Abstract
Pheochromocytomas are catecholamine-producing tumors presenting with various clinical symptoms, but mostly with headache, sweating, palpitations and hypertension. If not properly diagnosed, secretion of catecholamines may lead to fatal cardiovascular consequences. Biochemical testing for pheochromocytoma should be performed not only in symptomatic subjects or in subjects with adrenal incidentaloma but also in subjects with a genetic predisposition for pheochromocytoma (multiple endocrine neoplasia type 2, Von Hippel–Lindau (VHL) syndrome, neurofibromatosis type 1 (NF 1)and mutations of succinate dehydrogenase (SDH) genes). Once a pheochromocytoma is proven, computed tomography (CT), magnetic resonance imaging (MRI) and functional imaging with [123I]-MIBG may be used for tumor localization. Adequate medical pre-treatment is essential for successful operation which is performed in most cases by laparoscopy. After tumor removal, further follow-up is necessary due to possible recurrence. Although prognosis after tumor resection is excellent, a significant proportion of pheochromocytomas recur, some as metastases. Thus, appropriate follow-up is mandatory.
Pheochromocytomas are tumors arising from chromaffin cells of the adrenal medulla that synthesize, store, metabolize and usually but not always secrete catecholamines. Paragangliomas are tumors arising from extra-adrenal chromaffin cells and can originate either from sympathetic nervous system associated chromaffin tissue (mainly abdomen and pelvis, less frequently thorax) or parasympathetic associated chromaffin tissue (head and neck). Sympathetic paragangliomas are usually hormonally active and are sometimes called extra-adrenal pheochromocytomas. They occur less frequently than adrenal pheochromocytomas (Lenders et al. Citation2005).
Pheochromocytoma is a rare cause of secondary hypertension, with a prevalence of about 0.2–0.6% (Ariton et al. Citation2000; Omura et al. Citation2004). This contrasts with primary aldosteronism, which has a prevalence among unselected populations of hypertensives of 5–10% (Gordon et al. Citation1994; Rossi et al. Citation2006).
In contrast to the low prevalence of pheochromocytoma in hypertension, diagnosis of pheochromocytoma should always be ruled out in subjects with incidentally discovered adrenal tumors, since in this clinical condition the prevalence of pheochromocytoma may reach 4–5% (Mannelli et al. Citation1999; Mantero et al. Citation2000; Kasperlik-Zaluska et al. Citation2006). The prevalence of adrenal incidentalomas increases with age and may reach 7% in subjects more than 70 years old (Grumbach et al. Citation2003).
The prevalence of pheochromocytoma in autopsy studies (∼0.05%) also indicates that many tumors are missed, resulting in premature mortality (McNeil et al. Citation2000; Khorram-Manesh et al. Citation2006). Unlike other cancers, the typical age for diagnosis of sporadic pheochromocytoma is middle age. Pheochromocytomas with genetic syndromes are diagnosed earlier (Bravo and Tagle Citation2003). When diagnosed in children, pheochromocytoma often presents with extra-adrenal and multifocal involvement (Barontini et al. Citation2006).
Clinical presentation
The clinical presentation of pheochromocytoma is highly variable. Diagnosis may therefore be delayed. The vast majority of symptoms and signs are attributable to the excess of catecholamines which may be released by tumors continuously or paroxysmally. Clinical presentation may also depend on desensitization of catecholamine receptors as demonstrated in a study using a rat model of pheochromocytoma (Tsujimoto et al. Citation1987). Sulfoconjugation of adrenal catecholamines is the other mechanism by which the action of catecholamines is modulated (Kuchel et al. Citation1986). This may lead to lack of correlation between levels of released catecholamines and clinical symptoms, even during spells (Bravo and Tagle Citation2003).
The most common signs of catecholamine excess are hypertension, palpitations and tachycardia, headache, pallor, sweating and feelings of panic or anxiety. Less common signs are nausea, flushing, fever and constipation.
Headache, palpitations and sweating constitute a typical triad for pheochromocytoma and together with hypertension should always arouse suspicion of pheochromocytoma. The metabolic action of catecholamines may lead to weight loss, disturbances in glucose metabolism, including diabetes mellitus or lactic acidosis. Co-incidence of hypertension and diabetes mellitus type 2 in lean young or middle aged subjects is also suspicious for pheochromocytoma (Batide-Alanore et al. Citation2003). The frequency of clinical symptoms and signs is lower in hereditary forms of pheochromocytoma (), presumably due to active screening (Pacak et al. Citation2005). Subjects with initial presentation of pheochromocytoma as incidentaloma usually show mild symptoms and today comprise up to 25% of all subjects with diagnosed pheochromocytoma (Amar et al. Citation2005b).
Table I. Signs and symptoms in subjects with familial and sporadic pheochromocytoma.
Paroxysmal signs and symptoms, a consequence of episodic secretion of catecholamines, provide compelling clues for a pheochromocytoma. Surgical anesthesia and tumor manipulation are the most well known stimuli to elicit a catecholaminergic crisis. Food, micturition (urinary bladder pheochromocytoma) and various chemical compounds or drugs (e.g. glucagon, histamine, radiographic contrast agents, tyramine, metoclopramide, tricyclic antidepressants and β-blockers) may also elicit paroxysms (Lenders et al. Citation2005). Spells are usually unpredictable with various intervals, but for individual patients stereotypic in nature, usually lasting from anything between several minutes and 1 h. However, the most typical complaints such as headache, palpitations and sweating are nonspecific and often result in delay of diagnosis.
Hypertension in pheochromocytoma occurs in up to 80–90% of patients with tumors detected because of signs and symptoms. High blood pressure may present in sustained or paroxysmal forms, the latter either on a background of normotension or hypertension (Kaplan Citation2006). In up to 10–20% of subjects with pheochromocytoma, orthostatic hypotension may occur in those with sustained hypertension (Streeten and Anderson Citation1996). Contrasting with other forms of secondary hypertension where only one circulating hormone is responsible for the development of hypertension, pheochromocytoma may by characterized by the overproduction of three circulating catecholamines—norepinephrine, epinephrine and in a minority of cases, dopamine. The catecholamines exhibit different effects on different catecholamine receptors—typically norepinephrine-mediated stimulation of α-receptors leads to vasoconstriction whereas epinephrine also stimulates β2-receptors, causing vasodilatation. Subjects with predominantly norepinephrine-secreting pheochromocytoma (noradrenergic phenotype) develop hypertension more frequently than subjects with predominantly epinephrine-producing pheochromocytomas (adrenergic phenotype) who present more often with paroxysmal symptoms (Ito et al. Citation1992). Particularly adrenal pheochromocytomas often produce neuropeptide Y and this compound potentiates norepinephrine-induced vasoconstriction (deS Senanayake et al. Citation1995). Dopamine-producing tumors are relatively rare and often present with normotension (Proye et al. Citation1986; Eisenhofer et al. Citation2005). Blood pressure in subjects with pheochromocytoma is prone to abrupt fluctuations (in some cases from hypotension to severe hypertension) (Pešek et al. Citation2005). Twenty four hour blood pressure monitoring in subjects with pheochromocytoma may often show an elevation rather than a decrease in nocturnal blood pressure compared to the daytime blood pressure and occurs more frequently even in comparison with other forms of endocrine hypertension such as primary aldosteronism or Cushing's syndrome (Zelinka et al. Citation2004). Prompt blood pressure changes in pheochromocytoma result in higher blood pressure variability during 24-h ambulatory blood pressure monitoring compared to subjects with essential hypertension, in particular during the daytime (Zelinka et al. Citation2005).
Other cardiovascular complications of pheochromocytoma include sudden death, arrhythmias (brady- and tachyarrhythmias), myocardial infarction without preexisting coronary atherosclerosis, heart failure due to toxic catecholamine cardiomyopathy, dissecting aortic aneurysm, hypertensive encephalopathy, cerebrovascular accidents and noncardiogenic pulmonary edema or shock. Pheochromocytoma may also present with diabetic ketoacidosis, seizures, bowel pseudo-obstruction or multisystem crisis often with lactic acidosis (Brouwers et al. Citation2003). Other symptoms may be attributable to the co-secretion of other hormones or substances such as corticotropin-releasing hormone, adrenocorticotropin or interleukin six. Pheochromocytoma may also occur during pregnancy and thus may be misdiagnosed as pre-eclampsia. Accordingly, pheochromocytoma has been called the “Great Mimic” since its more than 80 manifestations can resemble so many other conditions that can confuse clinicians (Manger Citation2005).
Genetics of pheochromocytoma
To date, germ-line mutations in five different genes have been identified as causes of pheochromocytoma and functional paraganglioma (). Hereditary pheochromocytoma is associated with multiple endocrine neoplasia type 2 (MEN 2A or MEN 2B), NF 1, VHL syndrome and familial functional paraganglioma and pheochromocytoma due to germ-line mutations of genes encoding SDH subunits B and D (SDHB and SDHD; Tables and ). In general, the traits are inherited in an autosomal dominant pattern. Epidemiological studies among subjects with sporadic pheochromocytoma and functional paraganglioma have disclosed carriers of previously unrecognized germ-line mutations of four different genes (VHL, RET, SDHB and SDHD) in up to 24% of patients and thus challenging the traditional “rule of 10%” (Bornstein and Gimenez-Roqueplo Citation2006).
Table II. Main clinical features of syndromes associated with pheochromocytoma and functional paraganglioma.
Table III. Hereditary pheochromocytoma and paraganglioma: facts and figures.
Patients with MEN 2-related pheochromocytoma often lack hypertension or other symptoms (occur only in about 40 and 50%, respectively; ). In most cases, medullary carcinoma of the thyroid is the first presentation of this syndrome. MEN 2-related pheochromocytomas are characterized by production of epinephrine and norepinephrine and are, therefore, best detected by elevations of plasma or urinary metanephrine, usually but not always in association with elevations of normetanephrine and parent catecholamines (Eisenhofer et al. Citation2001). MEN 2-related pheochromocytomas are almost always intra-adrenal, often bilateral (in 50–80%) and rarely malignant ( < 5%) (Pacak et al. Citation2005; ). In addition, as in most epinephrine-secreting pheochromocytomas, hypertension is more likely to be paroxysmal than sustained if present ().
Overall about 20% of patients with VHL germ-line mutations develop pheochromocytoma (Ong et al. Citation2007). Unlike MEN 2, pheochromocytoma may be the first and also the sole presentation of VHL syndrome (type 2C of VHL—). Pheochromocytomas in VHL syndrome have an exclusively noradrenergic phenotype, reflecting lack of production of epinephrine and thus present more with sustained than paroxysmal complaints (Eisenhofer et al. Citation2001). These tumors are often asymptomatic when diagnosed during regular screening in this syndrome (). Biochemical diagnosis is best achieved from elevations of plasma or urinary normetanephrine (Eisenhofer et al. Citation2001). VHL-related tumors are mainly located intra-adrenally and occur in about 50% of patients bilaterally with a less than 5% incidence of metastases ().
In NF 1, pheochromocytoma is relatively rare ( < 5%). Pheochromocytomas in NF 1 usually produce epinephrine and norepinephrine, may present with bilateral adrenal involvement and are also rarely malignant (). Since genetic testing of the NF 1 gene is not routinely performed, only one study has been published about prevalence of this disease among subjects with apparently sporadic pheochromocytoma (Bausch et al. Citation2006; ).
Mutations of SDHB and SDHD genes predispose their carriers to extra-adrenal (SDHB and SDHD) and multifocal disease (more frequently in SDHD) (Neumann et al. Citation2004; Benn et al. Citation2006). Mutations of the SDHD gene are associated also with non-functional head and neck paragangliomas. Only head and neck paragangliomas have been described in SDHC mutations (Schiavi et al. Citation2005). Mitochondrial SDH functions in the Krebs cycle in the oxidation of succinate to fumarate. Since pheochromocytomas/paragangliomas appear to represent the sole clinical presentation of this syndrome, SDH mutations can often be diagnosed among subjects with apparently sporadic pheochromocytoma, particularly for those tumors with an extra-adrenal location (). Carriers of SDHB mutations have a high risk of malignant disease (up to 70%; ; Gimenez-Roqueplo et al. Citation2003; Amar et al. Citation2005a; Benn et al. Citation2006; Brouwers et al. Citation2006). These tumors produce predominantly norepinephrine and SDHB-related malignant tumors also produce dopamine (Timmers et al. Citation2007). Subjects with SDHB mutation may present with more clinical symptoms and signs than subjects with other hereditary pheochromocytoma (MEN 2 and VHL syndromes; ).
Although RET gene acts as a proto-oncogene and the other remaining genes as tumor suppressor genes (SDH and VHL genes are involved in regulation of hypoxia-inducible genes), a unifying hypothesis about involvement of these genes in development of pheochromocytoma/paraganglioma has been proposed recently. All these genes induce apoptosis during normal development of neuronal precursor cells as nerve growth factor becomes limiting. Germ-line mutations of NF 1, RET, SDH and VHL genes then allow sympathetic progenitors to escape from developmental apoptosis and so lead to their neoplastic transformation (Lee et al. Citation2005).
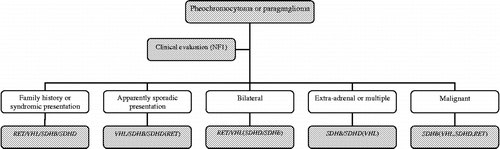
Should genetic testing be offered to all subjects with apparently sporadic pheochromocytoma? Although there are reasonable arguments for more widespread genetic testing, it is neither appropriate nor currently cost-effective to test every disease-causing gene in each patient with a pheochromocytoma or paraganglioma. Rather it seems to be more efficient to decide which genetic test should be performed according to the clinical presentation, including family history and physical examination (age at presentation, a single, bilateral, or extra-adrenal tumor or evidence of malignancy, and type of released catecholamine) as shown in and (Bornstein and Gimenez-Roqueplo Citation2006; Gimenez-Roqueplo et al. Citation2006).
Figure 1 Algorithm for genetic testing for genes associated with pheochromocytoma or paraganglioma. Abbreviations: RET, rearranged during transfection; SDHB, succinate dehydrogenase subunit B gene; SDHD, succinate dehydrogenase subunit D gene; VHL, Von Hippel–Lindau gene; NF 1, neurofibromatosis 1. Genes in parenthesis: second choice testing. Adapted from Bornstein and Gimenez-Roqueplo (Citation2006) and Gimenez-Roqueplo et al. (Citation2006).
Biochemical testing
Tests with appropriately high sensitivity and specificity are needed for screening (Lenders et al. Citation2005). Catecholamines may be released by chromaffin cells only intermittently or at very low rates and thus can not serve as the method of choice to diagnose pheochromocytoma. In contrast, the metanephrines, O-methylated metabolites of catecholamines (norepinephrine to normetanephrine and epinephrine to metanephrine) are constantly produced within the chromaffin cells independently of catecholamine release (Eisenhofer et al. Citation2004). Measurements of fractionated metanephrines (i.e. normetanephrine and metanephrine measured separately) in plasma or in urine are now well established to prove superior diagnostic sensitivity and specificity over plasma or urinary catecholamines (; Lenders et al. Citation2002; Sawka et al. Citation2003; Grossman et al. Citation2006). Plasma metanephrines are most often measured in their free form as produced within tumors, whereas urinary metanephrines are commonly measured after a deconjugation step and largely represent sulfate-conjugates produced by an enzyme localized mainly to gastrointestinal tissues (Eisenhofer et al. Citation2004b). This may explain higher diagnostic sensitivity of plasma metanephrines compared to urinary catecholamine metabolites (Lenders et al. Citation2002).
Table IV. Sensitivity and specificity of biochemical tests for diagnosis of pheochromocytoma.
In certain cases, measurement of plasma dopamine or methoxytyramine may provide additional clinical and diagnostic information (Eisenhofer et al. Citation2005). Overproduction of dopamine may indicate malignant tumor potential (John et al. Citation1999).
As with all biochemical tests of catecholamine excess, a remaining problem is that a positive result for urinary or plasma metanephrines does not always reliably indicate a pheochromocytoma. To distinguish true positive from false positive results, it is advocated to take into account the extent of the elevation in biochemical test results (Grossman et al. Citation2006). While an elevation of plasma or urinary normetanephrine slightly above the upper reference intervals may only marginally increase the post-test probability of pheochromocytoma, an elevation of more than four-fold above those intervals is associated with close to 100% probability of the tumor (Eisenhofer et al. Citation2003). The actual level of the abnormal result should, therefore, be used to determine the need for immediate tumor localization studies vs. additional biochemical investigations. To accurately interpret results in the so called grey-zone, sample conditions should also be taken into consideration (e.g. medications, seated or recumbent position during venepuncture, patient's compliance during urine collection, associate clinical conditions). Additional help to distinguish true positive from false positive results may be afforded using the clonidine suppression test. Failure to suppress plasma norepinephrine (a decrease of < 50% from basal or a persistent increased basal plasma norepinephrine of >3.00 nmol/l) after clonidine is highly predictive for pheochromocytoma (97%) (Bravo et al. Citation1981). In contrast, the negative predictive value of a normal test result is only 75%. If plasma normetanephrine is used (failure to suppress defined as a decrease of < 40% from basal or a persistent increased basal plasma normetanephrine of >0.60 nmol/l) instead of plasma norepinephrine, the positive and negative predictive values of this test improve to 100 and 96%, respectively (Eisenhofer et al. Citation2003).
Imaging
Imaging studies for detecting pheochromocytoma are usually not indicated unless the biochemical diagnosis is strongly positive, primarily in subjects with suspected sporadic pheochromocytoma. However, in patients with a hereditary predisposition or a previous history of the tumor, where the pre-test probability of a tumor is higher, less-compelling biochemical evidence might justify imaging studies (Grossman et al. Citation2006).
Since the vast majority of suspected tumors arise from adrenals or chromaffin tissue in the abdomen, methods of choice are magnetic resonance imaging (MRI) or computed tomography (CT) of the entire abdomen including pelvis with and without contrast (Ilias and Pacak Citation2004). Advantages of CT compared to MRI are better availability, lower cost and shorter scanning time but MRI (T2-weighted sequences) is preferred in children, pregnant females and in subjects with allergy to contrast. MRI is also superior to CT for detecting extra-adrenal tumors (better sensitivity for small tumors), especially in chest and neck (Sahdev et al. Citation2005) and postoperatively because MRI unlike CT is not interfered with by artifacts caused by metal surgical clips. Both methods possess good sensitivity for detecting tumors but they do not provide sufficient specificity to differentiate pheochromocytoma from other possible pathologies, particularly in the adrenals.
To confirm the diagnosis of pheochromocytoma and to exclude multilocular or malignant involvement, functional imaging should be performed, in particular in tumors with higher potential of malignancy or multifocal involvement (epinephrine-secreting adrenal pheochromocytomas >5 cm, all norepinephrine-secreting pheochromocytomas and functional paragangliomas) (Pacak et al. Citation2005). Scintigraphy using preferentially [123I]-metaiodobenzylguanidine [123I]-MIBG) offers excellent specificity (95–100%) and good sensitivity (83–100%) for detecting adrenal tumors (Nielsen et al. Citation1996; van der Harst et al. Citation2001; Miskulin et al. Citation2003), but sensitivity for paragangliomas or malignant tumors (71 and 56%, respectively) is less than optimal (Erickson et al. Citation2001; Ilias et al. Citation2003).
For subjects with negative [123I]-MIBG results, [111In]-octreotide scanning or positron emission tomography with [18F]-fluorodeoxyglucose may be useful (Shulkin et al. Citation1999; van der Harst et al. Citation2001). The latter modality is superior to [123I]-MIBG for malignant and rapidly growing poorly-differentiated tumors. Other imaging agents for positron emission tomography include as [18F]-fluorodopamine, [18F]-fluorodopa, [11C]-hydroxyephedrine, [18F]-dihydroxyphenylalanine and [11C]-epinephrine. All these imaging modalities are highly specific for pheochromocytoma (Grossman et al. Citation2006).
Management of pheochromocytoma
The correct clinical management of patients with pheochromocytoma relies on close collaboration between different specialists. In most patients, the tumor is cured by surgery. To minimize perioperative and postoperative morbidity and mortality, appropriate medical pretreatment is necessary at least 10–14 days before surgery. Most used drugs for medical pretreatment of pheochromocytoma are α- and β-adrenoceptor antagonists and calcium channel blockers (Prys-Robert Citation2000; van der Horst-Schrivers et al. Citation2006). Use of the non-competitive α-adrenoceptor antagonist pheonoxybenzamine may offer the advantage over competitive α-adrenoceptor antagonists (e.g. doxazosine or prazosin) by avoiding any possibility of drug displacement from α-adrenoceptors by excessive increases in catecholamines during surgery. However, use of phenoxybenzamine must be counterbalanced with higher risk of postoperative hypotension (Bravo and Tagle Citation2003). Although the maximal tolerable doses of α-adrenoceptor antagonists are recommended for the surgical pretreatment, clinical evidence that this strategy results in improvement of cardiovascular stability during surgery is still lacking (Weismann et al. Citation2006). Use of β-blockers is indicated in subjects with tachycardia but only while on treatment with α-adrenoceptor antagonists. To avoid or to lessen the risk of orthostatic hypotension during treatment with α-adrenoceptor antagonists, salt and fluid intake should be increased. The additional advantage of this approach is that it reduces the risk of postoperative hypotension.
The surgical method of choice for pheochromocytoma removal is laparoscopy with transperitoneal or retroperitoneal approach (Walz et al. Citation2006). The choice of procedure depends mainly on surgical experience and secondly on tumor dimensions (Wilhelm et al. Citation2006). In cases of bilateral or hereditary adrenal tumors, adrenal cortex-sparing surgery should be performed to minimize the risk of subsequent hypocortisolism. Advantages of laparoscopy over the conventional surgical approach are lower postoperative morbidity, and reduced hospital stay and expense. For treatment of hypertensive peaks and tachyarrhythmias during surgery, infusion of short acting drugs such as phentolamine, nicardipine, or sodium nitroprusside for blood pressure control and esmolol for tachycardias are used when necessary. Abrupt decrease of catecholamine levels after clamping off the venous drainage of the tumor to the general circulation is often accompanied by hypotension. Treatment requires fluid replacement and occasionally pressor agents (e.g. norepinephrine or phenylephrine) (Mannelli Citation2006). The other complication resulting from abrupt decrease of circulating catecholamines is hypoglycemia due to unopposed high insulin levels and low glycogen stores in liver (Kinney et al. Citation2002). This can be avoided by starting an IV infusion of 5% dextrose for 3–4 h immediately following tumor removal.
Currently, after proper medical preparation, operative mortality is less than 1% if performed by an experienced anesthesiologists and a skilful surgeon.
After the tumor removal, biochemical testing should be performed after about 14 days from surgery in order to check for remaining disease. If results of biochemical testing are entirely normal, resection is probably complete and those subjects with sporadic disease are likely cured. However, these patients should be followed-up for a risk of tumor recurrence or subsequent malignancy in at least annual intervals (Plouin et al. Citation1997). Those subjects at highest risk of subsequent malignancy (carriers of SDHB mutations, patients with functional paragangliomas and large adrenal tumors) and with hereditary pheochromocytomas require closer follow-up including clinical and biochemical assessment every 6–12 months (Mannelli Citation2006).
Malignant pheochromocytoma
Currently, the only criterion for malignancy is the presence of chromaffin cells at tissues sites where no chromaffin tissue should occur. To predict the potency of malignancy, several criteria and scoring systems have been developed on the basis of histopathology but perhaps the most predictive value are extra-adrenal tumor location, tumor size and presence of SDHB mutations. The prevalence of malignant pheochromocytoma is about 10% (5–26%); higher rates (up to 36%) have been reported in subjects with extra-adrenal tumors (O'Riordain et al. Citation1996; Ahlman Citation2006). The most frequent distant sites of metastatic involvement are bones (up to 70%), liver and lung. Those subjects with liver metastases may show shorter survival than those with solitary bone metastases. Overall, the 5-year survival rate in malignant pheochromocytoma is about 50% (Lehnert et al. Citation2004).
Currently, there is no cure for malignant pheochromocytoma or paraganglioma. Surgical treatment may help to obtain tumor reduction and to control hypertension and other clinical signs and symptoms. However, there are no randomized trials demonstrating any survival advantage of tumor debulking, but reduced tumor volume may facilitate subsequent radio- or chemotherapy (Ahlman Citation2006). Subjects with lesions avid for [121/131I]-MIBG scintigraphy may benefit from treatment with [131I]-MIBG, particularly with high doses (Fitzgerald et al. Citation2006). Only a few subjects develop complete remission after chemotherapy (mostly cyclophosphamide, dacarbazine and vincristine) (Averbuch et al. Citation1988). In some cases, radiofrequency ablation or external beam radiation may be beneficial. Therapy with somatostatin analogs does not seem to offer any benefit (Lamarre-Cliche et al. Citation2002). To define the role of therapeutic doses of [177Lu-DOTA0]Octreotate in the treatment of metastatic paraganglioma, larger numbers of subjects treated with this radiopharmaceutical are needed (van Essen et al. Citation2006).
Acknowledgements
This work was supported by the Intramural Research Program of the National Institute of Child Health and Human Development and in part by Research Projects of Czech Ministry of Education 0021620807, 0021620808 and 0021620817. The authors have no conflict of interest to disclose.
Related Research Data
References
- Ahlman H. Malignant pheochromocytoma: State of the field with future projections. Ann NY Acad Sci 2006; 1073: 449–464
- Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Munat A, Niccoli-Sire P, Richard S, Rohmer V, Sadoul JL, Strompf L, Schlumberger M, Bertagna X, Plouin PF, Jeunemaitre X, Gimenez-Roqueplo AP. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol 2005a; 23: 8812–8818
- Amar L, Servais A, Gimenez-Roqueplo AP, Zinzindohoue F, Chatellier G, Plouin PF. Year of diagnosis, features at presentation, and risk of recurrence in patients with pheochromocytoma or secreting paraganglioma. J Clin Endocrinol Metab 2005b; 90: 2110–2116
- Ariton M, Juan CS, AvRuskin TW. Pheochromocytoma: Clinical observations from a Brooklyn tertiary hospital. Endocr Pract 2000; 6: 249–252
- Averbuch SD, Steakley CS, Young RC, Gelmann EP, Goldstein DS, Stull R, Keiser HR. Malignant pheochromocytoma: Effective treatment with combination of cyclophosphamide, vincristine and dacarbazine. Ann Intern Med 1988; 109: 267–273
- Barontini M, Levin G, Sanso G. Characteristics of pheochromocytoma in a 4- to 20-year-old population. Ann NY Acad Sci 2006; 1073: 30–37
- Batide-Alanore A, Chatellier G, Plouin PF. Diabetes as a marker of pheochromocytoma in hypertensive patients. J Hypertens 2003; 21: 1703–1707
- Bausch B, Borozdin W, Neumann HP. European–American Pheochromocytoma Study Group. Clinical and genetic characteristics of patients with neurofibromatosis type 1 and pheochromocytoma. N Engl J Med 2006; 354: 2729–2731
- Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, Croxson M, Dahia PL, Elston M, Gimm O, Henley D, Herman P, Murday V, Niccoli-Sire P, Pasieka JL, Rohmer V, Tucker K, Jeunemaitre X, Marsh DJ, Plouin PF, Robinson BG. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab 2006; 91: 827–836
- Bornstein SR, Gimenez-Roqueplo AP. Genetic testing in pheochromocytoma. Increasing importance for clinical decision making. Ann NY Acad Sci 2006; 1073: 94–103
- Bravo EL, Tagle R. Pheochromocytoma: State-of-the-art and future prospects. Endocr Rev 2003; 24: 539–553
- Bravo EL, Tarazi RC, Fouad FM, Vidt DG, Gifford RW, Jr. Clonidine-suppression test: A useful aid in the diagnosis of pheochromocytoma. N Engl J Med 1981; 305: 623–626
- Brouwers FM, Eisenhofer G, Tao JT, Kant JA, Adams KT, Linehan WM, Pacak K. High frequency of SDHB germline mutations in patients with malignant catecholamine-producing paragangliomas: Implications for genetic testing. J Clin Endocrinol Metab 2006; 91: 4505–4509
- Brouwers FM, Lenders JW, Eisenhofer G, Pacak K. Pheochromocytoma as an endocrine emergency. Rev Endocr Metab Disord 2003; 4: 121–128
- Eisenhofer G, Walther MM, Huynh TT, Li ST, Bornstein SR, Vortmeyer A, Mannelli M, Goldstein DS, Linehan WM, Lenders JW, Pacak K. Pheochromocytomas in Von Hippel–Lindau syndrome and multiple endocrine neoplasia type 2 display distinct biochemical and clinical phenotypes. J Clin Endocrinol Metab 2001; 86: 1999–2008
- Eisenhofer G, Goldstein DS, Walther MM, Friberg P, Lenders JW, Keiser HR, Pacak K. Biochemical diagnosis of pheochromocytoma: How to distinguish true- from false-positive test results. J Clin Endocrinol Metab 2003; 88: 2656–2666
- Eisenhofer G, Bornstein SR, Brouwers FM, Cheung NK, Dahia PL, de Krijger RR, Giordano TJ, Greene LA, Goldstein DS, Lehnert H, Manger WM, Maris JM, Neumann HP, Pacak K, Shulkin BL, Smith DI, Tischler AS, Young WF, Jr. Malignant pheochromocytoma: Current status and initiatives for future progress. Endocr Relat Cancer 2004a; 11: 423–436
- Eisenhofer G, Kopin IJ, Goldstein DS. Catecholamine metabolism: A contemporary view with implications for physiology and medicine. Pharmacol Rev 2004b; 56: 331–349
- Eisenhofer G, Goldstein DS, Sullivan P, Csako G, Brouwers FM, Lain EW, Adams KT, Pacak K. Biochemical and clinical manifestations of dopamine-producing paragangliomas: Utility of plasma methoxytyramine. J Clin Endocrinol Metab 2005; 90: 2068–2075
- Erickson D, Kudva YC, Ebersold MJ, Thompson GB, Grant CS, van Heerden JA, Young WF, Jr. Benign paragangliomas: Clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab 2001; 86: 5210–5216
- Fitzgerald PA, Goldsby RE, Huberty JP, Price DC, Hawkins RA, Veatch JJ, Dela Cruz F, Jahan TM, Linker CA, Damon L, Matthay KK. Malignant pheochromocytomas and paragangliomas: A phase II study of therapy with high-dose 131I-metaiodobenzylguanidine (131I-MIBG). Ann NY Acad Sci 2006; 1073: 465–490
- Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, Khau Van Kien P, Corvol P, Plouin PF, Jeunemaitre X. COMETE Network. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res 2003; 63: 5615–5621
- Gimenez-Roqueplo AP, Lehnert H, Mannelli M, Neumann HP, Opocher G, Maher ER, Plouin PF. European Network for the study of Adrenal Tumors (ENS@T) Pheochromocytoma Working Group Phaechromocytoma, new genes and screening strategies. Clin Endocrinol (Oxf) 2006; 65: 699–705
- Gordon RD, Stowasser M, Tunny TJ, Klemm SA, Rutherford JC. High incidence of primary aldosteronism in 199 patients referred with hypertension. Clin Exp Pharmacol Physiol 1994; 21: 315–318
- Grossman A, Pacak K, Sawka A, Lenders JW, Harlander D, Peaston RT, Reznek R, Sisson J, Eisenhofer G. Biochemical diagnosis and localization of pheochromocytoma: Can we reach a consensus?. Ann NY Acad Sci 2006; 1073: 332–347
- Grumbach MM, Biller BM, Braunstein GD, Campbell KK, Carney JA, Godley PA, Harris EL, Lee JK, Oertel YC, Posner MC, Schlechte JA, Wieand HS. Management of the clinically inapparent adrenal mass (“incidentaloma”). Ann Intern Med 2003; 138: 424–429
- Ilias I, Pacak K. Current approaches and recommended algorithm for the diagnostic localization of pheochromocytoma. J Clin Endocrinol Metab 2004; 89: 479–491
- Ilias I, Yu J, Carrasquillo JA, Chen CC, Eisenhofer G, Whatley M, McElroy B, Pacak K. Superiority of 6-[18F]-fluorodopamine positron emission tomography versus [131I]-metaiodobenzylguanidine scintigraphy in the localization of metastatic pheochromocytoma. J Clin Endocrinol Metab 2003; 88: 4083–4087
- Ito Y, Fujimoto Y, Obara T. The role of epinephrine, norepinephrine, and dopamine in blood pressure disturbances in patients with pheochromocytoma. World J Surg 1992; 16: 759–764
- John H, Ziegler WH, Hauri D, Jaeger P. Pheochromocytomas: Can malignant potential be predicted?. Urology 1999; 53: 679–683
- Kaplan NM. Pheochromocytoma (with a preface about incidental adrenal masses). Kaplan's clinical hypertension, NM Kaplan. Lipincott Williams & Wilkins, Philadelphia 2006; 389–409
- Kasperlik-Zaluska AA, Roslonowska E, Slowinska-Srzednicka J, Otto M, Cichocki A, Cwikla J, Slapa R, Eisenhofer G. 1111 Patients with adrenal incidentalomas observed at a single endocrinological center: Incidence of chromaffin tumors. Ann NY Acad Sci 2006; 1073: 38–46
- Khorram-Manesh A, Jansson S, Wangberg B, Nilsson O, Tisell LE, Ahlman H. Mortality associated with pheochromocytoma: Increased risk for additional tumors. Ann NY Acad Sci 2006; 1073: 444–448
- Kinney MA, Narr BJ, Warner MA. Perioperative management of pheochromocytoma. J Cardiothorac Vasc Anesth 2002; 16: 359–369
- Kuchel O, Buu NT, Racz K, De Lean A, Serri O, Kyncl J. Role of sulfate conjugation of catecholamines in blood pressure regulation. Fed Proc 1986; 45: 2254–2259
- Lamarre-Cliche M, Gimenez-Roqueplo AP, Billaud E, Baudin E, Luton JP, Plouin PF. Effects of slow-release octreotide on urinary metanephrine excretion and plasma chromogranin A and catecholamine levels in patients with malignant or recurrent phaeochromocytoma. Clin Endocrinol (Oxf) 2002; 57: 629–634
- Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG, Jr., Schlisio S. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: Developmental culling and cancer. Cancer Cell 2005; 8: 155–167
- Lehnert H, Mundschenk J, Hahn K. Malignant pheochromocytoma. Front Horm Res 2004; 31: 155–162
- Lenders JW, Pacak K, Walther MM, Linehan WM, Mannelli M, Friberg P, Keiser HR, Goldstein DS, Eisenhofer G. Biochemical diagnosis of pheochromocytoma: Which test is best?. JAMA 2002; 287: 1427–1434
- Lenders JVM, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005; 366: 665–675
- Manger WM. The vagaries of pheochromocytoma. Am J Hypertens 2005; 18: 1266–1270
- Mannelli M. Management and treatment of pheochromocytomas and paragangliomas. Ann NY Acad Sci 2006; 1073: 405–416
- Mannelli M, Ianni L, Cilotti A, Conti A. Pheochromocytoma in Italy: A multicentric retrospective study. Eur J Endocrinol 1999; 141: 619–624
- Mantero F, Terzolo M, Arnaldi G, Osella G, Masini AM, Ali A, Giovagnetti M, Opocher G, Angeli A. A survey on adrenal incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab 2000; 85: 637–644
- McNeil AR, Blok BH, Koelmeyer TD, Burke MP, Hilton JM. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust NZ J Med 2000; 30: 648–652
- Miskulin J, Shulkin BL, Doherty GM, Sisson JC, Burney RE, Gauger PG. Is preoperative iodine 123 meta-iodobenzylguanidine scintigraphy routinely necessary before initial adrenalectomy for pheochromocytoma?. Surgery 2003; 134: 918–922
- Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, Hoegerle S, Boedeker CC, Opocher G, Schipper J, Januszewicz A, Eng C, European–American Paraganglioma Study Group. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004; 292: 943–951
- Nielsen JT, Nielsen BV, Rehling M. Location of adrenal medullary pheochromocytoma by I-123 metaiodobenzylguanidine SPECT. Clin Nucl Med 1996; 21: 695–699
- O'Riordain DS, Young WF, Jr., Grant CS, Carney JA, van Heerden JA. Clinical spectrum and outcome of functional extraadrenal paraganglioma. World J Surg 1996; 20: 916–921
- Omura M, Saito J, Yamaguchi K, Kakuta Y, Nishikawa T. Prospective study on the prevalence of secondary hypertension among hypertensive patients visiting a general outpatient clinic in Japan. Hypertens Res 2004; 27: 193–202
- Ong KR, Woodward ER, Killick P, Lim C, Macdonald F, Maher ER. Genotype-phenotype correlations in Von Hippel–Lindau disease. Hum Mutat 2007; 28: 143–149
- Pacak K, Ilias I, Adams KT, Eisenhofer G. Biochemical diagnosis, localization and management of pheochromocytoma: Focus on multiple endocrine neoplasia type 2 in relation to other hereditary syndromes and sporadic forms of the tumour. J Intern Med 2005; 257: 60–68
- Pešek J, Treška V, Ferda J, Mukenšnábl P. Images in cardiovascular medicine. Unusual case of pheochromocytoma with a surprising response to the intravenous administration of norepinephrine. Circulation 2005; 22: e327–e328
- Plouin PF, Chatellier G, Fofol I, Corvol P. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension 1997; 29: 1133–1139
- Proye C, Fossati P, Fontaine P, Lefebvre J, Decoulx M, Wemeau JL, Dewailly D, Rwamasirabo E, Cecat P. Dopamine-secreting pheochromocytoma: An unrecognized entity? Classification of pheochromocytomas according their type of secretion. Surgery 1986; 100: 1155–1162
- Prys-Roberts C. Phaeochromocytoma—recent progress in its management. Br J Anaesth 2000; 85: 44–57
- Rossi GP, Bernini G, Caliumi C, Desideri G, Fabris B, Ferri C, Ganzaroli C, Giacchetti G, Letizia C, Maccario M, Mallamaci F, Mannelli M, Mattarello MJ, Moretti A, Palumbo G, Parenti G, Porteri E, Semplicini A, Rizzoni D, Rossi E, Boscaro M, Pessina AC, Mantero F, PAPY Study Investigators. A prospective study of the prevalence of primary aldosteronism in 1,125 hypertensive patients. J Am Coll Cardiol 2006; 48: 2293–2300
- Sahdev A, Sohaib A, Monson JP, Grossman AB, Chew SL, Reznek RH. CT and MR imaging of unusual locations of extra-adrenal paragangliomas (pheochromocytomas). Eur Radiol 2005; 15: 85–92
- Sawka AM, Jaeschke R, Singh RJ, Young WF, Jr. A comparison of biochemical tests for pheochromocytoma: Measurement of fractionated plasma metanephrines compared with the combination of 24-hour urinary metanephrines and catecholamines. J Clin Endocrinol Metab 2003; 88: 553–558
- Schiavi F, Boedeker CC, Bausch B, Peczkowska M, Gomez CF, Strassburg T, Pawlu C, Buchta M, Salzmann M, Hoffmann MM, Berlis A, Brink I, Cybulla M, Muresan M, Walter MA, Forrer F, Valimaki M, Kawecki A, Szutkowski Z, Schipper J, Walz MK, Pigny P, Bauters C, Willet-Brozick JE, Baysal BE, Januszewicz A, Eng C, Opocher G, European–American Paraganglioma Study Group. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA 2005; 294: 2057–2063, Erratum in: JAMA 2006. 295. p. 628
- Shulkin BL, Thompson NW, Shapiro B, Francis IR, Sisson JC. Pheochromocytomas: Imaging with Pheochromocytomas: Imaging with 2-[fluorine-18]fluoro-2-deoxy-d-glucose PET. Radiology 1999; 212: 35–41
- Streeten DHP, Anderson GH, Jr. Mechanisms of orthostatic hypotension and tachycardia in patients with pheochromocytoma. Am J Hypertens 1996; 9: 760–769
- Timmers HJ, Kozupa A, Eisenhofer G, Raygada M, Adams KT, Solis D, Lenders JW, Pacak K. Clinical presentations, biochemical phenotypes, and genotype–phenotype correlations in patients with SDHB-associated pheochromocytomas and paragangliomas. J Clin Endocrinol Metab 2007; 2006–2315, doi: 10.1210/jc
- Tsujimoto G, Honda K, Hoffman BB, Hashimoto K. Desensitization of postjunctional alpha 1- and alpha 2-adrenergic receptor-mediated vasopressor responses in rat harboring pheochromocytoma. Circ Res 1987; 61: 86–98
- Walz MK, Alesina PF, Wenger FA, Koch JA, Neumann HP, Petersenn S, Schmid KW, Mann K. Laparoscopic and retroperitoneoscopic treatment of pheochromocytomas and retroperitoneal paragangliomas: Results of 161 tumors in 126 patients. World J Surg 2006; 30: 899–908
- Weismann D, Fassnacht M, Weinberger F, Hamelmann W, Diehl S, Lorenz K, Baerlehner E, Reincke M, Beuschlein F, Knoefel W, Nies C, Hahner S, Allolio B. Intraoperative haemodynamic stability in patients with phaeochromocytoma-minimally invasive vs. conventional open surgery. Clin Endocrinol (Oxf) 2006; 65: 352–358
- Wilhelm SM, Prinz RA, Onders RP, Solorzano CC. Analysis of large versus small pheochromocytomas: Operative approaches and patients outcomes. Surgery 2006; 140: 553–560
- Zelinka T, Štrauch B, Pecen L, Widimský J, Jr. Diurnal blood pressure variation in pheochromocytoma, primary aldosteronism and Cushing's syndrome. J Hum Hypertens 2004; 18: 107–111
- Zelinka T, Štrauch B, Petrák O, Holaj R, Vranková A, Weisserová H, Pacák K, Widimský J, Jr. Increased blood pressure variability in pheochromocytoma compared to essential hypertension patients. J Hypertens 2005; 23: 2033–2039
- deS Senanayake P, Denker J, Bravo EL, Graham RM. Production, characterization, and expression of neuropeptide Y by human pheochromocytoma. J Clin Invest 1995; 96: 2503–2509
- van Essen M, Krenning EP, Kooij PP, Bakker WH, Feelders RA, de Herder WW, Wolbers JG, Kwekkeboom DJ. Effects of therapy with [177Lu-DOTA0, Tyr3]octreotate in patients with paraganglioma, meningioma, small cell lung carcinoma, and melanoma. J Nucl Med 2006; 47: 1599–1606
- van der Harst E, de Herder WW, Bruining HA, Bonjer HJ, de Krijger RR, Lamberts SW, van de Meiracker AH, Boomsma F, Stijnen T, Krenning EP, Bosman FT, Kwekkeboom DJ. [(123)I]metaiodobenzylguanidine and [(111)In]octreotide uptake in begnign and malignant pheochromocytomas. J Clin Endocrinol Metab 2001; 86: 685–693
- van der Horst-Schrivers AN, Kerstens MN, Wolffenbuttel BH. Preoperative pharmacological management of phaeochromocytoma. Neth J Med 2006; 64: 290–295