
ABSTRACT
Objective: To investigate the cause of recessive congenital methemoglobinemia (RCM) in Indian families and to identify molecular defect associated with RCM.
Methods: Eight cases of RCM have been addressed to our laboratory in order to investigate the cause of cyanosis associated with genetic disorders. NADH-cytochrome b5 reductase (cytb5r) enzyme activities were measured by standard methods, and molecular analysis was performed by polymerase chain reaction (PCR) followed by DNA sequencing. The interpretation of mutation effect and the molecular modeling were performed by using specific software DEEP VIEW SWISS-PDB VIEWER and Pymol molecular graphics program.
Results and Discussion: Eight index cases from four unrelated families were referred for the cause of cyanosis. All patients showed mild to moderate cyanosis without mental retardation or any neurologic abnormalities. The methemoglobin levels were in the range of 11.5–22.41% with 50–70% reduction in CYTB5R activity. Spectroscopic analysis of the hemolysate showed normal peaks suggesting the absence of Hb-M. Molecular characterization showed a novel homozygous mutation p.Arg192Cys in CYB5R3 gene is an evolutionarily conserved position located in exon 7 in all eight index cases. The substitution of Cys is located on the interface of two domains of NADH-binding domain and is close proximity to the adenosine moiety would preclude the reciprocal ionic interaction (salt bridge) between Arg192 and Ile97 and may influence binding of the NADH coenzyme is hypothesized to cause disruption of hydrogen bonding and instability. Our study indicated that novel homozygous mutation p.Arg192Cys in CYB5R3 gene present in eight cases and the possibility of high prevalence of heterozygous in Indian population causing Type I RCM.
Introduction
Recessive congenital methemoglobinemia (RCM) is a very rare recessive genetic disorder (OMIM 250800) caused by a deficiency of NADH-cytochrome b5 reductase (CYTB5R). There are two types of methemoglobinemia. In type I, the defect affects the soluble form of the enzyme, is restricted to red blood cells and causes well-tolerated methemoglobinemia. In type II, the defect affects both the soluble and microsomal forms of the enzyme and is thus generalized, affecting red cells, leukocytes and all body tissues. Type II methemoglobinemia is associated with mental retardation and other neurologic symptoms. The soluble erythrocytic CYTB5R isoenzyme is involved in cytochrome b5 reduction and in erythrocyte methemoglobin reduction, whereas the membrane bound is a microsomal enzyme that participates in a fatty acid desaturation complex and in drug metabolism. The CYTB5R isoforms are the product of a single gene called DIA1 or CYB5R3), consisting of nine exons and localized on chromosome 22q13-qter [Citation1]. A total of 58 different mutations which cause RCM type I and II have been reported to date [Citation2], the majority are missense mutations and are associated with mild type I methemoglobinemia. The genotype–phenotype relationship is extensively discussed in recent papers [Citation2,Citation3]. Disruptive mutations, such as splicing, stop codon or frameshift, and mutation greatly affecting catalytic activity result in a global loss of both the membrane-bound and soluble forms, and are associated with the severe Type II disease [Citation4,Citation5]. In contrast, missense mutations reducing the stability of the enzyme cause an accelerated decline in the enzyme activity of erythroid cells, and therefore result in Type I disease. The genetic study suggested that some mutations have been identified in both Type-I and II congenital methemoglobinemia, which suggests that the clinical manifestations in compound heterozygotes are determined by the combination of CYTB5R enzymatic activities of the two variants. Mutations causing enzyme instability lead to type I RCM, whereas mutations that inactivate the enzyme are associated with type II RCM [Citation6,Citation7]. Here, we report a novel p.Arg192Cys mutation in CYB5R3 gene associated with RCM type I, in eight Indian patients. Structural modeling of the novel mutation was performed on human CYTB5R crystal structure and discussed the functional effect of the detected mutation on the structure of the enzyme.
Patients, materials and methods
The four index cases had a history of persistent slate-gray cyanosis since his birth. Cyanosis was noted at birth without evidence of cardiopulmonary abnormalities. All index cases were adults with both the sex were equally affected. Initially, blood samples were tested for the methemoglobin level and found in the range of 11.5–22.41% before ascorbic acid and 300 mg IV ascorbic acid was slowly administered in 24 h. After 24 hours of administration, methemoglobin levels were reduced to normal level. Their previous health history and family history were unremarkable. On physical examination, temperature, blood pressure, heart rate of 120–130 beats/min with normal pulses and respiratory rate of 40–44 beats/min with normal breathing. Oxygen saturation levels were decreased to 65–82% (with pulse oximeter) on room air while they had prominent cyanosis on perioral region and the extremities. The rest of the physical examination was normal. So far, all are maintaining a normal life. With this history, patients were referred to National Institute of Immunohaematology (NIIH) for further work up. The blood samples of all index cases, their siblings and family members were further investigated for causes of methemoglobinemia. Written informed consent in Hindi was obtained from the patients as per NIIH Institutional ethical committee guideline. Venous bloodsamples were drawn from normal individuals, as well as from patients with methemoglobinemia, their siblings and all other blood-related family members. The hematological and biochemical parameters were described in the . Hemoglobin electrophoresis was normal. A specific test for cyanosis investigation like methemoglobin level and NADH-CYTB5R enzyme activity was performed as per the standard method. The absence of Hemoglobin-M was ruled by a spectroscopic analysis of methemoglobin, which showed normal peaks at 632 and 502 nm [Citation8,Citation9].
Table 1. Hematological and biochemical parameters of four NADH-Cytb5R deficient Indian families.
Genomic DNA amplification and PCR/restriction analysis
The peripheral lymphocytes of the propositus, their family members, and an unrelated normal control were washed twice with phosphate-buffered saline, and genomic DNA was extracted using QiAamp Blood Kit (Qiagen). The genomic DNA was amplified with a forward and reverse primers using previously reported protocol [Citation10,Citation11]. The PCR reaction volume was 20 μL, containing 2.0 μL of 10 × PCR buffer, 2.0 μL of 2 mmol/L of each dNTP, 1 μL of 10-mmol/L of each primer, and 2U of Taq polymerase (Takara). Thirty cycles of denaturation at 94°C for 20 s, annealing at 58°C for 20 s, and elongation at 72°C for 20 s were carried out, followed by an extended incubation at 72°C for 5 min. Routine screening for the most common (p.Arg50Trp, p.Gly76Ser, p.Met177Val and p.Ala179Thr) Indian mutations were carried out by respective restriction endonuclease digestion at 37°C [Citation2]. These endonucleases are unable to detect reported common mutations in these cases; so we have done individual exon sequencing ABI PRISM 3130xl automated sequencer (Applied Biosystems, Foster City, CA) using BigDye terminator cycle sequencing chemistry. A restriction site analysis was then undertaken to identify the mutation that would permit to establish a rapid screening method to detect the novel mutation. BstUI was found to be an appropriate restriction enzyme digestion site identified for R192C mutation by NEB cutter V2.0, and BstUI restriction endonuclease digestion was subsequently performed at 60°C. BstUI recognizes a single restriction site in the wild-type sequence of exon 7 that is destroyed by the R192C.
Molecular modeling and molecular mechanics
The NADH-CYTB5R (Protein Data Bank (PDB) code – 1 UMK [Citation12,Citation13]] was used for the current study to construct a template for modeling. A ribbon plot of the structure of human erythrocyte NADH-CYTB5R structure was defined by X-ray crystallography. The model of the mutant protein was obtained by side chain substitution using DEEP VIEW SWISS-PDB VIEWER (http://expasy.org/spdbv/), and the new mutation was substituted at the corresponding position in the structure of the human CYTB5R protein by using the graphics program PYMOL (http://pymol.sourceforge.net).
Results
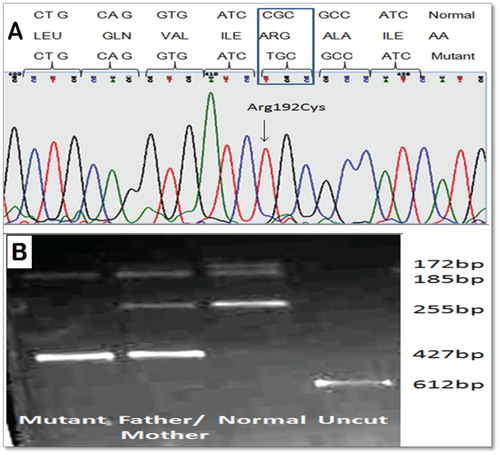
All the propositus are diagnosed with RCM Type I disease due to NADH-CYTB5R enzyme deficiency. All these index cases were presented no abnormalities except for a slight cyanosis of the lips and bluish discoloration of eyes sclera and tongue. Since the methemoglobinemia was suspected to have a familial background, the level of methemoglobin was determined in all sibling and parents were found to be elevated as shown in the . Before therapy with vitamins B2 and Ascorbic acid (Vitamin C), the Hb parameters were as follows: Hb concentration between 10.2 and 14.9 g/dL; and methemoglobin 11.5–22.41%. NADH-CYTB5R enzyme activities were determine by using the NADH ferricyanide method showed 30–50% reduced activity (10.92–16.6 IU/g Hb), whereas the normal control showed within 30–40 IU/g Hb. In the family 1, Elder sister (2.1-Proband II) and brother (2.3-Proband III) are also having mild complaints of cyanosis but they never required hospitalization and not tested in the past but also showed enzyme deficiency. In Family 2, Elder daughter (17 years/female) (2.1-Proband III) and son (12 years/male) (2.2-Proband III) are also having mild complaints of cyanosis and also detected enzyme deficiency ( and ). The enzyme activities of their parents were also reduced to 60–75% of normal. We investigated first the most common CYB5R3 gene mutation (Arg50Trp, Gly76Ser, Met177Val and Ala179Thr) reported earlier in Indian patients [Citation2]. No common mutations were detected in this patient and then entire coding sequence of the CYB5R3 gene was sequenced. In the index cases, we identified a single C> T substitution at nt 22490 of the CYB5R3 gene, thereby changing an arginine to cysteine amino acid substitution in codon 192 of the mature NADH-binding domain ((A) and ). We also confirm this mutation by restriction fragment length polymorphism using BstUI. The C to T transition eliminates a single BstUI site in the PCR fragment of exon 7, which permits rapid screening for the new R192C mutation by a simple BstUI restriction digest without the need of DNA sequencing ((B)).
Figure 1. Pedigree of the four families with RCM Type I disease due to a deficiency of the NADH-cytb5r enzyme associated with novel p.Arg192 > Cys mutation.

Figure 2. (A) Electropherogram of the exon 7 sequence indicating the T to C transition in position 22490 of the wild-type CYB5R3 sequence (OMIN:250800) and, top, the Box amino acid exchange in codon 192. (B) Ethidium bromide-stained 2% agarose gel of PCR fragments from all family members. The T→C transition eliminates a single BstUI site in the PCR fragment of exon 7 (612 bp), Normal (255, 185, 172 bp), Mutant (427 and 185 bp) and heterozygous (427, 255, 185, 172 bp), which permits rapid screening for the new R192C mutation in CYB5R3 gene by a simple BstUI restriction digest without the need of DNA sequencing.
The crystal structure of the NADH-binding domain of CYTB5R has been elucidated and is extremely useful for understanding the relationship between CYTB5R structure and function. The NADH-binding domain of CYTB5R displays an α/β fold, with a central β-sheet formed by five parallel and one anti-parallel β-strands flanked by Nα1-helices on each side. The arrangement of the α−helices and the β strands in the NADH-binding domain is to form three β1−α−β2 motifs: Νβ1−Να1−Νβ2 (Ser173-Asn209), Νβ2−Να3−Νβ3 (Val202-Leu238) and Νβ5−Να6−Νβ6 (Leu269-Phe300) [Citation13]. The first motif is typical of the NAD+-binding motif, in which the glycine-rich loop (Gly179–Gly182) forms a 310-turn and is in close contact with the ADP part of NAD+ in human CYTB5R. The codon 192 is localized in the β-strand at the amino edge of the β-sheet and forms a salt bridge with the buried isoleucine (I) codon 97 localized at the bottom of the Nβ2-strand (). This contributes to the stabilization of the salt-bridge network elaborated between some of the charged amino acids of the NADH-binding domain. Within extracellular proteins, cysteine is frequently involved in disulfide bonds, where pairs of cysteine are oxidized to form a covalent bond. If one half of a disulfide bond pair is lost, then the protein may not fold properly. As suggested by Bando et al. [Citation13], some of these salt bridges are important elements of folding, and therefore the mutation of R192C involved in a salt bridge may induce the abnormal folding of the Nβ2-strand.
Figure 3. (A) Three-dimensional view of human NADH-cytochrome b5 reductase according to the crystallographic structure (7–8) In relation to the active site and the activation domain, the arginine in codon 192 (yellow) is found at the opposing surface or front side of the molecule and in close proximity to the NADH-binding domain. A cofactor FAD is non-covalently bound in a large and wide boundary cleft between the two domains. (B) and (C) show that Normal and the mutation of Arg-192 to Cys would be predicted to change the hydrogen bonds’ pattern between the regions of codon Ile 97 in the FAD-binding domain.

Discussion
We have previously reported, clinical spectrum and molecular basis of RCM in India in which we have described 13 different CYTB5R mutations [Citation2,Citation13] and here we have diagnosed eight patients of NADH cytochrome b5 reductase deficiency causing RCM Type I disease due to novel p.Arg192Cys mutation and also thoroughly studied structure-function analysis using PDB file crystal structure elucidated by Bando et al. [Citation13]. Most of the patients with Type I disease present with mild visible cyanosis and do not become symptomatic until their methemoglobin levels exceed 25% of the total hemoglobin [Citation14]. These patients had only mild central cyanosis and no other clinical symptoms. For differential diagnosis of central cyanosis, some of the possibilities like sepsis, congenital heart disease with right to left shunt, congestive heart failure, cardiovascular collapse, pulmonary disorders, low oxygen affinity hemoglobin variants and methemoglobinemia (either congenital due to hemoglobin-M, congenital CYTB5R enzyme deficiency or acquired due to drugs or toxins) should be considered [Citation15]. These disorders can be differentiated by history, physical examination and appropriate laboratory tests [Citation16]. Methemoglobinemia is an uncommon clinical problem in infants and when present, it is usually caused by environmental toxicity from strong oxidizing agents and only very rarely from an inherited disorder of hemoglobin metabolism. Administration of ascorbic acid directly reduces methemoglobin, but the rate of reaction is slower. The level of methemoglobin can be drastically reduced by the administration of methylene blue [Citation17]. In severe methemoglobinemia, hyperbaric oxygen and exchange transfusions are alternative therapies if the combination therapy of intravenous methylene blue and ascorbic acid fails to reduce the methemoglobin level. It should be borne in mind that methylene blue should not be used in individuals with glucose-6-phosphate dehydrogenase (G6PD) deficiency because it is ineffective and may cause acute hemolysis. Our patient was started with only ascorbic acid treatment because they all had mild cyanosis with no other clinical symptoms.
In order to calculate the effects of the novel p.Arg192Cys mutation on the structure and stability of the mutant, molecular modeling was carried out. These missense mutations were not predicted to affect the enzymatic activity of the subunit, since the Cys192 residue is located in distance from the three active sites, Cys314, His373 and Asp396; 18.7, 20.1 and 12.2 Å for the former mutation and 13.0, 11.4 and 6.4 Å for the latter, respectively. In order to clarify how the two amino acid substitutions affect the conformation of the subunit, a possible structural alteration of the mutant proteins was calculated employing the coordinates obtained from X-ray crystallography [Citation13], since amino acid residue 192 is located on the interface of two domains () a dimer was subjected to examination for potential energy. Energy optimization was not carried out for the Arg192Cys dimer since it was thought to create an artificial deviation from the crystallographic structure. The side chain of Arg192 in the wild-type forms van der Waals bonds with the surrounding atoms. These bonds would be expected to be destroyed because the mutant Cys192 has a shorter side chain than Arginine. The reduced van der Waals force reflected a decrease in the number of atoms in the side chain. Furthermore, the loss of a positively charged Arginine residue led to a reduction in the electrostatic interaction (salt bridge) between the two monomers. If the mutant monomer could fold into a three-dimensional structure similar to that of the wild-type, the binding energy for dimerization of the two monomers would be reduced by the amino acid substitution. These circumstances may in turn disrupt the normal conformation of the A subunit molecule and destabilize it.
All patients showed a mild to moderate cyanosis and after ascorbic acid treatment, both cyanosis and methemoglobin levels were markedly decreased. In both the affected individual and parents NADH-CYTB5R activity were significantly reduced. Our patients thus have characteristics of type I RCM. The c.576C> T mutation causing the p.Arg192Cys substitution is located in exon 7, which encodes the protein fragment K154-G181. This structural abnormality is associated with a mild loss of function of CYTB5R protein as determined by the activity of enzyme in the patient blood sample. Previously described mutations in this region affected two residues only: Arg160 and Ala179. Of those, only p.Arg160X combined with p.Gln77X results in methemoglobinemia type II, and this phenotype can be related to very low catalytic activity or to a loss of protein expression of both mutated protein chains [Citation18,Citation19]. Other mutations in exon 6, homozygous or compound heterozygous with the mutations outside exon 6, cause RCM type I. Examples known to date include p.Ala179Val, p.Ala179Thr, and double mutants p.Arg160X/p.Asp240Gly and p.Ala179Thr/c.547 + 1G > A. The mutation (R192C) described here also falls in this class [Citation20].
We have identified a novel p.Arg192Cys mutation in CYB5R3 gene associated with type I RCM in the eight Indian cases in a four unrelated families and results of molecular modeling of the mutated enzyme correlated with the clinical phenotype, Hence, we conclude that presence of homozygous novel p.Arg192Cys mutation in CYB5R3 gene in eight cases suggesting a high prevalence in Indian population causing type I RCM.
Acknowledgments
The authors thank the families for participation in this study.
Disclosure Statement
No potential conflict of interest was reported by the authors.
ORCID
Vinod Gupta http://orcid.org/0000-0002-2400-014X
Additional information
Funding
References
- Percy MJ, Lappin TR. Recessive congenital methaemoglobinaemia: cytochrome b5 reductase deficiency. Br J Haematol. 2008;141:298–308.
- Warang PP, Kedar PS, Shanmukaiah C, et al. Clinical spectrum and molecular basis of recessive congenital methemoglobinemia in India. Clin Genet. 2015;87:62–67. doi: 10.1111/cge.12326
- Aslan D, Türköz-Sucak G, Percy MJ. Recessive congenital methemoglobinemia in immediate generations. Turk J Pediatr. 2016;58:113–115. doi: 10.24953/turkjped.2016.01.019
- Ewenczyk C, Leroux A, Roubergue A, et al. Recessive hereditary methaemoglobinaemia, type II: delineation of the clinical spectrum. Brain. 2008;131:760–761. doi: 10.1093/brain/awm337
- Davis CA, Crowley LJ, Barber MJ. Cytochrome b5 reductase: the roles of the recessive congenital methemoglobinemia mutants P144L, L148P and R159. Arch Biochem Biophys. 2004;431:233–244. doi: 10.1016/j.abb.2004.08.005
- Leroux A, Leturcq F, Deburgrave N, et al. Prenatal diagnosis of recessive congenital methaemoglobinaemia type II: novel mutation in the NADH-cytochrome b5 reductase gene leading to stop codon read-through. Eur J Haematol. 2005;74:389–395. doi: 10.1111/j.1600-0609.2004.00388.x
- Aalfs CM, Salieb-Beugelaar GB, Wanders RJ, et al. A case of methemoglobinemia type II due to NADH-cytochrome b5 reductase deficiency: determination of the molecular basis. Hum Mut. 2000;16:18–22. doi: 10.1002/1098-1004(200007)16:1<18::AID-HUMU4>3.0.CO;2-N
- Kaplan JC, Beutler E. Electrophoresis of red cell NADH- and NADPH-diaphoreses in normal subjects and patients with congenital methemoglobinemia. Biochem Biophys Res Commun. 1965;29:605. doi: 10.1016/0006-291X(67)90529-3
- Evelyn KA, Malloy HT. Microdetermination of oxyhemoglobin, methemoglobin, and sulfhemoglobin in a single sample of blood. J Biol Chem. 1938;126:655.
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: a laboratory manual. 2nd ed. Cold Spring Harbor (NY): Cold Spring Harbor Laboratory; 1989.
- Percy MJ, Gillespie MJS, Savage G, et al. Familial idiopathic methemoglobinemia revisited: original cases reveal 2 novel mutations in NADH-cytochrome b5 reductase. Blood. 2002;100:3447–3449. doi: 10.1182/blood-2002-05-1405
- Kedar PS, Warang PP, Ghosh K, et al. Severe mental retardation and recessive congenital methemoglobinemia in three Indian patient’s compound heterozygous for NADH-cytochrome b5 reductase gene mutations. Am J Hematol. 2011;86:327–329. doi: 10.1002/ajh.21956
- Bando S, Takano T, Yubisui T, et al. Structure of human erythrocytic NADH-cytochrome b5 reductase. Acta Crystallogr D Biol Crystallogr. 2004;60:1929–1934. doi: 10.1107/S0907444904020645
- Katsube T, Sakamoto N, Kobayashi Y, et al. Exonic point mutations in NADH-cytochrome B5 reductase genes of homozygotes for hereditary methemoglobinemia, types I and III: putative mechanisms of tissue-dependent enzyme deficiency. Am J Hum Genet. 1991;48:799–808.
- Percy MJ, McFerran NV, Lappin TR. Disorders of oxidised haemoglobin. Blood Rev. 2004;19:61–68.
- Wang Y, Wu Y-S, Zheng P-Z, et al. A novel mutation in the NADH-cytochrome b5 reductase gene of a Chinese patient with recessive congenital methemoglobinemia. Blood. 2000 May;95:3250–3255.
- Gibson QH. The reduction of methaemoglobin in red blood cells and studies on the cause of idiopathic methaemoglobinaemia. Biochem J. 1948;42:13–23.
- Forestier A, Pissard S, Cretet J, et al. Congenital recessive methemoglobinemia revealed in adulthood: description of a new mutation in cytochrome b5 reductase gene. Hemoglobin. 2015;39:438–441. doi: 10.3109/03630269.2015.1065882
- Gregg XT, Prchal JT. Red cell enzymopathies. Hematol Basic Princ Pract Chapter. 2009;45:611–623.
- Percy MJ, Crowley LJ, Davis CA, et al. Recessive congenital methaemoglobinaemia: functional characterization of the novel D239G mutation in the NADH-binding lobe of cytochrome b5 reductase. Br J Haematol. 2005;129:847–853. doi: 10.1111/j.1365-2141.2005.05526.x