
ABSTRACT
Objective: To identify ways that provision of hemophilia care can be maximized at the local level, irrespective of available resources or cultural or geographic challenges.
Methods: The SHIELD group used its multinational experience to share examples of local initiatives that have been employed to deliver optimal hemophilia care.
Results: The examples were reviewed and categorized into four key themes: guidelines and algorithms for delivery of care; collaboration with patients and allied groups for care and education; registries for the monitoring of treatment and outcomes and health care planning and delivery; and opportunities for personalization of care. These themes were then incorporated into a road map for collaborative care in hemophilia that reflected the contribution of best practice.
Discussion: Differing healthcare reimbursement systems, budgetary constraints, and geographical and cultural factors make it difficult for any country to fully deliver ideal care for people with hemophilia. The SHIELD approach for collaborative care provides illustrative examples of how four key themes can be used to optimize hemophilia care in any setting.
Abbreviations: AHCDC: Association of Hemophilia Clinic Directors of Canada; AICE: Italian Association of Hemophilia Centres; ATHN: American Thrombosis and Hemostasis Network; EAHAD: European Association for Haemophilia and Allied Disorders; EHC: European Hemophilia Consortium; FIX: Coagulation Factor IX; FVIII: Coagulation Factor VIII; HAL: Haemophilia Activity List; HJHS: Haemophilia Joint Health Score; HTC: Hemophilia Treatment Centre; HTCCNC: Hemophilia Treatment Centre Collaborative Network of China; MASAC: Medical and Scientific Advisory Council; MDT: Multidisciplinary team; NHD: National Haemophilia Database; NHF: National Hemophilia Foundation; PK: Pharmacokinetics; POCUS: Point of care ultrasound; PWH: People with haemophilia; SHIELD: Supporting Hemophilia through International Education, Learning and Development; WFH: World Federation of Hemophilia
Hemophilia is a rare X-linked bleeding disorder affecting approximately 400,000 people globally, caused by a deficiency of coagulation factor VIII (FVIII) (in hemophilia A) or factor IX (FIX) (in hemophilia B) [Citation1,Citation2].Footnote1 Current management of hemophilia involves treatment with either plasma-derived or recombinant factor VIII or IX products, ideally on a prophylactic basis for moderate and severe patients [Citation2–4]. The World Federation of Hemophilia (WFH) guidelines, first introduced in 2005 and updated in 2012, offer practical recommendations for the diagnosis and management of hemophilia and guidance on the management of complications, such as musculoskeletal issues, inhibitors and transfusion-related infections [Citation2]. However, there are multiple challenges to the funding and implementation of high quality, guideline driven, evidence based hemophilia care [Citation4]. The rarity of hemophilia may limit recognition and availability of tools and resources to confirm a diagnosis as well as restrict options for management of the disease and its complications. Funding may be inequitable, inconsistent, incomplete or absent, and further complicated by political and bureaucratic processes in place for its administration. Adherence to treatment recommendations and follow up is an ongoing challenge common to long-term medical conditions [Citation5]. Data suggest that up to 20% of people with hemophilia (PWH) do not follow their prescribed treatment [Citation6,Citation7].
Differing healthcare reimbursement systems, budgetary constraints, and geographical and cultural factors, make it difficult for any country to fully deliver ideal care. Some regions, mainly developed countries, may have sufficient treatment product available but then have a higher expectation of outcomes and often insufficient supportive care experience and personnel to monitor adherence and outcomes and implement regular follow-up [Citation8]. In contrast, developing regions may not have ready access to factor replacement but have developed excellent physiotherapy and rehabilitation programs. As a result, it is impossible to compare and contrast care quality and create models of care according to resources alone. Is it time therefore to move away from the pursuit of one perfect model and, instead, learn from global similarities and differences in the delivery of hemophilia care and focus on building the best approach tailored to each healthcare system and patient population?
Working together to deliver ideal care
The SHIELD group: Supporting Hemophilia through International Education, Learning and Development is an international panel of independent experts dedicated to the sharing of best practice and education in the management of hemophilia. A key element of SHIELD’s work is to share its multinational experience of hemophilia care across a variety of different settings and discuss the respective constraints to delivery of ideal care. This manuscript summarizes that discussion within a framework of four key themes – the availability and use of guidelines and algorithms for hemophilia care; how organizations can work together in the delivery of care; the role of patient registries; and personalization of care. The goal is to provide both specialist and non-specialist health care professionals who treat these patients with examples of evidence based best practice to enhance their approach to care.
Themed discussions are accompanied by summary recommendations, developed by the SHIELD group during the development of this manuscript and other educational materials, based on individual clinical experience and published evidence. These are incorporated into a collaborative road map () with the specific aim of guiding delivery of care regardless of setting and resources. Consistent feedback demonstrates how ongoing experience and new evidence, at both a global and local level, facilitates continuous improvements in care delivery.
Figure 1. A road map for collaborative care in hemophilia summarizing the SHIELD recommendations, potential outcomes and examples of how resources and best practice can contribute within a themed framework.
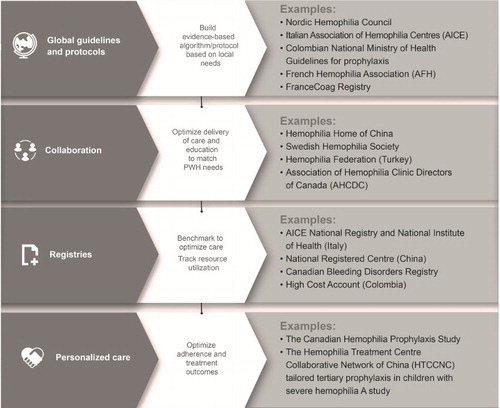
Key themes in the delivery of ideal hemophilia care
I - Guidelines and algorithms for hemophilia care
Guidelines aim to summarize published literature and expert consensus in order to harmonize the principles of care delivery around the world. The US Institute of Medicine states that clinical practice guidelines should be ‘ … informed by a systematic review of evidence and an assessment of the benefits and harms of alternative care options’ [Citation9]. Whilst this may be possible when developing clinical guidelines for diseases affecting large patient populations, the reality for rare diseases like hemophilia is that large, randomized studies to inform evidence-based recommendations are limited. As a result, alternative trial designs are needed to optimize the use of available patient data [Citation10].
A standardized approach to the management of rare diseases is important to enable faster initiation of treatment, improve cost-effectiveness of treatment and facilitate the tracking of treatment utilization. Whilst guidelines are usually based on global recommendations for models of care, algorithms can be country-specific. In some cases, they may even be centre-specific and reflect local needs of the patient population and localization of skills and resources to meet those needs.
Global guidelines have a key role to play in highlighting new knowledge that can advance the comprehensive management of hemophilia care. The availability of high-quality data establishing the efficacy and superiority of prophylactic factor replacement over episodic treatment of hemophilia and the need for greater use of validated, disease-specific assessment of care outcomes was a driving factor for the WFH to update its guidelines in 2012 [Citation2]. These guidelines are typically adapted for use at the national or even local (institutional) level, a process that can lead to a duplication of effort across regions. Their broad content allows for selection of relevant recommendations and adaptation to meet the specific needs of PWH at this local level. However, the extent to which global guidelines are reflected in local models of hemophilia care varies not only according to available resources but also to geography, regional alignment and sociocultural and political factors, often resulting in a ‘blending’ of global WFH and local guidelines. The Nordic Haemophilia Council (www.nordhemophilia.org), a body of hemophilia experts from the five Nordic countries (Denmark, Sweden, Norway, Finland and Iceland) regularly publishes and renews regional guidelines for hemophilia [Citation11]. The Italian Association of Hemophilia Centres’ (AICE) treatment guidelines for Hemophilia Treatment Centres (HTCs), based on published literature and scientific evidence, are usually updated every three years.
Global guidelines can even be adapted for those PWH living some distance away from treatment centers. In Colombia, treatment centers design home self-infusion protocols (based on the WFH guidelines) for patients who are receiving prophylaxis and living in remote areas. In 2015, the country’s National Ministry of Health issued its own guideline for prophylaxis in patients with severe hemophilia without inhibitors, resulting in 82% of severe patients now receiving prophylaxis [Citation12].
In recent years, there has been a large body of data supporting the value of a collaborative multidisciplinary team (MDT) in delivering effective and optimal care for patients with bleeding disorders [Citation13]. It builds on the WFH recommendations around provision of comprehensive care to address the wide-ranging needs of PWH and their families via a MDT of healthcare professionals with expertise and experience to attend to the relevant physical and psychosocial health issues, including but not limited to physicians, nurses, physiotherapists, and social workers [Citation2]. This model of care is relevant, irrespective of the availability of factor concentrate. In the United States, to be considered as federally qualified and be eligible for federal funding, an HTC is required to have a core team of specialists in place [Citation14]. A commitment ‘ … to treat the whole person and the family, through continuous supervision of all the medical and psychosocial aspects of bleeding disorders’ is mandated within the establishment of a comprehensive HTC [Citation15]. Recommendations from the Medical and Scientific Advisory Council (MASAC) of the National Hemophillia Foundation (NHF) list the social worker as a primary member of this MDT with a remit to ‘ … assist in the issues of daily living, such as adjusting to hemophilia and locating resources (e.g. insurance, transportation and housing)’ [Citation16]. Other organizations, such as the European Association for Haemophilia and Allied Disorders (EAHAD), support guidelines aimed at specific disciplines, for example, those published recently for nurses working in hemophilia care [Citation17] and a planned curriculum for physiotherapists. The organization also contributes to the development of basic principles of hemophilia care [Citation18]. Many HTCs regularly interact with local emergency departments to develop and update treatment algorithms that help in the initiation of immediate, appropriate therapy while waiting for advice from the HTC.
SHIELD recommendations:
Global guidelines provide a useful overview of the ideal standard of care. Identify the key priorities for the PWH in your region and select the elements of the available guidelines that are relevant and achievable.
Review other national or regional guidelines for models of local adaptation.
Identify other HTCs with similar hemophilia populations and tailor their examples of algorithms of care, derived from global guideline elements, to the needs of local PWH.
Work within HTCs to maximize resources and available personnel to optimize delivery of care in accordance with these adopted guidelines and algorithms.
II - Organizations working together in the delivery of care
When PWH, their families and healthcare providers collaborate, it has a positive impact on care and outcomes. There are two emblematic examples of the importance of such alliances in hemophilia care. The first, the WFH, was conceived as a joint venture between PWH and physicians; it was founded by a patient and its president is a patient. The second, the European Hemophilia Consortium (EHC; www.ehc.eu), is a non-profit organization that represents 45 national patient organizations from 27 member states of the European Union. It is involved in supporting both patient associations and medical research. Comprehensive care, as delivered by HTC networks, is an important illustration of how organizations can work together to address and deliver the needs of local PWH. Studies have shown the importance of a dedicated HTC in helping patients with inhibitors and increasing patient adherence to treatment [Citation19].
The size and complexity of care delivered by these HTCs can vary from region to region. In Italy, 57 HTCs within the AICE national network aim to harmonize patient management across 21 regions. A small number are classed as comprehensive care centers and offer specialized care including orthopaedic surgery and management of complex cases, such as inhibitor patients. In Colombia, management of inpatient care, surgery and emergency care is divided across 25 large centers situated in the main cities, all staffed by physicians specialized in hemophilia care. These centers also coordinate the home-delivery of factor for prophylaxis and on-demand treatment for patients living in remote or hard to access areas.
Working with patient organizations can help build awareness of hemophilia, influence healthcare policy, coordinate care standards and deliver standardized education to healthcare providers as well as PWH and their carers [Citation17,Citation20]. The mechanisms by which these organizations work together varies widely. In Canada, medical directors of the 25 HTCs make up the Association of Hemophilia Clinic Directors of Canada (AHCDC). The organization works to establish national standards of care in conjunction with the different associations comprising nurses, physiotherapists or social workers, as well as patients via the Canadian Hemophilia Society. This cooperation has been crucial to the introduction of a new national registry and ongoing negotiation with government for support of the HTC network and funding for current and novel treatment products.
There are other country-specific examples of collaboration. In China, rapid growth in the number of HTCs – from six to 50 between 2004 and 2016 – and the work of the organization, Hemophilia Home of China, has increased public awareness of hemophilia and led to greater coverage of the cost of concentrates by health insurance companies. In Germany, training camps set up by patient organizations supplement the work of HTCs in instructing patients around self-infusion and guiding treatment. In Sweden, healthcare providers work with the Swedish Hemophilia Society to develop patient information that is distributed via leaflets or booklets, online, at conferences and via summer camps. In Turkey, the first local Hemophilia Association was established in İzmir 20 years ago. Within 10 years, a Hemophilia Federation, comprising 12 regional associations throughout Turkey, was achieved. Since then, the organization has conducted educational meetings for clinicians, nurses and patients and parents, some in distant cities bordering with Syria, Iraq and Iran. In France, a similar collaboration led to an online training program dedicated to patients with mild hemophilia (www.hemomooc.fr), as well as an outreach program for females from families with hemophilia regarding their genetic and bleeding risks, diagnostic approaches and reproductive options. The United Kingdom Haemophilia Centre Doctors Organisation, established in 1968, oversees guideline publications covering topics such as joint bleeds and immune tolerance induction [Citation21,Citation22] and maintains a comprehensive national registry and online patient treatment record (Haemtrack; www.haemtrack.mdsas.com).
SHIELD recommendations
Partner with national and local patient organizations to deliver education programs to healthcare providers, PWH and their families using innovative formats such as summer camps as well as standardized web-based education programs.
Partner with patient organizations to increase hemophilia awareness and advocacy.
If there is no patient organization, encourage the setting up of one that can work at local level with a HTC or at national level with a network of HTCs.
III - Disease registries
The expression ‘You have to know where you are to know where you are going’ is directly relevant to the role of disease registries. These databases, comprising either individual or aggregate patient data, can further the understanding around variations in treatment; describe care patterns, including appropriateness and disparities in the delivery and quality of care; examine factors that influence prognosis and quality of life; and provide evidence on resource utilization [Citation23]. In addition, data regarding treatment outcomes may serve to inform guideline content in the absence of large-scale randomized controlled trials.
When looking at hemophilia specifically, national registries provide valuable insights into the variability of the PWH population in terms of prevalence, demographics and management from country to country. When linked to global guidelines for hemophilia, they can help to improve the diagnostic approach, treatment planning and delivery of care, and government resource allocation based on WFH priorities and population characteristics for that country. For example, details of therapy and bleeds for all patients with hemophilia are continuously updated in the Swedish Hemophilia Registry (www.kvalitetsregister.se). In Italy, all HTCs contribute to the National Registry, sending local epidemiological data and details of disease severity and treatment regiments to the AICE database via an electronic platform. The National Institute of Health (ISS) is responsible for the analysis and dissemination of this data, and AICE uses the database as a research tool for clinical studies [Citation24]. In China, patient members of the National Registered Center have now reached more than 13,000 and the data are used to improve rates of diagnosis and hemophilia knowledge, as well as explore China’s national treatment environment. In the UK, an individual’s longitudinal ‘steady state’ treatment on-line record (Haemtrack), musculoskeletal health measures (Haemophilia Joint Health Score; HJHS) and, increasingly, the Haemophilia Activity List (HAL) all inform this process. They also enrich clinic visits and also contribute to national epidemiology via the National Haemophilia Database (NHD). In France, the publicly-funded FranceCoag registry is a comprehensive, national project incorporating clinicians, patients and government authorities. It facilitates links within the community, harmonizes hemophilia care and permits specific research studies based on real world data [Citation25].
Registries can provide information on the use of treatment products, including the monitoring of prophylaxis and immune tolerance induction (ITI), data which are not always gathered by healthcare providers within the formal setting of the HTC. In Germany, home treatment is monitored by the HTC and patient entry of each infusion (time, reason, amount, batch number) in a diary, either in paper or electronic form, is mandatory according to the German transfusion law. Canada has recently introduced a national registry system that monitors home infusions. Information is entered electronically by patients and then analyzed to determine the appropriateness of the treatment regimen and track product inventories and utilization. Most HTCs in the United States participate in a registry through the American Thrombosis and Hemostasis Network (ATHN), a non-profit organization dedicated to collecting and collating national patient data. In addition to information entered by the HTCs, patients can also self-enter bleeding and treatment data into the registry using Advoy, a web-based electronic bleeding and infusion log (https://athnadvoy.athn.org), which can then be used to evaluate effectiveness of treatments and outcomes. The demands placed on these diverse registries will continue to evolve in parallel with the ambition to share meaningful outcomes data and ensure close pharmacovigilance as new products come to market and inform the different national procurement processes.
Collaboration by all organizations involved in hemophilia care can contribute to data collection around complications, such as emergency room visits, hospitalization, inhibitors, comorbidities and burden of disease. Such data can help health care systems follow trends and improve knowledge of both the national and local care situation and track respective outcome measures. For example, in 2015, the National Healthcare System in Colombia created a mandatory annual registry, the High Cost Account [Citation12], generating a complete clinical and administrative report which allows for patient follow-up and identifies strengths and weaknesses in the assistance process. The registry includes 2059 patients with hemophilia of which almost half are under 20 years of age. The High Cost Account shows the risk management indicators and motivating factors involved in patient care that improve interventions performed at all levels and progressively achieves substantial improvements in healthcare and quality of life.
SHIELD recommendations
If there is no national registry available, look at the data gathered by other regions and explore opportunities to share methodologies.
If there is a registry, use the real-time patient data provided to raise awareness of hemophilia and the burden of disease, particularly in terms of quality of life, and establish key epidemiological benchmarks.
Use treatment and outcomes data to develop cost utility arguments to drive improvements in resourcing hemophilia care.
IV - Tailored personalized care
The concept of personalized medicine results from the recognition that specialized diagnostic tests can help in the prescription of treatment that involves ‘the right person, the right drug at the right dose, and right time’ [Citation26]. In hemophilia, tailored personalized care involves the adaptation of treatment to life stage, bleeding frequency and lifestyle [Citation2,Citation27]. More specifically, the dose and frequency of infusions depend on pharmacokinetic (PK) properties of the specific product in addition to patient factors such as age, joint status, activity profile, lifestyle, access to homecare, vascular access, and parental and patient capabilities and understanding of the disease [Citation28]. The future availability of new therapeutic approaches for hemophilia may enhance the need for personalization.
Accurate diagnosis of hemophilia guides appropriate personalized care [Citation2], but diagnostic certainty about a bleed and subsequent recovery can be challenging; both PWH and clinicians can find it difficult to be certain if pain is attributable to an acute bleed [Citation29]. Studies suggest that the correct, clinical diagnosis is made in fewer than 50% of cases in the absence of imaging [Citation30,Citation31]. Utilization of point of care ultrasound (POCUS) is increasingly helpful in confirming a bleed and determining whether it is intra-or extra-articular [Citation32]. However, in many of the countries where PWH receive home treatment, the current principle of care is that the patient determines whether the symptoms warrant early intervention to manage a bleed.
Prophylaxis with regular infusion of either plasma-derived or recombinant factor VIII or IX products is the cornerstone of modern hemophilia care based on evidence of effectiveness in the prevention of bleeding and joint destruction in patients with moderate and severe disease [Citation2,Citation33]. The timing of initiation of prophylaxis i.e. whether at the first bleeding episode or before the age of two, and how long it continues, is subject to the constraints of local guidelines, population needs and budget availability [Citation34]. Dose and frequency should be adjusted according to individual PK, bleed frequency and changing needs during growth [Citation28]. However, the optimal regimen continues to be explored. The Canadian Hemophilia Prophylaxis Study examined escalating prophylaxis from one to three-times weekly, depending on a patient’s bleeding pattern [Citation35,Citation36]. In Sweden, all patients with severe and moderate hemophilia (and in special circumstances even mild) are offered prophylactic therapy with mainly recombinant FVIII and FIX products in accordance with the WFH guidelines. In Italy, prophylaxis is mainly implemented in severe and moderate patients at an average of three infusions per week in severe hemophilia A and twice weekly in severe hemophilia B. Although the majority are treated with recombinant products, there is still a group of patients treated with plasma-derived products. Prophylaxis for children in the United States with severe hemophilia typically starts around 1 to 2 years of age (or after the first major bleed), with a goal of alternate daily infusions for hemophilia A and once to twice weekly for hemophilia B. Standard half-life recombinant factor products are predominantly used but extended half-life products are gaining popularity and a small subset of patients, usually those with high concern for inhibitor development, are electing to use plasma-derived products. Treatment individualization by means of PK assessment and careful evaluation of bleeding phenotype, including ultrasound detection of early arthropathy, has been increasingly adopted over the last few years.
The tailoring of prophylactic regimens to lifestyle means that participation in physical activity and regular attendance at school or work can be encouraged. The benefits of regular exercise, in terms of fitness and improvements in joint, bone and muscle health, are well-recognized [Citation2,Citation37]. Working with other members of the MDT, physiotherapists play a key role in helping PWH to identify a suitable type and level of physical activity in which to participate [Citation38]. The value of close MDT involvement and support is demonstrated by the higher than expected adherence to prophylaxis in adolescents and young adult PWH in a recent UK study by van-Os et al. [Citation39].
Ultimately, PK measurements may help to define the best dosing schedule for each patient [Citation28] and a population PK evaluation makes this more practical in the clinical setting [Citation40]. An approach where FVIII trough levels are maintained above 1% has been shown to reduce the risk of a bleed when compared with the time spent with FVIII plasma levels below 1% [Citation41]. However, prophylaxis must consider individual PK differences that affect the duration of action of replacement factor (FVIII recovery, half-life and clearance) in the hemophilia population [Citation27,Citation28]. In China, the search for an individualized, cost effective and affordable prophylaxis regimen to reduce joint bleeding to an acceptable level remains a work in progress [Citation42-44]. Preliminary efficacy results with PK-tailored tertiary prophylaxis, started after a bleed has occurred in children with severe hemophilia A, show that it may be the most cost-effective individual prophylaxis approach [Citation45]. In response to the lack of affordability of standard full-dose prophylaxis for most families in China, the Hemophilia Treatment Centre Collaborative Network of China (HTCCNC) has begun studies evaluating the delivery of low-dose prophylaxis in boys with severe hemophilia A (10 IU/kg twice a week) and hemophilia B (20IU/kg weekly) [Citation46].
Once an appropriate prophylactic regimen has been established, management of PWH should take place in a home therapy setting where appropriate and the age and activity of the patient and other family lifestyle issues have been considered. Home therapy facilitates immediate access to clotting factor and optimal treatment, and has been shown to decrease pain, dysfunction and long-term disability as well as significantly decrease hospital admissions due to complications [Citation2]. Treatment effectiveness is also improved when the time between bleeding onset and infusion is minimized, especially for patients with inhibitors [Citation47]. Education around the management of infusions and close supervision is essential for successful home therapy, and patients need to be able to manage an emergency promptly. For some PWH, lack of skills and confidence in self-management may prove to be a barrier. Similarly, parents may also lack confidence in caring for a child with a bleeding disorder. Global initiatives such as the Parents Empowering Parents program (www.pepprogram.org) provide training to bolster parents’ confidence and professional knowledge-base regarding issues related to parenting and family management for parents of children with a bleeding disorder.
Beyond its role in acute care, the MDT is crucial following an acute joint or muscle bleed or after any surgical procedure. Rehabilitation is important for functional improvement and recovery after musculoskeletal bleeds and for those PWH with established haemophilic arthropathy [Citation2]. In the UK, the HJHS and HAL responses provide the MDT, and the physiotherapist in particular, with a baseline for the rehabilitation process. Recently, some centers have added routine POCUS assessment of key joints (elbows, knees and ankles) to identify demonstrable architectural change that is not yet symptomatic and to assist in treatment and rehabilitation. The physiotherapy team then agrees on a realistic muscle strengthening and joint stabilizing regimen, which may require several weeks or months for a serious bleed (e.g. psoas).
SHIELD recommendations
Educate PWH and their carers to improve recognition of a bleed and achieve prompt initiation of treatment.
Identify barriers to optimal personalized care, for example, lack of confidence to manage home therapy or concerns around participating in physical activity, and look at programs that help to address these.
Use patient data around infusions, bleeds and lifestyle to identify effective strategies that could be used on a wider basis.
Areas for further thinking and research
Careful examination of these overarching themes in the successful comprehensive care of PWH illuminates many of the barriers to achieving that ideal model of care. Many have been discussed already and some, like funding inequities and administrative processes, result from political and systemic factors that are beyond the remedy of the hemophilia care provider. However, other challenges may be amenable to change at the level of the HTC or local organization and should be considered in future research and resource planning. Many barriers to home therapy might be addressed by the local community, which could, in turn, impact on strategies for improved adherence to therapy, particularly among adolescent PWH. We have presented some different strategies for prophylaxis, but ultimately these regimens must be maximized within the unique local context to ensure both cost and clinical effectiveness. Finally, as registries develop and hemophilia treaters rely increasingly on the evidence available to inform clinical decisions, the definition of the minimal data set and key outcome measures is critical to facilitate both regional and international evaluation of care and outcomes to drive best practice.
Conclusion
Provision of primary prophylaxis for all moderate and severe PWH under the careful and regular monitoring of a comprehensive multidisciplinary team, is an admirable and reasonable goal. The members of SHIELD recognize that this ideal may not be fully achievable in any setting. However, multiple elements of best practice can contribute to the delivery of the best possible care, as listed in the recommendations and illustrated in . Patient communities are highly variable between countries and patient needs and expectations are also evolving at a rapid pace. Care and support infrastructure may be at very different stages in different regions. Finally, new therapeutic options, including non-replacement therapies and gene therapy, are rapidly changing the treatment paradigm.
Capturing the diversity of experience from around the world and sharing examples of best practice in collaboration between clinics, regions, and countries, as illustrated in , is a founding principle of the SHIELD group. Ultimately, we hope that this will contribute to the creation of an adaptable model of care which is grounded in global evidence but able to adapt organically to these different influences and lead to better treatment outcomes for all persons with hemophilia.
Acknowledgements
The authors are members of the SHIELD (Supporting Hemophilia through International Education, Learning and Development) group, an independent panel of physicians with expert interest in the treatment of haemophilia. Formation of the SHIELD group and its meetings were supported by Bayer Pharma Plc. Members received honoraria for attendance at meetings but no honoraria were paid for contributions to this manuscript. This publication and its content are solely the responsibility of the authors. Medical writing assistance was provided by Clark Health Communications under the direction of the authors and paid for by Bayer Pharma Plc.
Disclosure statement
Dr Nadine Andersson has participated in advisory boards for Bayer Pharma plc, Boehringer Ingelheim and SOBI and received speaker fees from Sobi, Bayer Pharma plc and CSL Behring. Dr Katharine Batt acts as a consultant to Bayer Pharma plc, NovoNordisk and Shire Pharmaceuticals and as a scientific advisor to Precision Health Economics. Dr Brian Blanchford has participated in a global advisory board for Bayer Pharma plc and ad hoc advisory boards for CSL Behring, Shire Pharmaceuticals, BPL and Grifols. Dr Roseline d’Oiron has received personal fees for lectures or advisory boards and received non-financial support for attending scientific meetings from Shire Pharmaceuticals/Baxalta, NovoNordisk, Bayer Pharma plc, Pfizer, Sobi, LFB, CSL Behring, Roche and Octapharma. Dr Carmen Escuriola-Ettingshausen has received honoraria, acted as a consultant or received travel funding from Alnylam Pharmaceuticals, Bayer Healthcare, CSL Behring, Biotest, Grifols, LFB Biopharmaceuticals, Freeline Therapeutics, Pfizer, Sobi, Roche, Octapharma and Shire Pharmaceuticals. Dr Dan Hart has received research income from Shire Pharmaceuticals and Octapharma and acted as a consultant/speaker for Pfizer, Shire Pharmaceuticals, Novo Nordisk, Octapharma, Roche, uniQure and Biotest Pharmaceuticals Corporation. Dr Kaan Kavakli has participated in advisory boards, acted as a speaker and provided consultancy and scientific advice to Bayer Pharma plc, Shire Pharmaceuticals, Pfizer and Novo Nordisk. Dr Víctor Jiménez Yuste has received reimbursement for attending symposia/congresses and/or honoraria for speaking, consulting and/or research funding from Shire Pharmaceuticals, Bayer Pharma plc, CSL-Behring, Grifols, Novo Nordisk, Sobi, Octapharma and Pfizer. Dr Maria Elisa Mancuso has acted as a consultant to Bayer Healthcare, CSL Behring, Pfizer, Novo Nordisk, Sobi/Biogen, Baxalta/Shire Pharmaceuticals and Kedrion Biopharma and has acted as a speaker for Bayer Healthcare, CSL Behring, Pfizer, Novo Nordisk, Sobi/Biogen, Octapharma and Biotest Pharmaceuticals Corporation. Dr Keiji Nogami has participated in a global advisory board for Bayer Pharma plc. Professor Carlos Ramírez has no conflicts of interest. Dr Jayson Stoffman has acted as a consultant to Bayer Pharma plc and Hoffman-La Roche Ltd. Professor Runhui Wu has no conflicts of interest.
Notes on contributors
Dr J. Stoffman is Medical Director of the Winnipeg Regional Health Authority Manitoba Bleeding Disorders Program and a Pediatric Hematologist/Oncologist at the Winnipeg Children’s Hospital and CancerCare Manitoba in Canada. He is also Program Director, Subspecialty and Fellowship Programs in the Section of Pediatric Hematology/Oncology/BMT at the University of Manitoba.
Dr N. G. Andersson is a pediatric hematologist and senior consultant at the Malmö Coagulation Centre in Sweden, and is responsible for Paediatric Coagulation.
Dr B. Branchford is Assistant Professor in the Department of Pediatrics, Division of Hematology/Oncology at the University of Colorado School of Medicine, the University of Colorado Hemophilia and Thrombosis Center, and Children’s Hospital Colorado, in the USA.
Dr K. Batt is Assistant Professor in Wake Forest University Baptist Medical Center’s Department of Internal Medicine, Section of Hematology/Oncology where she maintains a predominantly non-malignant hematology practice.
Dr R. D’Oiron is Clinical Investigator and Associate Director at the Reference Centre for Haemophilia and Other Congenital Rare Bleeding Disorders, Congenital Platelets Disorders and von Willebrand Disease at Bicêtre Hospital AP-HP, University Paris XI, Le Kremlin–Bicêtre, France.
Dr C. Escuriola Ettingshausen is the Director of the Haemophilia Centre Rhein Main - HZRM, Frankfurt-Mörfelden, Germany.
Dr D. P. Hart is a senior lecturer and honorary consultant Haematologist specialising in inherited bleeding disorders for one of the most ethnically diverse communities in Europe at The Royal London Hospital Haemophilia Centre, Barts and The London School of Medicine and Dentistry, London.
Dr V. Jiménez Yuste is Associate Professor in the Haematology Department at the Autónoma University of Madrid, Spain, and Head of the Haematology Department at La Paz University Hospital, Madrid, Spain.
Dr K. Kavakli is a Professor of Pediatrics and Pediatric Hematology at Ege University Children’s Hospital in Izmir, Turkey. He held the position of Chairman of the Department of Paediatric Haematology at Ege University Children’s Hospital in Izmir, Turkey for 10 years until 2014. In 2000, he was also appointed Director of the Ege Haemophilia Centre and Ege Hemophilia Association.
Dr M. E. Mancuso is a Hematologist with a PhD in Clinical Methodology and works as a Clinical Assistant at the Angelo Bianchi Bonomi Hemophilia and Thrombosis Center, Fondazione IRCCS Ca’ Granda, Ospedale Maggiore Policlinico in Milan, Italy.
Dr K. Nogami is Associate Professor in the department of Pediatrics at Nara Medical University in Kashihara, Japan.
Professor C. Ramírez is a Professor of Medicine at the Fundación Universitaria Sanitas in Bogotá, Colombia. He is also the Medical Director of the Department of Hematology at the Organización Sanitas Internacional in Bogotá and the Medical Director of the Haemophilia Center- Clinica Colsanitas.
Professor R. Wu is a professor in the pediatric department and Deputy Director of the Hematology Department of Capital Medical University in Beijing, China. She is a clinical Paediatrician Hematologist in the Hematology Center of Beijing Children’s Hospital, where she focusses on children’s bleeding disorders. Professor Wu is also the hemophilia paediatrician and director of the Hemophilia Comprehensive Care Programme at Beijing Children’s Hospital.
ORCID
J. Stoffman http://orcid.org/0000-0002-2096-8671
N. G. Andersson http://orcid.org/0000-0001-6058-8350
B. Branchford http://orcid.org/0000-0002-4076-270X
R. D'Oiron http://orcid.org/0000-0002-4843-7805
K. Kavakli http://orcid.org/0000-0003-1174-1958
M. E. Mancuso http://orcid.org/0000-0002-7113-4028
Notes
1 Supporting Hemophilia through International Education, Learning and Development
Related Research Data
References
- Stonebraker JS, Bolton-Maggs PH, Soucie JM, et al. A study of variations in the reported haemophilia A prevalence around the world. Haemophilia. 2010;16(1):20–32. doi: 10.1111/j.1365-2516.2009.02127.x
- Srivastava A, Brewer AK, Mauser-Bunschoten EP, et al. Treatment guidelines working group on behalf of World Federation of Hemophilia. Guidelines for the management of hemophilia. Haemophilia. 2013;19:e1–e47. doi: 10.1111/j.1365-2516.2012.02909.x
- Poonnoose P, Carneiro JDA, Cruickshank AL, et al. MUSFIH study group. Episodic replacement of clotting factor concentrates does not prevent bleeding or musculoskeletal damage - the MUSFIH study. Haemophilia. 2017;23(4):538–546. doi: 10.1111/hae.13242
- van den Berg HM. From treatment to prevention of bleeds: what more evidence do we need? Haemophilia. 2017;23(4):494–496. doi: 10.1111/hae.13256
- World Health Organization. Adherence to long-term therapies: Evidence for action. Available from: http://www.who.int/chp/knowledge/publications/adherence_full_report.pdf.
- Gringeri A, Doralt J, Valentino LA, et al. An innovative outcome-based care and procurement model of hemophilia management. Expert Rev Pharmacoecon Outcomes Res. 2016;16(3):337–345. doi: 10.1080/14737167.2016.1178066
- Zappa S, McDaniel M, Marandola J, et al. Treatment trends for haemophilia A and haemophilia B in the United States: results from the 2010 practice patterns survey. Haemophilia. 2012;18:e140–e153. doi: 10.1111/j.1365-2516.2012.02770.x
- Page D, Crymble S, Lawday K, et al. Penny wise, pound foolish: An assessment of Canadian Hemophilia/inherited bleeding disorder comprehensive care program services and resources. Haemophilia. 2016;22(4):531–536. doi: 10.1111/hae.12913
- Institute of Medicine (US) Committee on Standards for Developing Trustworthy Clinical Practice Guidelines, Graham R, Mancher M, et al. Clinical practice guidelines we can trust. Washington (DC): National Academies Press (US); 2011; 1, Introduction. Available from: https://www.ncbi.nlm.nih.gov/books/NBK209546/.
- Pai M, Key NS, Skinner M, et al. NHF-McMaster Guideline on care models for haemophilia management. Haemophilia. 2016;22(Suppl 3):6–16. doi: 10.1111/hae.13008
- Nordic Hemophilia Guidelines. Available from: http://nordhemophilia.org.
- National Ministry of Health and Social Protection (Colombia). Situación de la Hemofilia en Colombia, 2016. Registro de información para el año 2016. Cuenta de alto costo Ministerio de Salud. Available from: https://cuentadealtocosto.org/site/index.php/publicaciones#hemofilia
- Pipe SW, Kessler CM. Evidence-based guidelines support integrated disease management as the optimal model of haemophilia care. Hemophilia. 2016;22(Suppl 3):3–5. doi: 10.1111/hae.12997
- Skinner MW, Soucie JM, McLaughlin K. The national haemophilia program standards, evaluation and oversight systems in the United States of America. Blood Transfusion. 2014;12(Suppl 3):e542–e548.
- National Hemophilia Foundation. Comprehensive Medical Care Hemophilia Treatment 2017. Available from: https://www.hemophilia.org/Researchers-Healthcare-Providers/Comprehensive-Medical-Care-Hemophilia-Treatment-Centers.
- Medical and Scientific Advisory Council (MASAC) of the National Hemophilia Foundation. Standards and criteria for the care of persons with congenital bleeding disorders (revised April 2002). Available from: https://www.hemophilia.org/Researchers-Healthcare-Providers/Medical-and-Scientific-Advisory-Council-MASAC/MASAC-Recommendations/Standards-and-Criteria-for-the-Care-of-Persons-with-Congenital-Bleeding-Disorders.
- EAHAD Nurses Committee, Harrington C, Bedford M, et al. A European curriculum for nurses working in haemophilia. Haemophilia. 2016;22(1):103–109. doi: 10.1111/hae.12785
- Colvin BT, Astermark J, Fischer K, et al. Inter Disciplinary Working Group. European principles of haemophilia care. Haemophilia. 2008;14(2):361–374. doi: 10.1111/j.1365-2516.2007.01625.x
- Lindvall K, von Mackensen S, Elmståhl S, et al. Knowledge of disease and adherence in adult patients with haemophilia. Haemophilia. 2010;16(4):592–596.
- O’Mahony B, Skinner MW, Noone D, et al. Assessments of outcome in haemophilia - a patient perspective. Haemophilia. 2016;22(3):e208–e209. doi: 10.1111/hae.12922
- Hanley J, McKernan A, Creagh MD, et al. Musculoskeletal working party of the UKHCDO. Guidelines for the management of acute joint bleeds and chronic synovitis in haemophilia: A United Kingdom Haemophilia Centre Doctors' Organisation (UKHCDO) guideline. Haemophilia. 2017;23(4):511–520. doi: 10.1111/hae.13201
- Collins PW, Blanchette VS, Fischer K, et al. rAHF-PFM Study Group. Break-through bleeding in relation to predicted factor VIII levels in patients receiving prophylactic treatment for severe haemophilia A. J Thromb Haemost. 2009;7(3):413–420. doi: 10.1111/j.1538-7836.2008.03270.x
- LaBresh KA, Gliklich R, Liljestrand J, et al. Using “get with the guidelines” to improve cardiovascular secondary prevention. J Comm J Qual Saf. 2003;29(10):539–550.
- Hassan HJ, Morfini M, Taruscio D, et al. Current status of Italian registries on inherited bleeding disorders. Blood Transf. 2014;12(Suppl 3):s576–s581.
- Calvez T, Chambost H, Claeyssens-Donadel S, et al. Francecoag Network. Recombinant factor VIII products and inhibitor development in previously untreated boys with severe hemophilia A. Blood. 2014;124(23):3398–3408. doi: 10.1182/blood-2014-07-586347
- Hamburg MA, Collins FS. The path to personalized medicine. N Engl J Med. 2010;363:301–304. doi: 10.1056/NEJMp1006304
- Reininger AJ, Chehadeh HE. The principles of PK-tailored prophylaxis. Hämostaseologie. 2013;33(Suppl 1):S32–S35.
- Ljung R, Fischer K, Carcao M, et al. INPH group. Practical considerations in choosing a factor VIII prophylaxis regimen: role of clinical phenotype and trough levels. Thromb Haemost. 2016;115(5):913–920. doi: 10.1160/TH15-08-0664
- Auerswald G, Dolan G, Duffy A, et al. Pain and pain management in haemophilia. Blood Coagul Fibrinolysis. 2016;27:845–854. doi: 10.1097/MBC.0000000000000571
- Stephensen D, Drechsler WI, Scott OM. Biomechanics of lower limb haemophilic arthropathy. Blood Rev. 2012;26(5):213–221. doi: 10.1016/j.blre.2012.06.003
- Ceponis A, Wong-Sefidan I, Glass CS, et al. Rapid musculoskeletal ultrasound for painful episodes in adult haemophilia patients. Haemophilia. 2013;19(5):790–798. doi: 10.1111/hae.12175
- Strike KL, Iorio A, Jackson S, et al. Point of care ultrasonography in haemophilia care: recommendations for training and competency evaluation. Haemophilia. 2015;21(6):828–831. doi: 10.1111/hae.12767
- Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. New Engl J Med. 2007;357(6):535–544. doi: 10.1056/NEJMoa067659
- National Hemophilia Foundation. MASAC Recommendations concerning prophylaxis (regular administration of clotting factor concentrate to prevent bleeding) 2016. Document 179. Available from: http://www.hemophilia.org/NHFWeb/MainPgs/MainNHF.aspx?menuid=57&contentid=1007.
- Feldman BM, Pai M, Rivard GE, et al. Association of Hemophilia Clinic Directors of Canada Prophylaxis Study Group. Tailored prophylaxis in severe hemophilia A: interim results from the first 5 years of the Canadian Hemophilia primary prophylaxis study. J Thromb Haemost. 2006;4:1228–1236. doi: 10.1111/j.1538-7836.2006.01953.x
- Hang MX, Blanchette VS, Pullenayegum E, et al. On behalf of the Canadian Hemophilia Primary Prophylaxis Study Group. Age at first joint bleed and bleeding severity in boys with severe hemophilia A: Canadian hemophilia primary prophylaxis study. J Thromb Haemost. 2011;9:1067–1069. doi: 10.1111/j.1538-7836.2011.04228.x
- Wang M, Alvarez-Román MT, Chowdary P, et al. Physical activity in individuals with haemophilia and experience with recombinant factor VIII Fc fusion protein and recombinant factor IX Fc fusion protein for the treatment of active patients: a literature review and case reports. Blood Coagul Fibrinolysis. 2016;27:737–744. doi: 10.1097/MBC.0000000000000565
- Goto M, Takedani H, Yokota K, et al. Strategies to encourage physical activity in patients with hemophilia to improve quality of life. J Blood Medicine. 2016;8:85–98. doi: 10.2147/JBM.S84848
- van Os SB, Troop NA, Sullivan KR, et al. Adherence to prophylaxis in adolescents and young adults with severe haemophilia: A quantitative study with patients. PLoS ONE. 2017;12(1):e0169880, doi:10.1371/journal.pone.0169880.
- Iorio A, Keepanasseril A, Foster G, et al. Development of a Web-Accessible Population Pharmacokinetic Service-Hemophilia (WAPPS-Hemo): Study Protocol. JMIR Res Protoc. 2016;5(4):e239. doi: 10.2196/resprot.6558
- Collins PW, Quon DVK, Makris M, et al. Pharmacokinetics, safety and efficacy of a recombinant factor IX product, trenonacog alfa in previously treated haemophilia B patients. Haemophilia. 2017 Aug 17;24(1):104–112. [Epub ahead of print]. doi:10.1111/hae.13324.
- Wu R, Luke KH. The benefit of low dose prophylaxis in the treatment of hemophilia: A focus on China. Expert Rev Hematol. 2017;10(11):995–1004. doi: 10.1080/17474086.2017.1386096
- Wu R, Sun J, Xiao J, et al. A prospective study of health-related quality of life of boys with severe haemophilia A in China: comparing on-demand to prophylaxis treatment. Haemophilia. 2017;23(3):430–436. doi: 10.1111/hae.13198
- Tang L, Wu R, Sun J, et al. Short-term low-dose secondary prophylaxis for severe/moderate haemophilia A children is beneficial to reduce bleed and improve daily activity, but there are obstacles in its execution: a multi-centre pilot study in China. Haemophilia. 2013;19(1):27–34. doi: 10.1111/j.1365-2516.2012.02926.x
- Wu R, Luke KH, Poon MC, et al. Low dose secondary prophylaxis reduces joint bleeding in severe and moderate haemophilic children: a pilot study in China. Haemophilia. 2011;17(1):70–74. doi: 10.1111/j.1365-2516.2010.02348.x
- Yao W, Xiao J, Cheng X, et al. The efficacy of recombinant FVIII low-dose prophylaxis in Chinese pediatric patients with severe hemophilia A. A retrospective analysis from the ReCARE Study. Clin Appl Thromb Haemost. 2017;23(7):851–858. doi: 10.1177/1076029616679507
- Kavakli K, Yesilipek A, Antmen B, et al. The value of early treatment in patients with haemophilia and inhibitors. Haemophilia. 2010;16(3):487–494.