Abstract
Corticotropin releasing factor (CRF) coordinates behavioral, autonomic and hormonal responses to stress. Activation of the hypothalamic pituitary adrenal (HPA) axis with stimulation of CRF and vasopressin (VP) release from hypothalamic parvocellular neurons, and consequent secretion of ACTH from the anterior pituitary and glucocorticoid from the adrenal cortex, is the major endocrine response to stress. Current evidence indicates that the main regulator of ACTH secretion in acute and chronic conditions is CRF, in spite of the fact that the selective increases in expression of parvocellular VP and pituitary VP V1b receptors observed during prolonged activation of the HPA axis have suggested that VP becomes the predominant regulator. Following CRF release, activation of CRF transcription is required to restore mRNA and peptide levels, but termination of the response is essential to prevent pathology associated with chronic elevation of CRF and glucocorticoid production. While glucocorticoid feedback plays an important role in regulating CRF expression, the relative importance of direct transcriptional repression of the CRF gene by glucocorticoids in the overall feedback mechanism is not clear. In addition to glucocorticoids, intracellular feedback mechanisms in the CRF neuron, involving induction of repressor forms of cAMP response element modulator (CREM) limit CRF transcriptional responses by competing with the positive regulator, phospho-CREB. Rapid repression of CRF transcription following stress-induced activation is likely to contribute to limiting the stress response and to preventing disorders associated with excessive CRF production.
Preservation of homeostasis requires continuous behavioral, autonomic and endocrine adaptation to control perturbations caused by internal or external threats or stressors. The major endocrine response to stress is the activation of the hypothalamic pituitary adrenal (HPA) axis, leading to rapid secretion of ACTH by the pituitary corticotroph resulting in increased circulating concentrations of glucocorticoids from the adrenal cortex, which are critical for stress adaptation (Munck et al. Citation1984; Dallman et al. Citation1994; McEwen Citation1998). The major regulator of ACTH secretion is the 41-amino acid peptide, corticotropin releasing factor (CRF). The peptide, produced in parvocellular neurons of the hypothalamic paraventricular nucleus is released into the hypothalamo-pituitary portal circulation, and stimulates ACTH secretion by acting upon type 1 CRF receptors located in the plasma membrane of the pituitary corticotroph (Aguilera et al. Citation2004; Bale and Vale Citation2004). In addition, CRF released within the brain mediates behavioral and autonomic responses to stress (Heinrichs and Koob Citation2004; de Kloet et al. Citation2005), acting through CRF receptors distributed in limbic areas of the brain (Aguilera et al. Citation2004; Bale and Vale Citation2004). While CRF release is required for stress responses and maintenance of homeostasis during prolonged stress (Vale et al. Citation1983; Aguilera Citation1998), equally important are mechanisms responsible for limitation of stress responses, since chronic elevations in circulating glucocorticoids, or excessive CRF levels in the brain can lead to pathology, such as depression, immune and metabolic disorders (Munck et al. Citation1984; McEwen Citation1998; Stiedl et al. Citation2005). Thus, elucidation of the mechanisms controlling CRF expression is essential for a better understanding of the pathogenesis and therapeutic approaches to stress related disease. After a brief characterization of the mechanisms controlling the activation of hypothalamic CRF neurons, this article discusses the roles of glucocorticoids and intracellular feedback mechanisms mediating repression of CRF transcription during stress.
The hypothalamic CRF neuron and the stress response
Studies in the rat have shown that about 50% of the medio-dorsal hypophysiotropic CRF neurons also express VP in basal conditions, and this proportion increases markedly after adrenalectomy and during chronic stress paradigms associated with hyper-responsiveness of the HPA axis to a novel stress (Whitnall Citation1988; de Goeij et al. Citation1991; Aguilera Citation1994). In humans, variable proportions of CRF neurons co-localizing VP have been shown in brains from elderly and depressed patients but not in the young (Raadsheer et al. Citation1993, Citation1994), while in mice it appears that all CRF neurons co-express VP (Whitnall Citation1993). Acute stress leads to rapid release of both CRF and VP into the hypothalamo-pituitary portal circulation (Plotsky et al. Citation1985; Canny et al. Citation1989; Tannahill et al. Citation1991). As shown in , activation of the CRF neuron by a stressor results in immediate peptide release, within seconds of stimulation, as shown by measurements in pituitary portal blood and immunohistochemical studies showing depletion of storage pools in the external zone of the median eminence. Secretion is followed by consecutive increases in primary transcript (within minutes of stimulation), and mRNA levels within hours (). Transcriptional activation is required for restoration of the releasable pool of CRF (and VP) in the median eminence, and appropriate control of transcription levels of these peptides is essential for the long term regulation of HPA axis activity.
Figure 1 Diagram depicting the sequence of events in the regulation of CRF expression in the PVN. Following release of stimulatory neurotransmitters in the PVN in response to stress, there is a rapid release of CRF peptide from the median eminence into the pituitary portal circulation, as evidenced by a rapid disappearance of immunoreactive CRH from the median eminence (I). The image shows immunostaining for CRF in the median eminence of rats subjected to intracerebroventricular colchicine injection 18 h before ip injection of normal saline (controls) or HS (stress). This is followed by transient increases in CRF primary transcript (hnRNA) within minutes (II), and more sustained increases in CRF mRNA, within hours (III), shown by the PVN in situ hybridization images and time curves. Restoration of CRF peptide storage pools through mRNA translation occurs within hours or days (IV).
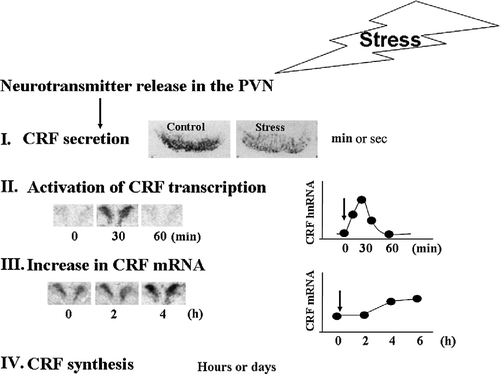
The pattern of transcriptional activation for CRF and vasopressin (VP) varies according to the stress paradigm, but for most acute stressors activation of CRF transcription precedes and is of shorter duration than that of VP (Herman et al. Citation1992; Kovacs and Sawchenko Citation1996; Ma et al. Citation1997; Ma and Aguilera Citation1999b). Usually, the time course of changes in the CRF primary transcript (and presumably transcription) parallels that of activation of the CRF neuron, as reflected by changes in early expression genes and activation of the HPA axis. It should be noted that repeated or sustained stress often results in habituation of the CRF responses, CRF mRNA levels returning to basal levels and there is no primary transcript response to the homotypic stressor, suggesting rapid limitation of the transcriptional response. In contrast, VP expression (reflected by increases in VP mRNA and primary transcript) remains elevated during chronic stress (Ma et al. Citation1999). This preferential increase in VP over CRF gene expression led to the hypothesis that during chronic stress VP becomes the major regulator of HPA axis activity, and that VP is responsible for the hyper-responsiveness of the HPA axis to a novel stress, observed in these conditions (Aguilera Citation1998). However, evidence based on the use of genetically modified animal models and pharmacological tools indicates that while CRH is required for most acute an chronic HPA axis responses to stress, VP seems to have a modulatory role, being necessary for a full ACTH response in some stress paradigms (Rivier and Plotsky Citation1986; Smith et al. Citation1998; Muglia et al. Citation2000; Tanoue et al. Citation2004; Zelena et al. Citation2004; Lolait et al. Citation2007).
Mechanisms regulating CRF expression in the PVN
CRF expression is under the influence of stimulatory and inhibitory neural pathways and several circulating hormones. Depending on the stress paradigm, the stress response in the PVN is initiated through activation of ascending noradrenergic pathways from the brain stem with direct connections to the PVN, or forebrain multi-synaptic circuitry involving a number of limbic structures, such as the amygdala, bed nucleus of the stria terminalis, lateral septum, subiculum and several hypothalamic nuclei. Most forebrain pathways involve inhibitory actions on GABAergic neurons exerting tonic inhibition on the CRF neuron. As indicated earlier, uncontrolled activation of stress circuits leading to excessive glucocorticoid secretion can result in homeostatic imbalance. Therefore, maintenance of appropriate levels of HPA axis activity requires efficient mechanisms for inhibiting stress-responsive PVN neurons. This is achieved by complex neural pathways, as well as glucocorticoid feedback. For a detailed description of stimulatory and inhibitory circuitry influencing the CRF neurons see reviews by Herman and Cullinan (Citation1997).
Role of glucocorticoids in the regulation of CRF transcription
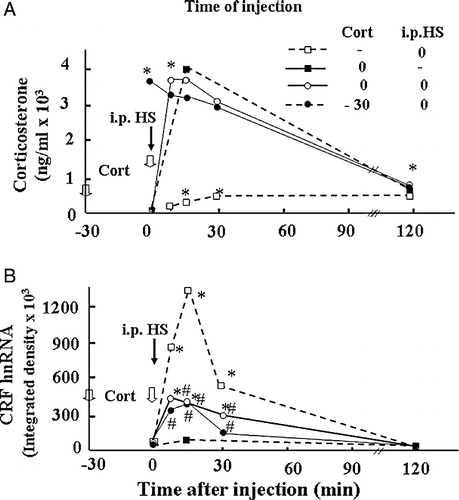
Glucocorticoids, the end product of stimulation of the HPA axis, are a major component of the negative feedback mechanism reducing CRF responses to stress. Glucocorticoids are clearly involved in the regulation of CRF expression in vivo as shown by the potentiation of stress responses following adrenalectomy and the converse effect of glucocorticoid administration (Lightman and Young Citation1989; Dallman et al. Citation1994). Injection of glucocorticoid into the PVN region decreases CRF mRNA expression and ACTH secretion, suggesting that glucocorticoid exert its negative feedback effects directly at the level of CRH neurons (Kovacs et al. Citation1986; Sawchenko Citation1987; Harbuz and Lightman Citation1989). The expression of glucocorticoid receptors in CRF neurons in the PVN also supports a direct action (Liposits et al. Citation1987; Fenoglio et al. Citation2004). In addition, glucocorticoids modulate the responsiveness of the parvocellular neurons to stress (Makino et al. Citation1995; Ma and Aguilera Citation1999a). For example, a minor stressor, such as intraperitoneal (ip) vehicle injection, causes marked increases in CRF and VP primary transcript expression in adrenalectomized rats, while having no effect in intact rats (Ma and Aguilera Citation1999a). On the other hand, administration of high doses of corticosterone increasing plasma levels by about 100-fold stress levels in these adrenalectomized rats was unable to significantly affect the increase in CRF primary transcript in response to the injection (Ma and Aguilera Citation1999a, ). These observations in adrenalectomized rats suggest that under normal conditions, low prevailing levels of glucocorticoids reduce the responsiveness of parvocellular neurons to minor stressors, but that the steroid has no direct inhibitory effect on stress-induced CRF transcription in adrenalectomized rats. In contrast to the inability of glucocorticoids to inhibit stress-induced CRF transcription in adrenalectomized rats, a similar injection of corticosterone (2.5 mg/100 g BW) in intact rats, markedly inhibited the increase in CRF primary transcript in response to the severe painful stressor of ip hypertonic saline (HS) injection (). Although the mechanism of the differential sensitivity of the CRF primary transcript to glucocorticoid inhibition in intact and adrenalectomized rats remain to be elucidated, it is clear that regulation of CRF transcription by glucocorticoids is complex, probably involving indirect effects in addition to actions of the steroid at the CRF promoter level. Microdialysis studies have shown that glucocorticoids inhibit stress-induced norepinephrine release from the PVN (Pacak et al. Citation1995). In addition, adrenalectomy increases and glucocorticoid administration decreases the content of alpha adrenergic receptors in the PVN (Day et al. Citation1999), and in vitro studies have shown switching in adrenergic receptor α1–α2 in hypothalamic sections after incubation with glucocorticoids (Feuvrier et al. Citation1998). This supports the possibility that a major mechanism by which glucocorticoids exert feedback inhibition of CRF transcription is by inhibition of afferent pathways to the PVN or by modulating the content of neurotransmitter receptors in the CRF neuron. Several investigators have reported direct effects of glucocorticoids on CRF promoter activity. There is no classical glucocorticoid response element (GRE) in the CRF promoter but there is evidence from in vitro experiments, mostly in cell lines transfected with CRF promoter-driven reporter genes, that glucocorticoids can regulate CRF gene expression through protein–protein interaction or by binding directly to response elements (Nicholson et al. Citation2004). Glucocorticoids can have stimulatory or inhibitory effects on CRF promoter activity depending on the cell type (Nicholson et al. Citation2004). Although there is evidence suggesting that glucocorticoids can inhibit CRF transcription (Guardiola-Diaz et al. Citation1996; Malkowski et al. Citation1997; Nicholson et al. Citation2004), the lack of a cell line representative of parvocellular CRF neurons, has made it difficult to elucidate the relative importance of direct transcriptional repression and indirect actions on afferent pathways.
Figure 2 Adrenalectomy (ADX) sensitizes CRF hnRNA responses to the minor stressor of ip injection of 100 μl of 50% ethanol in normal saline (used as vehicle for corticosterone (Cort) injections). (A) Time course of the changes in plasma corticosterone levels following injection of corticosterone (2.8 mg/100 g BW, ip) or vehicle (Veh) in 48-h adrenalectomized or sham-operated rats. Data points represent the mean and SE of values obtained in six rats per experimental group. *, p < 0.01 vs. ADX or sham basal (0 min). (B) Time course of the changes in CRF hnRNA after injection of corticosterone (2.8 mg/100 g BW, ip) or vehicle in 48-h adrenalectomized or sham-operated rats. Data points are the mean and SE of the optical density values obtained from film autoradiograms in six rats per experimental group. *, p < 0.01 vs. sham or ADX basal (0 min). From Ma and Aguilera (Citation1999a).
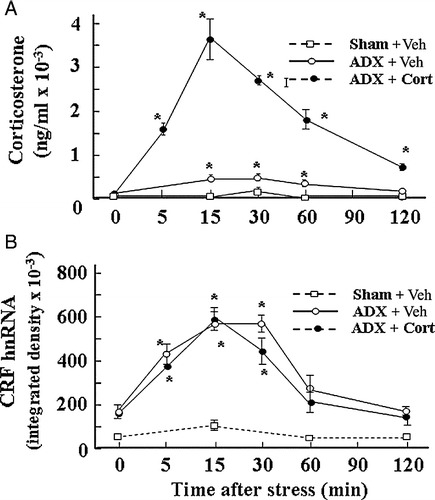
Figure 3 Effect of glucocorticoids on plasma corticosterone levels and CRF hnRNA responses to the painful stress of ip HS injection in intact rats. Time course of the changes in plasma corticosterone levels (A) or CRF hnRNA in the PVN (B), following administration of corticosterone (2.8 mg/100 g BW, ip) injected either at − 30 min or at 0 min before ip HS injection. Data points represent the mean and SE of values obtained in six rats per experimental group. *, p < 0.01 vs. ADX or sham basal (0 min). (B) Time course of the changes in CRF hnRNA after injection of corticosterone (2.8 mg/100 g BW, ip) or vehicle in 48-h adrenalectomized (ADX) or sham-operated rats. Data points are the mean and SE of the optical density values obtained from film autoradiograms in six rats per experimental group. *, p < 0.01 vs. sham or ADX basal (0 min).
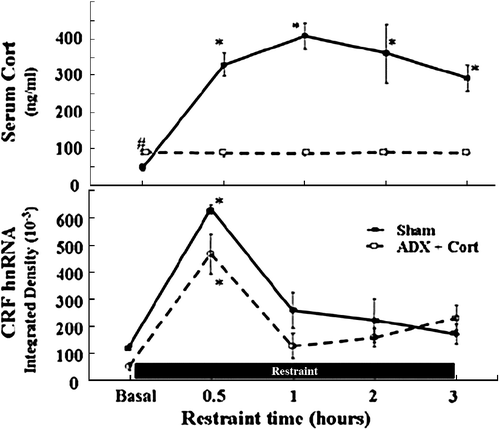
A number of studies have examined the time course of the changes in CRF primary transcript (heteronuclear RNA, hnRNA) during stress and shown that the increases are transient, even in the presence of sustained stimulation. As illustrated in , levels of CRF hnRNA increased in response to restraint stress decline by 60 min and returned to near basal levels by 90 min, in spite of the continuous presence of the restraint stress for 4 h (Shepard et al. Citation2005). In the same report, to determine the involvement of the stress-induced glucocorticoid surge in the decline of the CRF transcriptional response, CRF hnRNA responses to prolonged restraint were measured in adrenalectomized rats receiving glucocorticoid replacement. Sham-operated rats showed marked and sustained increases in plasma corticosterone in response to restraint, and as expected, adrenalectomized rats had low levels provided by the replacement and no increases following stress (). In spite of the lack of a glucocorticoid surge in adrenalectomized rats, CRF hnRNA responses in the PVN showed a pattern similar to sham-operated rats, returning to near basal levels by 1 h (). It is evident from the above data that the decline of restraint stress-induced CRF hnRNA levels is independent of glucocorticoids and that transcriptional activation can be transient in spite of the lack of a glucocorticoid surge. Since in the above report rats were maintained with low levels of glucocorticoid replacement, it was not possible to rule out that basal steroid levels have a permissive role in limiting CRF transcriptional responses by interacting with an additional repressor. Nevertheless, it is clear that stress-induced increases in circulating glucocorticoids are not responsible for limiting CRF transcriptional responses.
Figure 4 Glucocorticoids do not mediate the rapid decline of CRF gene transcription during stress. Plasma corticosterone levels (A) and CRF hnRNA levels in the PVN (B) during the course of 3 h restraint stress in sham-operated and adrenalectomized (ADX) rats receiving constant levels of corticosterone (Cort) via subcutaneous implants. Rats were killed by decapitation at the indicated time points. Data points are the mean and SE of the values obtained in five rats per experimental group. * p < 0.001 compared to basal; # p < 0.05 compared to basal sham-operated rats. From Shepard et al. (Citation2005).
Role of inducible cAMP early repressor (ICER) in regulating CRF transcription
Since increases in circulating glucocorticoids could not explain the self limitation of CRF transcription during stress, we investigated the possibility of an intracellular feedback mechanism involving inducible cAMP early repressor (ICER), a repressor isoform of the cAMP response element modulator (CREM). ICER is generated by cAMP-dependent activation of a second promoter located in the intron of the CREM gene (Foulkes et al. Citation1991; Molina et al. Citation1993), and it has been shown to mediate transcriptional repression in a number of neuroendocrine systems (Foulkes et al. Citation1996; Monaco et al. Citation1997; Borsook et al. Citation1999). ICER contains the DNA binding domain but not the transactivation domain of CREM, and acts as a competitive inhibitor of cAMP response element (CRE) dependent transcription (Foulkes et al. Citation1991; Lamas et al. Citation1996; Monaco et al. Citation1997). The CRF promoter contains a functional CRE at − 229 (Seasholtz et al. Citation1988) and several studies have shown that this element is important not only for activation of CRF transcription, but also for the repressor effect of glucocorticoids (Guardiola-Diaz et al. Citation1996; Nicholson et al. Citation2004).
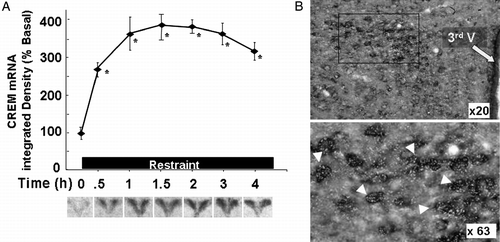
Stress leads to induction of ICER mRNA in the PVN within 30 min (equally detectable with CREM riboprobes or ICER specific oligoprobes), and this is followed by progressive increases in ICER protein, reaching a maximum by 3 h (Shepard et al. Citation2005). The same study shows that stress-induced ICER expression occurs in CRF neurons, as shown by double staining in situ hybridization, using digoxigenin-labeled CRF and 35S-labeled CREM riboprobes in hypothalamic sections of rats subjected to 3 h of restraint stress (). The ability of stress to induce CREM expression is not influenced by glucocorticoids since the pattern of expression and levels were similar in sham-operated and adrenalectomized rats (Shepard et al. Citation2005). The changes in CREM mRNA were accompanied by increases in low molecular weight bands in a western blot of approximately 18, 15 and 13.5 kDa, corresponding to the molecular size of the ICER protein. ICER bands were first detectable by 1 h and increased markedly following 3 h of restraint stress, at which time CRH hnRNA levels had returned to basal.
Figure 5 Stress increases CREM mRNA levels in the PVN. (A) Changes in CREM mRNA measured by in situ hybridization using 35S-labeled cRNA probes. Data points are the mean and SE of the pooled data. *, p < 0.001 compared with basal. Representative images from film autoradiography of 1 of 5 rats per experimental group are shown under the respective time points. Almost identical results were shown using a 48-mer oligoprobe specific for ICER. (B) Cellular localization of CREM mRNA in PVN sections of rats subjected to restraint stress for 3 h. Double labeling in situ hybridization using 35S-labeled CREM cRNA probes (bright green grains), and digoxigenin-labeled CRF cRNA probes, shows 35S-CREM staining (bright grains) overlaying digoxigenin-labeled parvicellular CRF neurons. Third V, 3rd ventricle; the lower panel shows higher magnification of the area enclosed in the square. The white arrowheads indicate some of the CRF neurons labeled with CREM. CREM staining in control non-stressed rats was no different from background staining (12.3 ± 1.8 and 9.6 ± 1.4 grains/cell, not shown). Adapted from Shepard et al. (Citation2005).
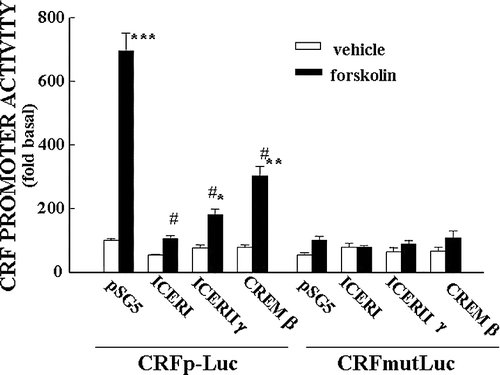
The ability of phospho-CREB and CREM to interact with the CRF–CRE has been shown by electrophoresis mobility shift assay (EMSA) using hypothalamic nuclear extracts from control and stressed rats, and 33P-labeled double stranded oligonucleotide corresponding to the rat CRE sequence (Shepard et al. Citation2005; Liu et al. Citation2006). More compelling evidence has been provided by chromatin immunoprecipitation assays showing parallel recruitment of CREM by the CRF promoter and decreased RNA polymerase II recruitment, in cross-linked chromatin extracted from hypothalami of rats stressed for 3 h. As reported by Shepard et al. (Citation2005) (), CRF promoter immunoprecipitation with the phospho-CREB antibody increased by stress for 15 min, corresponding with the increase in Pol II, but the recruitment of Pol II decreased by 3 h in spite of a progressive increase in phospho-CREB. On the other hand, the amount of CRF promoter immunoprecipitated with the CREM antibody increased by 3 h in association with the decrease in Pol II recruitment. The correlation between increases in ICER and decreases in both CRF hnRNA levels and RNA polymerase II recruitment by the CRF promoter strongly suggests that ICER inhibits CRF transcription. Supporting this possibility, in vitro experiments using the hypothalamic cell line 4B and a CRF promoter-driven luciferase reporter gene have shown marked decreases in CRF promoter activity when the cells are co-transfected with ICER expression vectors (; Liu et al. Citation2006).
Figure 6 Restraint stress induces recruitment of phospho-CREB and CREM by the CRF promoter in the hypothalamic PVN region. (A) Chromatin immunoprecipitation (ChIP) assays using phospho-CREB, CREM and Pol II antibodies and cross-linked DNA from microdissected hypothalamic PVN region of control rats and rats subjected to restraint stress for 30 min or 3 h. (B) PCR for the CRF coding region, performed on the same immunoprecipitants used in (A), showed non-specific bands, which were not changed by stress. Gel images are representative of the results in three experiments. Ab, antibody; pCREB, phospho-CREB; Pol II, RNA polymerase II. Adapted from Shepard et al. (Citation2005).
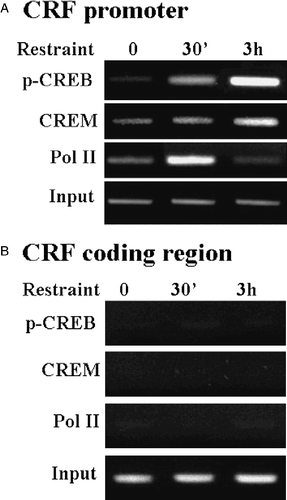
Figure 7 Repressor isoforms of CREM inhibit CRF promoter activity in a CRE dependent manner. Effect of co-transfection of the expression vector, pSG5, empty or containing ICER I, ICER II or CREMβ on basal and forskolin-stimulated CRF promoter activity in the hypothalamic cell line, H32 simultaneously transfected with CRF promoter–luciferase reporter genes. Cells were co-transfected with CRF promoter–luciferase reporter plasmids, without (CRFp-Luc) or with inactivating mutations of the CRE (CRFmut-Luc) and the expression vector, and 18 h later incubated with forskolin for 6 h before measurement of luciferase activity. Bars represent the mean and SE of the data obtained in three experiments. * p < 0.01 vs. respective vehicle; #, p < 0.01 compared to forskolin-stimulated pSG5. From Liu et al. (Citation2006).
It is noteworthy that the major neurotransmitters mediating activation of CRF transcription during stress, such as norepinephrine (acting upon alpha adrenergic receptors in CRF neurons, Day et al. Citation1999) or glutamate, do not stimulate cAMP production directly (Guarino et al. Citation1996; Moldrich and Beart Citation2003). However, previous studies using the hypothalamic cell line 4B, have shown that only minimal elevations in intracellular cAMP are sufficient to elicit full activation of a CRF promoter–luciferase reporter (Nikodemova et al. Citation2003). Thus, it is possible that small increases in cAMP as a consequence of stimulation of adenylyl cyclase secondary to calcium mobilization mediate activation of the CRF promoter (Cooper et al. Citation1995). Also, norepinephrine, a preferential stimulator of α-adrenergic receptors, could cause small increases in cAMP by activation of β-adrenergic receptors (Collins et al. Citation1991). In this regard, β-adrenergic receptors are present in the hypothalamus but there is no reported evidence of co-localization in CRF neurons. Therefore, stimulation of calcium–phospholipid-dependent pathways by stress may potentiate basal levels of cAMP-dependent transcription. In addition, it is well recognized that regulators mediated by calcium–phospholipid-dependent signaling can lead to CREB phosphorylation through alternative pathways such as transactivation of the MAP kinase cascade (Smith and Luttrell Citation2006). In this regard, studies in progress in our laboratory using 4B cells have shown that phorbol esters, which have no effect on ICER production on their own, increase the sensitivity of ICER responses to low concentrations of forskolin (Lui and Aguilera, Citation2006). An additional mechanism stimulating cAMP signaling in the CRF neuron could involve the CRF peptide itself. In this regard, stress causes marked increases in the expression of CRF1 receptor mRNA and CRF binding in CRF neurons of the parvocellular PVN (Luo et al. Citation1994). In addition, electron microscopic studies have shown local CRF-immunoreactive neuronal circuits in the paraventricular nucleus of the rat hypothalamus, suggesting that the peptide can access CRF1 receptors in CRF neurons through somatodendritic synaptic contacts in the PVN (Liposits et al. Citation1985). Thus, increases in cAMP in CRF neurons as the result of CRF release and stimulation of CRF1 receptors within the PVN, could have a dual effect first facilitating CRF transcription, through CREB phosphorylation, and later inhibiting it through induction of the repressor, ICER. Overall, the available evidence indicates that ICER and other inhibitory forms of CREM inhibit CRF transcription in a CRE-dependent manner. The induction of ICER expression and recruitment of CREM by the CRF promoter following prolonged stimulation of cells, together with the ability of ICER to reduce basal and cAMP-induced CRF promoter activation suggest that ICER plays a regulatory role in the regulation of CRF transcription. Induction of ICER could serve as a protective mechanism which contributes to limiting transcription of the CRF gene in the hypothalamus and avoid the pathological consequences of excessive expression of the neuropeptide.
Concluding remarks
There is increasing evidence that CRF and not VP secreted by parvocellular neurons of the hypothalamic paraventricular nucleus is the major regulator of the HPA axis during acute and chronic stimulation. Stress causes rapid release of CRF into the pituitary portal circulation followed by transient increases in CRF transcription. Induction of CRF gene transcription is required to restore CRF mRNA and peptide levels following HPA axis activation, but termination of the response is essential to prevent pathology associated with chronic elevation of CRF and hence glucocorticoid production. The mechanisms contributing to limiting stress responses involve changes in stimulatory and inhibitory neurocircuitry activity, glucocorticoid feedback, as well as cAMP-mediated intracellular feedback in the CRF neuron. Glucocorticoid feedback plays an important role restraining HPA axis activity, but direct inhibition of CRF transcription appears to play a minor role compared with inhibition at the pituitary or neural circuitry levels. Intracellular feedback mechanisms mediated by induction of repressor forms of CREM, limit CRF transcriptional responses by competing with the positive regulator, phospho-CREB, at the CRE at position − 229 in the CRF promoter. Although the exact mechanism by which glucocorticoids modulate neural pathways regulating the CRF neuron and the relative importance of direct glucocorticoid- and CREM-dependent repression on CRF transcription remains to be elucidated, rapid repression of CRF transcription is likely to contribute to limiting the stress response, thus preventing behavioral, metabolic and other disorders associated with chronic stress and excessive CRF production.
Acknowledgements
This work was supported by the Intramural Research Program of the NIH/NICHD.
Related Research Data
References
- Aguilera G. Regulation of pituitary ACTH secretion during chronic stress. Front Neuroendocrinol 1994; 15: 321–350
- Aguilera G. Corticotropin releasing hormone, receptor regulation and the stress response. TEM 1998; 9: 329–336
- Aguilera G, Nikodemova M, Wynn PC, Catt KJ. Corticotropin releasing hormone receptors: Two decades later. Peptides 2004; 25: 319–329
- Bale TL, Vale WW. CRF and CRF receptors: Role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol 2004; 44: 525–557
- Borsook D, Smirnova O, Behar O, Lewis S, Kobierski LA. PhosphoCREB and CREM/ICER: Positive and negative regulation of proenkephalin gene expression in the paraventricular nucleus of the hypothalamus. J Mol Neurosci 1999; 12: 35–51
- Canny BJ, Funder JW, Clarke IJ. Glucocorticoids regulate ovine hypophysial portal levels of corticotropin-releasing factor and arginine vasopressin in a stress-specific manner. Endocrinology 1989; 125: 2532–2539
- Collins S, Lohse MJ, O'Dowd B, Caron MG, Lefkowitz RJ. Structure and regulation of G protein-coupled receptors: The beta 2-adrenergic receptor as a model. Vitam Horm 1991; 46: 1–39
- Cooper DMF, Mons N, Karpen JW. Adenylyl cyclases and the interaction between calcium and cAMP signalling. Nature 1995; 374: 421–424
- Dallman MF, Akana SF, Levin N, Walker C-D, Bradbury MJ, Suemaru S, Scribner KS. Corticosteroids and the control of function in the hypothalamo-pituitary–adrenal (HPA) axis. Ann N Y Acad Sci 1994; 746: 22–28
- Day HEW, Campeau S, Watson SJ, Jr., Akil H. Expression of alpha 1b adrenoceptor mRNA in corticotropin-releasing hormone-containing cells of the rat hypothalamus and its regulation by corticosterone. J Neurosci 1999; 19: 10098–10106
- de Goeij DC, Kvetnansky R, Whitnall MH, Jezova D, Berkenbosch F, Tilders FJ. Repeated stress-induced activation of corticotropin-releasing factor neurons enhances vasopressin stores and colocalization with corticotropin-releasing factor in the median eminence of rats. Neuroendocrinology 1991; 53: 150–159
- de Kloet ER, Joels M, Holsboer F. Stress and the brain: From adaptation to disease. Nat Rev Neurosci 2005; 6: 463–475
- Fenoglio KA, Brunson KL, Avishai-Eliner S, Chen Y, Baram TZ. Region-specific onset of handling-induced changes in corticotropin-releasing factor and glucocorticoid receptor expression. Endocrinology 2004; 145: 2702–2706
- Feuvrier E, Aubert M, Mausset AL, Alonso G, Gaillet S, Malaval F, Szafarczyk A. Glucocorticoids provoke a shift from alpha2- to alpha1-adrenoreceptor activities in cultured hypothalamic slices leading to opposite noradrenaline effect on corticotropin-releasing hormone release. J Neurochem 1998; 70: 1199–1209
- Foulkes NS, Borrelli E, Sassone-Corsi P. CREM gene: Use of alternative DNA-binding domains generates multiple antagonists of cAMP-induced transcription. Cell 1991; 64: 739–749
- Foulkes NS, Borjigin J, Snyder SH, Sassone-Corsi P. Transcriptional control of circadian hormone synthesis via the CREM feedback loop. Proc Natl Acad Sci USA 1996; 93: 14140–14145
- Guardiola-Diaz HM, Kolinske JS, Gates LH, Seasholtz AF. Negative glucorticoid regulation of cyclic adenosine 3′, 5′-monophosphate-stimulated corticotropin-releasing hormone-reporter expression in AtT-20 cells. Mol Endocrinol 1996; 10: 317–329
- Guarino RD, Perez DM, Piascik MT. Recent advances in the molecular pharmacology of the [alpha]1-adrenergic receptors. Cell Signal 1996; 8: 323–333
- Harbuz MS, Lightman SL. Glucocorticoid inhibition of stress-induced changes in hypothalamic corticotrophin-releasing factor messenger RNA and proenkephalin A messenger RNA. Neuropeptides 1989; 14: 17–20
- Heinrichs SC, Koob GF. Corticotropin-releasing factor in brain: A role in activation, arousal, and affect regulation. J Pharmacol Exp Ther 2004; 311: 427–440
- Herman JP, Cullinan WE. Neurocircuitry of stress: Central control of the hypothalamo-pituitary–adrenocortical axis. Trends Neurosci 1997; 20: 78–84
- Herman JP, Schafer MK, Thompson RC, Watson SJ. Rapid regulation of corticotropin-releasing hormone gene transcription in vivo. Mol Endocrinol 1992; 6: 1061–1069
- Kovacs KJ, Sawchenko PE. Regulation of stress-induced transcriptional changes in the hypothalamic neurosecretory neurons. J Mol Neurosci 1996; 7: 125–133
- Kovacs K, Kiss JZ, Makara GB. Glucocorticoid implants around the hypothalamic paraventricular nucleus prevent the increase of corticotropin-releasing factor and arginine vasopressin immunostaining induced by adrenalectomy. Neuroendocrinology 1986; 44: 229–234
- Lamas M, Monaco L, Zazopoulos E, Lalli E, Tamai K, Penna L, Mazzucchelli C, Nantel F, Foulkes NS, Sassone-Corsi P. CREM: A master-switch in the transcriptional response to cAMP. Philos Trans R Soc Lond B Biol Sci 1996; 351: 561–567
- Lightman SL, Young WS. Influence of steroids on the hypothalamic corticotropin-releasing factor and preproenkephalin mRNA responses to stress. Proc Natl Acad Sci USA 1989; 86: 4306–4310
- Liposits Z, Paull WK, Setalo G, Vigh S. Evidence for local corticotropin releasing factor (CRF) immunoreactive neuronal circuits in the paraventricular nucleus of the rat hypothalamus. An electron microscopic immunocytochemical analysis. Histochemistry 1985; 83: 5–16
- Liposits Z, Uht RM, Harrison RW, Gibbs FP, Paull WK, Bohn MC. Ultrastructural localization of glucocorticoid receptor (GR) in hypothalamic paraventricular neurons synthesizing corticotropin releasing factor (CRF). Histochemistry 1987; 87: 407–412
- Lui Y, Aguilera G. Increases in endogenous CAMP inducible early repressor (ICER) inhibit corticot-ropin releasing hormone transcription in hypothalamic, 4B, cells and inprimary hypothalamic cell cultures. 2006, The Endocrine Society 88th Annnual Meeting, 2006, p 171, abst
- Liu Y, Kalintchenko N, Sassone-Corsi P, Aguilera G. Inhibition of corticotrophin-releasing hormone transcription by inducible cAMP-early repressor in the hypothalamic cell line, 4B. J Neuroendocrinol 2006; 18: 42–49
- Lolait SJ, Stewart LQ, Jessop DS, Young WS, 3rd, O'Carroll AM. The hypothalamic–pituitary–adrenal axis response to stress in mice lacking functional vasopressin V1b receptors. Endocrinology 2007; 148: 849–856
- Luo X, Kiss A, Makara G, Lolait SJ, Aguilera G. Stress-specific regulation of corticotropin releasing hormone receptor expression in the paraventricular and supraoptic nuclei of the hypothalamus in the rat. J Neuroendocrinol 1994; 6: 689–696
- Ma XM, Levy A, Lightman SL. Emergence of an isolated arginine vasopressin (AVP) response to stress after repeated restraint: A study of both AVP and corticotropin-releasing hormone messenger ribonucleic acid (RNA) and heteronuclear RNA. Endocrinology 1997; 138: 4351–4357
- Ma XM, Aguilera G. Differential regulation of corticotropin-releasing hormone and vasopressin transcription by glucocorticoids. Endocrinology 1999a; 140: 5642–5650
- Ma XM, Aguilera G. Transcriptional responses of the vasopressin and corticotropin-releasing hormone genes to acute and repeated intraperitoneal hypertonic saline injection in rats. Brain Res Mol Brain Res 1999b; 68: 129–140
- Ma XM, Lightman SL, Aguilera G. Vasopressin and corticotropin-releasing hormone gene responses to novel stress in rats adapted to repeated restraint. Endocrinology 1999; 140: 3623–3632
- Makino S, Schulkin J, Smith MA, Pacak K, Palkovits M, Gold PW. Regulation of corticotropin-releasing hormone receptor messenger ribonucleic acid in the rat brain and pituitary by glucocorticoids and stress. Endocrinology 1995; 136: 4517–4525
- Malkowski SP, Handanos CM, Dorin RI. Localization of a negative glucocorticoid response element of the human corticotropin-releasing hormone gene. Mol Cell Endocrinol 1997; 127: 189–199
- McEwen BS. Protective and damaging effects of stress mediators. N Engl J Med 1998; 338: 171–179
- Moldrich RX, Beart PM. Emerging signalling and protein interactions mediated via metabotropic glutamate receptors. Curr Drug Targets CNS Neurol Disord 2003; 2: 109–122
- Molina CA, Foulkes NS, Lalli E, Sassone-Corsi P. Inducibility and negative autoregulation of CREM: An alternative promoter directs the expression of ICER, an early response repressor. Cell 1993; 75: 875–886
- Monaco L, Lamas M, Tamai K, Lalli E, Zazopoulos E, Penna L, Nantel F, Foulkes NS, Mazzucchelli C, Sassone-Corsi P. Coupling transcription to signaling pathways: cAMP and nuclear factor cAMP-responsive element modulator. Adv Second Messenger Phosphoprotein Res 1997; 31: 63–74
- Muglia LJ, Bethin KE, Jacobson L, Vogt SK, Majzoub JA. Pituitary-adrenal axis regulation in CRH-deficient mice. Endocr Res 2000; 26: 1057–1066
- Munck A, Guyre PM, Holbrook NJ. Physiological functions of glucocorticoids in stress and their relation to pharmacological actions. Endocrinol Rev 1984; 5: 25–44
- Nicholson RC, King BR, Smith R. Complex regulatory interactions control CRH gene expression. Front Biosci 2004; 9: 32–39
- Nikodemova M, Kasckow J, Liu H, Manganiello V, Aguilera G. Cyclic adenosine 3′,5′-monophosphate regulation of corticotropin-releasing hormone promoter activity in AtT-20 cells and in a transformed hypothalamic cell line. Endocrinology 2003; 144: 1292–1300
- Pacak K, Palkovits M, Kvetnansky R, Matern P, Hart C, Kopin IJ, Goldstein DS. Catecholaminergic inhibition by hypercortisolemia in the paraventricular nucleus of conscious rats. Endocrinology 1995; 136: 4814–4819
- Plotsky PM, Bruhn TO, Vale W. Hypophysiotropic regulation of adrenocorticotropin secretion in response to insulin-induced hypoglycemia. Endocrinology 1985; 117: 323–329
- Raadsheer FC, Sluiter AA, Ravid R, Tilders FJ, Swaab DF. Localization of corticotropin-releasing hormone (CRH) neurons in the paraventricular nucleus of the human hypothalamus; age-dependent colocalization with vasopressin. Brain Res 1993; 615: 50–62
- Raadsheer FC, Hoogendijk WJ, Stam FC, Tilders FJ, Swaab DF. Increased numbers of corticotropin-releasing hormone expressing neurons in the hypothalamic paraventricular nucleus of depressed patients. Neuroendocrinology 1994; 60: 436–444
- Rivier CL, Plotsky PM. Mediation by corticotropin releasing factor (CRF) of adenohypophysial hormone secretion. Annu Rev Physiol 1986; 48: 475–494
- Sawchenko PE. Evidence for a local site of action for glucocorticoids in inhibiting CRF and vasopressin expression in the paraventricular nucleus. Brain Res 1987; 403: 213–224
- Seasholtz AF, Thompson RC, Douglass JO. Identification of a cyclic adenosine monophosphate-responsive element in the rat corticotropin-releasing hormone gene. Mol Endocrinol 1988; 2: 1311–1319
- Shepard JD, Liu Y, Sassone-Corsi P, Aguilera G. Role of glucocorticoids and cAMP-mediated repression in limiting corticotropin-releasing hormone transcription during stress. J Neurosci 2005; 25: 4073–4081
- Smith NJ, Luttrell LM. Signal switching, crosstalk, and arrestin scaffolds: Novel G protein-coupled receptor signaling in cardiovascular disease. Hypertension 2006; 48: 173–179
- Smith GW, Aubry JM, Dellu F, Contarino A, Bilezikjian LM, Gold LH, Chen R, Marchuk Y, Hauser C, Bentley CA, Sawchenko PE, Koob GF, Vale W, Lee KF. Corticotropin releasing factor receptor 1-deficient mice display decreased anxiety, impaired stress response, and aberrant neuroendocrine development. Neuron 1998; 20: 1093–1102
- Stiedl O, Meyer M, Jahn O, Ogren SO, Spiess J. Corticotropin-releasing factor receptor 1 and central heart rate regulation in mice during expression of conditioned fear. J Pharmacol Exp Ther 2005; 312: 905–916
- Tannahill LA, Sheward WJ, Robinson IC, Fink G. Corticotrophin-releasing factor-41, vasopressin and oxytocin release into hypophysial portal blood in the rat: Effects of electrical stimulation of the hypothalamus, amygdala and hippocampus. J Endocrinol 1991; 129: 99–107
- Tanoue A, Ito S, Honda K, Oshikawa S, Kitagawa Y, Koshimizu TA, Mori T, Tsujimoto G. The vasopressin V1b receptor critically regulates hypothalamic–pituitary–adrenal axis activity under both stress and resting conditions. J Clin Invest 2004; 113: 302–309
- Vale W, Rivier C, Brown MR, Spiess J, Koob G, Swanson L, Bilezikjian L, Bloom F, Rivier J. Chemical and biological characterization of corticotropin releasing factor. Recent Prog Horm Res 1983; 39: 245–270
- Whitnall MH. Major pro-vasopressin-expressing and pro-vasopressin-deficient subpopulations of corticotropin-releasing hormone neurons in normal rats. Differential distributions within the paraventricular nucleus. Neuroendocrinology 1988; 47: 176–180
- Whitnall MH. Regulation of the hypothalamic corticotropin-releasing hormone neurosecretory system. Prog Neurobiol 1993; 40: 573–629
- Zelena D, Foldes A, Mergl Z, Barna I, Kovacs KJ, Makara GB. Effects of repeated restraint stress on hypothalamo-pituitary–adrenocortical function in vasopressin deficient Brattleboro rats. Brain Res Bull 2004; 63: 521–530