
Abstract
This study investigated epigenetic risk factors that may contribute to stress-related cardiac disease in a rodent model. Experiment 1 was designed to evaluate the expression of microRNA-34a (miR-34a), a known modulator of both stress responses and cardiac pathophysiology, in the heart of male adult rats exposed to a single or repeated episodes of social defeat stress. Moreover, RNA sequencing was conducted to identify transcriptomic profile changes in the heart of repeatedly stressed rats. Experiment 2 was designed to assess cardiac electromechanical changes induced by repeated social defeat stress that may predispose rats to cardiac dysfunction. Results indicated a larger cardiac miR-34a expression after repeated social defeat stress compared to a control condition. This molecular modification was associated with increased vulnerability to pharmacologically induced arrhythmias and signs of systolic left ventricular dysfunction. Gene expression analysis identified clusters of differentially expressed genes in the heart of repeatedly stressed rats that are mainly associated with morphological and functional properties of the mitochondria and may be directly regulated by miR-34a. These results suggest the presence of an association between miR-34a overexpression and signs of adverse electromechanical remodeling in the heart of rats exposed to repeated social defeat stress, and point to compromised mitochondria efficiency as a potential mediator of this link. This rat model may provide a useful tool for investigating the causal relationship between miR-34a expression, mitochondrial (dys)function, and cardiac alterations under stressful conditions, which could have important implications in the context of stress-related cardiac disease.
Introduction
Extensive research has shown that chronic psychosocial stress can favor the onset and progression of cardiovascular disease (CVD) in both patients with established disease and individuals with no prior history (Bairey Merz et al., Citation2002; Rozanski et al., Citation1999). Notably, the magnitude of psychosocial stress-related CVD risk has been reported to be comparable with that attributed to traditional CVD risk factors (Rosengren et al., Citation2004). Potential autonomic, hormonal, and inflammatory mediators of this link have been identified (e.g. Black & Garbutt, Citation2002; Girod & Brotman, Citation2004). However, current scientific knowledge does not completely explain the complex pathophysiology underlying psychosocial stress-induced CVD.
It is becoming increasingly clear that epigenetic modulators might constitute an additional pathway leading to adverse cardiac remodeling under psychosocial stress conditions and therefore contribute to disease (Muka et al., Citation2016; Saban et al., Citation2014). Thus, a better understanding of these players may help identify novel targets for the prevention and timely treatment of psychosocial stress-related CVD. Cardiac transcriptional pathways are intimately regulated by microRNAs (miRNAs) (Small & Olson, Citation2011). MiRNAs are small noncoding RNAs, approximately 18–25 nucleotides in length at mature stage, which can mediate post-transcriptional gene repression by inhibiting translation and/or promoting degradation of target protein-coding mRNA by base pairing (Humphreys et al., Citation2005). Importantly, miRNA profiling in cardiac tissue of mouse models of CVD and human biopsies has revealed signature patterns of miRNAs dysregulation in myocardial infarction (Lin et al., Citation2010; Sun et al., Citation2017; van Rooij et al., Citation2008), end-stage heart failure (Ikeda et al., Citation2007), and cardiomyopathy (Quattrocelli et al., Citation2013; Sucharov et al., Citation2008), opening up a new field of investigation to understand the molecular mechanisms controlling gene expression also in the context of psychosocial stress-related CVD. Indeed, studies have shown that stressful events are associated with alterations in miRNAs biogenesis, which in turn could influence gene expression in response to stress (Leung et al., Citation2011; Malan-Muller et al., Citation2013).
In the current study, we focused on one specific family of miRNAs, the miR-34 family, and evaluated whether psychosocial stress exposure influences the cardiac expression of one of its members – the miR-34a isoform – in a rodent model. The rationale behind the choice of miR-34a as a key target of our investigation was based on the following pieces of evidence: (i) miR-34a expression is elevated in cardiac tissue of patients with heart disease or cardiac injury (Greco et al., Citation2012; Thum et al., Citation2007); (ii) likewise, miR-34a cardiac expression is augmented in a mouse model of myocardial infarction (Yang et al., Citation2015); (iii) inhibition of miR-34a through anti-miR-34a therapy improves cardiac function and reverses cardiac remodeling in mouse models of myocardial infarction or preexisting pathological hypertrophy (Bernardo et al., Citation2014; Yang et al., Citation2015), and in a rat model of doxorubicin cardiotoxicity (Piegari et al., Citation2020); (iv) miR-34a expression is elevated in the medial prefrontal cortex of mice exposed to acute stress exposure (Andolina et al., Citation2016); (v) knockout of the miR-34 family fosters behavioral resilience after acute and chronic stress exposure (Andolina et al., Citation2016, Citation2018).
However, despite such accumulating evidence on the potential role of miR-34a in cardiac pathophysiology (Boon et al., Citation2013) as well as in mediating neurochemical and behavioral stress responses (Andolina et al., Citation2017), to the best of our knowledge, no studies have investigated the effects of stress exposure on miR-34a cardiac expression. Here, we adopted a rodent model of repeated social defeat stress, a highly translational, naturalistic experimental paradigm which leads to severe consequences on the electrical stability and morpho-structural properties of the heart that are indicative of enhanced CVD risk (reviewed in Sgoifo et al., Citation2014). Anticipating signs of elevated miR-34a expression in the heart of rats exposed to repeated social defeat, we sought to characterize changes in putative miR34-a gene targets, to identify epigenetic risk factors of stress-induced CVD. Moreover, we aimed to investigate whether elevated miR-34a cardiac expression following repeated social defeat was coupled with specific alterations in (i) arrhythmia vulnerability, (ii) hemodynamic parameters, and (iii) myocardial weights.
Materials and methods
Animals
Experiments were conducted on 3-month-old male wild-type Groningen rats. This rat population, originally derived from the University of Groningen (The Netherlands) and currently bred in our laboratory under standard conditions, shows considerable individual differences in cardiovascular responses to environmental challenges (Sgoifo et al., Citation2005). Experimental rats (n = 17 for Experiment 1 and n = 14 for Experiment 2) were housed individually 1 week before the beginning of the procedures. Additional older (10–12 month-old) male wild-type Groningen rats (n = 8) were pair-housed in a different room with an oviduct-ligated female partner and used as residents in the resident-intruder paradigm (see below for details). All animals were kept in climate-controlled rooms, with a 12-h light cycle (lights on at 7 pm). Food and water were available ad libitum. Experiments were performed in accordance with the European Community Council Directive 2010/63/UE and approved by the Italian legislation on animal experimentation (D.L. 04/04/2014, n. 26, authorization n. 449/2017-PR). All efforts were made to reduce sample size and minimize animal suffering.
Social defeat stress
Social defeat stress was based on a classical “resident–intruder” paradigm (Miczek, Citation1979). Resident rats were screened for their level of aggression prior to inclusion in the study. Experimental rats were randomly assigned to the “intruder” or “control” group. Briefly, each intruder was transferred to the cage of a resident rat for 30 min after the removal of the resident’s female partner. Intruders were threatened and physically assaulted by the resident rat. After exhibiting signs of subordination and social defeat (i.e. when the intruder rat assumed a supine posture that was held for at least 5 s) or after 15 min, whichever came first, intruders were confined behind a Plexiglas partition within the resident’s cage for the remainder of the 30-min period, allowing for psychogenic exposure to aggressive threats without physical harm. At the end of the social defeat session, which took place between 10:00 and 11:00 am, intruders were returned to their home cages. Intruders were exposed to either one episode of social defeat stress (Experiment 1) or eight episodes on consecutive days (Experiments 1 and 2). In case of repeated episodes, intruders were exposed each day to a different resident, with a rotational design. Control rats were not present in the room during the social defeat procedure and remained in their home cages. Control manipulation consisted of handling for 60 s by the same experimenter and occurred in correspondence of each social defeat episode. Cage cleaning was done weekly for all animals by the same experimenter at the same time of the day.
Experiment 1
This experiment was designed to evaluate changes in miR-34a levels and gene expression in the heart of rats exposed to a single or repeated social defeat episodes. Rats were randomly assigned to the single social defeat (SSD, n = 5), repeated social defeat (RSD, n = 6), or control (CTR = 6) groups as described above. The CTR group served as a reference group for both the SSD and RSD groups. Twenty-four hours after the first social defeat (SSD group) or 24 h after the last social defeat/control procedure (RSD and CTR groups, respectively), rats were anesthetized using inhaled isoflurane (2% in 100% oxygen) (Zoetis, Italy). The hearts were quickly excised and perfused with a 0.9% NaCl solution to flush the residual blood from the tissue, immediately flash frozen in liquid nitrogen, and stored at −80 °C until molecular analysis.
RNA purification
Punches of the left ventricle were dissected using stainless-steel tubes (inside diameter: 2 mm). Total RNA was extracted using total RNA purification Kit (NorgenBiotek, Thorold, Canada) according to the manufacturer’s protocols. RNA quantity was determined by absorbance at 260 nm using a NanoDrop UV–Vis spectrophotometer. The same RNA samples were used both for the quantification of miR-34a expression via quantitative real time PCR analysis and for Next Generation Quant-Seq RNA analysis. Samples showing as outliers in a PCA (n = 1 for the RSD and n = 1 for the CTR group, Supplementary Figure 1) were excluded from these analyses.
Quantitative real-time RT-PCR (qPCR)
Total RNA (10 ng) from each punch sample was reverse transcribed to cDNA for miR-34a and the endogenous control sno135, using the TaqMan MicroRNA Reverse Transcription kit (Taqman Assay ID 000426 and 001230, Thermo Fisher Scientific, Waltham, MA, USA). cDNA templates (around 0.7 ng per sample) were amplified by qPCR, using the Thermo Fisher QuantStudio 3 thermal cycler apparatus. Cycle threshold (CT) miR-34a values were normalized to sno135 CT values. All data were run in triplicate and expressed as fold changes according to the ΔΔC(t) method (Schmittgen & Livak, Citation2008).
RNA library preparation, sequencing, and data analysis
mRNA sequencing was performed by NGD technology service (Pozzuoli, Italy). Total RNA from each heart sample was quantified using the Qubit 2.0 fluorimetric Assay (Thermo Fisher Scientific, Waltham, MA) and its quality was evaluated by Agilent 2100 Bioanalyzer RNA assay (Agilent Technologies, Santa Clara, CA, USA). Only RNA samples with RIN value >7.0 were included in the study. Libraries were prepared from 100 ng of total RNA using the QuantSeq 3′ mRNA-Seq Library Prep Kit FWD for Illumina (Lexogen GmbH). Quality of libraries was assessed by using screen tape High sentisivity DNA D1000 (Agilent Technologies, Santa Clara, CA, USA). Libraries were sequenced on a NovaSeq 6000 S1 system (flow cell: 100 cycles; Illumina Inc., San Diego, CA, USA). Illumina novaSeq base call (BCL) files were converted in fastq file through bcl2fastq (version v2.20.0.422). Sequence reads were trimmed using bbduk software (bbmap suite 37.31) to remove adapter sequences, poly-A tails, and low-quality end bases (regions with an average quality below 6). Alignment was performed with STAR 2.6.0a (Dobin et al., Citation2013) on rn6 assembly downloaded from UCSC Genome Browser website. Gene expression levels were determined with htseq-count 0.9.1 (Anders et al., Citation2015) by using Ensembl genes annotations (Version 6.0). Expression data were analyzed by Rosalind (https://rosalind.onramp.bio/), with a HyperScale architecture developed by OnRamp BioInformatics, Inc. (San Diego, CA, USA). Individual sample counts were normalized by Relative Log Expression (RLE) using DESeq2 R library (Love et al., Citation2014). Read Distribution percentages, violin plots, identity heatmaps, and sample MDS plots were generated as part of the QC step. Gene Ontology and pathway enrichments analyses were performed on those genes whose fold changes corrected p values were <.10, using the DAVID web-based tool [http://david.abcc.ncifcrf.gov/] (Huang et al., Citation2009).
The Quant-seq dataset is available from the GEO database under the accession number GSE169181.
Experiment 2
This experiment was designed to evaluate potential alterations in (i) vulnerability to arrhythmias, (ii) hemodynamic parameters, and (iii) myocardial weights in rats exposed to repeated episodes of social defeat. Rats were randomly assigned to RSD (n = 7) or CTR (n = 7) conditions as described above. Two weeks before the beginning of the social defeat stress/control procedure, rats were implanted, under isoflurane anesthesia (2% in 100% oxygen) (Zoetis, Italy), with radiotelemetric transmitters (TA11CTA-F40, DataSciences International, St. Paul, MN) for ECG recordings (sampling frequency 1000 Hz). The transmitter body was placed in the abdominal cavity; one electrode was fixed to the dorsal surface of the xyphoid process and another electrode was placed in the anterior mediastinum close to the right atrium, according to a previously described procedure (Sgoifo et al., Citation1996). Animals were housed individually post-surgery and were prophylactically injected for 2 days with gentamicine sulfate (Aagent, Fatro, Italy, 0.2 mL/kg, s.c.) and flunixin (Megluflen, Izo, Italy, 0.2 mL/kg, s.c.).
Vulnerability to cardiac arrhythmias
Twenty-four hours after the last social defeat/control procedure, rats were injected with the drug isoproterenol (β-adrenoceptor agonist, Sigma, St. Louis, MO, USA) at a dose (0.2 mg/kg, i.p.) known to induce arrhythmias in this rat strain (Carnevali et al., Citation2019). ECG signals were recorded during the hour that preceded and the hour that followed drug administration. The occurrence of arrhythmic events was determined offline via visual inspection of ECG traces using ChartPro 5.0 software by a trained experimenter according to (Curtis et al., Citation2013). Specifically, arrhythmic events were assigned the following point values according to (Grippo et al., Citation2012): (a) 1 point for isolated supraventricular events (premature atrial, junctional, or atrioventricular block) or an isolated premature ventricular or ventricular escape complex; (b) 2 points for supraventricular tachycardia or two or three consecutive premature ventricular complexes (salvo); (c) 3 points for premature ventricular beats in a trigeminal or bigeminal pattern or non-sustained ventricular tachycardia (defined as ≥3 and ≤10 consecutive premature ventricular contractions); (d) 4 points for sustained ventricular tachycardia (defined as >10 consecutive premature ventricular contractions) or asystole (absence of electrical activity during the loss of two or more consecutive cardiac cycles as defined by the length of the two preceding cardiac cycles). Using these categories, an arrhythmic burden was calculated for each rat, defined as the sum of the scored arrhythmic events during the hour that followed isoprotorenol injection. Moreover, we calculated the mean heart rate (reported in beats per minute, bpm) before and after isoproterenol injection.
Hemodynamic assessment
Three days after the isoproterenol challenge, hemodynamic data were recorded in RSD and CTR rats under ketamine chloride (Imalgene, Merial, Milan, Italy; 40 mg/kg i.p.) plus medetomidine hydrochloride (Domitor, Pfizer Italia S.r.l., Latina, Italy; 0.15 mg/kg i.p.) anesthesia. A microtip pressure transducer catheter (Millar SPC-320, Millar Instruments, Houston, TX) was inserted into the right carotid artery and connected to a recording system (Power Laboratory ML 845/4 channels, 2Biological Instruments, Besozzo, Italy) to collect systolic and diastolic blood pressures and heart rate. The catheter was then advanced into the left ventricle to measure left ventricular (LV) systolic pressure (LVSP), LV end-diastolic pressure (LVEDP), the peak rate of rise (+dP/dtmax) and decline (−dP/dtmax) of LV pressure (indexes of myocardial efficiency), isovolumic contraction time (IVCT), ejection time (ET; approximated as the time interval between aortic valve opening and time of − dP/dtmax), and LV relaxation time (LVRT, computed from − dP/dtmax to 5 mmHg above LVEDP and taken as index of isovolumic relaxation) using the software package AcqKnowledge 3.9 (Biopac Systems, Goleta, CA, USA). Finally, we computed the myocardial performance index (MPI) as the ratio between total time spent in isovolumic activity (IVCT + isovolumic relaxation time) and ET (Salemi et al., Citation2004). MPI is considered to reflect global cardiac function and possesses prognostic value in several pathologic conditions such as myocardial hypertrophy and infarction (Salemi et al., Citation2004).
Myocardial weights
After hemodynamic assessment, the heart of RSD and CTR rats was rapidly excised, perfused with a 0.9% NaCl solution to flush the residual blood from the tissue and weighed. The left and right ventricles were then separated and weighed. Myocardial weights were corrected for body weight and heart weight.
Statistical analysis
All statistical analyses were performed using the IBM SPSS statistical package (International Business Machines Corporation, Armonk, NY, USA, version 25). Normal distribution of variables was checked by means of the Kolmogorov–Smirnov test. To test for the effects of stress exposure on cardiac miR-34a expression (Experiment 1), a one-way ANOVA was applied (between subject factor “stress exposure,” three levels: CTR, SSD, and RSD) followed by Bonferroni's post-hoc comparisons. To test for the effects of stress exposure on cardiovascular data (Experiment 2), a series of unpaired Student’s t tests (RSD versus CTR rats) were applied. Data are reported as means ± standard error of the mean (SEM). The statistical significance was set at p<.05.
Results
Experiment 1
Repeated social defeat stress promotes the expression of miR-34a in cardiac tissue
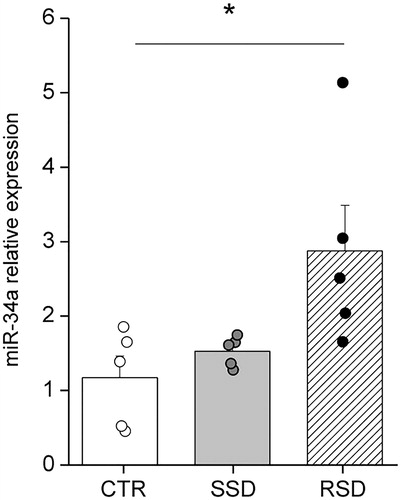
First, we tested whether the expression of miR-34a in the left ventricle was altered by a single episode and/or repeated episodes of social defeat stress. One factor ANOVA revealed a significant effect of stress exposure on miR-34a expression (F(2,13)=4.167, p<.05). Specifically, no significant changes were found in the cardiac expression of miR-34a between SSD and CTR rats. However, RSD rats showed a significantly larger expression of miR-34a compared to CTR rats (p<.05) ().
Figure 1. Relative expression of miR-34a in the left ventricle of control (CTR, n = 5), single social defeat (SSD, n = 5), and repeated social defeat (RSD, n = 5) rats. The CTR group served as a reference group for both the SSD and RSD groups. Values are reported as means ± SEM. *Indicates a significant difference (p<.05, Bonferroni post-hoc test).
mRNA-sequencing revealed different molecular targets potentially regulated by miR-34a in the cardiac tissue of rats exposed to repeated social defeat
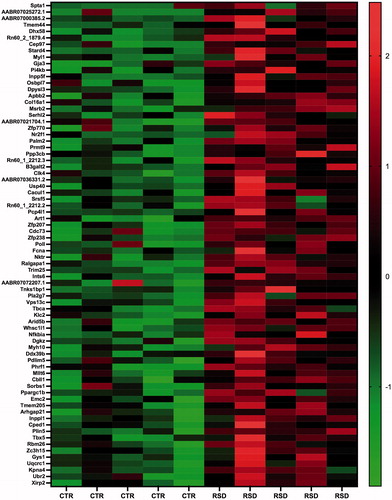
We aimed at evaluating transcriptional alterations induced by repeated social defeat stress in the left ventricle and at extracting genes that may be putatively targeted by miR-34a. Thus, we performed genome-wide Quant-Seq mRNA sequencing on the same samples used for miR-34a measures. We found 378 differentially expressed genes (DEGs) (adjusted p<.10) between RSD and CTR rats, of which 214 were down- and 164 were up-regulated, respectively, in RSD rats (Supplementary Tables 1 and 2). A subset of down- and up-regulated genes in RSD rats with higher statistical significance (adjusted p<.05) and their fold changes are illustrated in the heatmaps of and , respectively.
Figure 2. Heatmap showing a subset of down-regulated genes between control (CTR, n = 5) and repeated social defeat stress (RSD, n = 5) rats with the highest statistical significance (adjusted p<.05). Color intensity is proportional to the individual level of expression (normalized reads count).

Figure 3. Heatmap showing a subset of up-regulated genes between control (CTR, n = 5) and repeated social defeat stress (RSD, n = 5) rats with the highest statistical significance (adjusted p<.05). Color intensity is proportional to the individual level of expression (normalized reads count).
To unravel the physiological functions that are mostly represented by these altered transcripts, we performed Gene Ontology analysis and found several significantly enriched biological functions (adjusted p<.05) that were grouped in three main annotation clusters (). The first cluster included the “mitochondrion” as the main keyword, the second cluster comprised several neurodegenerative diseases and mitochondrial-related components or pathways like oxidative phosphorylation, mitochondrial respiratory chain complex I, mitochondrial inner membrane and cytochrome-c oxidase activity, and the third cluster included the cellular main signaling component phosphatidylinositol triphosphate.
Table 1. Gene Ontology analysis performed on the 378 differentially expressed genes between repeated social defeat stress rats and control rats.
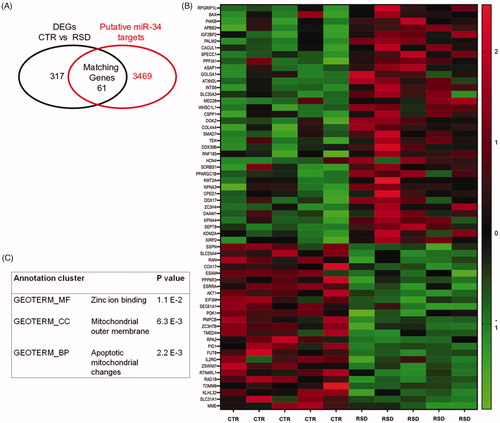
Furthermore, among the 378 DEGs, we aimed at identifying transcripts that may be directly regulated by miR-34a. With the aid of bioinformatic web-based predictor tool “Target Scan” (http://www.targetscan.org/vert_72/), we overlapped the list of DEGs between RSD and CTR rats with web-predicted miR-34s targets and found 61 matching genes (), which are reported in the heatmap of . Interestingly, Gene Ontology analysis performed on these 61 genes revealed several significantly enriched biological functions (p<.05), including the molecular function “zinc ion binding,” the cellular component “mitochondrial outer membrane,” and the biological process “apoptotic mitochondrial changes” ().
Figure 4. Panel A reports the number of matching genes (n = 61) that resulted from the comparison between the 378 differentially expressed (DEGs) found in control (CTR, n = 5) versus repeated social defeated (RSD, n = 5) rats, and those genes (n = 3530) that were predicted as putative miR-34 targets by TargetScan. Panel B shows the heatmap with the list of the 61 matching genes; color intensity is proportional to the individual level of expression (normalized reads count). Panel C reports the results of the Gene Ontology analysis on the 61 matching genes.
Experiment 2
Repeated social defeat promotes adverse electromechanical remodeling of the heart
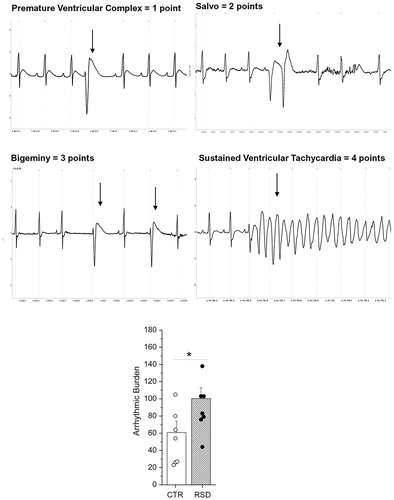
In this experiment, we aimed at documenting the effects of repeated social defeat stress on cardiac function. Following potent sympathetic stimulation with isoproterenol, RSD rats displayed a significantly higher arrhythmic burden compared with CTR rats (+65%, p<.05) (). No group differences were observed in mean heart rate values after isoproterenol administration (RSD: 475 ± 8 bpm versus CTR: 466 ± 10 bpm).
Figure 5. Top and middle panels report electrocardiographic examples of the most common forms of arrhythmias observed and classified in the rat. Classification criteria are defined in the methods according to Grippo et al. (Citation2012). Arrows denote arrhythmic events. Bottom panel represents the arrhythmic burden following isoproterenol injection in rats exposed to repeated social defeat (RSD, n = 7) or control procedure (CTR, n = 7). Values are reported as means ± SEM. *Indicates a significant difference (p<.05, unpaired Student’s t test).
We could not assess hemodynamic parameters in one RSD rat, which died just after the insertion of the catheter into the right carotid artery. RSD and CTR rats showed similar mean heart rate values under anesthesia (RSD: 242 ± 9 bpm versus CTR: 227 ± 8 bpm). Signs of systolic ventricular dysfunction were found in RSD rats compared with CTRs, as indicated by the significant reduction in the maximal rate of LV pressure rise during isovolumic contraction (+dP/dtmax) (−12%; p<.05), the significant prolongation of IVCT (+17%; p<.05), and the significant increase in MPI (+20%, p<.05) (). In addition, a decrease in ET (−9%) was observed in RSD rats compared with CTRs, although the difference did not reach statistical significance (p=.09) (). No significant differences between the two groups were found for − dP/dtmax (), systolic, and diastolic blood pressures, LVSP, LVEDP, and LVRT ().
Figure 6. Hemodynamic parameters in rats exposed to repeated social defeat (RSD, n = 6) or control procedure (CTR, n = 7). Values are reported as means ± SEM. +dP/dtmax: maximal rate of ventricular pressure rise; −dP/dtmax: maximal rate of ventricular pressure decline; IVCT: isovolumic contraction time; MPI: myocardial performance index. *Indicates a significant difference (p<.05, unpaired Student’s t test).
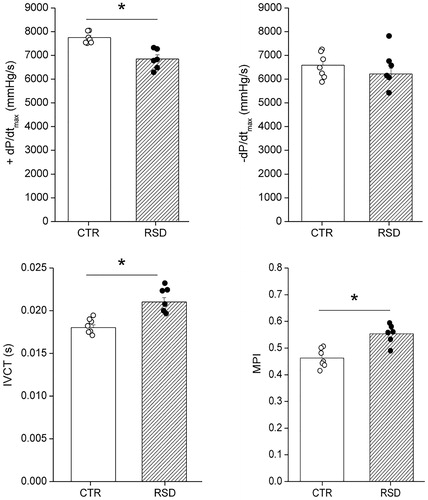
Table 2. Hemodynamic parameters in rats exposed to repeated social defeat (RSD) or control (CTR) condition.
RSD and CTR rats had similar body weight prior to the first social defeat/control procedure (RSD: 348 ± 7 g versus CTR: 355 ± 7 g). However, RSD rats gained significantly less weight than CTR rats during the experimental protocol (RSD: + 15 ± 3 g versus CTR: + 22 ± 2 g, p<.05). No differences in heart weights were observed between RSD and CTR rats ().
Table 3. Myocardial weights in rats exposed to repeated social defeat (RSD) or control (CTR) condition.
Discussion
In this work, we explored the molecular and functional consequences of repeated social defeat stress in the rat heart, with a specific focus on miRs-34a as known modulators of both neurochemical and behavioral stress responses and cardiac pathophysiology. The major finding of this study is that repeated social defeat stress increases miR-34a expression and alters transcriptomic pattern in the left ventricular tissue. These molecular modifications are associated with increased vulnerability to pharmacologically induced arrhythmias and signs of systolic left ventricular dysfunction. In the next sections, we will discuss the molecular and functional effects observed in the socially stressed rat heart as well as potential pathophysiological pathways linking miR-34a up-regulation to adverse cardiac remodeling.
Up-regulation of miR-34a expression and transcriptomic profile changes in the socially stressed heart
The miR-34 family has recently been implicated in the regulation of stress responses (Andolina et al., Citation2016, Citation2018; Lo Iacono et al., Citation2020). Specifically, we have shown in mice that the expression of the miR-34a isoform is augmented upon chronic stress exposure in the raphe nuclei, the primary brain region for the production of the neurotransmitter serotonin, and that miRs-34a in this brain region play a key role in mediating chronic stress-induced adverse behavioral effects (Andolina et al., Citation2018; Lo Iacono et al., Citation2020). To the best of our knowledge, this is the first study to document a larger relative expression of miRs-34a in the heart of rats exposed to repeated social defeat stress. Of note, clear signs of cardiac miR-34a overexpression were found well beyond (24 h after) the last defeat, but not after a single defeat episode. Notably, several studies have implicated miR-34a regulatory activity in the pathogenesis of CVD (Fan et al., Citation2013; Hu et al., Citation2019; Toshima et al., Citation2020; Zhang et al., Citation2018). For example, miR-34a expression is elevated in cardiac tissue from patients with diabetic ischemic heart and heart failure (Dehaini et al., Citation2019; Greco et al., Citation2012; Thum et al., Citation2007), and following cardiac damage in preclinical models (Bernardo et al., Citation2012, Citation2014; Boon et al., Citation2013; Lin et al., Citation2010; Yang et al., Citation2015). Consistent with the pathogenic role of miR-34-mediated regulation in the heart, inhibition of the miR-34 family improved cardiac function in preclinical models of cardiac pathology (Bernardo et al., Citation2012; Ooi et al., Citation2017). Based on these premises, increased expression of miR-34a in the heart of rats exposed to repeated social defeat stress might be interpreted as a first molecular sign of an altered homeostasis that ultimately leads to stress-related cardiac disorders. Supporting this view, Gene Ontology analysis on over 300 differently expressed genes upon repeated social defeat revealed clusters of genes associated with morphological (e.g. mitochondrial inner membrane) and functional (e.g. oxidative phosphorylation) properties of the mitochondria, the predominant energy generators in the heart. This is in line with the view that psychosocial stressors may affect cardiac mitochondria, altering their morphology and decreasing their ability to produce energy via oxidative phosphorylation (Picard & McEwen, Citation2018). A recent study reported changes in gene expression associated with endothelial cell migration, mesenchymal development, and extracellular matrix organization in rats exposed to a predator model of PTSD, which were coupled with multifocal cardiac lesions characterized by myofiber necrosis, fibrosis, and infiltration by mononucleated immune cells (Rorabaugh et al., Citation2020). Differences in gene expression profile of stressed rats from the study by Rorabaugh et al. (Citation2020) and the current investigation may be ascribed to the different nature of the stress protocols, cat exposure versus repeated social defeat, respectively. The latter, as we will also detail below, is known to induce severe alterations in the structural and functional properties of the heart compared to other stressors (Sgoifo et al., Citation2014). Nevertheless, we must acknowledge that in studies of social defeat, the control group is often placed into novel cages, whereas in this study control rats were handled and remained in their home cages. This could magnify the social defeat effect which includes novel cage exposure.
MiRNAs act through complementary binding with mRNAs to interfere with the translation process. Due to the short size of the complementary sequence, a single miRNA can potentially bind to hundreds of target mRNAs, resulting in the fine-tuning of gene expression profile. Therefore, we searched within the list of differentially expressed genes for those which represent putative targets of miR-34a. Similar to what was observed with the entire list of genes, many of the gene targets of miR-34a are linked to “mitochondrial outer membrane” and “mitochondrial apoptotic changes.” Of note, out of several hundreds of miRs expressed in all cell types, only 41 miRs (mitomiRs) have been detected in mitochondria, including the miR-34a isoform (Rippo et al., Citation2014). Based on this and the results of our study, we hypothesize that repeated social defeat stress may induce morphological and/or functional alterations in cardiac mitochondria and that miR-34a regulatory activity may be, at least partially, responsible for this effect.
Adverse electromechanical remodeling in the socially stressed heart: is compromised mitochondrial efficiency involved?
Rats exposed to repeated social defeat showed signs of both electrical and mechanical alterations that may predispose to cardiac dysfunction. Specifically, we found a larger vulnerability to cardiac arrhythmias following β-adrenergic stimulation with isoproterenol and signs of compromised ventricular contractility. Of note, heart rate under isoproterenol was similar between the two groups, suggesting that repeated social defeat did not affect β-adrenergic receptor sensitivity. This finding is in line with previous studies showing that repeated exposure to social defeat alters the electrophysiological properties of the ventricular myocardium, lowering the threshold for cardiac arrhythmias (Carnevali et al., Citation2013; Sgoifo et al., Citation2014). Notably, the increased susceptibility to arrhythmias and the reduced hemodynamic efficiency found in repeatedly stressed rats are consistent with the molecular data indicating a potential impairment of cardiac mitochondrial efficiency associated with miR-34a up-regulation. Indeed, studies have reported links between miR-34a expression levels and mitochondrial content, morphology and function. Specifically, miR-34a over-expression has been reported to (i) alter mitochondrial morphology and decrease mitochondrial content (Wen et al., Citation2018), (ii) favor mitochondrial-mediated increase in ROS production and cell apoptosis (Rippo et al., Citation2014), and (iii) reduce mitochondrial oxidative phosphorylation (Bukeirat et al., Citation2016). In contrast, inhibition of miR-34a was found to significantly restore the mitochondrial transmembrane potential (Wen et al., Citation2018).
The normal functioning heart requires coordinated electrical activity and contractile action. The energy demand to meet this unceasing action is satisfied by ATP predominantly produced by the mitochondria through oxidative phosphorylation (Harris & Das, Citation1991). Thus, cardiac function is particularly dependent on the efficiency of mitochondria performance. Mitochondria are also critically involved in the homeostatic regulation of cellular cations such as Ca2+, Na+, and K+, which is essential for normal electrical activity and cardiac contractility (Murphy & Eisner, Citation2009; Williams et al., Citation2013). Emerging evidence indicates that mitochondrial dysfunction can adversely impact cardiac electrical functioning by impairing intracellular ion homeostasis and membrane excitability through reduced ATP production and excessive reactive oxidative species (ROS) generation, resulting in increased propensity to cardiac arrhythmias (Yang et al., Citation2014). Relatedly, mitochondrial dysfunction is prevalent in arrhythmogenic cardiac diseases including cardiac hypertrophy, heart failure, and myocardial ischemia (Yang et al., Citation2014). Reduced ATP synthesis and increased ROS production associated with mitochondrial dysfunction, besides promoting apoptotic cell death (Dehaini et al., Citation2019), can also affect several cardiomyocyte molecular mechanisms, leading to impaired cardiac contractility (Bocchi et al., Citation2019). In addition, mitochondrial dysfunction has a profibrotic activity via TGF-β1-WNT signaling, resulting in fibroblast differentiation into myofibroblasts with collagen accumulation in the extracellular matrix (Yousefi et al., Citation2020). All these processes can lead to the development of an arrhythmogenic substrate and worsen hemodynamic performance. Therefore, based on the results of our study, we speculate that overexpression of miRs-34a in the heart of rats exposed to repeated social defeat may contribute to impaired mitochondrial efficiency, thereby favoring a larger vulnerability to pharmacologically-induced arrhythmias and signs of systolic left ventricular dysfunction.
Conclusion
miRNAs have emerged as one of the most promising areas for earlier prevention and novel treatment of stress-related disorders (Dwivedi, Citation2014; Issler & Chen, Citation2017; Scott et al., Citation2015). Our results suggest the presence of an association between miR-34a overexpression and signs of adverse electromechanical remodeling in the heart of rats exposed to repeated social defeat, and point to compromised mitochondrial efficiency as a potential mediator of this link.
In interpreting these results, we must acknowledge several limitations: first, this study provides indirect correlations between molecular (miR-34a expression) and cardiac functional data, and does not investigate the events in a dependent manner; second, no experiments have been conducted to assess the effects of repeated social defeat on cardiac mitochondrial function. Therefore, future studies are required to detail the causal relationship (for instance by antimiR-34 treatment) between miR-34a expression, mitochondrial (dys)function and cardiac alterations under stressful conditions, which could have important implications in the context of stress-related CVD. Moreover, given the documented role of miR34-a in mediating the adverse behavioral sequelae of chronic stress in mice (Andolina et al., Citation2016, Citation2018; Lo Iacono et al., Citation2020), it would be interesting to test whether miR-34a up-regulation could represent a common epigenetic mechanism underlying the comorbidity between stress-related psychopathologies and CVD.
Supplemental Material
Download MS Word (149.2 KB)Acknowledgments
The authors would like to thank Rhiannon Elizabeth Baird for her careful English language revision of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes on contributors
Diego Andolina
Diego Andolina Ph.D. in Psychobiology and Psychopharmacology and Assistant Professor at the Sapienza University of Rome.
Monia Savi
Monia Savi Ph.D. in Systemic Physiopathology and Assistant Professor at the University of Parma.
Donald Ielpo
Donald Ielpo Ph.D. student in Behavioral Neuroscience at the Sapienza University of Rome.
Margherita Barbetti
Margherita Barbetti Ph.D. student in Neuroscience at the University of Parma.
Leonardo Bocchi
Leonardo Bocchi Ph.D. in Systemic Physiopathology and technician at the University of Parma.
Donatella Stilli
Donatella Stilli Associate Professor at the University of Parma.
Rossella Ventura
Rossella Ventura Ph.D. in Psychobiology and Psychopharmacology and Associate Professor at the Sapienza University of Rome.
Luisa Lo Iacono
Luisa Lo Iacono Ph.D. in Neurobiology and Post-doctorate at the Sapienza University of Rome.
Andrea Sgoifo
Andrea Sgoifo Ph.D. in Cardiovascular Pathophysiology and Full Professor at the University of Parma.
Luca Carnevali
Luca Carnevali Ph.D. in Systemic Physiopathology and Assistant Professor at the University of Parma.
References
- Anders, S., Pyl, P. T., & Huber, W. (2015). HTSeq – A Python framework to work with high-throughput sequencing data. Bioinformatics, 31(2), 166–169. https://doi.org/10.1093/bioinformatics/btu638
- Andolina, D., Di Segni, M., Accoto, A., Lo Iacono, L., Borreca, A., Ielpo, D., Berretta, N., Perlas, E., Puglisi-Allegra, S., & Ventura, R. (2018). MicroRNA-34 contributes to the stress-related behavior and affects 5-HT prefrontal/GABA Amygdalar system through regulation of corticotropin-releasing factor receptor 1. Molecular Neurobiology, 55(9), 7401–7412. https://doi.org/10.1007/s12035-018-0925-z
- Andolina, D., Di Segni, M., Bisicchia, E., D'Alessandro, F., Cestari, V., Ventura, A., Concepcion, C., Puglisi-Allegra, S., & Ventura, R. (2016). Effects of lack of microRNA-34 on the neural circuitry underlying the stress response and anxiety. Neuropharmacology, 107, 305–316. https://doi.org/10.1016/j.neuropharm.2016.03.044
- Andolina, D., Di Segni, M., & Ventura, R. (2017). MiRNA-34 and stress response. Oncotarget, 8(4), 5658–5659. https://doi.org/10.18632/oncotarget.13923
- Bairey Merz, C. N., Dwyer, J., Nordstrom, C. K., Walton, K. G., Salerno, J. W., & Schneider, R. H. (2002). Psychosocial stress and cardiovascular disease: Pathophysiological links. Behavioral Medicine, 27(4), 141–147. https://doi.org/10.1080/08964280209596039
- Bernardo, B. C., Gao, X. M., Tham, Y. K., Kiriazis, H., Winbanks, C. E., Ooi, J. Y., Boey, E. J., Obad, S., Kauppinen, S., Gregorevic, P., Du, X. J., Lin, R. C., & McMullen, J. R. (2014). Silencing of miR-34a attenuates cardiac dysfunction in a setting of moderate, but not severe, hypertrophic cardiomyopathy. PLoS One., 9(2), e90337. https://doi.org/10.1371/journal.pone.0090337
- Bernardo, B. C., Gao, X. M., Winbanks, C. E., Boey, E. J., Tham, Y. K., Kiriazis, H., Gregorevic, P., Obad, S., Kauppinen, S., Du, X. J., Lin, R. C., McMullen, J. R. (2012). Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proceedings of the National Academy of Sciences of the United States of America, 109, 17615–17620. https://doi.org/10.1073/pnas.1206432109
- Black, P. H., & Garbutt, L. D. (2002). Stress, inflammation and cardiovascular disease. Journal of Psychosomatic Research, 52(1), 1–23. https://doi.org/10.1016/s0022-3999(01)00302-6
- Bocchi, L., Motta, B. M., Savi, M., Vilella, R., Meraviglia, V., Rizzi, F., Galati, S., Buschini, A., Lazzaretti, M., Pramstaller, P. P., Rossini, A., & Stilli, D. (2019). The histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) restores cardiomyocyte contractility in a rat model of early diabetes. International Journal of Molecular Sciences, 20(8), 1873. https://doi.org/10.3390/ijms20081873
- Boon, R. A., Iekushi, K., Lechner, S., Seeger, T., Fischer, A., Heydt, S., Kaluza, D., Treguer, K., Carmona, G., Bonauer, A., Horrevoets, A. J., Didier, N., Girmatsion, Z., Biliczki, P., Ehrlich, J. R., Katus, H. A., Muller, O. J., Potente, M., Zeiher, A. M., Hermeking, H., & Dimmeler, S. (2013). MicroRNA-34a regulates cardiac ageing and function. Nature, 495(7439), 107–110. https://doi.org/10.1038/nature11919
- Bukeirat, M., Sarkar, S. N., Hu, H., Quintana, D. D., Simpkins, J. W., & Ren, X. (2016). MiR-34a regulates blood-brain barrier permeability and mitochondrial function by targeting cytochrome c. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism, 36(2), 387–392. https://doi.org/10.1177/0271678X15606147
- Carnevali, L., Statello, R., & Sgoifo, A. (2019). Resting heart rate variability predicts vulnerability to pharmacologically-induced ventricular arrhythmias in male rats. Journal of Clinical Medicine, 8(5), 655. https://doi.org/10.3390/jcm8050655
- Carnevali, L., Trombini, M., Rossi, S., Graiani, G., Manghi, M., Koolhaas, J. M., Quaini, F., Macchi, E., Nalivaiko, E., & Sgoifo, A. (2013). Structural and electrical myocardial remodeling in a rodent model of depression. Psychosomatic Medicine, 75(1), 42–51. https://doi.org/10.1097/PSY.0b013e318276cb0d
- Curtis, M. J., Hancox, J. C., Farkas, A., Wainwright, C. L., Stables, C. L., Saint, D. A., Clements-Jewery, H., Lambiase, P. D., Billman, G. E., Janse, M. J., Pugsley, M. K., Ng, G. A., Roden, D. M., Camm, A. J., & Walker, M. J. (2013). The Lambeth Conventions (II): Guidelines for the study of animal and human ventricular and supraventricular arrhythmias. Pharmacology & Therapeutics, 139(2), 213–248. https://doi.org/10.1016/j.pharmthera.2013.04.008
- Dehaini, H., Awada, H., El-Yazbi, A., Zouein, F. A., Issa, K., Eid, A. A., Ibrahim, M., Badran, A., Baydoun, E., Pintus, G., & Eid, A. H. (2019). MicroRNAs as potential pharmaco-targets in ischemia-reperfusion injury compounded by diabetes. Cells, 8(2), 152. https://doi.org/10.3390/cells8020152
- Dobin, A., Davis, C. A., Schlesinger, F., Drenkow, J., Zaleski, C., Jha, S., Batut, P., Chaisson, M., & Gingeras, T. R. (2013). STAR: Ultrafast universal RNA-seq aligner. Bioinformatics, 29(1), 15–21. https://doi.org/10.1093/bioinformatics/bts635
- Dwivedi, Y. (2014). Emerging role of microRNAs in major depressive disorder: diagnosis and therapeutic implications. Dialogues in Clinical Neuroscience, 16(1), 43–61.
- Fan, F., Sun, A., Zhao, H., Liu, X., Zhang, W., Jin, X., Wang, C., Ma, X., Shen, C., Zou, Y., Hu, K., & Ge, J. (2013). MicroRNA-34a promotes cardiomyocyte apoptosis post myocardial infarction through down-regulating aldehyde dehydrogenase 2. Current Pharmaceutical Design, 19(27), 4865–4873. https://doi.org/10.2174/13816128113199990325
- Girod, J. P., & Brotman, D. J. (2004). Does altered glucocorticoid homeostasis increase cardiovascular risk? Cardiovascular Research, 64(2), 217–226. https://doi.org/10.1016/j.cardiores.2004.07.006
- Greco, S., Fasanaro, P., Castelvecchio, S., D'Alessandra, Y., Arcelli, D., Di Donato, M., Malavazos, A., Capogrossi, M. C., Menicanti, L., & Martelli, F. (2012). MicroRNA dysregulation in diabetic ischemic heart failure patients. Diabetes, 61(6), 1633–1641. https://doi.org/10.2337/db11-0952
- Grippo, A. J., Moffitt, J. A., Sgoifo, A., Jepson, A. J., Bates, S. L., Chandler, D. L., McNeal, N., & Preihs, K. (2012). The integration of depressive behaviors and cardiac dysfunction during an operational measure of depression: Investigating the role of negative social experiences in an animal model. Psychosomatic Medicine, 74(6), 612–619. https://doi.org/10.1097/PSY.0b013e31825ca8e5
- Harris, D. A., & Das, A. M. (1991). Control of mitochondrial ATP synthesis in the heart. Biochemical Journal, 280 (3), 561–573. https://doi.org/10.1042/bj2800561
- Hu, J., Gao, C., Wei, C., Xue, Y., Shao, C., Hao, Y., Gou, L. T., Zhou, Y., Zhang, J., Ren, S., Chen, J., Wang, Y., Fu, X. D. (2019). RBFox2-miR-34a-Jph2 axis contributes to cardiac decompensation during heart failure. Proceedings of the National Academy of Sciences of the United States of America, 116, 6172–6180. https://doi.org/10.1073/pnas.1822176116
- Huang, D. W., Sherman, B. T., Zheng, X., Yang, J., Imaminchi, T., Stephens, R., Lempicki, R. A. (2009). Extracting biological meaning from large gene lists with DAVID. Current Protocol Bioinformatics, 37, 1–13.
- Humphreys, D. T., Westman, B. J., Martin, D. I., Preiss, T. (2005). MicroRNAs control translation initiation by inhibiting eukaryotic initiation factor 4E/cap and poly(A) tail function. Proceedings of the National Academy of Sciences of the United States of America, 102, 16961–16966. https://doi.org/10.1073/pnas.0506482102
- Ikeda, S., Kong, S. W., Lu, J., Bisping, E., Zhang, H., Allen, P. D., Golub, T. R., Pieske, B., & Pu, W. T. (2007). Altered microRNA expression in human heart disease. Physiological Genomics, 31(3), 367–373. https://doi.org/10.1152/physiolgenomics.00144.2007
- Issler, O., & Chen, A. (2017). The role of microRNAs in stress-induced psychopathologies A2. In Fink, G. (Ed.) Stress: Neuroendocrinology and neurobiology. Academic Press.
- Leung, A. K., Vyas, S., Rood, J. E., Bhutkar, A., Sharp, P. A., & Chang, P. (2011). Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Molecular Cell, 42(4), 489–499. https://doi.org/10.1016/j.molcel.2011.04.015
- Lin, R. C., Weeks, K. L., Gao, X. M., Williams, R. B., Bernardo, B. C., Kiriazis, H., Matthews, V. B., Woodcock, E. A., Bouwman, R. D., Mollica, J. P., Speirs, H. J., Dawes, I. W., Daly, R. J., Shioi, T., Izumo, S., Febbraio, M. A., Du, X. J., & McMullen, J. R. (2010). PI3K(p110 alpha) protects against myocardial infarction-induced heart failure: Identification of PI3K-regulated miRNA and mRNA. Arteriosclerosis, Thrombosis, and Vascular Biology, 30(4), 724–732. https://doi.org/10.1161/ATVBAHA.109.201988
- Lo Iacono, L., Ielpo, D., Accoto, A., Di Segni, M., Babicola, L., D'Addario, S. L., Ferlazzo, F., Pascucci, T., Ventura, R., & Andolina, D. (2020). MicroRNA-34a regulates the depression-like behavior in mice by modulating the expression of target genes in the dorsal Raphè. Molecular Neurobiology, 57(2), 823–836. https://doi.org/10.1007/s12035-019-01750-2
- Love, M. I., Huber, W., & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology, 15(12), 550. https://doi.org/10.1186/s13059-014-0550-8
- Malan-Muller, S., Hemmings, S. M., & Seedat, S. (2013). Big effects of small RNAs: a review of microRNAs in anxiety. Molecular Neurobiology, 47(2), 726–739. https://doi.org/10.1007/s12035-012-8374-6
- Miczek, K. A. (1979). A new test for aggression in rats without aversive stimulation: differential effects of d-amphetamine and cocaine. Psychopharmacology, 60(3), 253–259. https://doi.org/10.1007/BF00426664
- Muka, T., Koromani, F., Portilla, E., O'Connor, A., Bramer, W. M., Troup, J., Chowdhury, R., Dehghan, A., & Franco, O. H. (2016). The role of epigenetic modifications in cardiovascular disease: A systematic review. International Journal of Cardiology, 212, 174–183. https://doi.org/10.1016/j.ijcard.2016.03.062
- Murphy, E., & Eisner, D. A. (2009). Regulation of intracellular and mitochondrial sodium in health and disease. Circulation Research, 104(3), 292–303. https://doi.org/10.1161/CIRCRESAHA.108.189050
- Ooi, J. Y. Y., Bernardo, B. C., Singla, S., Patterson, N. L., Lin, R. C. Y., & McMullen, J. R. (2017). Identification of miR-34 regulatory networks in settings of disease and antimiR-therapy: Implications for treating cardiac pathology and other diseases. RNA Biology, 14(5), 500–513. https://doi.org/10.1080/15476286.2016.1181251
- Picard, M., & McEwen, B. S. (2018). Psychological stress and mitochondria: A conceptual framework. Psychosomatic Medicine, 80(2), 126–140. https://doi.org/10.1097/PSY.0000000000000544
- Piegari, E., Cozzolino, A., Ciuffreda, L. P., Cappetta, D., De Angelis, A., Urbanek, K., Rossi, F., & Berrino, L. (2020). Cardioprotective effects of miR-34a silencing in a rat model of doxorubicin toxicity. Scientific Reports, 10(1), 12250. https://doi.org/10.1038/s41598-020-69038-3
- Quattrocelli, M., Crippa, S., Montecchiani, C., Camps, J., Cornaglia, A. I., Boldrin, L., Morgan, J., Calligaro, A., Casasco, A., Orlacchio, A., Gijsbers, R., D'Hooge, J., Toelen, J., Janssens, S., & Sampaolesi, M. (2013). Long-term miR-669a therapy alleviates chronic dilated cardiomyopathy in dystrophic mice. Journal of the American Heart Association, 2(4), e000284. https://doi.org/10.1161/JAHA.113.000284
- Rippo, M. R., Olivieri, F., Monsurro, V., Prattichizzo, F., Albertini, M. C., & Procopio, A. D. (2014). MitomiRs in human inflamm-aging: A hypothesis involving miR-181a, miR-34a and miR-146a. Experimental Gerontology, 56, 154–163. https://doi.org/10.1016/j.exger.2014.03.002
- Rorabaugh, B. R., Mabe, N. W., Seeley, S. L., Stoops, T. S., Mucher, K. E., Ney, C. P., Goodman, C. S., Hertenstein, B. J., Rush, A. E., Kasler, C. D., Sargeant, A. M., & Zoladz, P. R. (2020). Myocardial fibrosis, inflammation, and altered cardiac gene expression profiles in rats exposed to a predator-based model of posttraumatic stress disorder. Stress, 23(2), 125–135. https://doi.org/10.1080/10253890.2019.1641081
- Rosengren, A., Hawken, S., Ounpuu, S., Sliwa, K., Zubaid, M., Almahmeed, W. A., Blackett, K. N., Sitthi-Amorn, C., Sato, H., & Yusuf, S. (2004). Association of psychosocial risk factors with risk of acute myocardial infarction in 11119 cases and 13648 controls from 52 countries (the INTERHEART study): Case-control study. Lancet, 364(9438), 953–962. https://doi.org/10.1016/S0140-6736(04)17019-0
- Rozanski, A., Blumenthal, J. A., & Kaplan, J. (1999). Impact of psychological factors on the pathogenesis of cardiovascular disease and implications for therapy. Circulation, 99(16), 2192–2217. https://doi.org/10.1161/01.CIR.99.16.2192
- Saban, K. L., Mathews, H. L., DeVon, H. A., & Janusek, L. W. (2014). Epigenetics and social context: Implications for disparity in cardiovascular disease. Aging and Disease, 5(5), 346–355. https://doi.org/10.14336/AD.2014.0500346
- Salemi, V. M., Pires, M. D., Cestari, I. N., Cestari, I. A., Picard, M. H., Leirner, A. A., & Mady, C. (2004). Echocardiographic assessment of global ventricular function using the myocardial performance index in rats with hypertrophy. Artificial Organs, 28(4), 332–337. https://doi.org/10.1111/j.1525-1594.2004.47350.x
- Schmittgen, T. D., & Livak, K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nature Protocols, 3(6), 1101–1108. https://doi.org/10.1038/nprot.2008.73
- Scott, K. A., Hoban, A. E., Clarke, G., Moloney, G. M., Dinan, T. G., & Cryan, J. F. (2015). Thinking small: Towards microRNA-based therapeutics for anxiety disorders. Expert Opinion on Investigational Drugs, 24(4), 529–542. https://doi.org/10.1517/13543784.2014.997873
- Sgoifo, A., Carnevali, L., & Grippo, A. J. (2014). The socially stressed heart. Insights from studies in rodents. Neuroscience and Biobehavioral Reviews, 39, 51–60. https://doi.org/10.1016/j.neubiorev.2013.12.005
- Sgoifo, A., Costoli, T., Meerlo, P., Buwalda, B., Pico'-Alfonso, M. A., De Boer, S., Musso, E., & Koolhaas, J. (2005). Individual differences in cardiovascular response to social challenge. Neuroscience and Biobehavioral Reviews, 29(1), 59–66. https://doi.org/10.1016/j.neubiorev.2004.07.001
- Sgoifo, A., Stilli, D., Medici, D., Gallo, P., Aimi, B., & Musso, E. (1996). Electrode positioning for reliable telemetry ECG recordings during social stress in unrestrained rats. Physiology & Behavior, 60(6), 1397–1401. https://doi.org/10.1016/S0031-9384(96)00228-4
- Small, E. M., & Olson, E. N. (2011). Pervasive roles of microRNAs in cardiovascular biology. Nature, 469(7330), 336–342. https://doi.org/10.1038/nature09783
- Sucharov, C., Bristow, M. R., & Port, J. D. (2008). miRNA expression in the failing human heart: functional correlates. Journal of Molecular and Cellular Cardiology, 45(2), 185–192. https://doi.org/10.1016/j.yjmcc.2008.04.014
- Sun, T., Dong, Y. H., Du, W., Shi, C. Y., Wang, K., Tariq, M. A., Wang, J. X., & Li, P. F. (2017). The role of MicroRNAs in myocardial infarction: From molecular mechanism to clinical application. International Journal of Molecular Sciences, 18(4), 745. https://doi.org/10.3390/ijms18040745
- Thum, T., Galuppo, P., Wolf, C., Fiedler, J., Kneitz, S., van Laake, L. W., Doevendans, P. A., Mummery, C. L., Borlak, J., Haverich, A., Gross, C., Engelhardt, S., Ertl, G., & Bauersachs, J. (2007). MicroRNAs in the human heart: A clue to fetal gene reprogramming in heart failure. Circulation, 116(3), 258–267. https://doi.org/10.1161/CIRCULATIONAHA.107.687947
- Toshima, T., Watanabe, T., Narumi, T., Otaki, Y., Shishido, T., Aono, T., Goto, J., Watanabe, K., Sugai, T., Takahashi, T., Yokoyama, M., Kinoshita, D., Tamura, H., Kato, S., Nishiyama, S., Arimoto, T., Takahashi, H., Miyamoto, T., Sadahiro, M., & Watanabe, M. (2020). Therapeutic inhibition of microRNA-34a ameliorates aortic valve calcification via modulation of Notch1-Runx2 signalling. Cardiovascular Research, 116(5), 983–994. https://doi.org/10.1093/cvr/cvz210
- van Rooij, E., Sutherland, L. B., Thatcher, J. E., DiMaio, J. M., Naseem, R. H., Marshall, W. S., Hill, J. A., Olson, E. N. (2008). Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proceedings of the National Academy of Sciences of the United States of America, 105, 13027–13032. https://doi.org/10.1073/pnas.0805038105
- Wen, F., An, C., Wu, X., Yang, Y., Xu, J., Liu, Y., Wang, C., Nie, L., Fang, H., & Yang, Z. (2018). MiR-34a regulates mitochondrial content and fat ectopic deposition induced by resistin through the AMPK/PPARα pathway in HepG2 cells. International Journal of Biochemistry & Cell Biology, 94, 133–145. https://doi.org/10.1016/j.biocel.2017.11.008
- Williams, G. S., Boyman, L., Chikando, A. C., Khairallah, R. J., Lederer, W. J. (2013). Mitochondrial calcium uptake. Proceedings of the National Academy of Sciences of the United States of America, 110, 10479–10486. https://doi.org/10.1073/pnas.1300410110
- Yang, K. C., Yamada, K. A., Patel, A. Y., Topkara, V. K., George, I., Cheema, F. H., Ewald, G. A., Mann, D. L., & Nerbonne, J. M. (2014). Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation, 129(9), 1009–1021. https://doi.org/10.1161/CIRCULATIONAHA.113.003863
- Yang, Y., Cheng, H. W., Qiu, Y., Dupee, D., Noonan, M., Lin, Y. D., Fisch, S., Unno, K., Sereti, K. I., & Liao, R. (2015). MicroRNA-34a plays a key role in cardiac repair and regeneration following myocardial infarction. Circulation Research, 117(5), 450–459. https://doi.org/10.1161/CIRCRESAHA.117.305962
- Yousefi, F., Shabaninejad, Z., Vakili, S., Derakhshan, M., Movahedpour, A., Dabiri, H., Ghasemi, Y., Mahjoubin-Tehran, M., Nikoozadeh, A., Savardashtaki, A., Mirzaei, H., & Hamblin, M. R. (2020). TGF-beta and WNT signaling pathways in cardiac fibrosis: Non-coding RNAs come into focus. Cell Communication and Signaling, 18(1), 87. https://doi.org/10.1186/s12964-020-00555-4
- Zhang, C., Zhang, Y., Zhu, H., Hu, J., & Xie, Z. (2018). MiR-34a/miR-93 target c-Ski to modulate the proliferation of rat cardiac fibroblasts and extracellular matrix deposition in vivo and in vitro. Cellular Signalling, 46, 145–153. https://doi.org/10.1016/j.cellsig.2018.03.005