
ABSTRACT
Objectives: To report the hematological and molecular features as well as diagnostic aspects of the hitherto un-described interactions of two rare α-globin chain variants with α0-thalassemia commonly found among Southeast Asian populations.
Methods: The study was done on two adult Thai patients (P1 and P2) who had hypochromic microcytic anemia. Hb analysis was carried out using high performance liquid chromatography (HPLC) and capillary electrophoresis (CE). Mutations were identified by PCR and related techniques.
Results: Hb analysis of P1 using HPLC showed a normal Hb pattern, but CE demonstrated an abnormal peak at zone 7. DNA sequencing identified a CCG-CTG mutation at codon 95 of the α2 globin gene corresponding to the Hb G-Georgia [α95(G2)Pro → Leu(α2)] previously undescribed in the Thai population. In contrast, Hb analysis of P2 demonstrated an abnormal peak not fully separated from Hb A on HPLC, but not on CE. DNA analysis identified the rarely described Hb Nakhon Ratchasima [α63(E12)Ala → Val(α2)] mutation. Routine DNA analysis detected the SEA deletion α0-thalassemia in trans to the Hb variants in both cases. Hematological parameters were compared with those of patients with compound heterozygote for other α-globin variants and α0-thalassemia previously documented.
Conclusions: Identification of the patients confirmed that interaction of these rare Hb variants with α0-thalassemia does not lead to the Hb H disease. Differentiation of these two Hb variants from other clinically relevant hemoglobinopathies in a routine setting is, however, necessary. This can be accomplished using a combined Hb-HPLC and CE analysis followed by PCR-RFLP assays.
Introduction
Thalassemia and hemoglobinopathies are common and heterogeneous in Southeast Asia. In Thailand, for example, the prevalence of α-thalassemia is about 20–30% and that of β-thalassemia is 3.0–9.0%. The frequency of hemoglobin (Hb) E [26(B6)Glu → Lys, GAG > AAG] is about 30–40%; that of Hb Constant Spring or Hb Paksé is 1.0–8.0% [Citation1]. In addition, more than 30 Hb variants have been documented [Citation2]. With this variety of hemoglobinopathic alleles, interactions between them are commonly encountered. These can lead to complex thalassemia syndromes with clinical manifestations or clinically harmless but complicated routine thalassemia diagnostics [Citation3–6]. Interaction of Hb Constant Spring or Hb Pakse’ or α+-thalassemia with α0-thalassemia leads to severe Hb H disease, commonly encountered in the region [Citation7]. In contrast, the interaction of α0-thalassemia with other α-globin chain variants has rarely been documented. Here, we report, for the first time, the interaction of two rare α-globin chain variants, the Hb G-Georgia [α95(G2)Pro → Leu(α2)(HBA2:c287 C > T)] and Hb Nakhon Ratchasima [α63(E12)Ala → Val(α2)] with α0-thalassemia commonly found among the Southeast Asian population. Hematological features, Hb analytical profiles on HPLC and capillary electrophoresis (CE) of this Hb variant, as well as simple molecular diagnostics based on PCR-restriction fragment length polymorphism (RFLP) are also presented.
Materials and methods
Subject and hematological analysis
Ethical approval of the study protocol was obtained from our Institutional Review Board (IRB) at Khon Kaen University, Thailand (HE552035). Blood specimens of two Thai subjects were referred to our routine diagnostic laboratory at Khon Kaen University, Thailand for thalassemia investigation. Hematological parameters were recorded on a standard blood cell counter. Hb analysis was done using HPLC (Variant II; Bio-Rad Laboratories, Hercules, CA, U.S.A.) and CE system (Capillarys2 Flex Piercing; Sebia, Lisses, France) ().
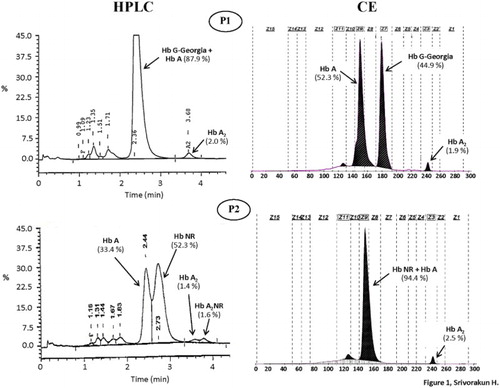
Figure 1. Hemoglobin analysis of the two patients (P1 and P2) who were respectively compound heterozygotes for Hb G-Georgia/α0-thalassemia and Hb Nakhon Ratchasima/α0-thalassemia using automated HPLC and CE systems. The separated profiles of Hb G-Georgia, Hb Nakhon Ratchasima (NR), Hb A, Hb A2 and Hb A2NR are depicted.
Routine DNA analysis
Identifications of α0-thalassemia (SEA/THAI deletions), α+-thalassemia (−α3.7 and −α4.2), αConstant Spring and αPaksé are routinely performed in our laboratory using PCR and related techniques as has been described [Citation8]. Identification of Hb Tak (β146+AC, Ter–Thr) and Hb Q-Thailand (α74GAC-CAC, Asp-His) with similar electrophoretic mobilities with those of Hb variants herein described were done using allele-specific PCR assays described elsewhere [Citation9,Citation10]. Direct DNA sequencing of the amplified α-globin gene was done using an ABI PRISM™ 3130 XL analyzer (Applied Biosystems Foster City, CA, U.S.A.).
Identification of the αG-Georgia and αNakhon Ratchasima mutations
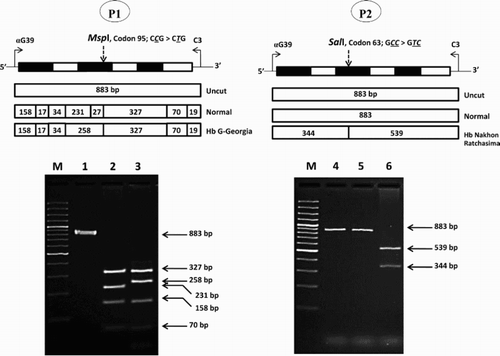
To establish rapid methods for the identification of Hb G-Georgia and Hb Nakhon Ratchasima in a routine setting, methods based on PCR-RFLP assay were developed. Selective amplification of an α2-globin gene was done using specific primers αG39 (5'-ACTCTTCTGGTCCCCACAGAC-3′) and C3 (5′-CCATTGTTGGCACATTCCGG-3′) [Citation11]. The amplified α2-globin gene fragment (883 bp) was then digested to completion with Msp I (5′-C▾CGG-3′) and Sal I (5′-G▾TCGAC-3′) (New England Biolabs Inc., Beverly, MA, U.S.A.) for Hb G-Georgia and Hb Nakhon Ratchasima mutations, respectively. The expected fragments were (327, 258 and 158 instead of 327, 231 and 158 bps) for Hb G-Georgia and (539 and 344 instead of 883 bp) for Hb Nakhon Ratchasima, as shown in .
Figure 2. Identification of the Hb G-Georgia and Hb Nakhon Ratchasima mutations by PCR-RFLP assays using MspI and SalI digestion, respectively. The locations and orientations of primers αG39 and C3 and the size of amplified α2-globin gene fragment (883 bp) are illustrated. The sizes of MspI and SalI-digested fragments specific for αG-Georgia (258 bp) and αNakhon Ratchasima (539 bp and 344 bp) and their normal counterparts are indicated. In agarose gel electrophoresis, M represents the GeneRuler 50 bp DNA ladders. 1 and 4: undigested amplified DNA, 2: MspI-digested amplified DNA of normal subject, 3: MspI-digested amplified DNA of the P1 with Hb G-Georgia/α0-thalassemia, 5: SalI-digested amplified DNA of normal subject, 6: SalI-digested amplified DNA of the P2 with Hb Nakhon Ratchasima/α0-thalassemia.
Results
P1 was an adult male who had mild hypochromic microcytic anemia with Hb 11.4 g/dl, MCV 57.8 fl and MCH 18.2 pg, compatible with being a thalassemia carrier commonly found in the region. As shown in , initial Hb-HPLC analysis revealed a normal Hb pattern with 2.0% Hb A2. However, analysis using a CE system identified, in addition to Hb A (52.3%) and Hb A2 (1.9%), an abnormal Hb separation at zone 7 (44.9%). This indicated that he was a carrier of an unknown Hb variant co-separating with Hb A on HPLC. Routine DNA analysis of α-thalassemia revealed as expected that he was also a carrier of α0-thalassemia (SEA deletion). Identification of Hbs Tak and Q-Thailand, the two common Hb variants detected at zone 7 of CE by PCR [Citation9,Citation10] yielded negative results. Since the amount of abnormal Hb found in P1 pointed to a β-globin chain variant, DNA sequencing of the β-globin gene was carried out. No mutation was detected (data not shown). However, further DNA sequencing of an α2-globin gene [Citation11] revealed unexpectedly a CCG (Pro) → CTG (Leu) transition at codon 95, corresponding to the Hb G-Georgia [Citation12]. As this mutation eliminates an MspI restriction site (5′-C▾CGG-3′) on the α2-globin gene, it could be confirmed by PCR-RFLP assay using MspI digestion as shown in . These DNA analyses confirmed that P1 was a compound heterozygote for a previously undescribed condition of α0-thalassemia/Hb G-Georgia.
The analysis of P2 indicated that he was suffering from severe anemia with Hb 5.3 g/dl, MCV 69.0 fl and MCH 22.7 pg. However, a relatively low number of Rbc and reduced Hb and Hct values indicated that this could be due to other underlining defects. Hb analysis identified an abnormal Hb not fully separated from Hb A on HPLC but co-migrating with Hb A on CE (). Hb A2 level was normal in both analyzers. PCR-RFLP analysis using Sal I indicated that he was a carrier of Hb Nakhon Ratchasima [α63(E12)Ala → Val(α2)], an α2-globin chain variant previously documented in Thailand [Citation13]. Routine α-thalassemia screening identified that he also carried an α0-thalassemia (SEA deletion). Accordingly, he was finally diagnosed as a compound heterozygote for α0-thalassemia/Hb Nakhon Ratchasima, another undescribed condition.
As for other α-globin chain variants including Hb Thailand [α56(E5)Lys → Thr(α1)], Hb Phnom Penh [α117(GH5)-Ile-α118(H1)(α1)] [Citation5] and Hb Hekinan [α27(B8)Glu → Asp(α2)] [Citation11] found in our laboratory, the association of Hb G-Georgia and Hb Nakhon Ratchasima with α0-thalassemia in the two patients did not lead to Hb H disease. lists hematological findings of these Thai patients with multiple globin gene interactions comparatively.
Table 1. Hematological data and globin genotypes of Thai patients with Hb G-Georgia/α0-thalassemia and Hb Nakhon Ratchasima/α0-thalassemia as compared to other α-globin chain variants with similar genotypes including Hbs Thailand [Citation5], Phnom Penh [Citation5] and Hekinan [Citation11] previously found in our laboratory. GG = Hb G-Georgia, NR = Hb Nakhon Ratchasima.
Discussion
Hb variants are common in Thailand where thalassemia is prevalent and heterogeneous. Interactions between them, resulting in complex syndromes with complicated laboratory diagnostics, are occasionally encountered in routine practices. Accurate diagnosis of the cases usually requires multiple testing. Using a combination of Hb-HPLC, Hb-CE followed by DNA analysis, we have now reported the hitherto undescribed conditions caused by interactions of two α-globin chain variants with α0-thalassemia (SEA deletion) in two Thai individuals.
In P1, we detected the interaction of Hb G-Georgia and α0-thalassemia. Hb G-Georgia is a rare variant found in heterozygote and double heterozygote with Hb C and Hb S [Citation14,Citation15]. The globin gene interaction observed in P1 is the first time this Hb variant has been found in association with α0-thalassemia. As shown in , HPLC could not identify this Hb variant with a substitution of Leucine for Prolein at amino acid number 95 of the α-globin chain. However, CE could demonstrate it as an unknown Hb variant excellently, separating at zone 7 before Hb A at zone 9. It has been demonstrated that Hb G-Georgia moved slightly faster than Hb S on alkaline cellulose acetate electrophoresis [Citation15]. However, with the amount of 44.9% Hb G-Georgia with no Hb A2 variant (α2GGδ2), Hb G-Georgia could be misinterpreted as Hb F or another β-globin chain variant. The level of Hb G-Georgia in the heterozygote state has been noted to be 23.4%, approximately one-fourth of the total Hb [Citation12]. A relatively higher proportion of Hb G-Georgia (44.9%) in P1 indicates as for Hb Hekinan, as previously described [Citation11], that with the limited availability of α-globin chain in α0-thalassemia, the formation of Hb G-Georgia is preferable. It is noteworthy that a Thai patient with Hb G-Georgia/α0-thalassemia did not develop Hb H disease [Citation16], the data indicating that this Hb G Georgia is not an α-thalassemic allele. However, a positive result for a dichlorophenolindophenol screening test for Hb E [Citation17] indicated instability of the Hb G-Georgia molecule (data not shown). This could be due to an alteration of α1β2 contact of the Hb molecule, though this should not have much effect on the Hb function. The hypochromic microcytosis observed in the patient could be explained by the α0-thalassemia allele. A Thai individual with the Hb St. Luke’s–Thailand [α95(G2)Pro-Arg], another Hb variant with a mutation at the same position, with Hb G-Georgia also presented with no clinical symptoms [Citation18].
Hb Nakhon Ratchasima found in association with α0-thalassemia in P2 is another rare α-globin chain variant. It has only been reported in combination with Hb E and α+-thalassemia in another Thai patient with mild anemia, but with otherwise normal Rbc indices [Citation13]. Hb Nakhon Ratchasima is likely a non-pathological α-globin chain variant. An amino acid substitution of Valine to Alanine at codon 63 of the α-globin chain of this Hb variant is located at the external surface, and appears to have no influence on the function of the Hb molecule. Accordingly, as for Hb G-Georgia, the association of Hb Nakhon Ratchasima with α0-thalassemia in P2 did not lead to the Hb H disease. Although P2 did have low MCV and MCH characteristics of an α0-thalassemia carrier, a relatively low number of Rbc and dramatically reduced Hb and Hct values pointed to other underlying defects, rather than the interaction of Hb Nakhon Ratchasima and α0-thalassemia. It is noteworthy, as shown in , that unlike Hb G-Georgia, Hb Nakhon Ratchasima co-migrates with Hb A on CE, but can be separated from Hb A on HPLC. However, the best way to confirm this is PCR-RFLP using Sal I digestion, as shown in .
Although both Hb G-Georgia and Hb Nakhon Ratchasima are rarely encountered and should be considered as benign abnormalities in both heterozygote and compound heterozygote forms, differentiation from other clinically relevant Hb variants in a routine setting is necessary. We demonstrated that a combination of Hb analysis using both HPLC and CE followed by PCR-RFLP assays is helpful in the identification of cases with the two Hb variants. This should facilitate genetic counseling and a prevention and control program for hemoglobinopathy in the region.
Acknowledgments
We thank Mr Ian Thomas for the helpful comments on the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Fucharoen S, Winichagoon P. Hemoglobinopathies in Southeast Asia. Hemoglobin. 1987;11:65–88. doi: 10.3109/03630268709036587
- Srivorakun H, Singha K, Fucharoen G, et al. A large cohort of hemoglobin variants in Thailand: molecular epidemiological study and diagnostic consideration. PLoS ONE. 2014;9:e108365. doi: 10.1371/journal.pone.0108365
- Sanchaisuriya K, Fucharoen G, Sae-ung N, et al. Molecular and hematologic features of hemoglobin E heterozygotes with different forms of alpha-thalassemia in Thailand. Ann Hematol. 2003;82:612–616. doi: 10.1007/s00277-003-0689-y
- Singsanan S, Srivorakun H, Fucharoen G, et al. Hb Phimai [beta72(E16)Ser→Thr]: a novel beta-globin structural variant found in association with Hb Constant Spring in pregnancy. Hemoglobin. 2011;35:103–110. doi: 10.3109/03630269.2011.557171
- Singha K, Srivorakun H, Fucharoen G, et al. Association of Hb Thailand [alpha56(E5)Lys-->Thr] and Hb Phnom Penh [alpha117(GH5)-Ile-alpha118(H1)] with alpha(0)-thalassemia: molecular and hematological features and differential diagnosis. Hemoglobin. 2013;37:37–47. doi: 10.3109/03630269.2012.747964
- Singha K, Fucharoen G, Fucharoen S. Interaction of Hb Grey Lynn (Vientiane) [α91(FG3)Leu > Phe (α1)] with Hb E [β26(B8) Glu > Lys] and α+-thalassemia: molecular and hematological analysis. Clin Lab. 2015;61:631–635. doi: 10.7754/Clin.Lab.2014.141112
- Boonsa S, Sanchaisuriya K, Fucharoen G, et al. The diverse molecular basis and hematological features of Hb H and AEBart's diseases in Northeast Thailand. Acta Haematol. 2004;111:149–154. doi: 10.1159/000076523
- Yamsri S, Sanchaisuriya K, Fucharoen G, et al. Prevention of severe thalassemia in northeast Thailand: 16 years of experience at a single university center. Prenat Diagn. 2010;30:540–546.
- Sanchaisuriya K, Chunpanich S, Fucharoen G, et al. Multiplex allele-specific PCR assay for differential diagnosis of Hb S, Hb D-Punjab and Hb Tak. Clin Chim Acta. 2004;343:129–134. doi: 10.1016/j.cccn.2003.12.029
- Singsanan S, Karnpean R, Fucharoen G, et al. Hemoglobin Q-Thailand related disorders: origin, molecular, hematological and diagnostic aspects. Blood Cells Mol Dis. 2010;45:210–214. doi: 10.1016/j.bcmd.2010.06.001
- Fucharoen S, Changtrakun Y, Ratanasiri T, et al. Complex interaction of Hb Hekinan [alpha27(B8) Glu-Asp] and Hb E [beta26(B8) Glu-Lys] with a deletional alpha-thalassemia 1 in a Thai family. Eur J Haematol. 2003;70:304–309. doi: 10.1034/j.1600-0609.2003.00049.x
- Huisman TH, Adams HR, Wilson JB, et al. Hemoglobin G Georgia or alpha 2-95 Leu (G2) beta-2. Biochim Biophys Acta. 1970;200:578–580 doi: 10.1016/0005-2795(70)90117-0
- Srivorakun H, Fucharoen G, Puangplruk R, et al. Complex interaction of Hb Nakhon Ratchasima [α63(E12)Ala-Val], a novel α2-globin chain variant with Hb E [β26(B8)Glu-Lys] and a deletional α+–thalassemia. Eur J Haematol. 2011;87:68–72. doi: 10.1111/j.1600-0609.2011.01616.x
- Reynolds S, Miller C, King R, et al. Hemoglobin C-G Georgia double heterozygosity: a case report. Ann Clin Lab Sci. 1992;22:414–416.
- Wrightstone RN, Hubbard M, Huisman TH., et al. Hemoglobin S-Galpha Georgia disease: a case report. Acta Haematol. 1974;51:315–320 doi: 10.1159/000208312
- Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood. 2003;101:791–800. doi: 10.1182/blood-2002-07-1975
- Fucharoen G, Sanchaisuriya K, Sae-ung N, et al. A simplified screening strategy for thalassaemia and haemoglobin E in rural communities in South-East Asia. Bull World Health Organ. 2004;82:364–372.
- Singha K, Fucharoen G, Jetsrisuparb A, et al. Molecular and hematological characteristics of a novel form of α-globin gene triplication: The hemoglobin St.Luke's-Thailand [α95(G2)Pro → Arg] or Hb St. Luke's [A2] HBA2. Clin Biochem. 2013;46:675–680. doi: 10.1016/j.clinbiochem.2013.01.022