
ABSTRACT
Background and aim of work: Sickle cell disease (SCD) is an inherited disease of the beta globin gene. The βS globin gene haplotypes are Senegal, Benin, Bantu, Cameroon, Arab-Indian and atypical haplotypes. In SCD, stroke is a life-threatening event in both adults and children. In light of paucity of studies on βS globin gene haplotypes in Egypt, we aimed to determine βS globin gene haplotypes in children with SCD and study their impact on stroke risk.
Methods: Fifty-two SCD patients were included in the study, they were 26 males and 26 females with age range from 3 to 18 years old. The PCR-RFLP technique was used for the determination of βS globin gene haplotypes. Transcranial Doppler (TCD) was done to identify patients at risk of stroke.
Results: Benin/Benin was the most prevalent haplotype detected in 50% followed by Benin/Bantu in 30.8% of studied patients. TCD study showed that 14/52 (26.9%) patients had abnormally high TCD flow velocities (TCD velocities ≥170 cm/s) and thus considered high stroke risk group, whereas 38/52 (73.1%) patients had TCD flow velocities <170 cm/s and are considered low stroke risk group. Stroke risk was not found to be associated with βS globin gene haplotype (p = .532).
Conclusion: This study provides a relevant contribution to our understanding of the anthropological and historical background of the population in Egypt where Benin haplotype is the commonest βS globin gene haplotype and homozygous Benin/Benin is associated with higher stroke risk than other haplotypes.
Introduction
Sickle cell disease (SCD) is a genetic hemoglobin disorder, caused by amino acid substitution in the β globin gene [Citation1]. Different βS globin gene haplotypes have been identified and are named according to the geographic region or ethnic group in which they were originally identified, i.e. Senegal (SEN), Benin (BEN), Bantu or Central African Republic (CAR), Cameroon (CAM) and Arab-Indian (ARAB) [Citation2]. Other less common haplotypes, known as atypical haplotypes (ATP), are also identified [Citation3].
SCD presentation and severity are variable among patients. This individual heterogeneity may be related to βS globin gene haplotypes. Senegal and Arab-Indian haplotypes are characterized by mild disease, lower occurrence of organ damage. Benin and Cameroon haplotypes are associated with intermediate disease severity. While Bantu haplotype is associated with severe clinical course [Citation2,Citation4,Citation5].
Stroke caused by vaso-occlusion is a serious complication of SCD [Citation6]. The risk of stroke in SCD patients is the highest during the first decade, and is most significant at the age of two to five years [Citation7]. Patients with SCD have 250 times higher risk of developing stroke than for those without SCD [Citation8]. Almost 11% of SCD patients have clinically apparent stroke before the age of 20 years and the risk increases to 24% by the age of 45 years [Citation7]. Some cases may have silent infarctions which often go unnoticed but can cause significant neurological damage and disability and are reported to be present in a further 17% to 25% of patients with SCD [Citation9]. In Africa, the rates are even higher due to added risk factors including prevalence of the Bantu βS globin gene haplotype, low hemoglobin level and leucocytosis [Citation8].
Stroke risk in SCD patients can be assessed by Transcranial Doppler (TCD) screening. TCD measures blood flow velocity in the arteries supplying the brain. High blood flow velocity in one or more major arteries indicates vessel narrowing and thus can predict increased stroke risk, and therefore preventive treatment could be started prior to first stroke [Citation10].
In 2009, SCD has been recognized by the United Nations General Assembly as a global public health concern being associated with high rates of morbidity and mortality and due the social and economic burden of the disease [Citation11].
Worldwide, 20–25 million people are affected with SCD; 50–60% of whom live in Africa [Citation12]. In Egypt, SCD distribution varies across different parts of the country where variable rates of 0.38% in the coastal areas to 9.0% in the New Valley oases have been reported, HbS carrier rates vary from 9% to 22% [Citation13]. Previous studies were done to determine the βS globin gene haplotypes in a limited number of Egyptian patients, Benin βS globin gene haplotype is the most common haplotype identified in the previous two studies, including 25 and 14 patients [Citation14,Citation15].
Data regarding βS globin gene haplotypes in Egyptian SCD patients are scarce requiring more studies to validate previously published data. In the present study, we determined βS globin gene haplotypes in 52 children with SCD and evaluated its impact on stroke risk.
Subjects and methods
The study included 52 SCD patients regularly attending the Pediatric Hematology Clinic, New Children Hospital, Cairo University. They were 26 males (50%) and 26 females (50%). Their ages ranged between 3 and 18 years (median 11.5 years). Diagnosis of SCD was established using hemoglobin electrophoresis and/or high-performance liquid chromatography; 22/52 cases (42.3%) had Hb S/S, 30/52 cases (57.7%) had Hb S/β. Clinical and hematological data were obtained from the patients’ medical records. Patients with sickle cell trait (HbA/S) or any risk factor of pediatric stroke including congenital heart disease, prothrombotic disorders, vascular malformation and autoimmune disease were excluded. All participants and/or their guardians gave informed consent before recruitment. The study was approved by the Ethics Committee, Cairo University and is conducted in accordance with the ethics guidelines of the Declaration of Helsinki.
Studied patients were subjected to TCD screening to assess stroke risk, and 2 mL of EDTA-anticoagulated venous blood samples were collected from each patient for the determination of βS globin gene haplotypes.
Transcranial Doppler
TCD examination was done using 1.5 Tesla MRI. Stroke risk was determined according to mean blood flow velocity in cerebral arteries. Patients with blood flow velocity <170 cm/s were considered at low risk of stroke [Citation16].
Determination of βS globin gene haplotypes
Genomic DNA was extracted from whole blood samples using the Gene JET Whole Blood Genomic DNA purification Mini kit (Thermo Scientific, Waltham, MA) according to the manufacturer’s instructions. Isolated DNA was stored at −70°C until used for polymerase chain reaction (PCR) amplification.
Six regions around and within the β globin gene cluster were amplified by PCR. PCR reactions were composed of total volume of 25 μL containing 150 ng genomic DNA, 2X Taq Green PCR Master Mix, 25 pM each of forward and reverse primers (Biosearch Technologies, Novata, CA). PCR amplification was carried out in the DNA thermal cycler (PTC programmable thermal controller; MJ Research, Watertown, MA). Amplification conditions were initial denaturation at 92°C for 5 min followed by 30 cycles of denaturation at 93°C for 1 min, annealing at * (*shown in ) for 1 min, and extension at 72°C for 1.5 min, with final extension for 3 min at 72°C [Citation17]. The amplified PCR products were visualized by 3% agarose gel electrophoresis under ultraviolet light. The studied regions within the β globin gene cluster and primers’ sequence used are shown in .
Table 1. The studied regions within the β globin gene cluster, primers’ sequence and annealing temperature used in PCR.
Digestion of the amplified products by specific restriction enzyme was done as follows: 10 μL of the amplified product was mixed with 1 μL restriction enzyme (Thermo Scientific, Waltham, MA) and the mixture was incubated for 10 min at 37°C. The product was analyzed by gel electrophoresis using 3% agarose gel (Promega, Madison, WI). The separated fragments were stained with ethidium bromide and visualized along with a 50-base pair (bp) ladder (MBI Fermentas, Vilnius, Lithuania) as a size marker using transilluminator (Bio-Rad). The restriction enzymes used and the resulting fragments bp lengths are shown in .
Table 2. Restriction enzymes used and RFLP product size [Citation19].
The obtained fragments lengths were interpreted as shown in to determine the βS globin gene haplotype. Common haplotypes are: Senegal (SEN), Benin (BEN), Bantu or Central African Republic (CAR), Cameroon (CAM) and Arab-Indian (ARAB) and the less common haplotypes are known as atypical haplotypes (ATP) [Citation18,Citation19].
Table 3. Determination of the βS globin gene haplotype.
Statistical analysis
Analysis was done using the statistical package SPSS (Statistical Package for the Social Science) version 23. Data were summarized using mean, standard deviation, median, minimum and maximum in quantitative data and using frequency (count) and relative frequency (percentage) for categorical data. Comparisons between quantitative variables were done using the non-parametric Kruskal–Wallis and Mann–Whitney tests). For comparing categorical data, the Chi square (χ2) test was performed. The Exact test was used instead when the expected frequency is less than 5. p-Values less than .05 were considered as statistically significant.
Results
Benin βS globin gene haplotype whether homozygous or heterozygous was found in 43/52 (82.6%) of patients. Homozygous Benin/Benin βS globin gene haplotype was the most common haplotype detected in half of the studied patients 26/52 (50%) followed by heterozygous Benin/Bantu in 16/52 (30.8%) patients, while other βS globin gene haplotype represented a minority in the studied patients, as shown in ( and ).
Figure 1. Homozygous Benin/Benin βS haplotype ----++/----++. An agarose gel electrophoresis of RFLP analysis by Hind II 5′ to ε gene demonstrates homozygous negative result (−/−) in number 1, XmnI 5′ to Gγ gene demonstrates homozygous negative result (−/−) in number 2, Hind III within IVS II Gγ gene demonstrates homozygous negative result (−/−) in number 3, Hind III within IVS II Aγ gene demonstrates homozygous negative result (−/−) in number 4, Hind II 3′ to ψβ gene demonstrates homozygous positive result (+/+) in number 5, Ava II within IVS II β gene demonstrates homozygous positive result (+/+) in number 6.
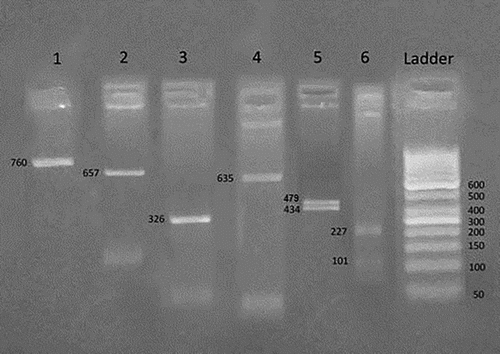
Figure 2. Heterozygous Benin/Bantu βS haplotype (----++)/(+----+). An agarose gel electrophoresis of RFLP analysis by Hind II 5′ to ε gene demonstrates positive heterozygous result (+/−) in number 1, XmnI 5′ to Gγ gene demonstrates homozygous negative result (−/−) in number 2, Hind III within IVS II Gγ gene demonstrates homozygous negative result (−/−) in number 3, Hind III within IVS II Aγ gene demonstrates homozygous negative result (−/−) in number 4, Hind II 3′ to ψβ gene demonstrates positive heterozygous positive result (+/−) in number 5, Ava II within IVS II β gene demonstrates homozygous positive result (+/+) in number 6.
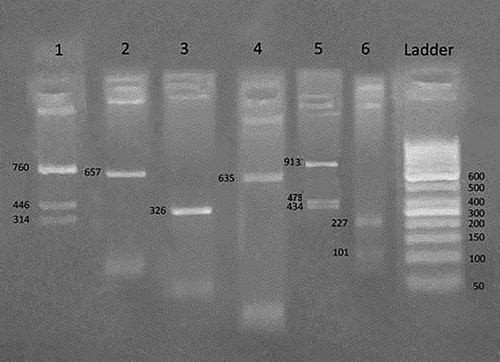
Table 4. Frequency of βS globin gene haplotypes in studied SCD patients.
TCD screening results showed that 38/52 (73.1%) patients were at low risk of developing stroke while 14/52 (26.9%) patients were high stroke risk group ().
Table 5. Stroke risk in the studied SCD patients.
No correlation was found between different βS globin gene haplotype and stroke in SCD as shown in .
Table 6. Stroke risk among different βS globin gene haplotypes.
No statistically significant difference was found between S/S patients and S/β patients as regards distribution of βS globin gene haplotype or stroke risk ().
Table 7. Stroke risk among Benin and non-Benin/Benin βS globin gene haplotypes in different patient groups.
Forty-four patients were receiving blood transfusion (84.9%). The volume of blood transfusion ranged from 20 to 180 mL/kg/year with a median of 120 mL/kg/year. Stroke risk was significantly lower among patients receiving blood transfusion compared to the non-transfused patients (p < .001).
Forty-one out of 52 studied patients (78.8%) were treated with hydroxyurea (HU) therapy. HU administration was not found to affect stroke risk (p = .460).
Other clinical (age, jaundice, vaso-occlusive crisis, hepatomegaly, splenectomy, pulmonary complications, bone complications, infection) and laboratory parameters (degree of anemia, bilirubin, HbF, HbS) were not found to be related to stroke risk (data not shown).
Discussion
βS globin gene analysis revealed that homozygous Benin/Benin haplotype was the most common haplotype detected in half of the studied patients 26/52 (50%) followed by heterozygous Benin/Bantu in 16/52 (30.8%) patients, while other βS globin gene haplotypes represented a minority among the studied patients. Our results are further validating previous studies that reported Benin haplotype as the predominant haplotype in Egyptian patients [Citation14,Citation15]. Other studies on SCD patients from several Middle East countries; in Lebanon, homozygous Benin/Benin was the most common haplotype (60%) followed by Benin/Bantu (22%) detected among 50 SCD patients [Citation21]. and in the West Bank of Palestine Benin haplotype was found predominant with a frequency of 88.1% [Citation22]. Predominance of Benin haplotype was also reported in Tunisia, another North African Country with a frequency of 60.54% among the studied Tunisian patients [Citation23]. The high frequency of Benin haplotype in Tunisian patients was explained by slave trade from the Benin region or by a trans-Saharian population migration [Citation24], which may also apply to Egypt.
Predominance of Benin haplotype in Egyptian patients can be explained by the high rate of positive consanguinity. Consanguinity has been identified as one of the main factors which are believed to play a major role in the increased frequencies of the HbS [Citation23]. The tradition of consanguineous marriage goes far back in history and has been known in the Middle Eastern Arab countries from biblical times, where such marriages are not necessarily limited to geographic or religious isolates or ethnic minorities [Citation25].
Although statistically non-significant (p = .532), stroke risk was found to be higher in homozygous Benin/Benin haplotype 8/26 (30.8%) than non-Benin/Benin 6/26 (23.1%).The βS globin gene haplotype is considered one of the factors influencing the severity of SCD in patients from different populations [Citation26–28]. A recent study compared the β-globin gene haplotype with the incidence of stroke in pediatric SCD patients and showed that Benin is the strongest haplotype associated with higher stroke risk (about 59%), followed by Bantu (about 22.3%) then Senegal (about 9.4%) [Citation29]. However, previous study on Egyptian patients agrees with our findings where no correlation was observed between clinical severity and βS globin gene haplotype [Citation14]. Another more recent Brazilian studies also stated that there was no correlation between βS globin gene haplotype and stroke in SCD [Citation30,Citation31]. These controversial results are probably related to the small sample size in each study. However, each population has its peculiarities that characterize it from other populations [Citation32].
Stroke risk was significantly lower among patients receiving blood transfusion compared to the non-transfused patients (p < .001). Chronic prophylactic blood transfusion therapies are widely known procedures that reduce the gravity of SCD and decrease the risk of stroke development [Citation33–37]. In STrOke Prevention (STOP) I trial in SCD, the consequent introduction of chronic transfusions for primary stroke prevention had an immediate impact on stroke rates in the state of California. Within 2 years after the results of the STOP I trial were published, SCD stroke rates dropped by a factor of 5 [Citation38].
HU is speculated to be effective in stroke prevention because it increases the HbF level [Citation39], and may also inhibit phagocyte production and thereby reduce the inflammation and red cell adherence to endothelial cells [Citation40]. In our study, HU administration was not found to affect stroke risk.
In conclusion, stroke is a major cause of morbidity in children and adults with SCD [Citation41]. Although studies of several different ethnic groups of patients with SCD suggested that the βS globin gene haplotype may be a useful predictor of disease severity, the association of βS globin gene haplotype with the severity of SCD must be cautiously interpreted as data are controversial. Several factors may influence the disease course in addition to βS globin gene haplotype [Citation32], as in our study where most of the patients were on chronic blood transfusion and/or HU therapy. Nevertheless, the lack of statistically significant correlations in our study could be due to the small sample size. More multicenter studies are necessary to better understand SCD and to identify genetic modulators influencing the diverse clinical presentations of SCD patients in our population that could guide prognosis to determine preventive measures and the best treatment for acute and chronic organ damage.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Heba H. Abou-Elew http://orcid.org/0000-0003-4154-0399
Rania A. Zayed http://orcid.org/0000-0001-7920-7060
References
- Dover GJ, Platt OS. Sickle cell disease. In: Nathan DG, Orkin SH, editors. Nathan and Oski’s hematology of infancy and childhood Vol. 1. Philadelphia: WB Saunders Company; 1998.
- Powars DR, Weiss JN, Chan LS, et al. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood. 1984;63:921–926.
- Zago MA, Silva WAJr, Dalle B, et al. Atypical βS haplotypes are generated by diverse genetic mechanisms. Am J Hematol. 2000;63:79–84. doi: 10.1002/(SICI)1096-8652(200002)63:2<79::AID-AJH4>3.0.CO;2-D
- Zago MA, Figueiredo MS, Ogo SH. Bantu βS cluster haplotype predominates among Brazilian blacks. Am J Phys Anthropol. 1992;88:295–298. doi: 10.1002/ajpa.1330880304
- Bortolini MC, Salzano FM. βs haplotype diversity in afro-Americans, Africans, and Euro-Asiatics – an attempt at a synthesis. Cienc Cult. 1999;51:175–180.
- Kolapo KO, Vento S. Stroke: A realistic approach to a growing problem in sub-Saharan Africa is urgently needed. Trop Med Int Health. 2011;16(6):707–710. doi: 10.1111/j.1365-3156.2011.02759.x
- Ohene-Frempong K, Weiner SJ, Sleeper LA, et al. Cerebrovascular accidents in sickle cell disease: rates and risk factors. Blood. 1998;91:288–294.
- Makani J, Williams TN, Marsh K. Sickle cell disease in Africa: burden and research priorities. Ann Trop Med Parasitol. 2007;101(1):3–14. doi: 10.1179/136485907X154638
- Kinney TR, Sleeper LA, Wang WC, et al. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. Pediatrics. 1999;103:640–645. doi: 10.1542/peds.103.3.640
- Adams RJ, McKie VC, Brambilla D, et al. Stroke prevention trial in sickle cell anemia. Control Clin Trials. 1998;19:110–129. doi: 10.1016/S0197-2456(97)00099-8
- United Nations General Assembly. Recognition of sickle-cell anaemia as a public health problem 2009.
- Aliyu Z, Kato G J, Taylor Jt, et al. Sickle cell disease and pulmonary hypertension in Africa: A global perspective and review of epidemiology, pathophysiology, and management. Am J Hematol. 2008;83(1): 63–70. doi: 10.1002/ajh.21057
- El-Beshlawy A, Youssry I. Prevention of hemoglobinopathies in Egypt. Hemoglobin. 2009;33(Suppl 1):S14–S20. doi: 10.3109/03630260903346395
- Shawky RM, Khalifa AS, Elkholy MS, et al. Genotype phenotype correlation and hormone profile in sickle cell anaemia. Egypt J Med Hum Genet. 2003;4(2):63–75.
- El-Hazmi MA, Warsy AS, Bashir N, et al. Haplotypes of the beta-globin gene as prognostic factors in sickle-cell disease. East Mediterr Health J. 1999;5(6):1154–1158.
- Adams RJ, Brambilla DJ, Granger S, et al. STOP study stroke and conversion to high risk in children screened with transcranial Doppler ultrasound during the STOP study. J Child Neuro. 2004;15:344–349. doi: 10.1177/088307380001500511
- Little S, et al. Amplification-Refractory mutation system (ARMS) analysis of point mutations. In: Dracopoli NC, editor. Current protocols in human molecular genetics 1994. New York: John Wiley & Sons, p. 9.8.1.– 9.8.12.
- Powars DR. βS-gene-cluster haplotypes in sickle cell anemia. Hematol Oncol Clin North Am. 1991;5:475–493. doi: 10.1016/S0889-8588(18)30426-X
- Rahimi Z, Karimi M, Haghshenass M, et al. Beta-globin gene cluster haplotypes in sickle cell patients from southwest Iran. Am J Hematol. 2003;74(3):156–160. doi: 10.1002/ajh.10422
- Old JM. Hemoglobinopathies. In: Elles R, editor. Methods in molecular medicine: molecular diagnosis of genetic disease. Totowa, NJ: Humana Press; 1996. p. 169–183.
- Inati A, Taher A, Bou Alawi W, et al. β-globin gene cluster haplotypes and HbF levels are not the only modulators of sickle cell disease in Lebanon. Eur J Haematol. 2003;70:79–83. doi: 10.1034/j.1600-0609.2003.00016.x
- Samarah F, Ayesh S, Athanasiou M, et al. Beta(S)-globin gene cluster haplotypes in the west bank of palestine. Hemoglobin. 2009;33:143–149. doi: 10.1080/03630260902861873
- El-Hazmi MA, Al-Hazmi AM, Warsy AS. Sickle cell disease in Middle East Arab countries. Indian J Med Res. 2011;134:597–610. doi: 10.4103/0971-5916.90984
- Pagnier J, Mears JG, Dunda-Belkhodja O, et al. Evidence for the multicentric origin of the sickle cell hemoglobin gene in Africa. Proc Natl Acad Sci USA. 1984;81:1771–1773. doi: 10.1073/pnas.81.6.1771
- Kamal H. Dictionary of pharaonic medicine. 1st ed. Vol. 19, Cairo: National; 1967. p. 286–288.
- Serjeant GR. The clinical features of sickle-cell disease. Amsterdam: North-Holland; 1974.
- Wainscoat JS, Thein SL, Higgs DR, et al. A genetic marker for elevated levels of haemoglobin F in homozygous sickle-cell disease? Br. J. Haematol.. 1985;60:261–268. doi: 10.1111/j.1365-2141.1985.tb07412.x
- Miller BA, Olivieri N, Salameh M, et al. Molecular analysis of the high-hemoglobin-F phenotype in Saudi Arabian sickle-cell anemia. New Engl J Med. 1987;316:244–250. doi: 10.1056/NEJM198701293160504
- Menaa F. Stroke in sickle cell anemia patients: A need for multidisciplinary approaches. Atherosclerosis. 2013;229(2):496–503. doi: 10.1016/j.atherosclerosis.2013.05.006
- Loggetto SR. Sickle cell anemia: clinical diversity and beta S-globin haplotypes. Rev Bras Hematol Hemoter. 2013;35(3):153–162.
- Rodrigues DO, Ribeiro LC, Sudário LC, et al. Genetic determinants and stroke in children with sickle cell disease. J Pediatr (Rio J). 2016;92(6):602–608. doi: 10.1016/j.jped.2016.01.010
- Silva Filho IL, Leite AC, Moura PG, et al. Reply: genetic polymorphisms and cerebrovascular disease in children with sickle cell anemia from Rio de Janeiro, Brazil. Arq Neuropsiquiatr. 2012;70:648–649. doi: 10.1590/S0004-282X2012000800023
- Steinberg MH, Barton F, Castro O, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645–1651. doi: 10.1001/jama.289.13.1645
- Lezcano NE, Odo N, Kutlar A, et al. Regular transfusion lowers plasma free hemoglobin in children with sickle-cell disease at risk for stroke. Stroke. 2006;37:1424–1426. doi: 10.1161/01.STR.0000221173.97108.01
- Lanzkron S, Strouse JJ, Wilson R, et al. Systematic review: hydroxyurea for the treatment of adults with sickle cell disease. Ann Intern Med. 2008;148:939–955. doi: 10.7326/0003-4819-148-12-200806170-00221
- Brawley OW, Cornelius LJ, Edwards LR, et al. National institutes of health consensus development conference statement: hydroxyurea treatment for sickle cell disease. Ann Intern Med. 2008;148:932–938. doi: 10.7326/0003-4819-148-12-200806170-00220
- Steinberg MH, McCarthy WF, Castro O, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5-year follow-up. Am J Hematol. 2010;85:403–408.
- Fullerton HJ, Adams RJ, Zhao S, et al. Declining stroke rates in Californian children with sickle cell disease. Blood. 2004;104:336–339. doi: 10.1182/blood-2004-02-0636
- Vicari P, Barretto de Mello A, Figueiredo MS. Effects of hydroxyurea in a population of Brazilian patients with sickle cell anemia. Am J Hematol. 2005;78:243–244. doi: 10.1002/ajh.20293
- Ware RE, Helms RW. Stroke with transfusions changing to hydroxyurea (SWiTCH). Blood. 2012;119(17):3925–3932. doi: 10.1182/blood-2011-11-392340
- Kassim AA, Galadanci NA, Pruthi S, et al. How I treat and manage strokes in sickle cell disease. Blood. 2015;125(22):3401–3410. doi: 10.1182/blood-2014-09-551564