
ABSTRACT
Objectives: Trifluoperazine (TFP) has potential anticancer activity that was demonstrated in different types of cancer cells. However, little is known about its effects in the T-cell ALK+ anaplastic large cell lymphoma (ALCL). In this study, we investigated the effects of TFP in this aggressive type of cancer.
Methods: The cytotoxicities of TFP on DEL and SUP-M2 cells were measured by trypan blue staining. The effects of TFP on cell cycle and apoptosis in DEL and SUP-M2 cells were determined by flow cytometry. The underlying anticancer mechanism of TFP was investigated by real-time PCR, Western Blotting, and nucleophosmin-ALK (NPM-ALK) tyrosine kinase assay in DEL cells.
Results: Our results show that TFP did not significantly affect cell viability of human normal lymphocytes, whereas it reduced the cell viability of the ALK+ T-cell lymphoma cells, DEL and SUP-M2, in concentration- and time-dependent manners. TFP also induced cell cycle arrest at G0/G1 phase, and caused cell apoptosis in these cells. These effects could be explained by the involvement of NPM-ALK and its downstream cell survival regulatory proteins.
Conclusions: Our results demonstrate for the first time that TFP is capable of inducing degradation and inhibition of kinase activity of NPM-ALK, as well as the increased level of phospho-NPM-ALK, leading to the inhibition of cell growth in ALK+ ALCL cells. TFP may have a potential to be utilized to treat this aggressive lymphoma.
Introduction
Anaplastic lymphoma kinase-expressing anaplastic large cell lymphoma (ALK+ ALCL) is a rare type of cancer [Citation1]. It is characterized by the expression of the chimeric protein nucleophosmin-ALK (NPM-ALK) that results from the t(2; 5)(p23; q35) chromosomal translocation [Citation2,Citation3]. ALK+ ALCL is an aggressive type of malignant T-cell lymphoma which frequently affects children and young adults. The combination chemotherapy cyclophosphomide, adriamycin, vincristine, and prednisone (CHOP)-based therapy has been commonly utilized to treat ALK+ ALCL. Alas, several studies demonstrated that this approach was associated with 30–40% relapse rate and subsequent death [Citation4–6]. For another, inhibitors of ALK such as Crizotinib and AP2611 have been applied naturally but with resistance occurred gradually [Citation7,Citation8]. These unsatisfactory treatments indicate that new therapeutics is still needed urgently.
Trifluoperazine (TFP), a phenothiazine derivative antipsychotic drug [Citation9], has been clinically utilized to treat schizophrenia and other mental disorders related to blockade of the dopamine receptor [Citation10]. In addition to the antipsychotic drug, TFP is also a calmodulin antagonist that interferes with Ca2+-calmodulin interactions, thereby blocking Ca2+-calmodulin-dependent cellular events. Recently, it has been demonstrated that TFP can be a potential antitumor agent [Citation11] because it is capable of inhibiting DNA synthesis and cell proliferation in a concentration- and time-dependent manner [Citation11–13]. Since TFP has potential anticancer activity in different types of cancer cells, whether TFP could affect ALK+ ALCL cell survival is not clear. In the present study, we investigated the effects of TFP in ALK+ ALCL. We found that TFP suppressed cell proliferation, and caused cell apoptosis and G0/G1-phase cell cycle arrest. These findings could be explained by TFP-induced degradation and inhibition of kinase activity of NPM-ALK, resulting in alterations of cell survival regulatory proteins downstream of NPM-ALK. These findings suggest that TFP could be potentially utilized for the treatment of this aggressive malignancy.
Materials and methods
Antibodies, cells, and reagents
Primary antibodies against p21, p27, AKT, pAKT, ERK, pERK, STAT3, pSTAT3, Bax, Bcl-2, Bcl-XL (Abcam, Cambridge, MA, U.S.A.), CDK2 (Cell Signaling Technology, Beverly, MA, U.S.A.), ALK (Dako, Carpinteria, CA, U.S.A.), pALK (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) were used. Secondary horseradish peroxidase-conjugated antibodies used were goat-rabbit (Jackson Immunoresearch Laboratories, West Grove, PA, U.S.A.).
The ALK+ T-cell ALCL cell lines DEL and SUP-M2 were cultured in humidified 5% CO2 atmosphere at 37°C and grown in RPMI-1640 (Gibco Laboratories, Grand Island, NY, U.S.A.) supplemented with 10% fetal bovine serum (FBS, Hyclone, Logan, UT, U.S.A.), and penicillin–streptomycin (Sigma-Aldrich, St. Louis, MO, U.S.A.).
Cell culture and separation of normal lymphocytes from human peripheral blood
The PBMCs were separated by Ficoll-Paque Plus reagent (GE Healthcare Biosciences, Piscataway, NJ, U.S.A.) with density gradient centrifugation from peripheral blood collected from healthy donors into sterile tubes, according to the manufacturer’s instructions after approval of the local ethic committee at the East China University of Science and Technology. Lymphocytes obtained were suspended in RPMI-1640 medium supplemented with 10% FBS, and cultured at 37°C in humidified 5% CO2 atmosphere.
Cell viability assay
Trypan blue (TB) staining was performed to determine the cell growth effects after treatment with TFP. Cells were harvested and seeded in 96-well culture plate at a concentration of 1.5 × 105 cells/ml in 100 μl cell culture medium with 0, 5, and 10 μM TFP. After incubation at 37°C for 24, 48, and 72 hours, 10 μl TB was added to each well. Then, total cells and stained cells were counted within the grids on the hemacytometer in 5 minutes, and the survival rate of the cells was calculated as the number of viable cells divided by the total number of cells. If cells take up TB, they are considered non-viable. Results were expressed as percentage of the controls, which were arbitrarily assigned 100% viability.
Cell cycle analysis
DEL and SUP-M2 cells (3 × 105 cells/ml) were plated in 6-well culture plate, treated for 24 and 48 hours with different concentrations of TFP, and then harvested in 15 ml centrifugal tubes. Cells were washed twice with cold PBS and then resuspended in 1.0 ml of 75% (v/v) ethanol after 100 μl PBS was added into the tube, and left overnight at −20°C. After that, the cells were washed twice with cold PBS and resuspended in cold PI solution (50 μg/ml) containing RNase A (0.1 mg/ml) in PBS (pH 7.4) for 30 minutes in the dark. Cell cycle was determined by a FACSCalibur Flow Cytometer (BD Biosciences, San Jose, CA, U.S.A.) with ModFit LT software (Verity Software House, Topsham, ME, U.S.A.).
Apoptosis assay
DEL and SUP-M2 cells were plated at a concentration of 2 × 105 cells/well in 12-well plate in RPMI-1640 medium for 24 or 48 hours and treated with different concentrations of TFP as described above. Thereafter, cells were harvested in tubes and Annexin V-FITC/PI Apoptosis Detection Kit (Dojindo Laboratories, Tokyo, Japan) was used according to the manufacturer’s protocol. Data acquisition and analysis were performed by means of a FACSCalibur Flow Cytometer with FlowJo software (http://www.flowjochina.com/).
Western blot analysis
To examine the effects of TFP on cell cycle and apoptosis-related protein, DEL cells were plated at a density of 3 × 105 cells/ml. After 24 and 48 hours treatment of TFP, respectively, the cells were harvested as described above. The cells were washed twice with cold PBS and then lysed in RIPA lysis buffer (Beyotime Institute of Biotechnology, Changzhou, China). Protein concentrations were measured using Quick Start™ Bradford Dye Reagent (Bio-Rad, Hercules, CA, U.S.A.). A quantity of 50 μg total protein per lane was separated by 8–12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Amersham Hybond-P polyvinylidene fluoride membranes (GE Healthcare, Buckinghamshire, U.K.). The membranes were blocked with 5% milk powder in 0.05% Tween-TBS, incubated with the specific antibodies, including p21, p27, CDK2, Bax, Bcl-XL, and Bcl-2 rabbit monoclonal antibodies. Detection of the target proteins on the membranes was performed using the ECL Western Blotting Detection Reagents. Signal density was detected using the MF-ChemiBIS Family including gel capture software (DNR Bio-Image system, Ltd., Jerusalem, Israel) and quantitatively analyzed by densitometry using the Image J software.
Reverse transcriptase polymerase chain reaction (RT-PCR) and real-time quantitative PCR (Q-PCR)
DEL cells were treated with TFP, and then total RNA was extracted by RNAiso Plus Kit (Takara Bio Inc., Otsu, Japan) according to instructions. By TaKaRa PrimeScript® RT reagent Kit with gDNA Eraser (Takara Bio, Inc., Shiga, Japan), the total RNA was reverse transcribed into cDNA after quantitation. The PCR reaction was performed according to the manufacturer’s instructions with the use of the following primer pairs: (1) CDK2 forward, 5′-TCTATGCCTGATTACAAGCCAA-3′; reverse, 5′-ATAACAAGCTCCGTCCATC-3′. (2) p21 forward, 5′-CGATGGAACTTCGACTTTGTCA-3′; reverse, 5′-GCACAAGGGTACAAGACAGTG-3′. (3) p27 forward, 5′-AACGTGCGAGTGTCTAACGG-3′; reverse, 5′-CCCTCTAGGGGTTTGTGATTCT-3′. (4) 18S forward, 5′-CGGCTACCACATCCAAGGAAG-3′; reverse, 5′-AGCTGGAATTACCGCGGCT-3′. All of these primer pairs can be used for both RT-PCR and Q-PCR. Results of Q-PCR were measured by BioRadCFXManager software and expressed as percentage of the controls, which were arbitrarily assigned 100% viability, and results of RT-PCR were quantified by gray level.
NPM-ALK tyrosine kinase activity assay
NPM-ALK tyrosine kinase activity was detected by a commercially available kit (Takara Bio, Otsu, Shiga, Japan) [Citation3]. DEL cell lysates were prepared and ALK antibody was used for immunoprecipitation. Agarose beads conjugated with protein A/G were added. Kinase reactions were initiated by adding ATP-2Na into immobilized wells coated with peptide substrate. Then, HRP-conjugated antiphosphotyrosine (PY20) antibody was added to each well and developed by the addition of horseradish peroxidase and TMBZ. The reaction was stopped with 1 N H2SO4 and absorbance was measured at 450 nm in a microplate reader (Tecan GENios Pro, Tecan Group Ltd. Maennedorf, Switzerland). The ATP-free reaction was used as a negative control.
Statistical analysis
Results were expressed as means ± SD. Statistical analysis was performed using the Student’s t-test. Values of p < 0.05 were considered to be statistically significant.
Results
TFP decreases cell viability in ALK+ ALCL cells.
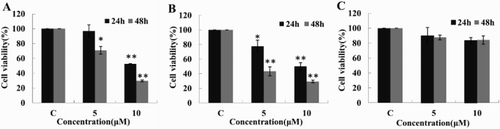
Using TB staining, we measured changes in cell viability. As shown in (A,B), TFP induced a time- and concentration-dependent decrease in the viability of DEL and SUP-M2 cells. At 48 hours, IC50 was 7.5 μM for DEL and 4.5 μM for SUP-M2 cells. Of important note is that TFP did not induce a significant decrease in the viability of human normal lymphocyte cells ((C)). Therefore, TFP obviously reduced cell viability in the two ALK+ ALCL cells.
Figure 1. The effect of TFP on ALK+ ALCL cell viability. (A) Cell viability of DEL cells with or without TFP treatment at 24 and 48 hours. (B) Cell viability of SUP-M2 cells with or without TFP treatment at 24 and 48 hours. (C) Cell viability of human normal lymphocyte cells with TFP or without TFP treatment at 24 and 48 hours. Cell survival was determined by TB assay. The experiments were repeated three times with consistent results. *p < 0.05. **p < 0.01.
TFP inhibits cell cycle progression at G0/G1 phase and induces apoptosis in ALK+ ALCL cells
To investigate whether the decrease in cell viability was due to the cell cycle arrest, flow cytometric analysis was performed after treatment with TFP. As shown in (A,B), TFP could affect the distribution of the cell cycle in SUP-M2 and DEL cells. Although changes in the cell cycle progression were not detected at 24 hours, the proportion of G0/G1 phase was significantly increased to 74.39% while that of control cells decreased to 43.52% at 48 hours in SUP-M2 cells. Similarly, the proportion of cells in the G0/G1 phase increased from 25.58 to 45.58% in DEL cells ((A,B)). The data showed that TFP could induce G0/G1 arrest in the two ALK+ ALCL cells.
Figure 2. Cell cycle arrest and apoptosis induced by TFP. (A, B) Cell cycle distribution of DEL and SUP-M2 cells with or without TFP treatment at 24 and 48 hours. The two cell lines after treated with TFP exhibited cell proportions in the G0/G1 phase increased significantly at 48 hours while no change at 24 hours. Representative results of three separate experiments are shown. (C, D) Cell apoptosis was assessed by Annexin V-FITC/PI Apoptosis Detection Kit. The average percent of AnnexinV-FTIC positive cells is shown to increase gradually in time- and concentration-dependent manners. *Significant difference at p < 0.05. **Extremely significant difference at p < 0.01. The experiments were repeated three times.
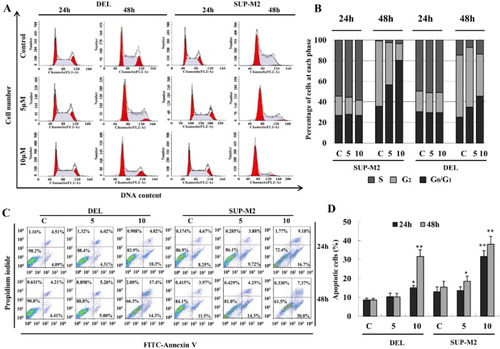
To examine the effect of TFP on cell apoptosis, flow cytometric analysis of Annexin V binding was performed. Cells were considered apoptotic when stained for Annexin V only or when stained simultaneously for Annexin V and PI. As shown in (C,D), apoptotic cells increased to 15.12 and 31.7% in DEL cells treated with TFP at 24 and 48 hours, respectively. Likewise, the apoptotic cells in SUP-M2 cells increased to 25.88 and 38.17% at 24 and 48 hours, respectively, after treatment with TFP.
Taken together, the data shows that the decrease in cell viability of ALK+ ALCL cells could have resulted from cell cycle arrest and apoptosis after treatment with TFP.
TFP induces cell cycle arrest and apoptosis via the regulation of NPM-ALK signaling
Thereafter, we investigated the effects of TFP on G0/G1 phase checkpoint molecules such as p21, p27, CDK2, CDK4, and Cyclin E1. Cells were harvested for RT-PCR and real-time PCR analysis at 24 hours after treated with TFP. P21 and p27 mRNA levels were increased while CDK2, CDK4 and Cyclin E1 mRNA levels were decreased significantly ((A,B)). In addition, Western Blot analysis demonstrated that TFP-induced upregulation of p21 and p27, and downregulation of CDK2 protein levels at 24 and 48 hours after treatment of TFP, respectively. ((C)). These alterations are consistent with the occurrence of G0/G1 phase cell cycle arrest. The members of the Bcl-2 family play an important role in the regulation of apoptosis. The ratios Bcl-XL/Bax and Bcl-2/Bax determine the fate of a cell rather than the absolute concentration of either [Citation14]. At 24 and 48 hours after treated with TFP, Bcl-XL expression was significantly reduced. The levels of Bcl-2 was decreased and then returned to the control level. While Bax expression had no obvious change and then was markedly increased ((D)). Altogether, the treatment of TFP originated a significant descent in the Bcl-XL/Bax and Bcl-2/Bax ratios. Thus, the imbalance between Bcl-XL, Bcl-2, and Bax expressions is believed to play a significant role in TFP-induced apoptosis.
Figure 3. Effects of TFP on cell cycle and apoptosis regulatory molecules. (A, B) Cells were harvested at indicated time points for RT-PCR and Q-PCR reactions. Both p21 and p27 mRNA levels were increased significantly while CDK2, CDK4 and Cyclin E1 were decreased at 24 hours after treatment with TFP. The data represent results from one of three independent experiments. (C) Western Blot of cell cycle checkpoint proteins after treatment with TFP. P21 and p27 protein levels were increased at both 24 and 48 hours, whereas CDK2 was decreased remarkably at 48 hours. (D) Western blot of apoptosis-related proteins. Bax demonstrated a relatively stable expression while Bcl-XL was decreased continually at both 24 and 48 hours, and Bcl-2 was reduced to a lower level at 24 hours but was increased significantly at 48 hours. (E) Western blot of NPM-ALK and related downstream survival proteins after treatment with TFP. At 24 and 48 hours, NPM-ALK was degraded while phosphorylated NPM-ALK showed an elevation. Phospho-AKT had no change with phospho-ERK was increased and phospho-STAT3 was decreased at 24 hours. At 48 hours, phospho-ERK, AKT and STAT3 were increased significantly.
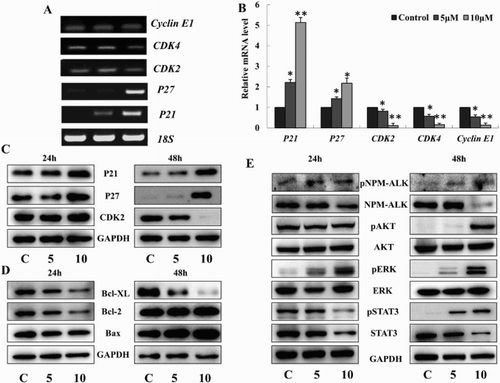
It is remarkable that the tyrosine kinase activity of NPM-ALK reduced gradually with the increased concentration of TFP (). However, as shown in (E), TFP increased the level of phosphorylated ERK with no effect of basal level of ERK at 24 and 48 hours. Neither phospho-AKT nor basal level of AKT changed significantly at 24 hour after treatment of TFP while the levels of phospho-AKT were increased compared to the control at 48 hours. In contrast, the basal levels of STAT3 were reduced at 24 and 48 hours after treated with TFP, while phospho-STAT3 was decreased and then increased compared to the corresponding controls. Altogether, TFP-induced degradation of STAT3 at both 24 and 48 hours and increased the levels of phospho-AKT and phospho-STAT3 at 48 hours ((E)). Most important of all, NPM-ALK phosphorylation was significantly increased while the basal level of NPM-ALK was decreased in a concentration- and time-dependent manner accompanied with the decrease of the basal level of STAT3 ((E)). It appears that TFP-induced cell cycle arrest and apoptosis of ALK+ ALCL cells via degradation and inhibition of kinase activity of NPM-ALK despite increased NPM-ALK, STAT3, and AKT phosphorylation ().
Figure 4. Decreased tyrosine kinase activity of NPM-ALK induced by TFP. Tyrosine kinase activity of NPM-ALK shows a gradual decrease after treatment of TFP for 48 hours. *Significant difference at p < 0.05. The experiments were repeated three times.
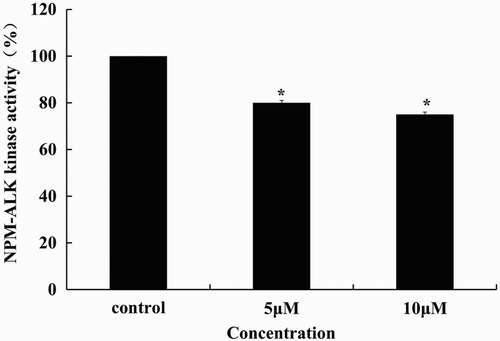
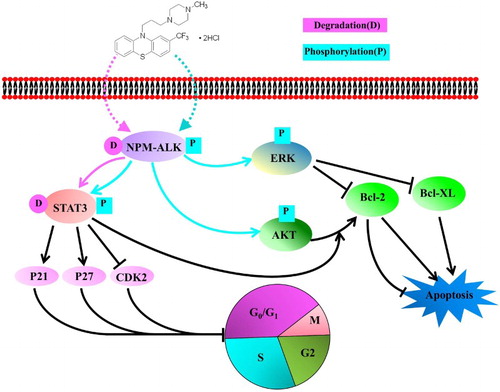
Figure 5. The mode of action of TFP in ALK+ ALCL cells. TFP inhibited the cell viability via induction of the degradation and decrease of tyrosine kinase activity of NPM-ALK. The cell apoptosis occurs through the activation of ERK pathway while G0/G1 cell cycle was arrested through the degradation of STAT3.
Discussion
The previous study has shown that phenothiazines suppress proliferation and induce apoptosis in cultured leukemic cells without any influence on the viability of normal lymphocytes [Citation15]. TFP, as one of the phenothiazine derivates, exhibits potential anticancer activity in different types of cancer cells. For instance, it induces cell apoptosis in A549 cells [Citation16], blocks the cell cycle progression in mouse leukemia cells [Citation17,Citation18]. In the present study, we investigated the effects of TFP on cell viability of ALK+ ALCL cells. Our results show that TFP has no effect on human normal lymphocyte cells while exhibits obvious cell viability inhibition in ALK+ ALCL. It is the first time to report that TFP could induce cell cycle arrest at G0/G1 phase and cell apoptosis in ALK+ ALCL cells.
NPM-ALK is a constitutively activated protein tyrosine kinase with significant oncogenic potential [Citation3]. To induce its oncogenic effects, NPM-ALK interacts with several proteins that regulate cell survival and growth [Citation1,Citation3]. From our results, the expression and tyrosine kinase activity of NPM-ALK was reduced dramatically while phosphorylation of NPM-ALK was markedly increased after treatment of TFP. Subsequently, the downstream ERK signaling pathway is activated in response to increased phosphorylation of NPM-ALK. The previous study has shown that the activation of ERK signaling suppressed the expression of Bcl-2 and Bcl-XL [Citation19]. Bcl-2, Bcl-XL and Bax proteins are very important regulators of apoptosis [Citation20]. Our results indicated that the treatment of TFP originated a significant descent in the Bcl-XL/Bax and Bcl-2/Bax ratios. Since the ratios Bcl-XL/Bax and Bcl-2/Bax determine the fate of a cell rather than the absolute concentration of either [Citation14], which suggests that TFP induces apoptosis via the altering these apoptosis-related proteins. It was noted that TFP treatment increased the levels of phospho-AKT and phospho-STAT3 at 48 hours ((E)). Generally, activation of STAT3 and AKT phosphorylation is thought to be determining factors in suppressing apoptosis [Citation21–24]. Therefore, from our data, the cell apoptosis induced by TFP was dominantly regulated by ERK pathway. For another, NPM-ALK causes enhanced transcription and/or stabilization of STAT3 mRNA levels, as well as increased phospho-STAT3 and total STAT3 levels in ALK+ ALCL cells [Citation25]. In our study, treatment of TFP-induced degradation of STAT3 at both 24 and 48 hours ((E)). We cannot exclude the possibility that the decreased levels of STAT3 were mediated by the degradation of NPM-ALK induced by TFP. Additionally, the cell cycle regulatory proteins p21, p27, and CDK2 are directly regulated by STAT3 [Citation26–28]. Our data demonstrated that the cell cycle arrest induced by TFP was related to the degradation of STAT3. Taken together, it is likely that TFP inhibited the cell viability in ALK+ ALCL cells via the involvement of NPM-ALK and its downstream cell survival regulatory proteins ().
In conclusion, we explored the inhibitory effect of TFP on ALK+ ALCL cells. Our results indicate that TFP inhibits the cell viability in ALK+ ALCL cells via induction of the degradation and decrease of tyrosine kinase activity of NPM-ALK, as well as the increased level of phospho-NPM-ALK. Moreover, TFP induces apoptosis through the activation of ERK while G0/G1 cell cycle arrest through the degradation of STAT3, thus leading to cellular growth retardation. Our results suggest that TFP may have a potential to be utilized to treat this aggressive lymphoma.
Acknowledgements
L.H. and P.S. designed research; L.H., X.Z., J.W., S.W., H.M.A. and P.S. performed research and collected data; L.H. and P.S. interpreted, analyzed data and wrote the paper. The contents of this paper are solely the responsibility of the authors and do not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Amin HM, Lai R. Pathobiology of ALK+ anaplastic large-cell lymphoma. Blood. 2007;110:2259–2267. doi: 10.1182/blood-2007-04-060715
- Greenland C, Touriol C, Chevillard G, et al. Expression of the oncogenic NPM-ALK chimeric protein in human lymphoid T-cells inhibits drug-induced, but not Fas-induced apoptosis. Oncogene. 2001;20:7386–7397. doi: 10.1038/sj.onc.1204870
- Shi P, Lai R, Lin Q, et al. IGF-IR tyrosine kinase interacts with NPM-ALK oncogene to induce survival of T-cell ALK + anaplastic large-cell lymphoma cells. Blood. 2009;114:360–370. doi: 10.1182/blood-2007-11-125658
- Brugieres L, Deley MC, Pacquenment H, et al. CD30+ anaplastic large-cell lymphoma in children: analysis of 82 patients enrolled in two consecutive studies of the French society of pediatric oncology. Blood. 1998;92:3591–3598.
- Brugières L, Quartier P, Le Deley MC, et al. Relapses of childhood anaplastic large-cell lymphoma: treatment results in a series of 41 children – a report from the French society of pediatric oncology. Ann Oncol. 2000;11:53–58. doi: 10.1023/A:1008352726155
- Laver JH, Kraveka JM, Hutchison RE, et al. Advanced-stage large-cell lymphoma in children and adolescents: results of a randomized trial incorporating intermediate-dose methotrexate and high-dose cytarabine in the maintenance phase of the APO regimen: a pediatric oncology group phase Ш trial. J Clin Oncol. 2005;23:541–547. doi: 10.1200/JCO.2005.11.075
- Ceccon M, Mologni L, Giudici G, et al. Treatment efficacy and resistance mechanisms using the second-generation ALK inhibitor AP26113 in human NPM-ALK-positive anaplastic large cell lymphoma. Mol Cancer Res. 2015;13:775–783. doi: 10.1158/1541-7786.MCR-14-0157
- Ceccon M, Mologni L, Bisson W, et al. Crizotinib-resistant NPM-ALK mutants confer differential sensitivity to unrelated Alk inhibitors. Mol Cancer Res. 2013;11:122–132. doi: 10.1158/1541-7786.MCR-12-0569
- Shin SY, Kim SY, Kim JH, et al. Induction of early growth response-1 gene expression by calmodulin antagonist trifluoperazine through the activation of Elk-1 in human fibrosarcoma HT1080 cells. J Biol Chem. 2001;276:7797–7805. doi: 10.1074/jbc.M009465200
- Feldkamp MD, Gakhar L, Pandey N, et al. Opposing orientations of the anti-psychotic drug trifluoperazine selected by alternate conformations of M144 in calmodulin. Proteins. 2015;83:989–996. doi: 10.1002/prot.24781
- Kang S, Hong J, Lee JM, et al. Trifluoperazine, a well-known antipsychotic, inhibits glioblastoma invasion by binding to calmodulin and disinhibiting calcium release channel IP3R. Mol Cancer Ther. 2017;16:217–227. doi: 10.1158/1535-7163.MCT-16-0169-T
- Shin SY, Choi BH, Kim JR, et al. Suppression of P-glycoprotein expression by antipsychotics trifluoperazine in Adriamycin-resistant L1210 mouse leukemia cells. Eur J Pharm Sci. 2006;28:300–306. doi: 10.1016/j.ejps.2006.03.002
- Gangopadhyay S, Karmakar P, Dasgupta U, et al. Trifluoperazine stimulates ionizing radiation induced cell killing through inhibition of DNA repair. Mutat Res. 2007;633:117–125. doi: 10.1016/j.mrgentox.2007.05.011
- Gross A. Bcl-2 proteins: regulators of the mitochondrial apoptotic program. IUBMB Life. 2001;52:231–236. doi: 10.1080/15216540152846046
- Zhelev Z, Ohba H, Bakalova R, et al. Phenothiazines suppress proliferation and induce apoptosis in cultured leukemic cells without any influence on the viability of normal lymphocytes. Cancer Chemoth Pharm. 2004;53:267–275. doi: 10.1007/s00280-003-0738-1
- Chen Q, Wu LJ, Wu YQ, et al. Molecular mechanism of trifluoperazine induces apoptosis in human A549 lung adenocarcinoma cell lines. Mol Med Rep. 2009;2:811–817.
- Hait HN, Grais L, Benz C, et al. Inhibition of growth of leukemic cells by inhibitors of calmodulin: phenothiazines and melittin, cancer. Chemoth Pharm. 1985;14:202–205.
- Ganapathi R, Yen A, Grabowski D, et al. Role of the calmodulin inhibitor trifluoperazine on the induction and expression of cell cycle traverse perturbations and cytotoxicity of daunorubicin and doxorubicin (Adriamycin) in doxorubicin-resistant P388 mouse leukaemia cells. Br J Cancer. 1986;53:561–566. doi: 10.1038/bjc.1986.88
- Wang Z, Nishikawa Y, Wang M, et al. Induction of apoptosis via mitogen-activated protein kinase pathway by a K vitamin analog in rat hepatocytes. J Hepatol. 2002;36:85–92. doi: 10.1016/S0168-8278(01)00230-6
- Xu HY Y, Guan XL, et al. Expression of regulating apoptosis gene and apoptosis index in primary liver cancer. World J Gastroenterol. 2000;6:721–724. doi: 10.3748/wjg.v6.i5.721
- Staber PB, Vesely P, Haq N, et al. The oncoprotein NPM-ALK of anaplastic large cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood. 2007;110:3374–3383. doi: 10.1182/blood-2007-02-071258
- Hamedani FS, Cinar M, Mo Z, et al. Crizotinib (PF-2341066) induces apoptosis due to downregulation of pSTAT3 and BCL-2 family proteins in NPM-ALK+ anaplastic large cell lymphoma. Leukmia Res. 2014;38:503–508. doi: 10.1016/j.leukres.2013.12.027
- Mochizuki T, Furuta S, Mitsushita J, et al. Inhibition of NADPH oxidase 4 activates apoptosis via the AKT/apoptosis signal-regulating kinase 1 pathway in pancreatic cancer PANC-1 cells. Oncogene. 2006;25:3699–3707. doi: 10.1038/sj.onc.1209406
- Ruchatz H, Coluccia AML, Stano P, et al. Constitutive activation of Jak2 contributes to proliferation and resistance to apoptosis in NPM/ALK-transformed cells. Ex Hematol. 2003;31:309–315. doi: 10.1016/S0301-472X(03)00007-9
- Zamo A, Chiarle R, Piva R, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene. 2002;21:1038–1047. doi: 10.1038/sj.onc.1205152
- Humbert L, Ghozlan M, Canaff L, et al. The leukemia inhibitory factor (LIF) and p21 mediate the TGFβ tumor suppressive effects in human cutaneous melanoma. BMC Cancer. 2015;15:3635–3616. doi: 10.1186/s12885-015-1177-1
- Zhao D, Besser AH, Wander SA, et al. Cytoplasmic p27 promotes epithelial–mesenchymal transition and tumor metastasis via STAT3-mediated Twist1 upregulation. Oncogene. 2015;34:5447–5459. doi: 10.1038/onc.2014.473
- Zhao J, Lin W, Cao Z, et al. Total alkaloids of Rubus aleaefolius poir. inhibit the STAT3 signaling pathway leading to suppression of proliferation and cell cycle arrest in a mouse model of hepatocellular carcinoma. Oncol Rep. 2013;30:1309–1314. doi: 10.3892/or.2013.2585