
ABSTRACT
Objectives: The aim of this study was to determine the anthropometric measurements in transfusion-dependent β-thalassemia children in Pakistan. The secondary aim was to correlate serum ferritin with the physical growth.
Methods: We enrolled 367 children (aged 5–17 years) with transfusion-dependent beta-thalassemia major in the study. Anthropometric measurements, serum ferritin levels, and pre-transfusion hemoglobin levels were measured. Serum ferritin was correlated with the height z-score for age.
Results: Laboratory evaluation showed that patients had significantly low mean pre-transfusion hemoglobin of 7.66 ± 1.34 g/dl (range 2.5–10.5) and high median (Q3–Q1) serum ferritin of 5012 ng/ml (6829–3532). The median (Q3–Q1) height-for-age z-score of children was low at −2.69 and (−1.46 to −3.80) and 65.4% children had stunted growth (height for age z-score <−2). There was a significant negative correlation between height for age z-score and serum ferritin levels (p < 0.000). Stunting of growth began early during 5–10 years of age but increased markedly with the progress of time.
Conclusions: The study showed that children with beta thalassemia major had delayed physical growth possibly secondary to iron overload. Effective and early iron chelation is needed for preventing growth failure in transfusion-dependent beta thalassemia.
Background
Thalassemia major is one of the most common genetically transmitted diseases in the world and is associated with reduced synthesis of structurally normal hemoglobin. With improved control of malnutrition and communicable diseases, β-thalassemia major patients who earlier died young are now surviving long enough to seek medical attention. There are 9.8 million thalassemia carriers in our country with an approximate carrier rate of 5–7% [Citation1]. At a human development index of 146, Pakistan has limited health-care services with thalassemia posing serious challenges on the scarce health resources. Definitive cure of transfusion-dependent thalassemia is bone marrow transplantation. However, this is not a cost-effective option for the majority of patients in an under privilege society. Their survival, therefore, depends on regular blood transfusions and iron chelating therapy [Citation2]. Despite being an effective treatment there are many serious long-term complications. Iron toxicity (hemosiderosis) is one of these devastating complications with consequential end-organ damage and stunted growth [Citation3]. Physical growth is relatively normal up until 9–10 years of age but the majority of thalassemia patients fail to achieve the same final height as their normal peers [Citation4]. Growth retardation in thalassemia is a well-observed phenomenon with a frequency between 25–66% globally [Citation3,Citation5–8] and 20–57% nationally [Citation9–11]. Growth retardation is multifactorial and the etiology varies with age. Chronic hypoxia, nutritional deficiencies (of zinc and folate), iron toxicity are the main contributors in early childhood [Citation4]. Iron as well as chelation toxicity, hepatitis, psychosocial issues and endocrinopathies (hypogonadism, hypothyroidism, and growth hormone failure) are the etiological factors for delayed or absent pre-pubertal growth spurt [Citation6,Citation12]. Recently, anemia, iron, and chelation toxicities have been analyzed at a molecular level. Chronic anemia increases erythropoietin secretion, marrow expansion and thinning of bony cortices as well as increases RANKL with a negative effect on bone formation [Citation13]. Excessive iron interferes with maturation of osteoid and deposits in hydroxyapatite crystals thus interfering with normal bone metabolism. Similarly desferoxamine (iron chelator) inhibits DNA synthesis, proliferation, and maturation of fibroblasts and osteoblasts [Citation13]. These factors have detrimental effects on bone mineralization and can also compromise physical growth in thalassemia.
Substantial systematic data for physical growth in beta thalassemia is not available nationally with limited information published in only three reports [Citation9–11]. We therefore designed a study to determine the anthropometric measurements in pediatric age group suffering from transfusion-dependent β-thalassemia. The secondary aim was to relate physical growth with pre-transfusion hemoglobin and serum ferritin in the same group of patients.
Material and methods
Setting
The study was conducted at the Sections of Hematology and Clinical Chemistry in the Department of Pathology and Laboratory Medicine, Aga Khan University, Karachi, Pakistan during January 2013 to December 2014. Patient recruitment was conducted at a non-profit organization named Fatimid Foundation Karachi (FFK), which provides free of cost blood components to the patients with various blood disorders. There are more than 4000 registered patients of thalassemia and hemophilia at FFK.
Patient selection
After informed consent from their parents, 367 pediatric patients (age 5–17 year) with transfusion-dependent β-thalassemia were recruited. All patients were diagnosed based on hemoglobin electrophoresis or high-performance liquid chromatography.
Data collection
Detail history was collected by a trained research officer on a predetermined questionnaire. The following information was collected from medical records of the patients: demographics (age, gender, age at the time of diagnosis and at first transfusion), anthropometrics (height and weight), and clinical details (blood transfusion history, iron chelation, last pre-transfusion hemoglobin, and last serum ferritin levels).
Sample collection
Five milliliter of blood samples were collected in two tubes (ethylenediaminetetraacetic acid and gel) at Fatimid foundation by a trained phlebotomist prior to blood transfusion to determine hemoglobin and serum ferritin.
Laboratory methodology
Serum ferritin was measured by chemi-luminescent micro-particle immunoassay on Cobos® E-601 and hemoglobin was measured using Sysmex® XP-100.
Anthropometry
World health organization (WHO) 2007 growth charts were used for boys and girls to assess their physical growth. Body mass index (BMI) was computed as weight in kg/height in square meters. z-Score of height (h-SDS), weight (w-SDS), and BMI (BMI-SDS) were calculated by x−µ/ᵟ [Citation14]. According to WHO reference 2007, a z-score of <−2 for height was considered as stunted growth and BMI of <−2 was considered as underweight or thin for age.
Statistical analysis
Data were collected, managed, edited, entered, and analyzed by statistical package of social sciences (SPSS) version 22 and STATA 13 (Stata Corp, College Station, TX, U.S.A.). A descriptive analysis of all patients in the study was performed. In order to analyze the profile of the sample according to the variables studied, frequency tables for categorical variables like (gender, age group) were created. Male: female ratio was also computed. The mean ± standard deviation, minimum and maximum values were calculated for parametric and median and interquartile range (IQR; Q3–Q1) for non-parametric variables. Spearman rho correlation was used to test relationships between the non-parametric variables. The level of significance taken for all the statistical tests was a p-value of <0.05. The association of h-SDS with median serum ferritin levels was further confirmed by comparing these laboratory parameters in children with height z-scores <2 with those with the z-score ≥2 using the Mann–Whitney test.
Results
Demographics (Table 1)
Three hundred and sixty-seven children (196 males and 171 females) with the mean age of 10.6 years (SD ± 3.3) were enrolled. At the time of study, 231 or 63% patients had a monthly transfusion requirement of two units of red cells. Though 96% patients were receiving iron chelation therapy, only one-third had compliance for regular treatment.
Table 1. Clinical and laboratory parameters of children with transfusion-dependent thalassemia (n = 367).
Laboratory parameters (Table 1)
Laboratory evaluation showed that patients had significantly low mean pre-transfusion hemoglobin which was slightly higher in females (7.86 g/dl) than in males (7.49 g/dl) (t = −2.65, p < 0.0084). The majority (83%) of the patients had pre-transfusion hemoglobin level less than Thalassemia International Federation (TIF) recommended values of 9–10.5 g/dl. [Citation15] Only 3% of patients had serum ferritin level less than 2000 ng/ml. There was no significant difference in serum ferritin level between female and male.
Anthropometry (Table1)
Anthropometric measurements showed that 40% patients were underweight (w-SDS <−2), 65% were stunted (h-SDS <−2), and 42% were malnourished (BMI-SDS <−2) including 23% thin and 19% severely thin patients. Obesity was not observed in any of the patients. demonstrated the progression of growth impairment by advancing age groups in those with stunted growth (65% of patients) and even in children with apparently normal h-SDS. There was a significant negative correlation between age and h-SDS (correlation coefficient r = −0.698, p < 0.000) as well as between age and BMI-SDS (r = −0.344, p < 0.000).
Table 2. Age wise comparison of height for age z-score (HZS) in children with stunted growth (n = 240) vs. normal growing children (n = 127).
Physical growth of patients and serum ferritin
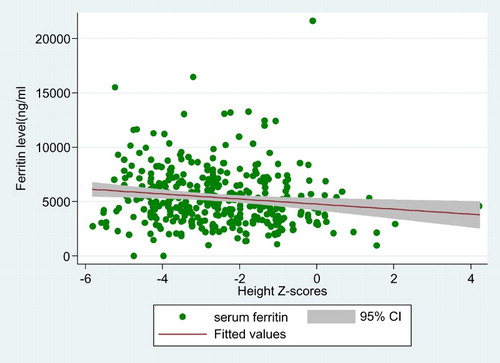
There was a significant negative correlation between h-SDS and serum ferritin levels (Spearman’s rho correlation r = −0.186, p < 0.000) ().The correlation between BMI-SDS and w-SDS with serum ferritin was inverse but insignificant (r = −0.066, p = 0.210 and r = −0.031, p = 0.683). Median (Q3–Q1) serum ferritin was higher at 5225.68 ng/ml (7019.25–3857.14) in those with stunted growth compared to 4445 ng/ml (6293–3116) in children with normal growth (p-value = 0.002).
Figure 1. Regression fit between mean serum ferritin levels (ng/ml) and height z-scores in children with transfusion-dependent thalassemia.
Discussion
The limited anthropometric data from this study demonstrated significant growth failure in transfusion-dependent thalassemia secondary to iron overload. There was progression of growth impairment by advancing age group in all children. The study showed sub-optimally managed patients reflected by low mean pre-transfusion hemoglobin of 7.6 ± 1.3 g/dl and high median serum ferritin level of 5012 ng/ml, which is almost five times the desired level of 1000 ng/ml. Low transfusion policy was observed as a patient’s management approach. Though 96% (n = 352) patients were receiving one or the other form of iron chelation therapy, only 123 (34%) patients were on regular iron chelation.
Despite major therapeutic progress in the last couple of decades growth failure still is a significant challenge in beta thalassemia major, often affecting the social adjustment and hence quality of life. Literature review revealed scarcity of data addressing this issue in thalassemia major from this part of the world. About two-third (65%) of our patients had short stature, and 42% of the patients were malnourished (BMI z-score <−2). An Iranian cross-sectional survey evaluated endocrine and metabolic dysfunction in 56 thalassemia patients; with 70% prevalence of stunted growth, mean serum ferritin level was 2888 ± 948 in this analysis [Citation16]. In our study, h-SDS was inversely related to median ferritin levels. This relation was maintained in patients even when they were stratified based on their stunted growth. This fact has previously been documented in other studies as well [Citation3,Citation7]. Pemde et al. reported significant association (p < 0.000) of higher mean ferritin levels (3720 ± 1512 ng/ml vs. 2570 ± 1196 ng/ml) in patients with short-stature than in patients with normal height [Citation7]. Similarly in another study, mean serum ferritin levels were better (2271.0 ng/ml) in patients with height over the third percentile than in patients with height lower than the third percentile (4567.0 ng/ml, p = 0.01) [Citation12].
According to WHO standards, less than 5% children should have height and BMI z-score of <−2. In our study, approximately half of our patients were below this acceptable cut-off. A study from Egypt reported, height and weight z-scores of <−2 in 49 and 47% patients, respectively [Citation5]. Although growth stunting commences in early years of life in thalassemia, growth retardation becomes more apparent with their advancing age [Citation17,Citation18]. In our study, albeit on the lower side, mean BMI z-score was acceptable (−1.2 ± 1.3) for the 5–10 year age group but decreased to below reference limits in the next two five year groups. No impact of gender was observed in the study population as height z-score of <−2 was observed in 20% males and 23% females; 25% males and females had a BMI z-score <−2).
The pathogenesis of growth failure in thalassemia major is multifactorial. One of the main factors is iron overload. Frequent blood transfusions can control pre-transfusion hemoglobin levels, but if serum ferritin levels are greater than the desired levels, patients’ physical growth can be affected. Pituitary MRI studies demonstrated that severe pituitary iron accumulation may develop as you as 4 years of age in thalassemia major [Citation19,Citation20]. The pubertal delay is apparent 10 years later when tissue changes often become irreversible. Beside iron-induced damage and chronic hypoxia (low pre-transfusion hemoglobin) other factors contributing to final adult height include ethnicity, genetic composition, hormonal status, nutritional deficiencies, degree of chelating agents utilization, emotional factors, endocrinopathies, [Citation21] and chronic liver disease. We did not evaluate the concomitant role of these factors in our study; however, inclusion of a reasonably larger number of patients in this study emphasizes the importance of optimally controlling iron homeostasis in this population.
Strengths and limitations
This anthropometric study remains the first to be conducted in our region, where there is an increased prevalence of thalassemia. The patients were not examined for their sexual development. Since hypogonadism is the major determinant of growth failure in beta thalassemia major, therefore, progression of stunting of growth with advancing age could have been due to hypogonadism. Only a single pre-transfusion hemoglobin was measured which could not be related to growth failure. The effects of other factors such as hormonal status and endocrinopathies on growth and their exact association with serum ferritin levels need to be determined in a meticulous manner. This could not be done in this study due to the expensive nature of hormonal testing.
Conclusion and recommendation
Limited data from this study indicated iron overload as the most likely contributory factor for growth failure. Early intervention by appropriate chelation therapy may help for normal growth progression.
Acknowledgements
Authors are thankful to all the thalassemia children and their parents who participated in this study. This study was approved by institutional review board of Aga Khan University ethical review committee (AKU_ERC) ERC no 2305-Pat-ERC-12 and the data was collected by the researcher after getting a written consent form from patient and guardian.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Bushra Moiz http://orcid.org/0000-0003-0777-3690
Additional information
Funding
References
- Ansari SH, Shamsi TS, Ashraf M, et al. Molecular epidemiology of β-thalassemia in Pakistan: Far reaching implications. Indian J Hum Genet. 2012;18(2):193–197.
- Valizadeh N, Farrokhi F, Alinejad V, et al. Bone density in transfusion dependent thalassemia patients in Urmia, Iran. Iran J Ped Hematol Oncol. 2014;4(2):68–71.
- Hashemi A, Ghilian R, Golestan M, et al. The study of growth in thalassemic patients and its correlation with Serum ferritin level. SSUJ. 2011;1(4):147–151.
- Skordis N, Kyriakou A. The multifactorial origin of growth failure in thalassaemia. Pediatr Endocrinol Rev. 2011;8(Suppl. 2):271–277.
- Fahim FM, Saad K, Askar EA, et al. Growth parameters and vitamin D status in children with thalassemia Major in Upper Egypt. Int J Hematol Oncol Stem Cell Res. 2013;7(4):10–14.
- Shalitin S, Carmi D, Weintrob N, et al. Serum ferritin level as a predictor of impaired growth and puberty in thalassemia major patients. Eur J Haematol. 2005;74(2):93–100.
- Pemde H, Chandra J, Singh V, et al. Physical growth in children with transfusion-dependent thalassemia. Pediatr Health Med Ther. 2011;2011:13–19.
- Vogiatzi MG, Macklin EA, Trachtenberg FL, et al. Differences in the prevalence of growth, endocrine and vitamin D abnormalities among the various thalassaemia syndromes in North America. Br J Haematol. 2009;146(5):546–556.
- Aslam MS, Roshan E, Iqbal A, et al. Frequency of short stature in ß - thalassemia major patients. Pakistan Armed Forces Med J. 2013;63(4):1–4.
- El Missiry M, Hamed Hussein M, Khalid S, et al. Assessment of serum zinc levels of patients with thalassemia compared to their siblings. Anemia. 2014;2014: 1–6.
- Ahmad A, Saeed K, Ahmad F, et al. Short stature and truncal shortening in iron overloaded non-chelated transfusion dependent thalassemic patients. Pak Paediatr J. 2010;34(1):23–27.
- Hamidah A, Arini MI, Zarina AL, et al. Growth velocity in transfusion dependent prepubertal thalassemia patients: results from a thalassemia center in Malaysia. Southeast Asian J Trop Med Public Health. 2008;39(5):900–905.
- Haidar R, Musallam KM, Taher AT. Bone disease and skeletal complications in patients with beta thalassemia major. Bone. 2011;48(3):425–432.
- Preedy V. Handbook of anthropometry. New York (NY): Springer; 2012.
- Trompeter S, Cohen A., et al. Blood transfusion. In: Cappellini MD, Cohen A, Porter J, editors. Guidelines for the management of transfusion dependent thalassaemia (TDT). Nicosia (CY): Thalassemia International Federation; 2014.
- Najafipour F, Aliasgarzadeh A, Aghamohamadzadeh N, et al. A cross-sectional study of metabolic and endocrine complications in beta-thalassemia major. Ann Saudi Med. 2008;28(5):361.
- Saxena A. Growth retardation in thalassemia major patients. Int J Hum Genet. 2003;3(4):237.
- Hattab FN. Patterns of physical growth and dental development in Jordanian children and adolescents with thalassemia major. J Oral Sci. 2013;55(1):71–77.
- Noetzli LJ, Panigrahy A, Mittelman SD, et al. Pituitary iron and volume predict hypogonadism in transfusional iron overload. Am J Hematol. 2012;87(2):167–171.
- Wood JC, Noetzl L, Hyderi A, et al. Predicting pituitary iron and endocrine dysfunction. Ann N Y Acad Sci. 2010;1202:123–128.
- Chrysis DC, Alexandrides TK, Koromantzou E, et al. Novel application of IGF-1 and IGFBP-3 generation tests in the diagnosis of the growth hormone axis disturbances in children with beta-thalassemia. Clin Endocrinol. 2001;54:253–259.