
ABSTRACT
Objective: Burkitt's lymphoma (BL) is a highly aggressive malignant lymphoma, its molecular biological mechanism has not been fully investigated. The construction of protein-protein interaction (PPI) networks and the identification of complexes through a cluster analysis are important research directions in the post-genome era. However, different cluster analysis algorithms have their own characteristics, and a single analysis has some limitations. In this study, we obtained the target and pathway information of BL using different clustering analyses.
Material and Methods: First, we obtained 50 BL genes by screening the Online Mendelian Inheritance in Man (OMIM) database; their related genes were further extracted from the literature. The PPI network was constructed with the Search Tool for Retrieval of Interacting Genes/Proteins (STRING). Afterward, the interaction data were input in Cytoscape3.4.0 software and related plug-ins were used to implement topology analysis and clustering analysis. Functional analysis based on the Kyoto Encyclopedia of Genes and Genomes (KEGG) database were used to characterize the biological importance of the clusters.
Results: We constructed a PPI network consisting of 459 nodes (proteins) and 1399 sides (interactions), 12 genes and 8 signaling pathways were found to be closely related to BL.
Conclusion: In this study, the use of combined algorithms to analyse gene interactions provides a new perspective for network-based analysis. The results of this study reveal new insights into the molecular mechanisms underlying BL, which may be novel therapeutic targets for disease management and may provide a bioinformatic basis for the further understanding of BL.
Burkitt’s lymphoma (BL) is a highly malignant tumour originating from follicular germinal centre cells and is a special subtype of B-cell non-Hodgkin’s lymphoma, which has a short tumour cell doubling time, high aggressiveness and poor prognosis characteristics. The main molecular characteristics of BL are CD19+, CD20+, BCL6+, CD10+, BCL2- and translocation of c-myc [Citation1]. Its pathogenesis is not clear; intensive chemotherapy is the main treatment at the present stage, but the considerable side effects of drugs indicate the urgency of research on the new low toxicity and effective methods [Citation2]. In recent years, with the rapid development of molecular biology, information such as cell cycle protein, apoptosis protein, oncogene expression and cell signal pathways of BL has been gradually revealed, but the exact pathogenesis remains to be further studied [Citation3].
The protein interaction network is both the product of the biological development and the interaction data of large-scale complexity [Citation4], for its research, now focused on the detection of protein complexes and functional modules [Citation5]. Protein interaction networks are usually composed of a large number of high-density protein nodes but also have sparsely connected nodes, which indicates that clustering is the primary tool for data mining in protein interaction networks [Citation6]. However, different clustering algorithms are based on different mathematical models, there are certain limitations [Citation7]; therefore, using a single clustering method to analyze the network will generate large or small errors and impact research results.
In this study, based on the existing methods, BL-related gene information were analyzed by using a series of bioinformatic methods, including protein–protein interaction (PPI) network construction, topology analysis, multiple cluster analysis and pathway enrichment analysis. The results we obtained may facilitate the understanding of the molecular mechanisms underlying BL.
Materials and methods
Searching for BL-related genes
BL-related genes were extracted from the Online Mendelian Inheritance in Man (OMIM) database, using the search term ‘Burkitt lymphoma’ in the ‘Gene’ section (Burkard et al. [Citation12]). Sixty-eight BL-related genes were identified (up to 2016-07-26); By reading the related articles one by one, miscellaneous data were screened out. Finally, 55 genes (up to 2016-07-26) related to the clinical, immuno and molecular definitions of BL were reserved, as shown in .
Table 1. Fifty genes associated with BL.
PPI network construction and topology attribute analysis
The Search Tool for the Retrieval of Interacting Genes/Protein (STRING) database was used to retrieve the predicted interactions for the identified genes and allows visualizing complex networks [Citation8]. Version 10.0 of STRING, which contains information on approximately 9.6 million proteins from more than 2000 organisms is a biological database and web resource of known and predicted PPIs. We set the required interaction score at >0.4 as the threshold used for analysis. Subsequently, the protein interaction was read into Cytoscape 3.4.0 (http://cytoscape.org/), which allows the network to be integrated into any type of attribute data and implement topology analysis [Citation9].
Mining function modules and implementing topology analysis
Different clustering analyses (OH-PIN, IPCA, EAGLE, MCOMD) were used to detect molecular complexes in the network through plug-ins CentiScaPe 2.1 and the Cytocluster 1.0.1 in Cytoscape, whereas ClusterViz 1.0.3 was used to achieve CML PPI network topology analysis. The parameters and the applied thresholds were shown in the notes of .
Pathway enrichment analysis
The proteins included in the eight molecular complexes were submitted to the online DAVID system (https://david.ncifcrf.gov/) (parameters: count = 2, EASE = 0.1, ‘species and background’ choosing ‘Homo sapiens’), which consists of an integrated biological knowledge base and analytical tools aimed at systematically extracting biological meaning from large gene or protein lists [Citation10,Citation11]; a relative search of the Kyoto Encyclopaedia of Genes and Genomes (KEGG), BBID, PANTHER, BIOCARTA and REACTOME_PATHWAY databases helped identify the biological pathways regulated by BL-related genes. A P-value ≤ 0.05 was used as the cut-off criterion.
Results
PPI network and molecular complexes
A gene/protein interaction network, consisting of 459 nodes (protein) and 1399 sides (interactions), was constructed after a comprehensive literature search of 50 related genes using the STRING (). Then, through OH-PIN, IPCA, EAGLE, MCOMD algorithm analysis, the first two clusters with the highest degree of association were obtained respectively for further analysis ().
Figure 1. PPI network of BL (Overall + partial). The yellow triangles indicate OMIM genetic disease-related proteins, while the blue polygons indicate proteins obtained from text mining. PPI: protein–protein interaction. BL: Burkitt’s lymphoma.
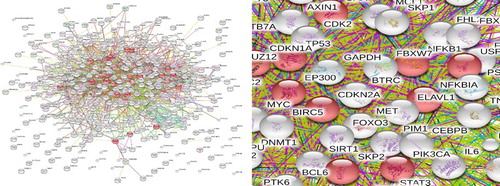
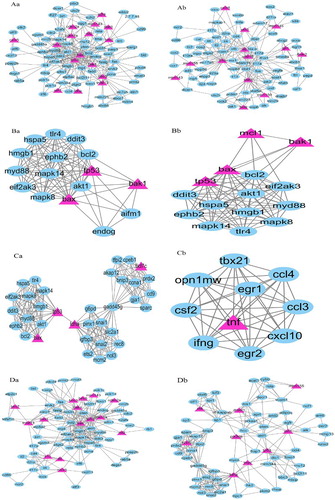
Figure 2. Molecular complexes of the PPI network. The yellow triangles indicate OMIM genetic disease-related proteins, while the blue polygons indicate proteins obtained from text mining. Complex Aa and Ab were gotten through EAGLE clustering analysis. Cut off-values: Clique size = 3, Complex size = 2(Complex Aa:Nodes:92, Edges:310, Modularity:1.84; Complex Ab:Nodes:89, Edges:230, Modularity:1.422). Complex Ba and Bb were gotten through IPCA clustering analysis. Cut off-values:Complex size = 2, Shortest path length = 2(Complex Ba:Nodes:16, Edges:91; Complex Bb: Nodes:15, Edges:84). Complex Ca and Cb were gotten through MCODE clustering analysis. Cut off-values:Node score = 0.2, degree = 4, k-core = 4, Maximum depth = 100(Complex Ca:Nodes:35, Edges:200, Score:11.765; Complex Cb:Nodes:10, Edges:45, Score:10). Complex Da and Db were gotten through OH-PIN clustering analysis. Cut off-values:Overlapping score = 0.5(Complex Da:Nodes:75, Edges:279; Complex Db: Nodes:70, Edges:248).
Results of network topology attribute analysis
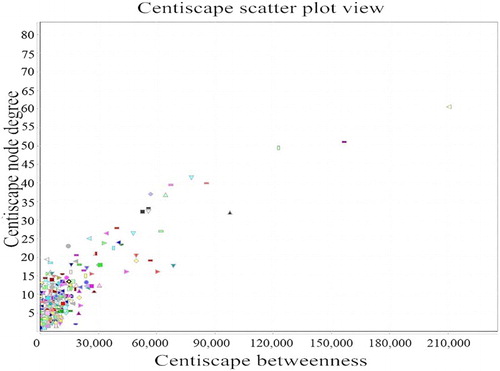
The network topology attribute analysis showed that the connectivity of nodes in the network (the number of nodes in the network) follows a descending distribution. That is, as the number of connected nodes increases, the number of corresponding nodes decreases. Thus, the gene-protein interaction networks are scale-free networks [Citation12]. We found that a ≥ 35 degree of nodes in the network corresponded to a sharp reduction in the number of odes; therefore, nodes with a ≥ 35 connectivity in the network were selected as the key nodes (hub), including tp53, bcl2, akt1, myc, bax, mdm2, mapk8, stat1, jak1, fos, il4, myd88, which totals 12 genes ().
Figure 3. Connectivity degree of each node degree and betweenness comparison. The horizontal axis represents betweenness, and the ordinate represents the connectivity degree. The graphic in the table represents each node in the network.
Molecular complex pathway enrichment
The enriched terms obtained from the Pathway enrichment analysis of BL are shown in the following table (). Only the first five enrichment pathways of each molecular complex are listed in the table, but we took all meaningful data for analysis. Finally, we took the same channel repeat number of ≥ 4 as the key path, a total of the following eight pathways: toll-like receptor signalling pathway, Jak-STAT signalling pathway, apoptosis signalling pathway, p53 signalling pathway, MAPK signalling pathway, colorectal cancer signalling pathway, pancreatic cancer signalling pathway and neurotrophin signalling pathway.
Table 2. Most significant pathways of eight molecular complexes were enriched (P ≤ 0.05).
Discussion
Protein is not only the material basis of life but also the executor of life activities; they rarely achieve their assigned functions in an individual way, such as genetic material replication, gene expression regulation, cell signal transduction, cell proliferation, apoptosis, metabolism and other processes, and activities are dependent on the interaction between proteins. Therefore, the study and analysis of PPI networks have naturally become the foundation to understanding cellular organizations, processes and functions [Citation13]. The PPI network, which has a modular structure, is made up of relatively independent subnets on the topology or biological function. Therefore, a clustering analysis of a PPI network can reveal its structure and predict the function of unknown proteins in the cluster [Citation14]. For example, Nanda Kumar Yellapu, etc., through the analysis of the PPI network found that PAK1 is an important target gene of breast cancer [Citation15]; Ma [Citation16] used microarray and system biology methods to describe the genetic background of cold syndrome in the TCM system. Based on the protein interaction network, Harriet Keane constructed the MPP+ model to analyze and predict the crosstalk between network proteins, and four target nodes/proteins (P62, GABARAP, GBRL1 and GBRL2) with a high betweenness were identified by the intervention of mitochondrial function and autophagy to influence Parkinson’s disease [Citation17]. Gu et al. [Citation18] studied inflammation and the angiogenesis gene network module response, predicting that adam17, cd40, ets1, foxo1, smad3 and tlr4 were the target genes of miR-145; Pan [Citation19] predicted the biomarkers that distinguish HCC from MHCC by identifying DEMs through the use of a network biological approach.
Clustering analysis, which is based on a certain algorithm and a given rule mining the protein network dense subgraph is a type of important method of mining complex data [Citation20].Traditional clustering algorithms are divided into hierarchical-based methods, graph-based methods, density-based methods and so on [Citation21]. However, there is no reliable evidence that the results of these methods have significant biological implications. Different types of clustering algorithms have their advantages and disadvantages: the biggest advantage of graph-based methods is simple and efficient, the advantage of the hierarchical-based method is that it can make the hierarchical modules of the whole protein network to be presented in a tree structure. However, the deficiency of the above two methods is that each protein node can only belong to one cluster. The density-based method allows a protein to repeatedly emerge in the process of expanding the search, which enables achieving the goal that the same protein belongs to different clusters, but this method cannot identify the non-dense sub-graph structure in the protein network [Citation22]. In the actual protein network, each protein node may belong to a number of protein complexes or functional modules, with multiple functions, involved in a number of different biological processes [Citation23].
Therefore, comprehensively considered, we used OH-PIN, IPCA, MCODE and EAGLE, which were four different clustering algorithm analyses, aimed at reducing the error and improve the authenticity of the results. OH-PIN is an agglomerative hierarchical algorithm that can distinguish overlapping protein functional modules. IPCA, improved by the DPClus [Citation24] algorithm, is based on the density of the algorithm; the process is to select the largest node as the seed, followed by an extended analysis, when all the nodes that may have been joined are detected as an output cluster [Citation25]. MCOMD is a non-overlapping clustering algorithm based on density [Citation26], but because nodes with large weights do not necessarily have a high degree of connectivity with other nodes, there is no guarantee that the predicted complexes will be tightly connected to each other; furthermore, in practical applications, the coverage rate of the complexes based on the density algorithm is not high [Citation27]. EAGLE is a type of agglomerative hierarchical algorithm based on the maximum clique, which can identify overlapping function modules [Citation28], by merging the two clusters with the largest similarity; this algorithm finds the optimal partitioning method to realize the identification of functional modules [Citation29]. In this study, we will analyze and compare the results of four types of cluster analyses, the high correlation degree of gene nodes and the common role of the signalling pathway is more biologically significant.
Twelve important target genes associated with BL were identified in our study. Related studies have shown that the c-myc gene in t (8; 14) translocation is recognized as the main molecular genetics of BL. Dave et al. compared the gene expression profiling of BL and diffuse large B-cell lymphoma with gene chip technology and found that the c-myc target gene and the germinal centre subgroup genes were overexpressed [Citation30]. However, studies have shown that the translocation of c-myc/Ig may also occur in normal precancerous cells [Citation31]. Further study found that c-myc can be used as a transcriptional active gene amplifier through the upregulation of cyclin D and E to drive cell proliferation [Citation32]. Epstein–Barr virus, as an important factor in the occurrence of BL, exists in almost all endemic BL. It can cause abnormal expression of apoptosis inhibitor gene BCL2 or the apoptosis of the Fas-mediated pathway in lymphocyte system to be suppressed, which leads to the suppression of lymphocyte apoptosis and the occurrence of lymphoma [Citation33]. The over expression of MDM2 can inhibit the expression of p53 protein, and its abnormal expression can disrupt cell cycle regulation in BL [Citation34], and the high expression of MDM2 and MDM4 may be the main mechanism in some cases. TP53 (encoding p53) is an important tumour suppressor gene that plays an important biological role in cell cycle regulation, DNA damage repair, cell differentiation, apoptosis and senescence and the transcriptional regulation of key genes such as p21, Bax, etc., which prevent the occurrence and development of tumour [Citation35,Citation36]. The findings of Pervez S confirmed this view [Citation37]; the Fos gene belongs to the immediate early response gene, also known as the ‘molecular switch’, and it plays a dual role in tumour cell apoptosis, invasive growth and tumour angiogenesis, which depends on the cell type and apoptotic stimulus signal types [Citation38]. IL-4 is one of the key cytokines in the process of tumour immunity, and its effect on tumours is also positive and negative, but more studies have shown that IL-4 has a growth-promoting effect on tumour, which can protect the death receptor and chemotherapy-induced apoptosis of tumour cells. As studies have shown, IL-4 significantly inhibits Fas/APO-I and chemotherapeutic drug-induced tumour cell apoptosis in prostate cancer, breast cancer and bladder tumour cells [Citation39] and can induce the differentiation of monocytes from blood into M2 macrophages that promote tumour growth and angiogenesis [Citation40]. Mapk8, stat1, jak1 and myd88 are important transduction proteins that play a key role in MAPK, Jak-STAT and toll-like signalling pathways, respectively.
According to the literature, we found that the key genes of this study are consistent with the high degree of target genes in traditional research, which proves that the gene/protein interaction network constructed by us has a certain value.
Using the pathway enrichment analysis of the eight molecular complexes, we mined eight important pathways. Among them, toll-like receptor is a pattern recognition receptor family; after stimulation, it activates AP-1, NF-κB and κ interferon regulatory factor and plays a regulatory role in the production of interferon, pro-inflammatory cytokines and effector cytokine immune responses through the toll-like receptor pathway. Myd88 (myeloid differentiation factor 88), a TIR domain-specific adapter protein, is the most important, and it participates in all TLRs-mediated signalling pathways except TLR3 [Citation41], which is consistent with the target obtained in this study. By studying the ibrutinib antilymphatic system tumour mechanism, Chen et al. [Citation42] found that targeted inhibition of Bruton tyrosine kinase can change the MAPK signalling pathway and inhibit the proliferation of BL. In addition, the traditional signalling pathway of the present study, such as the p53 signalling pathway, apoptosis signalling pathway and Jak-STAT signalling pathway also focused on the immune response, immune regulation and cell apoptosis. Analysis of the gene in the colorectal cancer signalling pathway and the pancreatic cancer signalling pathway showed the complexity of Burkitt lymphoma biological behaviour and extensive correlation with other tumours.
Conclusions
In this study, 12 important target genes (tp53, bcl2, akt1, myc, bax, mdm2, mapk8, stat1, jak1, fos, il4 and myd88) and 8 molecular complex pathways (toll-like receptor signaling pathway, Jak-STAT signaling pathway, apoptosis signaling pathway, p53 signaling pathway, MAPK signaling pathway, colorectal cancer signaling pathway, pancreatic cancer signaling pathway and neurotrophin signaling pathway) associated with BL were identified. These genes and pathways might play important roles in the biological progression and become new targets for treating BL. Additionally, we apply new ideas that combine multiple algorithms to analyze the interaction between genes, which is more reliable than a single method. However, further investigation is required to verify these results and the proposed mechanisms of action in BL.
Disclosure statement
No potential conflict of interest was reported by the authors.
ORCID
Additional information
Funding
References
- Yustein JT, Dang CV. Biology and treatment of Burkitt's lymphoma. Curr Opin Hematol. 2007;14(4):375–381. PMID:17534164. doi: 10.1097/MOH.0b013e3281bccdee
- Trzeciecka A, Klossowski S, Bajor M, et al. Dimeric peroxiredoxins are druggable targets in human Burkitt lymphoma. Oncotarget. 2016;7(2):1717–1731. PMID:26636537. doi: 10.18632/oncotarget.6435
- Campanero MR. Mechanisms involved in Burkitt’s lymphoma tumor formation. Clin Transl Oncol. 2008;10(5):250–255. PMID:18490240. doi: 10.1007/s12094-008-0193-x
- Xenarios I, Salwínski L, Duan XJ, et al. DIP, the database of interacting proteins: a research tool for studying cellular networks of protein interactions. Nucleic Acids Res. 2002;30(1):303–305. PMID:11752321. doi: 10.1093/nar/30.1.303
- Yu L, Gao L, Sun PG. Research on algorithms for complexes and functional modules prediction in protein-protein interaction networks. Chin J Comput. 2011;34(7):1239–1251. doi: 10.3724/SP.J.1016.2011.01239
- Liu H, Liao B, Peng LH. Research of clustering algorithms in protein-protein interaction network. Comput Eng Applic. 2008;44(30):142–144.
- Ramadan E, Naef A, Ahmed M. Protein complexes predictions within protein interaction networks using genetic algorithms. BMC Bioinformatics. 2016;17(Suppl 7):269, PMID:27454228. doi: 10.1186/s12859-016-1096-4
- Szklarczyk D, Franceschini A, Kuhn M, et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–D568. PMID:21045058. doi: 10.1093/nar/gkq973
- Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. PMID:14597658. doi: 10.1101/gr.1239303
- Huang DA, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. PMID:19131956. doi: 10.1038/nprot.2008.211
- Huang DW, Sherman BT, Tan Q, et al. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8(9):R183, PMID:17784955. doi: 10.1186/gb-2007-8-9-r183
- Burkard TR, Rix U, Breitwieser FP, et al. A computational approach to analyze the mechanism of action of the kinase inhibitor bafetinib. PLoS Comput Biol. 2010;6(11):e1001001, PMID:21124949. doi: 10.1371/journal.pcbi.1001001
- Stelzl U, Worm U, Lalowski M. A human protein-protein interaction network: a resource for annotating the proteome. Cell. 2005;122(6):957–968. PMID:16169070. doi: 10.1016/j.cell.2005.08.029
- Bosque G, Folch-Fortuny A, Picó J, et al. Topology analysis and visualization of potyvirus protein-protein interaction network. BMC Syst Biol. 2014;8:205. PMID: 25409737. doi: 10.1186/s12918-014-0129-8
- Yellapu NK, Pulaganti M, Pakala SB. Bioinformatics exploration of PAK1 (P21-activated kinase-1) revealed potential network gene elements in breast invasive carcinoma. J Biomol Struct Dyn. 2017;35(10):2269–2279. PMID:27456030. doi: 10.1080/07391102.2016.1216894
- Ma T, Tan C, Zhang H, et al. Bridging the gap between traditional Chinese medicine and systems biology: the connection of cold syndrome and NEI network. Mol Biosyst. 2010;6(4):613–619. PMID:20237638. doi: 10.1039/b914024g
- Keane H, Ryan BJ, Jackson B, et al. Protein-protein interaction networks identify targets which rescue the MPP+ cellular model of Parkinson’s disease. Sci Rep. 2015;5:17004, PMID:26608097. doi: 10.1038/srep17004
- Gu J, Chen Y, Li S, et al. Identification of responsive gene modules by network-based gene clustering and extending: application to inflammation and angiogenesis. BMC Syst Biol. 2010;4:47. PMID:20406493. doi: 10.1186/1752-0509-4-47
- Pan J, Cong Z, Zhong M, et al. Analysis of hepatocellular carcinoma and metastatic hepatic carcinoma via functional modules in a protein–protein interaction network. J Cancer Res Ther. 2014;10 Suppl(7):C186–C194.
- Wang J, Zhong J, Chen G, et al. Clusterviz: a cytoscape APP for cluster analysis of biological network. IEEE/ACM Trans Comput Biol Bioinform. 2015;12(4):815–822. PMID:26357321. doi: 10.1109/TCBB.2014.2361348
- Wang ZH, Dong YY, Wang XY. Review on several clustering methods in protein-protein interaction network. J Nat Univ Def Technol. 2009;31(4):81–86.
- Ji JZ, Liu ZJ, Liu HX, et al. An overview of research on functional module detection for protein-protein interaction networks. Acta Autom Sin. 2014;40(4):577–593.
- Li J, Zhao PX. Mining functional modules in heterogeneous biological networks using multiplex pagerank approach. Front Plant Sci. 2016;7:903. PMID:27446133.
- Altafuiamin M, Shinbo Y, Mihara K, et al. Development and implementation of an algorithm for detection of protein complexes in large interaction networks. BMC Bioinformatics. 2006;7:207. PMID:16613608. doi: 10.1186/1471-2105-7-207
- Tang Y, Li M. A cytoscape plugin for visualization and clustering analysis of protein interaction networks. Chin J Bioinform. 2014;12(1):38–45.
- Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4:2. PMID:12525261. doi: 10.1186/1471-2105-4-2
- Wang J, Li M, Deng Y, et al. Recent advances in clustering methods for protein interaction networks. BMC Genomics. 2010;11(Suppl 3):S10. PMID:21143777. doi: 10.1186/1471-2164-11-S3-S10
- Shen HW, Cheng XQ, Cai K, et al. Detect overlapping and hierarchical community structure in networks. Physica A. 2009;388(8):1706–1712. doi: 10.1016/j.physa.2008.12.021
- Cai J, Wang JX, Li M, et al. Clusterviz: a cytoscape plugin for clustering visualization. China J Bioinform. 2011;9(3):185–188.
- Yang LM, Ott G, Jaffe E, et al. Molecular diagnosis of Burkitt’s lymphoma. N Engl J Med. 2006;354(23):2431–2442. PMID:16760443. doi: 10.1056/NEJMoa055759
- Müller JR, Janz S, Goedert JJ, et al. Persistence of immunoglobulin heavy chain/c-myc recombination-positive lymphocyte clones in the blood of human immunodeficiency virus-infected homosexual men. Proc Natl Acad Sci U S A. 1995;92(14):6577–6581. PMID:7604036. doi: 10.1073/pnas.92.14.6577
- Pelengaris S, Khan M, Evan G. c-MYC: more than just a matter of life and death. Nat Rev Cancer. 2002;2(10):764–776. PMID:12360279. doi: 10.1038/nrc904
- Rowe M, Fitzsimmons L, Bell AI. Epstein–Barr virus and Burkitt lymphoma. Chin J Cancer. 2014;33(12):609–619. PMID:25418195.
- Hasegawa H, Yamada Y, Iha H, et al. Activation of p53 by Nutlin-3a, an antagonist of MDM2, induces apoptosis and cellular senescence in adult T-cell leukemia cells. Leukemia. 2009;23(11):2090–2101. PMID:19710698. doi: 10.1038/leu.2009.171
- Feng Z, Levine A. The regulation of energy metabolism and the IGF-1/mTOR pathways by the p53 protein. Trends Cell Biol. 2010;20(7):427–434. PMID:20399660. doi: 10.1016/j.tcb.2010.03.004
- Li Z, Gonzalez CL, Wang B, et al. Cdkn2a suppresses metastasis in squamous cell carcinomas induced by the gain-of-function mutant p53 R172H. J Pathol. 2016;240(2):224–234. PMID:27447534. doi: 10.1002/path.4770
- Pervez S, Nasir MI, Moatter T, et al. Characterization of genetic lesions inapoptosis-regulating and proliferation control genes in diffuse large B-cell non-Hodgkin’s lymphoma. J Cancer Res Ther. 2009;5(4):254–262. PMID: 20160358. doi: 10.4103/0973-1482.59901
- Tu DP, Ma XJ, Mo CM, et al. Progress and bioinformatics analysis of FOS protein. Hubei Agr Sci. 2015;54(7):1537–1542.
- Conticello C, Pedini F, Zeuner A, et al. IL-4 protects tumor cells from anti-CD95 and chemotherapeutic agents via up-regulation of antiapoptotic proteins. J Immunol. 2004;172(9):5467–5477. PMID:15100288. doi: 10.4049/jimmunol.172.9.5467
- Palma MD. Partners in crime: VEGF and IL-4 conscript tumour-promoting macrophages. J Pathol. 2012;227(1):4–7. PMID:22367978. doi: 10.1002/path.4008
- Sun B, Han DS. Negative regulation of toll-like receptors signaling pathways. Prog Biochem Biophys. 2009;36(12):1516–1522. doi: 10.3724/SP.J.1206.2009.00213
- Chen LL, Shi WY, You XF. Study on the role and its mechanism of Ibrutinib in inhibiting proliferation and inducing apoptosis in Burkitt’s lymphoma cell line. Med J Commun. 2016;30(1):24–28.