
ABSTRACT
Objectives: Hemoglobin (Hb) disorders consist of thalassemia and Hb structural variants, of which the major forms are associated with severe anemia and/or vascular occlusion. Current diagnostic techniques are highly accurate and mostly based on isoelectric focusing, high-performance liquid chromatography or mass spectrometry, which often require advanced laboratory equipment. In sub-Saharan Africa, the Hb disorders are mainly associated to the pathological variants hemoglobin S (HbS) and HbC. Unfortunately, until now, it is not easy to get a diagnosis of these disorders in this area. In this study, we tested the performance of a new molecular diagnostic tests on qualified samples.
Methods: The Human Hb S/C Lamp assay is a new polymerase chain reaction test able to detect HbS, HbC and HbA alleles without DNA extraction, directly on fresh or frozen blood samples, or on dried blood spots (DBS). In this study, we compared the genotyping of 248 blood samples (56 whole blood and 192 DBS) with this LAMP assay to the routine diagnostic methods performed in the genetics lab at the university hospital of Liège.
Results: Our results show that the LAMP method can detect HbS and HbC with an accuracy of 100%. Moreover, this test can be used for the neonatal screening because we did not observe any interference with fetal Hb.
Discussion: To our knowledge, this method is the only molecular assay that can be performed directly on dried blood cards without DNA extraction, lowering handling, turnaround time and costs.
Introduction
Hemoglobin (Hb) is a tetramer of two alpha- and two beta-globin chains. Hb disorders refer to a group of genetic diseases caused by mutations or deletions of the normal globin gene. These disorders are characterized by a reduced synthesis of one or more of the globin chains (thalassemia), or by the synthesis of structurally abnormal Hb. Both types of disorders can occur at the same time and are not mutually exclusive [Citation1,Citation2]. Up to date, more than 1000 mutations have been documented [Citation3]. The most significant variant, hemoglobin S (HbS), is characterized by the substitution of a glutamic acid by a valine, in the sixth position of the β-globin chain. Other structurally abnormal variants have lower clinical significance in the heterozygote or homozygote state. However, their combination with the HbS allele, or with thalassemia can lead to a clinically severe phenotype.
Sickle cell disorders encompass a wide range of genetic anomalies, including not only sickle cell anemia (homozygous HbSS), but also combination of HbS with other β-globin variants (such as HbC, HbD Punjab, HbE or HbO) or with β-thalassemia (HbS/β+thal or HbS/β0thal) [Citation4–7]. Sickle cell disorders are clinically marked by the polymerization of HbS in red blood cells, leading to anemia, and acute and chronic tissue damage secondary to blood vessel occlusions.
Hb disorders represent the most common single-gene disorders in the world, with very high prevalence in some tropical and sub-tropical regions [Citation8]. In Africa, sickle cell disease is the most prevalent genetic disease, as more than 75% of all global sickle cell disease births occur in sub-Saharan Africa. The Hb disorders account for about 3.4% of deaths in children under 5 years worldwide and 6.4% in Africa [Citation4]. Recently, frequencies of Hb disorders have risen significantly in industrialized western countries due to population movements and immigration over the past decades.
In most industrialized countries, newborn screening programs are now widespread and have significantly reduced early childhood mortality due to sickle cell disease. The World Health Organization recommends newborn screening for reducing pediatric mortality in Africa, however these national screening programs are logistically and economically challenging to implement [Citation1]. Overall, screening is usually performed by high-performance liquid chromatography (HPLC), isoelectric focusing or mass spectrometry as initial testing method [Citation9]. When electrophoretic or HPLC techniques are used as first-line method, and since some variants can migrate at the same time as HbS, a second-tier test is always necessary to confirm the diagnosis. Tandem mass spectrometry has shown to be a reliable technique in comparison with current methods for neonatal screening [Citation10,Citation11]. Molecular techniques are in general more specific and sensitive, however they are generally more expensive and often need special equipment and experienced lab technicians [Citation5,Citation12]. Isothermal nucleic acid amplification technologies, like loop mediated isothermal amplification (LAMP), have a significant advantage over other polymerase chain reaction (PCR) techniques as they can be implemented in a single step process at a constant temperature. Removing the constraint for rapid thermal cycling enables diagnostics to be conducted in small, simple and low-power instruments [Citation13]. Additionally, PCR usually needs extracted DNA to initiate amplification. By working with a polymerase resistant to blood PCR-inhibitors, we developed an assay using LAMP for the detection of HbS and HbC without DNA extraction. So, this molecular assay can be performed in a single tube directly on fresh or frozen whole blood, and on dried blood spots (DBS). In this paper we describe the clinical performance of Hb S/C detection with this LAMP method.
Material and methods
Design of the technique
The Hb S/C Kit (LaCAR MDx Technologies S.A., Liège, Belgium), is based on the LAMP method in which the target is amplified at a constant temperature. In this kit, the target sequence is amplified at 68°C by using six different primers in combination with a polymerase with high-strand displacement activity and replication activity [Citation14]. The six different primers attach to different regions on the target gene, adding to the specificity of the assay. Due to the loop amplification, the amount of DNA produced in LAMP is considerably higher in comparison to traditional PCR methods [Citation15]. Detection is done using a melting curve analysis for which an HbS mutation specific probe is used. Due to the mismatch with the HbA allele and the two mismatches with the HbC allele, the melting and corresponding change in fluorescence will occur at a different temperature for the different alleles. The change in fluorescence is measured and the melting curve with corresponding peaks are presented as result.
Samples
Fifty-six fresh EDTA blood samples and 192 dried blood cards were included in this study. All samples were initially routinely tested with the diagnostic methods (considered as reference methods, see below) used in the genetics center at the University of Liège. Afterwards samples were analyzed with the Hb S/C Lamp method. Fresh blood samples were conserved at 4°C until testing, while dried blood cards were stored at room temperature.
Reference methods
Routine diagnostic methods used in the genetics center at the University of Liège were used as reference methods. All blood samples were analyzed using capillary electrophoresis (Sebia, France), while all DBS were tested using tandem mass spectrometry [Citation11,Citation16].
Hemoglobin S/C kit
A schematic representation of the workflow is presented in .
Figure 1. Human Hb S/C LAMP test procedure. RT, room temperature.

For blood samples, 5 µl of fresh blood was added to 1 ml of lysis buffer. This suspension was mixed and left at room temperature for 10 min. For the DBS, 2 spots of 3 mm diameter were punched and added to 1 ml of lysis buffer. This suspension was thoroughly mixed and left at room temperature for 10 min. The suspension for the dried blood cards was additionally mixed 2–3 times during incubation. Twenty microliters of ready-to-use reaction buffer was dispensed in the PCR strip, before addition of 5 µl of lysed sample. Each run contains six samples and a positive and negative control. The strip was spinned down to avoid air bubbles and was put in the GENIE®III machine (Optigene Ltd, Horsham, UK), on which the HbS/C program was launched. Isothermal amplification is performed during 30 min, followed by a melting curve analysis from 40 to 90°C with a ramp rate of 0.085°C/s. The positive control is a plasmid control, containing the three different alleles (HbA, HbS and HbC). Interpretation of the results is done with the Genie Explorer software version 2.0.4.2 (Optigene Ltd, Horsham, UK). Results are interpreted using the melting temperatures and the graphical presentation of the melting curves.
Results
To test all samples with the Hb S/C kit, a total of 44 runs were performed. Results were accepted only if a correct melting temperature was obtained for the three peaks of the positive control and if the negative control did not show any peak (). Only one run had to be repeated as the positive control was not within the fixed limits, and the re-run generated correct results.
Table 1. Interpretation criteria for the Hb S/C kit.
Result interpretation was achieved by comparing the melting temperatures of the samples to those presented in . Additionally, the results should also be verified on the melting plot and the run should be repeated when curves present abnormal profiles. An example of the graphical result is shown in .
Figure 2. Result obtained with the Human Hb S/C LAMP method. (a) Graphical presentation of a result. The HbC, HbA and HbS peak are indicated on the graph. The melting curve for the positive plasmid control (ctl+), with three peaks, is presented in bold blue. The negative control (ctl−) is presented in pink. (b) Graphical presentation of the doubtful HbAS sample, indicated in bold blue. A small HbS peak is visible but is not detected by the software due to the limited peak deepness.
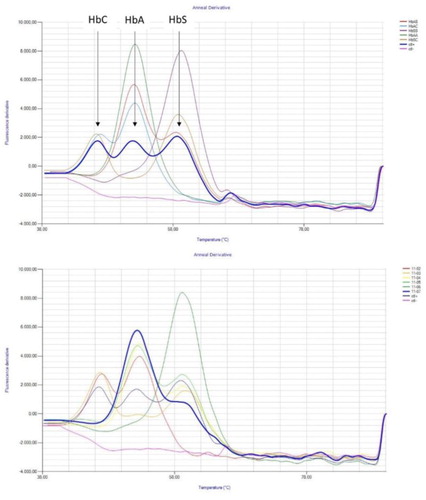
For the DBS samples, mean melting temperatures were 46.70°C for the C-peak, 52.16°C for the A-peak and 58.93°C for the S-peak, with standard deviation values of 0.16, 0.26 and 0.22, respectively. For blood samples, an average melting temperature of 46.92°C was found for the C-peak, 52.09°C for the A-peak and 58.81°C for the S-peak, with standard deviations of 0.30, 0.39 and 0.93, respectively. The coefficient of variation of melting temperatures for the positive control included in each run is 0.38% for the C-peak, 0.44% for the A peak and 0.43% for the S peak.
Considering the 56 blood samples, all showed similar results to the reference method for the detection of HbS and HbC genotypes. Our method did not detect mutations or deletions corresponding to β-thalassemia. We tested one sample diagnosed as HbS/β-thalassemia with the reference technique and, as expected, this sample was identified as HbAS with the Hb S/C kit. We also did not detect HbAD nor hereditary persistence of fetal hemoglobin (HPFH) ().
Table 2. Results from the Hb S/C LAMP method and standard procedures.
For the DBS, all samples included in our study were in complete agreement with the reference method, considering the specific selectivity of the Hb S/C kit (). Due to low fluorescence levels, two samples were identified as invalid in a first run but were correctly identified when repeated. Samples showing discrepant results were phenotyped with the reference technique as HbAE and HbAD. Indeed, the HbS/C assay does not detect the Hb D or E variant, so these samples were identified as HbAA. An overview of the results is presented in .
A total accuracy for all the variants of the cohorts of 92.3% was obtained, as 229 of the 248 tested samples were genotyped in complete correspondence with the reference techniques. However the HbS/C assay has been designed to only detect HbS and HbC. Consequently, if only the HbAA, HbAS, HbSS, HbSC and HbAC genotypes are considered, a perfect correlation with the routine methods was obtained and the sensitivity and the specificity of the new test were 100%. ).
Discussion
Our study shows that the simple LAMP method can detect HbA, HbS and HbC alleles with an accuracy of 100%. High sensitivity and specificity were obtained. Two invalid samples needed retesting, but finally the expected result was obtained.
The Hb S/C assay is not able, neither intended, to identify all the mutations associated with Hb disorders, but was specifically designed to only identify HbS and HbC variants. As expected, it was thus shown that β-thalassemia and other Hb variants such as HbE or HbD are not detected. As Hb D and E variants are mainly found respectively in southeast Asia and in the Punjab region of India, the Hb S/C kit alone is insufficient in Asian populations as a first-line screening tool, since two of the most common variants in that regions would be missed [Citation1,Citation17,Citation18]. However, in Africa, where HbS and HbC are common variants and where thalassemia mutations are very rare, the Hb S/C kit could be a very useful first-line screening tool to detect individuals with sickle cell disease and people carrying the sickle cell trait [Citation4].
It should be recommended that any patient with a homozygous (or a composite heterozygous C/S) mutation will be called back for a confirmation procedure and management. The results of the heterozygous patient (A/S) will also need to be transmitted to the reference clinician. Indeed, the technique might miss a second mutation, either a beta-thalassemia mutation or another rare Hb variant. Therefore, if a child with a heterozygous mutation presents specific symptoms, a more complete Hb study should be performed. Alternatively, confirmation of heterozygous results with a secondary method remains mandatory, to rule out any associated rare variant or thalassemia trait. Such a two-step protocol is in agreement with most other screening and diagnostic procedures, such as for cystic fibrosis, which also requires a second assay to confirm the diagnosis or to identify some rare variants. The Hb S/C kit could also be used in a newborn screening context, as all newborn samples were correctly genotyped. The presence of fetal hemoglobin (HbF) does not influence the result of this molecular assay, in contrast to phenotype screening methods, where HbF might interfere with accurate diagnosis of newborn blood samples.
The Hb S/C kit performs well on fresh blood samples but also on dried blood cards, which is the sample type of choice in rural areas as these can be easily transported and stored. To our knowledge, this method is one of the only molecular assays that can be performed directly on dried blood cards without DNA extraction, lowering handling and turnaround time, and costs. The robustness of the assay was shown by the limited variation in melting temperature, on the different patient samples and by the low coefficient of variation for the positive control. The amplification is running at constant temperature, eliminating the need for expensive thermocyclers. As amplification and detection are performed in the same device, no post-amplification manipulations are needed, greatly reducing the contamination risk, with minimal hands-on-time. The GENIE®III machine is small, portable, with an internal battery and allows decentralized testing. However, in countries where the option of a centralized newborn screening program is favored, a scaling-up is feasible with classical Q-PCR equipment.
Finally, we demonstrated here for first time the reliability of the LAMP technology in the screening of sickle cell and HbC disorders. Hb S/C LAMP method was shown to be fast, with only 40 min from sampling to result, and sensitive. This assay represents therefore a valuable first-line screening tool, as it only needs limited laboratory equipment and training. Indeed, recent tests performed in a small center in Africa, after a very basic training, indicate the robustness of the procedure in local conditions (data not shown). Such affordable test can therefore be easily implemented in small decentralized laboratories allowing diagnosis of patients in low resource areas, and enhancing subsequently preventive care.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Liselot Detemmerman: Master in Biochemistry, she is the R&D manager at LaCAR MDX Technologies. She has a broad expertise in clinical laboratory. Indeed, she was previously employed in the department of microbiology from the UZ Brussel.
Sabine Olivier: PhD in pharmaceutical Sciences, she is the innovation manager at LaCAR MDX technologies. She has a complementary expertise in scientific research and valorization of the medical sciences.
Vincent Bours: Medical doctor and university professor of Genetics at the university of Liège, he is the head of the department of Medical genetics at the university hospital of Liège and he is also a shareholder from the LaCAR company.
François Boemer: PhD in pharmaceutical Sciences, he is the head of the biochemical genetics lab at the university hospital of Liège.
Additional information
Funding
References
- Weatherall DJ, Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001;79:704–712.
- Ryan K, Bain BJ, Worthington D, et al. Significant haemoglobinopathies: guidelines for screening and diagnosis. Br J Haematol. 2010;149:35–49. doi: 10.1111/j.1365-2141.2009.08054.x
- Giardine B, Borg J, Viennas E, et al. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014;42:D1063–D1069. doi: 10.1093/nar/gkt911
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86:480–487. doi: 10.2471/BLT.06.036673
- Clark BE, Thein SL. Molecular diagnosis of haemoglobin disorders. Clin Lab Haematol. 2004;26:159–176. doi: 10.1111/j.1365-2257.2004.00607.x
- Kutlar F. Diagnostic approach to hemoglobinopathies. Hemoglobin. 2007;31:243–250. doi: 10.1080/03630260701297071
- Bender MA, Douthitt Seibel G. Sickle cell disease. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. Genereviews(R). Seattle (WA) 1993; Available from: https://www.ncbi.nlm.nih.gov/sites/books/NBK1377/
- Flint J, Harding RM, Boyce AJ, et al. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol. 1998;11:1–51. doi: 10.1016/S0950-3536(98)80069-3
- Campbell TA, Ware RE, Mason M. Detection of hemoglobin variants in erythrocytes by flow cytometry. Cytometry. 1999;35:242–248. doi: 10.1002/(SICI)1097-0320(19990301)35:3<242::AID-CYTO7>3.0.CO;2-Y
- Boemer F, Cornet Y, Libioulle C, et al. 3-years experience review of neonatal screening for hemoglobin disorders using tandem mass spectrometry. Clin Chim Acta. 2011;412:1476–1479. doi: 10.1016/j.cca.2011.04.031
- Boemer F, Ketelslegers O, Minon JM, et al. Newborn screening for sickle cell disease using tandem mass spectrometry. Clin Chem. 2008;54:2036–2041. doi: 10.1373/clinchem.2008.106369
- Yue L, Lin M, Chen JT, et al. Rapid screening for sickle cell disease by polymerase chain reaction-high resolution melting analysis. Mol Med Rep. 2014;9:2479–2484. doi: 10.3892/mmr.2014.2130
- Notomi T, Mori Y, Tomita N, et al. Loop-mediated isothermal amplification (LAMP): principle, features, and future prospects. J Microbiol. 2015;53:1–5. doi: 10.1007/s12275-015-4656-9
- Notomi T, Okayama H, Masubuchi H, et al. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000;28:63e. doi: 10.1093/nar/28.12.e63
- Nagamine K, Hase T, Notomi T. Accelerated reaction by loop-mediated isothermal amplification using loop primers. Mol Cell Probes. 2002;16:223–229. doi: 10.1006/mcpr.2002.0415
- Louahabi A, Philippe M, Lali S, et al. Evaluation of a new Sebia kit for analysis of hemoglobin fractions and variants on the capillarys system. Clin Chem Lab Med. 2006;44:307–345. doi: 10.1515/CCLM.2006.059
- Vichinsky E. Hemoglobin e syndromes. Hematology Am Soc Hematol Educ Program. 2007;1:79–83
- Itano HA. A third abnormal hemoglobin associated with hereditary hemolytic anemia. Proc Natl Acad Sci U S A. 1951;37:775–784. doi: 10.1073/pnas.37.12.775