Abstract
Alteration of tryptophan (TRP) metabolism elicited by proinflammatory cytokines has gained attention as a new concept to explain the etiological and pathophysiological mechanisms of major depression. The kynurenine (KYN) pathway, which is initiated by indoleamine 2,3-dioxygenase (IDO), is the main TRP metabolic pathway. It shares TRP with the serotonin (5-HT) pathway. Proinflammatory cytokines induce IDO under stress, promote the KYN pathway, deprive the 5-HT pathway of TRP, and reduce 5-HT synthesis. The resultant decrease in 5-HT production may relate to the monoamine hypothesis of major depression. Furthermore, metabolites of the KYN pathway have neurotoxic/neuroprotective activities; 3-hydroxykynurenine and quinolinic acid are neurotoxic, whereas kynurenic acid is neuroprotective. The hippocampal atrophy that appears in chronic depression may be associated with imbalances in neurotoxic/neuroprotective activities. Because proinflammatory cytokines also activate the hypothalamo-pituitary-adrenal (HPA) axis, these imbalances may inhibit the hippocampal negative feedback system. Thus, changes in the TRP metabolism may also relate to the HPA axis-hyperactivity hypothesis of major depression. In this article, we review the changes in TRP metabolism by proinflammatory cytokines under stress, which is assumed to be a risk factor for major depression, and the relationship between physiological risk factors for major depression and proinflammatory cytokines.
Introduction
Changes in the metabolism of the essential amino acid tryptophan (TRP), play an important role in the brain–endocrine–immune system interaction that is hypothesized to be involved in the pathophysiology of major depression. Two main pathways metabolize TRP: one is the kynurenine (KYN) pathway, which is initiated by the enzyme indoleamine 2,3-dioxygenase (IDO) (Hirata et al. Citation1974), and the other is the serotonin (5-HT) pathway, initiated by the enzyme tryptophan 5-monooxygenase (Ichiyama et al. Citation1970; tryptophan hydroxylase (TPH); ). Most TRP, i.e., ∼99% of dietary intake, is metabolized by the former pathway (Stone and Darlington Citation2002). Immunological challenges such as proinflammatory cytokines (IL-1, IFN-α, IFN-γ, and TNF-α) induce IDO activity (Darlington Citation2002). Because proinflammatory cytokines can induce depressive symptoms, cytokine-induced IDO activation may be associated with the etiology of major depression (Konsman et al. Citation2002, Maes Citation2004). Activated IDO metabolizes TRP to KYN, and may thereby deprive TPH of its substrate, TRP (). Thus, the activated KYN pathway may further inhibit the 5-HT pathway (Konsman et al. Citation2002, Kim Citation2003). The monoamine hypothesis, one of the main etiological hypotheses of depression, proposes that a decrease in monoamine levels in the central nervous system (CNS) is related to the onset and symptoms of depression (Delgado Citation2004; Owens Citation2004). Indeed, lowering 5-HT function by TRP depletion has a mood-lowering effect in the subgroups of recovered depressed patients, patients with seasonal affective disorder and vulnerable healthy subjects (Van der Does Citation2001). In the 1970s and 1980s, to clarify the relationship between the amount of KYN metabolites and symptoms of depression, urine levels of xanthurenic acid (XA), which is metabolized from 3-OH KYN (), was examined in depressed patients. The excretion of XA after ingestion of TRP was increased in depression, and improvement in XA excretion preceded clinical improvement (Hoes and Sijben Citation1981). Thus, the induction of IDO activity elicited by proinflammatory cytokines that inhibits 5-HT synthesis may be closely related to the etiological monoamine hypothesis of depression.
Figure 1 Kynurenine and serotonin pathways of tryptophan metabolism. ATP, Adenosine triphosphate; 5-HT, serotonin; 5-HIAA, 5-hydroxyindoleacetic acid; NAD, nicotinamide adenine dinucleotide.
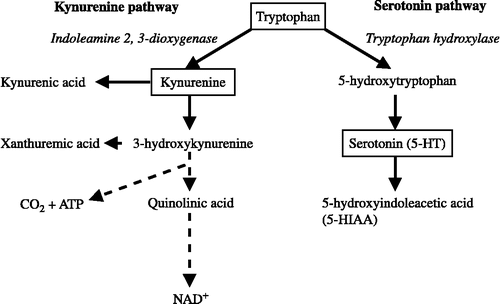
Figure 2 Immunological activation and tryptophan metabolism in the brain. In the macrophages and/or microglia, activated indoleamine 2,3-dioxygenase metabolizes tryptophan. Thus, the kynurenine pathway deprives the serotonin pathway of tryptophan. +, activation; − , inhibition. 3-OH KYN, 3-hydroxykynurenine; 5-HTP, 5-hydroxy tryptophan; 5-HT, serotonin; 5-HIAA, 5-hydroxyindoleacetic acid; IDO, indoleamine 2,3-dioxygenase; IFN, interferon; IL, interleukin; KYN, kynurenine; KYNA, kynurenic acid; NMDA, N-methyl-d-aspartate; NO, nitric oxide; QUIN, quinolinic acid; TNF, tumor necrosis factor; TRP, tryptophan.
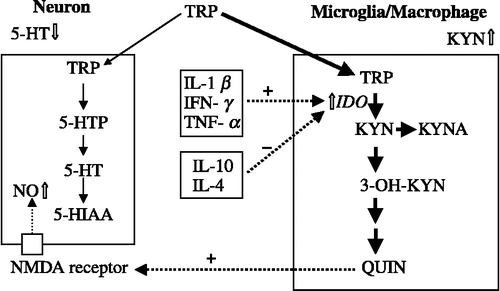
The monocyte–T-lymphocyte hypothesis has been proposed to explain the relationship between major depression and immunological activities (Smith Citation1991; Maes et al. Citation1995). This theory suggests that the high levels of IL-1 released from activated macrophages directly stimulate the release of corticotrophin-releasing factor (CRF) from the paraventricular nucleus (PVN) in the hypothalamus. Furthermore, IL-6 can also account for hypothalamo-pituitary-adrenal (HPA)-axis abnormalities, due to its direct action on the hypothalamus, again stimulating the release of CRF (Maes et al. Citation1995). Thus, the HPA-axis hyperactivity hypothesis, which proposes a relationship between HPA-axis hyperactivity and depression (Schule Citation2007), is closely related to the above-mentioned changes elicited by immunological activation. Both the monoamine and the HPA-axis hyperactivity hypotheses are central to etiological and pathophysiological hypotheses of major depression (Maes et al. Citation1995, Kim Citation2003).
Of the metabolites in the KYN pathway (), 3-hydroxy kynurenine (3-OH KYN) and quinolinic acid (QUIN) have neurotoxic effects while kynurenic acid (KYNA) has a neuroprotective effect (Kim Citation2003). Imbalance in the neurodegenerative and neuroprotective activities of KYN pathway metabolites may be linked to hippocampal atrophy in chronic depression, which plausibly relates to HPA-axis hyperactivity in depression. In gerbils, systemic immune activation induces brain IDO and increased brain and CSF QUIN, whereas brain activity of kynurenine aminotransferase (KAT), which converts KYN to KYNA, and the CSF KYNA level are unchanged. Consequently, the QUIN/KYNA ratio in CSF is increased (Saito et al. Citation1993). D retrovirus infection increases the QUIN/KYNA ratio in the CSF macaques with simian acquired immune deficiency syndrome (SAIDS) (Heyes et al. Citation1990). In humans, IFN-α therapy revealed that an increase in the serum KYN/KYNA ratio is significantly associated with the severity of depression (Wichers et al. Citation2005). In patients with major depression, plasma KYNA and the 1000 × KYNA/KYN ratio are decreased (Myint et al. Citation2007). Thus, the neurodegeneration process under stress may be more pronounced due to reinforcement by the neurotoxic effect of high QUIN levels (Myint and Kim Citation2003).
Although the activation of the KYN pathway has been reported in depression elicited by IFN-α therapy (Bonaccorso et al. 2002; Capuron et al. 2002, 2003; Wichers et al. 2005), the therapy is not the typical cause of clinical depression. The onset of clinical depression is more often elicited by social environment and psychological stress (Kendler et al. Citation1993; Paykel Citation1994).
In this article, we review the changes in TRP metabolism under physiological conditions and immunological challenge. An interaction between TRP metabolism and neuroendocrine/immune systems is proposed in relation to the etiological hypotheses of depression. To establish and test this hypothesis, it is important to examine changes in TRP metabolism under physiological conditions which are known to be risk factors for depression and to compare them with those elicited by IFN-α therapy.
Stress and brain tryptophan metabolism
An alteration of TRP metabolism is a good indicator of clinical depression elicited by immunological challenges (Bonaccorso et al. 2002; Capuron et al. 2002, 2003; Wichers et al. 2005) but stress also alters brain TRP metabolism.
Brain tryptophan uptake in response to acute stress and immunological challenge
Stress as well as immune challenges are known to elevate brain TRP levels. In animals, immobilization stress, food deprivation, exercise, and footshock increase brain TRP levels. Furthermore, immunological challenges such as IL-1 and lipopolysaccharide (LPS), which are known to activate the HPA axis, increase the brain TRP in mice (Dunn Citation1988a) and rats (Kabiersch et al. Citation1988). Because stress-related changes in brain TRP occur even in adrenalectomized rats (Curzon et al. Citation1972) and mice (Dunn Citation1988b), they are not dependent on adrenocortical activation. Activation of the sympathetic nervous system is responsible for the increase in brain TRP (Dunn and Welch Citation1991).
Two related mechanisms increase TRP level in the brain (). TRP is the only essential amino acid that binds to albumin (Pardridge Citation1998). In plasma, most TRP binds to albumin, so free TRP is only a small fraction (McMenamy Citation1965). One mechanism regulating brain TRP levels is the level of circulating free fatty acid (FFA), under control of the sympathetic nervous system. Stress and physical exercise (Chaouloff Citation1997) increase plasma FFA level via the sympathetic outflow, and then FFA binds to albumin. Because this binding reduces TRP affinity for albumin (McMenamy Citation1965), the increase in FFA levels inhibits TRP binding to albumin and increases free TRP levels in the plasma (Curzon et al. 1973; Strüder et al. 1999; ). The other mechanism regulating brain TRP levels is the competition between TRP and the so-called “large neutral” amino acids (LNAAs), which include aromatic amino acids and branched-chain amino acids, at the uptake site of the blood–brain barrier (BBB). The plasma TRP/LNAA ratio, rather than the TRP level, regulates TRP uptake activity at the BBB (Wurtman Citation1972; ). Thus, elevation of the TRP/LNAA ratio increases the brain uptake of TRP, and thereby increases brain TRP level.
Figure 3 Regulation of plasma level and brain uptake of tryptophan. Stress and immune challenges elevate the brain TRP level. Stimulation of the β-adrenergic system increases FFAs. FFAs bind to albumin, releasing TRP from albumin. Increased plasma TRP elevates the TRP/LNAAs ratio. Thus, the transporter system in the BBB for the uptake into the brain selects TRP rather than LNAAs. Another plasma TRP modulation relates to inflammation. The IRS activates the HPA axis to increase glucocorticoids, consuming TRP and other peripheral amino acids to produce plasma-positive APPs and to reduce plasma negative APPs such as albumin that bind TRP. Thus, HPA axis activity may also play a role in the modulation of plasma TRP levels. APPs, acute-phase proteins; BBB, blood–brain barrier; FFA, free fatty acid; HPA axis, hypothalamo-pituitary-adrenal axis; IRS, inflammatory response system; LNAA, large neutral amino acid; TRP, tryptophan.
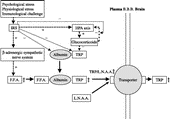
Finally, stress may increase the brain TRP level. Our recent data (CitationMiura et al. in press) suggest that 20-min novelty stress in mice, namely, exposure to an environment that the animal has not experienced before, elevates brain TRP levels. Thus, psychological stress possibly increases the brain TRP uptake in humans.
Acute stress and alteration of brain tryptophan metabolism
Acute psychological as well as physiological stress may alter brain TRP metabolism to activate the brain KYN pathway by induction of IDO.
IDO activity in the brain is present in neurons as well as in microglia and astrocytes (Guillemin et al. Citation2004; Roy et al. Citation2005).
In rats, physiological stress such as footshock increases brain TRP, KYN, KYNA and 3-OH KYN levels of rats (Pawlak et al. Citation2000). Brain TRP levels range from about 10 to 40 nmol/g, whereas those of KYN range from about 400 to 1200 pmol/g. Further, brain levels of KYNA range from about 20 to 40 pmol/g, and those of 3-OH KYN from about 40 to 80 pmol/g (Pawlak et al. Citation2000). Thus, brain TRP levels are far greater than those of KYN, KYNA, and 3-OH KYN. As physiological stress is able to increase brain TRP levels via increase of TRP uptake across the BBB, the increase in TRP transfer with stress may compensate for the decrease in TRP elicited by the conversion of TRP to KYN. Thus, brain IDO activity may be effective in physiological stress conditions without apparent immunological challenge. Our recent data (Miura et al. in press) suggest that 20-min novelty stress, i.e., exposure of animals to a condition they have never experienced before, elevated brain KYN levels as well as TRP levels without changing the KYN/TRP ratio. Thus, IDO may play a role in the elevation of the KYN level, accompanied by an increased TRP level elicited by acute psychological stress. Further, novelty stress decreased the 5-HT/TRP ratio. Thus, novelty stress shifted the balance between the KYN and the 5-HT pathways away from 5-HT.
However, influences of chronic and/or repeated stress on brain TRP metabolism, which may relate to the etiology of human depression, should be examined further.
Stress and proinflammatory cytokines
Exposure to stressors such as psychological stress activates the production of proinflammatory cytokines in humans. Psychological stress, specifically, an academic examination for medical students, significantly increases blood proinflammatory cytokines (Maes et al. Citation1998).
The brain–immune system interaction forms a bidirectional communication network, i.e., from the peripheral immune system to the brain and vice versa (Black Citation1994; Maier Citation2003). Peripherally activated proinflammatory cytokines released from macrophages signal to the brain via both humoral and neural routes. Because proinflammatory cytokines are relatively large and hydrophilic molecules, they are unlikely to cross the BBB by passive diffusion. They pass through the circumventricular organs (CVOs) and choroid plexus, and enter the brain parenchyma. At the CVOs, IL-1β induces, as a second messenger, the formation of prostaglandin E2, a small lipophilic molecule that is able to diffuse freely (Maier Citation2003). Furthermore, in addition to passive diffusion across the BBB, proinflammatory cytokines may enter the brain by active transport mechanisms (Banks et al. Citation1995; Watkins et al. Citation1995; Schiepers et al. Citation2005). Saturable transport mechanisms have already been identified for IL-1β, IL-1α, IL-6, and TNF-α (Banks et al. Citation1995). IL-1β also acts on glial cells in the CVOs, meninges, and choroid plexus to stimulate the production of IL-1β. Thus, IL-1β further induces de novo IL-1β production by glial cells in a paracrine manner in the brain. This process can propagate the IL-1β signal to distant regions (Maier Citation2003).
Cytokine signaling to the brain by neural routes involves the vagus nerve. IL-1β released from cells located near vagal afferent terminals can activate nerve impulses. However, the vagus may not be the only nerve that can provide this communication function (Maier Citation2003).
In addition, the newly synthesized IL-1β in the brain activates both the HPA axis and the sympathetic nervous system in response to peripheral immune stimulation (Maier Citation2003). Furthermore, cytokines that are produced in the brain enter the peripheral circulation.
An overlap between neurochemical changes elicited by stressors and immune challenges has been frequently noted. Stressors may activate peripheral host-defensive responses, if they elicit bidirectional, immune–brain and brain–immune communication. Stressors such as inescapable shock activate macrophage functions for several days. Novel environmental stress such as exposure to an “open field” increases IL-6 release from macrophages in rats (LeMay et al. Citation1990). Psychological stress increases IL-1, IL-6, sIL-2R, and cortisol concentrations in human subjects (Schulte et al. Citation1994). In the brain, acute stress indeed increases the IL-1β content (Minami et al. Citation1991; Shintani et al. Citation1995; Nguyen et al. Citation1998). Moreover, central and peripheral proinflammatory cytokine expression elicited by acute stress is region- and cytokine-specific (O'Connor et al. Citation2003). Previous exposure to a stressor increases LPS-stimulated IL-1β production (Johnson et al. Citation2002). Thus, stress may sensitize microglia to respond in a heightened fashion to a subsequent peripheral immune stimulus (Frank et al. Citation2007).
Furthermore, CRF may play a role in the interaction between a stressor and the production of proinflammatory cytokines (Leonard Citation2005). As CRF receptors are present on most immune cells such as the resident macrophages of the mouse spleen, human monocytes, macrophages, and T-lymphocytes (Schäfer et al. Citation1997), stressors may increase proinflammatory cytokines via CRF receptor stimulation (Leonard Citation2005).
Proinflammatory cytokines and tryptophan metabolism
Proinflammatory cytokines induce IDO activity. Subsequent activation of the KYN pathway results in TRP depletion that inhibits cell growth and proliferation, and the 5-HT biosynthesis ().
As described above, metabolites in the KYN pathway have a neurotoxic or neuroprotective activity (). 3-OH KYN and QUIN have a neurotoxic effect, whereas KYNA has a neuroprotective effect (Kim Citation2003). Some of the destructive properties of 3-OH KYN might be caused by its metabolism to 3-hydroxyanthranilic acid, which can undergo auto-oxidation with the formation of superoxide anions (Okuda et al. Citation1996; Stone and Darlington Citation2002; Wichers and Maes Citation2004). QUIN has the property of being a selective N-methyl-d-aspartate (NMDA) receptor agonist (Darlington Citation2002). A recent in vitro study reported that QUIN reduces antioxidant defenses in the cerebral cortex (Leipnitz et al. Citation2005). In contrast, KYNA may have neuroprotective properties because it has the properties of a NMDA receptor antagonist and could have agonist properties on nicotinic acetylcholine (nACh) receptors such as the α7-nACh receptor (Darlington Citation2002). The ratio of QUIN/KYNA is increased in the CSF of macaques with SAIDS (Heyes et al. Citation1990). Systemic immune activation in gerbils induces brain IDO, with no change in KAT activity, so the ratio of QUIN/KYNA in CSF increases (Saito et al. Citation1993). Human microglia as well as macrophages produced KYN and QUIN in cultures activated by IFN-γ (Heyes et al. Citation1996). These findings suggest that immunological challenges including a direct application of IFN-γ activate the KYN pathway and QUIN production in the CNS, but do not affect KYNA production.
Thus, the balance of neurotoxic and neuroprotective activities of these metabolites of TRP may play an important role in the pathogenesis of neurodegenerative disease involving depression.
Proinflammatory cytokines and the HPA axis
As described above, proinflammatory cytokines activate the HPA axis (Maier Citation2003; ). IL-1 is a potent stimulus of CRF synthesis, which supports the activation of the HPA axis (Black Citation1994; Leonard Citation2001). The production of prostaglandins (PGs) of the E2 series (PGE2s) at the level of the BBB transmits messages from the cytokines (Konsman et al. Citation2002). Synthesis of PGE2 is dependent on the induction of cyclooxygenase 2 (COX-2) and PGE synthase expressed in endothelial cells of the cerebral blood vessels and possibly perivascular macrophages. PGE2 activates the CRF neurons of the PVN. These processes activate the HPA axis (Konsman et al. Citation2002).
Figure 4 Regulation of brain KYN pathway and HPA axis by stress and immunological challenges. Stress and immune challenges activate the KYN pathway and the HPA axis. Proinflammatory cytokines activate the KYN pathway via induction of IDO, whereas they stimulate the HPA axis. Furthermore, proinflammatory cytokines attenuate the negative feedback control of the HPA axis. ACTH, adrenocorticotropic hormone; CRF, corticotropin releasing factor; 5-HIAA, 5-hydroxyindoleacetic acid; HPA axis, hypothalamo-pituitary-adrenal axis; 5-HT, serotonin; 5-HTTP, 5-hydroxy tryptophan; IDO, indoleamine 2,3-dioxygenase; IRS, inflammatory response system; KYN, kynurenine; KYNA, kynurenic acid; 3-OH KYN, 3-hydroxykynurenine; QUIN, quinolinic acid; TRP, tryptophan; TPH, tryptophan hydroxylase.
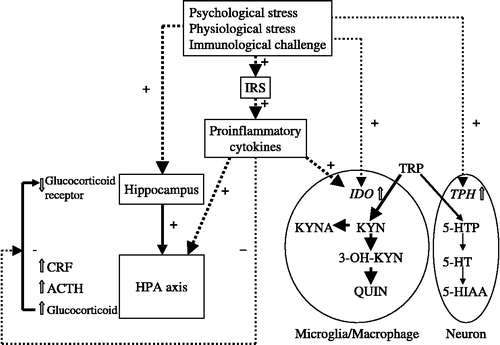
Further, the proinflammatory cytokine IFN-γ induces IDO expression in hypothalamic and pituitary cells (Tu et al. Citation2005). The activated IDO in these cells may produce 3-OH KYN and QUIN, which may result in neurodegenerative changes in the HPA axis. Hippocampal atrophy and subsequent inhibition of the negative feedback activity in the HPA axis in chronic depression might be associated with chronic exposure to these neurotoxic products of TRP.
Proinflammatory cytokines and brain monoamines
Proinflammatory cytokines act on brain monoamine systems in a cytokine- and region-specific manner in a complex way (Zalcman et al. Citation1994). As described above, proinflammatory cytokines may reduce the activity of 5-HT pathway by promoting the KYN pathway due to activation of IDO. Although a single i.p. INF-α injection has no significant effect on the levels of monoamines and their metabolites or monoamine turnover, repeated i.p. injection significantly decreases both dopamine (DA) and 3,4-dihydroxyphenylacetic acid levels in the mouse brain (Shuto et al. Citation1997). Acute intracerebroventricular infusion of INF-α increases 5-HT turnover in the prefrontal cortex and increases DA turnover in the hippocampus, while pre-treatment with diclofenac, a non-steroidal anti-inflammatory drug prevents these neurochemical responses in rats (De La Garza II et al. Citation2003). Chronic i.p. administration of INF-α to rats reduces DA levels in the prefrontal cortex, decreases 5-HT levels, and increases 5-HT turnover in the amygdala (De La Garza II et al. Citation2005). In other experiments, although chronic i.p. administration of INF-α to rats increases 5-HT turnover in the frontal cortex, hippocampus, amygdala, thalamus, hypothalamus, and brainstem, these treatments do not alter 5-HT transporter mRNA level (Sato et al. Citation2006). Acute i.p. injection of IL-1α to rats increases 5-HT release from the CA1 region of the hippocampus, whereas this treatment decreases norepinephrine (NE) release (Broderick Citation2002). IL-2 increases hypothalamic and hippocampal NE utilization and DA turnover in the prefrontal cortex, whereas IL-6 induces profound elevations of 5-HT and mesocortical DA activity in the hippocampus and prefrontal cortex (Zalcman et al. Citation1994).
Thus, although the effects of cytokines on brain monoamines are complex, these changes in the activities of brain monoamine systems elicited by proinflammatory cytokines may be related to the etiology and pathophysiology of depression.
Proinflammatory cytokines and depression
It is now generally accepted that psychological stress and psychiatric illness can compromise immune functions (Leonard Citation2001). There is strong evidence to suggest that changes in cytokine levels outside the brain cause changes in cytokine expression and activity in the brain. Conversely, cytokine changes in the brain elicit responses outside the brain (Dantzer et al. Citation1999). Proinflammatory cytokines induce neuroendocrine and behavioral changes similar to depression in animal models (Dantzer et al. Citation1999), and immune therapy using proinflammatory cytokines causes depressive symptoms in human subjects (Bonaccorso et al. 2002; Capuron et al. 2002, 2003; Wichers et al. 2005). In addition, changes in the immune system activity, especially the increased levels in proinflammatory cytokines, are found in human depressive patients (Smith Citation1991; Maes et al. Citation1995). Thus, depression may now be considered a disorder that is closely related to dysfunction in the immune–endocrine system (Leonard Citation2001).
Proinflammatory cytokines induce neuroendocrine and behavioral changes in animals that appear similar to those in depressed patients (Dantzer et al. Citation1999). In terms of neurochemical changes, proinflammatory cytokines increase the production of CRF, which can escape glucocorticoid negative feedback. In terms of behavioral changes, the anhedonia and helplessness that may be produced by proinflammatory cytokines are considered two essential features of a major depressive episode with melancholia.
An in vitro study has shown that the antidepressants clomipramine, sertraline, and trazodon at concentrations of 10− 6 M (similar to the plasma concentrations during clinical treatment) markedly reduce the proinflammatory cytokine IFN-γ, but increase the immune-inhibiting cytokine IL-10 release from human blood (Song Citation2000). Other studies showed that different antidepressants significantly inhibit proinflammatory cytokine secretion from human monocytes activated by LPS and increase the production of anti-inflammatory cytokines (Xia et al. Citation1996; Kubera et al. Citation2000). In contrast, ex vivo data collected in depressed patients are much less demonstrative, probably because of the heterogeneity of the patients and their clinical conditions (Haack et al. Citation1999). However, these actions of antidepressants in in vitro studies may help to explain the suppressive effects of antidepressants on proinflammatory cytokine production in vivo and on the immune function in depression (Song Citation2000; Castanon et al. Citation2002). The existence of 5-HT receptors on human immunocytes suggests a potential mechanism for the immune activity of antidepressants (Maes Citation2001). 5-HT significantly suppresses the production of IFN-γ, whereas the common activity of other antidepressants is to increase IL-10 secretion (Maes Citation2001). Thus, the suppressive effects of antidepressive drugs on the IFN-γ/IL-10 ratio are probably not related to the direct effects of 5-HT. Another possible mechanism of 5-HT revolves around cAMP. 5-HT stimulates 5-HT receptors on immunocytes and increases the intracellular levels of cAMP by a direct activation of adenylyl cyclase. Increased intracellular cAMP levels are considered to play a critical role in the production of IL-10 and IFN-γ (Maes Citation2001). Indeed, tricyclic antidepressants (TCAs) as well as selective serotonin reuptake inhibitors (SSRIs) significantly elevate intracellular cAMP levels in T-lymphocytes and monocytes (Xia et al. Citation1996). However, the exact mechanisms by which antidepressants exert their effects on proinflammatory cytokine production are still unknown.
Proinflammatory cytokines and the monoamine hypothesis of depression
As described above, the increase in proinflammatory cytokines in depression and the depressive symptoms following their administration may change brain monoamine system activity. Proinflammatory cytokines have been implicated in the alteration of presynaptic 5-HT as well as NE activities in the brain regions assumed to be involved in major depression, including the prefrontal cortex, hypothalamus, hippocampus, and amygdala (Zalcman et al. 1994; Broderick 2002; De La Garza II et al. 2003, 2005; Schiepers et al. 2005; Sato et al. 2006). Thus, possible explanations for the association between proinflammatory cytokines and dysfunction of monoaminergic systems in depression have been proposed. Proinflammatory cytokines regulate 5-HT transporter activity (Ramamoorthy et al. 1995; Mössner et al. 1998) and/or the number or sensitivity of postsynaptic 5-HT receptors, including the 5-HT1A and 5-HT2A receptors (Abe et al. Citation1999). In addition, proinflammatory cytokines stimulate COX-2 activity and thereby increase the synthesis of PGE2. Thus, an increase in the plasma and cerebrospinal fluid PGE2 levels reported in depressed patients may contribute to dysfunction in the release and uptake of brain monoamines by impeding the release of the amines (Leonard Citation2001).
Thus, activation of the inflammatory response system (IRS) and administration of proinflammatory cytokines may induce depressive symptoms through, among other mechanisms, their modulation of the 5-HT system (Bonaccorso et al. Citation2000).
Proinflammatory cytokines and HPA axis-hyperactivity hypothesis of depression
The best-documented changes in biological psychiatry are the increase in the activity of the HPA axis and glucocorticoid resistance in depressed patients (Dantzer et al. Citation1999). Proinflammatory cytokines might play a causal role in depression mainly because they provide another stimulatory input to the CRF system. The monocyte–T-lymphocyte hypothesis (Smith Citation1991; Maes et al. Citation1995) proposes that activated macrophages play a role in the clinical onset and pathophysiology of major depression, and suggests that high levels of IL-1 released from activated macrophages directly stimulate CRF release by the PVN neurones in the hypothalamus. According to this theory, cytokines would increase the reactivity of the HPA axis to external stressors by inducing the expression of vasopressin, another ACTH secretagogue, in parvocellular CRF neurons of the PVN. The chronically increased CRF release can by itself alter the production and action of brain cytokines (Dantzer et al. Citation1999). Proinflammatory cytokines activate the HPA axis and increase the release of glucocorticoids, which have strong immunosuppressive effects. Thus, the immunosuppression observed in major depression may be due to HPA axis hyperactivity elicited by proinflammatory cytokines and/or stress. The reciprocal relationships between HPA axis activity and the immune system may participate in the pathophysiology of major depression (Maes et al. Citation1995).
Depression and tryptophan metabolism
HPA axis activity and TRP availability, including plasma TRP level and TRP entry into the brain
The synthesis of 5-HT in the brain is directly dependent on brain TRP levels. In animals, tryptophan 2,3-dioxygenase (TDO; Hayaishi et al. Citation1957), another metabolizing enzyme of TRP, is induced by hydrocortisone. Although there is no direct evidence of TDO induction by steroids in humans, this effect may mediate a reduction in TRP availability in response to hypercortisolaemia (Porter et al. Citation2004; ). As described above, brain TRP levels are mainly regulated by two mechanisms, competitive binding to plasma albumin with FFA and competition with LNAA at the BBB (). Because glucocorticoid treatment induces hepatic and brain tyrosine aminotransferase, which is the major tyrosine-degrading enzyme (Hirota et al. Citation1985), tyrosine levels may also be altered by glucocorticoid levels (Porter et al. Citation2004). Further, tyrosine is one of the competing LNAAs at the BBB. Thus, glucocorticoid levels may regulate plasma TRP levels and TRP entry into the brain in a complex manner.
Glucocorticoids are known to attenuate the plasma TRP level and the TRP/LNAA ratio. However, the effects of glucocorticoids on TRP availability in healthy volunteers remain controversial. In healthy volunteers, dexamethasone significantly decreases plasma TRP and tyrosine levels as well as the plasma TRP/LNAA ratio (Maes et al. Citation1990a). One study reported that dexamethasone does not alter plasma TRP levels (Porter et al. Citation1999), whereas another study revealed that hydrocortisone significantly decreases plasma TRP (Bhagwagar et al. Citation2002). In depressive patients, dexamethasone also significantly decreases plasma TPP levels and the plasma TRP/LNAA ratio (Maes et al. Citation1990a,Citationb). In recovered depression patients, hydrocortisone also suppresses plasma TRP levels (Bhagwagar et al. Citation2002). Furthermore, patients with major depression and melancholia exhibit significantly lower TRP availability than patients with minor depression (Maes et al. Citation1990b). Thus, acute administration of glucocorticoids may suppress the availability of TRP ().
In addition, cortisol responses to dexamethasone are significantly and negatively associated with TRP availability (Maes et al. Citation1987, Citation1990c). However, there is no correlation between TRP/LNAA ratios and basal cortisol output (Maes et al. 1990b,c; Porter et al. 2004). Some other studies suggested that there is no significant difference in the plasma basal TRP levels between controls and depressed subjects (Cowen and Charig Citation1987; Price et al. Citation1991), although these studies did not measure LNAA levels or the TRP/LNAA ratio. Thus, the suppression of TRP availability in depression may relate to a dysfunction of the HPA axis feedback mechanism rather than to the basal level of glucocorticoids (Porter et al. Citation2004).
The mechanism underlying the attenuation in TRP availability in depression remains unknown. However, the direction of alteration in TRP availability reveals a decrease contrary to the increase in response to acute stress (). Acute stress and immunological challenges elevate plasma TRP and the TRP/LNAA ratio, whereas glucocorticoid treatment and depression suppress these measures. These findings suggest that peripheral TRP availability in response to acute stress and immunological challenge causes changes via the β-adrenergic system in the normal physiological condition but via the HPA axis in depression, although the possibility that the TRP changes caused by stress or proinflammatory cytokines have nothing to do with the onset of depression remains to be examined. Furthermore, these changes in depression may be associated with HPA axis feedback mechanism dysfunction rather than a deficiency in the basal level of glucocorticoids (Porter Citation2004). Stress-induced peripheral immune activation, or IRS, may induce peripheral IDO and/or TDO, and may consume TRP and other peripheral amino acids to produce positive acute-phase proteins (APPs) such as haptoglobin and C-reactive protein (Schiepers et al. Citation2005). In addition, the inflammation process reduces plasma-negative APPs such as albumin, which bind to TRP (). These alterations may modulate the increase in TRP availability in response to acute stress.
Prolonged and/or repeated exposure to stressful life events often precedes the onset of clinical depression. Thus, prolonged adaptation to these stresses may have a consumed ability to protect against new stress on the body and brain, i.e., allostatic load (McEwen Citation2004). Breakdown of the physiological adjustment system which increases the plasma TRP level, TRP/LNAA ratio, and brain TRP uptake in response to acute stress may result in the suppression of TRP availability.
Clinical major depression and tryptophan metabolism
All the above-described results suggest that proinflammatory cytokines induce depression. Some studies have indicated a correlation between the severity of depressive symptoms and serum TRP decrease and/or KYN increase in depression induced by cytokine therapy (Bonaccorso et al. 2002; Capuron et al. 2002, 2003). In the study of women in pregnancy and delivery, depression and anxiety symptoms in the early puerperium are causally related to an increased catabolism of TRP into KYN (Maes et al. Citation2002; Kohl et al. Citation2005). These changes in the plasma TRP and KYN/TRP ratio were significantly related to those in the IL-6 level (Maes et al. Citation2002).
In patients with major depression, the secretion of proinflammatory cytokines from activated macrophages and the synthesis of APPs from the liver are increased (Leonard Citation2001). In studies evaluating plasma TRP and LNAA together with indices of immune functions, patients with depression show decreased plasma TRP levels and TRP/LNAA ratios. Plasma TRP levels negatively correlate to IL-6 production (Maes et al. Citation1993). Furthermore, depressed subjects have significantly lower serum TRP and albumin levels (Maes et al. Citation1996). These studies suggest that lower TRP availability to the brain in depression is related to immune responses and lower serum albumin (Maes et al. Citation1993, Citation1996).
In recent years, imbalance in the neurodegenerative and neuroprotective activities of KYN pathway metabolites has been revealed as an aspect of depression. IFN-α therapy revealed that a long-term increase of the serum KYN/KYNA ratio, reflecting neurotoxic challenge, is significantly associated with a higher total Montgomery Asberg Depression Rating Scale (MADRS) score (Wichers et al. Citation2005). Further, plasma KYNA and the neuroprotective ratio, 1000 × KYNA/KYN, are decreased in patients with major depression (Myint et al. Citation2007). These studies suggest that the amount and ratio of the neuroprotective metabolite, KYNA, of the KYN pathway are lower in depressive patients. Indeed, patients with major depression who experienced no apparent immunological challenge as well as depressive patients induced by IFN-α showed a decrease in the KYNA level. Thus, a shift in the balance between the neurotoxic and neuroprotective activities of the KYN pathway toward the former may be associated with the etiology of depression.
Effects of antidepressants on the altered tryptophan metabolism
Although clinical findings support the alteration of TRP metabolism in depression, there is only scant evidence directly indicating the effects of antidepressants in restoring these changes in TRP metabolism. One study reported that paroxetine, an SSRI, did not suppress changes in KYN and neopterin levels elicited by IFN-α therapy in patients with malignant melanoma (Capuron et al. Citation2003). Further studies evaluating the responses of TRP metabolism to different types of antidepressants in patients with depression as well as in animal models are needed to clarify the role of TRP metabolism in the etiology and/or pathophysiology of depression.
Summary
In the present review, we explained how changes in TRP metabolism may be involved in the etiology and pathophysiology of depression. Because physiological and/or psychological stress as well as immunological challenges activate the IRS, and then induce proinflammatory cytokine expression both in the periphery and in the brain, these cytokines may induce and activate peripheral and brain IDO. The activated IDO metabolizes TRP into KYN, and shifts the balance between the KYN and 5-HT pathways of TRP metabolism to the former. Thus, the TRP available for 5-HT synthesis is reduced, and the subsequent 5-HT depletion in the brain may result in depression. The 5-HT shortage caused by IDO activation as well as presynaptic and postsynaptic modulation of the 5-HT system by proinflammatory cytokines may be associated with the monoamine hypothesis of depression. Furthermore, downstream of the activated KYN pathway, the balance of neuroprotective/neurotoxic metabolites may be important; 3-OH KYN and QUIN are neurotoxic, whereas KYNA is neuroprotective. Thus, an imbalance in these metabolites may cause neurodegenerative changes such as hippocampal atrophy in depression. Also, the activation of the HPA axis elicited by proinflammatory cytokines may be closely related to the HPA axis-hyperactivity hypothesis of depression. IRS activation elicited by stress and immunological challenges may thus provide the link between the monoamine and HPA axis-hyperactivity hypotheses of depression via proinflammatory cytokine-induced changes in TRP metabolism. Further accumulation of evidence that directly reveals that IDO induction, a shift in the balance between the KYN and 5-HT systems toward the former, and/or a shift in the balance between the neurotoxic and neuroprotective activities of the KYN pathway toward the former, elicited by physiological and psychological risk factors for depression such as prolonged and repeated exposure to stressful life events would provide support for the importance of the activation of the KYN pathway in the etiology of depression.
References
- Abe S, Hori T, Suzuki T, Baba A, Shiraishi H, Yamamoto T. Effects of chronic administration of interferon alpha A/D on serotonergic receptors in rat brain. Neurochem Res 1999; 24: 359–363
- Banks WA, Kastin AJ, Broadwell RD. Passage of cytokines across the blood–brain barrier. Neuroimmunomodulation 1995; 2: 241–248
- Bhagwagar Z, Hafizi S, Cowen PJ. Cortisol modulation of 5-HT-mediated growth hormone release in recovered depressed patients. J Affect Disord 2002; 72: 249–255
- Black PH. Immune system–central nervous system interactions: Effect and immunomodulatory consequences of immune system mediators on the brain. Antimicrob Agents Chemother 1994; 38: 7–12
- Bonaccorso S, Meltzer H, Maes M. Psychological and behavioural effects of interferons. Curr Opin Psychiatry 2000; 13: 673–677
- Bonaccorso S, Marino V, Puzella A, Pasquini M, Biondi M, Artini M, Almerighi C, Verkerk R, Meltzer H, Maes M. Increased depressive ratings in patients with hepatitis C receiving interferon-alpha-based immunotherapy are related to interferon-alpha-induced changes in the serotonergic system. J Clin Psychopharmacol 2002; 22: 86–90
- Broderick PA. Interleukin 1alpha alters hippocampal serotonin and norepinephrine release during open-field behavior in Sprague–Dawley animals: Differences from the Fawn-Hooded animal model of depression. Prog Neuropsychopharmacol Biol Psychiatry 2002; 26: 1355–1372
- Capuron L, Ravaud A, Neveu PJ, Miller AH, Maes M, Dantzer R. Association between decreased serum tryptophan concentrations and depressive symptoms in cancer patients undergoing cytokine therapy. Mol Psychiatry 2002; 7: 468–473
- Capuron L, Neurauter G, Musselman DL, Lawson DH, Nemeroff CB, Fuchs D, Miller AH. Interferon-alpha-induced changes in tryptophan metabolism. relationship to depression and paroxetine treatment. Biol Psychiatry 2003; 54: 906–914
- Castanon N, Leonard BE, Neveu PJ, Yirmiya R. Effects of antidepressants on cytokine production and actions. Brain Behav Immun 2002; 16: 569–574
- Chaouloff F. Effects of acute physical exercise on central serotonergic systems. Med Sci Sports Exerc 1997; 29: 58–62
- Cowen PJ, Charig EM. Neuroendocrine responses to intravenous tryptophan in major depression. Arch Gen Psychiatry 1987; 44: 958–966
- Curzon G, Joseph MH, Knott PJ. Effects of immobilization and food deprivation on rat brain tryptophan metabolism. J Neurochem 1972; 19: 1967–1974
- Curzon G, Friedel J, Knott PJ. The effect of fatty acids on the binding of tryptophan to plasma protein. Nature 1973; 242: 198–200
- Dantzer R, Wollman E, Vitkovic L, Yirmiya R. Cytokines and depression: Fortuitous or causative association?. Mol Psychiatry 1999; 4: 328–332
- De La Garza R, II, Anis GM. The non-steroidal anti-inflammatory drug diclofenac sodium attenuates IFN-alpha induced alterations to monoamine turnover in prefrontal cortex and hippocampus. Brain Res 2003; 977: 70–79
- De La Garza R, II, Asnis GM, Pedrosa E, Stearns C, Migdal AL, Reinus JF, Paladugu R, Vemulapalli S. Recombinant human interferon-alpha does not alter reward behavior, or neuroimmune and neuroendocrine activation in rats. Prog Neuropsychopharmacol Biol Psychiatry 2005; 29: 781–792
- Delgado PL. How antidepressants help depression: Mechanisms of action and clinical response. J Clin Psychiatry 2004; 65(Suppl 4)25–30
- Dunn AJ. Systemic interleukin-1 administration stimulates hypothalamic norepinephrine metabolism parallelling the increased plasma corticosterone. Life Sci 1988a; 43: 429–435
- Dunn AJ. Stress-related changes in cerebral catecholamine and indoleamine metabolism: Lack of effect of adrenalectomy and corticosterone. J Neurochem 1988b; 51: 406–412
- Dunn AJ, Welch J. Stress- and endotoxin-induced increases in brain tryptophan and serotonin metabolism depend on sympathetic nervous system activity. J Neurochem 1991; 57: 1615–1622
- Fernstrom JD, Wurtman RJ. Brain serotonin content: Physiological regulation by plasma neutral amino acids. Science 1972; 178: 414–416
- Frank MG, Baratta MV, Sprunger DB, Watkins LR, Maier SF. Microglia serve as a neuroimmune substrate for stress-induced potentiation of CNS pro-inflammatory cytokine responses. Brain Behav Immun 2007; 21: 47–59
- Guillemin GJ, Smythe G, Takikawa O, Brew BJ. Expression of indoleamine 2,3-dioxygenase and production of quinolinic acid by human microglia, astrocytes, and neurons. Glia 2004; 49: 15–23
- Haack M, Hinze-Selch D, Fenzel T, Kraus T, Kuhn M, Schuld A, Pollmacher T. Plasma levels of cytokines and soluble cytokine receptors in psychiatric patients upon hospital admission: Effects of confounding factors and diagnosis. J Psychiatr Res 1999; 33: 407–418
- Hayaishi O, Rothberg S, Mehler AH, Saito Y. Studies on oxygenases; enzymatic formation of kynurenine from tryptophan. J Biol Chem 1957; 229: 889–896
- Heyes MP, Mefford IN, Quearry BJ, Dedhia M, Lackner A. Increased ratio of quinolinic acid to kynurenic acid in cerebrospinal fluid of D retrovirus-infected rhesus macaques: Relationship to clinical and viral status. Ann Neurol 1990; 27: 666–675
- Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, Markey SP. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem J 1996; 320(Pt 2)595–597
- Hirata F, Hayaishi O, Tokuyama T, Senoh S. In vitro and in vivo formation of two new metabolites of melatonin. J Biol Chem 1974; 249: 1311–1313
- Hirota T, Hirota K, Sanno Y, Tanaka T. A new glucocorticoid receptor species: Relation to induction of tryptophan dioxygenase by glucocorticoids. Endocrinology 1985; 117: 1788–1795
- Hoes MJ, Sijben N. The clinical significance of disordered renal excretion of xanthurenic acid in depressive patients. Psychopharmacology (Berl) 1981; 75: 346–349
- Ichiyama A, Nakamura S, Nishizuka Y, Hayaishi O. Enzymic studies on the biosynthesis of serotonin in mammalian brain. J Biol Chem 1970; 245: 1699–1709
- Johnson JD, O'Connor KA, Deak T, Stark M, Watkins LR, Maier SF. Prior stressor exposure sensitizes LPS-induced cytokine production. Brain Behav Immun 2002; 16: 461–476
- Kabiersch A, del Rey A, Honegger CG, Besedovsky HO. Interleukin-1 induces changes in norepinephrine metabolism in the rat brain. Brain Behav Immun 1988; 2: 267–274
- Kendler KS, Kessler RC, Neale MC, Heath AC, Eaves LJ. The prediction of major depression in women: Toward an integrated etiologic model. Am J Psychiatry 1993; 150: 1139–1148
- Kohl C, Walch T, Huber R, Kemmler G, Neurauter G, Fuchs D, Solder E, Schrocksnadel H, Sperner-Unterweger B. Measurement of tryptophan, kynurenine and neopterin in women with and without postpartum blues. J Affect Disord 2005; 86: 135–142
- Konsman JP, Parnet P, Dantzer R. Cytokine-induced sickness behaviour: Mechanisms and implications. Trends Neurosci 2002; 25: 154–159
- Kubera M, Kenis G, Bosmans E, Scharpe S, Maes M. Effects of serotonin and serotonergic agonists and antagonists on the production of interferon-gamma and interleukin-10. Neuropsychopharmacology 2000; 23: 89–98
- LeMay LG, Vander AJ, Kluger MJ. The effects of psychological stress on plasma interleukin-6 activity in rats. Physiol Behav 1990; 47: 957–961
- Leipnitz G, Schumacher C, Scussiato K, Dalcin KB, Wannmacher CM, Wyse AT, Dutra-Filho CS, Wajner M, Latini A. Quinolinic acid reduces the antioxidant defenses in cerebral cortex of young rats. Int J Dev Neurosci 2005; 23: 695–701
- Leonard BE. Changes in the immune system in depression and dementia: Causal or co-incidental effects?. Int J Dev Neurosci. 2001; 19: 305–312
- Leonard BE. The HPA and immune axes in stress: The involvement of the serotonergic system. Eur Psychiatry 2005; 20(Suppl 3)S302–S306
- Maes M, De Ruyter M, Hobin P, Suy E. Relationship between the dexamethasone suppression test and the l-tryptophan/competing amino acids ratio in depression. Psychiatry Res 1987; 21: 323–335
- Maes M, Jacobs MP, Suy E, Minner B, Leclercq C, Christiaens F, Raus J. Suppressant effects of dexamethasone on the availability of plasma l-tryptophan and tyrosine in healthy controls and in depressed patients. Acta Psychiatr Scand 1990a; 81: 19–23
- Maes M, Schotte C, Scharpe S, Martin M, Blockx P. The effects of glucocorticoids on the availability of l-tryptophan and tyrosine in the plasma of depressed patients. J Affect Disord 1990b; 18: 121–127
- Maes M, Vandewoude M, Schotte C, Maes L, Martin M, Scharpe S, Blockx P. The relationships between the cortisol responses to dexamethasone and to l-5-HTP, and the availability of l-tryptophan in depressed females. Biol Psychiatry 1990c; 27: 601–608
- Maes M, Meltzer HY, Scharpe S, Bosmans E, Suy E, De Meester I, Calabrese J, Cosyns P. Relationships between lower plasma l-tryptophan levels and immune-inflammatory variables in depression. Psychiatry Res 1993; 49: 151–165
- Maes M, Smith R, Scharpe S. The monocyte-T-lymphocyte hypothesis of major depression. Psychoneuroendocrinology 1995; 20: 111–116
- Maes M, Wauters A, Verkerk R, Demedts P, Neels H, Van Gastel A, Cosyns P, Scharpe S, Desnyder R. Lower serum l-tryptophan availability in depression as a marker of a more generalized disorder in protein metabolism. Neuropsychopharmacology 1996; 15: 243–251
- Maes M, Song C, Lin A, De Jongh R, Van Gastel A, Kenis G, Bosmans E, De Meester I, Benoy I, Neels H, Demedts P, Janca A, Scharpe S, Smith RS. The effects of psychological stress on humans: Increased production of pro-inflammatory cytokines and a Th1-like response in stress-induced anxiety. Cytokine 1998; 10: 313–318
- Maes M. The immunoregulatory effects of antidepressants. Hum Psychopharmacol 2001; 16: 95–103
- Maes M, Verkerk R, Bonaccorso S, Ombelet W, Bosmans E, Scharpe S. Depressive and anxiety symptoms in the early puerperium are related to increased degradation of tryptophan into kynurenine, a phenomenon which is related to immune activation. Life Sci 2002; 71: 1837–1848
- Maier SF. Bi-directional immune–brain communication: Implications for understanding stress, pain, and cognition. Brain Behav Immun 2003; 17: 69–85
- McEwen BS. Protection and damage from acute and chronic stress: Allostasis and allostatic overload and relevance to the pathophysiology of psychiatric disorders. Ann NY Acad Sci 2004; 1032: 1–7
- McMenamy RH. Binding of indole analogues to human serum albumin. Effects of fatty acids. J Biol Chem 1965; 240: 4235–4243
- Minami M, Kuraishi Y, Yamaguchi T, Nakai S, Hirai Y, Satoh M. Immobilization stress induces interleukin-1 beta mRNA in the rat hypothalamus. Neurosci Lett 1991; 123: 254–256
- Miura H, Ozaki N, Shirokawa T, Isobe K. Changes in brain tryptophan metabolism elicited by age, social environment, and psychological stress in mice. Stress 2007, in press
- Moffett JR, Namboodiri MA. Tryptophan and the immune response. Immunol Cell Biol 2003; 81: 247–265
- Mössner R, Heils A, Stöber G, Okladnova O, Daniel S, Lesch KP. Enhancement of serotonin transporter function by tumor necrosis factor alpha but not by interleukin-6. Neurochem Int 1998; 33: 251–254
- Myint AM, Kim YK. Cytokine–serotonin interaction through IDO: A neurodegeneration hypothesis of depression. Med Hypotheses 2003; 61: 519–525
- Myint AM, Kim YK, Verkerk R, Scharpe S, Steinbusch H, Leonard B. Kynurenine pathway in major depression: Evidence of impaired neuroprotection. J Affect Disord 2007; 98: 143–151
- Nguyen KT, Deak T, Owens SM, Kohno T, Fleshner M, Watkins LR, Maier SF. Exposure to acute stress induces brain interleukin-1beta protein in the rat. J Neurosci 1998; 18: 2239–2246
- O'Connor KA, Johnson JD, Hansen MK, Wieseler Frank JL, Maksimova E, Watkins LR, Maier SF. Peripheral and central proinflammatory cytokine response to a severe acute stressor. Brain Res 2003; 991: 123–132
- Okuda S, Nishiyama N, Saito H, Katsuki H. Hydrogen peroxide-mediated neuronal cell death induced by an endogenous neurotoxin, 3-hydroxykynurenine. Proc Natl Acad Sci USA 1996; 93: 12553–12558
- Owens MJ. Selectivity of antidepressants: From the monoamine hypothesis of depression to the SSRI revolution and beyond. J Clin Psychiatry 2004; 65(Suppl 4)5–10
- Pardridge WM. Blood–brain barrier carrier-mediated transport and brain metabolism of amino acids. Neurochem Res 1998; 23: 635–644
- Pawlak D, Takada Y, Urano T, Takada A. Serotonergic and kynurenic pathways in rats exposed to foot shock. Brain Res Bull 2000; 52: 197–205
- Paykel ES. Life events, social support and depression. Acta Psychiatr Scand Suppl 1994; 377: 50–58
- Porter RJ, McAllister-Williams RH, Jones S, Young AH. Effects of dexamethasone on neuroendocrine and psychological responses to l-tryptophan infusion. Psychopharmacology (Berl) 1999; 143: 64–71
- Porter RJ, Gallagher P, Watson S, Young AH. Corticosteroid-serotonin interactions in depression: A review of the human evidence. Psychopharmacology (Berl) 2004; 173: 1–17
- Price LH, Charney DS, Delgado PL, Heninger GR. Serotonin function and depression: Neuroendocrine and mood responses to intravenous l-tryptophan in depressed patients and healthy comparison subjects. Am J Psychiatry 1991; 148: 1518–1525
- Ramamoorthy S, Ramamoorthy JD, Prasad PD, Bhat GK, Mahesh VB, Leibach FH, Ganapathy V. Regulation of the human serotonin transporter by interleukin-1 beta. Biochem Biophys Res Commun 1995; 216: 560–567
- Roy EJ, Takikawa O, Kranz DM, Brown AR, Thomas DL. Neuronal localization of indoleamine 2,3-dioxygenase in mice. Neurosci Lett 2005; 387: 95–99
- Saito K, Crowley JS, Markey SP, Heyes MP. A mechanism for increased quinolinic acid formation following acute systemic immune stimulation. J Biol Chem 1993; 268: 15496–15503
- Sato T, Suzuki E, Yokoyama M, Semba J, Watanabe S, Miyaoka H. Chronic intraperitoneal injection of interferon-alpha reduces serotonin levels in various regions of rat brain, but does not change levels of serotonin transporter mRNA, nitrite or nitrate. Psychiatry Clin Neurosci 2006; 60: 499–506
- Schäfer M, Mousa SA, Stein C. Corticotropin-releasing factor in antinociception and inflammation. Eur J Pharmacol 1997; 323: 1–10
- Schiepers OJ, Wichers MC, Maes M. Cytokines and major depression. Prog Neuropsychopharmacol Biol Psychiatry 2005; 29: 201–217
- Schule C. Neuroendocrinological mechanisms of actions of antidepressant drugs. J Neuroendocrinol 2007; 19: 213–226
- Schulte HM, Bamberger CM, Elsen H, Herrmann G, Bamberger AM, Barth J. Systemic interleukin-1 alpha and interleukin-2 secretion in response to acute stress and to corticotropin-releasing hormone in humans. Eur J Clin Invest 1994; 24: 773–777
- Shintani F, Nakaki T, Kanba S, Sato K, Yagi G, Shiozawa M, Aiso S, Kato R, Asai M. Involvement of interleukin-1 in immobilization stress-induced increase in plasma adrenocorticotropic hormone and in release of hypothalamic monoamines in the rat. J Neurosci 1995; 15: 1961–1970
- Shuto H, Kataoka Y, Horikawa T, Fujihara N, Oishi R. Repeated interferon-alpha administration inhibits dopaminergic neural activity in the mouse brain. Brain Res 1997; 747: 348–351
- Smith RS. The macrophage theory of depression. Med Hypotheses 1991; 35: 298–306
- Song C. The interaction between cytokines and neurotransmitters in depression and stress: Possible mechanism of antidepressant treatments. Hum Psychopharmacol 2000; 15: 199–211
- Stone TW, Darlington LG. Endogenous kynurenines as targets for drug discovery and development. Nat Rev Drug Discov 2002; 1: 609–620
- Strüder HK, Hollmann W, Platen P, Wostmann R, Weicker H, Molderings GJ. Effect of acute and chronic exercise on plasma amino acids and prolactin concentrations and on [3H]ketanserin binding to serotonin2A receptors on human platelets. Eur J Appl Physiol Occup Physiol 1999; 79: 318–324
- Tu H, Rady PL, Juelich T, Smith EM, Tyring SK, Hughes TK. Cytokine regulation of tryptophan metabolism in the hypothalamic-pituitary-adrenal (HPA) axis: Implications for protective and toxic consequences in neuroendocrine regulation. Cell Mol Neurobiol 2005; 25: 673–680
- Van der Does AJ. The effects of tryptophan depletion on mood and psychiatric symptoms. J Affect Disord 2001; 64: 107–119
- Watkins LR, Maier SF, Goehler LE. Cytokine-to-brain communication: A review & analysis of alternative mechanisms. Life Sci 1995; 57: 1011–1026
- Wichers MC, Maes M. The role of indoleamine 2,3-dioxygenase (IDO) in the pathophysiology of interferon-alpha-induced depression. J Psychiatry Neurosci 2004; 29: 11–17
- Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpe S, Maes M. IDO and interferon-alpha-induced depressive symptoms: A shift in hypothesis from tryptophan depletion to neurotoxicity. Mol Psychiatry 2005; 10: 538–544
- Widner B, Laich A, Sperner-Unterweger B, Ledochowski M, Fuchs D. Neopterin production, tryptophan degradation, and mental depression—what is the link?. Brain Behav Immun 2002; 16: 590–595
- Xia Z, DePierre JW, Nassberger L. Tricyclic antidepressants inhibit IL-6, IL-1 beta and TNF-alpha release in human blood monocytes and IL-2 and interferon-gamma in T cells. Immunopharmacology 1996; 34: 27–37
- Zalcman S, Green-Johnson JM, Murray L, Nance DM, Dyck D, Anisman H, Greenberg AH. Cytokine-specific central monoamine alterations induced by interleukin − 1, − 2 and − 6. Brain Res 1994; 643: 40–49