
ABSTRACT
Background and objectives: Coinheritance of δβ thalassemia and HPFH with inherited factors is sparsely documented and may affect treatment modalities. So, we screened the presence of α deletion and β mutations in δβ thalassemia and HPFH disorders in 52 cases with high Hb F concentration.
Material and methods: Fifty-two individuals with raised HbF levels were study subjects. CZE was done for quantitative assessment of hemoglobin variants. Asian Indian inversion deletion break point type A, B and HPFH-3 were done by GAP-PCR.
Results: 18/52 cases of δβ Gγ (Aγδβ)0 thalassemia and 28/52 cases of HPFH-3 deletion were characterized. 6/52 patients with raised HbF levels were negative for δβ Gγ (Aγδβ)0 and HPFH-3 deletion. 9/18 (50%) were heterozygous for Gγ(Aγδβ)0 break point type A, 6/18 (33%) were heterozygous for break point type B and 3/18 (17%) were homozygous. Of the nine patients heterozygous for Gγ(Aγδβ)0 break point type A, three (33%) patients were double heterozygous with alpha 3.7 kb deletion and two (22%) patients showed compound heterozygosity with IVS 1–5(G–C) mutation. 4/9 (45%) patients were Gγ(Aγδβ)0 heterozygous.
Discussion and conclusion: We found 5/18(27.β) δβ-thalassemia cases with co-inherited alpha 3.7 deletion and 3/18 (16β) cases with IVS 1–5(G–C) mutation. Patients showed features of thalassemia intermedia phenotype among which those with co-inherited IVS 1–5(G–C) mutation showed severe phenotype as compared to those with co-inherited alpha 3.7 deletion. So, we highlight importance of genotyping of patients with δβ thalassemia or HPFH and coinheritance with inherited factors which plays crucial role in clinicopathological profile and setting up prenatal diagnostic protocol.
Introduction
δβ thalassemia and hereditary persistence of fetal hemoglobin (HPFH) are the disorders of hematopoiesis, caused by large deletions in both δ and β globin genes and show raised fetal hemoglobin (HbF) levels in adult life [Citation1]. The thalassemia phenotype includes combinations and varying degrees of hypochromasia and microcytosis, anemia, reticulocytosis, splenomegaly and erythroid hyperplasia in bone marrow. The most prevalent and clinically important thalassemias are α and β-thalassemias with δβ thalassemia being rare disorders. These groups encompass a large and still growing number of molecular defects that result in suboptimal globin synthesis [Citation2]. Both δβ thalassemia and HPFH are frequently associated with variable-size deletions within the β-globin gene complex. Molecular studies can provide an excellent platform to investigate the regulation of gene expression and mechanism of hemoglobin (Hb) switching from HbF to adult Hb during the development. This Hb switching may play a crucial role in the pathogenesis of these disorders [Citation3]. Asian Indian inversion deletion Gγ (Aγδβ)0-thalassemia is one of the rare entities characterized by high HbF and occurs due to deletions or rearrangements that abolish expression from the Aγ, δ and β-globin genes while leaving the Gγ-globin gene intact. Ten different Gγ (Aγδβ)0-thalassaemia deletions and rearrangements have been reported in a wide range of ethnic groups [Citation4]. Due to its interaction with various inherited factors like α thalassemia deletions and β thalassemia mutations, patients showed clinical variability. The common molecular aberrations coinherited with δβ-thalassemia includes β-thalassemia mutations, e.g. IVS position 1–5 (G–C) and alpha 3.7 deletion. To the best of our knowledge, coinheritance of δβ thalassemia with α thalassemia is very sparsely documented till date. HPFH is characterized by large deletions involving both δ and β globin genes with raised HbF levels in adults [Citation5]. In literature, various forms of deletional HPFH have been reported, out of which, HPFH-3 causing 48.5 kb deletion in the β globin gene cluster is the commonest form, reported in India. This genetic trait, although rare, have been found in different parts of India [Citation6,Citation7].
The distinction between δβ thalassemia and HPFH is made, based on clinical and hematological profile and is of prime importance because the modalities of treatment are strikingly different for δβ thalassemia. While δβ thalassemia heterozygotes tend to have a modest elevation of HbF (5–20%) with microcytic hypochromic red cell indices, HPFH heterozygotes are characterized by higher HbF levels (up to 30%) with normal red cell indices [Citation8]. The presence of different forms of HPFH has been reported from Vietnam, China, Cambodia and Thailand. Worldwide, there is sparse data pertaining to the correlation of genotype and phenotype in δβ thalassemia and HPFH under the influence of inherited cofactors like α deletion and β mutation. In view of this, in the present study, we screened the presence of α deletion and β mutations in δβ thalassemia and HPFH disorders in 52 cases with a high HbF concentration.
Material and methods
A total of 52 individuals with raised HbF levels detected during routine screening at Clinical Hematology OPD, All India Institute of Medical sciences(AIIMS), New Delhi in the last 4 years (year 2013–2017) were the study subjects. Patients with XMN1 polymorphism were excluded from our study. Complete history which included the history of jaundice, mass abdomen (left side) and blood transfusion was taken along with written informed consent from the patients. This study was granted ethical approval from AIIMS, Institute ethical committee. About 10 mL of venous blood was drawn in EDTA vial and complete blood count was measured by automated cell analyser. Giemsa-stained peripheral blood smears were examined for red cell morphology. Capillary zone electrophoresis (CZE) (Sebia-Park Technology, Lisses, France) was used for the quantitative assessment of hemoglobin variants HbA, HbF, HbA2. Genomic DNA was isolated from whole blood using Bioserve DNA isolation kit. Asian Indian inversion deletion Gγ(Aγδβ)0-break point A (5′ deletion) and B (3′ deletion) and HPFH-3 were done by GAP-PCR in two tube reaction (one for mutant and one for wild type) using specific primers [Citation9] (). The primer sequences and PCR conditions for the diagnosis of α-deletion (GAP-PCR) and β-mutation (ARMS-PCR) were selected from the published literature [Citation10–13].
Table 1. Primer sequences for δβ Indian inversion deletion (type A and type B) and HPFH-3 deletion.
Results
Out of these 52 cases, 18 cases of δβ Gγ (Aγδβ)0 thalassemia and 28 cases of HPFH-3 deletion were characterized. The remaining 6/52 patients with raised HbF levels were negative for δβ Gγ (Aγδβ)0 and HPFH-3 deletion. Hematological parameters and capillary zone electrophoresis (CZE) findings of the δβ thalassemia and HPFH patients are summarized in .
Table 2. Hematological indices of δβ-thalassemia and HPFH patients (mean ± SD).
Nine of 18 (50%) were heterozygous for Gγ(Aγδβ)0 break point type A, 6/18 (33%) were heterozygous for break point type B and 3/18(17%) were homozygous. Of the nine patients heterozygous for Gγ(Aγδβ)0 break point type A, three (33%) patients were double heterozygous with alpha 3.7 kb deletion and two (22%) patients showed compound heterozygosity with IVS 1–5(G–C) mutation. Remaining four of nine (45%) patients were Gγ(Aγδβ)0 heterozygous. Of the six patients heterozygous for Gγ(Aγδβ)0 break point type B, two (33%) patients were double heterozygous with alpha 3.7 kb deletions and one (17%) compound heterozygous IVS 1–5(G–C) mutation, whereas remaining three of six (50%) patients were homozygous for Gγ(Aγδβ)0 inversion deletion. Clinico-hematological parameters and CZE findings of the δβ thalassemia patients with coinherited α or β mutation/deletion are summarized in .
Table 3. Phenotypic variables of δβ patients along with co-inherited factors α and β mutation.
Three out of 18 patients who were homozygous for Gγ(Aγδβ)0 Indian inversion deletion showed features of thalassemia intermedia phenotype, i.e. anemia, jaundice, mild hepatosplenomegaly and 1 patient had blood transfusion. Hemogram revealed Hb 9.0 g/dl, RBCs count 4.9 × 1012/l, MCV 78.8 fl, MCH 20.3 pg and RDW-CV 16%. Red cell morphology showed a microcytic hypochromic picture. On CZE, these patients had HbF 100% and HbA2 0%.
Among the 28/52 (54%) HPFH patients, HbF levels were raised (median 27.0, range 27 ± 7.22%) with normal to reduced Hb A2 levels.
Discussion
δβ thalassemia and HPFH are the heterogeneous disorders caused by large deletions involving both δ and β globin genes in the β-globin cluster and are characterized by increased HbF levels in adults [Citation14]. δβ thalassemia, a form of beta-thalassemia is characterized by decreased or absent synthesis of the δ and β globin chains with a compensatory increase in expression of fetal γ chain synthesis, resulting in the augmented production of HbF in adult life. δβ thalassemia and its several types have been characterized based on various molecular defects [Citation15]. As homozyzotes have no δ or β genes, they cannot synthesize HbA and HbA2 and have very high HbF, almost approaching 100% as compared to heterozygotes which tend to have a modest elevation of HbF (5–20%) with microcytic hypochromic red cell indices [Citation16]. In the present study, 3/18 were homozygous and 15/18 heterozygous δβ thalassemia patients. We found that all the δβ thalassemia patients had reduced RBC indices with mean HbF levels 100% and 19% in homozygotes and heterozygotes, respectively. As per our previous case report with JM Khunger et al. in the year 2014 from India, 10-year-old male child presented with features of thalassemia intermedia phenotype, i.e. microcytic hypochromic red cells, anemia, jaundice and mild hepatosplenomegaly. His hemogram revealed Hb 9.0 g/dl, MCV 78.8 fl, MCH 20.3 pg, RBCs count 4.9 × 109/l and RDW-CV 16%. CZE analysis revealed 100% HbF. These findings confirmed the diagnosis of δβ thalassemia [Citation17]. Our findings are comparable to the above-mentioned studies [Citation16,Citation17].
The HPFHs include a wide range of conditions characterized by deletional and non-deletional forms of this disorder. Deletional HPFH is more common and among these, HPFH-3 deletion is commonest in India. In HPFH-3, the deletion removes 48.5 kb of a DNA segment, starting from the 5′ end of the ψβ gene to a region 30 kb downstream of the β-globin gene in an L1 repetitive region. This rare genetic trait is known to result from more than 40 deletions worldwide and is characterized by normal red cell indices and is asymptomatic except for raised HbF [Citation18,Citation19]. In our study, among the 52 patients with raised HbF, all the 28 HPFH patients showed HPFH-3 deletion, and these patients were heterozygous. In our study, all the 28 heterozygous HPFH-3 Indian deletion cases had mean HbF 31.0% along with normal RBC indices. However, few cases had lower MCV, MCH and raised RDW, which was possibly due to associated iron deficiency anemia as confirmed by reduced iron stores (serum ferritin levels) on iron studies.
These findings are comparable to the study by A. Nadkarni et al. in Indian population who observed that high Hb F level was observed among Indian HPFH-3 heterozygotes (mean ± SD: 29.0 ± 5.33%) whereas in the patients with Vietnamese/Chinese deletion, the mean HbF level was 25.2 ± 3.61 [Citation7]. Ottolenghi et.al revealed the high HbF level among southern Italian HPFH heterozygous patients (Mean, Range: 30%, 29–32%) [Citation20]. These findings are in line with the present study.
Coinheritance of δβ thalassemia or HPFH with other factors like alpha and β-thalassemia is very sparsely documented in the literature till date. In our study, we found 5/18 (%) δβ-thalassemia cases with co-inherited alpha 3.7 deletion and 3/18(%) cases with IVS 1–5(G–C) mutation. These patients showed features of thalassemia intermedia phenotype among which those with co-inherited IVS 1–5(G–C) mutation showed severe phenotype (more anemic, jaundice and transfusion dependency) as compared to those with co-inherited alpha 3.7 deletion . As per Wood et al. in the year 1993 and Galanello et al. in the year 1998, patients with double heterozygous δβ thalassemia with β mutation had more reduced Hb and red cell indices like MCV, MCH, MCHC as compared to the δβ patients without β mutation. These patients had more propensity towards blood transfusion and higher splenomegaly [Citation14,Citation21]. Tan Jin Ai et al. also reported double heterozygosity for δβ and β-thalassemia in four Chinese families [Citation22]. Our results are quiet similar to above-mentioned studies [Citation14,Citation21,Citation23]. One case report from our institute (AIIMS, New Delhi) in year 2012 showed coexistence of δβ thalassemia with α and β which is quiet rare and interesting finding [Citation23].
Regarding the coinheritance of α and β thalassemia with HPFH, no double heterozygous condition was seen in our patients. However, Thein et al. in year 1989 described an Asian Indian family with a non-deletion form of HPFH and β0 thalassemia. The patient was homozygous beta zero thalassemia had an unusually mild form of the disease, which was attributed to the coinheritance of HPFH [Citation6].
In conclusion, we want to highlight the importance of genotyping of the patients with δβ thalassemia or HPFH who primarily have raised HbF levels, as though rare, these disorders are not so infrequent in India. Therefore, molecular characterization of these raised HbF determinants will play a pivotal role in providing the proper management, appropriate counseling and prenatal diagnosis for these disorders especially when associated with β/α thalassemia or other hemoglobinopathies. Co-inheritance of β/α thalassemia with δβ thalassemia and HPFH may play a crucial role in understanding the clinicopathological profile of these patients. δβ thalassemia patients with co-inherited α thalassemia may present clinically as thalassemia intermedia and co-expression with β thalassemia mutation may behave as thalassemia major. Although this study is our attempt to reveal the molecular frequencies of δβ-thalassemia and HPFH and their coinheritance with other factors in India, more studies in a greater subset of samples are needed to have a definitive conclusion for Indian population (–).
Figure 1. Gγ(Aγδβ)0-thalassemia break point A (5′ deletion). It was detected on 2% agarose gel. Explanation of the lanes is as follows; Lane 1AB, 4AB: Heterozygous; Lane 2AB, 3AB: Wild; Lane 5: 1 kb ladder.
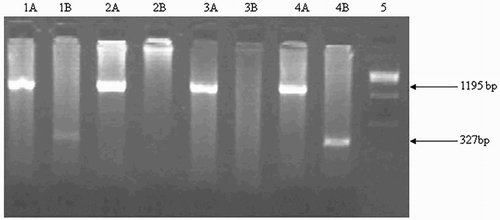
Figure 2. δβ-thalassemia break point B (3′ deletion). It was detected on 2% agarose gel. Explanation of the lanes is as follows; Lane 1AB and 2AB: Wild; Lane 3AB: Heterozygous; Lane 4: 100 bp ladder.
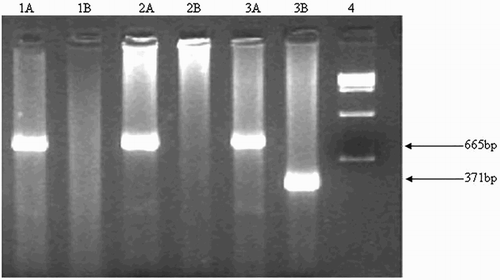
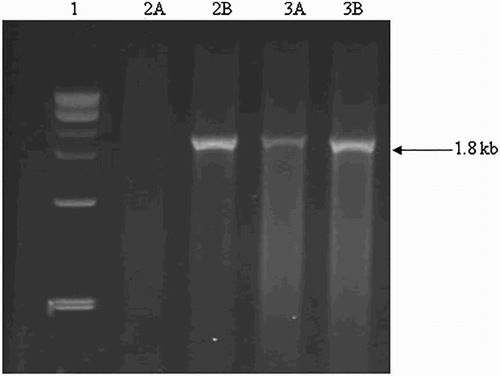
Figure 3. α-deletions in δβ thalassemia. It was detected on 2% agarose gel. Explanation of the lanes is as follows; Lane 1: 1 kb ladder; Lane 2A 2B: Wild; Lane 3A and 3B: Heterozygous.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Baysal E. HPFH and db-thalassemia conditions. Hemoglobin. 1993;17:569–589. doi: https://doi.org/10.3109/03630269309043499
- Weatherall DJ, Clegg JB. The thalassemia syndromes. 3rd ed. Oxford: Blackwell Scientific Publications; 1981. p. 875.
- Lim WF, Muniandi L, George E, et al. Hbf in HbE/β-thalassemia. a clinical and laboratory correlation. Hematology. 2015;20(6):349–353. doi: https://doi.org/10.1179/1607845414Y.0000000203
- Higgs DR, Thein SL, Wood WG, editors. The molecular pathology of the thalassemias. The thalassemia syndromes. 4th ed. Oxford: Blackwell Science; 2001, p. 133–191.
- Balgir RS. The burden of haemoglobinopathies in India and the challenges ahead. Curr Sci. 2000;79(11):1536–15347.
- Thein SL, Weatherall DJ. A non-deletion hereditary persistence of fetal hemoglobin (HPFH) determinant not linked to the beta-globin gene complex. Prog Clin Biol Res. 1989;316B:97–111.
- Nadkarni A, Wadia M, Gorakshakar A, et al. Molecular characterization of δβ-thalassemia and hereditary persistence of fetal haemoglobin in the Indian population. Haemoglobin. 2008;32(5):425–433. doi: https://doi.org/10.1080/03630260802341687
- Bollekens JA, Forget BC. Thalassemia and hereditary persistence of fetal hemoglobin. Hematol Oncol Clin North Am. 1991;5:399–422.
- Craig JE, Barnetson RA, Prior J, et al. Rapid detection of deletions causing delta beta thalassemia and hereditary persistence of fetal hemoglobin by enzymatic amplification. Blood. 1994;83(6):1673–1682.
- Baysal E, Huisman TH. Detection of common deletional alphathalassemia-2 determinants by PCR. Am J Hematol. 1994;46:208–213. doi: https://doi.org/10.1002/ajh.2830460309
- Smetanina NS, Huisman TH. Detection of alpha-thalassemia-2 (−3.7 kb) and its corresponding triplication ααα (anti-3.7 kb) by PCR: an improved technical change. Am J Hematol. 1996;53:202–203. doi: https://doi.org/10.1002/(SICI)1096-8652(199611)53:3<202::AID-AJH11>3.0.CO;2-F
- Chang JG, Lee LS, Lin CP, et al. Rapid diagnosis of alpha thalassemia-1 of Southeast Asia type and hydrops fetailis by polymerase chain reaction. Blood. 1991;78:853–854.
- Shaji RV, Eunice SE, Baidya S, et al. Determination of the breakpoint and molecular diagnosis of a common alpha-thalassaemia-1 deletion in the Indian population. Br J Haematol. 2003;123:942–947. doi: https://doi.org/10.1046/j.1365-141.2003.04704.x
- Wood WG. Hereditary persistence of fetal hemoglobin and δβ-thalassemia. In: Steinberg MH, Forget BG, Higgs DR, Nagel RL, editor. Disorders of hemoglobin: genetics, pathophysiology, and clinical management. Cambridge: Cambridge University Press; 2001. p. 356–388.
- Mayuranathan T, Rayabaram J, Das R, et al. Identification of rare and novel deletions that cause (δβ)0-thalassaemia and hereditary persistence of foetal haemoglobin in Indian population. Eur J Haematol. 2014;92(6):514–520. doi: https://doi.org/10.1111/ejh.12276
- Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann NY Acad Sci. 1998;850:38–44. doi: https://doi.org/10.1111/j.1749-6632.1998.tb10460.x
- Khunger JM, Gupta M, Singh R, et al. Haematological characterisation and molecular basis of Asian Indian inversion deletions delta beta thalassemia: a case report. J Clin Diagn Res. 2014 Sep;8(9):FD01–FD02.
- Supan F, Yutthana P, Satja S, et al. Molecular and hematological characterization of hereditary persistence of fetal hemoglobin-6/ Indian deletion – inversion Gγ(Aγδβ)0-thalassemia and Gγ(Aγδβ)0-thalassemia/HbE in Thai patients. Am J Hematol. 2002;71:109–113. doi: https://doi.org/10.1002/ajh.10202
- Carrocini GCS, Ondei LS, Zamaro PJA, et al. Evaluation of HPFH and δβ-thalassemia mutations in a Brazilian group with high Hb F levels. Mol Res. 2011;10(4):3213–3219. doi: https://doi.org/10.4238/2011.December.21.3
- Ottolenghi S, Giglioni B, Taramelli R, et al. Molecular comparison of δβ thalassemia and hereditary persistence of fetal hemoglobin DNAs: evidence of a regulatory area? Proc Natl Acad Sci USA. 1982;79(7):2347–2351. doi: https://doi.org/10.1073/pnas.79.7.2347
- Galanello R, Cao A. Relationship between genotype and phenotype; thalassemia intermedia. Ann NY Acad Sci. 1998;850(850):325–333. doi: https://doi.org/10.1111/j.1749-6632.1998.tb10489.x
- Tan Jin Ai MA, Yap SF, Tan KL, et al. Mild beta-thalassemia intermedia caused by compound heterozygosity for G(A)o/-thalassemia and molecular characterization of the defect in four Chinese families. Acta Haematol. 2003;109(4):169–175. doi: https://doi.org/10.1159/000070965
- Pandey S, Pandey S, Ranjan R, et al. Phenotypic heterogeneity of Asian Indian inversion deletions Gγ(aγδβ)0 breakpoint A and breakpoint B. Ind J Clin Biochem. 2013;28(1):98–101. doi: https://doi.org/10.1007/s12291-012-0232-9