
ABSTRACT
Objective: Factor VII deficiency is the commonest of the rare bleeding disorders with limited knowledge on clinical profile. The objective of this study was to study the prevalence and clinico-hematological profile of factor VII-deficient patients.
Methods: It is a retrospective observational study of probable inherited factor VII deficiency covering 18 months. Their clinical profile, family history, investigation and treatment records were studied in detail.
Results: The study group comprised of total 12 factor VII deficiency cases with mean age of 17.5 years of onset of symptoms. The commonest symptom was menorrhagia (41.6%) followed by epistaxis (25%) and easy bruisability (16.6%). These 12 patients when categorized according to bleeding severity: severe bleeding – 2, moderate bleeding – 3, mild bleeding – 6 and asymptomatic – 1. All cases had prolonged prothrombin time (PT) with mean PT of 35.4 seconds (range 18–50 seconds) and mean prolongation of PT from upper limit of normal – 19.4 seconds (range 2–34 seconds). Factor VII levels ranged from < 1–40% in these patients. Clinical symptoms were not in concordance with factor levels. Of 12 patients, required treatment other than local measures.
Discussion and Conclusion: Inherited factor VII deficiency is the commonest autosomally inherited factor deficiency with marked variation in the age of presentation and clinical symptoms. The laboratory results in form of PT and factor VII levels do not correlate with the severity of clinical presentation. A comprehensive evaluation to exclude acquired causes of factor VII deficiency, e.g. obesity, liver diseases, vitamin K deficiency and acquired inhibitors is required before labeling it as inherited in the absence of family history and molecular studies.
Introduction
Factor VII (F VII) deficiency is the commonest of the ‘rare inherited bleeding disorders’ with an estimated incidence of 1 case per 3,00,000 to 5,00,000 individuals [Citation1]. Due to the rarity of the disease, the current knowledge on clinical profile of such patients and their treatment is limited. In the recent times, two large population-based multicenter studies – the International registry on congenital FVII deficiency (IRF7) study and Seven treatment evaluation registry (STER) – have been collecting data on the same and have contributed significantly to a better understanding of this rare disorder [Citation2,Citation3]. However, until these studies come up with their final word on the pathogenesis, diagnosis and treatment of factor VII-deficient cases, such patients remain a mystery to the clinician and the diagnostician in routine clinical practice.
Factor VII is a Vitamin K-dependent glycoprotein, synthesized in the liver and having the shortest half-life (4–5 hours) among all coagulation proteins [Citation4]. It circulates in plasma in two forms – majority as an inactive zymogen (plasma concentration around 10 nmol L−1) and much smaller amount as the active form (plasma concentration around 10 pmol L−1) [Citation5]. It is the complex formed by activated factor VII and tissue factor (TF) which plays a leading role in initiating the extrinsic arm of coagulation cascade. Despite being a prime protein in the coagulation system, the factor VII-deficient patients mostly present with mucocutaneous bleeding which sometimes can be confused with platelet-related bleeding and hence require thorough evaluation [Citation6]. The clinical profile of factor VII-deficient cases varies widely and can range from asymptomatic to severe life-threatening bleeds. Paradoxically, even rare cases of thrombosis have been described in factor VII-deficient cases [Citation7].
Inherited factor VII deficiency is the only congenital bleeding disorder characterized by isolated prolongation of prothrombin time (PT) which can then be confirmed by one-stage PT-based assay for factor VII level. However, for reasons which are unclear hitherto, there is a poor correlation between factor VII levels and the bleeding symptoms, which may pose a diagnostic dilemma [Citation8].
The recent studies which have studied the mutational profile in factor VII-deficient cases have concluded that there is a marked phenotype–genotype disparity among these patients [Citation9,Citation10]. That means patients homozygous for the same mutation do not always belong to the same class of bleeding severity. This may be attributed to various genetic polymorphism of factor VII gene or the environmental factors affecting plasma levels of factor VII [Citation11,Citation12]. This further increases the confusion in patients where family members with identical mutations present with markedly different phenotype.
Management of these cases further adds to the uncertainties of this disorder as there are no generally agreed upon guidelines. The available options include fresh frozen plasma (FFP), factor concentrates and recombinant factor VII (RecFVIIa) [Citation13]. Factor VII deficiency is an FDA-approved indication for the use of RecFVIIa. STER study has been studying in detail the use of RecFVIIa in factor VII deficiency cases as treatment and prophylaxis modality [Citation3].
Till date, there have been few studies describing the clinical spectrum of patients presenting with factor VII deficiency. Most of these studies are from the west and data from developing countries are very sparse, bearing occasional case reports. This could be partly due to the biological heterogeneity of the disorder and partly due to difficulty in correct diagnosis and poor follow-up. The authors aim to study the prevalence and clinicohematological profile of factor VII-deficient patients in the Indian population.
Material and methods
This study was a retrospective observational study conducted in the Department of Hematology of a tertiary care center in India covering a period from March 2016 to August 2017. Twelve cases of probable inherited factor VII deficiency, presenting with prolonged PT and low factor VII levels, found during this period were included in the study. Cases suspected of Disseminated intravascular coagulation (DIC) and Vitamin K deficiency (those who responded to Inj Vitamin K trial) were excluded. The clinical profile, family history and treatment records of these cases were studied in detail. The minimum investigation profile obtained in all patients included screening coagulogram (platelet count, PT, activated prothrombin time, thrombin time and fibrinogen levels), inhibitor screen by mixing 1:1 with normal plasma, factor VII levels and liver function tests (serum bilirubin and liver enzymes). Factor VII level assay was performed in our laboratory by one stage PT-based assay on automated coagulometer (STA Compact by STA Diagnostics) using STA factor VII-deficient plasma. The samples for the same were collected in 3.2% sodium citrate vial as per standard guidelines and taking care to avoid cold activation of factor VII while processing [Citation14].
Results
Study population
During March 2016–August 2017, we came across 12 cases (38.5%, 7 females and 5 males) of factor VII deficiency out of the total 31 cases of rare bleeding disorders. All these 12 patients had prolonged PT and low factor VII levels. The mean age of the study group was 28 years (range 4–68 years) and the median age of onset of bleeding symptoms was 17.5 years (range 2–47 years). Out of these 12 cases, 5 patients presented in the pediatric age group, 4 in adolescence, 2 in adulthood and 1 case remained asymptomatic even at an elderly age. The detailed clinical and laboratory profile of these 12 patients is as shown in .
Table 1. Clinicohematological profile of patients with FVII deficiency.
Clinical profile
Spectrum of bleeding symptoms in our study group is given as per , which included gum bleedings (n = 1), epistaxis (n = 4), easy bruisability (n = 3), menorrhagia (n = 5), joint bleeds (n = 2), postoperative bleeding (n = 1), prolonged bleedings from wounds (n = 1) and hemoperitoneum (n = 1). According to severity criteria (as shown in ), three cases were in the severe category, two in moderate and six were in the mild category. One case was asymptomatic and was incidentally detected during preoperative evaluation.
Figure 1. Spectrum of bleeding symptoms in factor VII-deficient cases.
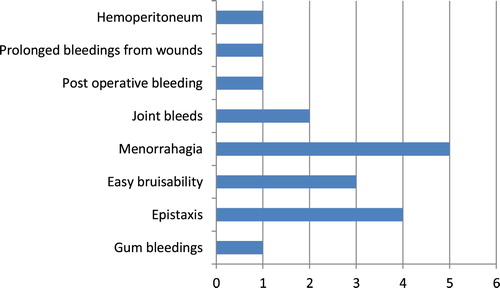
Table 2. Severity classification of factor VII-deficient patients (as adapted from [Citation15]).
No underlying apparent cause for factor VII deficiency was found in 11 out of 12 cases; however, 1 case (case no 12 –) on further evaluation showed the presence of systemic amyloidosis. History regarding drug intake, consanguineous marriage (positive history in 2 patients) and family history of bleeding in any first-degree relatives (negative in all 12 cases) was taken from clinical records.
Investigation profile
The screening coagulation profile of all 12 cases showed the presence of prolonged PT. The mean values of the screening profile in the study group were as follows: platelet count – 2.50 lakh (range 1.39–4.02 lakh), PT – 35.4 (range 18–50 seconds), activated PT – 30.7 (range 28–36 seconds), thrombin time – 16.4 (range 15–20 seconds), fibrinogen levels – 3.85 g dL−1 (range 2.8–4.5 g dL−1). The mean prolongation of PT from baseline in these patients was 19.4 seconds (range 2–34 seconds). In all these cases, baseline liver function tests were normal and inhibitor screen by mixing with normal plasma (1:1) was negative. Six out of 12 cases received vit K trial (inj Vit K 5 mg IV for 3 days) but their PT remained prolonged even after trial. Factor levels as assessed by one-stage PT-based factor VII assay showed low factor VII in all cases. Six out of 12 cases had factor VII levels less than 1%, 3 cases had factor VII level between 2 and 10% and 3 had factor VII levels above 20%. One case (case no 12) showed low factor X (2%) in addition to low factor VII (4%). This patient was found to have systemic amyloidosis on further evaluation with bone marrow examination and serum-free light chain assay.
Treatment profile
The treatment records of these patients revealed that 6 out of 12 cases did not receive any treatment other than local measures. Two patients out of five patients who presented with menorrhagia were given antifibrinolytics and responded well. One pregnant female patient (case no. 6) who presented with severe bleeding per vagina post-delivery and joint bleedings had severe anemia (Hb – 6.2 g dL−1) at presentation. This patient was transfused packed red cells in addition to FFP and a single dose of RecFVIIa. She responded well to RecFVIIa and the bleeding stopped after 24 hours. Four patients had a history of receiving FFP intermittently on demand. None of the patients received any prophylactic treatment.
Discussion
Among the rare bleeding disorders, the prevalence of factor VII deficiency is described as the highest (35–40%) by earlier studies [Citation15]. Our institutional data covering a period of 18 months also confirmed the same findings with 38.5% incidence. Inherited factor VII deficiency is an extremely heterogeneous disorder with regards to the clinical presentation, sites and severity of bleeding. This was evident in our study group where the patient ranged widely in terms of age of presentation (4–68 years) and severity of bleeding symptoms (severe – 3, moderate – 2 and mild – 6). A mild prominence of symptomatic female patients (7 out of 12 patients, 58%) was noted in our study. Similar observations were made by Mariani et al [Citation16]. The author described the increased incidence of female-specific symptoms in their study, e.g. menorrhagia, hemoperitoneum related to ovarian cyst, metrorrhagia and postpartum hemorrhage. The possible explanation for the same may be that hemostasis in the uterus is dependent mainly on the extrinsic pathway due to the abundance of TF natively. Hence, menorrhagia is a common presenting symptom in factor VII-deficient women. Other rare bleeding symptoms described in various studies include CNS bleeds, GI bleeds and thrombosis [Citation16]. The paradoxical association of thrombosis and inherited factor VII deficiency has been studied by Mariani et al. and Marty et al. , the mechanism of which remains unelucidated [Citation7,Citation16]. The possible factors could be co-existing thrombophilic state, therapy with RecVIIa or prothrombin complex concentrates which may adversely increase the levels of other vit K-dependent factors [Citation7]. However, we did not come across any case of thrombosis in this small study group.
Factor VII deficiency is associated with autosomal recessive inheritance and is more prevalent in communities practicing consanguineous marriages [Citation17]. However, history of consanguineous marriage was available only in two patients in our study group. Family history of bleeding in any first-degree relative was negative in all our patients. This could be due to marked phenotypic–genotypic disparity as described by Millar et al. in their studies [Citation18]. Mutational screening of patients and family members would have helped in identifying asymptomatic carriers of the similar mutation in the study group.
Factor VII levels in plasma are affected not only by genetic variations but also by a number of environmental factors, e.g. pregnancy, increasing age, obesity, underlying disorders, and vitamin K deficiency [Citation12]. Before considering a diagnosis of inherited factor VII deficiency, it is prudent to rule out any underlying disorders, vitamin k deficiency (using Vitamin K trial), liver disorders and rarely inhibitors. Also, it is essential to repeat factor VII levels on more than one occasion to rule out any transient/technical variations. Study for lupus anticoagulant may be taken up on a case to case basis.
The diagnosis of FVII deficiency is typically simple and patients present with isolated PT prolongation. Associated prolongation of APTT generally indicates towards either multiple factor deficiency or acquired causes like liver disorders and lupus anticoagulant. Only one patient in our study group presented with prolongation of PT and APTT who was later found to have multiple factor deficiency (F VII: 4% and FX: 2%) secondary to systemic amyloidosis. All other patients had isolated prolongation of PT thereby excluding other possibilities. In the absence of family history and mutation studies, it is difficult to label these cases as inherited factor VII deficiency though there are no apparent underlying disorders in 11 cases. The other rare possibility includes an acquired factor VII deficiency with non-neutralizing type of antibodies (which may cause factor VII deficiency by increased clearance from plasma and hence inhibitor screen may remain negative). A longer follow-up and repeat testing are required to identify such cases.
We observed that factor VII levels did not correlate with the prolongation of PT (above mean normal limit) and with the severity of the bleeding symptoms. Three out of six cases who presented with less than 1% factor VII levels had mild bleeding symptoms, while two had severe bleeding and one case was asymptomatic showing poor correlation between factor VII levels and bleeding severity. Three patients with factor VII levels above 20% and presence of bleeding symptoms indicate additional unknown mechanisms for bleeding. The disparity between the severity of bleeding and factor VII levels is previously reported by many studies [Citation2,Citation3,Citation9]. The other contributing variable to this disparity may be that the factor VII assay is influenced by a number of technical variables and different factor VII variants give variable results. For example type of thromboplastin, quality of calibrator or type of reference plasma may cause marked interlaboratory variations [Citation19]. There are no set standard recommendations as of now for laboratory diagnosis.
Laboratory diagnosis of FVII deficiency does not predict the individual risk of bleeding in the future. Matteo and Mariani studied the risk of future bleedings in patients of factor VII deficiency as per the clinical presentation at onset [Citation20]. They followed a cohort of 626 patients of factor VII deficiency for a period of 9.12 years and concluded that the first major bleeding symptom is an independent predictor of the risk of subsequent major bleeds [Citation20]. Our study being a retrospective one with a short study span could not derive any conclusion regarding future bleeding risks in these patients.
Treatment in our study group was mainly guided by the severity of symptoms which is the prevalent practice for lack of any standard guidelines. Therapeutic options available in our settings include antifibrinolytics, FFP and Rec VIIa in patients with severe bleeding phenotype. No patient was given any primary/secondary prophylaxis during the study period. Preliminary studies by STER have shown the benefit of secondary prophylaxis in patients with severe phenotype; however, long-term studies validating the same are lacking [Citation2,Citation3,Citation5].
Limitations of the study
This was a small study group with a limited number of cases. The genetic correlation by factor VII gene sequencing to assess patients phenotype and genotype correlation would have added more information to the study. However, due to limited availability of the technique and cost constraints, the same was not done in our cases.
Conclusion
Factor VII-deficient cases present with marked phenotypic heterogeneity. It is important to recognize these patients for their early referral, correct diagnosis and appropriate management. The current study highlights the uncertainties surrounding the diagnosis and treatment of inherited factor VII deficiency.
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes on contributors
Preeti Tripathi is senior resident in Department of Hematology at All India Institute of Medical Sciences (AIIMS), New Delhi.
Priyanka Mishra is senior resident in Department of Hematology at All India Institute of Medical Sciences (AIIMS), New Delhi.
Ravi Ranjan is scientist at Department of Hematology at All India Institute of Medical Sciences (AIIMS), New Delhi.
Seema Tyagi is professor in at Department of Hematology at All India Institute of Medical Sciences (AIIMS), New Delhi.
Tulika Seth is at Department of Hematology at All India Institute of Medical Sciences (AIIMS), New Delhi.
Renu Saxena is professor and head of Department of Hematology at All India Institute of Medical Sciences (AIIMS), New Delhi.
References
- Perry DJ. Factor VII deficiency – a review. Br J Haematol. 2002;118:689–700. doi: 10.1046/j.1365-2141.2002.03545.x
- Ingerslev J, Christiansen K, Sørensen B; International Registry on Factor VII Deficiency (IRF7) Steering Committee. Inhibitor to factor VII in severe factor VII deficiency: detection and course of the inhibitory response. J Thromb Haemost. 2005;3:799–800. doi: 10.1111/j.1538-7836.2005.01225.x
- Napolitano M, Giansily-Blaizot M, Dolce A, et al. Prophylaxis in congenital factor VII deficiency: indications, efficacy and safety. results from the Seven Treatment Evaluation Registry (STER). Haematologica. 2013;98:538–544. doi: 10.3324/haematol.2012.074039
- Bladbjerg EM, Gram J, Jespersen J. Plasma concentrations of blood coagulation factor VII measured by immunochemical and amidolytic methods. Scand J Clin Lab Invest. 2000;60:161–168. doi: 10.1080/003655100750044802
- Wildgoose P, Nemerson Y, Hansen LL, et al. Measurement of basal levels of factor VIIa in hemophilia A and B patients. Blood. 1992;80:25–28.
- Peyvandi F, Mannucci PM, Asti D, et al. Clinical manifestations in 28 Italian and Iranian patients with severe factor VII deficiency. Haemophilia. 1997;3:242–246. doi: 10.1046/j.1365-2516.1997.00137.x
- Marty S, Barro C, Chatelain B, et al. The paradoxical association between inherited factor VII deficiency and venous thrombosis. Haemophilia. 2008;14:564–570. doi: 10.1111/j.1365-2516.2007.01647.x
- Napolitano M, Siragusa S, Mariani G. Factor VII deficiency: clinical phenotype, genotype and therapy. J Clin Med. 2017;38:1–8.
- Herrmann FH, Wulff K, Auberger K, et al. Molecular biology and clinical manifestation of hereditary factor VII deficiency. Semin Thromb Hemost. 2000;26:393–400. doi: 10.1055/s-2000-8458
- Rao G, Viswabandya A, Nair SC, et al. Molecular basis of hereditary factor VII deficiency in India: five novel mutations including a double missense mutation (Ala191Glu; Trp364Cys) in 11 unrelated patients. Haematologica. 2007;92:1002–1003. doi: 10.3324/haematol.10835
- Bernardi F, Castaman G, Pinotti M, et al. Mutation pattern in clinically asymptomatic coagulation factor VII deficiency. Hum Mutat. 1996;8:108–115. doi: 10.1002/(SICI)1098-1004(1996)8:2<108::AID-HUMU2>3.0.CO;2-7
- Feng D, Tofler GH, Larson MG, et al. Factor VII gene polymorphism, factor VII levels, and prevalent cardiovascular disease: the Framingham Heart Study. Arterioscler Thromb Vasc Biol. 2000;20:593–600. doi: 10.1161/01.ATV.20.2.593
- Mariani G, Konkle BA, Ingerslev J. Congenital factor VII deficiency: therapy with recombinant activated factor VII – a critical appraisal. Haemophilia. 2006;12:19–27. doi: 10.1111/j.1365-2516.2006.01180.x
- Gralnick HR, Wilson OJ. Cold-promoted activation of factor VII and shortening of the prothrombin time. In: Wessler S, Becker CG, Nemerson Y, editors. The new dimensions of warfarin prophylaxis. Boston (MA): Springer; 1987. p. 113–129.
- Lapecorella M, Mariani G; International Registry on Congenital Factor VII Deficiency. Factor VII deficiency: defining the clinical picture and optimizing therapeutic options. Haemophilia. 2008;14(6):1170–1175. doi: 10.1111/j.1365-2516.2008.01844.x
- Mariani G, Herrmann FH, Dolce A, et al. Clinical phenotypes and factor VII genotype in congenital factor VII deficiency. Thromb Haemost. 2005;93(3):481–487.
- Palla R, Peyvandi F, Shapiro AD. Rare bleeding disorders: diagnosis and treatment. Blood. 2015;125:2052–2061. doi: 10.1182/blood-2014-08-532820
- Millar DS, Kemball-Cook G, McVey JH, et al. Molecular analysis of the genotype-phenotype relationship in factor VII deficiency. Hum Genet. 2000;107(4):327–342. doi: 10.1007/s004390000373
- Girolami A, Bertozzi I, Berti de Marinis G, et al. Activated FVII levels in factor VII Padua (Arg304Gln) coagulation disorder and in true factor VII deficiency: a study in homozygotes and heterozygotes. Hematology. 2011;16:308–312. doi: 10.1179/102453311X13085644680069
- Matteo NDM, Mariani G (on behalf of STER study group). Bleeding symptoms at disease presentation and prediction of ensuing bleeding in inherited FVII deficiency. Thromb Haemost. 2013;109:1051–1059. doi: 10.1160/TH12-10-0740