
Abstract
Psychiatric illnesses and cardiovascular disease (CVD) contribute to significant overall morbidity, mortality, and health care costs, and are predicted to reach epidemic proportions with the aging population. Within the Veterans Administration (VA) health care system, psychiatric illnesses such as post-traumatic stress disorder (PTSD) and CVD such as heart failure (HF), are leading causes of hospital admissions, prolonged hospital stays, and resource utilization. Numerous studies have demonstrated associations between PTSD symptoms and CVD endpoints, particularly in the Veteran population. Not only does PTSD increase the risk of HF, but this relationship is bi-directional. Accordingly, a VA-sponsored conference entitled “Cardiovascular Comorbidities in PTSD: The Brain-Heart Consortium” was convened to explore potential relationships and common biological pathways between PTSD and HF. The conference was framed around the hypothesis that specific common systems are dysregulated in both PTSD and HF, resulting in a synergistic acceleration and amplification of both disease processes. The conference was not intended to identify all independent pathways that give rise to PTSD and HF, but rather identify shared systems, pathways, and biological mediators that would be modifiable in both disease processes. The results from this conference identified specific endocrine, autonomic, immune, structural, genetic, and physiological changes that may contribute to shared PTSD-CVD pathophysiology and could represent unique opportunities to develop therapies for both PTSD and HF. Some recommendations from the group for future research opportunities are provided.
Introduction
Psychiatric illnesses and cardiovascular disease (CVD) contribute to significant overall morbidity, mortality, and health care costs, particularly within the Veterans Administration (VA) health care system. Specifically, by virtue of the demographics and background of the VA patient population, the co-existence of post-traumatic stress disorders (PTSDs) and heart failure (HF) is prevalent (Buckley & Kaloupek, Citation2001; Cohen, Marmar, Ren, Bertenthal, & Seal, Citation2009; Kubzansky, Koenen, Spiro, Vokonas, & Sparrow, Citation2007; Pole, Citation2007). Numerous studies have demonstrated associations between PTSD symptoms and cardiovascular endpoints. The presence of either self-reported or physician-rated PTSD is associated with increases in readmissions for cardiovascular events such as myocardial infarction (MI), angina, hypertensive complications, atrioventricular conduction deficits, and arterial disease, which can culminate in the development and progression of HF and cardiovascular-related mortality (Gander & von Kanel, Citation2006). Further studies continue to describe associations between PTSD and CVD, particularly in the Veteran population (Cohen et al., Citation2009; Crum-Cianflone et al., Citation2014; Jordan et al., Citation2013; Kibler, Tursich, Ma, Malcolm, & Greenbarg, Citation2014). In a meta-analysis of six studies, even after adjusting for comorbid depression, PTSD was an independent risk factor for incident coronary heart disease and cardiac-specific mortality (Edmondson, Kronish, Shaffer, Falzon, & Burg, Citation2013). Vietnam Veterans with PTSD were more likely to have evidence of atrioventricular conduction defects and a greater history of MI (Boscarino & Chang, Citation1999), while in a community-based sample, Veterans with PTSD had increased risk for developing HF compared with Veterans without PTSD (Roy, Foraker, Girton, & Mansfield, Citation2015), and PTSD is associated with a greater mortality in patients with HF (Fudim et al., Citation2018). Within the Veteran population, MI is one of the top five causes of cardiovascular hospital admission (Krishnamurthi, Francis, Fihn, Meyer, & Whooley, Citation2018) and Veterans are at higher risk of having new onset of CVD compared with non-Veterans (Assari, Citation2014). Even after controlling for all covariates, logistic regression confirmed the association between Veteran status and heart disease (adjusted relative risk = 1.483, 95% confidence interval = 1.176–1.871) (Assari, Citation2014). Thus in general, PTSD is associated with more than double the risk for ischemia and CVD events (Turner, Neylan, Schiller, Li, & Cohen, Citation2013).
In addition to PTSD-associated risks in developing CVD, the manifestation of CVD can in and of itself serve as a traumatic event, and studies suggest that the prevalence of PTSD is about 10–20% following acute coronary syndromes (Edmondson et al., Citation2012; Gander & von Kanel, Citation2006). More critically, PTSD symptoms after these cardiovascular events increased the risk of adverse cardiovascular outcomes (Edmondson & Cohen, Citation2013), predicted non-adherence to medication, and increased likelihood of CVD readmission over the first year (Shemesh et al., Citation2004). A recent study demonstrated lower coronary distensibility index (CDI) scores, which are associated with endothelial dependent plaque composition, in patients with PTSD, and PTSD was independently associated with an increased risk of major adverse cardiovascular events (MI or CVD death) over a 50 month follow-up (Ahmadi et al., Citation2018). Several past reviews have suggested a variety of factors might contribute to this comorbidity between PTSD and CVD, and particularly HF, including biologic factors such as dysregulation of the autonomic nervous system (ANS), hypothalamic-pituitary-adrenal (HPA) axis, oxidative stress, and inflammation (Cohen, Edmondson, & Kronish, Citation2015; Edmondson & Cohen, Citation2013; Gander & von Kanel, Citation2006). These complex neurophysiologically regulated systems may reflect and/or influence changes in psychological factors that interact with drug use, eating behaviors, maladaptive aging, and physiological processes such as metabolic disease (Wolf & Morrison, Citation2017).
Based on the existing comorbidity of PTSD and HF within the VA health system and in light of the associations briefly described in the preceding section, a VA sponsored conference entitled “Cardiovascular Comorbidities in PTSD: The Brain-Heart Consortium” was convened in July 2017 (Columbia, SC; attendee list presented in ) to explore potential relationships and common biological pathways between PTSD and HF. The theme of the conference was framed around the hypothesis that specific common systems are dysregulated in both PTSD and HF, resulting in a synergistic acceleration and amplification of both disease processes. This conference was not intended to identify all independent pathways that give rise to PTSD and HF, but rather identify shared systems, pathways, and biological mediators that would be modifiable in both disease processes. The purpose of this report is to summarize key points from this conference and frame common themes which emerged regarding the comorbidity between PTSD and HF, as well as the recommendations from the group for future research opportunities.
Table 1. Attendees at cardiovascular comorbidities in PTSD: The Brain-Heart Consortium.
Symptoms and etiology
PTSD symptoms and etiology
PTSD is a debilitating psychiatric disorder that develops in a subset of individuals after exposure to traumatic events and is associated with pervasive functional impairment and increased health problems, including CVD and sleep dysregulation (see sections ‘Introduction’ and ‘Brain systems involved in PTSD’). From the clinical perspective, PTSD is defined as a Trauma- and Stressor-Related Disorders with four distinct clusters of symptoms in the Diagnostic and Statistical Manual of Mental Disorders (DSM-5). Thus, PTSD requires exposure to a traumatic or stressful event as a diagnostic criterion (Criterion A), which is clarified as directly experiencing a traumatic event or witnessing the event in person, experiencing repeated or extreme exposure to aversive details in the aftermath of a traumatic event, or learning about the violent or unexpected death of a close facility member or friend. In DSM-5, the four symptom clusters defined for PTSD include reeexperiencing, avoidance, negative cognitions and mood, and arousal/reactivity. Reeexperiencing refers to having spontaneous memories, recurrent dreams, or flashbacks of the traumatic event, or psychological or physical distress associated with cues related to the event, while avoidance refers to avoiding the distressing memories, thoughts, feelings, or external reminders of the event. The cluster of negative alterations in cognitions and mood incorporates a broad spectrum of symptoms, including those that were originally referred to as “emotional numbing” associated with feelings of estrangement from others, diminished interest in activities, inability to experience positive emotions, or inability to recall key aspects of the event, in addition to having a generally negative emotional state with exaggerated negative expectations and distorted blame concerning both self and others. Finally, arousal can be associated with aggressive, angry, reckless, or self-destructive behavior, sleep disturbances, exaggerated startle, or hyper-vigilance, incorporating both the “flight” and the “fight” aspects of the stress response associated with PTSD (Wilson & Reagan, Citation2016).
While estimates suggest that somewhere between 50 and 84% of the general population will experience a traumatic event, most individuals are resilient to these stressors, with ∼10% of the population subsequently developing PTSD. Estimates of the lifetime or past 12-month prevalence of PTSD using the DSM-IV or DSM-5 criteria among adult Americans were 6.1–6.8% and 3.5–4.7%, respectively, and these values were higher for women than men (Goldstein et al., Citation2016; Kessler, Berglund, et al., Citation2005; Kessler, Chiu, Demler, Merikangas, & Walters, Citation2005). Importantly, rates among combat veterans are much higher, with estimates for the prevalence of PTSD among the total Gulf War Veteran population to be 10.1% (Kang, Natelson, Mahan, Lee, & Murphy, Citation2003) and for deployed Operation Enduring Freedom (OEF)/Operation Iraqi Freedom (OIF) (Afghanistan and Iraq) service members to be 13.8% (Tamelian & Jaycox, Citation2008).
Brain systems involved in PTSD
PTSD can be reflected by altered activity in multiple brain systems, including those that directly modify autonomic functions. Functional neuroimaging studies over the past two decades have implicated PTSD-specific changes in regional activation and functional connectivity. More specifically, hyper-activation of amygdala, insula, dorsal anterior cingulate cortex (dACC), as well as hypo-activation in ventral medial prefrontal cortex (vmPFC) and impaired hippocampal function have been consistently detected (Lebois, Wolff, & Ressler, Citation2016; Liberzon & Abelson, Citation2016; Pitman et al., Citation2012; Stark et al., Citation2015). Furthermore, PTSD has been associated with altered connectivity within different neural circuits (Liberzon & Abelson, Citation2016). These changes in brain function are consistent with the autonomic features of neurophysiological defense mechanisms associated with increased detection of threat in the environment. PTSD has been associated with attentional bias towards threat and, consequently, exaggerated threat detection, manifested in increased connectivity within salience network and heightened responsivity of insula, amygdala and dACC (Liberzon & Abelson, Citation2016). Autonomic neurophysiological states can be influenced by disruption in the hierarchically organized brain systems that comprise the central autonomic network (Williamson, Porges, Lamb, & Porges, Citation2014). Ventrolimbic (orbito-prefrontal cortex, anterior temporal lobe, amygdala) and thalamo-cortical pathways are important in the regulation of emotion and the intersection of emotion and autonomic responses. The dynamic activity between these autonomic brain systems is chronically modified by PTSD and diminishes the ability to shift to a safe neurophysiological state. Indeed, PTSD has been found to be associated with altered fear extinction (i.e. deficits in learning that cues once associated with trauma are now safe) and fear overgeneralization (i.e. diminished capacity to discriminate between dangerous and safe cues) (Jovanovic, Kazama, Bachevalier, & Davis, Citation2012). When an individual feels safe, bodily state is efficiently regulated; this is accomplished through the influence of myelinated vagal efferent pathways.
Beyond these behavioral implications of vagus nerve activation, vagal efferent pathways oppose fight or flight mechanisms (i.e. sympathetic nervous system (SNS)). Therefore, prefrontal-limbic brain circuits regulating behavior and terminating on vagal centers (dorsal motor nucleus of the vagus (DMV) or nucleus ambiguus) are poised to promote PTSD-like behaviors and cardiovascular dysfunction. For example, enhanced amygdalar activity is associated with increased SNS response to stress, as well as fear-conditioned cardiovascular responses (Powell et al., Citation1997), while PFC activity serves to suppress SNS activity (Verberne & Owens, Citation1998). Moreover, hyperactivity of the locus coeruleus (LC), a brain region that regulates both arousal and cardiac autonomic function has also been detected in PTSD (Krystal et al., Citation2018; Naegeli et al., Citation2018). Importantly, norepinephrine (NE) is released from the neurons of the LC projecting to various brain regions implicated in the stress response, including DMV, hippocampus, PFC, amygdala, hypothalamus, and thalamus (Hendrickson & Raskind, Citation2016). The coeruleo-vagal pathway (i.e. LC-DMV) provides a circuit by which the LC–NE system can inhibit activity of DMV neurons by binding to α2-adrenoceptors (Samuels & Szabadi, Citation2008). As such, activation of the LC not only produces arousal, but decreases vagal/increases sympathetic tone. There is mounting evidence for LC noradrenergic regulation of the circuit connecting the hippocampus with vmPFC; hypo-activation of the hippocampus and vmPFC, as well as reduced connectivity within this circuit, is likely to contribute to context processing deficits that could provoke dominance of fear over safety memories, inappropriate responsivity to trauma-associated cues, and hypervigilance in PTSD (Jin & Maren, Citation2015; Liberzon & Abelson, Citation2016). Additionally, PTSD-related biological abnormalities are likely to include a neural circuit that connects the hypothalamus and amygdala with the LC, in which corticotropin releasing factor (CRF) and NE interact to increase fear conditioning and encoding of emotional memories, enhance arousal and vigilance, and integrate endocrine responses to stress (Hendrickson & Raskind, Citation2016).
Animal models of PTSD
A comprehensive understanding of PTSD biomarkers, including neurocircuits, autonomic dysregulation, neurotransmitter abnormalities, and increased inflammation, requires mechanistic studies using animal models, as much of the experimental work is not feasible in humans (Pitman et al., Citation2012). There is a large array of potential animal models of PTSD (see ); however, most concentrate on inducing a severe trauma (i.e. shock, predator exposure, prolonged restraint, or multiple stressors within a 24–48 h period), allowing the animals to rest for at least seven days and then assessing enduring behavioral and physiological phenotypes (Deslauriers, Powell, & Risbrough, Citation2017; Deslauriers, Toth, Der-Avakian, & Risbrough, Citation2018). Models that adhere to relatively short severe trauma exposures are predator exposure (Deslauriers, Toth, et al., Citation2018; Zoladz & Diamond, Citation2016), single prolonged stress (SPS) (Lisieski, Eagle, Conti, Liberzon, & Perrine, Citation2018) and fear conditioning augmented by additional stressors (Perusini et al., Citation2015). Models that utilize chronic trauma exposure, often resulting in greater enduring depressive-like behavior are social defeat stress and chronic unpredictable stress (Deslauriers, Toth, et al., Citation2018; Finnell et al., Citation2017, Citation2018). Both the single severe trauma and the chronic trauma exposure models induce enduring anxiety-like responses; however, the chronic stress models tend to recapitulate aspects of depression that are not associated with PTSD, such as blunted HPA axis while single severe stressors are associated with higher arousal and HPA hypersensitivity observed in PTSD (Deslauriers, Toth, et al., Citation2018).
Table 2. Potential cardiovascular-related phenotypes in animal models of PTSD.
The animal models shown in have proven to be a useful tool for examining the molecular and cellular physiological mechanisms implicated in PTSD pathology that may be the targets for interventions (Deslauriers et al., Citation2017). For example, these models have been critical in showing that neurotrаnsmitter abnormalities and other molecular mechanisms induced by trauma occur within a specific anatomical context (i.e. within specific neurocircuits or cell types), which will be critical for development of potential interventions. Some models are also clearly associated with immune mechanisms that may also converge with CVD mechanisms (e.g. social defeat stress, predator stress and SPS, see ) (Deslauriers et al., Citation2017; Deslauriers, Toth, et al., Citation2018). Traumatic stress in animals has been shown to induce similar enduring effects on cardiovascular functions as those observed in PTSD patients, including reductions in heart rate variability (HRV), increased blood pressure, and heart damage () (Bruijnzeel, Stam, Croiset, & Wiegant, Citation2001; Carnevali et al., Citation2013; Crestani, Citation2016; Koresh et al., Citation2016; Laukova et al., Citation2014, 2018; Liu et al., Citation2016; Penna & Bassani, Citation2010; Rorabaugh et al., Citation2015; Vieira et al., Citation2018; Zoladz & Diamond, Citation2016). Importantly, however, effects on cardiovascular function can be highly dependent on type and duration of stressor (predictable vs. unpredictable, and chronicity) (Crestani, Citation2016) and sex or gonadal hormones (Finnell et al., Citation2017, Citation2018; Vieira et al., Citation2018), suggesting that careful consideration of the cardiovascular phenotypes under investigation must guide model selection.
HF definition and relation to PTSD
Similar to PTSD, HF is defined by a presentation of a constellation of symptoms rather than the underlying causality or etiology (Tanai & Frantz, Citation2015). HF consumes over 25% of total US health expenditures and is predicted to reach unsustainable levels by 2050 (Writing Group et al., Citation2016). The spectrum of HF symptomatology is usually one of increased fluid accumulation, reduced exercise tolerance, and as the disease progresses, activation of a number of neurohormonal systems and inflammatory cascades. One important development in the clinical management of HF was to recognize that HF is a generalized term and not a specific diagnosis. Thus, two distinct HF phenotypes have emerged which are defined not by symptom presentation, but rather by underlying pathophysiology. Specifically, patients presenting with HF and reduced left ventricular (LV) forward stroke volume, or ejection fraction, are defined as HF with reduced ejection fraction (HFrEF) whereas patients presenting with HF, a relatively normal LV ejection fraction but impaired LV filling are defined as HF with preserved ejection fraction (HFpEF). The predominant cause for HFrEF is ischemic heart disease such as following MI, whereas the predominant cause for HFpEF is prolonged LV afterload such as with hypertension; however, these CVD conditions are not mutually exclusive and are also influenced by age, body mass index, and other environmental factors (Ezekowitz et al., Citation2009). In a recent study, nearly 10% of the Veterans diagnosed with HFrEF had PTSD, and these patients had a higher burden of comorbidities (Fudim et al., Citation2018). These specific HF phenotypes are also important in terms of guiding therapy whereby device driven therapies such as cardiac resynchronization therapy and LV assist devices have demonstrated efficacy in HFrEF, but not in HFpEF. On the other hand, pharmacotherapies such as inhibition of sympathetic or renin–angiotensin pathways can be effective in both forms of HF. Thus, not dissimilar to PTSD, identifying the specific form and feature of HF as well as the activation of underlying pathways hold relevance as to therapeutic strategies.
While HF is due to impairments in LV functional performance, this results in systemic manifestations which affect all organ systems and the brain is not protected. Cerebral symptoms of HF include confusion, sleep disorders, dizziness, altered mood, and disorientation. There is also a link between impaired cognitive function and cardiac dysfunction that has been termed cardiogenic dementia. There are a number of shared risk factors between heart and brain, including perfusion abnormalities and rheological alterations. Manifestations of impaired cerebral physiology include fluctuating delirium precipitated by acute cardiac decompensation and chronic impaired cerebral function during periods of stable HF.
A major common feature between brain and heart is the limited capacity for the major cell types (neurons and cardiomyocytes) to repair and regenerate. In response to injury, cardiomyocyte responses include hypertrophy or cell death through a number of means including apoptosis and necrosis. Increased intracellular calcium concentrations and cyclic AMP formation is a common feature of cardiomyocyte responses to stress. While increased intracellular calcium has positive inotrope and negative lusitrope effects to increase contractility and reduce relaxation times, excess calcium entry can be a conduit for arrhythmias. In response to injury that induces neuron or cardiomyocyte loss, both brain and heart respond by stimulating an inflammatory response that initiates a wound healing cascade. The extracellular matrix (ECM) is a critical component, serving to both amplify and regulate inflammation as well as providing ECM proteins that serve as scar tissue (Lindsey, Citation2018; Spinale et al., Citation2016).
Animal models of heart failure
As with models of PTSD, there are no animal models of HF which completely recapitulate this complex disease process. However, the first consideration is the HF phenotype to be studied for inducing the initiating stimulus in the appropriate animal model. For HFrEF, the most common approach is to induce MI through coronary ligation in both small and large animals. For HFpEF, inducing LV pressure overload either mechanically (aortic constriction) or pharmacologically (infusion of vasopressor agents) are commonly employed. There are a number of reviews which identify the strengths and inherent weaknesses of these animal models. Depending upon the stimulus, there is every possibility of superimposing a PTSD stimulus and HF induction in the same animal, and is identified further in the future directions section. In terms of relevance to superimposing a form of PTSD and HF, rodent models would most likely be the exploratory start point (Horgan, Watson, Glezeva, & Baugh, Citation2014; Houser et al., Citation2012; Lindsey et al., Citation2018).
Autonomic nervous system in PTSD and HF
PTSD is an autonomic disorder: hyperarousal features
It has been established that PTSD is associated with ANS dysregulation such as diminished activity of the parasympathetic nervous system (PNS) linked to vegetative and restorative functioning, and elevated activity of the SNS related to mobilization of energy and stimulation of the fight or flight response (Blechert, Michael, Grossman, Lajtman, & Wilhelm, Citation2007; Brudey et al., Citation2015; Clausen, Aupperle, Sisante, Wilson, & Billinger, Citation2016; Green et al., Citation2016). Disorder-related alterations in PNS and SNS function in the setting of PTSD pathophysiology are reflected by an elevated heart rate and/or systemic arterial pressure, increased galvanic skin response (a pure measure of SNS activity) to startle, and increased ANS responses to trauma-related cues (Keary, Hughes, & Palmieri, Citation2009; McFall, Murburg, Ko, & Veith, Citation1990; Paulus, Argo, & Egge, Citation2013; Pole, Citation2007). Further, PNS disturbances have been reported including attenuated resting high frequency HRV or vagal tone as defined by respiratory sinus arrhythmia (RSA) (Sack, Hopper, & Lamprecht, Citation2004), since RSA represents a noninvasive index of parasympathetic control of heart rate. These associations are detectable even when controlling for depression and traumatic brain injury, two highly comorbid conditions in PTSD that are also associated with low HRV (Minassian et al., Citation2014). Twin studies have suggested that the reduced HRV associated with PTSD is not related to heritable factors, but may be related to symptom state (Shah et al., Citation2013), with resolution of RSA differences after successful treatment of PTSD. Prospective longitudinal studies, however, suggest that reduced HRV may be a risk factor for development of PTSD (Minassian et al., Citation2015), hence convergent risk factors in autonomic control could contribute to PTSD and CVD comorbidity. Consistent with SNS hyperactivity, exaggerated catecholamine responses to trauma-related stimuli have been reported in PTSD, as well as higher baseline levels of NE and epinephrine in CSF, plasma, and urine in patients with PTSD (Liberzon, Abelson, Flagel, Raz, & Young, Citation1999; Pitman et al., Citation2012; Southwick et al., Citation1999; Strawn & Geracioti, Citation2008; Yehuda et al., Citation1998). These autonomic features of PTSD are associated with the development of CVD even in subclinical populations. Thus, several features in the resting heart rate spectra are correlated with CVD mortality (Williamson et al., Citation2010), low frequency HRV is an independent predictor of death (Tsuji et al., Citation1994), and reduced low frequency HRV is linked to coronary artery disease and increased stroke risk (Binici, Mouridsen, Kober, & Sajadieh, Citation2011; Kotecha et al., Citation2012). Furthermore, disruption in neurophysiological regulation of emotion seen in PTSD may manifest as anger or depression or anxiety, and these changes in emotional state modify psychosocial behaviors including the ability to adaptively socially engage, resulting in loneliness. All of these emotional states are associated with modifications in ANS and the developmental of systemic diseases including CVD. Patients with PTSD have higher rates of aggression and anger (Novaco & Chemtob, Citation2002) and PTSD severity is a significant predictor of intermittent explosive disorder diagnosis (Reardon et al., Citation2014). Hostility is related to exaggerated autonomic reactivity to cognitive or pain stressors, reduced HRV, and CVD (Sloan et al., Citation2001; Williamson & Harrison, Citation2003). These co-morbid symptoms/traits rely on the same brain systems, supporting the idea that shifts in autonomic states impact aspects of mood/personality in a predictable manner.
Autonomic dysfunction in HF
The sympathetic system is similarly activated during the progression to HF, resulting in activation of pressure baroreceptors in the carotid sinus, aortic arch, and LV (Grassi, Seravalle, & Mancia, Citation2015). Afferent signals stimulate the central cardiovascular center in the brain to increase circulating blood volume. As a result of sympathetic activation, arginine vasopressin is released from the posterior pituitary gland and perfusion is redistributed systemically, including in the heart, kidney, vasculature, and skeletal muscles. Persistent sympathetic activation causes transcriptional and posttranscriptional changes at the level of the genome. Activation of the sympathetic system is a rapid response mechanism for adaptation to HF. Baroreceptors are stimulated in response to stretch activation, and the precise balance between low and high pressure systems shifts, such that baroreceptor inhibition falls and excitatory impulses rise. Like PTSD, the general increase in SNS activity is accompanied by a decrease in PNS activation, leading to reduced HRV, elevated blood pressure, and elevated peripheral resistance (Grassi et al., Citation2015; Parati & Esler, Citation2012). This in turn activates the renin–angiotensin–aldosterone system to regulate the salt–fluid balance. Plasma NE increases and correlates with mortality in patients with advanced HF (Slavikova, Kuncova, & Topolcan, Citation2007). Both β1 and α1-adrenergic receptors are often stimulated in HF, and the use of β adrenergic antagonists constitutes a standard of care (Najafi, Sequeira, Kuster, & van der Velden, Citation2016). Thus, in the context of PTSD and HF prolonged sympathetic activation can be particularly detrimental and exacerbate both disease processes.
Hyperarousal features of sleep and autonomic imbalance in PTSD and HF
Sleep disruption is amongst the most prevalent symptoms of PTSD and HF, with co-occurring sleep disorders diagnosable in approximately 50–70% of patients (Pak et al., Citation2018; Spoormaker & Montgomery, Citation2008). Sleep dysregulation is related to ANS activity, and the hyperarousal features of PTSD are particularly associated with sleep problems (van Wyk, Thomas, Solms, & Lipinska, Citation2016). In order to shift from a defensive disposition to one conducive to good sleep, the individual needs to determine safety and inhibit the more primitive limbic structures that control fight, flight, or freeze behaviors (Porges, Doussard-Roosevelt, Stifter, McClenny, & Riniolo, Citation1999). Although sleep is regulated through complicated interactions between multiple brain regions and neuromodulators, the vagally mediated components of the limbic system are critical to normal sleep function (Porges et al., Citation1999). Parasympathetic activity normally increases as people transition to sleep and is critical to sleep efficiency (Jung, Lee, Jeong, & Park, Citation2017; Woodward et al., Citation2009).
Common complaints in patients with PTSD include nightmares, distressed awakenings, sleep terrors, insomnia, and nocturnal panic attacks (Spoormaker & Montgomery, Citation2008). A meta-analysis of sleep quality in PTSD showed decreased slow wave sleep, and longer sleep latencies (Kobayashi, Boarts, & Delahanty, Citation2007). During slow wave sleep, there is a decline in mean arterial pressure in comparison to wakefulness, and a lack of reduction in mean arterial pressure is associated with CVD risk (Silvani, Bastianini, Berteotti, Lo Martire, & Zoccoli, Citation2013). The relationship of PTSD to sleep disruption is bidirectional, since PTSD increases the likelihood of sleep problems and PTSD is exacerbated by sleep problems. Further, sleep quality is a vulnerability factor for the development of PTSD, suggesting that sleep quality and PTSD are mechanistically linked (El-Solh, Riaz, & Roberts, Citation2018; van Liempt, Citation2012). This may involve dysregulation of the ANS, since nocturnal autonomic changes including lack of reduction in blood pressure are associated with hyperarousal symptoms of PTSD, greater sleep-related daytime dysfunction, poorer overall sleep quality, and more frequent use of sleep medication. This dysregulation of parasympathetic control during sleep may contribute to enhanced risk for CVD (Ulmer, Calhoun, Bosworth, Dennis, & Beckham, Citation2013; Ulmer, Hall, Dennis, Beckham, & Germain, Citation2018). People with PTSD show lower RSA during sleep (Woodward et al., Citation2009) which is linked to increased stroke risk (Binici et al., Citation2011), and treating sleep quality may improve symptoms of PTSD (Ho, Chan, & Tang, Citation2016).
A clear relationship has also been established with respect to CVD and sleep disturbances. Similar to PTSD, patients with HF often have disrupted sleep, and while there is an association between untreated obstructive sleep apnea and mortality in patients with HF, patients with HF often do not report excessive daytime sleepiness (Pak et al., Citation2018). Notably, sleep apnea both due to obstructive causes as well as centrally mediated (central sleep apnea, CSA), can contribute to the progression of CVD such as HF (Drager et al., Citation2017). For example, up to 40% of patients presenting with HF also have been identified to have sleep apnea (Wang et al., Citation2007). This specific sleep disorder likely contributes to the progression of the HF process due to the episodic hypoxia/hypercapnia leading to periods of sympathetic nervous stimulation, generation of reactive oxygen species and inflammation. Thus, clinical trials have been performed whereby airway management such as continuous positive airway pressure (CPAP) was instituted to modify CVD progression (Drager et al., Citation2017). Unfortunately, in the Sleep Apnea Cardiovascular Endpoints (SAVE) trial, in which over 2700 patients were randomized to standard of care with and without CPAP, there was no relative reduction in major cardiovascular events (Bradley et al., Citation2005). These findings suggested that more centrally mediated approaches for regulating CSA in the context of patients with CVD, particularly that of HF, may be more effective. Indeed, recent clinical trials using neurostimulation approaches, particularly of the phrenic nerve, have shown potential benefit in patients with CSA and HF (Costanzo et al., Citation2016). Since this form of neurostimulation may affect both afferent and efferent pathways, and thus modify sympathetic system outflow, then a synergistic effect may occur in patients with both CSA and HF. This observed interrelationship of sleep disturbances such as CSA to that of CVD, have promulgated the concept that sleep disorders are a top tier modifiable risk factor for cardiovascular events (Drager et al., Citation2017). Recent studies are also suggesting cognitive behavioral therapy might be beneficial to the sleep disturbances in patients with HF failure; although, the impact of this emerging therapeutic approach on cardiovascular function in HF remains to be determined (Redeker et al., Citation2018). Importantly, poor sleep quality is also associated with CVD factors independent of sleep apnea (Tosur et al., Citation2014). Since sleep disorders are a prevalent in patients with PTSD and HF, then the inter-relationship between pathways regulating sleep and wakefulness to that of CVD progression, and the role of dysregulation of the ANS in sleep disturbances, constitute important areas for future research.
Parallel cell signaling pathways in PTSD and HF
Although discrete in many ways, the brain and heart represent parallel systems in survival, response, and modulation. Physiologically, brain function and cardiac function are delicately balanced by sensitive ionic gradients and fluxes orchestrated in a coordinated fashion by numerous proteins, channels, and receptors. As a consequence, it is not surprising that these seemingly disparate tissues are both highly vulnerable to similar insults that challenge homeostasis, such as lack of oxygen, acid–base disturbances, oxidative stress, and inflammation. For example, reduced oxygen to both the brain and the heart yield irreversible damage to cell types that do not regenerate (neurons and cardiomyocytes), but can induce similar wound-healing and repair responses. Neurons and cardiomyocytes are also electrically excitable and communicate with other cells, making the heart and brain capable of coordinated rhythmic or oscillatory patterns that are fundamental for their function. Thus, an increased understanding of one system may provide valuable insight into the other system, as well as comorbidities between cardiovascular and brain disorders.
Based on the ability of heart and brain cells to communicate with one another, and for heart and brain to regulate function of the other, the integration of the complex microdomains and signaling pathways is critical to normal function of both organ systems. Several common systems impact both brain and heart, and have been suggested as possible underlying mediators that might induce comorbidity between HF and PTSD (see , and ). While beyond the scope of this review to document all potentially common changes, a few systems seem to be potential common mediators between PTSD and CVD in general and HF in particular. These changes are seen as not only differences in signaling cascades and mediator levels in PTSD or HF patients (), but also in genetic polymorphisms associated with these conditions (). Given the ANS dysregulation, it is not surprising the changes in catecholamines (especially NE and epinephrine) and their transporters, as well as α and β adrenergic receptors are seen in both conditions, as well as changes in the cholinergic system associated with reduced parasympathetic tone (see and ) (Brudey et al., Citation2015; Deslauriers, Acheson, et al., Citation2018; Laukkanen, Makikallio, Kauhanen, & Kurl, Citation2009; Liberzon et al., Citation2014; Marques et al., Citation2017; Mir et al., Citation2018; Pietrzak et al., Citation2015; Snapir et al., Citation2003). As stress-related disorders, changes are also seen in HPA axis function or cortisol levels and regulators of glucocorticoid receptor signaling such as FKBP5 (Gill, Vythilingam, & Page, Citation2008; Lebois et al., Citation2016; Lovallo et al., Citation2016; Miller, McKinney, Kanter, Korte, & Lovallo, Citation2011), as well as several aspects of the renin–angiotensin system (RAS) and polymorphisms in angiotensin converting enzyme, angiotensin levels, and angiotensin receptors (Cameron et al., Citation2006; Nylocks et al., Citation2015; Raynolds et al., Citation1993; Winkelmann et al., Citation1999; Wu et al., Citation2009). An emerging literature suggests the endocannabinoid system may be a common mediator for cardiovascular and PTSD related changes (Hill, Campolongo, Yehuda, & Patel, Citation2018; Pacher, Steffens, Hasko, Schindler, & Kunos, Citation2017). In HF, changes in proteolytic enzymes such as matrix metalloproteinases (MMPs), protein accumulation, and changes in the ECM are associated with anatomical changes in the heart (DeLeon-Pennell, Meschiari, Jung, & Lindsey, Citation2017; Frangogiannis, Citation2017; Lima et al., Citation2018; Spinale et al., Citation2016). Given the functional anatomical changes seen in PTSD discusses above, the role of these ECM changes in the brain remains to be elucidated; although, animal models suggest involvement of these signaling cascades in brain as well (Finnell et al., Citation2017).
Figure 1. Mediators and modulators of the co-morbidity between PTSD and heart failure. Diagram shows that stress, such as experiences during combat, influences multiple systems in the brain and heart that can serve to induce PTSD as well as cardiovascular diseases. Mounting evidence suggests that the co-morbidities between PTSD and heart failure might be due to similar autonomic nervous system dysregulation, similar genetic polymorphisms, and/or common changes in cell signaling, communication and remodeling, plus immune dysregulation. Sleep dysregulation, gender, aging, and metabolic disorders can all modify the severity and progression of PTSD and heart failure symptoms, but it is yet unknown how these modifiers contribute to the comorbidity between PTSD and heard failure.
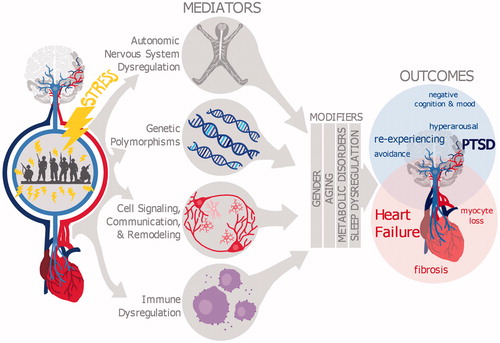
Table 3. Common mediators between PTSD and heart failure: cell signaling, communication, and remodeling.
Table 4. Genetic polymorphisms that may confer vulnerability to PTSD and CVD: evidence from gene variant association studies or links to disease endophenotypes.
Inflammation as a common mediator of PTSD and heart failure
A common biological cascade that is operative in most disease states, whereby PTSD and HF are no exception, is inflammation. While this is a broad term and what specifically defines inflammation is dependent upon location and context, there are common inflammatory signaling systems that appear to be in common. In PTSD, elevated levels of inflammatory markers like C reactive protein (CRP) and pro-inflammatory plasma cytokines, including interleukins (ILs)-1, IL-6, tumor necrosis factor (TNF), as well as decreased anti-inflammatory markers, have been found (Breen et al., Citation2018; Deslauriers et al., Citation2017; Lindqvist et al., Citation2017; Wang, Caughron, & Young, Citation2017), and the increased IL-6 and CRP have been shown to contribute to the increased CVD risk in PTSD (Boscarino, Citation2008). In advanced HF, an inflammatory cascade is similarly involved which can induce proteases such as MMPs. There is a strong relationship between inflammation and MMPs, as a number of MMPs proteolytically activate cytokines, chemokines, and growth factors (Lindsey, Citation2018). For example, pro-IL-1 is biologically activated by MMP-9 (Schonbeck, Mach, & Libby, Citation1998). Later, new ECM is produced by fibroblasts to form scar tissue. Importantly, several studies have linked ANS dysregulation, seen in both PTSD and HF, to alterations in the immune system and inflammation. For example, reduced HRV has been associated with increased inflammatory markers (Frasure-Smith, Lesperance, Irwin, Talajic, & Pollock, Citation2009) in depression and CVD. Further, a growing body of literature has indicated that parasympathetic activation via cholinergic activity inhibits inflammation, while sympathetic activation leads to the increased release of inflammatory cytokines (Olofsson, Rosas-Ballina, Levine, & Tracey, Citation2012; Tracey, Citation2007). In fact, cardiac arrest with cerebral ischemia leads to microglial activation, proinflammatory cytokines, and neural damage, which also compromises the ability of the cholinergic anti-inflammatory pathway to contain inflammation (Norman et al., Citation2011). These studies suggest that changes in different pathways mediating inflammatory processes for HF and PTSD might be inter-related, and interact via vagal afferent/efferent pathways and immune signaling. An important consideration in investigating the intersection of PTSD and HF will be the role of neuroinflammatory processes and microglial activation in the central nervous system, and how these central changes modulate neural plasticity or synaptic reorganization that help drive peripheral changes including ANS dysfunction and/or peripheral immune markers (see Deslauriers et al., Citation2017).
While activation of inflammatory pathways and the induction of proteases such as MMPS occur in both PTSD and HF, how these can affect critical remodeling process such as synaptic remodeling within the brain and ECM remodeling within the myocardium remains to be explored. Several aspects of these common pathways also represent means of communication, and feedback loops, between the heart and brain. As stress disorders, of course the responses and feedback loops in the HPA axis are critical. But other points of communication include immune signaling as well as vagal efferents/afferents and the ANS. The latter provide links to the functional anatomical changes seen in PTSD in regions controlling ANS function, such as the prefrontal cortex, amygdala, and LC. In addition, the circumventricular organs (CVOs) of the brain are a specialized group of structures that lack a blood–brain barrier and are thus uniquely positioned to monitor the systemic circulation (sensory CVOs) or being able to secrete substances directly into the circulation (secretory CVOs) (Ferguson, Citation2014). Neurons localized within the sensory CVOs are therefore ideally positioned at the blood–brain interface to relay information from changes in systemic milieu to the brain. The CVOs thus represent crucial gateways for body-to-brain communication. Of relevance to PTSD and cardiovascular comorbidity, sensory CVOs such as the subfornical organ (SFO) and vascular organ of the lamina terminalis (OVLT) have direct innervation to forebrain and hindbrain brain areas regulating stress, autonomic, and emotional responses. The CVOs have been shown to sense circulating concentrations of angiotensin II, sodium, calcium, as well as osmolality, and they control a variety of autonomic outputs through efferent projections to essential hypothalamic and medullary autonomic control centers, providing an avenue of communicating peripheral changes in many mediators seen in to the brain (Ferguson, Citation2014). Finally, an additional means of communication are the extracellular vesicles called exosomes, that have been implicated in cell-cell communication and potentially various disease states, but their potential role in the comorbidities between PTSD and HF remains unknown (Gill et al., Citation2018; Shanmuganathan, Vughs, Noseda, & Emanueli, Citation2018).
An example of a novel common mediator of PTSD and HF: the orexin system
An example of what might emerge as a novel therapeutic target from looking at commonalities between PTSD and HF is the orexin system. The orexin/hypocretin neuropeptides discovered in the late 1990s have a well-established role as a physiological integrator in the control of sleeping and homeostatic regulation, as well as attention, arousal, and stress responses (Berridge, Espana, & Vittoz, Citation2010; Carter, Schaich Borg, & de Lecea, Citation2009; Fadel & Burk, Citation2010; Flores, Saravia, Maldonado, & Berrendero, Citation2015; Mahler, Moorman, Smith, James, & Aston-Jones, Citation2014; Sakurai, Citation2007). Two peptides, orexinA/hypocretin1 (OxA) and orexinB/hypocretin2 (OxB) are produced by the preproorexin gene by orexin neurons that are restricted to the hypothalamus but project throughout the central nervous system, and the two G protein-coupled receptors (orexin/hypocretin 1 receptor (Ox1R/HcrtR1), orexin 2 receptor (Ox2R/HcrtR2)) are found throughout the brain (Abreu, Molosh, Johnson, & Shekhar, Citation2018; Fadel & Burk, Citation2010; Flores et al., Citation2015).
The orexin system is well poised to impact the neural systems underlying the four PTSD symptom clusters of reeexperiencing, avoidance, negative cognitions and mood, and arousal/reactivity, as well as the comorbidities of PTSD including sleep dysregulation, substance abuse, and CVD. The orexin system modulates sleep, homeostatic regulation, arousal, and attention, but also fear learning and extinction (Abreu et al., Citation2018; Berridge et al., Citation2010; Fadel & Burk, Citation2010; Flores et al., Citation2015; Mahler et al., Citation2014; Sakurai, Citation2007) as well as reward and motivated behaviors, including food and drug seeking behaviors (Mahler et al., Citation2014; Yeoh, Campbell, James, Graham, & Dayas, Citation2014). Studies have demonstrated low OxA levels in cerebrospinal fluid and plasma in PTSD patients compared to normal controls, and a negative association between orexin concentrations and more severe symptoms in these combat veterans with PTSD (Strawn et al., Citation2010). The positive correlation between CSF and blood levels of orexin suggest this may also serve as a biomarker for PTSD (Strawn et al., Citation2010). Preclinical studies have shown that intracerebroventricular administration of OxA or OxB induces stress-like behavioral responses, activation of the HPA axis, and cardiovascular responses associated with sympathetic activation (Abreu et al., Citation2018; Berridge et al., Citation2010; Carrive, Citation2017). Pharmacological studies, studies using neurotoxic lesions of the hypothalamus including orexin neurons, and studies in Ox1 or Ox2 knockout mice all suggest a role for orexin in both the behavioral and cardiovascular responses during fear learning and extinction (Carrive, Citation2017; Flores et al., Citation2015; Furlong & Carrive, Citation2007; Sears et al., Citation2013; Soya & Sakurai, Citation2018); although, unique roles for Ox1 and Ox2 receptors in different brain areas are seen. Interestingly, systemic administration of the dual Ox1R/Ox2R antagonist almorexant decreased cardiovascular measures, but not the behavioral responses during contextual fear (Furlong, Vianna, Liu, & Carrive, Citation2009).
The orexin system provides a provocative novel target and may provide a signaling pathway that helps explain why some individuals are susceptible to traumatic stress, and others remain resilient. While somewhere between 50 and 84% of the general population will experience a traumatic event, estimates suggest that only around 10% of the population will go on to develop PTSD (Goldstein et al., Citation2016; Kessler, Berglund, et al., Citation2005; Kessler, Chiu, et al., Citation2005). Thus, a better understanding of individual differences in orexinergic systems that lead to risk and resilience has implications for a variety of anxiety disorders, but particularly PTSD and panic disorders (Abreu et al., Citation2018; Flores et al., Citation2015; Yeoh et al., Citation2014). Divergent changes in the hypothalamic orexin system are associated with individual differences in behavioral fear extinction (Sharko, Fadel, Kaigler, & Wilson, Citation2017) and CO2-associated freezing (Monfils et al., Citation2018), plus the orexin system has been implicated in mediating the PTSD-like symptoms in the susceptible population using two different animal models of PTSD (Cohen et al., Citation2016; Grafe, Eacret, Dobkin, & Bhatnagar, Citation2018). Interestingly, many of these effects of orexin on fear behaviors involve the LC, providing a link to neural systems controlling noradrenergic outputs and sympathetic responses (Sears et al., Citation2013; Soya & Sakurai, Citation2018). Taken together, modulation of orexin receptors (Abreu et al., Citation2018; Flores et al., Citation2015; Soya & Sakurai, Citation2018; Yeoh et al., Citation2014) and/or intranasal administration of orexinergic peptides (Calva, Fayyaz, & Fadel, Citation2018) may represent novel pharmacological approaches for PTSD.
Orexins also modulate cardiovascular functions, through both central autonomic control and peripheral actions. The extensive projections of orexin neurons to brainstem areas such as the LC and rostral ventrolateral medulla (RVLM) anatomically support their role in the central regulation of autonomic responses, especially cardiovascular responses to stress (Carrive, Citation2017; Grimaldi, Silvani, Benarroch, & Cortelli, Citation2014) and sympathetic regulation associated with chronic hypertension (Abreu et al., Citation2018). In a rat model of MI and progressive HFrEF, a significant reduction in orexin mRNA levels was associated with the degree of cardiovascular compromise (Hayward et al., Citation2015), while in an Ox2R deficient transgenic mouse model cardiac function worsened with overstimulation of the sympathetic or angiotensin receptor pathways (Perez et al., Citation2015). In human studies, a specific single nucleotide polymorphism (SNP) within the OxR2 was identified in HFrEF patients (Perez et al., Citation2015). This orexin receptor gene variant was not only present in a subset of HFrEF patients using GWAS, but more importantly, those HFrEF patients appeared to be more refractory to conventional HF therapeutics. Further, higher OxA plasma levels in HFrEF patients were associated with greater functional improvements following conventional HF therapeutics when compared to HFrEF patients with low OxA plasma levels (Ibrahim et al., Citation2016). These studies continue to provide support for the postulate that a relative reduction in specific bioactive signaling pathways, such as that of orexin, can potentially accelerate the HF process as well as influence response to HF therapeutics.
Like PTSD, these preclinical and clinical studies provide for some provocative questions and new directions in terms of orexin in HF. Specifically, HF is a clinical syndrome and there are distinct phenotypes of this disease which include HFrEF, but also HFpEF. In HFpEF, which may constitute up to 50% of overall HF patients, whether and to what degree the SNP in the orexin receptor occurs and whether orexin plasma levels are altered remains unexplored. In light of the fact that OxR2 gene polymorphisms in HFrEF patients and transgenic ablation in mice were associated with HF progression (Perez et al., Citation2015), then exploration of the effects of these orexin receptor antagonists on cardiovascular outcomes may be warranted. Moreover, sleep disturbances such as insomnia is not an infrequent symptom in patients with PTSD which also has been shown to be associated with changes in orexin levels (Strawn et al., Citation2010). The positive correlation between CSF and blood levels of orexin (OxA) suggest this may also serve as a biomarker for PTSD and myocardial remodeling in HF patients (Ibrahim et al., Citation2016); although, it remains to be determined if there are genetic linkages between the orexin system and PTSD, as in HF. Despite therapeutic issues with targeting single neuropeptide systems, identifying critical intersections and relationships of orexin signaling to that of HF and PTSD syndromes would be an important translational and clinical research direction.
Recommendations for future research
The focus in this consortium on PTSD and CVD, but particularly HF, represents a distinct perspective, because interconnections have not historically been combined in one evaluation. Based on the existing comorbidity of PTSD and HF, particularly within the VA health system, the VA sponsored consortium identified several specific common systems that are dysregulated in both PTSD and HF, resulting in a synergistic acceleration and amplification of both disease processes. Further, we developed recommendations for further exploration of provocative new research directions and therapeutic targets, described below. These postulates are intended to help guide policy makers and/or funding agencies, as well as the research communities focused on PTSD and HF, to identify and bridge gaps in knowledge and further our understanding of the common mediators between these comorbid conditions, as well as identify novel therapeutic approaches.
Postulate one. Since PTSD accelerates/exacerbates HF and vice-versa, studies are needed to further explore the comorbidities between PTSD and HF. (1) Use Big Data approaches to define relative risk between PTSD and HF, including distinguishing PTSD comorbidity in patients with HFpEF vs. HFrEF. Studies are needed to examine if patients with PTSD (especially Veterans) have worse outcomes with HF, plus the factors contributing to PTSD symptoms in patients with CVD in general and HF in particular. The VA system in general, the Million Veteran Program, and VINCI represent excellent opportunities in this domain. One example is use of Mendelian randomization approaches with existing GWAS data in HF and PTSD populations. (2) Since an earlier cardiovascular event (such as MI) could serve as the traumatic stressor for HF and/or PTSD, additional studies expanding the heart-to-brain connection are needed. Such studies could explore if treatments for HF improve PTSD symptomology, and if sensory afferent and/or interoceptive mechanisms or the common pathways mentioned above are involved in the impact of HF treatments on PTSD. (3) Use imaging studies (both existing databases and prospective new studies) to better examine the relationship between HF and PTSD. (4) Enhance human studies through adding relatively simple cardiovascular measures to PTSD studies and basic PTSD assessments to HF studies. This approach would require more synergistic research teams combining cardiovascular and psychiatric expertise in large-scale clinical studies. (5) Enhance existing and develop new animal models to assess PTSD-like and HF phenotypes to further explore the common mediators between cardiovascular and behavioral phenotypes. These models will be important for determining common causation and identifying and testing novel therapeutic targets for comorbid PTSD and HF. Animal models would also be useful for exploring the impact of prior cardiovascular events on PTSD-like symptoms as well as cardiovascular changes due to traumatic stress.
Postulate two. Since concordant autonomic dysfunction occurs in PTSD and HF, studies are needed to explore if autonomic dysfunction plays a causative role in the comorbidity between PTSD and HF. (1) Determining the causative links between altered HRV, diminished parasympathetic tone, and enhanced sympathetic tone in PTSD with HF. Ascertain the most informative correlations between ANS changes in heart and brain in patients with PTSD and HF, or in animal models of PTSD or HF. (2) Human-based and preclinical studies using interventions like ANS stimulation to alter the balance between sympathetic and parasympathetic processes in PTSD and HF. (3) Studies interrogating the progression of autonomic dysfunction in both PTSD and HF in clinical and pre-clinical populations.
Postulate three. Examine the role of sleep dysregulation in PTSD and HF. (1) Studies evaluating if sleep dysregulation is a result or a cause of autonomic dysfunction, and if sleep dysregulation is one common mediator of the PTSD-HF comorbidity. (2) Studies elucidating if sleep dysregulation emerges after patients develop these conditions, or if sleep dysregulation predicts susceptibility to PTSD or HF. (3) Studies examining orexin as a key player in the connections between sleep dysregulation, PTSD, and HF.
Postulate four. Common cellular, inflammatory, and signaling pathways exist in both PTSD and HF, thus studies are needed to explore specific mediators of PTSD and HF, with the goal of identifying new therapeutic targets. (1) Studies identifying and exploiting common signaling cascades and molecules as interventional strategies in PTSD and HF. An example is the orexin/hypocretin system. (2) Studies modifying the common inflammatory pathways, such as MMP-9, IL-1β, and TGF, to determine whether there is an overlapping therapeutic strategy for treating patients with both HF and PTSD. A key question is if systemic inflammation is needed for adverse changes in the brain and heart. (3) The RAS is a common initiator for inflammation and behavioral changes, so is there a link between RAS-induced heart and brain inflammation and is this a therapeutic opportunity? (4) Studies determining the role of common changes in ECM, plaque formation, and tissue/synapse remodeling in PTSD and HF. (5) Studies exploring the role of points of communication and feedback loops between heart and brain in the progression of PTSD and HF. What is the role of vagal efferents/afferents in PTSD and HF? Are the CVOs or exosomes novel approaches for therapeutic strategies? (6) Develop new imaging paradigms that can detect protein buildup (such as plaques), remodeling, and common mediators in patients with both PTSD and HF.
Postulate five. Common modifiers exist in PTSD and HF progression that remain to be explored. (1) Studies examining the influence of age in PTSD and HF. How does age independently influence PTSD and HF progression, and their comorbidity? Are both PTSD and HF related to accelerated aging processes as a common mediator? (2) What are the influences of gender and gonadal hormones in the comorbidity and progression of PTSD and HF? (3) Many of the common mediators are impacted by obesity, diabetes, and metabolic disorders, so how do these conditions influence PTSD and HF?
Acknowledgements
We would like to thank Dr. Victoria Macht Preston for constructing the figure. Support for the meeting provided through a BLRD Veterans Administration Field Meeting Proposal awarded to the William Jennings Bryan Dorn VA Medicine Center (now the Columbia VA Health Care System, Columbia, SC).
Importantly for the field, many similar recommendations were recently posted from a workshop on “The Cardiovascular Consequences of Post-Traumatic Stress Disorder,” sponsored by the National Heart, Lung, and Blood Institute (NHLBI) in Bethesda, Maryland from November 13-14, 2018. See: https://www.nhlbi.nih.gov/events/2018/nhlbi-working-group-cardiovascular-consequences-post-traumatic-stress-disorder
Disclosure statement
Israel Liberzon – no COI to report. Government grants NIH, DoD, foundation grants – Cohen Veterans Bioscience, Industry – Sunovion Inc, ARMGO Pharm. Inc. None is related to cardiovascular/PTSD comorbidity. Other authors (MAW, MLL, YL, VBR, RS, SKW, JBW, FGS) have no disclosures to report.
Additional information
Funding
Notes on contributors
Marlene A. Wilson
Marlene A. Wilson is Chair and Professor of Pharmacology, Physiology and Neuroscience at the University of South Carolina School of Medicine and a Research Health Scientist in the Columbia VA Health Care System. She contributed her expertise in preclinical models of PTSD and other psychiatric disorders to this work, and served as PI of the VA Field Meeting Proposal that initiated this Brain-Heart Consortium meeting.
Israel Liberzon
Israel Liberzon is currently the Department Head of Psychiatry at Texas A&M College of Medicine, but previously founded the PTSD and neuroimaging research program at the University of Michigan and the VA Ann Arbor Medical Center. As a clinician and researcher, along with his student Yana Lokshina, he contributes his translational expertise in the neuroanatomy and neuroimaging PTSD and comorbid conditions.
Merry L. Lindsey
Merry L. Lindsey is the Stokes-Shackleford Professor and Chair of Cellular and Integrative Physiology at the University of Nebraska Medical Center. She is also Director of the Center for Heart and Vascular Research at UNMC and in the Research Service at the Omaha VA Medical Center. She contributed her extensive expertise in cardiac physiology, including the role of cardiac inflammation and extracellular matrix biology in heart failure and cardiovascular dysfunction.
Yana Lokshina
Merry L. Lindsey is the Stokes-Shackleford Professor and Chair of Cellular and Integrative Physiology at the University of Nebraska Medical Center. She is also Director of the Center for Heart and Vascular Research at UNMC and in the Research Service at the Omaha VA Medical Center. She contributed her extensive expertise in cardiac physiology, including the role of cardiac inflammation and extracellular matrix biology in heart failure and cardiovascular dysfunction.
Victoria B. Risbrough
Victoria B. Risbrough is a Professor in the Department of Psychiatry at the University of California San Diego, and serves as Co-Associate Director of the Neuroscience Research Unit at the VA Center of Excellence for Stress and Mental Health. She contributed her expertise on translational mechanisms and treatments of anxiety disorders using preclinical and clinical approaches, particular understanding mechanisms of risk and resilience to PTSD using preclinical models and clinical assessments in veterans.
Renu Sah
Renu Sah is an Associate Professor in the Department of Pharmacology and Systems Physiology at the University of Cincinnati College of Medicine and a Research Scientist at the Cincinnati VA Medical Center. She contributed expertise in the neurobiology of fear relevant to PTSD pathophysiology, particularly using translational approaches for investigating mechanisms underlying comorbidity, body-brain communications and contributory neuromodulators.
Susan K. Wood
Susan K. Wood is an Assistant Professor of Pharmacology, Physiology and Neuroscience at the University of South Carolina School of Medicine and is co-investigator on several VA Merit Awards. She contributed her expertise in cardiovascular dysregulation using preclinical models of PTSD and depression, particularly the impact of psychosocial stress in both males and females.
John B. Williamson
John B. Williamson is an Assistant Professor in the Departments of Psychiatry and Neuroscience at the University of Florida College of Medicine, and a Research Health Scientist at the North Florida-South Georgia VA Medical Center. He is an investigator in the Center for OCD and Anxiety Related Disorders, and serves as the Emotion Function Initiative Lead in the Brain Rehabilitation Research Center. He contributed expertise in the mechanisms of brain dysfunction in patients with PTSD and cerebrovascular disease, as well as sleep and autonomic functions.
Francis G. Spinale
Francis G. Spinale is the Associate Dean for Research and Graduate Education and Professor in the Department of Cell Biology and Anatomy at the University of South Carolina School of Medicine, as well as the Research Service at the Columbia VA Health Care System. He is a clinician scientist leading a translational research effort in cardiovascular remodeling, and contributed his expertise in heart failure and cardiovascular dysfunction, as well as co-organizing the Brain-Hearth Consortium meeting.
References
- Abreu, A.R., Molosh, A.I., Johnson, P.L., & Shekhar, A. (2018). Role of medial hypothalamic orexin system in panic, phobia and hypertension. Brain Research, doi:10.1016/j.brainres.2018.09.010
- Ahmadi, N., Hajsadeghi, F., Nabavi, V., Olango, G., Molla, M., Budoff, M., … Yehuda, R. (2018). The long-term clinical outcome of posttraumatic stress disorder with impaired coronary distensibility. Psychosomatic Medicine, 80, 294–300. doi:10.1097/PSY.0000000000000565
- Assari, S. (2014). Veterans and risk of heart disease in the United States: A cohort with 20 years of follow up. International Journal of Preventive Medicine, 5, 703–709.
- Berridge, C.W., Espana, R.A., & Vittoz, N.M. (2010). Hypocretin/orexin in arousal and stress. Brain Research, 1314, 91–102. doi:10.1016/j.brainres.2009.09.019
- Binici, Z., Mouridsen, M.R., Kober, L., & Sajadieh, A. (2011). Decreased nighttime heart rate variability is associated with increased stroke risk. Stroke, 42, 3196–3201. doi:10.1161/STROKEAHA.110.607697
- Blechert, J., Michael, T., Grossman, P., Lajtman, M., & Wilhelm, F.H. (2007). Autonomic and respiratory characteristics of posttraumatic stress disorder and panic disorder. Psychosomatic Medicine, 69, 935–943. doi:10.1097/PSY.0b013e31815a8f6b
- Boscarino, J.A. (2004). Posttraumatic stress disorder and physical illness: Results from clinical and epidemiologic studies. Annals of the New York Academy of Sciences, 1032, 141–153. doi:10.1196/annals.1314.011
- Boscarino, J.A. (2008). A prospective study of PTSD and early-age heart disease mortality among Vietnam veterans: Implications for surveillance and prevention. Psychosomatic Medicine, 70, 668–676. doi:10.1097/PSY.0b013e31817bccaf
- Boscarino, J.A., & Chang, J. (1999). Electrocardiogram abnormalities among men with stress-related psychiatric disorders: Implications for coronary heart disease and clinical research. Annals of Behavioral Medicine, 21, 227–234. doi:10.1007/BF02884839
- Bradley, T.D., Logan, A.G., Kimoff, R.J., Sériès, F., Morrison, D., Ferguson, K., … Floras, J.S. (2005). Continuous positive airway pressure for central sleep apnea and heart failure. New England Journal of Medicine, 353, 2025–2033. doi:10.1056/NEJMoa051001
- Breen, M.S., Tylee, D.S., Maihofer, A.X., Neylan, T.C., Mehta, D., Binder, E.B., … Glatt, S.J. (2018). PTSD blood transcriptome mega-analysis: shared inflammatory pathways across biological sex and modes of trauma. Neuropsychopharmacology, 43, 469–481. doi:10.1038/npp.2017.220
- Brudey, C., Park, J., Wiaderkiewicz, J., Kobayashi, I., Mellman, T.A., & Marvar, P.J. (2015). Autonomic and inflammatory consequences of posttraumatic stress disorder and the link to cardiovascular disease. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology, 309, R315–R321. doi:10.1152/ajpregu.00343.2014
- Bruijnzeel, A.W., Stam, R., Croiset, G., & Wiegant, V.M. (2001). Long-term sensitization of cardiovascular stress responses after a single stressful experience. Physiology & Behavior, 73, 81–86. doi:10.1016/S0031-9384(01)00435-8
- Buckley, T.C., & Kaloupek, D.G. (2001). A meta-analytic examination of basal cardiovascular activity in posttraumatic stress disorder. Psychosomatic Medicine, 63, 585–594. doi:10.1097/00006842-200107000-00011
- Calva, C.B., Fayyaz, H., & Fadel, J.R. (2018). Increased acetylcholine and glutamate efflux in the prefrontal cortex following intranasal orexin-A (hypocretin-1). Journal of Neurochemistry, 145, 232–244. doi:10.1111/jnc.14279
- Cameron, V.A., Mocatta, T.J., Pilbrow, A.P., Frampton, C.M., Troughton, R.W., Richards, A.M., & Winterbourn, C.C. (2006). Angiotensin type-1 receptor A1166C gene polymorphism correlates with oxidative stress levels in human heart failure. Hypertension, 47, 1155–1161. doi:10.1161/01.HYP.0000222893.85662.cd
- Carnevali, L., Trombini, M., Rossi, S., Graiani, G., Manghi, M., Koolhaas, J.M., … Sgoifo, A. (2013). Structural and electrical myocardial remodeling in a rodent model of depression. Psychosomatic Medicine, 75, 42–51. doi:10.1097/PSY.0b013e318276cb0d
- Carrive, P. (2017). Orexin, stress and central cardiovascular control. A link with hypertension? Neuroscience & Biobehavioral Reviews, 74, 376–392. doi:10.1016/j.neubiorev.2016.06.044
- Carter, M.E., Schaich Borg, J., & de Lecea, L. (2009). The brain hypocretins and their receptors: Mediators of allostatic arousal. Current Opinion in Pharmacology, 9, 39–45. doi:10.1016/j.coph.2008.12.018
- Clausen, A.N., Aupperle, R.L., Sisante, J.F., Wilson, D.R., & Billinger, S.A. (2016). Pilot investigation of PTSD, autonomic reactivity, and cardiovascular health in physically healthy combat veterans. PLoS One, 11, e0162547. doi:10.1371/journal.pone.0162547
- Cohen, B.E., Edmondson, D., & Kronish, I.M. (2015). State of the art review: Depression, stress, anxiety, and cardiovascular disease. American Journal of Hypertension, doi:10.1093/ajh/hpv047
- Cohen, S., Ifergane, G., Vainer, E., Matar, M.A., Kaplan, Z., Zohar, J., … Cohen, H. (2016). The wake-promoting drug modafinil stimulates specific hypothalamic circuits to promote adaptive stress responses in an animal model of PTSD. Translational Psychiatry, 6, e917. doi:10.1038/tp.2016.172
- Cohen, B.E., Marmar, C., Ren, L., Bertenthal, D., & Seal, K.H. (2009). Association of cardiovascular risk factors with mental health diagnoses in Iraq and Afghanistan war veterans using VA health care. JAMA, 302, 489–492. doi:10.1001/jama.2009.1084
- Costanzo, M.R., Ponikowski, P., Javaheri, S., Augostini, R., Goldberg, L., Holcomb, R., … Abraham, W.T. (2016). Transvenous neurostimulation for central sleep apnoea: A randomised controlled trial. Lancet, 388, 974–982. doi:10.1016/S0140-6736(16)30961-8
- Crestani, C.C. (2016). Emotional stress and cardiovascular complications in animal models: A review of the influence of stress type. Frontiers in Physiology, 7, 251. doi:10.3389/fphys.2016.00251
- Crum-Cianflone, N.F., Bagnell, M.E., Schaller, E., Boyko, E.J., Smith, B., Maynard, C., … Smith, T.C. (2014). Impact of combat deployment and posttraumatic stress disorder on newly reported coronary heart disease among US active duty and reserve forces. Circulation, 129, 1813–1820. doi:10.1161/CIRCULATIONAHA.113.005407
- DeLeon-Pennell, K.Y., Meschiari, C.A., Jung, M., & Lindsey, M.L. (2017). Matrix metalloproteinases in myocardial infarction and heart failure. Progress in Molecular Biology and Translational Science, 147, 75–100. doi:10.1016/bs.pmbts.2017.02.001
- Deslauriers, J., Acheson, D.T., Maihofer, A.X., Nievergelt, C.M., Baker, D.G., Geyer, M.A., & Risbrough, V.B. (2018). COMT val158met polymorphism links to altered fear conditioning and extinction are modulated by PTSD and childhood trauma. Depression and Anxiety, 35, 32–42. doi:10.1002/da.22678
- Deslauriers, J., Powell, S., & Risbrough, V.B. (2017). Immune signaling mechanisms of PTSD risk and symptom development: Insights from animal models. Current Opinion in Behavioral Sciences, 14, 123–132. doi:10.1016/j.cobeha.2017.01.005
- Deslauriers, J., Toth, M., Der-Avakian, A., & Risbrough, V.B. (2018). Current status of animal models of posttraumatic stress disorder: Behavioral and biological phenotypes, and future challenges in improving translation. Biological Psychiatry, 83, 895–907. doi:10.1016/j.biopsych.2017.11.019
- Drager, L.F., McEvoy, R.D., Barbe, F., Lorenzi-Filho, G., Redline, S., & Initiative, I. (2017). Sleep apnea and cardiovascular disease: Lessons from recent trials and need for team science. Circulation, 136, 1840–1850. doi:10.1161/CIRCULATIONAHA.117.029400
- Edmondson, D., & Cohen, B.E. (2013). Posttraumatic stress disorder and cardiovascular disease. Progress in Cardiovascular Diseases, 55, 548–556. doi:10.1016/j.pcad.2013.03.004
- Edmondson, D., Kronish, I.M., Shaffer, J.A., Falzon, L., & Burg, M.M. (2013). Posttraumatic stress disorder and risk for coronary heart disease: A meta-analytic review. American Heart Journal, 166, 806–814. doi:10.1016/j.ahj.2013.07.031
- Edmondson, D., Richardson, S., Falzon, L., Davidson, K.W., Mills, M.A., & Neria, Y. (2012). Posttraumatic stress disorder prevalence and risk of recurrence in acute coronary syndrome patients: A meta-analytic review. PLoS One, 7, e38915. doi:10.1371/journal.pone.0038915
- El-Solh, A.A., Riaz, U., & Roberts, J. (2018). Sleep disorders in patients with posttraumatic stress disorder. Chest, 154, 427–439. doi:10.1016/j.chest.2018.04.007
- Eraly, S.A., Nievergelt, C.M., Maihofer, A.X., Barkauskas, D.A., Biswas, N., Agorastos, A., … Baker, D.G. (2014). Assessment of plasma C-reactive protein as a biomarker of posttraumatic stress disorder risk. JAMA Psychiatry, 71, 423–431. doi:10.1001/jamapsychiatry.2013.4374
- Ezekowitz, J.A., Kaul, P., Bakal, J.A., Armstrong, P.W., Welsh, R.C., & McAlister, F.A. (2009). Declining in-hospital mortality and increasing heart failure incidence in elderly patients with first myocardial infarction. Journal of the American College of Cardiology, 53, 13–20. doi:10.1016/j.jacc.2008.08.067
- Fadel, J., & Burk, J.A. (2010). Orexin/hypocretin modulation of the basal forebrain cholinergic system: Role in attention. Brain Research, 1314, 112–123. doi:10.1016/j.brainres.2009.08.046
- Ferguson, A.V. (2014). Circumventricular organs: Integrators of circulating signals controlling hydration, energy balance, and immune function. In L.A. De Luca, Jr., J.V. Menani, & A.K. Johnson (Eds.), Neurobiology of body fluid homeostasis: Transduction and integration. Boca Raton (FL): CRC Press.
- Finnell, J.E., Lombard, C.M., Padi, A.R., Moffitt, C.M., Wilson, L.B., Wood, C.S., & Wood, S.K. (2017). Physical versus psychological social stress in male rats reveals distinct cardiovascular, inflammatory and behavioral consequences. PLoS One, 12, e0172868. doi:10.1371/journal.pone.0172868
- Finnell, J.E., Muniz, B.L., Padi, A.R., Lombard, C.M., Moffitt, C.M., Wood, C.S., … Wood, S.K. (2018). Essential role of ovarian hormones in susceptibility to the consequences of witnessing social defeat in female rats. Biological Psychiatry, 84, 372–382. doi:10.1016/j.biopsych.2018.01.013
- Flores, A., Saravia, R., Maldonado, R., & Berrendero, F. (2015). Orexins and fear: Implications for the treatment of anxiety disorders. Trends in Neurosciences, 38, 550–559. doi:10.1016/j.tins.2015.06.005
- Frangogiannis, N.G. (2017). The extracellular matrix in myocardial injury, repair, and remodeling. Journal of Clinical Investigation, 127, 1600–1612. doi:10.1172/JCI87491
- Frasure-Smith, N., Lesperance, F., Irwin, M.R., Talajic, M., & Pollock, B.G. (2009). The relationships among heart rate variability, inflammatory markers and depression in coronary heart disease patients. Brain, Behavior, and Immunity, 23, 1140–1147. doi:10.1016/j.bbi.2009.07.005
- Fudim, M., Cerbin, L.P., Devaraj, S., Ajam, T., Rao, S.V., & Kamalesh, M. (2018). Post-traumatic stress disorder and heart failure in men within the veteran affairs health system. The American Journal of Cardiology, 122, 275–278. doi:10.1016/j.amjcard.2018.04.007
- Furlong, T., & Carrive, P. (2007). Neurotoxic lesions centered on the perifornical hypothalamus abolish the cardiovascular and behavioral responses of conditioned fear to context but not of restraint. Brain Research, 1128, 107–119. doi:10.1016/j.brainres.2006.10.058
- Furlong, T.M., Vianna, D.M., Liu, L., & Carrive, P. (2009). Hypocretin/orexin contributes to the expression of some but not all forms of stress and arousal. European Journal of Neuroscience, 30, 1603–1614. doi:10.1111/j.1460-9568.2009.06952.x
- Gander, M.L., & von Kanel, R. (2006). Myocardial infarction and post-traumatic stress disorder: Frequency, outcome, and atherosclerotic mechanisms. The European Journal of Cardiovascular Prevention & Rehabilitation, 13, 165–172. doi:10.1097/01.hjr.0000214606.60995.46
- Gill, J., Mustapic, M., Diaz-Arrastia, R., Lange, R., Gulyani, S., Diehl, T., & Kapogiannis, D. (2018). Higher exosomal tau, amyloid-beta 42 and IL-10 are associated with mild TBIs and chronic symptoms in military personnel. Brain Injury, 32, 1277–1284. doi:10.1080/02699052.2018.1471738
- Gill, J., Vythilingam, M., & Page, G.G. (2008). Low cortisol, high DHEA, and high levels of stimulated TNF-alpha, and IL-6 in women with PTSD. Journal of Traumatic Stress, 21, 530–539. doi:10.1002/jts.20372
- Goldstein, R.B., Smith, S.M., Chou, S.P., Saha, T.D., Jung, J., Zhang, H., & Grant, B.F. (2016). The epidemiology of DSM-5 posttraumatic stress disorder in the United States: Results from the National Epidemiologic Survey on Alcohol and Related Conditions-III. Social Psychiatry and Psychiatric Epidemiology, 51, 1137–1148. doi:10.1007/s00127-016-1208-5
- Grabe, H.J., Spitzer, C., Schwahn, C., Marcinek, A., Frahnow, A., Barnow, S., … Rosskopf, D. (2009). Serotonin transporter gene (SLC6A4) promoter polymorphisms and the susceptibility to posttraumatic stress disorder in the general population. American Journal of Psychiatry, 166, 926–933. doi:10.1176/appi.ajp.2009.08101542
- Grafe, L.A., Eacret, D., Dobkin, J., & Bhatnagar, S. (2018). Reduced orexin system function contributes to resilience to repeated social stress. eNeuro, 5. doi:10.1523/ENEURO.0273-17.2018
- Grassi, G., Seravalle, G., & Mancia, G. (2015). Sympathetic activation in cardiovascular disease: Evidence, clinical impact and therapeutic implications. European Journal of Clinical Investigation, 45, 1367–1375. doi:10.1111/eci.12553
- Green, K.T., Dennis, P.A., Neal, L.C., Hobkirk, A.L., Hicks, T.A., Watkins, L.L., … Beckham, J.C. (2016). Exploring the relationship between posttraumatic stress disorder symptoms and momentary heart rate variability. Journal of Psychosomatic Research, 82, 31–34. doi:10.1016/j.jpsychores.2016.01.003
- Grimaldi, D., Silvani, A., Benarroch, E.E., & Cortelli, P. (2014). Orexin/hypocretin system and autonomic control: New insights and clinical correlations. Neurology, 82, 271–278. doi:10.1212/WNL.0000000000000045
- Hayward, L.F., Hampton, E.E., Ferreira, L.F., Christou, D.D., Yoo, J.K., Hernandez, M.E., & Martin, E.J. (2015). Chronic heart failure alters orexin and melanin concentrating hormone but not corticotrophin releasing hormone-related gene expression in the brain of male Lewis rats. Neuropeptides, 52, 67–72. doi:10.1016/j.npep.2015.06.001
- Hendrickson, R.C., & Raskind, M.A. (2016). Noradrenergic dysregulation in the pathophysiology of PTSD. Experimental Neurology, 284, 181–195. doi:10.1016/j.expneurol.2016.05.014
- Hill, M.N., Campolongo, P., Yehuda, R., & Patel, S. (2018). Integrating endocannabinoid signaling and cannabinoids into the biology and treatment of posttraumatic stress disorder. Neuropsychopharmacology, 43, 80–102. doi:10.1038/npp.2017.162
- Ho, F.Y., Chan, C.S., & Tang, K.N. (2016). Cognitive-behavioral therapy for sleep disturbances in treating posttraumatic stress disorder symptoms: A meta-analysis of randomized controlled trials. Clinical Psychology Review, 43, 90–102. doi:10.1016/j.cpr.2015.09.005
- Horgan, S., Watson, C., Glezeva, N., & Baugh, J. (2014). Murine models of diastolic dysfunction and heart failure with preserved ejection fraction. Journal of Cardiac Failure, 20, 984–995. doi:10.1016/j.cardfail.2014.09.001
- Houser, S.R., Margulies, K.B., Murphy, A.M., Spinale, F.G., Francis, G.S., Prabhu, S.D., … Koch, W.J. (2012). Animal models of heart failure: A scientific statement from the American Heart Association. Circulation Research, 111, 131–150. doi:10.1161/RES.0b013e3182582523
- Ibrahim, N.E., Rabideau, D.J., Gaggin, H.K., Belcher, A.M., Conrad, M.J., Jarolim, P., & Januzzi, J.L., Jr. (2016). Circulating concentrations of orexin a predict left ventricular myocardial remodeling. Journal of the American College of Cardiology, 68, 2238–2240. doi:10.1016/j.jacc.2016.08.049
- Jiang, R., Babyak, M.A., Brummett, B.H., Hauser, E.R., Shah, S.H., Becker, R.C., … Williams, R.B. (2017). Brain-derived neurotrophic factor rs6265 (Val66Met) polymorphism is associated with disease severity and incidence of cardiovascular events in a patient cohort. American Heart Journal, 190, 40–45. doi:10.1016/j.ahj.2017.05.002
- Jin, J., & Maren, S. (2015). Prefrontal-hippocampal interactions in memory and emotion. Frontiers in Systems Neuroscience, 9, 170. doi:10.3389/fnsys.2015.00170
- Jordan, H.T., Stellman, S.D., Morabia, A., Miller-Archie, S.A., Alper, H., Laskaris, Z., & Cone, J.E. (2013). Cardiovascular disease hospitalizations in relation to exposure to the September 11, 2001 World Trade Center disaster and posttraumatic stress disorder. Journal of the American Heart Association, 2, e000431. doi:10.1161/JAHA.113.000431
- Jovanovic, T., Kazama, A., Bachevalier, J., & Davis, M. (2012). Impaired safety signal learning may be a biomarker of PTSD. Neuropharmacology, 62, 695–704. doi:10.1016/j.neuropharm.2011.02.023
- Jung, D.W., Lee, Y.J., Jeong, D.U., & Park, K.S. (2017). New predictors of sleep efficiency. Chronobiology International, 34, 93–104. doi:10.1080/07420528.2016.1241802
- Kang, H.K., Natelson, B.H., Mahan, C.M., Lee, K.Y., & Murphy, F.M. (2003). Post-traumatic stress disorder and chronic fatigue syndrome-like illness among Gulf War veterans: A population-based survey of 30,000 veterans. American Journal of Epidemiology, 157, 141–148. doi:10.1093/aje/kwf187
- Keary, T.A., Hughes, J.W., & Palmieri, P.A. (2009). Women with posttraumatic stress disorder have larger decreases in heart rate variability during stress tasks. International Journal of Psychophysiology, 73, 257–264. doi:10.1016/j.ijpsycho.2009.04.003
- Kessler, R.C., Berglund, P., Demler, O., Jin, R., Merikangas, K.R., & Walters, E.E. (2005). Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Archives of General Psychiatry, 62, 593–602. doi:10.1001/archpsyc.62.6.593
- Kessler, R.C., Chiu, W.T., Demler, O., Merikangas, K.R., & Walters, E.E. (2005). Prevalence, severity, and comorbidity of 12-month DSM-IV disorders in the National Comorbidity Survey Replication. Archives of General Psychiatry, 62, 617–627. doi:10.1001/archpsyc.62.6.617
- Kibler, J.L., Tursich, M., Ma, M., Malcolm, L., & Greenbarg, R. (2014). Metabolic, autonomic and immune markers for cardiovascular disease in posttraumatic stress disorder. World Journal of Cardiology, 6, 455–461. doi:10.4330/wjc.v6.i6.455
- Kobayashi, I., Boarts, J.M., & Delahanty, D.L. (2007). Polysomnographically measured sleep abnormalities in PTSD: A meta-analytic review. Psychophysiology, 44, 660–669. doi:10.1111/j.1469-8986.2007.537.x
- Koresh, O., Kaplan, Z., Zohar, J., Matar, M.A., Geva, A.B., & Cohen, H. (2016). Distinctive cardiac autonomic dysfunction following stress exposure in both sexes in an animal model of PTSD. Behavioural Brain Research, 308, 128–142. doi:10.1016/j.bbr.2016.04.024
- Kotecha, D., New, G., Flather, M.D., Eccleston, D., Pepper, J., & Krum, H. (2012). Five-minute heart rate variability can predict obstructive angiographic coronary disease. Heart, 98, 395–401. doi:10.1136/heartjnl-2011-300033
- Krishnamurthi, N., Francis, J., Fihn, S.D., Meyer, C.S., & Whooley, M.A. (2018). Leading causes of cardiovascular hospitalization in 8.45 million US veterans. PLoS One, 13, e0193996. doi:10.1371/journal.pone.0193996
- Krystal, J.H., Abdallah, C.G., Pietrzak, R.H., Averill, L.A., Harpaz-Rotem, I., Levy, I., … Southwick, S.M. (2018). Locus coeruleus hyperactivity in posttraumatic stress disorder: Answers and questions. Biological Psychiatry, 83, 197–199. doi:10.1016/j.biopsych.2017.09.027
- Kubzansky, L.D., Koenen, K.C., Spiro, A., III, Vokonas, P.S., & Sparrow, D. (2007). Prospective study of posttraumatic stress disorder symptoms and coronary heart disease in the Normative Aging Study. Archives of General Psychiatry, 64, 109–116. doi:10.1001/archpsyc.64.1.109
- Laukkanen, J.A., Makikallio, T.H., Kauhanen, J., & Kurl, S. (2009). Insertion/deletion polymorphism in alpha2-adrenergic receptor gene is a genetic risk factor for sudden cardiac death. American Heart Journal, 158, 615–621. doi:10.1016/j.ahj.2009.07.023
- Laukova, M., Tillinger, A., Novakova, M., Krizanova, O., Kvetnansky, R., & Myslivecek, J. (2014). Repeated immobilization stress increases expression of beta3-adrenoceptor in the left ventricle and atrium of the rat heart. Stress and Health, 30, 301–309. doi:10.1002/smi.2515
- Lazuko, S.S., Kuzhel, O.P., Belyaeva, L.E., Manukhina, E.B., Fred Downey, H., Tseilikman, O.B., … Tseilikman, V.E. (2018). Posttraumatic stress disorder disturbs coronary tone and its regulatory mechanisms. Cellular and Molecular Neurobiology, 38, 209–217. doi:10.1007/s10571-017-0517-x
- Lebois, L.A.M., Wolff, J.D., & Ressler, K.J. (2016). Neuroimaging genetic approaches to posttraumatic stress disorder. Experimental Neurology, 284, 141–152. doi:10.1016/j.expneurol.2016.04.019
- Liberzon, I., & Abelson, J.L. (2016). Context processing and the neurobiology of post-traumatic stress disorder. Neuron, 92, 14–30. doi:10.1016/j.neuron.2016.09.039
- Liberzon, I., Abelson, J.L., Flagel, S.B., Raz, J., & Young, E.A. (1999). Neuroendocrine and psychophysiologic responses in PTSD: A symptom provocation study. Neuropsychopharmacology, 21, 40–50. doi:10.1016/S0893-133X(98)00128-6
- Liberzon, I., King, A.P., Ressler, K.J., Almli, L.M., Zhang, P., Ma, S.T., … Galea, S. (2014). Interaction of the ADRB2 gene polymorphism with childhood trauma in predicting adult symptoms of posttraumatic stress disorder. JAMA Psychiatry, 71, 1174–1182. doi:10.1001/jamapsychiatry.2014.999
- Lima, B.B., Hammadah, M., Wilmot, K., Pearce, B.D., Shah, A., Levantsevych, O., & Vaccarino, V. (2018). Posttraumatic stress disorder is associated with enhanced interleukin-6 response to mental stress in subjects with a recent myocardial infarction. Brain, Behavior, and Immunity, 75, 26–33. doi:10.1016/j.bbi.2018.08.015
- Lindqvist, D., Dhabhar, F.S., Mellon, S.H., Yehuda, R., Grenon, S.M., Flory, J.D., … Wolkowitz, O.M. (2017). Increased pro-inflammatory milieu in combat related PTSD – A new cohort replication study. Brain, Behavior, and Immunity, 59, 260–264. doi:10.1016/j.bbi.2016.09.012
- Lindsey, M.L. (2018). Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nature Reviews Cardiology, 15, 471–479. doi:10.1038/s41569-018-0022-z
- Lindsey, M.L., Bolli, R., Canty, J.M., Du, X.-J., Frangogiannis, N.G., Frantz, S., … Heusch, G. (2018). Guidelines for experimental models of myocardial ischemia and infarction. American Journal of Physiology-Heart and Circulatory Physiology, 314, H812–H838. doi:10.1152/ajpheart.00335.2017
- Lisieski, M.J., Eagle, A.L., Conti, A.C., Liberzon, I., & Perrine, S.A. (2018). Single-prolonged stress: A review of two decades of progress in a rodent model of post-traumatic stress disorder. Front Psychiatry, 9, 196. doi:10.3389/fpsyt.2018.00196
- Liu, M., Xu, F., Tao, T., Song, D., Li, D., Li, Y., … Liu, X. (2016). Molecular mechanisms of stress-induced myocardial injury in a rat model simulating posttraumatic stress disorder. Psychosomatic Medicine, 78, 888–895. doi:10.1097/PSY.0000000000000353
- Lovallo, W.R., Enoch, M.-A., Acheson, A., Cohoon, A.J., Sorocco, K.H., Hodgkinson, C.A., … Goldman, D. (2016). Early-life adversity interacts with FKBP5 genotypes: altered working memory and cardiac stress reactivity in the Oklahoma Family Health Patterns Project. Neuropsychopharmacology, 41, 1724–1732. doi:10.1038/npp.2015.347
- Mahler, S.V., Moorman, D.E., Smith, R.J., James, M.H., & Aston-Jones, G. (2014). Motivational activation: A unifying hypothesis of orexin/hypocretin function. Nature Neuroscience, 17, 1298–1303. doi:10.1038/nn.3810
- Marques, F.Z., Eikelis, N., Bayles, R.G., Lambert, E.A., Straznicky, N.E., Hering, D., … Lambert, G.W. (2017). A polymorphism in the norepinephrine transporter gene is associated with affective and cardiovascular disease through a microRNA mechanism. Molecular Psychiatry, 22, 134–141. doi:10.1038/mp.2016.40
- McFall, M.E., Murburg, M.M., Ko, G.N., & Veith, R.C. (1990). Autonomic responses to stress in Vietnam combat veterans with posttraumatic stress disorder. Biological Psychiatry, 27, 1165–1175. doi:10.1016/0006-3223(90)90053-5
- Mehta, D., Voisey, J., Bruenig, D., Harvey, W., Morris, C.P., Lawford, B., & Young, R.M. (2018). Transcriptome analysis reveals novel genes and immune networks dysregulated in veterans with PTSD. Brain, Behavior, and Immunity, 74, 133–142. doi:10.1016/j.bbi.2018.08.014
- Miller, M.W., McKinney, A.E., Kanter, F.S., Korte, K.J., & Lovallo, W.R. (2011). Hydrocortisone suppression of the fear-potentiated startle response and posttraumatic stress disorder. Psychoneuroendocrinology, 36, 970–980. doi:10.1016/j.psyneuen.2010.12.009
- Minassian, A., Geyer, M.A., Baker, D.G., Nievergelt, C.M., O'Connor, D.T., Risbrough, V.B., & Marine Resiliency Study, T. (2014). Heart rate variability characteristics in a large group of active-duty marines and relationship to posttraumatic stress. Psychosomatic Medicine, 76, 292–301. doi:10.1097/PSY.0000000000000056
- Minassian, A., Maihofer, A.X., Baker, D.G., Nievergelt, C.M., Geyer, M.A., Risbrough, V.B., & Marine, R.S.T. (2015). Association of predeployment heart rate variability with risk of postdeployment posttraumatic stress disorder in active-duty marines. JAMA Psychiatry, 72, 979–986. doi:10.1001/jamapsychiatry.2015.0922
- Mir, R., Bhat, M., Javid, J., Jha, C., Saxena, A., & Banu, S. (2018). Potential impact of COMT-rs4680G > A gene polymorphism in coronary artery disease. Journal of Cardiovascular Development and Disease, 5, pii: E38. doi:10.3390/jcdd5030038
- Monfils, M.H., Lee, H.J., Keller, N.E., Roquet, R.F., Quevedo, S., Agee, L., & Shumake, J. (2018). Predicting extinction phenotype to optimize fear reduction. Psychopharmacology (Berlin), 236, 99–110. doi:10.1007/s00213-018-5005-6
- Naegeli, C., Zeffiro, T., Piccirelli, M., Jaillard, A., Weilenmann, A., Hassanpour, K., … Mueller-Pfeiffer, C. (2018). Locus coeruleus activity mediates hyperresponsiveness in posttraumatic stress disorder. Biological Psychiatry, 83, 254–262. doi:10.1016/j.biopsych.2017.08.021
- Najafi, A., Sequeira, V., Kuster, D.W., & van der Velden, J. (2016). Beta-adrenergic receptor signalling and its functional consequences in the diseased heart. European Journal of Clinical Investigation, 46, 362–374. doi:10.1111/eci.12598
- Norman, G.J., Morris, J.S., Karelina, K., Weil, Z.M., Zhang, N., Al-Abed, Y., … DeVries, A.C. (2011). Cardiopulmonary arrest and resuscitation disrupts cholinergic anti-inflammatory processes: A role for cholinergic alpha7 nicotinic receptors. The Journal of Neuroscience, 31, 3446–3452. doi:10.1523/JNEUROSCI.4558-10.2011
- Novaco, R.W., & Chemtob, C.M. (2002). Anger and combat-related posttraumatic stress disorder. Journal of Traumatic Stress, 15, 123–132. doi:10.1023/A:1014855924072
- Nylocks, K.M., Michopoulos, V., Rothbaum, A.O., Almli, L., Gillespie, C.F., Wingo, A., & Ressler, K.J. (2015). An angiotensin-converting enzyme (ACE) polymorphism may mitigate the effects of angiotensin-pathway medications on posttraumatic stress symptoms. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 168B, 307–315. doi:10.1002/ajmg.b.32313
- Olofsson, P.S., Rosas-Ballina, M., Levine, Y.A., & Tracey, K.J. (2012). Rethinking inflammation: Neural circuits in the regulation of immunity. Immunological Reviews, 248, 188–204. doi:10.1111/j.1600-065X.2012.01138.x
- Pacher, P., Steffens, S., Hasko, G., Schindler, T.H., & Kunos, G. (2017). Cardiovascular effects of marijuana and synthetic cannabinoids: The good, the bad, and the ugly. Nature Reviews Cardiology, 15, 151–166. doi:10.1038/nrcardio.2017.130
- Pak, V.M., Strouss, L., Yaggi, H.K., Redeker, N.S., Mohsenin, V., & Riegel, B. (2018). Mechanisms of reduced sleepiness symptoms in heart failure and obstructive sleep apnea. Journal of Sleep Research, e12778. doi:10.1111/jsr.12778
- Parati, G., & Esler, M. (2012). The human sympathetic nervous system: Its relevance in hypertension and heart failure. European Heart Journal, 33, 1058–1066. doi:10.1093/eurheartj/ehs041
- Paulus, E.J., Argo, T.R., & Egge, J.A. (2013). The impact of posttraumatic stress disorder on blood pressure and heart rate in a veteran population. Journal of Traumatic Stress, 26, 169–172. doi:10.1002/jts.21785
- Penna, L.B., & Bassani, R.A. (2010). Increased spontaneous activity and reduced inotropic response to catecholamines in ventricular myocytes from footshock-stressed rats. Stress, 13, 73–82. doi:10.3109/10253890902951778
- Perez, M.V., Pavlovic, A., Shang, C., Wheeler, M.T., Miller, C.L., Liu, J., … Ashley, E.A. (2015). Systems genomics identifies a key role for hypocretin/orexin receptor-2 in human heart failure. Journal of the American College of Cardiology, 66, 2522–2533. doi:10.1016/j.jacc.2015.09.061
- Perusini, J.N., Meyer, E.M., Long, V.A., Rau, V., Nocera, N., Avershal, J., & Fanselow, M.S. (2015). Induction and expression of fear sensitization caused by acute traumatic stress. Neuropsychopharmacology, 41, 45–57. doi:10.1038/npp.2015.224
- Pietrzak, R.H., Sumner, J.A., Aiello, A.E., Uddin, M., Neumeister, A., Guffanti, G., & Koenen, K.C. (2015). Association of the rs2242446 polymorphism in the norepinephrine transporter gene SLC6A2 and anxious arousal symptoms of posttraumatic stress disorder. The Journal of Clinical Psychiatry, 76, e537–e538. doi:10.4088/JCP.14l09346
- Pitman, R.K., Rasmusson, A.M., Koenen, K.C., Shin, L.M., Orr, S.P., Gilbertson, M.W., … Liberzon, I. (2012). Biological studies of post-traumatic stress disorder. Nature Reviews Neuroscience, 13, 769–787. doi:10.1038/nrn3339
- Pole, N. (2007). The psychophysiology of posttraumatic stress disorder: A meta-analysis. Psychological Bulletin, 133, 725–746. doi:10.1037/0033-2909.133.5.725
- Porges, S.W., Doussard-Roosevelt, J.A., Stifter, C.A., McClenny, B.D., & Riniolo, T.C. (1999). Sleep state and vagal regulation of heart period patterns in the human newborn: An extension of the polyvagal theory. Psychophysiology, 36, 14–21. doi:10.1017/S004857729997035X
- Powell, D.A., Chachich, M., Murphy, V., McLaughlin, J., Tebbutt, D., & Buchanan, S.L. (1997). Amygdala–prefrontal interactions and conditioned bradycardia in the rabbit. Behavioral Neuroscience, 111, 1056–1074. doi:10.1037//0735-7044.111.5.1056
- Raynolds, M.V., Bristow, M.R., Bush, E.W., Abraham, W.T., Lowes, B.D., Zisman, L.S., & Perryman, M.B. (1993). Angiotensin-converting enzyme DD genotype in patients with ischaemic or idiopathic dilated cardiomyopathy. Lancet, 342, 1073–1075. doi:10.1016/0140-6736(93)92061-W
- Reardon, A.F., Hein, C.L., Wolf, E.J., Prince, L.B., Ryabchenko, K., & Miller, M.W. (2014). Intermittent explosive disorder: Associations with PTSD and other axis I disorders in a US military veteran sample. Journal of Anxiety Disorders, 28, 488–494. doi:10.1016/j.janxdis.2014.05.001
- Redeker, N.S., Conley, S., Anderson, G., Cline, J., Andrews, L., Mohsenin, V., & Jeon, S. (2018). Effects of cognitive behavioral therapy for insomnia on sleep, symptoms, stress, and autonomic function among patients with heart failure. Behavioral Sleep Medicine, 21, 1–13. doi:10.1080/15402002.2018.1546709
- Rorabaugh, B.R., Krivenko, A., Eisenmann, E.D., Bui, A.D., Seeley, S., Fry, M.E., … Zoladz, P.R. (2015). Sex-dependent effects of chronic psychosocial stress on myocardial sensitivity to ischemic injury. Stress, 18, 645–653. doi:10.3109/10253890.2015.1087505
- Roy, S.S., Foraker, R.E., Girton, R.A., & Mansfield, A.J. (2015). Posttraumatic stress disorder and incident heart failure among a community-based sample of US veterans. American Journal of Public Health, 105, 757–763. doi:10.2105/AJPH.2014.302342
- Sack, M., Hopper, J.W., & Lamprecht, F. (2004). Low respiratory sinus arrhythmia and prolonged psychophysiological arousal in posttraumatic stress disorder: Heart rate dynamics and individual differences in arousal regulation. Biological Psychiatry, 55, 284–290. doi:10.1016/S0006-3223(03)00677-2
- Sakurai, T. (2007). The neural circuit of orexin (hypocretin): Maintaining sleep and wakefulness. Nature Reviews Neuroscience, 8, 171–181. doi:10.1038/nrn2092
- Samuels, E.R., & Szabadi, E. (2008). Functional neuroanatomy of the noradrenergic locus coeruleus: Its roles in the regulation of arousal and autonomic function. Part I: Principles of functional organisation. Current Neuropharmacology, 6, 235–253. doi:10.2174/157015908785777229
- Schonbeck, U., Mach, F., & Libby, P. (1998). Generation of biologically active IL-1 beta by matrix metalloproteinases: A novel caspase-1-independent pathway of IL-1 beta processing. Journal of Immunology, 161, 3340–3346.
- Sears, R.M., Fink, A.E., Wigestrand, M.B., Farb, C.R., de Lecea, L., & Ledoux, J.E. (2013). Orexin/hypocretin system modulates amygdala-dependent threat learning through the locus coeruleus. Proceedings of the National Academy of Sciences of the United States of America, 110, 20260–20265. doi:10.1073/pnas.1320325110
- Shah, A.J., Lampert, R., Goldberg, J., Veledar, E., Bremner, J.D., & Vaccarino, V. (2013). Posttraumatic stress disorder and impaired autonomic modulation in male twins. Biological Psychiatry, 73, 1103–1110. doi:10.1016/j.biopsych.2013.01.019
- Shanmuganathan, M., Vughs, J., Noseda, M., & Emanueli, C. (2018). Exosomes: Basic biology and technological advancements suggesting their potential as ischemic heart disease therapeutics. Frontiers in Physiology, 9, 1159. doi:10.3389/fphys.2018.01159
- Sharko, A.C., Fadel, J.R., Kaigler, K.F., & Wilson, M.A. (2017). Activation of orexin/hypocretin neurons is associated with individual differences in cued fear extinction. Physiology & Behavior, 178, 93–102. doi:10.1016/j.physbeh.2016.10.008
- Shemesh, E., Yehuda, R., Milo, O., Dinur, I., Rudnick, A., Vered, Z., & Cotter, G. (2004). Posttraumatic stress, nonadherence, and adverse outcome in survivors of a myocardial infarction. Psychosomatic Medicine, 66, 521–526. doi:10.1097/01.psy.0000126199.05189.86
- Silvani, A., Bastianini, S., Berteotti, C., Lo Martire, V., & Zoccoli, G. (2013). Treating hypertension by targeting orexin receptors: Potential effects on the sleep-related blood pressure dipping profile. The Journal of Physiology, 591, 6115–6116. doi:10.1113/jphysiol.2013.265504
- Slavikova, J., Kuncova, J., & Topolcan, O. (2007). Plasma catecholamines and ischemic heart disease. Clinical Cardiology, 30, 326–330. doi:10.1002/clc.20099
- Sloan, R.P., Bagiella, E., Shapiro, P.A., Kuhl, J.P., Chernikhova, D., Berg, J., & Myers, M.M. (2001). Hostility, gender, and cardiac autonomic control. Psychosomatic Medicine, 63, 434–440. doi:10.1097/00006842-200105000-00012
- Snapir, A., Mikkelsson, J., Perola, M., Penttila, A., Scheinin, M., & Karhunen, P.J. (2003). Variation in the alpha2B-adrenoceptor gene as a risk factor for prehospital fatal myocardial infarction and sudden cardiac death. Journal of the American College of Cardiology, 41, 190–194. doi:10.1016/S0735-1097(02)02702-X
- Southwick, S.M., Paige, S., Morgan, C.A., III, Bremner, J.D., Krystal, J.H., & Charney, D.S. (1999). Neurotransmitter alterations in PTSD: Catecholamines and serotonin. Seminars in Clinical Neuropsychiatry, 4, 242–248. doi:10.153/SCNP00400242
- Soya, S., & Sakurai, T. (2018). Orexin as a modulator of fear-related behavior: Hypothalamic control of noradrenaline circuit. Brain Research, doi:10.1016/j.brainres.2018.11.032
- Spinale, F.G., Frangogiannis, N.G., Hinz, B., Holmes, J.W., Kassiri, Z., & Lindsey, M.L. (2016). Crossing into the next frontier of cardiac extracellular matrix research. Circulation Research, 119, 1040–1045. doi:10.1161/CIRCRESAHA.116.309916
- Spoormaker, V.I., & Montgomery, P. (2008). Disturbed sleep in post-traumatic stress disorder: Secondary symptom or core feature? Sleep Medicine Reviews, 12, 169–184. doi:10.1016/j.smrv.2007.08.008
- Stark, E.A., Parsons, C.E., Van Hartevelt, T.J., Charquero-Ballester, M., McManners, H., Ehlers, A., … Kringelbach, M.L. (2015). Post-traumatic stress influences the brain even in the absence of symptoms: A systematic, quantitative meta-analysis of neuroimaging studies. Neuroscience & Biobehavioral Reviews, 56, 207–221. doi:10.1016/j.neubiorev.2015.07.007
- Strawn, J.R., & Geracioti, T.D. Jr. (2008). Noradrenergic dysfunction and the psychopharmacology of posttraumatic stress disorder. Depression and Anxiety, 25, 260–271. doi:10.1002/da.20292
- Strawn, J.R., Pyne-Geithman, G.J., Ekhator, N.N., Horn, P.S., Uhde, T.W., Shutter, L.A., … Geracioti, T.D. Jr. (2010). Low cerebrospinal fluid and plasma orexin-A (hypocretin-1) concentrations in combat-related posttraumatic stress disorder. Psychoneuroendocrinology, 35, 1001–1007. doi:10.1016/j.psyneuen.2010.01.001
- Tamelian, T.L., & Jaycox, L.H. (2008). Invisible wounds of war: Psychological and cognitive injuries, their consequences, and services to assist recovery. Retrieved from Santa Monica, CA.
- Tanai, E., & Frantz, S. (2015). Pathophysiology of heart failure. Comprehensive Physiology, 6, 187–214. doi:10.1002/cphy.c140055
- Tosur, Z., Green, D., De Chavez, P.J., Knutson, K.L., Goldberger, J.J., Zee, P., & Carnethon, M.R. (2014). The association between sleep characteristics and prothrombotic markers in a population-based sample: Chicago Area Sleep Study. Sleep Medicine, 15, 973–978. doi:10.1016/j.sleep.2014.04.005
- Tracey, K.J. (2007). Physiology and immunology of the cholinergic antiinflammatory pathway. Journal of Clinical Investigation, 117, 289–296. doi:10.1172/JCI30555
- Tsuji, H., Venditti, F.J., Jr., Manders, E.S., Evans, J.C., Larson, M.G., Feldman, C.L., & Levy, D. (1994). Reduced heart rate variability and mortality risk in an elderly cohort. The Framingham Heart Study. Circulation, 90, 878–883. doi:10.1161/01.CIR.90.2.878
- Turner, J.H., Neylan, T.C., Schiller, N.B., Li, Y., & Cohen, B.E. (2013). Objective evidence of myocardial ischemia in patients with posttraumatic stress disorder. Biological Psychiatry, 74, 861–866. doi:10.1016/j.biopsych.2013.07.012
- Ulmer, C.S., Calhoun, P.S., Bosworth, H.B., Dennis, M.F., & Beckham, J.C. (2013). Nocturnal blood pressure non-dipping, posttraumatic stress disorder, and sleep quality in women. Behavioral Medicine, 39, 111–121. doi:10.1080/08964289.2013.813434
- Ulmer, C.S., Hall, M.H., Dennis, P.A., Beckham, J.C., & Germain, A. (2018). Posttraumatic stress disorder diagnosis is associated with reduced parasympathetic activity during sleep in United States Veterans and Military Service Members of the Iraq and Afghanistan Wars. Sleep, 41. doi:10.1093/sleep/zsy174
- van Liempt, S. (2012). Sleep disturbances and PTSD: A perpetual circle? European Journal of Psychotraumatology, 3, 19142. doi:10.3402/ejpt.v3i0.19142
- van Wyk, M., Thomas, K.G., Solms, M., & Lipinska, G. (2016). Prominence of hyperarousal symptoms explains variability of sleep disruption in posttraumatic stress disorder. Psychological Trauma, 8, 688–696. doi:10.1037/tra0000115
- Verberne, A.J., & Owens, N.C. (1998). Cortical modulation of the cardiovascular system. Progress in Neurobiology, 54, 149–168. doi:10.1016/S0301-0082(97)00056-7
- Vieira, J.O., Duarte, J.O., Costa-Ferreira, W., Morais-Silva, G., Marin, M.T., & Crestani, C.C. (2018). Sex differences in cardiovascular, neuroendocrine and behavioral changes evoked by chronic stressors in rats. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 81, 426–437. doi:10.1016/j.pnpbp.2017.08.014
- von Kanel, R., Begre, S., Abbas, C.C., Saner, H., Gander, M.L., & Schmid, J.P. (2010). Inflammatory biomarkers in patients with posttraumatic stress disorder caused by myocardial infarction and the role of depressive symptoms. Neuroimmunomodulation, 17, 39–46. doi:10.1159/000243084
- von Kanel, R., Schmid, J.P., Abbas, C.C., Gander, M.L., Saner, H., & Begre, S. (2010). Stress hormones in patients with posttraumatic stress disorder caused by myocardial infarction and role of comorbid depression. Journal of Affective Disorders, 121, 73–79. doi:10.1016/j.jad.2009.05.016
- Wang, Z., Caughron, B., & Young, M.R.I. (2017). Posttraumatic stress disorder: An immunological disorder? Frontiers in Psychiatry, 8, 222. doi:10.3389/fpsyt.2017.00222
- Wang, H., Parker, J.D., Newton, G.E., Floras, J.S., Mak, S., Chiu, K.-L., … Bradley, T.D. (2007). Influence of obstructive sleep apnea on mortality in patients with heart failure. Journal of the American College of Cardiology, 49, 1625–1631. doi:10.1016/j.jacc.2006.12.046
- Way, B.M., & Taylor, S.E. (2011). A polymorphism in the serotonin transporter gene moderates cardiovascular reactivity to psychosocial stress. Psychosomatic Medicine, 73, 310–317. doi:10.1097/PSY.0b013e31821195ed
- Williamson, J.B., & Harrison, D.W. (2003). Functional cerebral asymmetry in hostility: A dual task approach with fluency and cardiovascular regulation. Brain and Cognition, 52, 167–174. doi:10.1016/S0278-2626(03)00038-1
- Williamson, J.B., Lewis, G., Grippo, A.J., Lamb, D., Harden, E., Handleman, M., & Porges, S.W. (2010). Autonomic predictors of recovery following surgery: A comparative study. Autonomic Neuroscience, 156, 60–66. doi:10.1016/j.autneu.2010.03.009
- Williamson, J.B., Porges, E.C., Lamb, D.G., & Porges, S.W. (2014). Maladaptive autonomic regulation in PTSD accelerates physiological aging. Frontiers in Psychology, 5, 1571. doi:10.3389/fpsyg.2014.01571
- Wilson, M.A., & Reagan, L.P. (2016). Special issue: New perspectives in PTSD. Experimental Neurology, 284, 115–118. doi:10.1016/j.expneurol.2016.09.011
- Winkelmann, B.R., Russ, A.P., Nauck, M., Klein, B., Böhm, B.O., Maier, V., … März, W. (1999). Angiotensinogen M235T polymorphism is associated with plasma angiotensinogen and cardiovascular disease. American Heart Journal, 137, 698–705. doi:10.1016/S0002-8703(99)70226-7
- Wolf, E.J., & Morrison, F.G. (2017). Traumatic stress and accelerated cellular aging: From epigenetics to cardiometabolic disease. Current Psychiatry Reports, 19, 75. doi:10.1007/s11920-017-0823-5
- Woodward, S.H., Arsenault, N.J., Voelker, K., Nguyen, T., Lynch, J., Skultety, K., … Sheikh, J.I. (2009). Autonomic activation during sleep in posttraumatic stress disorder and panic: A mattress actigraphic study. Biological Psychiatry, 66, 41–46. doi:10.1016/j.biopsych.2009.01.005
- Writing Group, M., Mozaffarian, D., Benjamin, E.J., Go, A.S., Arnett, D.K., Blaha, M.J., & Stroke Statistics, S. (2016). Executive summary: Heart disease and stroke statistics—2016 update: A report from the American Heart Association. Circulation, 133, 447–454. doi:10.1161/CIR.0000000000000366
- Wu, C.-K., Luo, J.-L., Wu, X.-M., Tsai, C.-T., Lin, J.-W., Hwang, J.-J., … Chiang, F.-T. (2009). A propensity score-based case-control study of renin–angiotensin system gene polymorphisms and diastolic heart failure. Atherosclerosis, 205, 497–502. doi:10.1016/j.atherosclerosis.2008.12.033
- Yehuda, R., Siever, L.J., Teicher, M.H., Levengood, R.A., Gerber, D.K., Schmeidler, J., & Yang, R.K. (1998). Plasma norepinephrine and 3-methoxy-4-hydroxyphenylglycol concentrations and severity of depression in combat posttraumatic stress disorder and major depressive disorder. Biological Psychiatry, 44, 56–63. doi:10.1016/S0006-3223(98)80007-3
- Yeoh, J.W., Campbell, E.J., James, M.H., Graham, B.A., & Dayas, C.V. (2014). Orexin antagonists for neuropsychiatric disease: Progress and potential pitfalls. Frontiers in Neuroscience, 8, 36. doi:10.3389/fnins.2014.00036
- Zoladz, P.R., & Diamond, D.M. (2016). Predator-based psychosocial stress animal model of PTSD: Preclinical assessment of traumatic stress at cognitive, hormonal, pharmacological, cardiovascular and epigenetic levels of analysis. Experimental Neurology, 284, 211–219. doi:10.1016/j.expneurol.2016.06.003