
ABSTRACT
Background: Myeloid sarcoma (MS) is characterized by extramedullary infiltration by immature myeloid cells. Owing to rarity of this disease, the clinical features and overall outcomes are yet to be clarified.
Objective: To define clinical characteristics, epidemiology, pathologic findings, treatment options and outcomes in MS.
Methods: We conducted a retrospective review of 23 patients diagnosed with MS at our institute over a period of 13 years (2002–2015).
Results: MS presented mostly as a manifestation of relapsed acute myeloid leukemia, seen in 39% of patients. Skin and subcutaneous soft tissues were the most common sites of anatomic involvement (69.5%). Ninety five percent (n = 19) were positive for classical myeloid markers with either cytochemical staining (chloracetate-esterase, MPO), flow-cytometry (CD33, CD34, CD13 and CD117), or immunohistochemistry (CD34, CD43, CD68 and lysozyme). Of these, 52% were positive for CD33 (n = 12), 35% for CD68 (n = 8), 30% for CD34 (n = 7), and 26% for lysozyme (n = 6). Cytogenetic abnormalities were seen in 63% (n = 12/19) patients on bone-marrow aspirate, with five patients displaying a complex (n = 3) or monosomal (n = 2) karyotype. Twenty seven percent patients with a normal karyotype had presence of deleterious mutations (FLT3, ASXL, STAG and JAK2) on further testing with myeloid mutation panel. The Median overall survival (OS) of the entire cohort was 15.9 months (95% CI, 7.4–24.4 months). The OS was significantly better for patients <65 years (24.6 vs. 3.4 months, p = 0.009) of age, and for those attaining a complete remission (CR) to induction therapy (25.7 vs. 0.8 months, p < 0.001). All patients who underwent allogeneic hematopoietic stem cell transplant attained long-term remissions, with a median follow-up of 54 (range 32–120) months.
Conclusion: Failure to achieve CR with induction therapy, and age >65 years are associated with poor outcomes in MS. Allogeneic stem-cell transplant in first remission appears to be the most effective modality for achieving long-term remissions.
Introduction
Myeloid sarcoma (MS), also known as chloroma or granulocytic sarcoma is characterized by extramedullary tumorous infiltration by immature myeloid cells [Citation1–3].These immature cells of myeloid lineage can have monoblastic, myelomonocytic or granulocytic differentiation [Citation4]. Predominant extramedullary infiltration with or without bone marrow involvement occurs in about 2–7% of patients with acute myeloid leukemia (AML) [Citation1–11]. MS can present in various forms – as de novo AML, as a manifestation of relapsed disease or as therapy related neoplasm. MS may also present as blastic transformation of other myeloid disorders like chronic myeloid leukemia, myeloproliferative neoplasms (MPNs), myelodysplastic syndrome (MDS) etc. Uncertainty exists regarding the significance of MS development during the natural history of AML. Different reviews have reported both favorable as well as unfavorable outcomes in patients with MS, when compared with AML [Citation11–13]. The aim of our study was to analyze the epidemiology, clinical feature, pathological characteristics, treatment outcomes and overall prognosis in patients with pathologically confirmed MS.
Materials and methods
Ethical approval
Study protocol was approved by the University of Arkansas for Medical Sciences, institutional review board (IRB). The IRB waived the requirement for an informed consent.
Study group
We searched the University of Arkansas for Medical Sciences (UAMS), Little Rock, Arkansas’s electronic medical database from 2002 to 2015 for patients with pathologically confirmed diagnosis of MS. Patient charts were reviewed retrospectively for data collection including age, gender, anatomic site, comorbidities, history of AML or other neoplastic disorders, pathologic characteristics, treatments employed, response to induction therapy and overall survival (OS). Survival data was also obtained from the UAMS tumor registry database.
Histology, immune-histochemistry and flow-cytometry
Initial diagnosis was made on core or surgical biopsy specimens. Hematoxylin and eosin stained slides and immune-histochemistry (IHC) on formalin fixed paraffin tissue sections from patients with MS were reviewed, along with wright-giemsa stained peripheral blood and bone marrow aspirate smears to confirm a pathologic diagnosis of MS. Immunohistochemistry stains for each MS slide included antibodies against cluster of differentiation (CD) 34, CD68, CD43, CD117 and lysozyme.
Flow-cytometry was performed using 8 color flow cytometer from Beckman- Dickson company, and included markers for CD20, CD117, CD34, CD33, CD10, CD45, HLA DR, CD19, CD15, CD13, CD56, CD7, CD36, CD14, CD3, CD2, CD4, CD5, and CD8.
Molecular studies
Conventional cytogenetics was available in 19 patients. Fluorescent in situ hybridization (FISH) panel included probes for deletion(5)(q31)/-5, deletion(7)(q31)/-7, t(8;21)(q22;q22.3), t(15;17)(q24;q21),11q23, and inversion(16)(p13.1q22)/t(16;16)(p13.1;q22).
Myeloid malignancies mutation panel by Next Generation Sequencing analyzing 57 genes including fms like tyrosine kinase 3 (FLT3) was sent to American Regional and University Pathologists Inc. (ARUP) laboratories. FLT3 testing was performed using multiplex polymerized chain reaction and capillary electrophoresis.
Statistical analysis
We evaluated the clinical features, pathology, molecular characteristics and treatment outcomes of all patients identified. Descriptive statistics were performed for clinical, pathologic and radiologic attributes. Median survival time was calculated as the time from diagnosis of MS until death, or censored on the date of last contact for living patients. The effect of age and complete remission (CR) on OS was analyzed using Kaplan–Meier curves and log-rank test. The data analysis for this paper was generated using SAS software, university version 3p.2, copyright © 2012–2015 SAS Institute Inc.
Findings
Clinical characteristics
A total of 23 individual cases with a pathologic diagnosis of MS were identified ( and ). The median age at diagnosis of MS was 58 years (range 36–84 years). Male to female ratio was 2:1. Twenty-one (91%) patients were white, one patient was Hispanic and one was of African-American descent. In 74% (n = 17) of patients, MS presented as a manifestation of progression or relapse of an underlying hematopoietic disorder, whereas only 26% (n = 6) patients presented as de novo MS, without a prior history hematopoietic disorder. Acute myeloid leukemia was the most common associated hematopoietic malignancy. Fifty-two percent (n = 13) patients had a history of prior or concomitant AML, with a median time from diagnosis of AML to MS of 8.8 months (range 0–60 months). Other associated diseases included T-cell prolymphocytic leukemia (n = 2), IgM monoclonal gammopathy of unknown significance (n = 1), MPN (n = 1) and MDS (n = 2). Two patients likely had therapy related MS, with MS occurring at 6 months and 10 months after initial diagnosis of MDS and MPN respectively; along with evidence of monosomy 7 on karyotyping.
Table 1. Epidemiology and laboratory characteristics of MS.
Table 2. Clinical and pathologic characteristics of myeloid sarcoma cases.
Sites of involvement
Skin was the most common site of involvement, seen in 69.5% (n = 16) patients (). Subcutaneous tissue and lymph node involvement was seen in 17% (n = 4) patients, with anatomic locations in chest wall, vulva, penis and axilla. Other sites of involvement included gingivae, spleen, small intestine and bone in one patient each.
Laboratory findings
The mean white blood cell count was 21.4 × 103/µl (range 0.6–103 × 103/µl). Thirty percent patients had leukocytosis (n = 7) and forty-three percent patients (n = 10) had circulating blasts on peripheral smear at MS diagnosis. Seventy eight percent of patients (n = 18) had anemia as well as thrombocytopenia at MS diagnosis. Mean hemoglobin level was 10 g/dl (range 7–15 g/dl), and mean platelet count was 122 000 × 103/µl (range 15 000–430 000 × 103/µl. Fifty-two percent of patients (n = 12) had an elevated lactate dehydrogenase at diagnosis, but creatinine and electrolytes were within normal range for all 23 patients.
Pathology findings
Pathologic characteristics of all 23 patients are presented in .
Histology
On hematoxylin and eosin (H&E) staining, MS was characterized by diffuse infiltration of tissues with a monomorphic population of intermediate to large sized mononuclear cells, with large nuclei, irregular nuclear contours, open chromatin, high N:C ratio and moderate amount of cytoplasm; mostly with myeloblastic, myelomonocytic or monocytic differentiation.
Immune-histochemistry and flow-cytometry findings
Upon immunohistochemistry, 78% (n = 18) samples stained positively with classic myeloid cytochemical stains – myeloperoxidase and chloroacetate. Similarly 78% (n = 18) were also positive for classic myeloid makers on flow-cytometry, including CD33 and CD117. Ninety five percent (n = 19) were positive for classical myeloid markers with either cytochemical staining (chloracetate-esterase, MPO, or) or on flow-cytometry CD33, CD34 and CD117. Fifty-two percent were positive for CD33 (n = 12), 35% for CD68 (n = 8), 30% for CD34 (n = 7), and 26% for lysozyme (n = 6). Other commonly expressed markers included CD13 (22%), CD43 (9%), CD45 (9%) and CD79a (9%).
Bone marrow biopsy findings
Bone marrow biopsy results at the time of MS diagnosis were available in 87% (n = 20) of patients. Majority of the bone marrow biopsy samples (n = 15) were notable for evidence of either AML or MDS. There were two notable outliers – one patient had evidence of mature T cell lymphoproliferative process with bone marrow infiltration by lymphoid cells showing co-expression of CD3, CD7, CD5, CD8, CD52, CD2, and lack of CD34; and one patient had hypercellular bone marrow for age with megakaryocytic hyperplasia with atypia, and fibrosis (increased reticulin fibrosis grade 2/3 and increased collagen deposition); findings that were most consistent with a differential of early primary myelofibrosis versus MDS/MPN unclassifiable versus MDS with fibrosis. Three patients had normal trilinear hematopoiesis without evidence of any hematopoietic abnormalities on bone marrow aspirate, biopsy or flow-cytometry. Bone marrow biopsy was not performed or not available for review in three patients.
Karyotype and molecular studies
Karyotyping results were available in 19 patients, and the results are shown in . Cytogenetic abnormalities were seen in 63% of patients. Cytogenetics on bone marrow aspirate specimens of five patients revealed either a complex (three patients) or monosomal (two patients) karyotype. Common chromosomal abnormalities included +9, t(6;9), t(8;16), t(15;17), del(5) and del(7). Additionally, FISH or ARUP laboratory myeloid mutation panel was obtained in 11 patients. Three patients were noted to have genetic mutations on molecular testing, despite normal karyotype, including FLT3 D835Y mutation in one patient; ASXL1 E635fs*15, STAG2 I872fs*23 and STAG2 T244fs*7 mutations in one patient, and JAK2 (c.1849G > T; p.V617F) and TP53 (c.613T > G; p.Y205D mutations in one patient.
Treatment and outcomes
Anthracycline-based induction therapy was employed in 60% (n = 12) of patients. Idarubicin plus cytarabine containing 7 plus 3 regimen was used as initial induction regimen for most of these patients [Citation8,Citation9]; except two patients-one received all-trans retinoic acid (ATRA) plus idarubicin, and one received high dose cytarabine plus mitoxantrone. Fifty percent (n = 6) of those who received 7 plus 3 induction regimen, failed to attain a bone marrow CR following first induction chemotherapy and required a second induction, with either 5 plus 2 regimen (cytarabine plus daunorubicin) or FLAG-Ida regimen (Fludarabine, cytarabine, idarubicin). Five patients were not considered candidates for anthracycline-based therapy, either due to impaired cardiac function or due to prior use of high cumulative doses anthracyclines. Three of these patients received palliative intent therapy with cytarabine-based combination (with either clofarabine or etoposide), and one patient each received cyclophosphamide plus cladribine, and gemtuzumab ozogamicin, respectively. In total, 60% (n = 16) patients achieved a CR after first or second induction therapy. Patients who achieved CR to induction, underwent consolidation therapy with either hematopoietic stem cell transplant (HSCT) or chemotherapy. Four patients underwent allogeneic HSCT (allo-HSCT) and one patient underwent autologous HSCT (auto-HSCT) due to unavailability of a matched unrelated or matched sibling donor. Those who were not considered candidates for HSCT due to poor performance status, age, co-morbidities or patient preference; received high dose cytarabine-based regimens, except one patient who received ATRA plus 6-mercaptopurine (6MP) plus methotrexate maintenance. Interestingly, one patient who was diagnosed with MS of spleen achieved a long-term remission with splenectomy alone. In addition to systemic therapy, two patients also received radiotherapy to the sites of involvement – one due to chemotherapy refractory symptomatic disease and second due to para-spinal involvement.
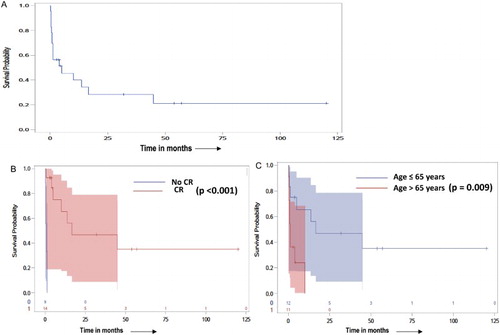
Of the 23 patients analyzed, seven were alive at last follow-up and 16 patients had died. Twelve patients died due to MS, with to failure to achieve remission to induction therapy, or experienced early relapse following a brief initial remission. There was one toxicity associated death, due to septicemia as a complication of prolonged neutropenia. Cause of death was thought to be unrelated to MS in one patient and unknown in two patients. The median OS was 15.9 months (95% CI, 7.4–24.4 months) for the whole group ((A)), with a five OS of 21% (95% CI 4–38%). The OS was significantly better for patients less than 65 years (24.6 vs. 3.4 months, p = 0.009) of age and for those attaining a CR to induction therapy (25.7 vs. 0.8 months, p < 0.001) ((B,C)). All four patients who underwent allo-HSCT were alive at last follow-up, with a median follow-up of 54 (range 32–120) months.
Figure 1. (A) Kaplan–Meier estimates for OS of entire cohort (A), by attainment of CR (B) and age at diagnosis of MS (C). (A) Overall survival in all 23 myeloid sarcoma patients analyzed; (B) Overall survival by achievement of CR; (C) Overall survival by age.
Discussion
The current WHO classification categorized MS as one of the major subcategories of AML [Citation2]. This study explored characteristics of adult MS. Mirroring the results of previous publications, the median age of diagnosis of MS in our review was 58 years and there was a slight male predominance [Citation1–12]. Anatomically, the most common sites were skin and subcutaneous tissues. Majority of patients with MS in this study had an underlying or simultaneous hematologic disorder including AML, CML, MDS, and MPN; whereas 26% presented with de novo MS. In our review, 91% of biopsy specimens were positive for CD 33, MPO or lysozyme. Other common markers included CD34 and CD117. Acknowledging the selection bias of a retrospective review, together the above data indicates that CD33, CD34, CD117 and MPO may be the most sensitive markers for MS. Additionally, similar to findings reported by Pileri et al. [Citation6], positivity for plasmacytoid marker CD68 was seen in 35% (n = 8) samples in our review. Other studies, by Al-Khateeb et al. and Alexiev et al., corroborate high sensitivity of CD34 and CD43, respectively, [Citation8,Citation14]. Upon karyotyping and molecular studies, chromosomal abnormalities were seen in 63% patients with MS, including previously infrequently reported t(6;9), t(8;16) and t(15;17). Genomic alterations were noted in all patients reviewed by Deeb et al. using genomic profiling, with the most common being chromosome 8 abnormalities [Citation15]. Frequent extramedullary invasion by immature myeloid cells was described by Byrd and Tallman et al. in core binding factor leukemias, especially those with t(8;21) [Citation10,Citation11]. Contrasting these observations and in keeping with review by Pileri et al., chromosomal 8 abnormalities were not frequently witnessed in our review [Citation6]. In our review, 20% patients with a normal karyotype and FISH panel, were noted to have genetic alterations in FLT3, ASXL1, STAG2 and JAK2 genes upon myeloid mutation panel testing with next generation sequencing. FLT3 mutations have previously been described in five patients by Mirza et al., using chromosomal microarray, and three out of nine patients by Ansari et al. [Citation16,Citation17]. Falini et al. have reported the presence of nucleophosmin (NPM) mutation in about 16 percent patients, however most of the cases were pediatric [Citation18]. In our analysis NPM mutations were not commonly witnessed.
If a diagnosis of MS is suspected, immunohistochemistry, flow cytometry, cytogenetic and molecular studies must be performed for diagnosis. Additionally, FISH studies on soft tissue biopsy are recommended [Citation6,Citation14,Citation19]. It should be noted that karyotype abnormalities on bone marrow and FISH studies on MS tissue are concordant in only about 70% of patients [Citation6]. Both should thus be performed whenever possible. Histologically, MS is composed of homogeneous population of immature myeloid cells, commonly myeloblasts, monoblasts or myelomonocytes; although rarely erythroblastic or megakaryoblastic infiltration has also been reported [Citation6,Citation14,Citation19,Citation20]. Diagnosis can be challenging, especially in cases of de novo and isolated MS without evidence of concomitant AML or MDS. Morphologically, there are several important diagnostic considerations, especially the ‘small blue round cell tumors’, non-Hodgkin lymphomas, melanoma and poorly differentiated carcinoma [Citation6,Citation21]. With the availability of IHC, a diagnosis can be successfully confirmed in a vast majority of patients. It is imperative to test for an array of markers including CD34, CD68, MPO, CD117, CD61, CD68, CD33, CD13, CD14, CD11c, Tdt and glycophorin [Citation1,Citation2]. B and T cell markers such as CD3, CD4, CD20, CD79a etc. should be included in the panel to rule out non-Hodgkin lymphomas. Imaging findings in MS can be non-specific on both computed tomography (CT) as well as magnetic resonance imaging (MRI) of the target organ/site [Citation22–24]. MRI has higher sensitivity compared to CT for MS involving central nervous system, musculo-skeletal system and abdomino-pelvic regions [Citation23,Citation24]. Positron emission tomography (PET-CT) is highly sensitive for detecting MS lesions, estimating tumor burden and assessing treatment responses [Citation25]. PET-CT sensitivity for MS detection approaches 90% [Citation26].
Pathophysiology of MS remains poorly understood, however altered homing mechanisms for myeloid lineage blasts have been implicated. Differential aberrant expression of chemokine receptors type 1 (CX3CR1), type 4 (CXCR4), type 5 (CCR5) and 7 (CXCR7) by MS blasts has been reported by Faaij et al. [Citation27]. Additionally, altered interaction between AML blasts and endothelial cells or extracellular matrix through proteases such as matrix metallopeptidase 9 (MMP9), MMP2, membrane type-1 metalloproteinase and inhibitor of metalloproteinase-2 may be responsible for heightened invasiveness of leukemic clones that display propensity for tissue invasion and homing [Citation28,Citation29] seen in MS.
As is the case with rare disease, there are no randomized clinical trials to help guide treatment approach. In most single institution series previously published, AML like induction therapy, followed by consolidation with either chemotherapy or allogeneic stem cell transplantation continues to be the current standard of care in fit patients [Citation6,Citation9]. Systemic therapy is superior to local therapy alone, even in cases with isolated MS [Citation30]. At our institute anthracycline-based induction regimen was employed for treatment of most cases, unless contraindications to anthracycline therapy prohibited their use. An initial remission was achieved in 60% of cases, indicating responsiveness of MS to cytotoxic therapy. Although only four patients successfully underwent allo-HSCT, long-term remissions were achieved in all four patients. Observation of such long-term remissions with allo-HSCT in our analysis is in concordance with outcomes noted in previous single institute series [Citation6,Citation8,Citation31]. Thus allo-HSCT in first CR should be considered for all eligible patients. There is a paucity of robust predictive or prognostic markers associated with MS. In one review by Lan et al., timely diagnosis and early implementation of chemotherapy were associated with improved outcomes [Citation20]. In our study, failure to achieve a CR after induction therapy and age >65 years were identified as poor prognostic factors. The number of patients in our review was too few to meaningfully study the influence of other potential prognostic factors such as histologic subtypes, cytogenetics, molecular signatures, underlying hematopoietic disorder etc. Owing to lack of data, uncertainty persists regarding the prognostic implications of molecular signatures in MS. It is however conceivable that negative prognostic signatures seen in AML, such as FLT3-ITD and myeloid/lymphoid or mixed-lineage leukemia 1 (MLL) mutations, will likely confer a poor prognosis in MS as well. Additional studies are needed to decipher implications of molecular signature upon prognosis and outcomes in MS.
This study presents a detailed review of MS cases diagnosed, treated and followed at our institute over a 13-year period. There are several limitations to this review. First, it is a retrospective study with potential shortcomings of missing data and confounding factors that could not have been controlled, such as smoking status, co-morbidities etc. Second, the number of patients was relatively small. Our work, however, provides helpful insight into clinical, pathologic and radiologic characteristics, and outcomes of this rare entity. In the absence of prospective randomized trials, our review not only adds valuable information to validate and update our current diagnostic and management approach to this rare disease, but offers hypothesis generating information about efficacy of allo-HSCT, and potential prognostic markers such as age and achievement of CR, that may serve as stratification criteria for future randomized trials for MS.
Conclusion
In summary, MS is a rare disease, commonly associated with an underlying hematological disorder. In our series, 27% patients with a normal karyotype had presence of deleterious mutations (FLT3, ASXL, STAG and JAK2) on myeloid mutation panel. Failure to achieve CR after induction and advanced age (>65 years) are associated with poor outcomes in MS. Upfront allogeneic stem-cell transplant is the most effective modality to achieve long-term remission.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Varinder Kaur http://orcid.org/0000-0002-6480-7204
References
- Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H. WHO classification of tumours of hematopoietic and lymphoid tissues, 4th ed. Lyon: IRAC; 2008. p. 140–141.
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. doi: 10.1182/blood-2009-03-209262
- Neiman RS, Barcos M, Berard C, et al. Granulocytic sarcoma: a clinicopathologic study of 61 biopsied cases. Cancer. 1981;48:1426–1437. [PubMed: 7023656]. doi: 10.1002/1097-0142(19810915)48:6<1426::AID-CNCR2820480626>3.0.CO;2-G
- Muss HB, Moloney WC. Chloroma and other myeloblastic tumors. Blood. 1973;42:721–728. [PubMed: 4518064].
- Menasce LP, Banerjee SS, Beckett E, et al. Extra-medullary myeloid tumour (granulocytic sarcoma) is often misdiagnosed: a study of 26 cases. Histopathology. 1999;34:391–398. [PubMed: 10231412]. doi: 10.1046/j.1365-2559.1999.00651.x
- Pileri SA, Ascani S, Cox MC, et al. Myeloid sarcoma: clinico-pathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia. 2007;21:340–350. doi: 10.1038/sj.leu.2404491
- Paydas S, Zorludemir S, Ergin M. Granulocytic sarcoma: 32 cases and review of the literature. Leuk Lymphoma. 2006;47(12):2527–2541. doi: 10.1080/10428190600967196
- Al-Khateeb H, Badheeb A, Haddad H, et al. Myeloid sarcoma: clinicopathologic, cytogenetic, and outcome analysis of 21 adult patients. Leuk Res Treatment. 2011;2011:523168.
- Yamauchi K, Yasuda M. Comparison in treatments of non-leukemic granulocytic sarcoma: report of two cases and a review of 72 cases in the literature. Cancer. 2002;94(6):1739–1746. doi: 10.1002/cncr.10399
- Tallman MS, Hakimian D, Shaw JM, et al. Granulocytic sarcoma is associated with the 8;21 translocation in acute myeloid leukemia. J Clin Oncol. 1993;11(4):690–697. doi: 10.1200/JCO.1993.11.4.690
- Byrd JC, Weiss RB, Arthur DC, et al. Extramedullary leukemia adversely affects hematologic complete remission rate and overall survival in patients with t(8;21)(q22;q22): Results from cancer and leukemia group B 8461. J Clin Oncol. 1997;15:466–475. doi: 10.1200/JCO.1997.15.2.466
- Tsimberidou AM, Kantarjian HM, Wen S, et al. Myeloid sarcoma is associated with superior event-free survival and overall survival compared with acute myeloid leukemia. Cancer. 2008;113(6):1370–1378. doi: 10.1002/cncr.23691
- Felice MS, Zubizarreta PA, Alfaro EM, et al. Good outcome of children with acute myeloid leukemia and t(8;21)(q22;q22), even when associated with granulocytic sarcoma: a report from a single institution in Argentina. Cancer. 2000;88:1939–1944. doi: 10.1002/(SICI)1097-0142(20000415)88:8<1939::AID-CNCR24>3.0.CO;2-Z
- Alexiev BA, Wang A, Ning Y, et al. Myeloid sarcomas: a histologic, immunohistochemical, and cytogenetic study. Diagn Pathol. 2007;2(1):42. doi: 10.1186/1746-1596-2-42
- Deeb G, Baer MR, Gaile DP, et al. Genomic profiling of myeloid sarcoma by array comparative genomic hybridization. Genes Chromosomes Cancer. 2005;44(4):373–383. doi: 10.1002/gcc.20239
- Mirza MK, Sukhanova M, Stozel F, et al. Genomic aberrations in myeloid sarcoma without blood or bone marrow involvement: characterization of formalin-fixed paraffin-embedded samples by chromosomal microarrays. Leuk Res. 2014;38(9):1092–1096. doi: 10.1016/j.leukres.2014.05.004
- Ansari-Lari MA, Yang CF, Tinawi-Aljundi R, et al. FLT3 mutations in myeloid sarcoma. Br J Haematol. 2004;126(6):785–91. doi: 10.1111/j.1365-2141.2004.05124.x
- Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352(3):254–266. doi: 10.1056/NEJMoa041974
- Seifert RP, Bulkeley W, Zhang L, et al. A practical approach to diagnose soft tissue myeloid sarcoma preceding or coinciding with acute myeloid leukemia. Ann Diagn Pathol. 2014;18(4):253–260. doi: 10.1016/j.anndiagpath.2014.06.001
- Lan TY, Lin DT, Tien HF, et al. Prognostic factors of treatment outcomes in patients with granulocytic sarcoma. Acta Haematol. 2009;122(4):238–246. doi: 10.1159/000253592
- Suh YK, Shin HJ. Fine-needle aspiration biopsy of granulocytic sarcoma: a clinicopathologic study of 27 cases. Cancer. 2000;90(6):364–372. doi: 10.1002/1097-0142(20001225)90:6<364::AID-CNCR7>3.0.CO;2-1
- Guermazi A, Feger C, Rousselot P, et al. Granulocytic sarcoma (chloroma): imaging findings in adults and children. AJR Am J Roentgenol. 2002;178(2):319–325. doi: 10.2214/ajr.178.2.1780319
- Shinagare AB, Krajewshi KM, Hornick JL, et al. MRI for evaluation of myeloid sarcoma in adults: a single institution 10 year experience. AJR Am J Roentgenol. 2012;199(6):1193–1198. doi: 10.2214/AJR.12.9057
- Ooci GC, Chim CS, Khong PL, et al. Radiologic manifestations of granulocytic sarcoma in adult leukemia. AJR Am J Roentgenol. 2001;176(6):1427–1431. doi: 10.2214/ajr.176.6.1761427
- Udea K, Ichikawa M, Takahashi M, et al. FDG-PET is effective in the detection of granulocytic sarcoma in patients with myeloid malignancy. Leuk Res. 2010;34(9):1239–1241. doi: 10.1016/j.leukres.2010.04.017
- Stolzel F, Christoph R, Radke J, et al. 18F-FDG-PET/CT for detection of extramedullary acute myeloid leukemia. Haematologica. 2011;96(10):1552–1556. doi: 10.3324/haematol.2011.045047
- Faaij CM, Willemze AJ, Revesz T, et al. Chemokine/chemokine receptor interactions in extramedullary leukaemia of the skin in childhood AML: differential roles of CCR2, CCR5, CXCR4 and CXCR7. Pediatr Blood Cancer. 2010;55(2):344–348. doi: 10.1002/pbc.22500
- Stefanidakis M, Karjalainen K, Jaalouk DE, et al. Roel of leukemia cell invadosome in extramedullay infiltration. Blood. 2009;114(14):3008–3017. doi: 10.1182/blood-2008-04-148643
- Wang C, Chen Z, Li Z, et al. The essential roles of matrix metalloproteinase-2, membrane type 1 metalloproteinase and tissue inhibitor of metalloproteinase-2 in the invasive capacity of acute monocytic leukemia SHI-1 cells. Leuk Res. 2010;34(8):1083–1090. doi: 10.1016/j.leukres.2010.01.016
- Cunningham I. Extramedullary sites of leukemia relapse after transplant. Leuk Lymhoma. 2006;47(9):1754–1767. doi: 10.1080/10428190600632857
- Avni BR, Rund D, Levin M, et al. Clinical implications of acute myeloid leukemia presenting as granulocyticsarcoma. Hematol Oncol. 2012;30(1):34–40. doi: 10.1002/hon.994