
ABSTRACT
Objectives: There are more than 200 known mutations found in patients with β-thalassemia, a possibility to identify an unknown or novel mutation becomes less possible. Here, we report a novel mutation in a patient from Thailand who presented with chronic hemolytic anemia.
Methods: A comprehensive hematology and DNA analysis was applied in the index patient and her mother.
Results: Hematological and hemoglobin analyses were consistent with the clinical diagnosis of Hb E/β0-thalassemia. However, we could find only Hb E heterozygous mutation using our common polymerase chain reaction-based mutation detection of the β-globin genes. Furthermore, the molecular analysis demonstrated a novel T-deletion at codon 42 of the second exon of the β-globin gene which we named ‘Hb Yala’ according to the origin of this index family.
Discussion: This mutation was assumed to generate a truncated β-globin chain terminating at codon 60 with possible unstable variant leading to a ‘null’ or β0-thalassemia. However, the clinical phenotype was surprisingly mild and no other ameliorating genetic factors, including co-inheritance of α-thalassemia and high propensity of Hb F by Xmn I polymorphism, were found.
Conclusion: This report has provided evidence that genotype–phenotype correlation in thalassemia syndromes is highly complex and a correct clinical severity classification of thalassemia should be mainly based on clinical evaluation.
Introduction
β-Thalassemia is an inherited disease, characterized by an imbalance in globin synthesis, due to the defective biosynthesis of the β-globin chain of hemoglobin (Hb) molecule. This defective biosynthesis results in reduced (β+) or absent (β0) production of the β-globin chain and a relative increase in the corresponding α-globin chain [Citation1–4]. Worldwide, thalassemia is considered to be the most common single-gene disorder, with over 200 β-thalassemia mutations described to date [Citation5]. These mutations can affect any point in the process from expression of the β-globin gene through its protein synthesis and are commonly observed in populations in the Mediterranean, Middle East, Indian subcontinent and Southeast Asia [Citation5]. Although the β-globin mutations are often population-specific with common mutations in each population, encounters with unusual or unknown mutations are not uncommon [Citation6,Citation7]. Clinically, severity varies from asymptomatic state to severe hemolytic anemia and ineffective erythropoiesis requiring regular blood transfusions.
Over 30 point mutations in the β-globin gene have now been identified to be common in Southeast Asia [Citation8–10], of which, our group have identified several patients with rare and unknown mutations in recent years [Citation11–13]. The co-inherited Hb E/β-thalassemia is the most common form of β-thalassemia in Thailand; it is a heterogeneous disease causing chronic health problems [Citation14]. This disease is characterized by the presence of both Hb E and F, with Hb E ranging from 35 to 75%. Clinically, the disease presents with a wide range of phenotypes from thalassemia intermedia or non-transfusion-dependent thalassemia (NTDT) to transfusion-dependent thalassemia (TDT) or thalassemia major [Citation14,Citation15].
In this report, we described a recently identified novel mutation, Hb Yala [HBB:c.129delT] in a Thai patient who presented with compound heterozygous Hb E/β-thalassemia. To understand genotype–phenotype correlation in the index patient, we further studied co-inheritance of α-thalassemia and the Xmn I polymorphism. A complete clinical history and genetic study of this index case is described herein.
Methods
Hematological study
Routine hematological study was conducted by an automated red blood cell counter (Sysmex F280, Kobe, Japan) and reticulocytes were detected by staining peripheral blood with methylene blue. Hemoglobin was analyzed by isoelectric focusing (Perkin Elmer Life Sciences, Zaventem, Belgium) and low pressure liquid chromatography on an automated hemoglobin analyzer (HB GOLD; Cumbria, Burrow-in-Furness, U.K.). This study was approved by a local ethical review board committee at Siriraj Hospital, Bangkok, Thailand and informed consent was obtained from the participants.
Iron assessment
Serum ferritin levels of patient were obtained using a standard ELISA technique and measured every 6 months in this patient. Liver iron concentration (LIC) was analyzed by magnetic resonance imaging-based technique, FerriScan® as previously described [Citation16].
Molecular characterization of the globin genes
DNA studies were performed on peripheral blood samples from the proband and her mother. DNA was isolated from peripheral leucocytes using a standard phenol chloroform technique. To determine the affected β-thalassemia allele, we screened 10 common β-thalassemia mutations in the Thai population as previously described [Citation13] with additional seven mutations (17 in total) using the reverse dot blot (RDB) hybridization technique (the sequences of all RDB oligonucleotide probes are available upon request). To identify unknown β-thalassemia mutation, we performed direct genomic DNA sequencing of the 2.48 kb fragment covering the whole β-globin gene, including the 5' and 3' untranslated regions in both DNA samples. The amplified polymerase chain reaction (PCR) fragments were subsequently sequenced by fluorescent-labelled dideoxy terminators (Perkin Elmer Biosystems, Foster City, CA, U.S.A.) and analyzed on a 3100-automated capillary sequencer (Applied Biosystems, Foster City, CA, U.S.A.) (primers sequences are available on request). The restriction fragment length polymorphism (RFLP) technique was performed using Sml I restriction enzyme for the confirmation of the mutation and also Xmn I restriction enzyme for the detection of the Xmn I polymorphism at the Gγ-globin gene, according to the manufacturer’s instructions (NEB, Beverly, MA, U.S.A.). The α-globin genotypes were analyzed for seven common α-thalassemia alleles; -- SEA, -- THAI, -- MED, -- FIL, -- 20.5, -α3.7, -α4.2 using a multiplex gap-PCR technique [Citation17] and direct genomic sequencing of both α2 and α1 globin genes, as described elsewhere [Citation18].
Results
Case history
The patient, an 11-year-old Thai girl from Yala, a province in the Southern part of Thailand was referred to our department at the Faculty of Medicine Siriraj Hospital, Bangkok for a definitive diagnosis. Anemia was detected at 7 years of age following a hemolytic crisis in which she required the transfusion of one unit of blood. During the following 4 years since, the patient has been well, without any further requirements for a blood transfusion and with baseline a hemoglobin (Hb) level of 8.8–9.5 g/dL. Physical examination showed mild pallor, mild hepatomegaly (2 cm below right costal margin) and splenomegaly (4 cm below left costal margin). The patient had a normal growth and development throughout her childhood and puberty. Her weight and height were within the 50th percentile throughout our 12-year follow-up period. Her serum ferritin levels were always between 250 and 400 µg/L, while the LIC was 2.8 mg Fe/g dry weight, as determined by FerriScan® (last performed in 2013). Based on her clinical presentation, she was classified as NTDT. Her peripheral blood smear showed marked anisopoikilocytosis with fragmented red blood cells and numerous target cells consistent with thalassemia disease (data not shown). Hemoglobin analyses revealed approximately 40% Hb E and 60% Hb F, which was consistent with Hb E/β0-thalassemia and no abnormal Hb species could be detected. The patient’s and her mother’s hematology are summarized in . During the course of the study, a blood sample from the father was not available.
Table 1. Hematological parameters in a Thai family with a frameshift mutations at codon 42 (-T) in the β-globin gene.
Identification of a novel β-globin mutation in the proband and the mother
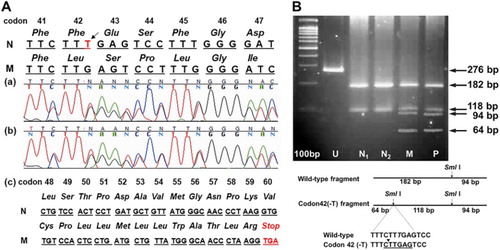
Using RDB analysis for 17 common β-globin mutations, only the Hb E mutation (codon 26 Glu > Lys; HBB:c.79G > A) was detected. Direct genomic sequencing of amplified PCR products of the whole β-globin gene was carried out and a T-deletion within exon-II (HBB:c.129delT), which we named ‘Hemoglobin Yala’ ((A)), was identified. This deletion causes a frameshift at codon 42 which results in a premature termination codon (PTC) at codon 60. This novel mutant was also found in the mother and further confirmed by RFLP using Sml I ((B)). Therefore, the proband is a compound heterozygote of Hb E and Hb Yala, while the mother is Hb Yala carrier.
Figure 1. Molecular identification of Hb Yala (c.129delT) in a Thai family, (A) Direct genomic sequencing reveals T-deletion (black arrow) at the codon 42 of the β-globin gene in patient (a) and her mother (b) resulting in a generation of new termination codon (TGA) at codon 60 ((c) and the lower panel). The protein sequences are shown in italic. N = normal genomic sequence; M = mutant genomic sequence. (B) Confirmation for the novel mutation using the PCR-based analysis. A schematic diagram shows the generation of a new restriction enzyme site of Sml I by codon 42 (-T) in the amplified β-globin gene fragment from both patient (P) and her mother (M). This PCR-RFLP fragment was not observed in normal controls (N1 and N2). U, undigested PCR products.
Owing to a milder-than-expected clinical phenotype in the patient, co-inheritance of α-thalassemia mutations, which could ameliorate clinical severity in Hb E/β-thalassemia, was excluded using the PCR-based analyses. The proband and the mother had normal α-globin genotypes. The Xmn I polymorphic site at −158 of the Gγ-globin promoter in the proband and the mother was ± which was not associated with a high propensity of γ-globin gene expression.
Discussion
This study was conducted in an attempt to report an unusual mutation in a patient diagnosed with Hb E/β0-thalassemia, namely Hb Yala (HBB:c.129delT). Previously, our group described this mutation as an abstract report in a local hematology meeting [Citation19]; however, it was not listed in available public databases [Citation20,Citation21] and as such we believe this is its first detailed clinical description.
The deletion of thymidine at position 129 of the coding sequence results in a frameshift and as a consequence creates a PTC at codon 60, which would produce a protein of less than half the expected size with altered amino acids from codons 42 to 60. Truncated globin chains are usually unstable and cause severe erythrocyte pathobiology with the physical properties leading to marked ineffective erythropoiesis and peripheral hemolysis. Therefore, this PTC mutation is expected to create a β0-thalassemia phenotype with no β-globin production and a more severe phenotype. This was also confirmed from hemoglobin analysis of the patient showing Hb E of 39.8% and Hb F of 60.2% () without detectable Hb A [Citation22]. Previously, heterozygotes with frameshift mutations of codon 44–59 of the β-globin gene, all resulting in the codon 60 PTC had mild anemia and marked microcytosis [Citation23–27]. These red blood cell phenotypes are comparable with those of the mother described in this study, suggesting β0-thalassemia heterozygote.
However, this patient in our study had a relatively mild non-transfusion-dependent phenotype, suggesting a less severe pathobiology to globin synthesis. Using anin vitro model, this mutation was analyzed for its impact on mutated β-globin gene transcription and translation [Citation28]. Consistently with a milder clinical phenotype in our patient, this nucleotide deletion caused a significant reduced mRNA probably due to non-sense mediated decay mechanism using a quantitative real-time polymerase chain reaction analysis. This suggests that an expected toxic truncated β-globin variant might not be generated leading to a less severe phenotype [Citation28]. This mutation has been reported as a functional study before [Citation28]; however, it is of interest to further describe clinical case presentation as a reference and a more detailed clinical history to provide further evidence that there is always a discrepancy between a functional cell and/or animal model and true clinical effects in human subjects. Such a difference highlights a complexity of genotype–phenotype correlation even within a so-called single-gene disorder, such as thalassemia.
Besides the aforementioned possible compensated mechanism for a milder β-thalassemia effect, other ameliorating factors such as α-thalassemia and increased Hb F production [Citation14,Citation29,Citation30] might cooperate and cause a more balanced α and β globin synthesis. However, this was not the case in this patient since she was found without ameliorating genetic determinants. However, the possibility of other genetic modifiers that have recently been identified through our genome wide association study cannot be ruled out [Citation31].
In conclusion, we have described herein an index Hb E/β-thalassemia patient due to a novel mutation producing a frameshift and an assumed PTC at codon 60 which might generate a truncated globin variant and Hb E mutation. Based on molecular prediction, it is expected that this mutation would result in the clinically severe β0-thalassemia phenotype. However, our patient’s presentation was milder than expected, providing further evidence for a complex genotype–phenotype correlation in Hb E/β-thalassemia as previously reviewed by Olivieri et al. [Citation32]. This result suggests a limitation of genetic analysis to predict clinical severity. Unless we have learnt more from whole genome and/or whole exome studies, clinical classifications of thalassemia patients, either TDT vs. NTDT, remain largely based on a careful clinical evaluation using several proposed clinical indicators [Citation33].
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Weatherall DJ, Clegg JB, editors. Chapter 4 The molecular pathology of the thalassaemias. In: The thassaemia syndromes. 4th ed. Oxford: Blackwell Science; 2001.
- Thein SL. Genetic modifiers of the beta-hemoglobinopathies. Br J Haematol. 2008;141(3):357–366. doi: 10.1111/j.1365-2141.2008.07084.x
- Higgs DR, Engel JD, Stamatoyannopoulos G. Thalassemia. Lancet. 2012;379(9813):373–383. doi: 10.1016/S0140-6736(11)60283-3
- Cao A, Galanello R. Beta-thalassemia. Genet Med. 2010;12(2):61–76. doi: 10.1097/GIM.0b013e3181cd68ed
- Steinberg MH. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. 2nd ed. New York (NY): Cambridge University Press; 2009.
- Fucharoen S, Winichagoon P. Hemoglobinopathies in Southeast Asia. Indian J Med Res. 2011;134:498–506.
- Nuntakarn L, Fucharoen S, Fucharoen G, et al. Molecular, hematological and clinical aspects of thalassemia major and thalassemia intermedia associated with Hb E-beta-thalassemia in Northeast Thailand. Blood Cells Mol Dis. 2009;42(1):32–35. doi: 10.1016/j.bcmd.2008.09.002
- Fucharoen S, Winichagoon P. Hemoglobinopathies in Southeast Asia: molecular biology and clinical medicine. Hemoglobin. 1997;21(4):299–319. doi: 10.3109/03630269709000664
- Fucharoen S, Fucharoen G, Sriroongrueng W, et al. Molecular basis of beta-thalassemia in Thailand: analysis of beta-thalassemia mutations using the polymerase chain reaction. Hum Genet. 1989;84(1):41–46. doi: 10.1007/BF00210668
- Thein SL, Hesketh C, Taylor P, et al. Molecular basis for dominantly inherited inclusion body beta-thalassemia. Proc Natl Acad Sci U S A. 1990;87(10):3924–3928. doi: 10.1073/pnas.87.10.3924
- Tanphaichitr VS, Viprakasit V, Veerakul G, et al. Homozygous hemoglobin Tak causes symptomatic secondary polycythemia in a Thai boy. J Pediatr Hematol Oncol. 2003;25(3):261–265. doi: 10.1097/00043426-200303000-00016
- Chinchang W, Viprakasit V, Pung-Amritt P, et al. Molecular analysis of unknown beta-globin gene mutations using polymerase chain reaction-single strand conformation polymorphism (PCR-SSCP) technique and its application in Thai families with beta-thalassemias and beta-globin variants. Clin Biochem. 2005;38(11):987–996. doi: 10.1016/j.clinbiochem.2005.07.013
- Chinchang W, Viprakasit V. Further identification of Hb G-Coushatta [beta22(B4)Glu-->Ala (GAA-->GCA)] in Thailand by the polymerase chain reaction-single-strand conformation polymorphism technique and by amplification refractory mutation system-polymerase chain reaction. Hemoglobin. 2007;31(1):93–99. doi: 10.1080/03630260601059225
- Viprakasit V, Tanphaichitr VS, Chinchang W, et al. Evaluation of alpha hemoglobin stabilizing protein (AHSP) as a genetic modifier in patients with beta thalassemia. Blood. 2004;103(9):3296–3299. doi: 10.1182/blood-2003-11-3957
- Winichagoon P, Thonglairoam V, Fucharoen S, et al. Severity differences in beta-thalassemia/hemoglobin E syndromes: implication of genetic factors. Br J Haematol. 1993;83(4):633–639. doi: 10.1111/j.1365-2141.1993.tb04702.x
- St Pierre TG, et al. Noninvasive measurement and imaging of liver iron concentrations using proton magnetic resonance. Blood. 2005;105(2):855–861. doi: 10.1182/blood-2004-01-0177
- Bier FF, Kleinjung F. Feature-size limitations of microarray technology–a critical review. Fresenius J Anal Chem. 2001;371(2):151–376. doi: 10.1007/s002160101003
- Viprakasit V, Tanphaichitr VS, Pung-Amritt P, et al. Clinical phenotypes and molecular characterization of Hb H-Pakse disease. Haematologica. 2002;87(2):117–125.
- Viprakasit V, Chinchang W. Identification of hemoglobin YALA; a novel β thalassemia mutation due to thymidine deletion of codon 42 (–T) causing β0 thalassemia and its interaction with hemoglobin E. Proceedings of the Annual Scientific Meetings of the HAA. Sydney, Australia, October 2011.
- Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308
- Patrinos GP, Giardine B, Riemer C, et al. Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucl Acids Res. 2004;32(Database issue:):D537–D541. doi: 10.1093/nar/gkh006
- Fucharoen S, Winichagoon P. Clinical and hematologic aspects of hemoglobin E beta-thalassemia. Curr Opin Hematol. 2000;7(2):106–112. doi: 10.1097/00062752-200003000-00006
- Kinniburgh AJ, Maquat LE, Schedl T, et al. mRNA-deficient beta o-thalassemia results from a single nucleotide deletion. Nucleic Acids Res. 1982;10(18):5421–5427. doi: 10.1093/nar/10.18.5421
- el-Kalla S, Mathews AR. A novel beta-thalassemia mutation [codon 45(-T)] in a Pakistani family. Hemoglobin. 1997;21(6):499–503. doi: 10.3109/03630269708999181
- Ringelhann B, Szelenyi JG, Horanyi M, et al. Molecular characterization of beta-thalassemia in Hungary. Hum Genet. 1993;92(4):385–387. doi: 10.1007/BF01247340
- Landin B, Berglund S. A novel mutation in the beta-globin gene causing beta-thalassemia in a Swedish family. Eur J Haematol. 1996;57(2):182–184. doi: 10.1111/j.1600-0609.1996.tb01359.x
- Meloni A, Demurtas M, Moi L, et al. A novel beta-thalassemia mutation: frameshift at codon 59 detected in an Italian carrier. Hum Mutat. 1994;3(3):309–311. doi: 10.1002/humu.1380030322
- Forster L, Ardakani RM, Qadah T, et al. The effect of nonsense mediated decay on transcriptional activity within the novel β-thalassemia mutation HBB: c.129delT. Hemoglobin. 2015;39(5):334–339.
- Laosombat V, Wongchanchailert M, Sattayasevana B, et al. Clinical and hematologic features of beta0-thalassemia (frameshift 41/42 mutation) in Thai patients. Haematologica. 2001;86(2):138–141.
- Gilman JG, Huisman TH. DNA sequence variation associated with elevated fetal G gamma globin production. Blood. 1985;66(4):783–787.
- Nuinoon M, Makarasara W, Mushiroda T, et al. A genome-wide association identified the common genetic variants influence disease severity in beta0-thalassemia/hemoglobin E. Hum Genet. 2010;127(3):303–314. doi: 10.1007/s00439-009-0770-2
- Olivieri NF, Pakbaz Z, Vichinsky E. Hb E/beta-thalassaemia: a common & clinically diverse disorder. Indian J Med Res. 2011;134: 522–531.
- Cappellini MD, Porter JB, Musallam KM, et al. Development of a new disease severity scoring system for patients with non-transfusion-dependent thalassemia. Eur J Intern Med. 2016;28:91–96. doi: 10.1016/j.ejim.2015.10.003