
Abstract
In an ischemic environment, brain tissue responds to oxygen deprivation with the initiation of rapid changes in bioenergetic metabolism to ensure ion and metabolic homeostasis. At the same time, the accelerated cleavage of membrane phospholipids changes membrane composition and increases free fatty acid concentration. Phospholipid breakdown also generates specific messengers that participate in signaling cascades that can either promote neuronal protection or cause injury. The net impact of signaling events affects the final outcome of the stroke. While reoxygenation is a life-saving intervention, it can exacerbate brain damage. Although compromised energy metabolism is restored shortly after reperfusion, alterations in membrane phospholipid composition with subsequent accumulation of lipid oxoderivates are neurotoxic, causing oxidative stress and ischemia–reperfusion (IR) injury. Thus, plasma and mitochondrial membranes are the first responders as well as mediators of IR-induced stress signals. In this review, we focus on ischemia-induced changes in brain energy metabolism and membrane functions as the causal agents of cell stress responses upon reoxygenation. The first part of the review deals with the specificities of neuronal bioenergetics during IR and their impact on metabolic processes. The second part is concentrated on involvement of both plasma and mitochondrial membranes in the production of messengers which can modulate neuroprotective pathways or participate in oxidative/electrophilic stress responses. Although the etiology of IR injury is multifactorial, deciphering the role of membrane and membrane-associated processes in brain damage will uncover new therapeutic agents with the ability to stabilize neuronal membranes and modulate their responses in favor of prosurvival pathways.
Introduction
Brain ischemia represents a cascade of neurochemical processes evolving in time and space, unleased by interruption or sudden restriction of cerebral blood flow. Although rapid reperfusion is the essential step in prevention of death and restoration of brain functions, it exacerbates brain injury and neurocognitive deficits in patients after stroke. Thus ischemia-reperfusion (IR) injury is a highly heterogeneous phenomenon with a multifactorial etiology. This serious stress event elicits stress responses stemming from initialization of numerous molecular processes, both in the brain and the periphery. In the periphery, IR-induced overactivation of the neuroendocrine system followed by an imbalance in hormone production/release causes a number of undesirable responses that can exacerbate neuronal damage and impact the final outcome.
Bioenergetic failure is considered to be a key event in ischemic episode, and membranes are the first respondents to ischemia-induced stress signals. Therefore, the aim of this review is to discuss the early stage of IR injury, particularly from the view of brain bioenergetics and participation of neuronal and mitochondrial membranes in stress signal propagation. The review starts with the brief insight into IR-induced molecular processes. In the next parts of the work we deal with peculiarities of brain energy metabolism and the processes driven by impaired cell bioenergetics, phospholipid breakdown, and disordered membrane functions that finally can result in cell dysfunction, oxidative stress, and initialization of cell death.
Key molecular processes in the ischemic-reperfusion cascade
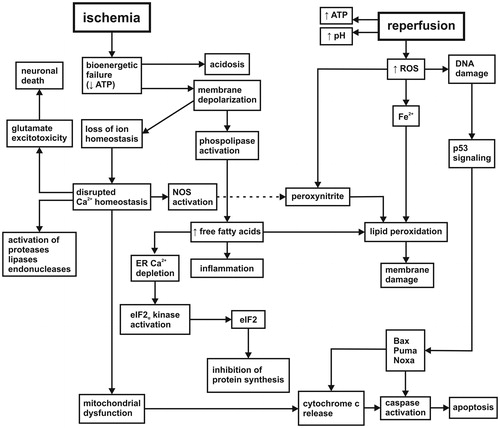
A pivotal event in the pathogenesis of brain ischemia is bioenergetic failure (). A drastic ATP decline leads to the failure of maintaining ion gradients and membrane depolarization. Membrane depolarization induces phospholipase activation followed by the breakdown of phospholipids and increase in free fatty acids (FFAs). Depolarization also leads to disturbance of Ca2+ homeostasis (Lipton, Citation1999) that triggers a lot of cellular processes such as excessive release of glutamate, activation of proteases, lipases, endonucleases or inhibition of protein synthesis. Mitochondrial Ca2+ overload results in mitochondrial dysfunction, increased membrane permeability, cytochrome c release, and activation of apoptosis (Racay et al., Citation2009). After reoxygenation xanthine/hypoxanthine oxidation, activation of NO synthase, oxidative metabolism of FFAs and altered mitochondrial function are the major sources of free radicals. They cause oxidative damage to many cellular constituents such as lipids, proteins, or DNA. In response to DNA damage, the tumor-supressor transcription factor p53 stops the cell cycle and activates expression of proapoptotic proteins (Bax, Puma, Noxa). These proteins suppress antiapoptotic regulation, reduce survival signal transduction, and initiate apoptosis (Culmsee & Mattson, Citation2005). The final outcome of IR injury is determined by the dominance of signals in both neurotoxic and neuroprotective pathways. Ultimately, the sensitivity and vulnerability of individual brain regions as well as cell-specific differences influence the extent, severity, and reversibility of brain damage.
Figure 1. Cascade of molecular events initialized by ischemia/reperfusion. The etiology of ischemic-reperfusion injury involves bioenergetic failure, the loss of intracellular ion homeostasis, membrane integrity, glutamate excitotoxicity, protein synthesis inhibition, progressive proteolysis, inflammation, oxidative stress, and reduced survival signal transduction (more details in the text). NOS: nitric oxide synthase; ER: endoplasmic reticulum; eIF2: eukaryotic initiation factor 2; ROS: reactive oxygen species; Bax: Bcl-2-associated X protein; Puma: p53 upregulated modulator of apoptosis; Noxa: PMA-induced protein.
Brain bioenergetics and its response to ischemia and reperfusion
Specificities of brain bioenergetics: a preferential utilization of glucose
The brain is extremely sensitive to ischemia due to its reliance on oxidative phosphorylation for energy production. Astrocytes and neurons, the principal structural elements of the brain tissue, are massive consumers of O2 and glucose. However, they exhibit a different preference for glucose utilization (Almeida et al., Citation2001). While astrocytes consume the majority of glucose and convert it to lactate, neurons contribute minimally to glucose consumption under resting conditions (Kasischke et al., Citation2004). This divergence in preferential utilization of the substrate between astrocytes and neurons is notably evident under energy crisis such as ischemia. Neurons and astrocytes respond to energetic stress by activation of AMP-activated protein kinase (AMPK), a key energy sensor in most eukaryotic cells (Manwani & McCullough, Citation2013). In astrocytes AMPK signaling upregulates glycolysis and enhances energy production (Pellerin & Magistretti, Citation2012). Glycolytically generated ATP is used in maintenance of mitochondrial membrane potential, which results in increased resistance of astrocytes to proapoptotic signaling (Almeida et al., Citation2001). However, in neurons upregulation of glycolysis is set by the level of phosphofructokinase 2 (PFK2). PFK2 generates fructose-2,6-bisphosphate, the allosteric activator of phosphofructokinase 1, a key regulator of glycolysis (). Neurons, in contrast to astrocytes, physiologically have low activity of PFK2 because of its continous degradation by the E3 ubiquitin ligase APCCdh1 (anaphase-promoting complex-Cdh1) and hence a low glycolytic rate (Herrero-Mendez et al., Citation2009; Rodriguez-Rodriguez et al., Citation2012). The reason for this metabolic program is a low expression of glutamate-cysteine ligase, the rate-limiting enzyme in gluthatione (GSH) biosynthesis. To compensate this deficit and ensure an appropriate antioxidant defense, neurons have an efficient GSH regenerative system where majority of glucose is directed to the pentose phosphate pathway (PPP) to regenerate reduced glutathione (Bolaños et al., Citation2010).
Figure 2. Preferential utilization of glucose in neurons. Neurons preferentially utilize glucose in the pentose phosphate pathway to produce reduced glutathione. Under energy crisis glucose is redirected into glycolysis to produce ATP resulting in weakened antioxidant defense (see the text for more details). TCA: tricarboxylic acid cycle; IF1: F1F0-ATPase inhibitory factor 1; DHAP: dihydroxyacetone phosphate.
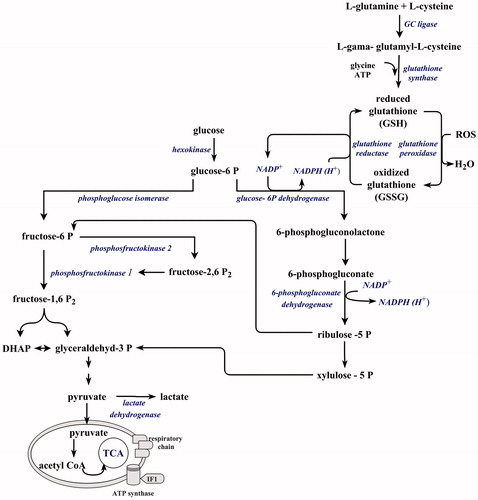
However, under an ischemic insult, glutamate-induced increase in Ca2+ flux increases glucose uptake. At the same time Ca2+ induced inactivation of APCCdh1 stabilizes PFK2 and glucose is diverted from the PPP to glycolysis (Rodriguez-Rodriguez et al., Citation2013). This metabolic reprogramming decreases NADPH production and impairs GSH regeneration. After reoxygenation when the burst of ROS is initiated, ischemia-induced changes in the redox state will have a profound impact on an antioxidant defense and will result in oxidative stress and augmentation of apoptic signaling. The redirection of glucose to the PPP and restoration of NADPH is dependent on the level of glucose 6-phosphate dehydrogenase, the rate-limiting enzyme in the PPP, that seem to be under the control of TIGAR (TP53-induced glycolysis and apoptosis regulator) (Li et al., Citation2014; Zhao et al., Citation2012).
Based on these findings, it is evident that a small cell-specific divergence in regulation of glycolysis can elicit profound changes in cellular metabolism under pathological conditions. It is possible that this difference is also implicated in increased sensitivity and vulnerability of neurons to ischemic attack. It seems that enhancement of glycolysis in neurons results in only limited neuroprotection, as the increased flow of glucose to the bioenergetic glycolytic pathway leads to unfavorable effect on the antioxidative defense system. This preferential utilization of glucose in the PPP also indicates the priority of antioxidative defense over bioenergetic requirements in neurons. The findings also support the hypothesis of lactate utilization for bioenergetic purposes via the astrocyte–neuron lactate shuttle as has been suggested previously (Pellerin & Magistretti, Citation2012).
Mitochondrial F1F0-ATPase inhibitory factor 1 (IF1) and its impact on brain bioenergetics
Given the essentiality of energy for life, the control of energy metabolism is ensured at several levels, including F1F0-ATP synthase (complex V, CV), which is considered to be a key control point. CV is a reversible engine that physiologically catalyzes the synthesis of ATP. An ischemic challenge creates a situation whereby the CV reverses its operating mode and starts ATP hydrolysis (Nicholls & Budd, Citation2000). This activity can deplete ATP and hasten cell death, but it is limited by the mitochondrial protein IF1, an endogenous CV inhibitor (Pullman & Monroy, Citation1963). Physiologically, IF1 inhibits CV hydrolase activity in situations that compromise oxidative phosphorylation, such as hypoxia/ischemia. IF1 can exist in two forms depending on its phosphorylation state. The active dephosphorylated form binds onto the CV and inhibits the hydrolase activity, while the inactive phosphorylated form prevents CV binding and subsequent inhibition of ATP hydrolysis (García-Bermúdez et al., Citation2015). Phosphorylation of IF1 probably involves cAMP/PKA signaling pathway (Acin-Perez et al., Citation2009). During ischemia, Ca2+ accumulation in mitochondria activates mitochondrial phosphatases (Hopper et al., Citation2006) that dephosphorylate many mitochondrial proteins, among them the respiratory chain members, most notably cytochrome c and cytochrome c oxidase (Hüttemann et al., Citation2012). IF1 can also be the target of mitochondrial phosphatases. Finally, the shift of IF1 toward the dephosphorylated state was observed in hypoxic and cancer cells. This shift correlated with enhanced binding of IF1 onto CV and rewiring energy metabolism to glycolysis (Sánchez-Cenizo et al., Citation2010; Wei et al., Citation2015). Thus, the change of biological activity of IF1 might represent a signal leading to redirection of glucose from the PPP pathway to glycolysis. Moreover, increased mitochondrial Ca2+ represents “a gas pedal” (Gellerich et al., Citation2013) for mitochondrial respiration, thus priming the respiratory chain for hyperactivation when reperfusion is initiated. A few minutes following reperfusion, ATP levels are restored nearly to control levels (Taylor et al., Citation2015). However, the dephosphorylated status of the respiratory chain members and the loss of the allosteric regulation of the respiratory chain by ATP at the level of cytochrome c oxidase (Bender & Kadenbach, Citation2000) lead to hyperpolarization of mitochondrial membrane followed by a significant and prolonged production of ROS (Starkov & Fiskum, Citation2003). Based on the redirection of glucose from the PPP to glycolysis, the potential of antioxidant defense to detoxify excessive amount of ROS is attenuated, which has profound consequences for survival of neuronal cells.
In summary, these findings support an assumption that IF1 may act as another regulator of energy metabolism due to its crucial role in mediating the metabolic switch under stress conditions. The regulatory function of IF1 is accomplished by the change of its biological activity that involves the alterations in the phosphorylation status of the protein and cAMP/PKA signaling pathway.
Brain ischemia/reperfusion and its impact on membranes
Involvement of neuronal membrane in ischemic-reperfusion injury
Lipid oxoderivatives produced in enzyme-catalyzed pathways
Neuronal membranes represent the first mediator in the stress responses evoked an ischemic attack. Due to the high amount of polyunsaturated fatty acids (PUFAs) incorporated in membrane phospholipids, low levels of antioxidants such as GSH, glutathione peroxidase, vitamin E, and a near absence of catalase, neuronal membranes can be a rich source of reactive radicals and signaling messengers.
Ischemia-induced activation of phospholipases (especially PLA2) triggers phospholipid backbone hydrolysis, releasing FFAs and lysophopholipids. Once released, FFAs can reincorporate into phospholipids, diffuse outside the cell or enter metabolism. However, in stroke they accumulate and undergo oxidative metabolism by non-enzymatic and enzymatic processes () catalyzed by cyclooxygenases (COX), lipoxygenases (LOX), cytochrome P450, and peroxidases. These pathways produce oxygenated compounds, among them arachidonic acid (AA) and docosahexaenoic acid (DHA) derivatives being most important. The COX enzymes generate increased amount of eicosanoids, with the major product prostaglandin H2. It is metabolized to other bioactive prostanoids (PGD2, PGE2, PGF2), tromboxane TXA2, and prostacyclin PGI2 (Yu et al., Citation2014). The AA derivatives represent a critical component of postischemic neuroinfammation, as evidenced by studies where genetic and pharmacological blockade of COX-2 elicits robust neuroprotection in laboratory animals subjected to focal or global ischemia (Nakayama et al., Citation1998). Conversely, neuronal overexpression of COX-2 in transgenic mice potentiated neuronal injury after global ischemic insult (Xiang et al., Citation2007).
Figure 3. Ischemia/reperfusion-induced breakdown of membrane phospholipids. Activation of phospholipases triggers phospholipid hydrolysis releasing lysophospholipids and free fatty acids. Free fatty acids undergo oxidative metabolism by enzyme and non-enzyme pathways. The enzyme-catalyzed pathways produce oxygenated derivatives such as prostaglandines, prostacyclines, tromboxanes, leukotriens, or epoxides from arachidonic acid. They are responsible for postischemic inflammation. DHA oxoderivatives can elicit antiinflammatory and protective effects. Oxoderivatives of non-enzyme pathways produce reactive lipid species that can be involved in both damaging and protective cell responses (more details in the text). PGD2, PGE2, PGF2: prostaglandins; NPD1: neuroprotectin 1; 4-HNE: 4-hydroxy-2-nonenal; MDA: malondialadehyde; GSH: glutathione.
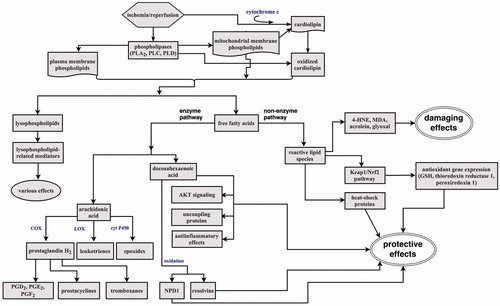
Recent advances in lipidomics show that oxygenases, including COX-2 and LOX can also mediate the formation of electrophilic lipid oxoderivatives from ω-3 FFAs, e.g. DHA, docosapentaenoic, docosatetraenoic, and eicosapentaenoic acid (Groeger et al., Citation2010). These derivatives have anti-inflammatory effects and promote resolution of inflammation by suppressing NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) activation, modulating cytokine expression, activating G-protein coupled receptors, and promoting protective responses (Serhan et al., Citation2008). Brain docosanoids were the first recognized lipid mediators of anti-inflammatory effects during experimental IR (Marcheselli et al., Citation2003). These stereospecific messengers are the products of two DHA-oxygenation pathways. The first oxygenation pathway results in the formation of neuroprotectin D1 (NPD1; 10,17S-docosatriene) and the second pathway, which is active in the presence of aspirin and involves an acetylated COX-2, leading to the formation of the resolvin-type messengers, 17-R resolvins. These mediators attenuated IR-triggered leukocyte infiltration, cytokine-mediated proinflammatory gene activation, and elicited neuroprotection by reducing the stroke infarct volume. Besides docosanoid formation, DHA is involved in a positive modulation of AKT signaling and the activation of prosurvival pathways (Akbar et al., Citation2005). A positive effect of DHA in ischemia also involves uncoupling proteins (UCPs), the mitochondrial transporter proteins that sustain the proton conductance across the inner mitochondroal membrane. Increased expression and activity of UCP2 correlated with neuronal survival after stroke (Mehta & Li, Citation2009). Thus, in the early period of IR episode the formation of DHA prosurvival derivatives can represent a hormetic response of a cell to cope with the pathological insult. These neuroprotective processes seem to be depend on unesterified DHA pool than the estrified DHA pool (Orr et al., Citation2013).
Oxoderivatives produced in non-enzyme pathways
While the production of specific lipid oxoderivatives is controlled by enzymatic pathways, nonspecific lipid peroxidation proceeds through chain reactions in an uncontrolled manner to produce reactive lipid species (RLS). In biological membranes, the presence of proteins can result in the tranfer of RLS to protein side chains and adduct formation (Fritz & Petersen, Citation2011). Nucleophilic amino acids cysteine, lysine, and histidine are particularly prone to modification with RLS. The most reactive and cytotoxic RLS are α,β-unsaturated aldehydes (4-hydroxy-2-nonenal, [4-HNE], acrolein), dialdehydes (malondialdehyde and glyoxal), and ketoaldehydes (4-oxo-trans-2-nonenal) (Grimsrud et al., Citation2008). 4-HNE is increased in ischemic brain and after its stabilization in the membrane bilayer can react with membrane proteins, e.g. transporters for glutamate, glucose, Na+/K+ ATPase (Urabe et al., Citation2000; Vazdar et al., Citation2012). It also suppresses ADP and ATP transport through mitochondrial inner membrane, inhibits cytochrome c oxidase activity and thus affects the energy-producing capacity of mitochondria in tissue already afflicted by stroke-induced energy crisis (Kaplan et al., Citation2007). As the free energy of 4-HNE stabilization in the membrane is relatively small, 4-HNE can leave the membrane and act both in intra- and extracellular space (Vazdar et al., Citation2012). Due to the high reactivity of C3 position, 4-HNE can react via Michael addition reaction with cellular thiols of glutathione, thioredoxin, and thioredoxin reductase and dysregulate the cellular redox status (Fang & Holmgren, Citation2006).
Although much is known about damaging effects, RLS can also trigger adaptive responses of the cell to oxidative/electrophilic stress via Keap1-Nrf2 (Kelch-like ECH-associated protein 1/NF-E2-related factor 2) pathway. The pathway upregulates transcription of genes responsible for the synthesis of antioxidant proteins such as GSH, thioredoxin reductase 1, peroxiredoxin, or heme oxygenase-1 (Taguchi et al., Citation2011). Electrophilic lipids can also increase the ability of the cell to cope with proteotoxic stress through upregulation of heat-shock proteins in cytosolic, nuclear as well as mitochondrial localization, e.g. Hsp110, Hsp90, Hsp70, Hsp60, Hsp40, Hsp10 (Tan et al., Citation2015). These protective responses are of particular interest in a view of molecular mechanisms triggered by ischemic preconditioning, the phenomenon of ischemic tolerance to lethal ischemic injury (Obrenovitch, Citation2008). The diverse biological effects of RLS are probably dependent on subcellular localization of protein targets. It is hypothesized that cytosolic targets drive the adaptive responses of the cell to oxidative/electrophilic stress, while targeting an electrophile to mitochondria governs mitochondrial signaling, ROS production, cellular respiration, and promotes mitochondria-dependent cell death (Higdon et al., Citation2012).
Mitochondrial membrane and its response to ischemic-reperfusion injury
Cardiolipin and its role in ischemic-reperfusion injury
Given the localization of the respiratory chain in the inner mitochondrial membrane, mitochondrial phospholipids are directly involved in the formation of RLS during IR episode. Although the major precursor of lipid mediators, phosphatidylserine is lacking in mitochondria (Vance & Tasseva, Citation2013), cardiolipin (CL), the unique phospholipid of the inner mitochondrial membrane with about 20 different, mostly PUFA residues is an exceptionally good source of lipid mediators. Unlike heart, muscle, liver, and kidney CLs with predominatly C18:2 residues, brain CLs display the extreme diversity of molecular species with long-chain PUFA residues such as AA, DHA, and docosatetraenoic (C22:4) acid. The high diversification of PUFAs in brain CLs predetermines the high diversity of lipid oxoderivatives formed under oxidative stress. Indeed, lipidomic analyzes identified 56 major molecular species of CLs in lipid extracts from mouse brain, of which 55 were highly oxidizable polyunsaturated CLs containing one to four PUFAs (Tyurina et al., Citation2014). The demand for a sufficient amounts of oxidizing equivalents, such as H2O2 or lipid hydroperoxides to trigger CL oxidation is accomplished by oxygen delivery to ischemia-impaired mitochondria. While the conventional view on the biogenesis of RLS in the CNS is based on phospholipid hydrolysis by Ca2+-dependent phospholipases followed by oxygenation of released FFAs, CL oxidation has been reported to be Ca2+-independent process that is strictly dependent on catalytic interactions between CL and cytochrome c (cyt c). These interactions are followed by hydrolysis of oxygenated CL species by two types of PLA2 with final accumulation of lysocardiolipins and oxidized polyunsaturated FFAs (Ji et al., Citation2015). The oxidative modification CLs as well as cyt c are required not only for the appearance of CLs in the outer mitochondrial membrane but also for release of proapoptotic factors and initiation of apoptosis (Gonzalvez & Gottlieb, Citation2007). Remarkably, elevated levels of CL were reported in lung fluid of patients with pneumonia, where the CL transport was mediated by a mutant type of the P-type ATPase transmembrane lipid pump, ATP8b1 (Ray et al., Citation2010). Another study by Wan et al. (Citation2014) reported that oxidized CL could trigger leukotriene B4 production in both human monocyte-derived macrophages and neutrophils. The fact that IR episode triggers mitochondrial dysfunction and cell death supports the idea, that CL, cyt c and oxidized CL derivatives might be released into extracellular space as a result of the loss of hydrophobicity and association with mitochondria. Subsequent hydrolysis by phospholipases might generate RLS as signaling mediators in extracellular space and contribute to inflammatory processes. These processes can be also potentiated by ceramide (the building blocks of membrane sphingolipids or glycosphingolipids containing C14–C16 FFAs) accumulation in mitochondria after IR due to their ability to release cyt c and other proapoptotic proteins (Novgorodov & Gudz, Citation2011).
Although the primary role of CL is closely related to mitochondrial function/dysfunction and intialization of apoptosis, recent findings suggest that this mitochondrial phospholipid might also participate in extracellular signaling pathways and thus exacerbate IR damage.
Cardiolipin and the mitochondrial respiratory chain
Oxidative damage to CL involves also another dimension of membrane pathology linked to defects in the functioning of respiratory chain complexes and efficiency of mitochondrial energy production (Chen & Lesnefsky, Citation2006). Decreased enzymatic activity of mitochondrial respiratory chain members I (NADH:ubiquinone oxidoreductase), III (coenzyme Q:cytochrome c-oxidoreductase), and IV (cytochrome c oxidase) in IR-injured mitochondria have been described in numerous studies (Chomova et al., Citation2012). Exogenous CL-liposomes are able to restore enzymatic activities to the level of control animals following IR, while other phospholipids and peroxidized CL-liposomes were ineffective (Paradies et al., Citation2004), demonstrating the requirement of CL for catalytic activities of mitochondrial respiratory complexes. CL also plays an important role in stabilization of the higher supramolecular organization of the respiratory chain in supercomplexes and respirasomes (Pfeiffer et al., Citation2003). Supercomplexes are structural entities composed of one to four monomers of individual respiratory complexes (e.g. I1III2IV4 and III2IV4). Their functional relevance is to support fast substrate channeling, catalytic enhancement, and sequestration of reactive intermediates (Acin-Perez & Enriquez, Citation2014). Interestingly, the study by Suthammarak et al. (Citation2013) reported interactions of mitochondrial superoxide dismutase with the supercomplex I:III:IV. However, other lines of evidence suggest that respirasomes are the building blocks for the higher helical structures called respiratory strings (Wittig & Schägger, Citation2009). If the purpose of supramolecular organization is fast substrate channeling, ROS sequestration and efficacy of the respiratory chain, it could be predicted that F1F0-ATP synthase also makes up the higher molecular structures. Indeed, F1F0-ATP synthase has been found to form dimeric and homooligomeric structures (Krause et al., Citation2005) and most importantly ATP synthasome, which is suggested to be composed of ATP synthase, ADP/ATP translocase and inorganic phosphate carrier (Nůsková et al., Citation2015). Thus, stable associations of ATP synthase to supramolecular structures could be essential to maintain bioenergetically fully competent mitochondria. Recently the study by Giorgio et al. (Citation2013) demonstrated that dimers of the F1F0-ATP synthase are responsible for the formation of the the mitochondrial permeability transition pore (MPTP), a key effector of cell death. It has been well reviewed elsewhere that the conditions that occur following IR are exactly those that would induce MPTP opening.
Because the supramolecular oganization of the respiratory chain strongly depends on membrane lipid amount and composition, undoubtedly IR attack will also affect this arrangement of the respiratory chain. Disturbances in the supermolecular arrangement of the respiratory chain as well as F1F0-ATP synthase will elicit responses at the level of energy efficacy, ROS production, cell metabolism, and survival/death signal propagation in neuronal cells afflicted by IR episode.
Conclusion
Despite the multimodal nature of IR injury, the status of cellular bioenergetics is the major determinant of the pathophysiological consequences manifested in the brain after ischemic episode. The rationale is that cell constituents and majority of physiological processes are directly or indirectly impacted by the loss of energy. Plasma and mitochondrial membranes are no exception to this rule, since the maintenance of their proper functions and a defined phospholipid composition depend on adequate amount of energy. Moreover, membrane phospholipids are a potent source of bioactive signal messengers and free radicals. A decline in energy supply and a disruption in energy homeostasis destabilize membrane integrity and prime membranes to be more susceptible to pathological events after reoxygenation. As a result, a pronounced disruption in membrane integrity and cellular milieu resulting from ischemia elicits multiple signals, some protective and some harmful, upon reestablishment of blood flow. Therefore, defining the molecular basis for cellular dysfunction resulting from disrupted membrane composition and membrane associated processes will uncover possible therapeutic strategies that may be able to stabilize neuronal and mitochondrial membranes and thus modulate their responses to IR-induced stress signals.
Funding information
The work was supported by grants VEGA 1/0349/16 and MZ SR-2012/10-UKBA-10.
Disclosure statement
The authors declare that have no conflicts of interest and have no financial, consulting and personal relationships with organizations that could influence their work.
Related Research Data
References
- Acin-Perez R, Enriquez JA. (2014). The function of the respiratory supercomplexes: the plasticity model. Biochim Biophys Acta 1837:444–50.
- Acin-Perez R, Salazar E, Kamenetsky M, Buck J, Levin LR, Manfredi G. (2009). Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab 9:265–76.
- Akbar M, Calderon F, Wen Z, Kim HY. (2005). Docosahexaenoic acid: a positive modulator of Akt signaling in neuronal survival. Proc Natl Acad Sci USA 102:10858–63.
- Almeida A, Almeida J, Bolaños JP, Moncada S. (2001). Different responses of astrocytes and neurons to nitric oxide: the role of glycolytically generated ATP in astrocyte protection. Proc Natl Acad Sci USA 98:15294–9.
- Bender E, Kadenbach B. (2000). The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett 466:130–4.
- Bolaños JP, Almeida A, Moncada S. (2010). Glycolysis: a bioenergetic or a survival pathway? Trends Biochem Sci 35:145–9.
- Chen Q, Lesnefsky EJ. (2006). Depletion of cardiolipin and cytochrome c during ischemia increases hydrogen peroxide production from the electron transport chain. Free Radic Biol Med 40:976–82.
- Chomova M, Tatarkova Z, Dobrota D, Racay P. (2012). Ischemia-induced inhibition of mitochondrial complex I in rat brain: effect of permeabilization method and electron acceptor. Neurochem Res 37:965–76.
- Culmsee C, Mattson MP. (2005). p53 in neuronal apoptosis. Biochem Biophys Res Commun 331:761–77.
- Fang J, Holmgren A. (2006). Inhibition of thioredoxin and thioredoxin reductase by 4-hydroxy-2-nonenal in vitro and in vivo. J Am Chem Soc 128:1879–85.
- Fritz KS, Petersen DR. (2011). Exploring the biology of lipid peroxidation-derived protein carbonylation. Chem Res Toxicol 24:1411–19.
- García-Bermúdez J, Sánchez-Aragó M, Soldevilla B, Del Arco A, Nuevo-Tapioles C, Cuezva JM. (2015). PKA phosphorylates the ATPase inhibitory factor 1 and inactivates its capacity to bind and inhibit the mitochondrial H(+)-ATP synthase. Cell Rep 12:2143–55.
- Gellerich FN, Gizatullina Z, Gainutdinov T, Muth K, Seppet E, Orynbayeva Z, Vielhaber S. (2013). The control of brain mitochondrial energization by cytosolic calcium: the mitochondrial gas pedal. IUBMB Life 65:180–90.
- Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, et al. (2013). Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA 110:5887–92.
- Gonzalvez F, Gottlieb E. (2007). Cardiolipin: setting the beat of apoptosis. Apoptosis 12:877–85.
- Grimsrud PA, Xie H, Griffin TJ, Bernlohr DA. (2008). Oxidative stress and covalent modification of protein with bioactive aldehydes. J Biol Chem 283:21837–41.
- Groeger AL, Cipollina C, Cole MP, Woodcock SR, Bonacci G, Rudolph TK, Rudolph V, et al. (2010). Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat Chem Biol 6:433–41.
- Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol 11:747–52.
- Higdon A, Diers AR, Oh JY, Landar A, Darley-Usmar VM. (2012). Cell signalling by reactive lipid species: new concepts and molecular mechanisms. Biochem J 442:453–64.
- Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, Witzmann FA, et al. (2006). Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry 45:2524–36.
- Hüttemann M, Lee I, Grossman LI, Doan JW, Sanderson TH. (2012). Phosphorylation of mammalian cytochrome c and cytochrome c oxidase in the regulation of cell destiny: respiration, apoptosis, and human disease. Adv Exp Med Biol 748:237–2364.
- Ji J, Baart S, Vikulina AS, Clark RS, Anthonymuthu TS, Tyurin VA, Du L, et al. (2015). Deciphering of mitochondrial cardiolipin oxidative signaling in cerebral ischemia-reperfusion. J Cereb Blood Flow Metab 35:319–28.
- Kaplan P, Tatarkova Z, Racay P, Lehotsky J, Pavlikova M, Dobrota D. (2007). Oxidative modifications of cardiac mitochondria and inhibition of cytochrome c oxidase activity by 4-hydroxynonenal. Redox Rep 12:211–18.
- Kasischke KA, Vishwasrao HD, Fisher PJ, Zipfel WR, Webb WW. (2004). Neural activity triggers neuronal oxidative metabolism followed by astrocytic glycolysis. Science 305:99–103.
- Krause F, Reifschneider NH, Goto S, Dencher NA. (2005). Active oligomeric ATP synthases in mammalian mitochondria. Biochem Biophys Res Commun 329:583–90.
- Li M, Sun M, Cao L, Gu JH, Ge J, Chen J, Han R, et al. (2014). A TIGAR-regulated metabolic pathway is critical for protection of brain ischemia. J Neurosci 34:7458–71.
- Lipton P. (1999). Ischemic cell death in brain neurons. Physiol Rev 79:1431–568.
- Manwani B, McCullough LD. (2013). Function of the master energy regulator adenosine monophosphate-activated protein kinase in stroke. J Neurosci Res 91:1018–29.
- Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, Hardy M, et al. (2003). Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem 278:43807–17.
- Mehta SL, Li PA. (2009). Neuroprotective role of mitochondrial uncoupling protein 2 in cerebral stroke. J Cereb Blood Flow Metab 29:1069–78.
- Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose ME, Stetler RA, Isakson PC, et al. (1998). Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc Natl Acad Sci USA 95:10954–9.
- Nicholls DG, Budd SL. (2000). Mitochondria and neuronal survival. Physiol Rev 80:315–60.
- Novgorodov SA, Gudz TI. (2011). Ceramide and mitochondria in ischemic brain injury. Int J Biochem Mol Biol 2:347–61.
- Nůsková H, Mráček T, Mikulová T, Vrbacký M, Kovářová N, Kovalčíková J, Pecina P, Houštěk J. (2015). Mitochondrial ATP synthasome: expression and structural interaction of its components. Biochem Biophys Res Commun 464:787–93.
- Obrenovitch TP. (2008). Molecular physiology of preconditioning-induced brain tolerance to ischemia. Physiol Rev 88:211–47.
- Orr SK, Palumbo S, Bosetti F, Mount HT, Kang JX, Greenwood CE, Ma DW, et al. (2013). Unesterified docosahexaenoic acid is protective in neuroinflammation. J Neurochem 127:378–93.
- Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM. (2004). Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res 94:53–9.
- Pellerin L, Magistretti PJ. (2012). Sweet sixteen for ANLS. J Cereb Blood Flow Metab 32:1152–66.
- Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, Schägger H. (2003). Cardiolipin stabilizes respiratory chain supercomplexes. J Biol Chem 278:52873–80.
- Pullman ME, Monroy GC. (1963). A naturally occurring inhibitor of mitochondrial adenosine triphosphatase. J Biol Chem 238:3762–9.
- Racay P, Tatarkova Z, Chomova M, Hatok J, Kaplan P, Dobrota D. (2009). Mitochondrial calcium transport and mitochondrial dysfunction after global brain ischemia in rat hippocampus. Neurochem Res 34:1469–78.
- Ray NB, Durairaj L, Chen BB, McVerry BJ, Ryan AJ, Donahoe M, Waltenbaugh AK, et al. (2010). Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat Med 16:1120–7.
- Rodriguez-Rodriguez P, Almeida A, Bolaños JP. (2013). Brain energy metabolism in glutamate-receptor activation and excitotoxicity: role for APC/C-Cdh1 in the balance glycolysis/pentose phosphate pathway. Neurochem Int 62:750–6.
- Rodriguez-Rodriguez P, Fernandez E, Almeida A, Bolaños JP. (2012). Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration. Cell Death Differ 19:1582–9.
- Sánchez-Cenizo L, Formentini L, Aldea M, Ortega AD, García-Huerta P, Sánchez-Aragó M, Cuezva JM. (2010). Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J Biol Chem 285:25308–13.
- Serhan CN, Chiang N, Van Dyke TE. (2008). Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol 8:349–61.
- Starkov AA, Fiskum G. (2003). Regulation of brain mitochondrial H2O2 production by membrane potential and NAD(P)H redox state. J Neurochem 86:1101–7.
- Suthammarak W, Somerlot BH, Opheim E, Sedensky M, Morgan PG. (2013). Novel interactions between mitochondrial superoxide dismutases and the electron transport chain. Aging Cell 12:1132–40.
- Taguchi K, Motohashi H, Yamamoto M. (2011). Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 16:123–40.
- Tan K, Fujimoto M, Takii R, Takaki E, Hayashida N, Nakai A. (2015). Mitochondrial SSBP1 protects cells from proteotoxic stresses by potentiating stress-induced HSF1 transcriptional activity. Nat Commun 6:6580.
- Taylor JM, Zhu XH, Zhang Y, Chen W. (2015). Dynamic correlations between hemodynamic, metabolic, and neuronal responses to acute whole-brain ischemia. NMR Biomed 28:1357–65.
- Tyurina YY, Poloyac SM, Tyurin VA, Kapralov AA, Jiang J, Anthonymuthu TS, Kapralova VI, et al. (2014). A mitochondrial pathway for biosynthesis of lipid mediators. Nat Chem 6:542–52.
- Urabe T, Yamasaki Y, Hattori N, Yoshikawa M, Uchida K, Mizuno Y. (2000). Accumulation of 4-hydroxynonenal-modified proteins in hippocampal CA1 pyramidal neurons precedes delayed neuronal damage in the gerbil brain. Neuroscience 100:241–50.
- Vance JE, Tasseva G. (2013). Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim Biophys Acta 1831:543–54.
- Vazdar M, Jurkiewicz P, Hof M, Jungwirth P, Cwiklik L. (2012). Behavior of 4-hydroxynonenal in phospholipid membranes. J Phys Chem B 116:6411–15.
- Wan M, Hua X, Su J, Thiagarajan D, Frostegård AG, Haeggström JZ, Frostegård J. (2014). Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by Annexin A5. Atherosclerosis 235:592–8.
- Wei S, Fukuhara H, Kawada C, Kurabayashi A, Furihata M, Ogura S, Inoue K, Shuin T. (2015). Silencing of ATPase inhibitory factor 1 inhibits cell growth via cell cycle arrest in bladder cancer. Pathobiology 82:224–32.
- Wittig I, Schägger H. (2009). Supramolecular organization of ATP synthase and respiratory chain in mitochondrial membranes. Biochim Biophys Acta 1787:672–80.
- Xiang Z, Thomas S, Pasinetti G. (2007). Increased neuronal injury in transgenic mice with neuronal overexpression of human cyclooxygenase-2 is reversed by hypothermia and rofecoxib treatment. Curr Neurovasc Res 4:274–9.
- Yu L, Yang B, Wang J, Zhao L, Luo W, Jiang Q, Yang J. (2014). Time course change of COX2-PGI2/TXA2 following global cerebral ischemia reperfusion injury in rat hippocampus. Behav Brain Funct 10:42.
- Zhao G, Zhao Y, Wang X, Xu Y. (2012). Knockdown of glucose-6-phosphate dehydrogenase (G6PD) following cerebral ischemic reperfusion: the pros and cons. Neurochem Int 61:146–55.