
ABSTRACT
Objectives: To summarize the effects of the bone marrow niche on hematopoiesis and leukemogenesis and discuss the chemotherapy resistance that can arise from interactions between the niche and leukemia stem cells.
Methods: We review the major roles of the bone marrow niche in cell proliferation, adhesion and drug resistance. The signaling pathways and major molecular participants in the niche are discussed. We also address potential niche-targeting strategies for the treatment of acute myeloid leukemia (AML).
Results: The bone marrow niche supports normal hematopoiesis and affects acute myeloid leukemia (AML) initiation, progression and chemotherapy resistance.
Discussion: AML is a group of heterogeneous malignant diseases characterized by the excessive proliferation of hematopoietic stem and/or progenitor cells. Even with intensive chemotherapy regimens and stem cell transplantation, the overall survival rate for AML is poor. The bone marrow niches of malignant cells are remodeled into a leukemia-permissive environment, and these reformed niches protect AML cells from chemotherapy-induced cell death. Inhibiting the cellular and molecular interactions between the niche and leukemia cells is a promising direction for targeted therapies for AML treatment.
Conclusions: Interactions between leukemia cells and the bone marrow niche influence hematopoiesis, leukemogenesis, and chemotherapy resistance in AML and require ongoing study. Understanding the mechanisms that underlie these interactions will help identify rational niche-targeting therapies to improve treatment outcomes in AML patients.
Introduction
Acute myeloid leukemia (AML) is a malignant clonal disease of hematopoietic stem and progenitor cells (HSPCs). Despite aggressive chemotherapy and stem cell transplantation, the prognosis for young patients with AML remains unsatisfactory: the 5-year overall survival rate is approximately 40%. The survival rate in adults more than 60 years old is also poor, and 60%–80% of patients eventually die of refractory and/or relapsed leukemia. In older adults aged 65–70, the 5-year survival rate is only 10% [Citation1]. Residual leukemic stem cells (LSCs) in the bone marrow niche are less sensitive to chemotherapy and are a major clinical factor in disease relapse [Citation2–4]. To prolong overall survival in patients with AML, new strategies other than epigenetic modification and immunomodulation are needed. Treatment strategies that attempt to eliminate leukemia cells by focusing on the interaction between LSCs and the bone marrow niche might be useful clinically [Citation5–7].
Bone marrow niche and its effects on hematopoiesis and leukemogenesis
The bone marrow microenvironment, also known as the bone marrow niche, was first proposed by Schofield in 1978 [Citation8]. It contains the bone marrow mesenchymal stem and progenitor cells, osteolineage cells, sinusoidal endothelium, perivascular stromal cells, adipocytes, unmyelinated Schwann cells, immune cells (mainly regulating T cells), and many other components, including a variety of adhesion factors, growth factors, and chemokines [Citation9]. The bone marrow niche influences the biological behavior of hematopoietic stem cells (HSCs) via different signaling cascades and maintains normal hematopoiesis. HSCs can be localized in the endosteal region adjacent to osteoblasts and in the vascular area, which is full of sinusoid endothelial cells (ECs) and perivascular cells. As shown in , these two zones are defined as the endosteal and vascular niches, respectively, and they act on HSCs synergistically to regulate cell self-renewal, proliferation and differentiation [Citation9].
Figure 1. Bone marrow niche and associated cellular interactions. HSCs interact with and are regulated by an intricate and vibrant multicellular bone marrow niche. They have close anatomical and functional relationships with cells in the bone marrow. The players in the bone marrow niche include cytokines, the extracellular matrix, adhesion factors, and the sympathetic nervous system. Osteolineage cells, immune cells, stromal cells, and neurons are collectively involved in several ligand–receptor interactions such as CXCL12/CXCR4, VLA-4/VCAM-1, and the Ang-1/Tie2 axis, which orchestrate the colonization, differentiation, and homing of HSCs. OB, osteoblast; CAR cell, CXCL12-abundant reticular cell; T-reg, regulatory T cell; Opn, osteopontin; G-CSF, granulocyte-colony stimulating factor; GM-CSF, granulocyte macrophage-colony stimulating factor; CXCL12, CXC chemokine ligand 12; CXCR4, receptor for CXCL12; VCAM-1, vascular cell adhesion molecule 1; VLA-4, very late antigen 4; Ang-1, Angiopoietin-1; IL, interleukin.
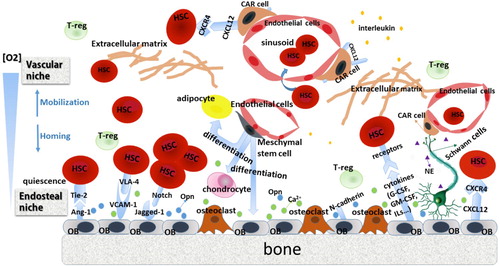
The endosteal niche is composed of sinusoidal endothelium, perivascular stromal cells, and osteolineage cells (osteoblasts and osteoclasts) [Citation3]. Osteoblasts, cover the endosteum and form an interface between the bone and the marrow. There are two types of osteoblasts: spindle-shaped N-cadherin+ osteoblasts (SNO) and oval-shaped osteoblasts [Citation10]. These cells provide a haven for quiescent HSCs. The binding of HSCs with osteoblasts, especially SNO cells, maintains cell dormancy and is mediated by adhesion factors involved in the Ang-1/Tie2 and TPO/MPL signaling pathways [Citation11,Citation12]. Osteoblast ablation can provoke quiescent cells to enter the proliferating cycle [Citation13]. Osteoblasts also play a vital role in regulating HSC content [Citation14]. Myriad studies have demonstrated the close relationship between osteoblasts and hematopoiesis. Osteoblast-deficient transgenic mice exhibits reduced bone formation and a hematopoietic failure in bone marrow accompanied by enhanced extramedullary hematopoiesis [Citation15]. A transgenic mouse model has been generated to produce osteoblast-specific, activated PTH/PTHrP receptors (PPRs) under the control of the alpha-1 collagen promoter [Citation16]. The osteoblastic cell count in the trabecular bone of the col1-caPPR mice was much higher than that of the controls, leading to hematopoietic cell growth through Notch activation. Schaniel et al. [Citation17] established a mouse model that constitutively expressed Wnt inhibitory factor 1 (Wif1, a pan Wnt inhibitor) on a specific promoter region of osteoblasts. They found that the numbers of HSPCs in bone marrow and the spleen increased sharply, along with an increase in the proportion of dividing HSPCs. The bone morphogenetic protein (BMP) signal regulated the HSC pool through the BMP receptor type IA (BMPRIA) [Citation10]. Osteoblast-enriched ALCAM+Sca-1− cells, a subgroup isolated from CD45−CD31−Ter119− endosteal cells, significantly promoted the long-term reconstitution activity of HSCs. The expression levels of osteoblastic markers and relevant adhesion molecules such as Runt-related transcription factor 2 (Runx2), CD44, VCAM-1, Cadherin 11, Cdh2, and Alcam were also comparatively increased [Citation18]. N-cadherin and β-catenin have been shown to be requisite adhesion molecules that mediated the interaction between SNO cells and long-term HSCs [Citation10]. Direct regulation of erythrocytes and an indirect effect on B lymphopoiesis by osteoblasts has also been demonstrated [Citation19,Citation20]. However, osteoblasts can also negatively affect the proliferation of HSCs. Increased bone marrow cellularity and the higher proportion of cycling hematopoietic progenitor cells in osteopontin-null (Opn−/−) mice compared to wild-type controls suggest that Opn has a suppressive effect on HSCs. The number of in vitro CD34+CD38− cells cultured with thrombin-cleaved bovine Opn (tcOpn) decreased at the trend level [Citation21]. In general, osteoblasts derived from multipotent mesenchymal stromal cells (MSCs) sustain long-term HSC survival, proliferation, and quiescence by secreting various cytokines, chemokines, and signaling molecules.
The vascular niche, also known as the endothelial niche, comprises arterioles, capillaries, and fenestrated sinusoidal vessels. It mainly consists of vascular ECs, and CXCL12 abundant reticular cells (CAR cells), which promote the differentiation and proliferation of short-term HSCs [Citation22,Citation23]. As soon as HSCs are mobilized, they leave the intima to reach the vascular area, which is associated with hematopoietic reconstitution. The HSCs have an intimate relationship with ECs. Specialized ECs support HSC maintenance, differentiation, and self-renewal via multiple angiocrine factors and play an active role in regulating the homing and trafficking of HSCs via the CXCR4/CXCL12 and RANK/RANKL pathways [Citation24]. The CAR cells overexpress vascular cell adhesion molecule-1 (VCAM-1), E-/P-selectin, CD44, interleukins (ILs), and platelet-derived growth factor (PDGF), all of which have regulatory roles in cell adhesion and angiogenesis [Citation25].
Alterations of niche cells can contribute to disease initiation or progression. For example, elevated numbers of granulocyte/macrophage progenitors in the spleen, peripheral blood, and bone marrow have been found in RARγ-deficient mice. When these bone marrow progenitors were engrafted onto wild-type mice, the wild-type mice did not develop a myeloproliferative-like disease. These results suggest that the myeloproliferative syndrome phenotype is at least partially caused by a defective bone marrow microenvironment, rather than by the mutation of hematopoietic cells [Citation26]. Genetic alterations in niche cells may also contribute to hematopoietic disorders. The loss of the miRNA processing endonuclease Dicer1 from osteoprogenitors in a mouse model resulted in many myelodysplastic features, but transplantation of these hematopoietic cells into wild-type mice did not result in any abnormalities [Citation27]. The mutation of β-catenin in osteoblasts stimulated the expression of Jagged-1, and the activation of the Notch signaling pathway induced AML development with chromosomal alterations [Citation28].
Similar to their healthy counterparts, malignant cells reside in a complicated, self-strengthening leukemic microenvironment. They alter the bone marrow niche at the expense of normal hematopoiesis to facilitate their own growth and reinforce neoplastic disease processes. The endothelial niche can be hijacked by leukemia cells and it has been demonstrated that AML cells integrate into the vascular endothelium by cell fusion or attachment and become dormant [Citation29]. Increased microvascular density and levels of angiogenesis have been found in AML compared to normal bone marrow samples [Citation30]. The ECs also support the proliferation of leukemia blasts and are actively involved in leukemogenesis. The co-culture of AML cells with microvascular ECs resulted in the proliferation of malignant cells and exacerbated the AML phenotype. The co-culture also established a chemoprotective niche, characterized by the high expression of soluble factors, including IL-3, IL-6, stem cell factor (SCF), granulocyte-colony stimulating factor (G-CSF), and granulocyte macrophage-colony stimulating factor (GM-CSF). In a complementary study, the abnormal secretion of these angiocrine factors potently inhibited the apoptosis of leukemia cells and promoted their growth via paracrine mechanisms [Citation31]. However, inhibition of angiocrine receptors abrogated chemotherapy-induced angiogenesis and successfully reversed the resistance of AML [Citation32].
Neural regulation also plays an important role in the development of myeloid malignancies. In a mouse model, damaging the sympathetic nervous system increased the infiltration of leukemia, increased adrenergic signaling transduced by Adrβ2-supported leukemogenesis and regulated the fate of LSCs [Citation33].
Taken together, these studies suggest that hematopoietic cells and niche cells work synergistically in healthy states and hematopoietic malignancies.
Role of the bone marrow niche in chemotherapy resistance
The bone marrow niche mediates cell resistance to chemotherapy via small molecules secreted by osteoblasts, ECs, and stromal cells and by direct cell-to-cell contact mediated by adhesion factors. Accordingly, drug resistance can be either soluble factor-mediated drug resistance (SM-DR) or cell adhesion-mediated drug resistance (CAM-DR). As noted above, soluble molecules in the bone marrow niche include CXCL12, VEGF, IL-6, FGF, G-CSF, and others. These molecules mediate chemotherapy resistance via different mechanisms. Vascular endothelial growth factor C (VEGF-C) facilitates leukemia progression with a higher ratio of Bcl-2 to Bax, which suggests suppressive effects on cell apoptosis. It has also been shown to rescue leukemia cells from chemotherapy-induced cell death [Citation34]. The contribution of osteoblasts to chemotherapy resistance in AML includes aspects of both SM-DR and CAM-DR. Osteoblasts have been shown to protect AML cells from CXCL12-induced cell death through a soluble factor-mediated mechanism in both CXCL12-expressing AML cell lines, and clinical patient samples [Citation35]. In addition, HSCs and leukemic cells tightly adhere to osteoblasts on the bone surface or stroma via a series of adhesion factors (integrins and cadherins) and extracellular matrix proteins (fibronectin and osteopontin). These cell-to-cell or cell-to-matrix interactions play a critical role in the process of mobilization and homing and in drug resistance and clinical recurrence [Citation3]. The bone marrow niche is a vital source of factors that facilitate the survival of malignant cells and enable the development of minimal residual disease (MRD). Several signal transduction pathways are highly associated with shielding the bone marrow niche from malignant cells. Next, we discuss recent concepts concerning how these networks impinge on leukemia cell proliferation, survival, and drug resistance ().
Figure 2. Crosstalk between leukemia cells and niche cells associated with drug resistance. Niche cells can be hijacked during leukemogenesis, and malignant cells depend upon the endosteal or stromal components in the microenvironment. The drug resistance of leukemia cells arises from their direct physical contact with the stroma and is conferred by soluble factors. Cell–cell adhesion and multiple cytokines or growth factors affect malignant cells, at least in part, by evoking diverse intracellular pro-survival cascades and by increasing ABC transporter-mediated drug efflux. FN, fibronectin; ECM, extracellular matrix; ABC transporter, ATP-binding cassette transporter; MRP1, multidrug resistance-associated protein 1.
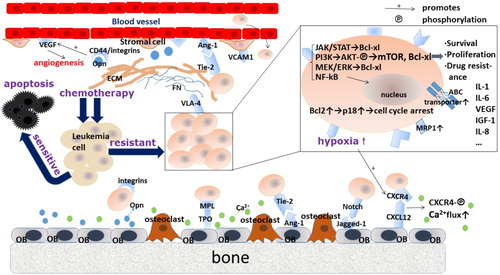
Interaction between VLA-4 and VCAM-1 or fibronectin
The adherence of AML cells to the bone marrow microenvironment via VLA-4 leads to CAM-DR. VLA-4 is an integrin dimer, α4β1, which is located on leukemic cells. Fibronectin (FN) is a major component of the extracellular matrix and is secreted by various cells. The VLA-4–FN interaction plays an instrumental role in chemokine-mediated homing and mobilization [Citation36] and is closely related to MRD. Matsunaga et al. [Citation37] found that AML cells coated with FN had lower apoptosis rates and higher survival rates than those cultured with BSA in vitro. AML patients who are VLA-4 negative have much higher overall survival rates than those with high VLA-4 expression. The negative prognostic value of VLA-4 in AML might imply that physical contact between fibronectin and VLA-4-positive AML cells reduces chemotherapy sensitivity and provides a sanctuary for MRD, ultimately resulting in a shorter survival time. With anti–VLA-4–specific antibodies, cells from VLA-4–positive AML patients co-cultured with stromal cells or FN tend to be more sensitive to chemotherapy than those without VLA-4 antibodies. Poulos et al. [Citation38] have demonstrated that ECs establish a fertile niche, activation of ECs by VEGF-A leads to the expansion of AML cells and increases the number of EC-adherent leukemia cells partly via VLA-4/VCAM-1 interaction. Furthermore, ECs stimulated by VEGF-A conferred chemotherapeutic protection on AML cells. In contrast, the inhibition of VEGFR2 led to a profound increase in chemosensitivity.
CXCL12/CXCR4 pathway
For CAM-DR, the CXCL12/CXCR4 axis is indispensable. Protected by the marrow stroma, residual leukemia cells show little sensitivity to traditional chemotherapy. This sensitivity is mainly mediated by the crosstalk between CXCL12 (also known as stromal cell-derived factor-1, SDF-1) and its receptor CXCR4. CXCL12 is widely expressed by CAR cells, osteoblasts, fibroblasts, and ECs [Citation39], and it usually binds with chemokine receptor CXCR4, which belongs to a G-protein coupled receptor. The binding of CXCL12 to CXCR4 can trigger the phosphorylation of CXCR4, promote calcium flux, and activate some downstream pro-survival signaling pathways, including the MEK/ERK, JAK/STAT, and PI3K/AKT axes [Citation40]. Rombouts et al. [Citation41] found that CXCR4 levels in AML patients with an FMS-like tyrosine kinase-3/internal tandem duplication (Flt3/ITD) mutation were markedly increased. A subsequent study demonstrated that CXCR4 might act as a useful indicator of an unfavorable prognosis even in AML with a normal karyotype [Citation42]. The CXCL12/CXCR4 interaction is highly involved in cell survival, adhesion, and migration [Citation43,Citation44]. CXCL12 is considered a potent chemoattractant for both normal hematopoietic cells and leukemic cells [Citation45]. CXCR4 activation has been shown to be a prerequisite for the migration of mobilized AML cells beneath marrow stromal cells [Citation46]. As such, malignant cells tend to use CXCR4 to access the protective niches that are usually restricted to normal HSCs and thereby reside in a safe haven that favors their survival and promotes resistance to conventional chemotherapies. CXCR4-deficient mice showed aberrant B-lymphopoiesis, defective myelopoiesis [Citation47], and restored sensitivity to the myelosuppressive antimetabolite 5-fluorouracil (5-FU) [Citation48]. Osteoblasts also had a protective effect on AML cells and induced cell cycle arrest in MV4–11 cell lines. The combination of G-CSF and AMD3100, a CXCR4 blocker, successfully inhibited cell adhesion and migration, promoted cell division in AML and disrupted the interaction between the endosteal niche and leukemia cells [Citation49]. Sison et al. [Citation50] demonstrated that the upregulation of surface CXCR4 on AML cells was a possible mechanism of drug resistance, and AMD3100 reversed the resistance and diminished stromal protection. Inhibition of the CXCR4 pathway can disturb cell–matrix communication and induce malignant cells to mobilize from the endosteal sanctuary. Such a perturbation might eventually overcome the protection of leukemic cells by niche cells and sensitize them to chemotherapeutic agents, ultimately decreasing the tumor burden. Another small-molecule reversible antagonist of CXCR4 is AMD3465, which also successfully abrogated protection against chemotherapy-induced cell apoptosis and enhanced the chemosensitivity of AML cells both in vivo and in vitro [Citation51,Citation52]. These results suggest a potential role for CXCR4-targeting molecules in the treatment of AML.
TPO/MPL signaling pathway
The thrombopoietin (TPO) receptor, also known as myeloproliferative leukemia protein (MPL), was first identified from the myeloproliferative leukemia virus (v-mpl). The TPO/MPL signaling pathway plays an important role in many physiologic and pathologic processes. TPO, which is also secreted by osteoblasts, is the ligand of MPL and has long been known to regulate the process of megakaryopoiesis. TPO/MPL signaling is vital for HSC hibernation and self-renewal. MPL+ AML cells exhibit clonogenic potential and are associated with drug resistance [Citation53]. Exogenous TPO maintains cell dormancy and promotes HSC proliferation [Citation54]. The neutralization of MPL significantly decreased the total HSC cell count and the number of dormant HSCs in the endosteal niche, together with down-regulation of cell-cycle-related genes such as p57kip2 and Tie2. Exogenous TPO also caused S phase entry, and the expression levels of p57kip2 and Tie2 genes increased at the trend level [Citation55]. TPO plays a prominent role in hematopoietic malignancies and regulates leukemogenesis. The levels of TPO and c-MPL were found to be notably increased in AML patients, and increased expression of TPO secreted by BMSC-derived osteoblasts from AML patients was also observed. After treatment with daunorubicin(DNR), the viability of an AML cell line HEL was the highest when they were co-cultured with osteoblasts originated from AML patients, while HEL-single cultured group showed the lowest survival rate [Citation12]. These results suggest that upregulation of TPO leads to DNR resistance, although the underlying mechanisms still need to be clarified. The TPO/c-MPL pathway participates in LSC stemness and cell dormancy, which can partially explain why malignant cells seem to escape chemotherapeutic cytotoxicity. This signaling pathway could be a promising therapeutic target for refractory and relapsed leukemia, and associated inhibitors or specific antibodies might enhance chemotherapy sensitivity in AML.
Ang-1/Tie-2 and associated molecules
The interaction between angiopoietin-1 (Ang-1) in osteoblasts and Tie-2 on HSCs also plays a pleiotropic role in cell adhesion, survival, and quiescence. Tie-2 is a unique receptor tyrosine kinase and can also be expressed by ECs and AML cells [Citation56]. Tie-2 dysfunction might induce cancer development, metastasis and drug resistance. In hematologic neoplasms, elevation of Ang-1 is related to higher disease risk and shorter survival time in AML or MDS patients. Once Ang-1 binds to ECs, Tie-2 is phosphorylated, which promotes the activation of the phosphatidylinositol 3-kinase (PI3-K)/Akt signaling pathway, and the knockdown of Tie-2 leads to decreased viability of malignant cells [Citation57]. Arai et al. [Citation11] found that Tie-2+ HSCs treated by exogenous Ang-1 became flattened and tended to cling tightly to the bone surface. Mice transplanted with Tie-2+ c-kit+Sca-1+Lin− (KSL)-side-population (SP) cells cultured with SCF, TPO, and Ang-1 retained much higher levels of donor cells than cells not cultured with Ang-1. The cells also showed an inhibitory effect on cell division, thus maintaining self-renewal. Those SP cells can be regarded as LSCs in the leukemic situation and may help malignant cells evade antileukemic therapy. Ang-1 was also found to promote cell cycle arrest in EVl1 high leukemia cells, via maintenance of the G0/G1 phase through p18 upregulation [Citation58]. Similar studies have also shown that the CDK inhibitors p57 and p18 increased after HSCs were treated with Ang-1, suggesting that Ang-1/Tie-2 signaling can be a potent cell cycle regulator [Citation59]. Considering that drug resistance largely depends on cell dormancy in the bone marrow niche, activation of the Ang-1/Tie-2 axis could be a feature of AML chemotherapy resistance and could contribute to adverse prognosis.
Akt pathway
Akt, also known as protein kinase B (PKB), was initially discovered as the oncogene in the transforming retrovirus AKT8 [Citation60]. It is known to suppress cell apoptosis, regulate proliferation, and participate in cancer invasion and metastasis. Akt activation promotes HSC proliferation and might induce myeloproliferative disease, T-cell lymphoma, and leukemic transformation [Citation61]. It can also phosphorylate FOXO transcription factors, which promote the translocation of FOXO proteins from the nucleus to the cytoplasm [Citation62]. This interesting phenomenon is an important mechanism to protect HSCs from noxious stresses, lower cell metabolism, and maintain a quiescent state [Citation63]. In AML, up-regulation of the Akt pathway might predict poor prognosis and shorter survival time by regulating the expression of proteins associated with chemotherapy resistance, such as multidrug resistance-associated protein 1 (MRP1) and the membrane ATP-binding cassette (ABC) transporter [Citation64]. Intriguingly, Tamburini et al. [Citation65] found that constitutive activation of the PI3K/Akt pathway led to a favorable outcome in de novo AML patients. A possible mechanism for these outcomes is that PI3K promotes entry of LSCs into the S phase, thus improving susceptibility to chemotherapy. A more comprehensive understanding of the PI3K/Akt/mTOR pathway should be sought.
Hypoxia and the HIF-1α pathway
Oxygen in cells is unevenly distributed with levels increasing on a gradient from the endosteum to sinusoids [Citation66]. The levels of hypoxia-inducible factor 1α (HIF-1α), considered a marker of hypoxia, show the opposite pattern. The role of hypoxia and HIF-1α in hematologic malignancies remains controversial. In some studies, leukemogenesis acceleration and enhancement of an even more aggressive MPN phenotype was observed in transgenic mice with a Hif-1α deletion [Citation67,Citation68]. In contrast, hypoxia also induced cell cycle arrest and weakened the chemotherapy sensitivity of AML cells, which might have been regulated by a downstream-activated PI3K/Akt pathway and some antiapoptotic proteins [Citation69]. Activation of the hypoxia/HIF-1α pathway also facilitated angiogenesis and enhanced cytokine secretion, both of which might induce chemotherapy resistance. Oxygen concentration is considered a determinant parameter in the regulation of the function and expression of CXCR4, which correlates with drug resistance and overall survival in AML patients. In a reduced pO2 environment (6% O2), the overexpression of CXCR4 was detected in patient samples and in AML cell lines, and increasing the O2 concentration resulted in decreased CXCR4 expression, alterations of lipid rafts by the depletion of cholesterol content in the cellular membrane, and increased shedding of CXCR4+ microparticles [Citation70]. Hypoxia regulates stem cell fates and plays an indirect but vital role in tumor progression, metastasis, and drug resistance in hematopoietic malignancies [Citation71].
Osteopontin
Osteopontin (Opn) is an extracellular matrix glycoprotein secreted by endosteal osteoblasts. It plays a key role in cell dormancy, invasion and the development of MRD. Diverse receptors expressed on the cell surface can interact with Opn, including CD44 and various integrins, including α4β1, α5β1, α9β1, and others. The β3 integrins play a vital role in leukemogenesis and the chemoresistance of AML [Citation72]. Retrospective studies have found a correlation between high levels of Opn expression and poor prognosis in AML patients [Citation73,Citation74]. Overexpression of Opn b and c isoforms led to angiopoiesis, and these isoforms might prevent the cell apoptosis induced by conventional chemotherapy in AML [Citation75]. A bone marrow niche established an Opn-rich area can specifically recruit Opn-expressing blasts. In turn, leukemic cells secrete Opn into the niche. Opn regulation probably entails positive feedback through host-tumor cell interactions. Opn can also expand the quiescent microenvironment and endow leukemic blasts with resistance to chemotherapy. A blockade of the Opn signaling pathway markedly increased the proportion of cycling cells and inhibited cell homing, which ultimately led to a higher tumor load. Injection of anti-Opn into engrafted mice followed by treatment with Ara-C decreased the MRD burden compared with Ara-C chemotherapy alone. The Opn neutralizer-treated mice might be vulnerable to cytotoxic stresses [Citation76].
The bone marrow microenvironment can support cell survival, differentiation, and proliferation. It can also maintain cell dormancy and mediate drug resistance via various signal transduction pathways, such as the interplay between VLA-4 and VCAM-1, the SDF-1/CXCR4 axis, and the TPO/c-MPL pathway. Many adhesion molecules, soluble factors and cell components in the bone marrow niche are interconnected and ultimately form a sophisticated network.
Drug resistance mediated by leukemic stem cells
We cannot ignore the role of LSCs in the bone marrow microenvironment and the pathogenesis of myeloid malignancies. LSCs share certain properties with HSCs, including strong self-renewal capacities and multilineage differentiation. They can escape from apoptosis due to the remodeling of osteoblasts. Dormant LSCs are believed to underlie disease relapse and chemotherapy resistance in AML [Citation77]. By interacting with hematopoietic bone marrow niches, LSCs maintain their quiescence and self-renewal capacity. They can tolerate many cell cycle specific cytotoxic agents. Saito demonstrated that pretreatment with G-CSF dramatically reduced the number of CD34+CD38− LSCs in the G0 phase and was accompanied by an increase in actively dividing LSCs, which induced cell apoptosis and enhanced chemotherapy sensitivity. This finding suggests new possibilities for AML treatment [Citation78]. Relapse after chemotherapy is also characterized by more heterogeneous and complicated LSC populations [Citation79].
Therapeutic targeting of the bone marrow niche
Enhancing the sensitivity of AML cells to chemotherapy continues to be an active area of clinical research. In addition to novel cytotoxic chemotherapy agents, epigenetic modifiers of transcription and cell cycle regulation are increasingly the focus of research. As knowledge about LSCs and the bone marrow microenvironment has progressed, the protective effect of bone marrow stromal cells on leukemia cells and how these cells and microenvironments regulate chemotaxis, adhesion, and homing have been increasingly well understood. Addressing the combined effect of chemotherapy and specific drugs targeting bone marrow niches on leukemia cells can be a potential therapeutic direction to diminish MRD.
The first preclinical trial focusing on the CXCL12/CXCR4 axis was conducted in 2009. It was designed to block the interactions of leukemic cells with niche cells using the CXCR4 inhibitor AMD3465 to overcome resistance to chemotherapy in AML [Citation51]. AMD3465 successfully inhibited the migration of primary AML cells and ameliorated the stroma-mediated cytoprotection against spontaneous apoptosis. In in vivo systems that mimic the physiologic microenvironment, AMD3465 promoted cell mobilization and enhanced antitumor effects, contributing to a reduced AML disease burden. Recently, the combination of plerixafor (AMD3100) with mitoxantrone, etoposide and AraC (MEC) was used in 52 AML patients. It achieved a relatively satisfactory complete remission (CR) rate of 39% compared with 21% for the MEC regimen (NCT00512252). The main pharmacological mechanism disrupted the CXCR4/CXCL12 interaction and blocked downstream signals such as the PI3K/AKT and MAPK pathways. The mobilization rate of leukemic blasts also doubled without leukostasis, which may benefit patients with refractory or relapsed AML by augmenting chemosensitivity [Citation80]. Another Phase 2a trial (NCT 01838395) focused on the CXCR4 antagonist BL-8040 (BKT140) and found an effect on the mobilization of AML blasts. When BL-8040 was combined with Ara-C in patients with relapsed or refractory AML, the differentiation of granulocytes increased, and the CR rates improved significantly [Citation81]. Other CXCR4 antagonists and antibodies, including LY2510924, CX-01, POL6326, and NOX-A12, also potently hampered cell growth and produced a sustained pharmacodynamic effect on mobilization [Citation82–85]. These trial results indicate that blockade of the CXCR4/CXCL12 axis could be used to supplement conventional chemotherapy. Other studies examining evolving targets that might help to restore chemotherapy sensitivity in AML are ongoing. Fogler et al. [Citation86] first reported that a dual CXCR4/E-selectin inhibitor, GMI-1359, significantly promoted mobilization in a mouse model of FLT3-ITD+ AML. The dual inhibitor functioned better than an antagonist aimed at E-selectin or CXCR4 alone, and is considered an innovative drug to prolong survival of FLT3-mutated AML. GMI-1271 is a specific E-selectin antagonist that targets interactions between the bone marrow niche and cancer cells. Its safety, efficacy, and pharmacokinetics in AML with a combination of MEC or idarubicin and cytarabine (DA 7 + 3; cytarabine for 7 days and idarubicin for 3 days) regimen have been studied (NCT 02306291) [Citation87]. In response to the critical role of the VLA4/FN interaction in CAM-DR, the FNIII14 peptide was synthesized. It restored chemotherapy sensitivity in AML and eradicated MRD with the help of a cytotoxic drug. No severe treatment-associated adverse effects were found [Citation88]. AS-101, a VLA-4 inhibitor, effectively abrogated AML chemoresistance by redox inactivation and might improve the survival rates of AML patients [Citation89]. CD47 is widely expressed on cancer cells, and its monoclonal antibody Hu5F9-G4 has been synthesized successfully. A Phase Ib trial (NCT03248479) will evaluate the tolerability and efficacy of Hu5F9-G4 alone or combined with azacitidine in relapsed/refractory AML and high-risk MDS [Citation90]. Finally, because of the hypoxic nature of the bone marrow niche, the hypoxia-activated prodrug (HAP) evofosfamide (TH-302) has been developed. TH-302 single-drug therapy has shown additive advantages in patients with advanced leukemia [Citation91] ().
Table 1. Summary of drugs that target the bone marrow niche in AML.
Discussion and outlook
AML remains a life-threatening hematopoietic malignancy, and disease relapse is a difficult problem for clinicians and patients. The DA protocol is the keystone for induction therapy. Personalized treatments for AML, especially after disease relapse, are rapidly developing. The role of the bone marrow microenvironment in leukemia relapse is garnering increased attention. Leukemia cells can remodel the niche and this remodeling is likely to favor malignant cell expansion over normal HSCs and induce leukemic transformation. Future studies on the interactions between LSCs and the bone marrow microenvironment might result in changes to leukemia therapy strategies from conventional cytotoxic chemotherapy to targeted therapies that include adhesion factors, chemokines, and their receptors. A niche-targeted therapeutic strategy is attractive but faces challenges to avoid eliminating normal HSCs, which would be an adverse outcome. In addition, whether a specific target is appropriate for all genetic subtypes of AML remains to be determined. Although new relevant niche-directed therapies have already been reported, the intrinsic mechanisms and regulatory networks require further investigation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Isidori A, Salvestrini V, Ciciarello M, et al. The role of the immunosuppressive microenvironment in acute myeloid leukemia development and treatment. Expert Rev Hematol. 2014;7(6):807–818. doi: 10.1586/17474086.2014.958464
- Riether C, Schurch CM, Ochsenbein AF. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. 2015;22(2):187–198. doi: 10.1038/cdd.2014.89
- Tabe Y, Konopleva M. Advances in understanding the leukaemia microenvironment. Br J Haematol. 2014;164(6):767–778. doi: 10.1111/bjh.12725
- Chiarini F, Lonetti A, Evangelisti C, et al. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: from biology to therapeutic targeting. Biochim Biophys Acta. 2016;1863(3):449–463. doi: 10.1016/j.bbamcr.2015.08.015
- Wouters BJ, Delwel R. Epigenetics and approaches to targeted epigenetic therapy in acute myeloid leukemia. Blood. 2016;127(1):42–52. doi: 10.1182/blood-2015-07-604512
- Kornblau SM, Womble M, Qiu YH, et al. Simultaneous activation of multiple signal transduction pathways confers poor prognosis in acute myelogenous leukemia. Blood. 2006;108(7):2358–2365. doi: 10.1182/blood-2006-02-003475
- Austin R, Smyth MJ, Lane SW. Harnessing the immune system in acute myeloid leukaemia. Crit Rev Oncol Hematol. 2016;103:62–77. doi: 10.1016/j.critrevonc.2016.04.020
- Schofield R. The relationship between the spleen colony-forming cell and the haemopoietic stem cell. Blood Cells. 1978;4(1–2):7–25.
- He N, Zhang L, Cui J, et al. Bone marrow vascular niche: home for hematopoietic stem cells. Bone Marrow Res. 2014;2014:128436. doi: 10.1155/2014/128436
- Zhang J, Niu C, Ye L, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425(6960):836–841. doi: 10.1038/nature02041
- Arai F, Hirao A, Ohmura M, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118(2):149–161. doi: 10.1016/j.cell.2004.07.004
- Dong-Feng Z, Ting L, Yong Z, et al. The TPO/c-MPL pathway in the bone marrow may protect leukemia cells from chemotherapy in AML patients. Path Onc Res. 2014;20(2):309–317. doi: 10.1007/s12253-013-9696-z
- Zhao M, Li L. Osteoblast ablation burns out functional stem cells. Blood. 2015;125(17):2590–2591. doi: 10.1182/blood-2015-03-633651
- Taichman RS, Reilly MJ, Emerson SG. The hematopoietic microenvironment: osteoblasts and the hematopoietic microenvironment. Hematology. 1999;4(5):421–426. doi: 10.1080/10245332.1999.11746468
- Visnjic D, Kalajzic Z, Rowe DW, et al. Hematopoiesis is severely altered in mice with an induced osteoblast deficiency. Blood. 2004;103(9):3258–3264. doi: 10.1182/blood-2003-11-4011
- Calvi LM, Adams GB, Weibrecht KW, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–846. doi: 10.1038/nature02040
- Schaniel C, Sirabella D, Qiu J, et al. Wnt-inhibitory factor 1 dysregulation of the bone marrow niche exhausts hematopoietic stem cells. Blood. 2011;118(9):2420–2429. doi: 10.1182/blood-2010-09-305664
- Nakamura Y, Arai F, Iwasaki H, et al. Isolation and characterization of endosteal niche cell populations that regulate hematopoietic stem cells. Blood. 2010;116(9):1422–1432. doi: 10.1182/blood-2009-08-239194
- Rankin EB, Wu C, Khatri R, et al. The HIF signaling pathway in osteoblasts directly modulates erythropoiesis through the production of EPO. Cell. 2012;149(1):63–74. doi: 10.1016/j.cell.2012.01.051
- Wu JY, Purton LE, Rodda SJ, et al. Osteoblastic regulation of B lymphopoiesis is mediated by Gsalpha-dependent signaling pathways. Proc Natl Acad Sci USA. 2008;105(44):16976–16981. doi: 10.1073/pnas.0802898105
- Nilsson SK, Johnston HM, Whitty GA, et al. Osteopontin, a key component of the hematopoietic stem cell niche and regulator of primitive hematopoietic progenitor cells. Blood. 2005;106(4):1232–1239. doi: 10.1182/blood-2004-11-4422
- Kopp HG, Avecilla ST, Hooper AT, et al. The bone marrow vascular niche: home of HSC differentiation and mobilization. Physiol (Bethesda). 2005;20:349–356.
- Konopleva MY, Jordan CT. Leukemia stem cells and microenvironment: biology and therapeutic targeting. J Clin Oncol. 2011;29(5):591–599. doi: 10.1200/JCO.2010.31.0904
- Rafii S, Shapiro F, Pettengell R, et al. Human bone marrow microvascular endothelial cells support long-term proliferation and differentiation of myeloid and megakaryocytic progenitors. Blood. 1995;86(9):3353–3363.
- Guerrouahen BS, Al-Hijji I, Tabrizi AR. Osteoblastic and vascular endothelial niches, their control on normal hematopoietic stem cells, and their consequences on the development of leukemia. Stem Cells Int. 2011;2011:1–8. doi: 10.4061/2011/375857
- Walkley CR, Olsen GH, Dworkin S, et al. A microenvironment-induced myeloproliferative syndrome caused by retinoic acid receptor gamma deficiency. Cell. 2007;129(6):1097–1110. doi: 10.1016/j.cell.2007.05.014
- Raaijmakers MHGP, Mukherjee S, Guo S, et al. Bone progenitor dysfunction induces myelodysplasia and secondary leukaemia. Nature. 2010;464(7290):852–857. doi: 10.1038/nature08851
- Kode A, Manavalan JS, Mosialou I, et al. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature. 2014;506(7487):240–244. doi: 10.1038/nature12883
- Cogle CR, Goldman DC, Madlambayan GJ, et al. Functional integration of acute myeloid leukemia into the vascular niche. Leukemia. 2014;28(10):1978–1987. doi: 10.1038/leu.2014.109
- Hussong JW, Rodgers GM, Shami PJ. Evidence of increased angiogenesis in patients with acute myeloid leukemia. Blood. 2000;95(1):309–313.
- Hatfield K, Ryningen A, Corbascio M, et al. Microvascular endothelial cells increase proliferation and inhibit apoptosis of native human acute myelogenous leukemia blasts. Int J Cancer. 2006;119(10):2313–2321. doi: 10.1002/ijc.22180
- Drusbosky L, Gars E, Trujillo A, et al. Endothelial cell derived angiocrine support of acute myeloid leukemia targeted by receptor tyrosine kinase inhibition. Leukemia Res. 2015;39(9):984–989. doi: 10.1016/j.leukres.2015.05.015
- Hanoun M, Zhang D, Mizoguchi T, et al. Acute myelogenous leukemia-induced sympathetic neuropathy promotes malignancy in an altered hematopoietic stem cell niche. Cell Stem Cell. 2014;15(3):365–375. doi: 10.1016/j.stem.2014.06.020
- Dias S, Choy M, Alitalo K, et al. Vascular endothelial growth factor (VEGF)–C signaling through FLT-4 (VEGFR-3) mediates leukemic cell proliferation, survival, and resistance to chemotherapy. Blood. 2002;99(6):2179–2184. doi: 10.1182/blood.V99.6.2179
- Kremer KN, Dudakovic A, McGee-Lawrence ME, et al. Osteoblasts protect AML cells from SDF-1-induced apoptosis. J Cell Biochem. 2014;115(6):1128–1137. doi: 10.1002/jcb.24755
- Papayannopoulou T, Craddock C, Nakamoto B, et al. The VLA4/VCAM-1 adhesion pathway defines contrasting mechanisms of lodgement of transplanted murine hemopoietic progenitors between bone marrow and spleen. Proc Natl Acad Sci USA. 1995;92(21):9647–9651. doi: 10.1073/pnas.92.21.9647
- Matsunaga T, Takemoto N, Sato T, et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat Med. 2003;9(9):1158–1165. doi: 10.1038/nm909
- Poulos MG, Gars EJ, Gutkin MC, et al. Activation of the vascular niche supports leukemic progression and resistance to chemotherapy. Exp Hematol. 2014;42(11):976–986.e3. doi: 10.1016/j.exphem.2014.08.003
- Ponomaryov T, Peled A, Petit I, et al. Induction of the chemokine stromal-derived factor-1 following DNA damage improves human stem cell function. J Clin Invest. 2000;106(11):1331–1339. doi: 10.1172/JCI10329
- de Lourdes Perim A, Amarante MK, Guembarovski RL, et al. CXCL12/CXCR4 axis in the pathogenesis of acute lymphoblastic leukemia (ALL): a possible therapeutic target. Cell Mol Life Sci. 2015;72(9):1715–1723. doi: 10.1007/s00018-014-1830-x
- Rombouts EJ, Pavic B, Lowenberg B, et al. Relation between CXCR-4 expression, Flt3 mutations, and unfavorable prognosis of adult acute myeloid leukemia. Blood. 2004;104(2):550–557. doi: 10.1182/blood-2004-02-0566
- Konoplev S, Rassidakis GZ, Estey E, et al. Overexpression of CXCR4 predicts adverse overall and event-free survival in patients with unmutated FLT3 acute myeloid leukemia with normal karyotype. Ann NY Acad Sci. 2007;109(6):1152–1156.
- Semerad CL, Christopher MJ, Liu F, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. 2005;106(9):3020–3027. doi: 10.1182/blood-2004-01-0272
- Tavor S, Petit I, Porozov S, et al. CXCR4 regulates migration and development of human acute myelogenous leukemia stem cells in transplanted NOD/SCID mice. Cancer Res. 2004;64(8):2817–2824. doi: 10.1158/0008-5472.CAN-03-3693
- Wright DE, Bowman EP, Wagers AJ, et al. Hematopoietic stem cells are uniquely selective in their migratory response to chemokines. J Exp Med. 2002;195(9):1145–1154. doi: 10.1084/jem.20011284
- Burger JA, Spoo A, Dwenger A, et al. CXCR4 chemokine receptors (CD184) and alpha4beta1 integrins mediate spontaneous migration of human CD34+ progenitors and acute myeloid leukaemia cells beneath marrow stromal cells (pseudoemperipolesis). Brit J Haematol. 2003;122(4):579–589. doi: 10.1046/j.1365-2141.2003.04466.x
- Ma Q, Jones D, Borghesani PR, et al. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc Natl Acad Sci USA. 1998;95(16):9448–9453. doi: 10.1073/pnas.95.16.9448
- Sugiyama T, Kohara H, Noda M, et al. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–988. doi: 10.1016/j.immuni.2006.10.016
- Shen ZH, Zeng DF, Kong PY, et al. AMD3100 and G-CSF disrupt the cross-talk between leukemia cells and the endosteal niche and enhance their sensitivity to chemotherapeutic drugs in biomimetic polystyrene scaffolds. Blood Cells Mol Dis. 2016;59:16–24. doi: 10.1016/j.bcmd.2016.03.009
- Sison EAR, McIntyre E, Magoon D, et al. Dynamic chemotherapy-induced upregulation of surface CXCR4 expression as a mechanism of chemotherapy resistance in pediatric acute myeloid leukemia. Mol Cancer Res. 2013;11(9):1004–1016. doi: 10.1158/1541-7786.MCR-13-0114
- Zeng Z, Shi YX, Samudio IJ, et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood. 2009;113(24):6215–6224. doi: 10.1182/blood-2008-05-158311
- Andreeff M, Konoplev S, Wang RY, et al. Massive mobilization of AML cells into circulation by disruption of leukemia/stroma cell interactions using CXCR4 antagonist AMD3100: first evidence in patients and potential for abolishing bone marrow microenvironment-mediated resistance. Blood. 2006;108(11):568.
- Li H, Rao Q, Yu P, et al. Expression of MPL in leukemia stem cells and its role in stemness maintainance. Blood. 2016;128(22):1723.
- Arai F, Yoshihara H, Hosokawa K, et al. Niche regulation of hematopoietic stem cells in the endosteum. Ann NY Acad Sci. 2009;1176(1):36–46. doi: 10.1111/j.1749-6632.2009.04561.x
- Yoshihara H, Arai F, Hosokawa K, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007;1(6):685–697. doi: 10.1016/j.stem.2007.10.020
- Reikvam H, Hatfield KJ, Lassalle P, et al. Targeting the angiopoietin (Ang)/Tie-2 pathway in the crosstalk between acute myeloid leukaemia and endothelial cells: studies of Tie-2 blocking antibodies, exogenous Ang-2 and inhibition of constitutive agonistic Ang-1 release. Expert Opin Investig Drug. 2010;19(2):169–183. doi: 10.1517/13543780903485659
- Bachegowda L, Morrone K, Winski SL, et al. Pexmetinib: a novel dual inhibitor of Tie2 and p38 MAPK with efficacy in preclinical models of myelodysplastic syndromes and acute myeloid leukemia. Cancer Res. 2016;76(16):4841–4849. doi: 10.1158/0008-5472.CAN-15-3062
- Ichihara E, Kaneda K, Saito Y, et al. Angiopoietin1 contributes to the maintenance of cell quiescence in EVI1(high) leukemia cells. Biochem Biophys Res Commun. 2011;416(3–4):239–245. doi: 10.1016/j.bbrc.2011.10.061
- Gomei Y, Nakamura Y, Yoshihara H, et al. Functional differences between two Tie2 ligands, angiopoietin-1 and -2, in regulation of adult bone marrow hematopoietic stem cells. Exp Hematol. 2010;38(2):82–89.e1. doi: 10.1016/j.exphem.2009.11.007
- Staal SP, Hartley JW, Rowe WP. Isolation of transforming murine leukemia viruses from mice with a high incidence of spontaneous lymphoma. Proc Natl Acad Sci USA. 1977;74(7):3065–3067. doi: 10.1073/pnas.74.7.3065
- Kharas MG, Okabe R, Ganis JJ, et al. Constitutively active AKT depletes hematopoietic stem cells and induces leukemia in mice. Blood. 2010;115(7):1406–1415. doi: 10.1182/blood-2009-06-229443
- Burgering BM, Kops GJ. Cell cycle and death control: long live Forkheads. Trends Biochem Sci. 2002;27(7):352–360. doi: 10.1016/S0968-0004(02)02113-8
- Suda T, Arai F, Shimmura S. Regulation of stem cells in the niche. Cornea. 2005;24(Suppl):S12–S17. doi: 10.1097/01.ico.0000178742.98716.65
- Schaich M, Soucek S, Thiede C, et al. MDR1 and MRP1 gene expression are independent predictors for treatment outcome in adult acute myeloid leukaemia. Br J Haematol. 2005;128(3):324–332. doi: 10.1111/j.1365-2141.2004.05319.x
- Tamburini J, Elie C, Bardet V, et al. Constitutive phosphoinositide 3-kinase/Akt activation represents a favorable prognostic factor in de novo acute myelogenous leukemia patients. Blood. 2007;110(3):1025–1028. doi: 10.1182/blood-2006-12-061283
- Parmar K, Mauch P, Vergilio JA, et al. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci USA. 2007;104(13):5431–5436. doi: 10.1073/pnas.0701152104
- Velasco-Hernandez T, Hyrenius-Wittsten A, Rehn M, et al. HIF-1α can act as a tumor suppressor gene in murine acute myeloid leukemia. Blood. 2014;124(24):3597–3607. doi: 10.1182/blood-2014-04-567065
- Velasco-Hernandez T, Tornero D, Cammenga J. Loss of HIF-1alpha accelerates murine FLT-3(ITD)-induced myeloproliferative neoplasia. Leukemia. 2015;29(12):2366–2374. doi: 10.1038/leu.2015.156
- Drolle H, Wagner M, Vasold J, et al. Hypoxia regulates proliferation of acute myeloid leukemia and sensitivity against chemotherapy. Leuk Res. 2015;39(7):779–785. doi: 10.1016/j.leukres.2015.04.019
- Fiegl M, Samudio I, Clise-Dwyer K, et al. CXCR4 expression and biologic activity in acute myeloid leukemia are dependent on oxygen partial pressure. Blood. 2008;113(7):1504–1512. doi: 10.1182/blood-2008-06-161539
- Muz B, de la Puente P, Azab F, et al. The role of hypoxia and exploitation of the hypoxic environment in hematologic malignancies. Mol Cancer Res. 2014;12(10):1347–1354. doi: 10.1158/1541-7786.MCR-14-0028
- Johansen S, Brenner AK, Bartaula-Brevik S, et al. The possible importance of beta3 integrins for leukemogenesis and chemoresistance in acute myeloid leukemia. Int J Mol Sci. 2018;19(1):251. doi: 10.3390/ijms19010251
- Powell JA, Thomas D, Barry EF, et al. Expression profiling of a hemopoietic cell survival transcriptome implicates osteopontin as a functional prognostic factor in AML. Blood. 2009;114(23):4859–4870. doi: 10.1182/blood-2009-02-204818
- Liersch R, Gerss J, Schliemann C, et al. Osteopontin is a prognostic factor for survival of acute myeloid leukemia patients. Blood. 2012;119(22):5215–5220. doi: 10.1182/blood-2011-11-389692
- Mirzaei A, Mohammadi S, Ghaffari SH, et al. Osteopontin b and c isoforms: molecular candidates associated with leukemic stem cell chemoresistance in acute myeloid leukemia. Asian Pac J Cancer Prev. 2017;18(6):1707–1715.
- Boyerinas B, Zafrir M, Yesilkanal AE, et al. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood. 2013;121(24):4821–4831. doi: 10.1182/blood-2012-12-475483
- Ishikawa F, Yoshida S, Saito Y, et al. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nat Biotechnol. 2007;25(11):1315–1321. doi: 10.1038/nbt1350
- Saito Y, Uchida N, Tanaka S, et al. Induction of cell cycle entry eliminates human leukemia stem cells in a mouse model of AML. Nat Biotechnol. 2010;28(3):275–280. doi: 10.1038/nbt.1607
- Ho T-C, LaMere M, Stevens BM, et al. Evolution of acute myelogenous leukemia stem cell properties after treatment and progression. Blood. 2016;128(13):1671–1678. doi: 10.1182/blood-2016-02-695312
- Uy GL, Rettig MP, Motabi IH, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119(17):3917–3924. doi: 10.1182/blood-2011-10-383406
- Borthakur G, Tallman MS, Ofran Y, et al. Clinical response in relapsed/refractory aml patients correlates with leukemic blast mobilization and differentiation induced by BL-8040, a potent CXCR4 antagonist; results of a phase IIa study. Proceedings of the 21st Congress of the European Hematology Association; 2016 Jun 9–12; Copenhagen, Denmark: Ferrata Storti Foundation; 2016. p. 43.
- Galsky MD, Vogelzang NJ, Conkling P, et al. A phase I trial of LY2510924, a CXCR4 peptide antagonist, in patients with advanced cancer. Clin Cancer Res. 2014;20(13):3581–3588. doi: 10.1158/1078-0432.CCR-13-2686
- Kovacsovics TJ, Mims AS, Salama ME, et al. CX-01, a low anticoagulant heparin, may enhance count recovery and treatment efficacy in acute myeloid leukemia. Blood. 2016;128(22):5220.
- Karpova D, Brauninger S, Wiercinska E, et al. Joint congress DGTI & DGI 2016, 7.-10. September 2016, Nürnberg, Abstracts. Transfus Med Hemoth. 2016;43(1):1–88.
- Becker PS, Foran JM, Altman JK, et al. Targeting the CXCR4 pathway: safety, tolerability and clinical activity of ulocuplumab (BMS-936564), an anti-CXCR4 antibody, in relapsed/refractory acute myeloid leukemia. Blood. 2014;124(21):386.
- Fogler WE, Flanner H, Wolfgang C, et al. Administration of the dual E-selectin/CXCR4 antagonist, GMI-1359, results in a unique profile of tumor mobilization from the bone marrow and facilitation of chemotherapy in a murine model of FLT3 ITD AML. Blood. 2016;128(22):2826.
- Devata S, Sood SL, Hemmer MV, et al. First in human phase 1 single dose escalation studies of the E-selectin antagonist GMI-1271 show a favorable safety, pharmacokinetic, and biomarker profile. Blood. 2015;126(23):1004.
- Matsunaga T, Fukai F, Miura S, et al. Combination therapy of an anticancer drug with the FNIII14 peptide of fibronectin effectively overcomes cell adhesion-mediated drug resistance of acute myelogenous leukemia. Leukemia. 2008;22(2):353–360. doi: 10.1038/sj.leu.2405017
- Layani-Bazar A, Skornick I, Berrebi A, et al. Redox modulation of adjacent thiols in VLA-4 by AS101 converts myeloid leukemia cells from a drug-resistant to drug-sensitive state. Cancer Res. 2014;74(11):3092–3103. doi: 10.1158/0008-5472.CAN-13-2159
- Liu J, Wang L, Zhao F, et al. Pre-clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PLoS ONE. 2015;10(9):e0137345. doi: 10.1371/journal.pone.0137345
- Badar T, Handisides DR, Benito JM, et al. Phase I study of evofosfamide, an investigational hypoxia-activated prodrug, in patients with advanced leukemia. Am J Hematol. 2016;91(8):800–805. doi: 10.1002/ajh.24415
- Andreeff M, Zeng ZH, Kelly MA, et al. Mobilization and elimination of FLT3-ITD+ acute myelogenous leukemia (AML) stem/progenitor cells by plerixafor/G-CSF/sorafenib: results from a phase I trial in relapsed/refractory AML patients. Blood. 2012;120(21):142.
- Borthakur G, Nagler A, Ofran Y, et al. BL-8040, a peptidic CXCR4 antagonist, induces leukemia cell death and specific leukemia cell mobilization in relapsed/refractory acute myeloid leukemia patients in an ongoing phase IIa clinical trial. Blood. 2014;124(21):950.