdavi MR, et al. The effect of folic acid supplementation in beta-thalassemia major: a randomized placebo-controlled clinical trial. Arch Iran Med. 2006;9(3):266–268.
- Puliyel M, Sposto R, Berdoukas VA, et al. Ferritin trends do not predict changes in total body iron in patients with transfusional iron overload. Am J Hematol. 2014;89(4):391–394.
- Musallam KM, Cappellini MD, Daar S, et al. Serum ferritin level and morbidity risk in transfusion-independent patients with beta-thalassemia intermedia: the ORIENT study. Haematologica. 2014;99(11):e218–e221.
- Taher A, El Rassi F, Isma’eel H, et al. Correlation of liver iron concentration determined by R2 magnetic resonance imaging with serum ferritin in patients with thalassemia intermedia. Haematologica. 2008;93(10):1584–1586.
- Pakbaz Z, Fischer R, Fung E, et al. Serum ferritin underestimates liver iron concentration in transfusion independent thalassemia patients as compared to regularly transfused thalassemia and sickle cell patients. Pediatr Blood Cancer. 2007;49(3):329–332.
- Angelucci E, Brittenham GM, McLaren CE, et al. Hepatic iron concentration and total body iron stores in thalassemia major. N Engl J Med. 2000;343(5):327–331.
- Musallam KM, Cappellini MD, Taher AT. Evaluation of the 5 mg/g liver iron concentration threshold and its association with morbidity in patients with beta-thalassemia intermedia. Blood Cells Mol Dis. 2013;51(1):35–38.
- Taher A, Musallam KM, El Rassi F, et al. Levels of non-transferrin-bound iron as an index of iron overload in patients with thalassaemia intermedia. Br J Haematol. 2009;146(5):569–572.
- Vichinsky E. Non-transfusion-dependent thalassemia and thalassemia intermedia: epidemiology, complications, and management. Curr Med Res Opin. 2016;32(1):191–204.
- Cappellini MD, Cohen A, Piga A, et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood. 2006;107(9):3455–3462.
- Cappellini MD, Cohen A, Porter J, et al. Guidelines for the management of transfusion dependent thalassaemia (TDT). TIF publication. 2014 (20).
- Musallam KM, Angastiniotis M, Eleftheriou A, et al. Cross-talk between available guidelines for the management of patients with beta-thalassemia major. Acta Haematol-Basel. 2013;130(2):64–73.
- Taher AT, Porter JB, Viprakasit V, et al. Deferasirox effectively reduces iron overload in non-transfusion-dependent thalassemia (NTDT) patients: 1-year extension results from the THALASSA study. Ann Hematol. 2013;92(11):1485–1493.
- Taher AT, Cappellini MD, Aydinok Y, et al. Optimising iron chelation therapy with deferasirox for non-transfusion-dependent thalassaemia patients: 1-year results from the THETIS study. Blood Cells Mol Dis. 2016;57:23–29.
- Taher AT, Origa R, Perrotta S, et al. New film-coated tablet formulation of deferasirox is well tolerated in patients with thalassemia or lower-risk MDS: results of the randomized, phase II ECLIPSE study. Am J Hematol. 2017;92(5):420–428.
- Saliba AN, Taher AT. Morbidities in non-transfusion-dependent thalassemia. Ann N Y Acad Sci. 2016;1368(1):82–94.
- Musallam KM, Taher AT, Cappellini MD, et al. Clinical experience with fetal hemoglobin induction therapy in patients with beta-thalassemia. Blood. 2013;121(12):2199–2212. quiz 372.
- Preza GC, Ruchala P, Pinon R, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest. 2011;121(12):4880–4888.
- Casu C, Goldberg A, Nemeth E, et al. Treatment with minihepcidin peptide improves anemia and iron overload in a mouse model of thalassemia intermedia. Blood. 2013;122(21):431.
- Casu C, Oikonomidau R, Shah Y, et al. Concurrent treatment with minhepcidin and deferiprone improves anemia and enhances reduction of spleen iron in a mouse model of non-transfusion dependent thalassemia. Blood. 2014;124(21):748.
- Gardenghi S, Ramos P, Roy CN, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemia. Blood. 2010;116(21):443–444.
- Ramos E, Ruchala P, Goodnough JB, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120(18):3829–3836.
- Sankaran VG, Weiss MJ. Anemia: progress in molecular mechanisms and therapies. Nat Med. 2015;21(3):221–230.
- Nai A, Pagani A, Mandelli G, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood. 2012;119(21):5021–5029.
- Guo S, Casu C, Gardenghi S, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest. 2013;123(4):1531–1541.
- Schmidt PJ, Toudjarska I, Sendamarai AK, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(-/-) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood. 2013;121(7):1200–1208.
- Casu C, Aghajan M, Oikonomidou PR, et al. Combination of Tmprss6- ASO and the iron chelator deferiprone improves erythropoiesis and reduces iron overload in a mouse model of beta-thalassemia intermedia. Haematologica. 2016;101(1):e8–e11.
- Libani IV, Guy EC, Melchiori L, et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood. 2008;112(3):875–885.
- Taher A, Hussain I, Cappellini MD. Thalassemia intermedia: revisited. Blood Cell Mol Dis. 2006;37(1):12–20.
- Aydinok Y, Karakas Z, Cassinerio E, et al. Efficacy and safety of ruxolitinib in regularly transfused patients with thalassemia: results from single-arm, multicenter, phase 2a truth study. Blood. 2016;128(22):852.
- Melchiori L, Gardenghi S, Guy EC, et al. Use of Jak2 inhibitors to limit ineffective erythropoiesis and iron absorption in mice affected by beta-thalassemia and other disorders of red cell production. Blood. 2009;114(22):798.
- Li HH, Rybicki AC, Suzuka SM, et al. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med. 2010;16(2):177–182.
- Ruckle J, Jacobs M, Kramer W, et al. Single-dose, randomized, double-blind, placebo-controlled study of ACE-011 (ActRIIA-IgG1) in postmenopausal women. J Bone Miner Res. 2009;24(4):744–752.
- Cappellini MD, Porter J, Origa R, et al. A phase 2a, open-label, dose-finding study to determine the safety and tolerability of sotatercept (ACE-011) in adults with beta (beta)-thalassemia: interim results. Blood. 2013;122(21):3448.
- Taher AT, Cappellini MD. Management of non-transfusion-dependent thalassemia: a practical guide. Drugs. 2014;74(15):1719–1729.
- Baronciani D, Angelucci E, Potschger U, et al. Hemopoietic stem cell transplantation in thalassemia: a report from the European Society for Blood and Bone Marrow Transplantation Hemoglobinopathy Registry, 2000-20. Bone Marrow Transpl. 2016;51(4):536–541.
- King A, Shenoy S. Evidence-based focused review of the status of hematopoietic stem cell transplantation as treatment of sickle cell disease and thalassemia. Blood. 2014;123(20):3089–3094.
- Locatelli F, Kabbara N, Ruggeri A, et al. Outcome of patients with hemoglobinopathies given either cord blood or bone marrow transplantation from an HLA-identical sibling. Blood. 2013;122(6):1072–1078.
- Lucarelli G, Galimberti M, Polchi P, et al. Marrow transplantation in patients with thalassemia responsive to iron chelation-therapy. New Engl J Med. 1993;329(12):840–844.
- Angelucci E, Matthes-Martin S, Baronciani D, et al. Hematopoietic stem cell transplantation in thalassemia major and sickle cell disease: indications and management recommendations from an international expert panel. Haematologica. 2014;99(5):811–820.
- Bank A, Dorazio R, Leboulch P. A phase I/II clinical trial of beta-globin gene therapy for beta-thalassemia. Ann NY Acad Sci. 2005;1054:308–316.
- Raja JV, Rachchh MA, Gokani RH. Recent advances in gene therapy for thalassemia. J Pharm Bioallied Sci. 2012;4(3):194–201.
- Rai P, Malik P. Gene therapy for hemoglobin disorders – a mini-review. J Rare Dis Res Treat. 2016;1(2):25–31.
- Cox DB, Platt RJ, Zhang F. Therapeutic genome editing: prospects and challenges. Nat Med. 2015;21(2):121–131.
- Cappellini MD, Porter JB, Musallam KM, et al. Development of a new disease severity scoring system for patients with non-transfusion-dependent thalassemia. Eur J Intern Med. 2016;28:91–96.