
ABSTRACT
Background: The phenotypic expression of sickle cell disease (SCD) is a complex pathophysiologic condition. However, sickle erythrocytes might be the cause for multiple sources of pro-oxidant processes with consequent linked to chronic and systemic oxidative stress. Herein, we explored the SCD phenomena could be the result in formation of oxidative stress as well as inflammation in children.
Material and methods: Blood samples of 147 SCD subjects were evaluated. A control group was formed of 156 individuals without SCD. Different oxidative stress markers and inflammatory mediators were measured by using various biochemical techniques. Plasma samples were collected from blood for the measurement of antioxidants and reactive oxygen species (ROS).
Results: The levels of plasma hydroxyl radical (HO•), and nitric oxide (NO) production were higher in SCD children in compared to control groups. The plasma antioxidants capacities such as superoxide dismutase (SOD), catalase (CAT), glutathione (GSH), glutathione peroxidase (GPx) and protein thiol levels were significantly reduced in SCD children. The plasma lipid peroxidation, protein carbonylation, DNA damage markers were significantly altered in different age groups of SCD children. Further, our results showed that SCD children have chronic inflammatory disease due to persistent alteration of haemoglobin content, reticulocyte, total bilirubin, platelet, creatinine, leukocytes, and altered expression of inflammatory mediators in compared to control groups.
Conclusion: SCD children have high oxidative stress, and conversely, decreased antioxidant activity. Decrease in antioxidant activity might explained the reduction in lipid peroxidation, protein carbonylation and increased inflammation, which in turn intensify the symptoms of SCD in children.
Introduction
Sickle cell disease (SCD) is a class of hemoglobinopathy in human and closely associated to inherit hemolytic anemia due to single base substitution in the β-globin gene that results in the formation of sickle hemoglobin (HbS) [Citation1]. The phenotypic expression of SCD is a complex pathophysiologic condition of multiple sources of pro-oxidant processes with consequent chronic and systemic oxidative stress. In healthy biological systems, erythrocytes have an environment of continuous free radical generation. However, reactive oxygen species (ROS) and the end-products of their oxidative reactions are potential markers of SCD severity [Citation2]. On the other hand, HbS molecules suffer from repeated polymerization and depolymerization generating greater amounts of ROS, which can lead to a cyclic cascade characterized by blood cell adhesion, hemolysis, increased susceptibility to infections, chronic inflammatory diseases, and microvascular damage in organs leading to reduced quality of life and life expectancy [Citation3,Citation4].
The major pro-oxidant sources in SCD subjects are sickle erythrocytes, where the unstable autoxidative HbS and increased metabolic turnover due to recurrent HbS polymerizations/depolymerizations cause increased ROS generation [Citation5–7]. Many lines of studies have reported that sickle erythrocytes generated two-fold greater extent of superoxide (O2•_), hydrogen peroxide (H2O2), HO• and lipid oxidation products compared with HbA-containing erythrocytes [Citation8,Citation9]. Among all these free radicals, O2•_ were converted to highly reactive HO• through the Fenton and Haber–Weiss reactions in the presence of H2O2 and transitional or redox-active metal ions like iron and copper [Citation10]. Therefore, iron released during hemolysis of the RBC was considered as one of the major factors to the enhanced ROS production in SCD subjects. In order to counteract ROS, erythrocytes have a self-sustaining activity of antioxidants such as superoxide dismutase (SOD), CAT, GPx, GR, GSH, and vitamins [Citation8,Citation11]. The overproduction ROS overwhelms the defenses in blood and caused damage to biological macromolecules such as proteins, lipids, and DNA leading to change the RBC membrane properties, alter membrane permeability, and lead to hemolysis [Citation12]. Earlier studies have demonstrated that compared to HbA-containing erythrocytes, sickle erythrocytes showed both an increased endogenous extent of oxidized lipid and an increased susceptibility for further lipid peroxidation (LPO) [Citation13]. More importantly, the chronic inflammatory condition in SCD subjects was characterized by endothelial damage, increased production of free radicals, hemolysis, increased expression of adhesion molecules by leukocytes, activation of granulocytes, monocytes, platelets, and increased production of proinflammatory cytokines [Citation14,Citation15]. Studies have reported that proinflammatory mediators derived from leukocytes, platelets, and endothelial cells, such as tumor necrosis factor alpha (TNF-α) and the interleukins (IL), IL-6, IL-1β, and IL-8, were usually higher in SCD subjects [Citation16,Citation17].
Hematological profiles of SCD subjects in different age groups are extremely variable in different geographical locations throughout the globe. Also, anemia is a common finding during the neonatal period and early childhood [Citation18]. Specifically, information on the hematological profile of children suffering from SCD and its complications is lacking. More importantly, the use of oxidative stress, as a potential marker of SCD in pediatric subjects and pathophysiologic severity, has not yet been fully studied. In this light, the present study was aimed to measure the levels of reactive oxygen/nitrogen species and the activities of antioxidants, biomolecule damages including proteins, lipids, and DNA on children suffering from SCD. Also, we measured the plasma concentrations of inflammatory mediators in SCD children.
Materials and methods
Trizol, L-Glutathione reduced, and 5,5-dithiobis(2-nitrobenzoic acid) (DTNB) were obtained from Sigma Aldrich (U.S.A.). Anti-OGG1 was purchased from Novus, while pH2A.X and secondary antibody were obtained from Cell Signaling Technology, U.S.A.
The SCD subjects and age-matched healthy controls who sought treatment from the physicians were recruited for this study. A standardized medical protocol was followed providing detailed demographic and clinical information about all the participants. The present study was carried out on children (N = 147) with age groups 6 months to 10 years of SCD and children (N = 156) without SCD, who served as control groups. All SCD children were screened using a questionnaire to their concerned parents. The patient history and concomitant illness like rheumatic heart disease, diabetes mellitus, hypertension, or any other complications were excluded from the study population. Also the patients who had taken medications, known to affect the analysis of the hematological as well as biochemical parameters within the 24 h prior to blood collection, were also excluded from the study groups. All the cases and controls chosen for the study gave their written consent in the language they understand and also the consent of their parents for participating in the study, after being explained the nature of the study. From each patient, 1 ml blood was collected with/without EDTA coated vial for biochemical parameter assessment. The blood was centrifuged at 1000 g for 10 min to separate the plasma and stored at −80°C until use. Protein thiol content was measured immediately after blood collection and plasma separation.
The plasma antioxidants, SOD, and CAT were measured by a spectrophotometer as in the protocol previously described [Citation19]. Briefly, for the SOD measurement, 80 µg plasma proteins were mixed with 1.4 ml of the reaction mixture (Tris buffer, L-methionine, Triton X-100, hydroxylamine hydrochloride, EDTA) and 80 µl of Riboflavin. Subsequently, 1 ml of Griess’ reagent was added to the mixture and the absorbance was recorded at 540 nm. The SOD activity was expressed as units per milligram of protein (U/mg). For the measurement of CAT, 80 µg plasma protein was dissolved in 800 µl of 50 µM of phosphate buffer. Freshly prepared 400 µl of 30 mM H2O2 was added to the solution and absorbance was recorded at 240 nm by rate kinetics. The CAT activity was expressed in Mcat/mg. Reduced GSH content was measured as described by Kapoor and Kakkar [Citation20]. Briefly, 80 µg of plasma protein was mixed with 500 µl of the reaction mixture (0.02 EDTA, dH2O, 50%TCA, and Tris buffer pH 8.8), and 100 µl of DTNB. Subsequently, the mixture was incubated at 37 °C for 30 min and the absorbance was recorded at 412 nm. The HO• production was determined by a spectrophotometer as described previously [Citation19]. Briefly, 300 µl plasma sample was mixed with 700 µl of phosphate buffered saline (pH 7.8), 0.1 M EDTA, 2 mM sodium salicylate, 40 µl 10 N HCl, and 0.25 g NaCl. Equal volume of ice-cold diethyl ether was added to the mixture. Absorbance was recorded at 510 nm after 30 min incubation at 25 °C. The generated HO• was expressed as absorbance at 510 nm/30 min. Furthermore, the level of NO was determined by measuring the nitrite production [Citation21]. Briefly, 100 µl of plasma was mixed with 100 µl Griess’ reagent and absorbance was recorded at 520 nm.
For the determination of LPO, 100 µl (80 µg) plasma protein was mixed with butylated hydroxytoluene (0.022%) and incubated at 37 °C for 1 h [Citation22]. Thiobarbituric acid solution (0.375%) was added to the above mixture followed by centrifugation at 800 g. The reaction mixture was kept in a boiling water bath for 15 min, chilled at room temperature, and absorbance was recorded at 532 nm. For the determination of protein carbonylation (PC), 450 µl (80 µg) plasma protein was mixed with 450 µl of the buffer, 100 µl of 10% streptomycin sulfate, and centrifuged at 1,000 g for 10 min. Then 400 µl of the supernatant was mixed with equal volume of 10 mM 2, 4-dinitrophenylhydrazine and kept in the dark for 30 min. Ice-cold trichloro acetic acid (20%) was added to it and then centrifuged at 1000 g. Pellet was dissolved in 6 M guanidine hydrochloride and absorbance was recorded at 365 nm. Total plasma thiol concentration or sulfhydryl groups (SH) was measured by the methods described by Hu [Citation23]. The 50 µl aliquot of plasma sample was mixed with 1 ml of the Tris-EDTA buffer, and absorbance (A1) was measured at 412 nm. Furthermore, 20 µl of 10 mM DTNB was added in the above solution and after 15 min at ambient temperature, the absorption (A2) was measured again together with a DTNB blank (B). Total SH group was calculated as follows: (A2−A1−B) × (1.07/0.05)/13.6 = (A2−A1−B) × 1.57 mM.
For the western blot analysis, 40 μg of protein sample was resolved in 12% Tris-glycine polyacrylamide gels, transferred to nitrocellulose membranes and blocked in 5% non-fat skimmed milk. Membranes were incubated overnight with the primary antibodies at 4 °C. After washing with 1XPBST (phosphate buffered saline with Tween-20), the membranes were incubated with appropriate horseradish peroxidase (HRP) conjugated secondary antibody for 2 h and the protein bands were detected by enhanced chemiluminescence (ECLTM Plus, Invitrogen, USA). For loading control, the same membrane was reprobed with anti-β-actin antibody. Next, to quantify the mRNA level, total RNA was isolated using Trizol (Sigma Aldrich, U.S.A.) and double-stranded cDNA was prepared using Revert Aid cDNA synthesis kit (Thermo scientific). Quantitative real-time PCR (qRT-PCR) was performed with a CFX connect Real-Time system (BIO-RAD, U.S.A.) using the KAPA SYBR® FAST qPCR Kit (KAPA Biosystems, Wilmington, MA, USA). Oligonucleotides used for qRT-PCR are given in . The 2-ΔΔCT method was used to quantify relative levels of target gene mRNA.
Table 1. Genes used in qPCR.
The data were expressed as the mean ± SD. The unpaired Student’s T test was used to evaluate the significance between controls and SCD subjects groups. All the statistical analyses were performed using Graphpad prism. P < 0.05 value was considered statistically significant. All experiments were performed three times.
Result
Blood samples of 147 SCD subjects (71 females and 76 males; age group: 6 months to 10 years) were evaluated. A control group of 156 individuals without SCD (72 females and 84 males; age group: 6 months to 10 years) was formed. All SCD children were screened using a questionnaire to their concerned parents about their education status and family history of SCD (). Hematological profile of study subjects is shown in , and values were compared between both the sexes. Hemoglobin and total leukocyte count (TLC) were significantly (p < 0.001) lower in SCD subjects compared to healthy male and female children. Similarly, the platelet count was lower in the SCD children (p < 0.001). To evaluate the hemolytic profile on SCD children, we measured the reticulocyte percentage, creatinine, and the circulating values of the total bilirubin. The plasma reticulocyte percentage, creatinine, and bilirubin levels were significantly (p < 0.001) higher in SCD subjects when compared with healthy male and female children.
Table 2. Sociodemographic characteristics of control and sickle cell disease (SCD) groups.
Table 3. Hematological parameters of control and sickle cell disease (SCD) group.
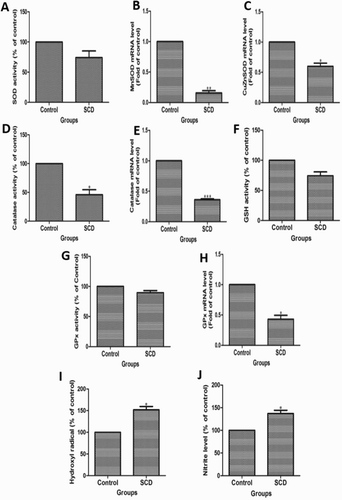
Many confusing reports on antioxidants, lipid, and protein oxidation were being presented differently in SCD subjects [Citation24,Citation25]. Earlier reports demonstrated that the significant increase in SOD was observed in homozygous sickle cell anemic patients [Citation24,Citation25]. Here, the level of SOD was lower in children suffering from SCD as compared to healthy children ((A)). Furthermore, we checked the mRNA expression of mitochondrial MnSOD and cytosolic CuZnSOD. As shown in (B and C), the MnSOD and CuZnSOD expressions were lower in SCD subjects. Next, we measured the CAT activity in SCD subjects. (D) shows that CAT activity was lower in SCD subjects as compared to healthy children. Similarly, we measured the mRNA expression of CAT in SCD children, and the expression of CAT was lower in SCD children as compared to healthy individuals ((E)). Furthermore, we measured the levels of GSH in SCD subjects. (F) observes that decreased activity of GSH in SCD subjects as compared to healthy individuals. Additionally, we measured the levels of GPx in SCD subjects. As shown in (G and H), GPx activity in both protein and mRNA was lower in SCD subjects as compared to healthy children. Furthermore, oxidative stress analysis was done in SCD children based on the detection of HO• and NO level in plasma. (I) shows that HO• production was significantly higher in SCD subjects as compared to healthy children. Similarly, the production of NO in SCD subjects was significantly higher as compared to healthy children ((J)).
Figure 1. Comparison of oxidative stress markers in children suffering from sickle cell disease (SCD) and healthy individuals. (A) Spetrophotometer analysis of superoxide dismutase (SOD) in SCD subjects. The qRT-PCR analysis of (B) MnSOD and (C) CuZnSOD gene expression in SCD subjects. (D) Spectrophotometer and (E) qRT-PCR analysis of CAT activity in SCD subjects. (F). Spetrophotometer analysis of GSH in SCD subjects. (G) Spetrophotometer and (H) qRT-PCR analysis of GPx activity in SCD subjects. Spetrophotometer analysis of (I) HO• and (J) NO in SCD subjects.
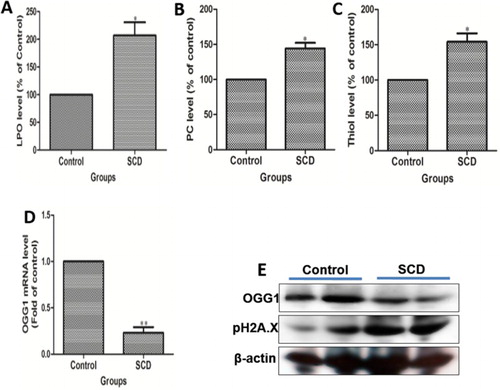
In SCD subjects, sickle hemoglobin polymerization leads to a remarkable spectrum of biochemical, molecular, and physiological pathology associated with many clinical complications. Here, we observed that the level of LPO and PC was significantly (p < 0.001) increased in SCD children as compared to healthy individuals ((A&B)). Also, the total thiol level was significantly (p < 0.001) higher in children with SCD as compared to healthy individuals ((C)). OGG1 is known as the DNA repair enzyme, hence acts as an important DNA damage sensor. (D) shows that the expression of OGG1 at mRNA level was decreasing in SCD children as compared to healthy ones. Again, we checked the OGG1 expression at the protein level and found the same pattern of results as in the case of mRNA expression in SCD children ((E)). Similarly, H2A.X is another DNA damage marker. As shown in (E), the expression of H2A.X was increased in SCD children as compared to the healthy one.
Figure 2. Comparison of biomolecule damage in children suffering from sickle cell disease (SCD) and healthy individuals. (A) Spetrophotometer analysis of lipid peroxidation (LPO), (B) protein carbonylation (PC) and (C) total thiol level in SCD subjects. (D) mRNA expression and (E) western blot analysis of OGG1 and H2A.X in SCD children as compared to healthy ones.
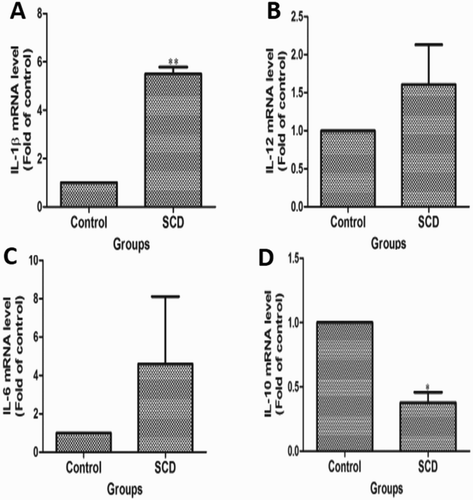
Recent studies have demonstrated that recurrent vaso-occlusions, hemolytic events, and endothelial cell activation induce a continuous inflammatory response in SCD subjects, which manifests as increased levels of inflammatory cytokines, decreased the bioavailability of NO, and oxidative stress [Citation26]. More importantly, many inflammatory cytokines were produced by activated endothelial cells in different pathophysiological conditions that played a significant role in the vaso-occlusive crisis in SCD subjects. Plasma cytokine levels were measured by qRT-PCR to check the inflammation in children suffering from SCD and healthy children. As shown in (A–D), the mRNA expressions of IL-1β, IL-6, and IL-12 were higher and IL-10 was lower in SCD subjects in comparison to healthy children.
Figure 3. Comparison of inflammatory mediators in children suffering from sickle cell disease (SCD) and healthy individuals. The qRT-PCR analysis of (A) IL-1β, (B) IL-12, (C) IL-6, and (D) IL-10 in children suffering from SCD and healthy children.
Discussion
Recently, studies have demonstrated that the polymerization of HbS was the key event in the SCD pathophysiology, ensuing to physicochemical changes in erythrocytes, which triggered hemolysis and vaso-occlusion [Citation27]. More importantly, hemolysis occurs with an early destruction of erythrocytes containing HbS by releasing hemoglobin and heme iron free in the plasma. On the other hand, vaso-occlusion is mediated by ischemic-reperfusion cycles, causing tissue damage resulting from oxidative stress, activation of endothelial cells, leukocytes and platelets, an increase of adhesion molecule expression, and the release of many inflammatory cytokines. In healthy biological compartments, erythrocytes in blood have an environment of continuous free radical generation due to the presence of hemoglobin. To counteract oxidative stress, erythrocytes have a self-sustaining antioxidant defense system, including SOD, CAT, GPx, and GR, along with low molecular weight antioxidants, such as GSH and vitamins [Citation11]. On the other hand, a disease like SCD has a higher potential for oxidative damage due to chronic redox imbalance in red cells often resulting in the clinical manifestation of mild to severe hemolysis in subjects with a genetic disorder. Recently, studies have demonstrated that SCD subjects have higher quantities of ROS. In the present study, we have evaluated the oxidative stress markers in children (age group 6 months to 10 years) suffering from SCD. Our study showed a significantly lower level of SOD activity in SCD subjects than the control groups. The decreased activity of SOD seen in SCD children may be due to excessive production of ROS and it was concluded that the decreased activity of SOD may be a defense mechanism in response to increased oxidative stress causing H2O2 build-up [Citation28,Citation29]. However, H2O2 was produced via two electron transfer or due to sickling and it was removed by two main antioxidants such as GPx and CAT [Citation30,Citation31]. The CAT is usually more important than GPx because of its ability to degrade H2O2 without consuming cellular reducing equivalents, which is the energy efficient way of removing H2O2. However, there is an inconsistency in studies on CAT level in SCD subjects. Some studies have shown that a decrease in CAT activity in SCD subjects. Maybe there were some possibilities that elevated antioxidant levels in sickle erythrocytes were the consequence of higher reticulocytes content in the blood of SCD subjects. Here, we observed that the level of CAT decreased in children suffering from SCD in all age groups. In contrast, several research reports observed an increase in CAT level in SCD subjects [Citation32,Citation33]. Increase in CAT might be a preventing effect to scavenge H2O2, whereas the decreased levels might be due to the devastating and prolonged oxidative stress. Furthermore, GSH, a cofactor for GPx to reduce H2O2, was easily oxidized to glutathione disulfide (GSSG) by oxidant compounds. In our study, GSH concentration was significantly reduced in children suffering from SCD, and the results correlate with the findings of adult SCD subjects. Also, GPx concentration was reduced in children suffering from SCD with a direct relationship to the disease severity.
Many studies have demonstrated that sickle erythrocytes in SCD subjects were the key pro-oxidant sources, where the unstable autoxidative HbS and increased metabolic turnover, due to recurrent HbS polymerizations and depolymerizations, increased ROS generation [Citation5–8]. Therefore, SCD subjects having sickle erythrocytes have been reported to generate two-fold greater extent of O2•_, H2O2, HO• and lipid oxidation products [Citation32,Citation33]. Our result showed an increase in HO• level in SCD children as compared to healthy individuals. Moreover, Chirico and Pialoux [Citation34] demonstrated that NO bioavailability can be reduced in several ways and through the uncoupling of endothelial nitric oxide synthase in SCD subjects. Here, we observed that the level of NO increased in SCD children as compared to healthy individuals. LPO occurs by a radical chain reaction involving oxidation and destruction of polyunsaturated lipid membrane structures leading to loss of normal cellular deformability and biological properties including the degree of fluidity, increased tissue permeability, hence making the RBC membranes more susceptible to hemolysis [Citation35]. In agreement with others, our study showed that the basal level of LPO was significantly higher in SCD children than in healthy individuals. Furthermore, PC was determined and observed that in SCD children PC was more as compared to healthy individuals. Similarly, the total plasma thiol concentration was determined and it was observed that in SCD children, the level was higher as compared to that of healthy individuals. Next, we observed the DNA damage process in SCD children and observed that the level of OGG1, a DNA repair enzyme expression, was less in SCD subjects as compared to healthy individuals. Hence, our study showed that in SCD subjects, the imbalance in antioxidant level leading to ROS generation followed by LPO, PC and DNA damage, results in oxidative stress.
Furthermore, a recent publication demonstrated that SCD was considered a chronic inflammatory disease due to the persistent activation of leukocytes, platelets, and endothelial cells, resulting from hemolysis and vaso-occlusion mediated by ischemia-reperfusion cycles. Studies also have demonstrated that the chronic inflammation was characterized by increased leukocyte count and activation of granulocytes, monocytes, and platelets in SCD subjects [Citation35]. Moreover, proinflammatory mediators derived from leukocytes, platelets, and endothelial cells, such as tumor necrosis factor alpha (TNF-α) and the interleukins (ILs), were usually higher in SCD subjects [Citation14,Citation15]. Interestingly, the increased production and release of proinflammatory mediators can favor the vaso-occlusive process due to endothelial activation, erythrocytes, and leukocyte adhesion to vascular endothelial and endothelial cell apoptosis in SCD subjects [Citation36,Citation37]. Our study also showed that the concentration of hemoglobin was lower in SCD children. Total bilirubin and reticulocyte levels were higher in SCD children as compared to healthy individuals. The platelet count and TLC were lower in SCD children; however, the creatinine concentration was higher in SCD children, which showed that inflammation was a key component in SCD pathophysiology. When subjected to a proinflammatory profile, higher plasma levels of IL-1β, IL-12, and IL-6 and lower plasma levels of IL-10 in SCD children compared to control individuals were observed. Importantly, IL-1β was the key cytokine that participated in the initial inflammatory response and able to promote the production and release of other important mediators that trigger inflammatory cascade SCD children.
Conclusion
The results of this study indicate that the presence of HbS in erythrocyte of SCD subjects not only causes biochemical alterations of biomolecules but also is responsible for increases in free radical production. The imbalance between overproduction of free radicals and inability of cellular content of antioxidants to eliminate leads to the oxidative stress and inflammation in children suffering from SCD. Moreover, the inflammatory profile in children suffering from SCD seems to be profoundly associated with oxidative stress and therefore further research is required on oxidative stress and inflammation of SCD subjects that in future may guide adjuvant treatment decisions. More importantly, to date there were scarce studies regarding oxidative stress and inflammation in children suffering from SCD and on the role of the erythrocytes in the free radical generation and the molecular strategies triggered by these cells that could be important for the attenuation of the general blood oxidative stress and inflammation. Therefore, more studies about oxidative stress biomarkers and antioxidant therapeutic strategies that reduce oxidative stress generation and abolish inflammation in the blood, remain a worthy and promising future goal. This oxidative stress biomarkers and antioxidant therapeutic strategies might lead to more specific and effective prognostic information of SCD subjects specifically in children that to development more efficacious therapeutic drugs for mitigating oxidative stress and inflammation in sickle erythrocytes, thus alleviating the disease outcomes.
Acknowledgments
Authors are thanked for skilful technical assistance at Paediatric Department, KIMS, Bhubaneswar, India.
Disclosure statement
No potential conflict of interest was reported by the authors.
Related Research Data
References
- Steinberg MH, Sebastiani P. Genetic modifiers of sickle cell disease. Am J Hematol. 2012;87:795–803. doi: 10.1002/ajh.23232
- Nur E, Biemond BJ, Otten HM, et al. Oxidative stress in sickle cell disease; pathophysiology and potential implications for disease management. Am J Hematol. 2011;86:484–489. doi: 10.1002/ajh.22012
- Silva DG, Belini Junior E, de Almeida EA, et al. Oxidative stress in sickle cell disease: an overview of erythrocyte redox metabolism and current antioxidant therapeutic strategies. Free Radic Biol Med. 2013;65:1101–1109. doi: 10.1016/j.freeradbiomed.2013.08.181
- Barabino GA, Platt MO, Kaul DK. Sickle cell biomechanics. Annu Rev Biomed Eng. 2010;12:345–367. doi: 10.1146/annurev-bioeng-070909-105339
- Banerjee T, Kuypers FA. Reactive oxygen species and phosphatidylserine externalization in murine sickle red cells. Br J Haematol. 2004;124:391–402. doi: 10.1046/j.1365-2141.2003.04781.x
- Hebbel RP, Morgan WT, Eaton JW, et al. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc Natl Acad Sci USA. 1988;85:237–241. doi: 10.1073/pnas.85.1.237
- Akohoue SA, Shankar S, Milne GL, et al. Energy expenditure, inflammation, and oxidative stress in steady-state adolescents with sickle cell anemia. Pediatr Res. 2007;61:233–238. doi: 10.1203/pdr.0b013e31802d7754
- Al-Naama LM, Hassan MK, Mehdi JK. Association of erythrocytes antioxidant enzymes and their cofactors with markers of oxidative stress in patients with sickle cell anemia. Qatar Med J. 2015;2015:14. doi: 10.5339/qmj.2015.14
- Klings ES, Farber HW. Role of free radicals in the pathogenesis of acute chest syndrome in sickle cell disease. Respir Res. 2001;2:280–285. doi: 10.1186/rr70
- Voskou S, Aslan M, Fanis P, et al. Oxidative stress in β-thalassaemia and sickle cell disease. Redox Biol. 2015;6:226–239. doi: 10.1016/j.redox.2015.07.018
- Perrone S, Tataranno ML, Stazzoni G, et al. Oxidative injury in neonatal erythrocytes. J Matern Fetal Neonatal Med. 2012;25:104–108. doi: 10.3109/14767058.2012.715471
- Ama Moor VJ, Pieme CA, Chetcha Chemegne B, et al. Oxidative profile of sickle cell patients in a Cameroonian urban hospital. BMC Clin Pathol. 2016;16:280. doi: 10.1186/s12907-016-0037-5
- Jain SK, Shohet SB. A novel phospholipid in irreversibly sickled cells: evidence for in vivo peroxidative membrane damage in sickle cell disease. Blood. 1984;63:362–367.
- Zhang D, Xu C, Manwani D, et al. Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood. 2016;127:801–809. doi: 10.1182/blood-2015-09-618538
- Bandeira IC, Rocha LB, Barbosa MC, et al. Chronic inflammatory state in sickle cell anemia patients is associated with HBB*S haplotype. Cytokine. 2014;65:217–221. doi: 10.1016/j.cyto.2013.10.009
- Qari MH, Dier U, Mousa SA. Biomarkers of inflammation, growth factor, and coagulation activation in patients with sickle cell disease. Clin Appl Thromb Hemost. 2012;18:195–200. doi: 10.1177/1076029611420992
- Sarray S, Saleh LR, Lisa Saldanha F, et al. Serum IL-6, IL-10, and TNFα levels in pediatric sickle cell disease patients during vasoocclusive crisis and steady state condition. Cytokine. 2015;72:43–47. doi: 10.1016/j.cyto.2014.11.030
- Steiner LA, Gallagher PG. Erythrocyte disorders in the perinatal period. Semin Perinatol. 2007;31:254–261. doi: 10.1053/j.semperi.2007.05.003
- Kumar P, Raman T, Swain MM, et al. Hyperglycemia-induced oxidative-nitrosative stress induces inflammation and neurodegeneration via augmented tuberous sclerosis complex-2 (TSC-2) activation in neuronal cells. Mol Neurobiol. 2017;54:238–254. doi: 10.1007/s12035-015-9667-3
- Kapoor R, Kakkar P. Protective role of morin, a flavonoid, against high glucose induced oxidative stress mediated apoptosis in primary rat hepatocytes. PLoS One. 2012;7:e41663. doi: 10.1371/journal.pone.0041663
- Tsikas D. Methods of quantitative analysis of the nitric oxide metabolites nitrite and nitrate in human biological fluids. Free Radic Res. 2005;39:797–815. doi: 10.1080/10715760500053651
- Sagar S, Kumar P, Behera RR, et al. Effects of CEES and LPS synergistically stimulate oxidative stress inactivates OGG1 signaling in macrophage cells. J Hazard Mater. 2014;278:236–249. doi: 10.1016/j.jhazmat.2014.05.096
- Hu ML. Measurement of protein thiol groups and glutathione in plasma. Methods Enzymol. 1994;233:380–385. doi: 10.1016/S0076-6879(94)33044-1
- Das SK, Nair RC. Superoxide dismutase, glutathione peroxidase, catalase and lipid peroxidation of normal and sickled erythrocytes. Br J Haematol. 1980;44:87–92. doi: 10.1111/j.1365-2141.1980.tb01186.x
- Beretta L, Gerli GC, Ferraresi R, et al. Antioxidant system in sickle red cells. Acta Haematol. 1983;70:194–197. doi: 10.1159/000206722
- Steinberg MH. Pathophysiologically based drug treatment of sickle cell disease. Trends Pharmacol Sci. 2006;27:204–210. doi: 10.1016/j.tips.2006.02.007
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376:2018–2031. doi: 10.1016/S0140-6736(10)61029-X
- Silva DG, Belini Junior E, Torres Lde S, et al. Relationship between oxidative stress, glutathione S-transferase polymorphisms and hydroxyurea treatment in sickle cell anemia. Blood Cells Mol Dis. 2011;47:23–28. doi: 10.1016/j.bcmd.2011.03.004
- Reid M, Badaloo A, Forrester T, et al. In vivo rates of erythrocyte glutathione synthesis in adults with sickle cell disease. Am J Physiol Endocrinol Metab. 2006;291:E73–E79. doi: 10.1152/ajpendo.00287.2005
- Morris CR, Suh JH, Hagar W, et al. Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood. 2008;111:402–410. doi: 10.1182/blood-2007-04-081703
- Hebbel RP. Beyond hemoglobin polymerization: the red blood cell membrane and sickle disease pathophysiology. Blood. 1991;77:214–237.
- Chirico EN, Pialoux V. Role of oxidative stress in the pathogenesis of sickle cell disease. IUBMB Life. 2012;64:72–80. doi: 10.1002/iub.584
- Pham-Huy LA, He H, Pham-Huy C. Free radicals, antioxidants in disease and health. Int J Biomed Sci. 2008;4:89–96.
- Hebbel RP. Ischemia-reperfusion injury in sickle cell anemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain. Hematol Oncol Clin North Am. 2014;28:181–198. doi: 10.1016/j.hoc.2013.11.005
- Hoppe CC. Inflammatory mediators of endothelial injury in sickle cell disease. Hematol Oncol Clin North Am. 2014;28:265–286. doi: 10.1016/j.hoc.2013.11.006
- Lanaro C, Franco-Penteado CF, Albuqueque DM, et al. Altered levels of cytokines and inflammatory mediators in plasma and leukocytes of sickle cell anemia patients and effects of hydroxyurea therapy. J Leukoc Biol. 2009;85:235–242. doi: 10.1189/jlb.0708445
- Conran N, Franco-Penteado CF, Costa FF. Newer aspects of the pathophysiology of sickle cell disease vaso-occlusion. Hemoglobin. 2009;33:1–16. doi: 10.1080/03630260802625709