
Abstract
There is evidence that plasma cortisol concentration can be either increased or decreased in patients with depression and related anxiety and stress-related disorders; the exact pathophysiological mechanisms of this state are not almost clear. Several distinct theories were proposed and mechanisms, which could lead to decreased glucocorticoid signaling and/or levels, were described. However, there is a possible drawback in almost all the theories proposed: insufficient attention to the inflammatory process, which is undoubtedly present in several stress-related disorders, including post-traumatic stress disorder (PTSD). Previous studies only briefly mentioned the presence of an inflammatory reaction’s signs in PTSD, without giving it due importance, although recognizing that it can affect the course of the disease. With that, the state of biochemical changes, characterized by the low glucocorticoids, glucocorticoid receptor’s resistance and the signs of the persistent inflammation (with the high levels of circulating cytokines) might be observed not only in PTSD but in coronary heart diseases and systemic chronic inflammatory diseases (rheumatoid arthritis) as well. That is why the present review aims to depict the pathophysiological mechanisms, which lead to a decrease in glucocorticoids in PTSD due to the action of inflammatory stimuli. We described changes in the glucocorticoid system and inflammatory reaction as parts of an integral system, where glucocorticoids and the glucocorticoid receptor reside at the apex of a regulatory network that blocks several inflammatory pathways, while decreased glucocorticoid signaling and/or level leads to unchecked inflammatory reactions to promote pathologies such as PTSD.
This review emphasizes the importance of inflammatory reaction in the development of puzzling conditions sometimes observed in severe diseases including post-traumatic stress disorder — the decreased levels of glucocorticoids in the blood. Following the classical concepts, one would expect an increase in glucocorticoid hormones, since they are part of the feedback mechanism in the immune system, which reduces stress and inflammation. However, low levels of glucocorticoid hormones are also observed. Thus, this review describes potential mechanisms, which can lead to the development of such a state.
LAY SUMMARY
Introduction
The earliest evidence that chronic stress was associated with reduced daily output of glucocorticoids (Bourne et al., Citation1967; Friedman et al., Citation1963) puzzled the stress research community since it contradicted a well-accepted paradigm that stress increases glucocorticoid secretion. Later studies, however, confirmed that chronic stress may be indeed associated with decreased glucocorticoid signaling (McEwen, Citation2000; Tseilikman et al., Citation2020). Blunted glucocorticoid signaling was also observed in patients with several stress-related disorders, such as post-traumatic stress disorder (PTSD; Smith et al., Citation1989; Ströhle et al., Citation2008; Zaba et al., Citation2015), depression (Lopes et al., Citation2012), high trait anxiety (Duncko et al., Citation2006; Jezova et al., Citation2004), and panic disorder (Jezova et al., Citation2010; Petrowski et al., Citation2010). Decreased circulating baseline glucocorticoid levels were observed in animal models of PTSD (Boero et al., Citation2018; Dremencov et al., Citation2019; Lazuko et al., Citation2018; Whitaker et al., Citation2016).
Several distinct theories were proposed and mechanisms, which could lead to the blunted glucocorticoid signaling were described. Among others, the development model whereby hypocortisolism develops via the hypoactivity of the HPA axis after prolonged periods of chronic stress have been proposed (Hellhammer & Wade, Citation1993). Thus, the frailty (Morrison et al., Citation2001; Oldehinkel et al., Citation2001) or overstimulation of the HPA axis (Fries et al., Citation2005; Perrin et al., Citation2019) were shown to drive the exhaustion of the HPA axis, which may lead to hypocortisolemia. An increase in the hypothalamic release of corticotropin-releasing factor (CRF), leading to subsequent adaptive downregulation of CFR receptors at the level of pituitary gland also has been proposed (Hauger et al., Citation1990). According to Yehuda, (Citation2001), impaired glucocorticoid signaling results from enhanced negative feedback by glucocorticoids, which is secondary to an increased sensitivity of glucocorticoid receptors (GR) in target tissues, while the duration, intensity, number and chronicity of stressors may further pronounce these effects (Fries et al., Citation2005; Miller et al., Citation2007). Later on, several mechanisms of the inadequate GR and monoamines signaling, metabolism and distribution typical for PTSD were described (Bierer et al., Citation2014; Geuze et al., Citation2012; Grinchii et al., Citation2018; Pivac et al., Citation2007), with the hypothesis, based on the presumption of the increased activity of 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) and CYP3A and decreased of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) leading to higher levels of 11-dehydrocorticosterone and lower levels of basal glucocorticoids being among them (Tseilikman et al., Citation2020). The peculiarities in neural interactions between prefrontal, amygdala, and hippocampal structures in PTSD were also studied (Morey et al., Citation2016; Woon & Hedges, Citation2008). Finally, the results of meta-analysis supported the evidence of prenatal programing of the axis, primarily on baseline cortisol measures and, disconcertingly, equivalent effect sizes for hyper and hypo-secretion (Pearson et al., Citation2015). Mounting evidence was emerging for prenatal development as a time during which programing of the axis might occur; however, whether those means elevations or suppressions were not completely clear (Koss & Gunnar, Citation2018).
However, there is a possible drawback in almost all the theories proposed: insufficient attention to the inflammatory process, which is undoubtedly present in PTSD. Previous studies only briefly mentioned the presence of an inflammatory reaction’s signs in PTSD, without giving it due to importance, although recognizing that it can affect the course of the disease or compound variabilities between various studies (Hori & Kim, Citation2019; Perrin et al., Citation2019; Tseilikman et al., Citation2020).
With that, numerous studies involving human subjects and animal experiments have revealed a possible linkage between inflammatory conditions and PTSD, as their results indicated, the signs of the inflammatory reaction manifested itself in increased levels of pro-inflammatory cytokines, such as Il-1, IL-6, TNF-a (reviewed in Gill et al., Citation2009; Wang & Young, Citation2016), elevations in RBC, WBC, platelets (Lindqvist et al., Citation2017), levels of CRP (Michopoulos et al., Citation2015) and pre-activating of PBMCs (Gola et al., Citation2013). Moreover, the higher pro-inflammatory scores in subjects with PTSD were revealed even after controlling for other factors (Lindqvist et al., Citation2014). While levels of anti-inflammatory cytokines have been less frequently measured in subjects with PTSD, there have been some studies indicating reduced levels of these anti-inflammatory mediators, including IL-4, IL-10 and TGF-β, number of lymphocytes, number of T cells, NK cell activity, and total amounts of IFN-gamma (Von et al., Citation2007; Teche et al., Citation2017). The imbalances in immune cell compositions in subjects with PTSD revealed itself in the increase in numbers of pro-inflammatory cells and the reduction of anti-inflammatory cells (Sommershof et al., Citation2009; Zhou et al., Citation2014).
That is why the present review aims to depict the pathophysiological mechanisms, which lead to the development of hypocortisolemia in PTSD due to the action of inflammatory stimuli.
11β-HSD activity
Cortisone is inactive and requires metabolism to cortisol catalyzed by 11β-HSD1. The liver is the major organ for converting cortisone to cortisol, but this also occurs in multiple other tissues including the brain. Conversion of cortisol to cortisone is mainly by the kidney, via 11β-HSD2.
With that, corticosteroid metabolism significantly changes in inflammatory conditions (Ichikawa et al., Citation1977), which suggests an increase in 11β-HSD1 activity, which increases cortisone metabolic clearance and the cortisol/cortisone ratio (Edwards, Citation2012). Hardy et al. (Citation2006) investigating local corticosteroid metabolism in fibroblasts from rheumatoid arthritis and osteoarthritis patients revealed that 11β-HSD1 expression has increased after TNF-α or IL-1β with an associated increase in enzyme activity (Hardy et al., Citation2006). Stress was also shown to increase the adrenal and hepatic (Quinkler et al., Citation2003) activities and macrophage expression of 11β-HSD1 (Sesti-Costa et al., Citation2012). Later on, chemical inhibitor studies confirmed that the increase in 11β-HSD1 expression with TNFα)/IL-1β occurred via the proximal HSD11B1 gene promoter and depended on NF-κB signaling (Ahasan et al., Citation2012). The situation might be different in other cell types such as peripheral blood mononuclear cells (Straub & Cutolo, Citation2016), but altogether this suggests that local glucocorticoid production is part of the normal response to inflammation (Edwards, Citation2012).
Inflammation causes the changes in 11β-HSD2 activity as well, and the changes in its activity were revealed not only in inflammatory diseases (Hardy et al., Citation2006), but also in stress and PTSD (Igarreta et al., Citation1999; Zallocchi et al., Citation2004; Bierer et al., Citation2014). There is evidence of both an increase (Bierer et al., Citation2014; Straub & Cutolo, Citation2016) and a decrease (Stegk et al., Citation2009; Tsugita et al., Citation2008) in 11β-HSD2 activity, depending on the tissues/organs where the activity was determined. Thus, the increase in 11β-HSD2 activity was detected in immune cells (Olsen et al., Citation2004) and mixed synovial cells (Schmidt et al., Citation2005), in kidneys (Igarreta et al., Citation1999) and brain (Alderman & Vijayan, Citation2012) of stressed animals and adrenal glands of animals with PTSD (Tselikman, Citation2019), which could result in lower cortisol levels at the inflammatory site.
The consequences of changes in 11β-HSD1/11β-HSD2 activity in PTSD due to the action of pro-inflammatory cytokines cannot be overestimated. 11β-HSD-1 amplifies glucocorticoid feedback on the HPA axis and is an important regulator of neuronal glucocorticoid exposure under both basal and stress conditions in vivo (Harris et al., Citation2001). Thus, 11β-HSD1-deficient mice showed elevated plasma corticosterone and ACTH levels, suggesting abnormal HPA control and exaggerated ACTH and corticosterone responses to restraint stress, suggesting diminished glucocorticoid feedback, while the 11β-HSD1 excess might have the reverse effect (lower cortisol, diminished response to stress, reduced 24-h glucocorticoid secretion, and relative adrenal atrophy) (Harris et al., Citation2001). Moreover, liver glucocorticoid metabolism (intensified in PTSD) may influence both basal and stress-associated HPA functions without any alteration in hypothalamic 11β-HSD1 activity (Paterson et al., Citation2007). Regeneration of glucocorticoids by 11β-HSD1 in the liver normalizes all aspects of HPA axis dysregulation in 11β-HSD1-deficient mice, without restoration of enzyme activity in key feedback areas of the forebrain, and therefore, hepatic glucocorticoid metabolism influences basal as well as stress-associated functions of the HPA axis (Paterson et al., Citation2007).
It should be noted, that not only cytokines but also the stress-induced change in the level of neurotransmitters could also modulate the activity of 11β-HSD. It has been demonstrated that an increase in the messenger cAMP of the β‐adrenergic signaling pathway inhibits both 11β‐HSD1 and 11β‐HSD2 (Hammami & Siiteri, Citation1991), while adenosine was shown to increase cortisone reactivation (Ferguson et al., Citation1999) and to inhibit (as isoproterenol, a β‐adrenergic receptor agonist) cortisol inactivation (Schmidt et al., Citation2005), while stress-mediated catecholamines induce rapid, reversible decreases in 11β-HSD2 mRNA expression (Sarkar et al., Citation2001). These results indicated that sympathetic neurotransmitters increase cortisol by inhibiting its inactivation or by increasing the reactivation of cortisone and by this, shift the balance of corticosteroids (Schmidt et al., Citation2005). Moreover, stress reaction, by enhancing intracellular active glucocorticoids, contributes to producing increased PEPCK activity, with the consequent rise in glycemia (Altuna et al., Citation2006), which in turn leads to the development of a whole cascade of pathological reactions.
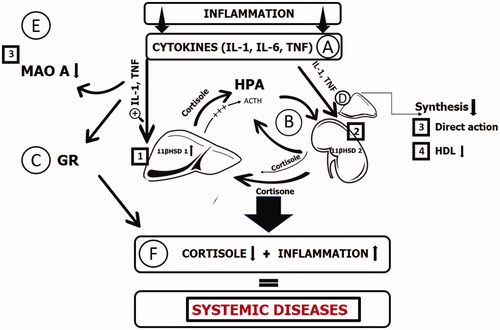
In general, with PTSD, the following sequence of changes may well occur (): the prolonged action of pro-inflammatory cytokines causes an increase in hepatic 11β-HSD-1 activity and tissue-depended changes in 11β-HSD-2 activity, with the hepatic production of cortisol higher than adrenal production and augmentation of active glucocorticoid levels, which lead to HPA axis dysregulation and decrease in adrenocorticotropic hormone (ACTH), and, due to the feedback mechanism to the appearance of hypocortisolemia. Thereby, cortisol production becomes more independent of central brain-derived hormonal regulation.
Figure 1. Pathophysiology of the decreased glucocorticoids in inflammatory state. Glucocorticoid level changes are shown in the example of cortisol. The prolonged action of pro-inflammatory cytokines (A) causes an increase in hepatic 11β-HSD-1 activity (1) and tissue-depended changes in 11β-HSD-2 activity (2), with the hepatic production of cortisol higher than adrenal production and augmentation of active glucocorticoid levels, which lead to HPA axis dysregulation and decrease in adrenocorticotropic hormone (ACTH), and, due to the feedback mechanism to the decreased levels of glucocorticoids (state F). Thereby, cortisol production (B) becomes more independent of central brain-derived hormonal regulation. Pro-inflammatory cytokines also cause the decrease in MAO-A activity (3) and lead to blunted glucocorticoid signaling in tissues (C) and, aside from the direct action on adrenal gland morphology and function (3), also causes the glucocorticoid substrate deficiency (4), lowing the HDL levels. Altogether, these lead to the pathological picture with decreased glucocorticoids and the signs of system inflammatory reaction, typical for several diseases (F).
Glucocorticoid signaling, GR receptors
Glucocorticoid signaling is mediated by the high-affinity mineralocorticoid receptor (MR) expressed predominantly in the limbic structures and the widely distributed low-affinity GR. MR in the limbic structures, particularly the hippocampus, is in charge of the maintenance of a basal state of stress system activity and is involved in the initial phase of the stress response (Douglas Bremner Citation2019). The GR facilitates the termination of the stress response by negative feedback, and thus is central to the regulation of glucocorticoids levels (Douglas Bremner Citation2019)
The changes in GR density, binding activity, and sensitivity, as well as changes in the GR isoform ratio, were described in inflammatory conditions, stress, and PTSD. The first studies in patients with PTSD revealed the higher density of GR in leukocytes, which correlated with the severity of PTSD symptoms (Yehuda et al., Citation1991; Yehuda et al., Citation1993). Later on, similar results were obtained in combat exposed veterans with PTSD which revealed the higher GR density in both combat-related PTSD patients and combat exposed controls compared to healthy controls (Yehuda et al., Citation1995). Subjects with diagnosed PTSD after being sexually abused also had a higher mean leukocyte GR density (Stein et al., Citation1997). However, other studies reported no difference in baseline and post DST leukocyte GR density between PTSD patients and controls (Yehuda et al., Citation2002). Moreover, in several studies, GR density was reported to be significantly lower in patients with PTSD and compared to healthy controls (Gotovac et al., Citation2003; de Kloet et al., Citation2007). The effect of inflammatory reaction which accompanies PTSD on GR density is also not clear: the reduced GRα expression was shown to be correlated to the upregulation of the inflammatory genes in monocytes (Carvalho et al., Citation2014), while similar GR numbers to controls were shown in patients with the systemic low-grade inflammatory state (Ysrraelit et al., Citation2008) and even animals with SIRS (Hoffman et al., Citation2015).
The data of GR binding activity and sensitivity also varies. Enhanced glucocorticoid sensitivity in peripheral mononuclear blood cells was described (Rohleder et al., Citation2004; Yehuda et al., Citation2004), although T cells in veterans with PTSD revealed the decreased glucocorticoid sensitivity (de Kloet et al., Citation2007). The decrease in binding affinity and glucocorticoid sensitivity was also observed in an inflammatory state or due to the action of inflammatory cytokines (Ysrraelit et al., Citation2008). Such abnormalities of GR function were demonstrated in in vitro studies of T cells incubated with a combination of IL‐2 and IL‐4, Il-13 (Almawi et al., Citation1991), IL‐1β, IL‐6, and interferon (IFN)‐γ (Kam et al., Citation1993) and in sepsis (Molijn et al., Citation1995). These results suggest that cytokines produced during an inflammatory response may induce GR resistance in relevant cell types by direct effects on the GR, thereby providing an additional pathway by which the immune system can influence the HPA axis (Pariante et al., Citation1999).
With that, among the factors contributing to the inconsistency of the data and contradiction in results may be the variations in the inflammatory syndrome’s intensity/severity, which could be different in the abovementioned studies and even in different time-points of each study. The dynamic pattern of the inflammatory reaction and, therefore, its action was well characterized by the results of GJ Molijn et al. (Citation1995), were the increased sensitivity of the peripheral mononuclear cells to dexamethasone during the period of sepsis have changed and normalized during the ensuing period of clinical recovery of those patients (Molijn et al., Citation1995).
Another mechanism of inflammation’s action on the GR receptor’s functions includes the inhibition of GR translocation and GR-mediated gene transcription and inducing the up-regulation of the cytosolic receptors (Pariante et al., Citation1999). The changes in the GR isoforms ratio can be responsible for glucocorticoid resistance as well. GR gene encodes two protein isoforms: a cytoplasmic alpha form (GRα), which binds hormone, translocates to the nucleus, and regulates gene transcription, and a nuclear-localized beta isoform (GRβ), which does not bind known ligands and attenuates GRα action (Kino et al., Citation2009). The GRβ isoform has been shown to have a dominant-negative effect on GRα‐induced transcriptional activity (Hoffman et al., Citation2015). Studies examining GR isoforms have shown an increase in both GRα and GRβ and a concomitant decrease in the GRα: GRβ ratio in inflammatory states (Hoffman et al., Citation2015; Longui et al., Citation2000; Webster, Oakley, Jewell, & Cidlowski, Citation2001) and depression (Carvalho et al., Citation2014). The accumulation of GRb isoform makes it an attractive candidate to explain the appearance of glucocorticoid resistance due to the action of inflammatory cytokines (Webster, Oakley, Jewell, & Cidlowski, 2001).
We should also note the possibility of triggering GR resistance by glucose, which could be mediated through both enhancing acetylation (via, among others, regulation of essential clock genes such as Per) and hindering deacetylation of GR (through possible regulation of sirtuin activity) (Kassi & Papavassiliou, Citation2012).
Moreover, the molecular mechanisms of GR resistance are very diverse and may involve also the changes in epigenetic modification’s patterns and components of the ultra-short negative loop of GC signaling in cells (Merkulov et al., Citation2017).
Substrate deficiency
Stress is known to influence adrenal gland morphology and function [reviewed in Berger et al., Citation2019]. Functional and structural signs of adrenal cortex degeneration were observed in PTSD, including decreased plasma concentration of corticosterone, decreased weight of adrenal glands, reduced thickness of the fasciculate zone, and hydropic degeneration of adrenal gland cells (Manukhina et al., Citation2018). The described stress-induced changes in the adrenal morphology could affect the synthesis of cortisol as well (Arlt & Stewart, Citation2005). About 80% of circulating cortisol is derived from plasma cholesterol, the remaining 20% is synthesized in situ from acetate and other precursors, while the high-density lipoprotein (HDL) is the preferred cholesterol source of the steroidogenic substrate in the adrenal gland (Yaguchi et al., Citation1998). HDL is substantially reduced in conditions, accompanied by the inflammatory reaction, including myocardial infarction, sepsis, and burns (Marik, Citation2007). Thus, in severe sepsis, lipoprotein concentrations rapidly change and can be reduced to 50% of recovery concentrations with the pattern of early rapid decline to be found primarily in the HDL (Marik, Citation2007).
The decrease in HDL is revealed not only in the abovementioned diseases, but also in patients with PTSD and depression as well [reviewed in Levine et al., Citation2014]. Thus, patients with full PTSD had lower HDL-C (Känel et al., Citation2010) and higher prevalence of HDL <1.0 mmol/l and TG >2.3 mmol/l (Walczewska et al., Citation2011) than patients without PTSD. Moreover, HDL-C levels were inversely associated with the severity of disease: the PTSD total symptoms, reeexperiencing symptoms, and avoidance symptoms (Känel et al., 2010) and the higher serum total cholesterol was associated with lower odds for having suicidal ideation, clinically significant aggression, and depressive symptoms in PTSD (Vilibić et al., Citation2014).
Finally, the role of substrate deficiency as a cause of adrenal insufficiency was confirmed in the study which demonstrated that HDL was the sole predictor of cortisol response in critically ill patients, associated with an attenuated response to ACTH (van der Voort et al., Citation2003). Thus, substrate deficiency due to the action of inflammatory cytokines (Jäättelä et al., Citation1991) in PTSD may contribute to the low GS levels.
MAO-A activity
MAO-A is a key enzyme in the degradation of biogenic amines such as serotonin and dopamine, and the changes in its activity in stress, depression, and PTSD were studied intensively over the last decades.
Glucocorticoid enhances monoamine oxidase A gene expression by 1) regulation of R1 translocation; 2) direct interaction of the GR with the third glucocorticoid/androgen response element; and 3) indirect interaction of GR with the Sp1 or R1 transcription factor on Sp1-binding sites of the MAO-A promoter (Manoli et al., Citation2005; Ou et al., Citation2006). Chronic stress and glucocorticoid exposures lasting 24 h or longer have been associated with greater MAO-A synthesis, levels, and activity (Soliman et al., Citation2012). It is of interest, that dexamethasone was more effective in activating MAO-A gene expression upon 24 h or longer time treatment but not 12 h (Ou et al., Citation2006). Moreover, continuous glucocorticoid administration over 26 d caused a 3-fold increase in frontal/parietal cortex MAO-A activity in rats (Slotkin et al., Citation1998), and chronic stress led to 2–3 times increased MAO-A-mRNA levels in the raphe nucleus of CBA/Lac mice (Filipenko et al., Citation2002). Similar changes were observed in depression (reviewed in Higuchi et al., Citation2017): depressed patients exhibited lower MAOA methylation than healthy controls. Moreover, MAO-A densities in several brain regions (e.g. prefrontal cortex, hippocampus, and midbrain) were higher in patients with depression even in the recovery phase and were associated with the recurrence of depressive symptoms (Meyer et al., Citation2009). On the contrary, a highly consistent relationship between acute stressors and glucocorticoid administration and decreased MAO-A binding, activity, and protein levels was revealed (Soliman et al., Citation2012). Exacerbation period in PTSD decreases MAO activity, thereby increasing the availability of catecholamines and the hypermethylation of 3 CpGs in the MAOA gene exonI/intronI region (Ziegler et al., Citation2018).
The inflammatory reaction, via pro-inflammatory cytokines, modulates the MAO-A activity as well. MAO-A expression is suppressed by the IL-6 signaling (Huang et al., Citation2012), while the inhibition of IL-6R signaling or IL-6R siRNA increased MAO-A activity (Bharti et al., Citation2018). With that, TNF‐α increases the Sp1 expression that in turn activates MAOA gene transcription (Gupta et al., Citation2015) and systemic LPS administration leads to longer-lasting changes in inhibitory network function and increase in MAO-A and ACh-E activities responsible for reduced neuromodulator actions (Ming et al., Citation2015).
Inflammation, CNS and PTSD
Fundamentally, there are two points of view on the mechanism of inflammatory reaction’s influence on CNS. From one point of view, in conditions under which the immune system is strongly activated by infection or injury, as well as by severe or chronic stressful conditions, glia and other brain immune cells change their morphology and functioning and secrete high levels of pro-inflammatory cytokines and prostaglandins, which disrupts the delicate balance needed for the neurophysiological actions of immune processes and produces direct detrimental effects (Yirmiya & Goshen, Citation2011). In this case, the endogenous IL-1 plays a critical role in HPA axis activation after stress (Goshen et al., Citation2003). It is produced by ameboid microglia, astrocytes, brain oligodendrocytes, while its receptors are shown to be highly concentrated in the dentate gyrus, in the choroid plexus at various levels of the brain, in the pituitary and the meninges (Ban et al., Citation1991). IL-1R mRNA was found in the hippocampus, choroid plexus, cerebellum, and in high levels in the endothelium of postcapillary venules and glial cells surrounding arterioles throughout the brain (Wong & Licinio, Citation1994), and, which is important, interleukin-1 type I receptor is expressed in human hypothalamus (Hammond et al., Citation1999). That is why the production of brain IL-1 is an important link in stress-induced activation of the HPA axis and secretion of glucocorticoids, which mediate the effects of stress on memory functioning and neural plasticity, exerting beneficial effects at low levels and detrimental effects at high levels (Goshen & Yirmiya, Citation2009).
Other researchers are focusing on the blood-derived cytokines. During the systemic immune challenge, the organum vasculosum laminae terminalis with its dense vascularization by fenestrated capillaries lacking blood–brain barrier (BBB) function allows direct access of circulating pyrogens to brain tissue located in close vicinity to the preoptic area of the hypothalamus (Ott et al., Citation2010). Moreover, it was shown, that interleukins could be transported from blood to brain across the BBB by a saturable system (Gutierrez et al., Citation1994). While the amount of blood-borne cytokines entering the brain is modest, it is comparable to that of other water-soluble compounds, known to cross the BBB in sufficient amounts to affect brain function (Banks et al., Citation1995).
Blood-derived cytokines also act on the CNS via endothelial cells, which are a critical component of the BBB. These cells are innately programed to respond to a myriad of inflammatory cytokines or other danger signals and their activation to produce local inflammatory mediators (including IL-1) contributes to the CNS activation in inflammatory conditions as well (O’Carroll et al., Citation2015). Moreover, brain endothelial cells are the major source of prostaglandin E2 under the inflammatory state, which influences the central neuronal and HPA axis activity (Matsumura & Kobayashi, Citation2004).
Generally, we fully agree with H. Hori and Kim (Citation2019) that a better understanding of the causes and consequences of the altered inflammatory system will aid in developing biologically oriented diagnostics and treatment for PTSD (Hori & Kim, Citation2019).
Conclusion
The described state of the biochemical changes, characterized by the low cortisol levels, GR resistance and the signs of the persistent inflammation (higher levels of IL-6) could be observed not only in PTSD but in coronary heart diseases and systemic chronic inflammatory diseases (RA) as well (Nikkheslat et al., Citation2015; Pariante, Citation2017; Straub & Cutolo, Citation2016). Thus, the depressed patients with coronary heart disease have higher inflammation (higher IL-6 mRNA and higher levels of c-reactive protein) together with reduced expression of GR mRNA, which was associated with HPA axis hypoactivity (Nikkheslat et al., Citation2015; Pariante, Citation2017).
The similarity of the clinical features of the distinct diseases makes one think about the common links in their pathogenesis and the inflammatory reaction could serve as one of these links. With that, there are several studies of Danese et al. (Citation2007–2009), which have direct associations with PTSD studies of R Yehuda (2005–2007). Thus, it was shown that the increased inflammation was present not only in subjects who were depressed and had a history of childhood maltreatment but also in those who had experienced maltreatment but were not depressed (Danese et al., Citation2007, Citation2008, Citation2009). Together, these findings indicate that the increased inflammation is a “biological scar” of the early exposure to high levels of stress (or possibly directly to inflammation), early in childhood or even in utero (Pariante, Citation2017; Plant et al., Citation2016; Preez et al., Citation2016), that leads to the typical picture of PTSD with hypocortisolemia and low-grade inflammation.
Author contribution
All authors contributed to the study concept and the study design. Alexey Sarapultsev, Eliyahu Dremencov, Petr Sarapultsev, Maria Komelkova, Olga Tselikman, and Vadim Tselikman drafted the manuscript, Petr Sarapultsev and Eliyahu Dremencov provided critical revisions. Maria Komelkova created the artwork. All authors approved the final version of the manuscript for submission.
Disclosure statement
The authors declare no conflict of interest and no financial interest in the publication of this manuscript.
Additional information
Funding
Notes on contributors
Alexey Sarapultsev
Alexey Sarapultsev, MD (Ural State Medical Academy, Ekaterinburg, Russia, 2005), PhD (Tymen State Medical Academy, Tymen, Russia, 2010), ScD (Institute of Immunology and Physiology, Ural Division of the Russian Academy of Sciences, Ekaterinburg, Russia, 2019). Senior researcher, Institute of Immunology and Physiology, Ural Division of the Russian Academy of Sciences, Ekaterinburg, Russia.
Petr Sarapultsev
Petr Sarapultsev, MD (Sverdlovsk State Medical Institute, Sverdlovsk, USSR, 1971), PhD (Sverdlovsk State Medical Institute, Sverdlovsk, USSR, 1983), ScD (Sverdlovsk State Medical Institute, Sverdlovsk, Russia, 1993), Professor (1996). Principal researcher, Institute of Immunology and Physiology, Ural Division of the Russian Academy of Sciences, Ekaterinburg, Russia.
Eliyahu Dremencov
Eliyahu Dremencov, MMedSc (Hebrew University, Jerusalem, Israel, 2000), PhD (Bar-Ilan University, Israel, 2004) is an Independent Research Fellow in the Institute of Molecular Physiology and Genetics (IMPG), Center for Biosciences, and Institute of Experimental Endocrinology, Biomedical Research Center, Slovak Academy of Science (since 2013), Head of the IMPG Neuropharmacological Laboratory (2016-), Head of the IMPG (2018-)
Maria Komelkova
Maria Komelkova, MD (Chelyabinsk state University, Chelyabinsk, Russia, 2005), PhD (South-Ural State Medical University, Chelyabinsk, Russia, 2015). Senior researcher, Institute of Immunology and Physiology, Ural Division of the Russian Academy of Sciences, Ekaterinburg, Russia; Senior researcher, School of Medical Biology in the South Ural State University
Olga Tseilikman
Olga Tseilikman, PhD (2001, South Ural State Medical University, Chelyabinsk, Russia), DSc (2005, Omsk State Medical University, Omsk, Russia), is a Professor in the School of Medical Biology, South Ural State University, Chelyabinsk, Russia.
Vadim Tseilikman
Vadim Tseilikman, MSc Chelyabinsk State Pedagogical University, Chelyabinsk Russia, 1982), PhD (Siberian State Medical University, Tomsk, Russia, 1992), DSc (Institute of General Pathology and Pathological Physiology, Moscow, Russia, 1999). Director of School of Medical Biology in the South Ural State University (2017-).
References
- Ahasan, M. M., Hardy, R., Jones, C., Kaur, K., Nanus, D., Juarez, M., Morgan, S. A., Hassan-Smith, Z., Bénézech, C., Caamaño, J. H., Hewison, M., Lavery, G., Rabbitt, E. H., Clark, A. R., Filer, A., Buckley, C. D., Raza, K., Stewart, P. M., & Cooper, M. S. (2012). Inflammatory regulation of glucocorticoid metabolism in mesenchymal stromal cells. Arthritis and Rheumatism, 64(7), 2404–2413. https://doi.org/10.1002/art.34414
- Alderman, S. L., & Vijayan, M. M. (2012). 11β-Hydroxysteroid dehydrogenase type 2 in zebrafish brain: A functional role in hypothalamus-pituitary-interrenal axis regulation. The Journal of Endocrinology, 215(3), 393–402. https://doi.org/10.1530/JOE-12-0379
- Almawi, W. Y., Lipman, M. L., Stevens, A. C., Zanker, B., Hadro, E. T., & Strom, T. B. (1991). Abrogation of glucocorticoid-mediated inhibition of T cell proliferation by the synergistic action of IL-1, IL-6, and IFN-gamma. Journal of Immunology (Baltimore, Md.: 1950), 146(10), 3523–3527.
- Altuna, M. E., Lelli, S. M., Martín de Viale, L. C. S., & Damasco, M. C. (2006). Effect of stress on hepatic 11beta-hydroxysteroid dehydrogenase activity and its influence on carbohydrate metabolism. Canadian Journal of Physiology and Pharmacology, 84(10), 977–984. https://doi.org/10.1139/y06-046
- Arlt, W., & Stewart, P. M. (2005). Adrenal corticosteroid biosynthesis, metabolism, and action. Endocrinology and Metabolism Clinics of North America, 34(2), 293–313. https://doi.org/10.1016/j.ecl.2005.01.002
- Ban, E., Milon, G., Prudhomme, N., Fillion, G., & Haour, F. (1991). Receptors for interleukin-1 (alpha and beta) in mouse brain: Mapping and neuronal localization in hippocampus. Neuroscience, 43(1), 21–30. https://doi.org/10.1016/0306-4522(91)90412-H
- Banks, W. A., Kastin, A. J., & Broadwell, R. D. (1995). Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation, 2(4), 241–248. https://doi.org/10.1159/000097202
- Berger, I., Werdermann, M., Bornstein, S. R., & Steenblock, C. (2019). The adrenal gland in stress - Adaptation on a cellular level. The Journal of Steroid Biochemistry and Molecular Biology, 190, 198–206. https://doi.org/10.1016/j.jsbmb.2019.04.006
- Bharti, R., Dey, G., Das, A. K., & Mandal, M. (2018). Differential expression of IL-6/IL-6R and MAO-A regulates invasion/angiogenesis in breast cancer. British Journal of Cancer, 118(11), 1442–1452. https://doi.org/10.1038/s41416-018-0078-x
- Bierer, L. M., Bader, H. N., Daskalakis, N. P., Lehrner, A. L., Makotkine, I., Seckl, J. R., & Yehuda, R. (2014). Elevation of 11β-hydroxysteroid dehydrogenase type 2 activity in Holocaust survivor offspring: Evidence for an intergenerational effect of maternal trauma exposure. Psychoneuroendocrinology, 48, 1–10. https://doi.org/10.1016/j.psyneuen.2014.06.001
- Boero, G., Pisu, M. G., Biggio, F., Muredda, L., Carta, G., Banni, S., Paci, E., Follesa, P., Concas, A., Porcu, P., & Serra, M. (2018). Impaired glucocorticoid-mediated HPA axis negative feedback induced by juvenile social isolation in male rats. Neuropharmacology, 133, 242–253. https://doi.org/10.1016/j.neuropharm.2018.01.045
- Bourne, P. G., Rose, R. M., & Mason, J. W. (1967). Urinary 17-OHCS levels. Data on seven helicopter ambulance medics in combat. Archives of General Psychiatry, 17(1), 104–110. https://doi.org/10.1001/archpsyc.1967.01730250106015
- Carvalho, L. A., Bergink, V., Sumaski, L., Wijkhuijs, J., Hoogendijk, W. J., Birkenhager, T. K., & Drexhage, H. A. (2014). Inflammatory activation is associated with a reduced glucocorticoid receptor alpha/beta expression ratio in monocytes of inpatients with melancholic major depressive disorder. Translational Psychiatry, 4, e344https://doi.org/10.1038/tp.2013.118
- Danese, A., Moffitt, T. E., Harrington, H., Milne, B. J., Polanczyk, G., Pariante, C. M., Poulton, R., & Caspi, A. (2009). Adverse childhood experiences and adult risk factors for age-related disease: Depression, inflammation, and clustering of metabolic risk markers. Archives of Pediatrics & Adolescent Medicine, 163(12), 1135–1143. https://doi.org/10.1001/archpediatrics.2009.214
- Danese, A., Moffitt, T. E., Pariante, C. M., Ambler, A., Poulton, R., & Caspi, A. (2008). Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Archives of General Psychiatry, 65(4), 409–415. https://doi.org/10.1001/archpsyc.65.4.409
- Danese, A., Pariante, C. M., Caspi, A., Taylor, A., & Poulton, R. (2007). Childhood maltreatment predicts adult inflammation in a life-course study. Proceedings of the National Academy of Sciences of the United States of America, 104(4), 1319–1324. https://doi.org/10.1073/pnas.0610362104
- de Kloet, C. S., Vermetten, E., Bikker, A., Meulman, E., Geuze, E., Kavelaars, A., Westenberg, H. G. M., & Heijnen, C. J. (2007). Leukocyte glucocorticoid receptor expression and immunoregulation in veterans with and without post-traumatic stress disorder. Molecular Psychiatry, 12(5), 443–453. https://doi.org/10.1038/sj.mp.4001934
- Douglas Bremner, J. (2019). Posttraumatic stress disorder: From neurobiology to treatment. Wiley. Retrieved September 19, 2019, from https://www.wiley.com/en-us/Posttraumatic+Stress+Disorder%3A+From+Neurobiology+to+Treatment-p-9781118356111
- Dremencov, E., Lapshin, M., Komelkova, M., Alliluev, A., Tseilikman, O., Karpenko, M., Pestereva, N., Manukhina, E., Downey, H. F., & Tseilikman, V. (2019). Chronic predator scent stress alters serotonin and dopamine levels in the rat thalamus and hypothalamus, respectively. General Physiology and Biophysics, 38(2), 187–190. https://doi.org/10.4149/gpb_2019003
- Duncko, R., Makatsori, A., Fickova, E., Selko, D., & Jezova, D. (2006). Altered coordination of the neuroendocrine response during psychosocial stress in subjects with high trait anxiety. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 30(6), 1058–1066. https://doi.org/10.1016/j.pnpbp.2006.04.002
- Edwards, C. (2012). Sixty years after Hench-corticosteroids and chronic inflammatory disease. The Journal of Clinical Endocrinology and Metabolism, 97(5), 1443–1451. https://doi.org/10.1210/jc.2011-2879
- Ferguson, S. E., Pallikaros, Z., Michael, A. E., & Cooke, B. A. (1999). The effects of different culture media, glucose, pyridine nucleotides and adenosine on the activity of 11beta-hydroxysteroid dehydrogenase in rat Leydig cells. Molecular and Cellular Endocrinology, 158(1–2), 37–44. https://doi.org/10.1016/S0303-7207(99)00186-0
- Filipenko, M. L., Beilina, A. G., Alekseyenko, O. V., Dolgov, V. V., & Kudryavtseva, N. N. (2002). Repeated experience of social defeats increases serotonin transporter and monoamine oxidase A mRNA levels in raphe nuclei of male mice. Neuroscience Letters, 321(1-2), 25–28. https://doi.org/10.1016/S0304-3940(01)02495-8
- Friedman, S. B., Mason, J. W., & Hamburg, D. A. (1963). Urinary 17-hydroxycorticosteroid levels in parents of children with neoplastic disease: A study of chronic psychological stress. Psychosomatic Medicine, 25, 364–376. https://doi.org/10.1097/00006842-196307000-00007
- Fries, E., Hesse, J., Hellhammer, J., & Hellhammer, D. H. (2005). A new view on hypocortisolism. Psychoneuroendocrinology, 30(10), 1010–1016. https://doi.org/10.1016/j.psyneuen.2005.04.006
- Geuze, E., van Wingen, G. A., van Zuiden, M., Rademaker, A. R., Vermetten, E., Kavelaars, A., Fernández, G., & Heijnen, C. J. (2012). Glucocorticoid receptor number predicts increase in amygdala activity after severe stress. Psychoneuroendocrinology, 37(11), 1837–1844. https://doi.org/10.1016/j.psyneuen.2012.03.017
- Gill, J. M., Saligan, L., Woods, S., & Page, G. (2009). PTSD is associated with an excess of inflammatory immune activities. Perspectives in Psychiatric Care, 45(4), 262–277. https://doi.org/10.1111/j.1744-6163.2009.00229.x
- Gola, H., Engler, H., Sommershof, A., Adenauer, H., Kolassa, S., Schedlowski, M., Groettrup, M., Elbert, T., & Kolassa, I.-T. (2013). Posttraumatic stress disorder is associated with an enhanced spontaneous production of pro-inflammatory cytokines by peripheral blood mononuclear cells. BMC Psychiatry, 13(1), 40https://doi.org/10.1186/1471-244X-13-40
- Goshen, I., & Yirmiya, R. (2009). Interleukin-1 (IL-1): A central regulator of stress responses. Frontiers in Neuroendocrinology, 30(1), 30–45. https://doi.org/10.1016/j.yfrne.2008.10.001
- Goshen, I., Yirmiya, R., Iverfeldt, K., & Weidenfeld, J. (2003). The role of endogenous interleukin-1 in stress-induced adrenal activation and adrenalectomy-induced adrenocorticotropic hormone hypersecretion. Endocrinology, 144(10), 4453–4458. https://doi.org/10.1210/en.2003-0338
- Gotovac, K., Sabioncello, A., RabatíC, S., Berki, T., & Dekaris, D. (2003). Flow cytometric determination of glucocorticoid receptor (GCR) expression in lymphocyte subpopulations: Lower quantity of GCR in patients with post-traumatic stress disorder (PTSD). Clinical and Experimental Immunology, 131(2), 335–339. https://doi.org/10.1046/j.1365-2249.2003.02075.x
- Grinchii, D., Paliokha, R., Tseilikman, V., & Dremencov, E. (2018). Inhibition of cytochrome P450 by proadifen diminishes the excitability of brain serotonin neurons in rats. General Physiology and Biophysics, 37(6), 711–713. https://doi.org/10.4149/gpb_2018040
- Gupta, V., Khan, A. A., Sasi, B. K., & Mahapatra, N. R. (2015). Molecular mechanism of monoamine oxidase A gene regulation under inflammation and ischemia-like conditions: Key roles of the transcription factors GATA2, Sp1 and TBP. Journal of Neurochemistry, 134(1), 21–38. https://doi.org/10.1111/jnc.13099
- Gutierrez, E. G., Banks, W. A., & Kastin, A. J. (1994). Blood-borne interleukin-1 receptor antagonist crosses the blood-brain barrier. Journal of Neuroimmunology, 55(2), 153–160. https://doi.org/10.1016/0165-5728(94)90005-1
- Hammami, M. M., & Siiteri, P. K. (1991). Regulation of 11 beta-hydroxysteroid dehydrogenase activity in human skin fibroblasts: Enzymatic modulation of glucocorticoid action . The Journal of Clinical Endocrinology and Metabolism, 73(2), 326–334. https://doi.org/10.1210/jcem-73-2-326
- Hammond, E. A., Smart, D., Toulmond, S., Suman-Chauhan, N., Hughes, J., & Hall, M. D. (1999). The interleukin-1 type I receptor is expressed in human hypothalamus. Brain, 122(9), 1697–1707. https://doi.org/10.1093/brain/122.9.1697
- Hardy, R. S., Filer, A., Cooper, M. S., Parsonage, G., Raza, K., Hardie, D. L., & Hewison, M. (2006). Differential expression, function and response to inflammatory stimuli of 11β-hydroxysteroid dehydrogenase type 1 in human fibroblasts: A mechanism for tissue-specific regulation of inflammation. Arthritis Research & Therapy, 8(4), R108. https://doi.org/10.1186/ar1993
- Harris, H. J., Kotelevtsev, Y., Mullins, J. J., Seckl, J. R., & Holmes, M. C. (2001). Intracellular regeneration of glucocorticoids by 11beta-hydroxysteroid dehydrogenase (11beta-HSD)-1 plays a key role in regulation of the hypothalamic-pituitary-adrenal axis: Analysis of 11beta-HSD-1-deficient mice. Endocrinology, 142(1), 114–120. https://doi.org/10.1210/endo.142.1.7887
- Hauger, R. L., Lorang, M., Irwin, M., & Aguilera, G. (1990). CRF receptor regulation and sensitization of ACTH responses to acute ether stress during chronic intermittent immobilization stress. Brain Research, 532(1-2), 34–40. https://doi.org/10.1016/0006-8993(90)91738-3
- Hellhammer, D. H., & Wade, S. (1993). Endocrine correlates of stress vulnerability. Psychotherapy and Psychosomatics, 60(1), 8–17. https://doi.org/10.1159/000288675
- Higuchi, Y., Soga, T., & Parhar, I. S. (2017). Regulatory pathways of monoamine oxidase A during social stress. Frontiers in Neuroscience, 11, 604. https://doi.org/10.3389/fnins.2017.00604
- Hoffman, C. J., McKenzie, H. C., Furr, M. O., & Desrochers, A. (2015). Glucocorticoid receptor density and binding affinity in healthy horses and horses with systemic inflammatory response syndrome. Journal of Veterinary Internal Medicine, 29(2), 626–635. https://doi.org/10.1111/jvim.12558
- Hori, H., & Kim, Y. (2019). Inflammation and post-traumatic stress disorder. Psychiatry and Clinical Neurosciences, 73(4), 143–153. https://doi.org/10.1111/pcn.12820
- Huang, L., Frampton, G., Rao, A., Zhang, K-s., Chen, W., Lai, J-m., Yin, X-y., Walker, K., Culbreath, B., Leyva-Illades, D., Quinn, M., McMillin, M., Bradley, M., Liang, L.-J., & DeMorrow, S. (2012). Monoamine oxidase A expression is suppressed in human cholangiocarcinoma via coordinated epigenetic and IL-6-driven events. Laboratory Investigation; A Journal of Technical Methods and Pathology, 92(10), 1451–1460. https://doi.org/10.1038/labinvest.2012.110
- Ichikawa, Y., Yoshida, K., Kawagoe, M., Saito, E., Abe, Y., Arikawa, K., & Homma, M. (1977). Altered equilibrium between cortisol and cortisone in plasma in thyroid dysfunction and inflammatory diseases. Metabolism: Clinical and Experimental, 26(9), 989–997. https://doi.org/10.1016/0026-0495(77)90016-6
- Igarreta, P., Calvo, J. C., & Damasco, M. C. (1999). Activity of renal 11βhydroxysteroid dehydrogenase 2 (11βHSD2) in stressed animals. Life Sciences, 64(24), 2285–2290. https://doi.org/10.1016/S0024-3205(99)00179-4
- Jäättelä, M., Ilvesmäki, V., Voutilainen, R., Stenman, U.-H., & Saksela, E. (1991). Tumor necrosis factor as a potent inhibitor of adrenocorticotropin-induced cortisol production and steroidogenic P450 enzyme gene expression in cultured human fetal adrenal cells. Endocrinology, 128(1), 623–629. https://doi.org/10.1210/endo-128-1-623
- Jezova, D., Makatsori, A., Duncko, R., Moncek, F., & Jakubek, M. (2004). High trait anxiety in healthy subjects is associated with low neuroendocrine activity during psychosocial stress. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 28(8), 1331–1336. https://doi.org/10.1016/j.pnpbp.2004.08.005
- Jezova, D., Vigas, M., Hlavacova, N., & Kukumberg, P. (2010). Attenuated neuroendocrine response to hypoglycemic stress in patients with panic disorder. Neuroendocrinology, 92(2), 112–119. https://doi.org/10.1159/000283560
- Kam, J. C., Szefler, S. J., Surs, W., Sher, E. R., & Leung, D. Y. (1993). Combination IL-2 and IL-4 reduces glucocorticoid receptor-binding affinity and T cell response to glucocorticoids. Journal of Immunology (Baltimore, Md.: 1950), 151((7), 3460–3466.
- Känel, R. V., Kraemer, B., Saner, H., Schmid, J.-P., Abbas, C. C., & Begré, S. (2010). Posttraumatic stress disorder and dyslipidemia: Previous research and novel findings from patients with PTSD caused by myocardial infarction. The World Journal of Biological Psychiatry : The Official Journal of the World Federation of Societies of Biological Psychiatry, 11(2), 141–147. https://doi.org/10.3109/15622970903449846
- Kassi, E., & Papavassiliou, A. G. (2012). Glucose can promote a glucocorticoid resistance state. Journal of Cellular and Molecular Medicine, 16(5), 1146–1149. https://doi.org/10.1111/j.1582-4934.2012.01532.x
- Kino, T., Su, Y. A., & Chrousos, G. P. (2009). Human glucocorticoid receptor isoform beta: Recent understanding of its potential implications in physiology and pathophysiology. Cellular and Molecular Life Sciences : Cmls, 66(21), 3435–3448. https://doi.org/10.1007/s00018-009-0098-z
- Koss, K. J., & Gunnar, M. R. (2018). Annual research review: Early adversity, the hypothalamic-pituitary-adrenocortical axis, and child psychopathology. Journal of Child Psychology and Psychiatry, and Allied Disciplines, 59(4), 327–346. https://doi.org/10.1111/jcpp.12784
- Lazuko, S. S., Kuzhel, O. P., Belyaeva, L. E., Manukhina, E. B., Downey, H. F., Fred Downey, H., Tseilikman, O. B., Komelkova, M. V., & Tseilikman, V. E. (2018). Posttraumatic Stress Disorder Disturbs Coronary Tone and Its Regulatory Mechanisms. Cellular and Molecular Neurobiology, 38(1), 209–217. https://doi.org/10.1007/s10571-017-0517-x
- Levine, A. B., Levine, L. M., & Levine, T. B. (2014). Posttraumatic Stress Disorder and Cardiometabolic Disease. Cardiology, 127(1), 1–19. https://doi.org/10.1159/000354910
- Lindqvist, D., Mellon, S. H., Dhabhar, F. S., Yehuda, R., Grenon, S. M., Flory, J. D., Bierer, L. M., Abu-Amara, D., Coy, M., Makotkine, I., Reus, V. I., Aschbacher, K., Bersani, F. S., Marmar, C. R., & Wolkowitz, O. M. (2017). Increased circulating blood cell counts in combat-related PTSD: Associations with inflammation and PTSD severity. Psychiatry Research, 258, 330–336. https://doi.org/10.1016/j.psychres.2017.08.052
- Lindqvist, D., Wolkowitz, O. M., Mellon, S., Yehuda, R., Flory, J. D., Henn-Haase, C., Bierer, L. M., Abu-Amara, D., Coy, M., Neylan, T. C., Makotkine, I., Reus, V. I., Yan, X., Taylor, N. M., Marmar, C. R., & Dhabhar, F. S. (2014). Proinflammatory milieu in combat-related PTSD is independent of depression and early life stress. Brain, Behavior, and Immunity, 42, 81–88. https://doi.org/10.1016/j.bbi.2014.06.003
- Longui, C. A., Vottero, A., Adamson, P. C., Cole, D. E., Kino, T., Monte, O., & Chrousos, G. P. (2000). Low glucocorticoid receptor α/β ratio in T-cell lymphoblastic leukemia. Hormone and Metabolic Research, 32(10), 401–406. https://doi.org/10.1055/s-2007-978661
- Lopes, R. P., Grassi-Oliveira, R., de Almeida, L. R., Stein, L. M., Luz, C., Teixeira, A. L., & Bauer, M. E. (2012). Neuroimmunoendocrine interactions in patients with recurrent major depression, increased early life stress and long-standing posttraumatic stress disorder symptoms. Neuroimmunomodulation, 19(1), 33–42. https://doi.org/10.1159/000327352
- Manoli, I., Le, H., Alesci, S., McFann, K. K., Su, Y. A., Kino, T., Chrousos, G. P., & Blackman, M. R. (2005). Monoamine oxidase-A is a major target gene for glucocorticoids in human skeletal muscle cells. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology, 19(10), 1359–1361. https://doi.org/10.1096/fj.04-3660fje
- Manukhina, E. B., Tseilikman, V. E., Tseilikman, O. B., Komelkova, M. V., Kondashevskaya, M. V., Goryacheva, A. V., Lapshin, M. S., Platkovskii, P. O., Alliluev, A. V., & Downey, H. F. (2018). Intermittent hypoxia improves behavioral and adrenal gland dysfunction induced by posttraumatic stress disorder in rats. Journal of Applied Physiology (Bethesda, Md.: 1985)), 125(3), 931–937. https://doi.org/10.1152/japplphysiol.01123.2017
- Marik, P. (2007). Mechanisms and clinical consequences of critical illness associated adrenal insufficiency. Current Opinion in Critical Care, 13(4), 363–369. https://doi.org/10.1097/MCC.0b013e32818a6d74
- Matsumura, K., & Kobayashi, S. (2004). Signaling the brain in inflammation: The role of endothelial cells. Frontiers in Bioscience : A Journal and Virtual Library, 9, 2819–2826. https://doi.org/10.2741/1439
- McEwen, B. S. (2000). Allostasis and allostatic load: Implications for neuropsychopharmacology. Neuropsychopharmacology : Official Publication of the American College of Neuropsychopharmacology, 22(2), 108–124. https://doi.org/10.1016/S0893-133X(99)00129-3
- Merkulov, V. M., Merkulova, T. I., & Bondar, N. P. (2017). Mechanisms of brain glucocorticoid resistance in stress-induced psychopathologies. Biochemistry. Biokhimiia, 82(3), 351–365. https://doi.org/10.1134/S0006297917030142
- Meyer, J. H., Wilson, A. A., Sagrati, S., Miler, L., Rusjan, P., Bloomfield, P. M., Clark, M., Sacher, J., Voineskos, A. N., & Houle, S. (2009). Brain monoamine oxidase A binding in major depressive disorder: Relationship to selective serotonin reuptake inhibitor treatment, recovery, and recurrence. Archives of General Psychiatry, 66(12), 1304–1312. https://doi.org/10.1001/archgenpsychiatry.2009.156
- Michopoulos, V., Rothbaum, A. O., Jovanovic, T., Almli, L. M., Bradley, B., Rothbaum, B. O., Gillespie, C. F., & Ressler, K. J. (2015). Association of CRP genetic variation and CRP level with elevated PTSD symptoms and physiological responses in a civilian population with high levels of trauma. The American Journal of Psychiatry, 172(4), 353–362. https://doi.org/10.1176/appi.ajp.2014.14020263
- Miller, G. E., Chen, E., & Zhou, E. S. (2007). If it goes up, must it come down? Chronic stress and the hypothalamic-pituitary-adrenocortical axis in humans. Psychological Bulletin, 133(1), 25–45. https://doi.org/10.1037/0033-2909.133.1.25
- Ming, Z., Wotton, C. A., Appleton, R. T., Ching, J. C., Loewen, M. E., Sawicki, G., & Bekar, L. K. (2015). Systemic lipopolysaccharide-mediated alteration of cortical neuromodulation involves increases in monoamine oxidase-A and acetylcholinesterase activity. Journal of Neuroinflammation, 12, 37https://doi.org/10.1186/s12974-015-0259-y
- Molijn, G. J., Spek, J. J., van Uffelen, J. C., de Jong, F. H., Brinkmann, A. O., Bruining, H. A., Lamberts, S. W., & Koper, J. W. (1995). Differential adaptation of glucocorticoid sensitivity of peripheral blood mononuclear leukocytes in patients with sepsis or septic shock. The Journal of Clinical Endocrinology & Metabolism, 80(6), 1799–1803. https://doi.org/10.1210/jcem.80.6.7775626
- Morey, R. A., Haswell, C. C., Hooper, S. R., & Bellis, M. D. D. (2016). Amygdala, hippocampus, and ventral medial prefrontal cortex volumes differ in maltreated youth with and without chronic posttraumatic stress disorder. Neuropsychopharmacology : Official Publication of the American College of Neuropsychopharmacology, 41(3), 791–801. https://doi.org/10.1038/npp.2015.205
- Morrison, M. F., Ten Have, T., Freeman, E. W., Sammel, M. D., & Grisso, J. A. (2001). DHEA-S levels and depressive symptoms in a cohort of African American and Caucasian women in the late reproductive years. Biological Psychiatry, 50(9), 705–711. https://doi.org/10.1016/S0006-3223(01)01169-6
- Nikkheslat, N., Zunszain, P. A., Horowitz, M. A., Barbosa, I. G., Parker, J. A., Myint, A.-M., Schwarz, M. J., Tylee, A. T., Carvalho, L. A., & Pariante, C. M. (2015). Insufficient glucocorticoid signaling and elevated inflammation in coronary heart disease patients with comorbid depression. Brain, Behavior, and Immunity, 48, 8–18. https://doi.org/10.1016/j.bbi.2015.02.002
- O’Carroll, S. J., Kho, D. T., Wiltshire, R., Nelson, V., Rotimi, O., Johnson, R., Angel, C. E., & Graham, E. S. (2015). Pro-inflammatory TNFα and IL-1β differentially regulate the inflammatory phenotype of brain microvascular endothelial cells. Journal of Neuroinflammation, 12, 131https://doi.org/10.1186/s12974-015-0346-0
- Oldehinkel, A. J., van den Berg, M. D., Flentge, F., Bouhuys, A. L., ter Horst, G. J., & Ormel, J. (2001). Urinary free cortisol excretion in elderly persons with minor and major depression. Psychiatry Research, 104(1), 39–47. https://doi.org/10.1016/S0165-1781(01)00300-6
- Olsen, N., Sokka, T., Seehorn, C. L., Kraft, B., Maas, K., Moore, J., & Aune, T. M. (2004). A gene expression signature for recent onset rheumatoid arthritis in peripheral blood mononuclear cells. Annals of the Rheumatic Diseases, 63(11), 1387–1392. https://doi.org/10.1136/ard.2003.017194
- Ott, D., Murgott, J., Rafalzik, S., Wuchert, F., Schmalenbeck, B., Roth, J., & Gerstberger, R. (2010). Neurons and glial cells of the rat organum vasculosum laminae terminalis directly respond to lipopolysaccharide and pyrogenic cytokines. Brain Research, 1363, 93–106. https://doi.org/10.1016/j.brainres.2010.09.083
- Ou, X.-M., Chen, K., & Shih, J. C. (2006). Glucocorticoid and androgen activation of monoamine oxidase A is regulated differently by R1 and Sp1. Journal of Biological Chemistry, 281(30), 21512–21525. https://doi.org/10.1074/jbc.M600250200
- Pariante, C. M. (2017). Why are depressed patients inflamed? A reflection on 20 years of research on depression, glucocorticoid resistance and inflammation. European Neuropsychopharmacology : The Journal of the European College of Neuropsychopharmacology, 27(6), 554–559. https://doi.org/10.1016/j.euroneuro.2017.04.001
- Pariante, C. M., Pearce, B. D., Pisell, T. L., Sanchez, C. I., Po, C., Su, C., & Miller, A. H. (1999). The Proinflammatory Cytokine, interleukin-1alpha, reduces glucocorticoid receptor translocation and function. Endocrinology, 140(9), 4359–4366. https://doi.org/10.1210/endo.140.9.6986
- Paterson, J. M., Holmes, M. C., Kenyon, C. J., Carter, R., Mullins, J. J., & Seckl, J. R. (2007). Liver-Selective transgene rescue of hypothalamic-pituitary-adrenal axis dysfunction in 11β-hydroxysteroid dehydrogenase type 1-deficient mice. Endocrinology, 148, 961–966. https://doi.org/10.1210/en.2006-0603
- Pearson, J., Tarabulsy, G. M., & Bussières, E.-L. (2015). Fetal programming and cortisol secretion in early childhood: A meta-analysis of different programming variables. Infant Behavior and Development, 40, 204–215. https://doi.org/10.1016/j.infbeh.2015.04.004
- Perrin, A. J., Horowitz, M. A., Roelofs, J., Zunszain, P. A., & Pariante, C. M. (2019). Glucocorticoid resistance: Is it a requisite for increased cytokine production in depression? A systematic review and meta-analysis. Frontiers in Psychiatry, 10, 423. https://doi.org/10.3389/fpsyt.2019.00423
- Petrowski, K., Herold, U., Joraschky, P., Wittchen, H.-U., & Kirschbaum, C. (2010). A striking pattern of cortisol non-responsiveness to psychosocial stress in patients with panic disorder with concurrent normal cortisol awakening responses. Psychoneuroendocrinology, 35(3), 414–421. https://doi.org/10.1016/j.psyneuen.2009.08.003
- Pivac, N., Knezevic, J., Kozaric-Kovacic, D., Dezeljin, M., Mustapic, M., Rak, D., Matijevic, T., Pavelic, J., & Muck-Seler, D. (2007). Monoamine oxidase (MAO) intron 13 polymorphism and platelet MAO-B activity in combat-related posttraumatic stress disorder. Journal of Affective Disorders, 103(1-3), 131–138. https://doi.org/10.1016/j.jad.2007.01.017
- Plant, D. T., Pawlby, S., Sharp, D., Zunszain, P. A., & Pariante, C. M. (2016). Prenatal maternal depression is associated with offspring inflammation at 25 years: A prospective longitudinal cohort study. Translational Psychiatry, 6(11), e936https://doi.org/10.1038/tp.2015.155
- Preez, A. D., Leveson, J., Zunszain, P. A., & Pariante, C. M. (2016). Inflammatory insults and mental health consequences: Does timing matter when it comes to depression? Psychological Medicine, 46(10), 2041–2057. https://doi.org/10.1017/S0033291716000672
- Quinkler, M., Troeger, H., Eigendorff, E., Maser-Gluth, C., Stiglic, A., Oelkers, W., Bähr, V., & Diederich, S. (2003). Enhanced 11beta-hydroxysteroid dehydrogenase type 1 activity in stress adaptation in the guinea pig. The Journal of Endocrinology, 176(2), 185–192. https://doi.org/10.1677/joe.0.1760185
- Rohleder, N., Joksimovic, L., Wolf, J. M., & Kirschbaum, C. (2004). Hypocortisolism and increased glucocorticoid sensitivity of pro-Inflammatory cytokine production in Bosnian war refugees with posttraumatic stress disorder. Biological Psychiatry, 55(7), 745–751. https://doi.org/10.1016/j.biopsych.2003.11.018
- Sarkar, S., Tsai, S.-W., Nguyen, T. T., Plevyak, M., Padbury, J. F., & Rubin, L. P. (2001). Inhibition of placental 11β-hydroxysteroid dehydrogenase type 2 by catecholamines via α-adrenergic signaling. American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, 281(6), R1966–R1974. https://doi.org/10.1152/ajpregu.2001.281.6.R1966
- Schmidt, M., Weidler, C., Naumann, H., Anders, S., Schölmerich, J., & Straub, R. H. (2005). Reduced capacity for the reactivation of glucocorticoids in rheumatoid arthritis synovial cells: Possible role of the sympathetic nervous system? Arthritis and Rheumatism, 52(6), 1711–1720. https://doi.org/10.1002/art.21091
- Sesti-Costa, R., Ignacchiti, M. D. C., Chedraoui-Silva, S., Marchi, L. F., & Mantovani, B. (2012). Chronic cold stress in mice induces a regulatory phenotype in macrophages: Correlation with increased 11β-hydroxysteroid dehydrogenase expression. Brain, Behavior, and Immunity, 26(1), 50–60. https://doi.org/10.1016/j.bbi.2011.07.234
- Slotkin, T. A., Seidler, F. J., & Ritchie, J. C. (1998). Effects of aging and glucocorticoid treatment on monoamine oxidase subtypes in rat cerebral cortex: Therapeutic implications. Brain Research Bulletin, 47(4), 345–348. https://doi.org/10.1016/S0361-9230(98)00111-7
- Smith, M. A., Davidson, J., Ritchie, J. C., Kudler, H., Lipper, S., Chappell, P., & Nemeroff, C. B. (1989). The corticotropin-releasing hormone test in patients with posttraumatic stress disorder. Biological Psychiatry, 26(4), 349–355. https://doi.org/10.1016/0006-3223(89)90050-4
- Soliman, A., Udemgba, C., Fan, I., Xu, X., Miler, L., Rusjan, P., Houle, S., Wilson, A. A., Pruessner, J., Ou, X.-M., & Meyer, J. H. (2012). Convergent effects of acute stress and glucocorticoid exposure upon MAO-A in humans. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 32(48), 17120–17127. https://doi.org/10.1523/JNEUROSCI.2091-12.2012
- Sommershof, A., Aichinger, H., Engler, H., Adenauer, H., Catani, C., Boneberg, E.-M., Elbert, T., Groettrup, M., & Kolassa, I.-T. (2009). Substantial reduction of naïve and regulatory T cells following traumatic stress. Brain, Behavior, and Immunity, 23(8), 1117–1124. https://doi.org/10.1016/j.bbi.2009.07.003
- Stegk, J. P., Ebert, B., Martin, H.-J., & Maser, E. (2009). Expression profiles of human 11beta-hydroxysteroid dehydrogenases type 1 and type 2 in inflammatory bowel diseases. Molecular and Cellular Endocrinology, 301(1-2), 104–108. https://doi.org/10.1016/j.mce.2008.10.030
- Stein, M. B., Yehuda, R., Koverola, C., & Hanna, C. (1997). Enhanced dexamethasone suppression of plasma cortisol in adult women traumatized by childhood sexual abuse. Biological Psychiatry, 42(8), 680–686. https://doi.org/10.1016/S0006-3223(96)00489-1
- Straub, R. H., & Cutolo, M. (2016). Glucocorticoids and chronic inflammation. Rheumatology (Oxford, England), 55(suppl 2), ii6–ii14. https://doi.org/10.1093/rheumatology/kew348
- Ströhle, A., Scheel, M., Modell, S., & Holsboer, F. (2008). Blunted ACTH response to dexamethasone suppression-CRH stimulation in posttraumatic stress disorder. Journal of Psychiatric Research, 42(14), 1185–1188. https://doi.org/10.1016/j.jpsychires.2008.01.015
- Teche, S. P., Rovaris, D. L., Aguiar, B. W., Hauck, S., Vitola, E. S., Bau, C. H. D., Freitas, L. H., & Grevet, E. H. (2017). Resilience to traumatic events related to urban violence and increased IL10 serum levels. Psychiatry Research, 250, 136–140. https://doi.org/10.1016/j.psychres.2017.01.072
- Tseilikman, V., Dremencov, E., Tseilikman, O., Pavlovicova, M., Lacinova, L., & Jezova, D. (2019). Role of glucocorticoid- and monoamine-metabolizing enzymes in stress-related psychopathological processes. Stress (Amsterdam, Netherlands), 1–12. https://doi.org/10.1080/10253890.2019.1641080
- Tseilikman, V., Dremencov, E., Tseilikman, O., Pavlovicova, M., Lacinova, L., & Jezova, D. (2020). Role of glucocorticoid- and monoamine-metabolizing enzymes in stress-related psychopathological processes. Stress, 23(1), 1–12. https://doi.org/10.1080/10253890.2019.1641080
- Tsugita, M., Iwasaki, Y., Nishiyama, M., Taguchi, T., Shinahara, M., Taniguchi, Y., Kambayashi, M., Terada, Y., & Hashimoto, K. (2008). Differential regulation of 11beta-hydroxysteroid dehydrogenase type-1 and -2 gene transcription by proinflammatory cytokines in vascular smooth muscle cells. Life Sciences, 83(11-12), 426–432. https://doi.org/10.1016/j.lfs.2008.07.005
- van der Voort, P. H. J., Gerritsen, R. T., Bakker, A. J., Boerma, E. C., Kuiper, M. A., & de Heide, L. (2003). HDL-cholesterol level and cortisol response to synacthen in critically ill patients. Intensive Care Med, 29, 2199–2203. https://doi.org/10.1007/s00134-003-2021-7
- Vilibić, M., Jukić, V., Pandžić-Sakoman, M., Bilić, P., & Milošević, M. (2014). Association between total serum cholesterol and depression, aggression, and suicidal ideations in war veterans with posttraumatic stress disorder: A cross-sectional study. Croatian Medical Journal, 55(5), 520–529. https://doi.org/10.3325/cmj.2014.55.520
- Von, R. K., Hepp, U., Kraemer, B., Traber, R., Keel, M., Mica, L., & Schnyder, U. (2007). Evidence for low-grade systemic proinflammatory activity in patients with posttraumatic stress disorder. Journal of Psychiatric Research, 41(9), 744–752. https://doi.org/10.1016/j.jpsychires.2006.06.009
- Walczewska, J., Rutkowski, K., Wizner, B., Cwynar, M., & Grodzicki, T. (2011). Stiffness of large arteries and cardiovascular risk in patients with post-traumatic stress disorder. European Heart Journal, 32(6), 730–736. https://doi.org/10.1093/eurheartj/ehq354
- Wang, Z., & Young, M. R. I. (2016). PTSD, a Disorder with an Immunological Component. Frontiers in Immunology, 7, 219https://doi.org/10.3389/fimmu.2016.00219
- Webster, J. C., Oakley, R. H., Jewell, C. M., & Cidlowski, J. A. (2001). Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: A mechanism for the generation of glucocorticoid resistance. Proceedings of the National Academy of Sciences of the United States of America, 98(12), 6865–6870. https://doi.org/10.1073/pnas.121455098
- Whitaker, A. M., Farooq, M. A., Edwards, S., & Gilpin, N. W. (2016). Post-traumatic stress avoidance is attenuated by corticosterone and associated with brain levels of steroid receptor co-activator-1 in rats. Stress (Amsterdam, Netherlands), 19(1), 69–77. https://doi.org/10.3109/10253890.2015.1094689
- Wong, M. L., & Licinio, J. (1994). Localization of interleukin 1 type I receptor mRNA in rat brain. Neuroimmunomodulation, 1(2), 110–115. https://doi.org/10.1159/000097143
- Woon, F. L., & Hedges, D. W. (2008). Hippocampal and amygdala volumes in children and adults with childhood maltreatment-related posttraumatic stress disorder: A meta-analysis. Hippocampus, 18(8), 729–736. https://doi.org/10.1002/hipo.20437
- Yaguchi, H., Tsutsumi, K., Shimono, K., Omura, M., Sasano, H., & Nishikawa, T. (1998). Involvement of high density lipoprotein as substrate cholesterol for steroidogenesis by bovine adrenal fasciculo-reticularis cells. Life Sciences, 62(16), 1387–1395. https://doi.org/10.1016/S0024-3205(98)00077-0
- Yehuda, R. (2001). Biology of posttraumatic stress disorder. The Journal of Clinical Psychiatry, 62 (Suppl 17), 41–46.
- Yehuda, R., Boisoneau, D., Lowy, M. T., & Giller, E. L. (1995). Dose-response changes in plasma cortisol and lymphocyte glucocorticoid receptors following dexamethasone administration in combat veterans with and without posttraumatic stress disorder. Archives of General Psychiatry, 52(7), 583–593. https://doi.org/10.1001/archpsyc.1995.03950190065010
- Yehuda, R., Golier, J. A., Yang, R. K., & Tischler, L. (2004). Enhanced sensitivity to glucocorticoids in peripheral mononuclear leukocytes in posttraumatic stress disorder. Biological Psychiatry, 55(11), 1110–1116. https://doi.org/10.1016/j.biopsych.2004.02.010
- Yehuda, R., Halligan, S. L., Grossman, R., Golier, J. A., & Wong, C. (2002). The cortisol and glucocorticoid receptor response to low dose dexamethasone administration in aging combat veterans and holocaust survivors with and without posttraumatic stress disorder. Biological Psychiatry, 52(5), 393–403. https://doi.org/10.1016/S0006-3223(02)01357-4
- Yehuda, R., Lowy, M. T., Southwick, S. M., Shaffer, D., & Giller, E. L. (1991). Lymphocyte glucocorticoid receptor number in posttraumatic stress disorder. The American Journal of Psychiatry, 148(4), 499–504. https://doi.org/10.1176/ajp.148.4.499
- Yehuda, R., Southwick, S. M., Krystal, J. H., Bremner, D., Charney, D. S., & Mason, J. W. (1993). Enhanced suppression of cortisol following dexamethasone administration in posttraumatic stress disorder. The American Journal of Psychiatry, 150(1), 83–86. https://doi.org/10.1176/ajp.150.1.83
- Yirmiya, R., & Goshen, I. (2011). Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain, Behavior, and Immunity, 25(2), 181–213. https://doi.org/10.1016/j.bbi.2010.10.015
- Ysrraelit, M. C., Gaitán, M. I., Lopez, A. S., & Correale, J. (2008). Impaired hypothalamic-pituitary-adrenal axis activity in patients with multiple sclerosis. Neurology, 71(24), 1948–1954. https://doi.org/10.1212/01.wnl.0000336918.32695.6b
- Zaba, M., Kirmeier, T., Ionescu, I. A., Wollweber, B., Buell, D. R., Gall-Kleebach, D. J., Schubert, C. F., Novak, B., Huber, C., Köhler, K., Holsboer, F., Pütz, B., Müller-Myhsok, B., Höhne, N., Uhr, M., Ising, M., Herrmann, L., & Schmidt, U. (2015). Identification and characterization of HPA-axis reactivity endophenotypes in a cohort of female PTSD patients. Psychoneuroendocrinology, 55, 102–115. https://doi.org/10.1016/j.psyneuen.2015.02.005
- Zallocchi, M., Matkovic, L., & Damasco, M. C. (2004). Adrenal 11-beta hydroxysteroid dehydrogenase activity in response to stress. Canadian Journal of Physiology and Pharmacology, 82(6), 422–425. https://doi.org/10.1139/y04-035
- Zhou, J., Nagarkatti, P., Zhong, Y., Ginsberg, J. P., Singh, N. P., Zhang, J., & Nagarkatti, M. (2014). Dysregulation in microRNA expression is associated with alterations in immune functions in combat veterans with post-traumatic stress disorder. PLOS One, 9(4), e94075https://doi.org/10.1371/journal.pone.0094075
- Ziegler, C., Wolf, C., Schiele, M. A., Feric Bojic, E., Kucukalic, S., Sabic Dzananovic, E., Goci Uka, A., Hoxha, B., Haxhibeqiri, V., Haxhibeqiri, S., Kravic, N., Muminovic Umihanic, M., Cima Franc, A., Jaksic, N., Babic, R., Pavlovic, M., Warrings, B., Bravo Mehmedbasic, A., Rudan, D., … Domschke, K. (2018). Monoamine oxidase A gene methylation and its role in posttraumatic stress disorder: First evidence from the South Eastern Europe (SEE)-PTSD Study. International Journal of Neuropsychopharmacology, 21(5), 423–432. https://doi.org/10.1093/ijnp/pyx111