
Abstract
Inherited platelet disorders are important conditions that often manifest with bleeding. These disorders have heterogeneous underlying pathologies. Some are syndromic disorders with non-blood phenotypic features, and others are associated with an increased predisposition to developing myelodysplasia and leukemia. Platelet disorders can present with thrombocytopenia, defects in platelet function, or both. As the underlying pathogenesis of inherited thrombocytopenias and platelet function disorders are quite diverse, their evaluation requires a thorough clinical assessment and specialized diagnostic tests, that often challenge diagnostic laboratories. At present, many of the commonly encountered, non-syndromic platelet disorders do not have a defined molecular cause. Nonetheless, significant progress has been made over the past few decades to improve the diagnostic evaluation of inherited platelet disorders, from the assessment of the bleeding history to improved standardization of light transmission aggregometry, which remains a “gold standard” test of platelet function. Some platelet disorder test findings are highly predictive of a bleeding disorder and some show association to symptoms of prolonged bleeding, surgical bleeding, and wound healing problems. Multiple assays can be required to diagnose common and rare platelet disorders, each requiring control of preanalytical, analytical, and post-analytical variables. The laboratory investigations of platelet disorders include evaluations of platelet counts, size, and morphology by light microscopy; assessments for aggregation defects; tests for dense granule deficiency; analyses of granule constituents and their release; platelet protein analysis by immunofluorescent staining or flow cytometry; tests of platelet procoagulant function; evaluations of platelet ultrastructure; high-throughput sequencing and other molecular diagnostic tests. The focus of this article is to review current methods for the diagnostic assessment of platelet function, with a focus on contemporary, best diagnostic laboratory practices, and relationships between clinical and laboratory findings.
Introduction
Inherited platelet disorders are important causes of abnormal bleeding. Among the known inherited causes of bleeding and thrombotic disorders, many are conditions that alter platelet numbers and/or function [Citation1]. The pathogenesis of inherited platelet function disorders (PFDs) and inherited thrombocytopenias (ITs) has emerged to be quite complex and heterogeneous. New causes of PFDs and ITs continue to emerge, and presently, many commonly encountered PFDs have an unknown molecular cause, even when new tools, such as next-generation exome sequencing are applied [Citation2–7]. The complex and heterogeneous pathogenesis of these disorders continues to challenge clinicians and the diagnostic laboratories that assess patients for potential platelet disorders.
Over the last few decades, there have been major efforts to improve and standardize the laboratory assessment for PFDs, including platelet aggregation studies. This had led to the publication of many guidelines, including a recent guidance document from the International Society on Thrombosis and Haemostasis (ISTH) on stepwise investigations for diagnosing inherited PFDs [Citation8–11]. The last decades have also seen the launch of external quality assessment (EQA) programs to assess components of PFD diagnosis, including the post-analytical interpretation of light transmittance aggregometry (LTA) tests, a “gold standard” test for PFD assessment [Citation12]. EQA programs have also been established for the evaluation of platelet dense (δ)-granule deficiency (DGD) by transmission electron microscopy (TEM), using air-dried, whole-mount (WM) preparations of platelet-rich plasma (PRP) to examine and quantify the average number of δ-granules per platelet [Citation12].
The focus of this article is to review current methods for the diagnostic assessment of suspected inherited defects in platelet function, with a focus on contemporary, best diagnostic laboratory practices. The use of platelet function tests for drug monitoring and other purposes will not be discussed in this review.
Current concepts on the causes of inherited platelet disorders
New causes of inherited PFDs continue to emerge and some of the genes that are mutated in these disorders can also be mutated in acquired PFDs (e.g. pathogenic RUNX1 mutations are seen in both congenital and acquired PFDs). summarizes current information on the genes underlying characterized, inherited platelet disorders, including those that cause PFDs and/or ITs [Citation13]. The genes that cause ITs are summarized separately in [Citation6,Citation7,Citation14–70,Citation72]. summarizes PFDs associated with a normal platelet count [Citation43,Citation73–93,Citation96–98]. There are excellent recent reviews on the causes and features of inherited PFDs and ITs [Citation1,Citation99–102].
Figure 1. Causes of platelet function disorders and inherited thrombocytopenias. The figure summarizes the genes and pathways that contribute to the pathogenesis of platelet disorders, which includes regulatory genes that have impacts on the early or later stages of megakaryopoiesis, as summarized in Lentaigne et al. [Citation13].
![Figure 1. Causes of platelet function disorders and inherited thrombocytopenias. The figure summarizes the genes and pathways that contribute to the pathogenesis of platelet disorders, which includes regulatory genes that have impacts on the early or later stages of megakaryopoiesis, as summarized in Lentaigne et al. [Citation13].](/cms/asset/44eda53c-64e4-47b8-baeb-a607297076de/ilab_a_2049199_f0001_c.jpg)
Table 1. Overview of different inherited thrombocytopenias.
Table 2. Overview of inherited platelet function disorders associated with a normal platelet count. The conditions listed have a normal platelet size.
Some disorders, such as congenital megakaryocytic thrombocytopenia (CAMT) result from mutations in the genes encoding thrombopoietin (THPO) or the thrombopoietin receptor (MPL) [Citation1,Citation49,Citation50,Citation72,Citation99–102]. An important group of PFDs is caused by pathogenic mutations in genes that regulate transcript in early megakaryopoiesis, and thus alter platelet numbers and/or functions; these include mutations in genes that also cause hereditary predisposition to other blood disorders, including myelodysplastic syndrome and acute leukemia (e.g. mutations in ANKRD26, ETV6, RUNX1) [Citation1,Citation7,Citation20,Citation21,Citation60,Citation99–102].
Pathogenic mutations in genes affecting platelet granule biogenesis and trafficking cause the α-granule protein disorders gray platelet syndrome (GPS) (e.g. NBEAL2) and arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome (VPS33B, VIPAS39), and the syndromic δ-granule deficiencies of Hermansky-Pudlak syndrome (HPS) (HPS1, HPS3-6, BLOC1S3, DTNBP1, PLDN, AP3D1, and AP3B1) and Chediak-Higashi syndrome (CHS) (LYST), and several syndromic disorders that impair platelet secretion (STXBP2, NBEAL2) [Citation1,Citation52,Citation82,Citation85,Citation97–104].
Mutations in genes for a variety of platelet cytoskeletal proteins are known causes of syndromic and non-syndromic ITs, including MYH9-related disorders, Wiskott-Aldrich syndrome (WAS) and the related, X-linked thrombocytopenia (XLT), and many other ITs, some of which are benign (e.g. ACTN1-related disorders) [Citation1,Citation18,Citation19,Citation70,Citation71,Citation99–102]. One interesting cause of a rather benign IT results from a mutation in CYCS that increases apoptosis during platelet formation [Citation24,Citation105,Citation106].
Several severe PFDs result from severe deficiencies or dysfunction of GPIbIX (Bernard-Soulier syndrome; BSS), αIIbβ3 (Glanzmann thrombasthenia; GT), or molecules that impair αIIbβ3 function through other mechanisms (e.g. deficiencies of Kindlin 3, also known as FERMT3, and calcium and diacylglycerol-regulated guanine nucleotide exchange factor I [CalDAG-GEFI]) [Citation1,Citation37,Citation42,Citation78,Citation92,Citation99–102].
Several platelet disorders result from mutations of platelet receptors that increase ligand binding, which interestingly, manifest with bleeding and not thrombosis. For example, gain-of-function mutations of GPIbα that increase von Willebrand factor (VWF) binding cause platelet-type von Willebrand disease (VWD) [Citation38,Citation107]. Activating mutations of the αIIb or β3 chain of αIIbβ3 cause ITGA2B/ITGB3-related thrombocytopenia, also known as variant GT, a dominant PFD that impairs platelet cytoskeletal rearrangement, reduces αIIbβ3 expression on platelets, and impairs platelet aggregation [Citation108].
A number of recessive PFDs affect the platelet transmembrane receptors (genes) for platelet-activating agonists, including the receptors for ADP (P2RY12, P2RX1) or thromboxane (TBXA2R), or impair thromboxane generation (e.g. PTGS1) [Citation1,Citation91,Citation94,Citation99–102,Citation109]. Others are caused by mutations in genes encoding proteins involved in key signaling and activating pathways, including the collagen receptors GPVI that are important for platelet activation and α2β1 that mediates adhesion [Citation1,Citation81,Citation99–102,Citation110,Citation111]. York and Stormorken syndromes are complex, syndromic PFDs with thrombocytopenia, non-blood phenotypic features, and defects in calcium channels caused by different mutations in the gene STIM1 [Citation1,Citation64,Citation65,Citation99–102,Citation112].
Some PFDs cause unique impairments in hemostasis. Scott syndrome is a rare, recessive PFD caused by mutations in ANO6, which encodes the calcium-dependent phospholipid scramblase anoctamin 6 [Citation74]. Scott syndrome severely impairs the activation-induced expression of procoagulant phospholipids on the outer surface of platelets and other cells [Citation74]. Quebec platelet disorder (QPD) is a unique α-granule disorder caused by a duplication mutation of PLAU that repositions a downstream, megakaryocyte-specific gene enhancer element that “rewires” PLAU; the consequence is a unique, platelet-dependent, gain-of-function defect in fibrinolysis, due to >100 increased levels of urokinase plasminogen activator (uPA) in platelets and megakaryocytes [Citation113]. QPD results in intraplatelet plasmin generation, which proteolyzes diverse α-granule proteins, and accelerates clot lysis when QPD platelets release uPA [Citation114].
Prevalence of platelet disorders
The prevalence of platelet disorders has not been examined in population-based studies, however, several studies report that amongst patients tested for bleeding problems, the prevalence of some PFDs, including DGD, is similar to that of VWD [Citation115–125]. A worldwide survey on the investigation of PFDs indicated that laboratories commonly find aggregation abnormalities amongst referred patients, particularly with ADP and/or epinephrine [Citation126]. In areas of the world where consanguinity is culturally accepted, rare autosomal recessive PFDs have a much higher prevalence. A recent genome database study, that looked for predicted loss-of-function variants in the exome sequences of 58 platelet-associated genes with known phenotypic correlates, estimated the prevalence of PFD at 0.329% of the general population [Citation127]. Accordingly, PFDs are considered common and important causes of bleeding.
Approach to the diagnosis of platelet disorders: clinical evaluation of the bleeding history
The first step in the evaluation of patients with a suspected disorder of platelet function and/or numbers is a thorough evaluation of the personal and family history of bleeding. Positive family history is a predictor of finding a bleeding disorder in index patients [Citation128], particularly for dominantly inherited disorders. Most platelet disorders are associated with symptoms of mucocutaneous bleeding, such as extensive bruising, epistaxis, and heavy menstrual bleeding. They can also manifest with abnormal bleeding with hemostatic challenges, including childbirth, surgeries, and invasive dental procedures (e.g. tooth extractions). Challenge-related bleeding from PFDs and ITs typically occurs at the time of or soon after (i.e. on the same day) a hemostatic challenge, although in QPD, the onset is delayed by 1–3 days, reflecting increased fibrinolysis [Citation129,Citation130]. In contrast to severe PFDs, mild PFDs can manifest with excessive bleeding after some but not all hemostatic challenges [Citation129,Citation131,Citation132].
Bleeding problems of PFDs and ITs have also been evaluated using bleeding assessment tools (BATs), which have the advantages of standardizing data collection and symptom severity grading as well as identifying when a person’s bleeding exceeds that expected for the general population [Citation130,Citation133–138]. Recent studies indicate that patients with inherited PFDs have a significantly higher ISTH-BAT score than healthy controls and a higher score than for patients with IT, although there is some overlap between each of these groups [Citation131]. Within each of these groups, including healthy controls, females have higher bleeding scores than males, so sex-specific cutoffs are needed for bleeding score assessment [Citation131]. Analyses of sex differences in bleeding symptoms indicate that both healthy females and females with inherited PFD report more mucocutaneous bleeding than males [Citation129,Citation139]. BAT scores for inherited PFDs are also predictive of future hemorrhages requiring treatment [Citation131,Citation132].
BATs have also been used to evaluate the bleeding risks for specific types of PFDs, using data for the bleeding symptoms and problems reported by: (1) persons with QPD vs. unaffected family members or (2) persons with commonly encountered PFD vs. general population controls [Citation129,Citation130,Citation140]. illustrates that for commonly encountered PFDs, there are significantly increased risks for experiencing bleeding symptoms and problems [Citation129]. The magnitude of these risks reflects that some patients with these disorders experience excessive bleeding with some but not all hemostatic challenges [Citation129].
A bleeding history evaluation for a PFD should also be mindful of syndromic features that suggest a specific disorder (see reviews [Citation1,Citation99–102]). For example, HPS is associated with oculocutaneous albinism, lung fibrosis, and colitis; Jacobsen/Paris-Trousseau syndrome (PTS) is associated with facial dysmorphism, cardiac defects, and mental retardation; MYH9-related disorders are associated with inclusion bodies in neutrophils (Döhle bodies) with or without sensorineural hearing defects, elevation of liver enzymes, nephritis, and/or cataracts; thrombocytopenia with absent radius (TAR) syndrome is associated with absent radii; and WAS is associated with microplatelets, eczema, and susceptibility to infections.
Associations between test findings and bleeding symptoms
Some interesting associations have been found between the findings for some platelet function disorder tests and bleeding symptoms. For example, impaired platelet aggregation with collagen is associated with bruising and wound healing problems, and more severe DGD is associated with surgical bleeding [Citation129]. Among persons with PFD, bleeding scores are similar for those with or without DGD [Citation141]. The ability of platelets to support thrombin generation (TG) may also influence bleeding as associations have been found between some PRP thrombin generation endpoints and prolonged bleeding from minor cuts and wound healing problems [Citation142].
Approach to diagnosis: laboratory investigations
Recent guidelines on laboratory investigations for inherited PFDs have recommended a multistep diagnostic workup, after exclusion of VWD [Citation9,Citation10]. Many laboratories perform only a limited number of the recommended investigations, such as LTA, a test for DGD, and glycoprotein analysis when GT or BSS is suspected [Citation143]. When LTA is included in the panel of tests for bleeding disorder diagnosis, the test panel sensitivity for detection of a bleeding disorder is improved, without sacrificing specificity [Citation123]. Diagnostic strategies need to consider that some common PFDs, such as DGD, may go undetected unless specific tests for DGD are included in the investigations [Citation141]. They also need to consider that abnormal findings for some PFD diagnostic tests (e.g. abnormal LTA with multiple agonists, confirmed-granule deficiency) are highly predictive of a bleeding disorder (). summarizes key recommendations from the most recent ISTH guidance document on investigating suspected PFD [Citation9,Citation131].
Table 3. Likelihood of experiencing bleeding for commonly encountered platelet function disorders.
Table 4. Likelihood of a bleeding disorder based on findings for different platelet function disorder tests [Citation9,Citation24,Citation141,Citation144].
Platelet count and morphology
An essential component of platelet disorder investigations is the assessment of peripheral blood platelet count and platelet size, which help to narrow the differential diagnosis. Among hematology analyzers, those that use a fluorescence-based method for platelet counting give more precise estimates of the platelet count [Citation145,Citation146]. Low platelet counts should be confirmed on a light microscopy review of the blood film to assess for clumps and pseudo-thrombocytopenia, which is caused by EDTA-dependent platelet agglutination or satellitism (i.e. a rosette distribution of platelets around neutrophils, and less commonly, around other white blood cells) [Citation147–149]. Pseudothrombocytopenia is reported in up to 17% of patients referred to hematology clinics for thrombocytopenia [Citation149–154] and it can be readily evaluated by a review of the blood smear [Citation155].
When assessing for hereditary macrothrombocytopenia, platelet size assessment by optical methods appear superior to impedance methods, and a mean platelet volume (MPV) >12.4 fL helps differentiate patients with MYH9-related disorders and biallelic BSS from those with immune thrombocytopenia (ITP) [Citation156]. Measurement of the mean platelet diameter (MPD) by optical microscopy, or software-assisted image analysis, is also helpful to distinguish inherited PFD with giant platelets from ITP as well as identify PFD associated with abnormally small platelets [Citation157], although such assessments have not been widely adopted.
The evaluation of the blood smear is also helpful to identify morphologic anomalies that are suggestive of some specific diagnoses. For example, large platelets are associated with some disorders, such as platelet-type VWD and GATA1 mutations, giant platelets are suggestive of MYH9-related disorders, PTS and BSS, whereas microplatelets are suggestive of WAS, TAR, or CAMT [Citation9]. Giant α-granules are suggestive of PTS, whereas absent or rare azurophilic α-granules in platelets examined by light microscopy are suggestive of GPS, which can be secondary to mutations in NBEAL2, GFI1B, or GATA1 as well as the VPS33B or VIPAS39 mutations that are associated with ARC syndrome [Citation9,Citation27,Citation34,Citation52,Citation158,Citation159]. δ-granules are not visible on standard Wright-Giemsa or May-Grünwald-Giemsa staining and need to be evaluated by other approaches (discussed later).
Morphologic abnormalities of other peripheral blood cells can also provide helpful clues to a PFD diagnosis. For example, Döhle-like inclusion bodies in neutrophils are typical of MYH9-related disorders, and abnormal red cell morphology is a manifestation of the dyserythropoiesis seen in PFDs caused by GATA1 mutations [Citation27,Citation51,Citation158].
Immunofluorescent staining of blood smears for platelet disorder assessment
Immunofluorescence staining of blood films to evaluate for a platelet disorder has not been widely adopted. However, it can help to characterize up to 27% of suspected inherited PFDs when performed with a standardized protocol to stain fixed, permeabilized blood cells with antibodies specific to platelet proteins of interest, in combination with control sample assessment and application of an algorithm that integrates MPV and immunofluorescent data [Citation160,Citation161]. This method can be used to evaluate for a variety of PFDs, including GT, BSS, PFDs with cytoskeleton abnormalities (such as MYH9-related disorders and PFDs with β1-tubulin and filamin A abnormalities), DGD, GPS, and PFDs caused by GATA1 and GFI1B mutations [Citation160,Citation161]. It requires very little blood, which is advantageous for testing children and can be done on shipped dried blood films to provide diagnostic support to resource-poor and remote settings.
Screening tests for platelet disorders and other primary hemostatic defects
Bleeding time
The bleeding time (BT) is no longer recommended as a laboratory test as it has poor sensitivity and specificity, even when the test is standardized (e.g. with control of venous pressure, temperature, and standardization of the site and the tool used to make an incision of standardized depth and length) [Citation162,Citation163]. Accordingly, the BT is not recommended for investigating PFDs or other disorders [Citation9,Citation10].
PFA-100/200 closure times
Closure times (CTs) are considered an optional test for PFDs that requires a PFA-100 or PFA-200 analyzer and sodium citrate anticoagulant whole blood (WB) [Citation164]. Briefly, WB samples are aspirated at high shear into disposable cartridges and encounter a membrane with an aperture that is coated with collagen and epinephrine or with collagen and ADP [Citation164]. The agonists trigger platelet activation and accumulation that closes the aperture, leading to the cessation of blood flow [Citation164]. A prolonged CT can reflect a defect in VWF or in platelet numbers or function [Citation165–167] but the test is also prolonged if there is a low hematocrit [Citation167–169]. The PFA-200 has improved ease of use compared to the PFA-100 but uses the same principle [Citation164]. An additional cartridge that is sensitive to the detection of P2Y receptor deficiency or blockade, is available, but its clinical utility for PFD diagnosis is uncertain [Citation170–173]. While the CT is more sensitive to VWD and PFDs than the BT, it does not have sufficient sensitivity to be used for PFD screening [Citation119,Citation167,Citation174–178].
Light transmission aggregometry
LTA was developed in the 1960s by Born and O’Brien, and it remains the gold standard test for PFD assessment [Citation179,Citation180]. Briefly, LTA measures the platelet aggregation responses of PRP to added agonists by measuring changes in light transmission that occur as platelets aggregate, which increases light transmission and are quantified, as illustrated by the normal LTA study shown in . summarizes the maximal aggregation (MA) findings for patient LTA results shown in the review () and includes findings for additional agonists.
Figure 2. Light transmission platelet aggregation findings for a control subject. The aggregation tracing illustrates that there is significant aggregation with all agonists, except with the lower concentration of ristocetin, which is expected. The maximal aggregation with each agonist falls within the reference interval. See for the reference interval for each agonist.
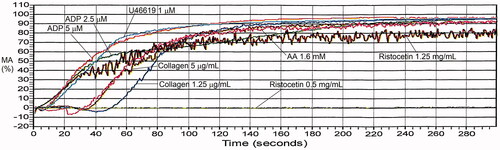
Figure 3. von Willebrand factor loss-of-function and gain-of-function defects, evaluated by light transmission platelet aggregometry. Panel (A) illustrates ristocetin-induced platelet aggregation findings for a person with type 2A von Willebrand disease, who had delayed aggregation with ristocetin due to their loss-of-function defect. The maximal aggregation attained was within the reference interval, despite the delayed response. Panel (B) illustrates the findings for a person with type 2B von Willebrand disease and significant thrombocytopenia, whose gain-of-function defect in von Willebrand factor resulted in increased aggregation with the lower concentration of ristocetin. The responses to other agonists were within the expected range for a platelet rich plasma sample with a very low platelet count. See for maximal aggregation data.
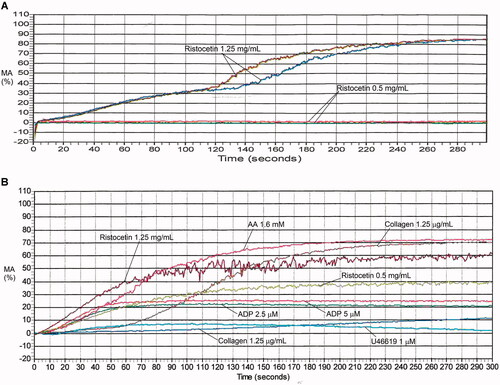
Figure 4. Aggregation findings for Bernard-Soulier syndrome. There is no agglutination or aggregation evident with ristocetin, whereas the responses to other agonists are within the expected range. See for maximal aggregation data.
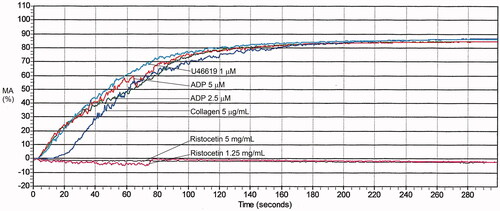
Figure 5. Aggregation findings for Quebec platelet disorder. There is reduced primary aggregation and no secondary aggregation with epinephrine, which is a hallmark feature of the disorder. This subject also had reduced maximal aggregation, and deaggregation, with thromboxane analogue U46619 as an incidental finding. See for maximal aggregation data.
Figure 6. Aggregation findings for Glanzmann thrombasthenia. A response is evident with ristocetin but there is no response, apart from shape change, with other agonists. With ristocetin, the maximal response achieved is reduced, reflecting that there is agglutination but no aggregation. See for maximal aggregation data.
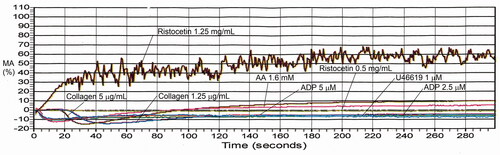
Figure 7. Example of aggregation findings for δ-granule deficiency. Findings are shown for representative subject with confirmed platelet δ-granule deficiency (average: 1.2 δ-granules/platelet; reference interval: 4.9–10.0 δ-granules/platelet). Aggregation findings in this disorder can range from non-diagnostic to showing multiple abnormalities, particularly with weak agonists. This person’s maximal aggregation responses were within the reference interval with all agonists except there was a reduced response to the lower concentration of collagen and reduced secondary aggregation with epinephrine. See for maximal aggregation values compared to the reference interval.
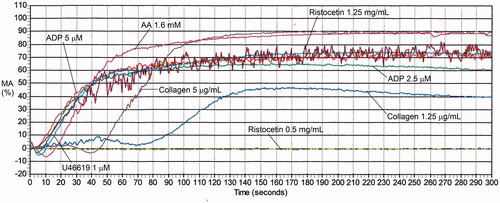
Figure 8. Examples of aggregation abnormalities with multiple agonists due to RUNX1 haploinsufficiency and unknown causes. Panel (A) shows the findings for a person with RUNX1 haploinsufficiency and mild thrombocytopenia, who had reduced aggregation with collagen and thromboxane analogue U46619, shared with other affected family members (not shown), who also had reduced maximal aggregation with arachidonic acid. Panel B shows the findings for an individual from a family with an autosomal dominant platelet disorder of unknown molecular cause, who had abnormal responses to multiple agonists that were particularly evident with arachidonic acid and thromboxane analogue U46619 as well as lower concentrations of collagen. See for the maximal aggregation data of these subjects.
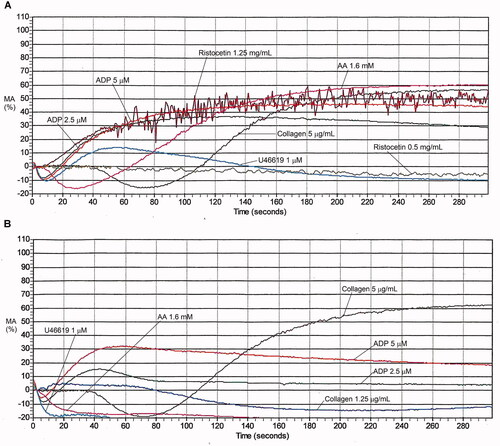
Figure 9. Aggregation abnormalities associated with aspirin or an aspirin-like defect. There is no aggregation with arachidonic acid whereas the aggregation response to thromboxane analogue U46619 is completely normal. Additionally, there is absent secondary aggregation with epinephrine (not shown) and reduced maximal aggregation with the lower concentration of collagen. See for maximal aggregation data.
Table 5. International Society on Thrombosis and Haemostasis recommended investigations for suspected inherited platelet function disorders.
Standardization of LTA has been a challenge as it requires control of many pre-analytical and analytical variables (). Several organizations have recommendations to standardize LTA, and the North American guideline includes recommendations on how to interpret the findings [Citation8,Citation10,Citation11,Citation181].
Table 6. Maximal aggregation findings for representative cases with platelet function disorders, evaluated by light transmission platelet aggregometry.
Table 7. International recommendations on preanalytical variables for platelet aggregation studies by light transmission aggregometry.
There has been limited application of EQA to LTA because the assay requires fresh PRP. Despite the recommendations to standardize agonist panels, no commercial supplier has developed a kit with all recommended agonists. The laboratories use different agonists, at different concentrations, which makes direct comparisons challenging [Citation182]. The THROMKID-Plus Study Group reported high variability in LTA findings between 15 centers in Germany and Austria (due to different protocols, reagents, agonist concentrations, cutoff values) until they standardized their agonists and procedures [Citation183]. The North American Specialized Coagulation Laboratory Association (NASCOLA) has collaborated with the External quality Control of diagnostic Assays and Tests (ECAT) Foundation to provide EQA assessments of the post-analytical interpretation of LTA findings, which has highlighted that there are opportunities to improve and standardize LTA interpretation practices [Citation12].
Method and preanalytical variables
LTA is performed using samples collected by venipuncture into collection tubes containing a calcium chelator, commonly 3.2% buffered sodium citrate, at a 9:1 ratio of blood to anticoagulant [Citation11]. Before drawing the sample, the collection of information on the patient’s medications (including non-prescription drugs), supplements, and natural products is helpful to limit preanalytical interferences [Citation11]. As aggregation responses are affected by exercise and smoking, a minimal period of abstinence and rest before sample collection is reasonable [Citation11,Citation184–187]. Hemolysis and lipemia, and potentially icterus, can interfere as LTA uses optical detection. To minimize ex vivo platelet activation, LTA samples should be hand transported and not sent by pneumatic tube transport [Citation188,Citation189]. As there are diurnal variations in platelet function (e.g. with epinephrine), and there are time-dependent changes in aggregation responses post-sample collection, many laboratories schedule LTA tests for the morning and complete the test within 4 h post-collection, as recommended [Citation11,Citation190]. Alternative anticoagulants have been explored to extend LTA sample viability but none permit long delays in shipment or testing [Citation191]. Blood and PRP collected for LTA and other platelet function studies should not be refrigerated because cold exposure alters platelet function, and PRP requires a brief preincubation at 37 °C immediately before testing [Citation192]. To limit pH changes that affect LTA responses, blood for aggregation tests is often collected into buffered anticoagulants, and the harvested PRP is kept in capped tubes to limit air exposure [Citation11]. PRP for LTA is prepared by low-speed centrifugation at 200 G for 10 min [Citation11]. Higher centrifugal forces reduce erythrocyte contamination but they also reduce platelet recovery, due to the loss of the larger platelets [Citation193,Citation194].
Some centers adjust the platelet count of the PRP to a standardized value with autologous platelet-poor plasma (PPP) before LTA, whereas the ISTH does not recommend an adjustment [Citation9,Citation11]. The adjustment predominantly affects weak agonist responses, with native non-adjusted PRP showing significantly higher maximal aggregation responses to epinephrine and ADP compared to samples diluted with PPP; these differences affect MA reference intervals (RIs) [Citation144,Citation195–197]. Although both native and platelet count-adjusted PRP detect abnormalities predictive of a bleeding disorder, a prospective, non-inferiority study found that platelet count-adjusted PRP was actually superior to native PRP for detecting impaired aggregation responses due to a bleeding disorder [Citation144].
Many laboratories are uncertain about how to evaluate LTA in patients with thrombocytopenia. Some have used diluted control PRP samples to establish RIs for low platelet count PRP, using regression to define the 95% confidence intervals for MA responses for a range of sample platelet counts [Citation198]. Laboratories should not refuse to test LTA when patients are thrombocytopenic as LTA is very helpful to diagnose several thrombocytopenic disorders, including BSS, type 2B VWD, platelet-type VWD, and ITGA2B/ITGB3-related thrombocytopenia [Citation11,Citation198].
RIs for LTA MA responses to agonists require data for a minimum of 40 unique general population control samples, and non-parametric statistical analysis is recommended as most agonist responses do not show a Gaussian distribution [Citation198]. Changes in procedures (e.g. new agonist lots) should be validated with a smaller number of samples [Citation11].
LTA is measured by monitoring turbidity changes in stirred PRP samples, tested at 37 °C, with the PRP and PPP used to set the limits for 0 and 100% MA, respectively. The LTA agonist panel should include weak and strong agonists, with consideration of guideline recommendations on the agonists and agonist concentrations to test () [Citation11]. To ensure that the testing can evaluate for common PFDs, and important rare defects in specific receptors and pathways, the testing needs to include: ADP to test for P2Y12 and P2Y1 defects; collagen to test for a variety of defects, including those specific to GPVI and α2β1, impaired MA due to COX-1 inhibitor drugs, and other PFDs (which can require a low concentration of collagen for optimal detection of many functional impairments); arachidonic acid (AA) to test for defects in thromboxane generation and impaired aggregation from common PFDs; thromboxane analog U46619 to help detect common PFDs and distinguish these conditions from aspirin-like defects; epinephrine, as it helps detect abnormalities from common PFDs; and ristocetin, which is tested at high and low concentrations to respectively assess for loss and gain of functions defects that affect VWF binding to platelets. After adding agonists, LTA is monitored for 5 min with most agonists and for 10 min with epinephrine to ensure that MA is reached and that deaggregation can be evaluated.
Table 8. International recommendations on analytical variables for platelet aggregation studies by light transmission aggregometry.
Interpretation
Adequate interpretation of LTAs requires careful assessment of multiple quantitative and qualitative parameters:
Shape change, which may not be evident with all agonists. With collagen, the “shape change” reflects the adhesion of platelets to collagen fibrils that reduces the light transmission before aggregation starts. P2Y1 defects are associated with a loss of ADP-induced shape change.
Latency of the reaction, which is usually longer with epinephrine.
The slope of the aggregation curve, which is often reduced when MA is reduced.
The presence of a secondary aggregation wave, which is particularly distinct with epinephrine, unless there is absent secondary aggregation. The primary and secondary aggregation responses are often fused with higher concentrations of ADP and with strong agonists.
The absence or presence of deaggregation, which may accompany reduced MA, particularly with some agonists (e.g. thromboxane analog U46619). Deaggregation with ADP is particularly striking with inherited P2Y12 defects, which are rare.
With ristocetin, the initial response reflects agglutination, which triggers platelet activation and aggregation. The agglutination and aggregation components may be fused, particularly when the MA response to ristocetin is normal. illustrates typical findings for loss of function and gain of function defects in VWF. In the loss of function defects in VWF (e.g. type 2A, type 2M, or type 1 VWD with very low VWF levels), the agglutination is often reduced and slow (as illustrated in ), which delays and often reduces the aggregation that follows agglutination. With a gain of function defects, due to type 2B VWD (), and platelet-type VWD, there is an increased aggregation response to lower concentrations of ristocetin, even if there is significant thrombocytopenia as was the case in this example (note: the responses to other agonists in are within the expected range for low platelet count samples). In BSS, platelet counts are low, the blood film shows megathrombocytes, and RIPA is absent ().
The North American guideline on LTA provides helpful guidance on how to interpret LTA findings [Citation8]. Test records should be examined for abnormalities in the blood count and platelet morphology, to verify that the PRP contained more platelet than the peripheral blood and had minimal red cell and leukocyte contamination, and to ensure that the testing was completed within the appropriate time limits (i.e. within 4 h of sample collection). Simultaneously tested control samples are important quality controls. MA responses are very important to the overall interpretation, and it is good practice to repeat the tests for any agonists that showed abnormal findings to confirm the observation. Next, tracings should be evaluated for abnormalities (e.g. very delayed aggregation, deaggregation, comparison of the response to different concentrations of the agonist that is abnormal), which are typically only present if MA is impaired. Finally, the pattern of abnormal responses should be carefully examined to determine which agonists are affected and if the pattern suggests specific types of defects, as outlined in our customized interpretation guide (), which considers recommendations in the North American guidelines [Citation8].
Table 9. Summary of recommended agonists for platelet aggregation studies by different international guidelines.
Table 10. Approach to interpreting platelet aggregation studies by light transmission aggregometry. The approach has been updated from published guidance [Citation8].
illustrate the typical abnormal LTA findings from different PFDs. Isolated, reduced aggregation with thromboxane analog U46619 is a fairly common, non-diagnostic finding, but it can also be seen in persons with PFD, as illustrated in , which shows both reduced MA and deaggregation.
Isolated, absent secondary aggregation with epinephrine is also a fairly common, non-diagnostic finding for platelet count adjusted PRP but it is an abnormal finding for native PRP [Citation144,Citation197,Citation199]. In QPD, epinephrine aggregation responses are reduced with absent secondary aggregation and often reduced primary aggregation, as shown in . QPD aggregation abnormalities with epinephrine are evident with both platelet count adjusted and native PRP samples, but the epinephrine aggregation response can be artifactually normalized if the reagent to simultaneously measure ATP release is added [Citation200]. Some but not all QPD aggregation tests show additional abnormalities, such as reduced aggregation with thromboxane (as in the example shown in ), ADP, and/or collagen [Citation201].
Conditions that dramatically impair MA responses to multiple agonists include acquired and inherited GT (), and ITGA2B/ITGB3-related thrombocytopenia. In GT and ITGA2B/ITGB3-related thrombocytopenia, shape change is normal, but aggregation is either absent or markedly reduced due to severe deficiency/dysfunction of the platelet fibrinogen receptor αIIbβ3. The response to ristocetin in GT and ITGA2B/ITGB3-related thrombocytopenia is quantitatively reduced, as agglutination occurs but it is not followed by aggregation ().
With DGD, platelet aggregation responses can be normal, non-diagnostic (e.g. absent secondary aggregation with epinephrine), or show abnormalities with multiple agonists (e.g. reduced MA with collagen, AA, and thromboxane analog U46619) [Citation141]. The aggregation findings for an illustrative DGD case are shown in . MA with ADP is normal in DGD, as the exogenous ADP overcomes the loss of platelet δ-granule ADP [Citation141].
Some PFDs, including conditions associated with pathogenic RUNX1 mutations (), impair responses to multiple agonists but there is more aggregation in response to some agonists than seen with GT () [Citation129,Citation140]. Some inherited PFDs of unknown cause have aggregation abnormalities that resemble those seen with pathogenic RUNX1 mutations ( vs. ) [Citation129].
A comparison of the findings with AA and thromboxane analog U46619 is important as it often helps to distinguish common PFDs with multiple abnormal agonist responses (e.g. from DGD, pathogenic RUNX1 mutations, and PFD due to other causes) from PFDs caused by aspirin or aspirin-like defects (). With aspirin or aspirin-like defect (), there is impaired aggregation with AA but normal aggregation with thromboxane analog U46619, which are typically accompanied by reduced aggregation with collagen (particularly with lower collagen concentrations), absent secondary aggregation with epinephrine, and sometimes, reduced aggregation with ristocetin.
Impedance aggregometry
Aggregation responses can also be evaluated by measuring changes to electrical impedance, either with PRP or WB. Impedance aggregometry tracings for PRP and WB look similar to PRP LTA tracings, although shape change is not detected by impedance changes. In impedance aggregometry, platelets in the sample form a coating on two submerged electrodes, with additional platelets attaching to the electrode after adding an agonist, to proportionately increase the electrical impedance. With WB aggregometry (WBA) with collagen, leukocytes also attach to the electrode, which contributes to the changes in electrical impedance [Citation202,Citation203]. Recent guidelines on testing for PFDs recommend assessing aggregation responses by LTA rather than WBA, as there is more extensive literature on LTA as a diagnostic test for a bleeding disorder [Citation9].
WBA can be measured using several devices, including a lumiaggregometer (CHRONO-LOG Corporation, Havertown, PA, USA) and a Multiplate analyzer (Roche Diagnostics, Mannheim, Germany). While WBA with the Multiplate analyzer detects severe aggregation defects, such as GT, it is inferior to LTA for detection of a PFD [Citation178,Citation204–208].
Some point-of-care devices test WBA by modified methods that are not validated for the investigation of an underlying PFD. The VerifyNow device (Werfen, Barcelona, Spain) tests aggregation with capture beads to assess for impaired platelet function due to drugs, such as P2Y12 inhibitors. The PlateletWorks test (Helena Laboratories, Beaumont, TX, USA) assesses the percentage of “functional platelets” by comparing differences in platelet counts for blood collected into anticoagulant, with or without an agonist to induce aggregation; it has been used, in combination with viscoelastic testing, to guide transfusion therapy after cardiac surgery [Citation209].
When performing WBA for PFD assessment, a smaller volume of sodium citrate anticoagulant blood is needed compared to LTA (typically 6–9 vs. 20–30 ml) and the agonist concentrations tested differ from LTA [Citation210]. WBA should be completed within 3 h from collection [Citation211]. WBA is not affected by icterus or lipemia, but hemolyzed specimens should not be tested as hemolyzed samples often show platelet activation [Citation210]. In WBA, the contribution of platelets of all sizes is evaluated because macrothrombocytes are not lost as there is no sample centrifugation [Citation181,Citation212]. Like LTA samples, WBA samples should be maintained at room temperature and not refrigerated. WB samples are diluted 1:1 with saline solution before WBA, unless the platelet count is <100 × 109/L [Citation181,Citation211]. However, procedures have been established and validated for the assessment of thrombocytopenic patients, using impedance aggregometry, that may encourage more laboratories to perform aggregation for thrombocytopenic patients [Citation213,Citation214].
Lumiaggregometry
Lumiaggregometry (Chrono-log, Havertown, PA USA) is a bioluminescent method that assesses platelet δ-granule ATP release, typically at the same time as performing LTA or WBA [Citation210,Citation215–217]. It is commonly done to assess for platelet secretion defects [Citation126]. The test uses a reagent and the luciferin-luciferase reaction principle to quantify secretion [Citation218]: briefly, when platelets are activated, δ-granules release their stored ATP and ADP. The released ATP combines with D-luciferin, which in the presence of luciferase, produces luciferyl adenylate that reacts with oxygen to produce light. The light emission can be quantified, relative to an ATP standard, to determine the amount of ATP secreted by the platelets. shows normal lumiaggregometry findings whereas shows abnormal findings. With strong agonists (e.g. collagen, ), ATP release begins at the same time as aggregation whereas, with weak agonists (e.g. epinephrine, ), the release is delayed until secondary aggregation commences. In , the findings with collagen are normal, whereas with AA, aggregation and ATP release are reduced, and with thromboxane analog U46619, aggregation and ATP release are completely absent.
Figure 10. Examples of normal and abnormal lumiaggregometry findings. Each tracing show simultaneous evaluation of light transmission platelet aggregation (upper part of each tracing) and ATP release (lower part of each tracing), evaluated by lumiaggregometry. In panel (A), the ATP release with strong agonists (e.g. collagen), begins at the same time as aggregation. In panel (B), the ATP release with epinephrine, which is a weak agonist, is delayed until the start of secondary aggregation. In panel (C), there are several abnormal findings: aggregation and ATP release are absent with thromboxane analogue and aggregation and ATP release are reduced with arachidonic acid. Panels (A,C) also show the normal, rapid ATP release with thrombin (aggregation was not monitored as thrombin clots the sample).
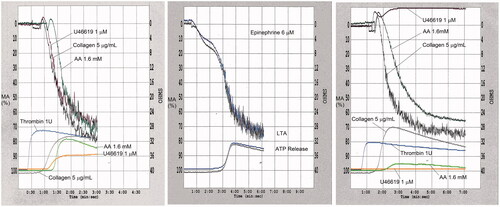
The measurement of ATP release cannot discern between abnormal test findings due to DGD or defective δ-granule content release. The clinical utility of testing ATP release by lumiaggregometry for diagnosing PFDs has been questioned as the test endpoints for individual agonists show significant intra- and inter-individual variability that negatively affect the test reproducibility and diagnostic usefulness [Citation217,Citation219,Citation220].
Transmission electron microscopy
TEM has been used to evaluate platelets, with a resolution of up to 0.2 nm [Citation221]. TEM can be done with thin sections of fixed platelets (with or without immunogold labeling) and with WM preparations of dried, unfixed platelets to visualize δ-granules (e.g. ).
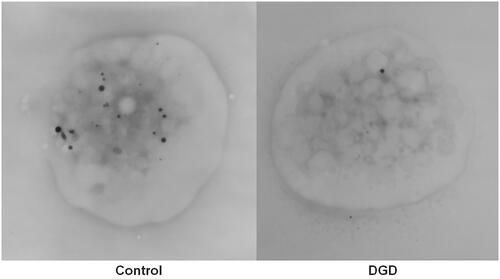
Figure 11. Transmission electron microscopy images of whole platelets in whole mount preparations of control and δ-granule deficient (DGD) samples. The image from the control platelet (left) shows numerous electron dense structures and the majority of these structures have smooth contours and a uniform electron density, which is typical of δ-granules. The platelet from the δ-granule deficient subject contains only one electron dense granule, consistent with their low δ-granule count and confirmed δ-granule deficiency.
TEM evaluation of platelet thin sections permits visualization of many platelet structures, including internal and external membranes, the surface-connected canalicular system, α-granules, mitochondria, the cytoskeleton, and glycogen stores. Recently, a standardized approach to platelet TEM was proposed to improve evaluations of platelet ultrastructure [Citation222]. Platelet TEM can be helpful to confirm some diagnoses, such as the α-granule deficiency of GPS when light microscopy suggests this diagnosis.
Platelet WM TEM is one of the accepted tests for quantifying platelet δ-granule numbers [Citation9,Citation141]. WM TEM is included in the initial workup for primary hemostatic defects as DGD has a prevalence similar to VWD, and LTA and lumiaggregometry are not sensitive enough to be used as a screening test for DGD [Citation124,Citation141,Citation220,Citation223–225]. Associations between DGD (diagnosed by WM TEM), bleeding, and elevated ISTH-BAT bleeding scores indicate that DGD diagnosed by WM TEM is associated with a bleeding disorder [Citation141].
WM TEM is a relatively simple method, described by James White, to assess for DGD [Citation226]. Briefly, blood is collected into acid-citrate-dextrose (ACD) or sodium citrate (as for LTA), spun to harvest PRP, and then kept at room temperature until a drop of the PRP is placed on a Formvar-coated grid, followed by a brief rinse with distilled water, cautious blotting with a paper filter, and air-drying of the sample before TEM [Citation141,Citation226–228]. As there is a significant reduction in the platelet δ-granule count within a day of sample collection, WM TEM should be done on the same day as the sample is drawn [Citation229]. Our preference is to prepare the grids within a few hours after sample collection. Because platelet δ-granules are rich in calcium and phosphorus, they appear dark (electron-dense) without any fixation or staining (). The average platelet δ-granule count is then determined by counting DG in 30–50 platelets (our RI is 4.8–10.0 δ-granules/platelet), with comparison to a control sample [Citation141]. compares δ-granules in a representative platelet from a control and DGD patient. Some suggest counting δ-granules in more than 80 platelets to reduce the coefficient of variation (CV), but in our experience, results are similar whether δ-granules in 30 or 100 platelets are evaluated [Citation141,Citation222].
The NASCOLA EQA program for assessment of δ-granules by WM TEM showed good agreement (>80%) between laboratories in distinguishing between normal and DGD samples as well as good consensus on the electron-dense structures within platelets that should be counted or not counted as δ-granules [Citation12,Citation230]. Typically, only the uniformly electron-dense structures, with smooth boundaries (including tadpole and purse-shaped granules) are counted, whereas less dense, irregular structures are not counted (both types of structures are evident in the control platelet shown in ). Some centers have published their methods to standardize what is counted as a δ-granule, but it is generally recommended that each laboratory established its own RI for platelet δ-granule counts, with a minimal number of dedicated interpreters to reduce inter-individual variability, as average δ-granule counts can vary significantly between centers [Citation124,Citation141,Citation222,Citation231,Citation232]. When the test was set up at our center, energy dispersive spectroscopy analysis of the electron-dense structures was performed to verify which structures had calcium and phosphorus content typical of δ-granules, but few laboratories have this capability. Some have proposed that assessments for DGD include a formal evaluation of the δ-granule diameter as some patients with a bleeding diathesis have normal numbers but smaller-sized δ-granules [Citation225]. Finally, reduced δ-granule counts should be verified on a second sample because mild reductions (4.0–4.8 DG/platelet compared to the RI of 4.8–6.0) are often false positive [Citation141].
Thin section (TS) TEM requires more complex sample preparation than WM TEM. It is important to limit platelet activation before fixation. Typically, blood for TS TEM is collected into ACD anticoagulant, followed by preparation of PRP, then fixation of the platelets with buffered glutaraldehyde for a fixed time, followed by serial dehydration with alcohol before embedding the sample in a resin for preparation of ultrathin sections with an ultramicrotome, as outlined in protocols [Citation233–236]. Samples should not be collected into EDTA as platelet structure is altered [Citation237]. TS TEM can be helpful to diagnose rare disorders (e.g. platelet spherocytosis, GPS, CHS, and PTS) [Citation28,Citation238–241]. As TS TEM examines thin cross-sections of platelets, it has been suggested that to diagnose α-granule deficiency, these granules need to be deficient in more than 15% of 100 platelet cross-sections [Citation222].
Other measurements of δ-granule content
Serotonin release
Virtually all circulating serotonin, also known as 5-HT, is contained within platelet δ-granules as platelets take up serotonin for storage in these granules via the serotonin transporter (SERT), and the vesicular monoamine transporter 2 (VMAT2); this makes serotonin useful for the assessment of platelet δ-granule serotonin uptake, content, and release [Citation242,Citation243]. When released, platelet serotonin is thought to promote platelet aggregation via binding to platelet 5-HT2a receptors [Citation244]. Serotonin release assays (SRAs) with radiolabeled 14C-5-HT have been the gold standard assay for evaluating δ-granule secretion for the diagnosis of heparin-induced thrombocytopenia (HIT) but are rarely used for inherited PFD assessment because the test requires a radioisotope [Citation126,Citation215,Citation245,Citation246]. SRA is performed by incubating PRP with the radioactive serotonin at 37 °C for 15 to 30 min to allow uptake, before adding an imipramine solution to stop the serotonin uptake. Next, platelets are incubated with agonists (with or without monitoring the response by LTA), before adding a formalin-EDTA solution to stop the further release. Then, total and released (sample supernatant) serotonin are quantified, using a β scintillation counter, to determine the serotonin uptake and release [Citation247,Citation248]. Application of the test to PFD diagnosis requires the determination of an RI for control samples [Citation248]. Serotonin uptake can also be assessed to help differentiate DGD from secretion defects.
Other techniques have been developed to evaluate platelet δ-granule contents and their release without radioisotopes, using either fluorimetry (with o-phtalaldehyde, a reagent that reacts with serotonin), flow cytometry with mepacrine uptake, serotonin antibodies to visualize δ-granules (discussed further in the flow cytometry section), high-performance liquid chromatography (HPLC) or enzyme-linked immunosorbent assay (ELISA) of DG contents [Citation249–256]. HPLC endpoints for serotonin can be measured either by electrochemical or fluorescent means [Citation254,Citation255]. The methods have been adapted to evaluate single platelets and to interpret dynamic results [Citation253,Citation257]. These methods offer many advantages including high sensitivity, good precision with a CV of <10%, a relatively low cost, but they require significant technical expertise [Citation252,Citation258]. For the evaluation of HIT, there is good concordance (92%) of the test findings to that of radioactive SRA, with samples showing lower serotonin release (e.g. estimated release of 20–49%) accounting for most of the discrepancies [Citation259]. A recent modification that used liquid chromatography in tandem with mass spectrometry to measure serotonin release showed excellent performance [Citation260]. However, few laboratories use such methods [Citation126].
While antibodies are available to quantify serotonin by ELISA [Citation261], there is limited information on their use for evaluating PFDs, and for the diagnosis of HIT, measurement of serotonin release by ELISA is inferior to HPLC [Citation262].
Platelet ATP-ADP measurement
Bioluminescent assays can also be used to measure the ATP-ADP content of platelet lysate and releasate [Citation263,Citation264]. With this approach, the enzyme adenyl kinase in samples is inhibited by an ethanol-EDTA solution to prevent the conversion of ADP to ATP, and the content of ATP in samples is determined by the luciferin-luciferase reaction. Next, the sample ADP is converted in ATP by the phosphoenolpyruvate-pyruvate kinase enzymatic reaction so that it can be measured by the luciferin-luciferase reaction. The difference between the results is used to calculate the platelet ADP content, and the ratio of ATP:ADP as a ratio of more than 2 in platelets is suggestive of DGD [Citation265]. The same testing can also be done with the supernatant of activated platelets, to differentiate between DGD and a secretion defect [Citation266,Citation267]. HPLC has also been used to quantify ADP and ATP content of δ-granules with good test performance characteristics [Citation268–270].
Measurement of α-granule content
The contents of α-granules include both granule membrane proteins and stored, soluble proteins. While many α-granule membrane proteins are shared with the outer platelet membrane, P-selectin is a marker of platelet activation that is sequestered in both platelet α- and δ-granules until granule release occurs [Citation271]. The soluble contents of α-granules include procoagulant and anticoagulant substances (fibrinogen, VWF, multimerin 1, coagulation factor V, PAI-1, α2-antiplasmin, uPA), growth and angiogenic factors (TGF-β, PDGF, VEGF), and chemokines, including PF4 [Citation271]. Some soluble α-granule constituents are present at undetectable or very low levels in the plasma (e.g. PF4, β-TG, multimerin 1) and have been tested as markers of α-granule release, mainly in research settings [Citation126,Citation272–276]. P-selectin expression on activated platelets is often evaluated by flow cytometry [Citation126].
Assays to assess platelet procoagulant function, including thrombin generation assays
Platelet activation leads to redistribution of negatively charged procoagulant phospholipids, with the movement of phosphatidyl serine (PS) and phosphatidyl ethanolamine (PE) from the inner to the outer platelet membrane; the release of polyphosphate from δ-granules; and the release of partially activated factor V from α-granules which enhances coagulation. Flow cytometry analysis of activated platelets suggests that there are subpopulations of platelets that express procoagulant PS and PE [Citation277]. Platelet activation through glycoprotein VI or platelet thrombin receptors leads to increases in intracellular calcium and PS and PE externalization, which facilitate tenase and prothrombinase complex formation and subsequent TG [Citation278]. TG is defective in Scott syndrome, a rare bleeding disorder caused by mutations in ANO6 (also named TMEM16F), which encodes the scramblase responsible for calcium-mediated PS and PE exposure; Scott syndrome results in significantly impaired TG on platelets and other cells [Citation75].
Increases in circulating procoagulant platelets are postulated to cause a pathogenic hypercoagulable state that complicates some malignancies and ischemic strokes [Citation279–284]. However, few laboratories offer tests to assess this.
DDAVP treatment for inherited PFDs increases platelet procoagulant activity and TG, without affecting P-selectin expression, δ-granule secretion, or platelet aggregation responses [Citation285].
Newer methods of assessing TG, including the assessment by calibrated automated thrombogram (CAT), with PRP, have been used to evaluate platelet procoagulant function in PFDs and other bleeding disorders, including severe hemophilia [Citation142,Citation286–294]. CAT is done using PRP (adjusted to 150 × 109 platelets/L), a fluorogenic substrate of thrombin (Z-GGR-AMC), with added calcium and recombinant tissue factor. This mixture is incubated at 37 °C in a fluorometer [Citation295], with a calibrator, to quantify TG, with measurement of the endogenous thrombin potential, peak thrombin generation, time to peak thrombin generation, and lag time [Citation295,Citation296]. In severe hemophilia and other severe factor deficiencies, PRP TG is more impaired in those that have more severe bleeding, suggesting platelet-dependent TG influences the phenotype of bleeding disorders [Citation286,Citation287,Citation291]. Not surprisingly, the most profound abnormalities in platelet-dependent TG have been observed in patients with Scott Syndrome and in mice lacking ANO6 [Citation73,Citation297].
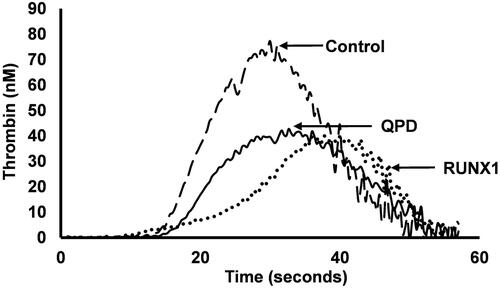
CAT has recently been used to study TG in commonly encountered PFDs that impair the ability of platelets to support coagulation, as illustrated by that compares thrombograms for a control subject to a δ-granule deficient subject to subjects with a pathogenic RUNX1 mutation and QPD [Citation142,Citation292]. However, the overlap between PRP TG findings for controls and PFD subjects limits potential diagnostic applications of CAT for PFD assessment [Citation142]. The defects in platelet-dependent thrombin generation in persons with pathogenic RUNX1 mutations likely reflect impaired platelet activation and the defects in DGD likely reflect impairments from the combined deficiencies of δ-granule ADP, which impairs platelets activation, and polyphosphate that enhances TG [Citation142]. PRP, but not PPP, TG is impaired in QPD (), proportionate to the loss of platelet factor V that results from intraplatelet plasmin generation, triggered by megakaryocyte-specific overexpression of PLAU, the urokinase plasminogen activator gene [Citation292]. Presently, CAT and other TG assays are mainly used to assess PFDs for research purposes [Citation298].
Figure 12. Thrombograms compare platelet rich plasma thrombin generation profiles for representative control and platelet function disorder subjects. The control sample generated more thrombin than the samples from the subjects with confirmed Quebec platelet disorder (QPD) or a familial platelet function disorder due to RUNX1 haploinsufficiency (RUNX1). The defects in these platelet function disorders include impairments in the ability of platelets to support thrombin generation.
Flow cytometry
While LTA is considered the gold standard in the investigation of PFDs, flow cytometry analysis can also provide important information. First, flow cytometry can be used to investigate: platelet glycoprotein deficiencies; abnormal platelet aggregation or secretion; and defects in platelet procoagulant function (e.g. by assessment of annexin V binding) [Citation250,Citation299–306]. Second, flow cytometry can be used to assess WB, even if there is significant thrombocytopenia or limited sample volume, which can be helpful for the investigation of PFDs in children [Citation307–310]. Nonetheless, flow cytometry tests for PFDs are expensive to run, and they need to be standardized and validated for PFD evaluation, as discussed in the recent guidelines from the ISTH [Citation306,Citation311–313]. Although flow cytometry is recommended by the ISTH for the investigation of inherited PFDs, only a minority of clinical laboratories use it for initial tests, as more use it for second step investigations [Citation9,Citation126]. Flow cytometry protocols have been developed that show a good correlation with LTA and detect abnormalities associated with bleeding [Citation306,Citation314–318].
Mass cytometry has potential advantages over flow cytometry analyses of platelets as it allows for multiplex assessment of single cells, with simultaneous evaluation of many different components (e.g. surface and intracellular proteins, markers of platelet activation) [Citation319], as well as the option of using barcodes to analyze results for individual cells or multiple samples simultaneously [Citation320,Citation321].
Protocols for performing flow cytometry analyses of platelets include a recent ISTH document [Citation311,Citation322–324]. Briefly, blood is collected in anticoagulant, similar to LTA, after excluding recent ingestion of potentially interfering medications that could affect platelet activation [Citation311,Citation325–327]. Sample centrifugation, and red cell lysis, should be avoided [Citation327]. The sample is then diluted in buffered saline, at a physiological pH, to limit platelet activation, and incubated with appropriate fluorochrome-conjugated monoclonal antibodies [Citation326]. Fixatives (e.g. paraformaldehyde or a commercially available preparation like PAMfix) can be used provided they do not interfere with recognizing the markers of interest [Citation317,Citation324,Citation327–330]. Ideally, the test should be completed within 4 h from sample collection, although some glycoprotein and functional analyses give acceptable results up to 24 h post-collection, with fixation permitting even longer delays or transport of the sample to a central laboratory [Citation311,Citation317,Citation331–333]. Samples for platelet flow cytometry are often kept at room temperature to limit activation, but there may be advantages to testing the samples at 37 °C, as this reduces the variability in findings [Citation333]. Local protocols require validation to determine optimal sample preparation (including fixation), appropriate conjugated antibody concentrations for sample incubations, and appropriate agonist concentrations if testing platelet activation [Citation312]. Control samples should be tested, and ideally, an RI should be established for the procedure to interpret the findings [Citation312].
Surface glycoprotein analysis
Flow cytometry can be helpful to quantify platelet surface glycoproteins with much smaller volumes of blood than required for LTA. It has been used successfully to diagnose the glycoprotein deficiencies of GT (congenital but also acquired forms), BSS, and collagen receptor deficiencies [Citation299–302,Citation334,Citation335]. The last ISTH guidelines recommend assessing αIIbβ3 (CD41/CD61) and GPIb/IX (CD42a/CD42b) in the initial investigations, and then α2β1 (CD49/CD29), GPIV (CD36), and GPVI as a second step [Citation9], although many laboratories only perform flow cytometry to confirm the certain diagnosis (e.g. GT and BSS, as shown in ) [Citation126]. Consensus ISTH recommendations on flow cytometry assessments of inherited and acquired PFDs and thrombocytopenias were recently published [Citation311] (key recommendations are summarized in ).
Figure 13. Examples of flow cytometry analysis to confirm the glycoprotein deficiencies for cases with Glanzmann thrombasthenia (GT) and Bernard-Soulier syndrome (BSS). The panels compare the relative mean fluorescent intensity (MFI) from flow cytometry analysis of control compared to patient samples, evaluated with fluorescent antibodies to the platelet glycoprotein (GP) receptor αIIbβ3 that is deficient in GT (upper panel) and to the GPIbα and GPIX components of the platelet GPIb/IX/V complex that is deficient in BSS (lower panel).
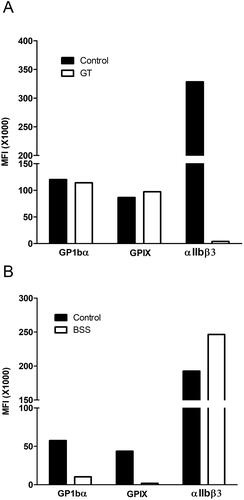
Table 11. Summary of key consensus recommendations from the International Society on Thrombosis and Haemostasis on using flow cytometry for the assessment of disorders of platelet number and function [Citation311].
Platelet function testing after agonist addition
Platelet activation is evaluated with antibodies that detect conformational changes in platelet membrane glycoproteins, such as αIIbβ3, or the secretion of P-selectin or CD63 onto the outer platelet membrane [Citation330,Citation336,Citation337]. The testing can be done with agonists that target different pathways, such as ADP, thrombin receptor-activating peptide (TRAP)-6, cross-linked collagen-related peptide (CRP-XL), AA, thromboxane analog U46619, and ristocetin [Citation306,Citation313,Citation315,Citation326,Citation338,Citation339].
Markers of platelet activation are typically used to assess disorders that impair platelet activation and signaling, which is outside the realm of many diagnostic laboratories [Citation304,Citation340–342]. P-selectin levels on the platelet surface should be low before platelet activation and a basal level of 15% is considered acceptable for proceeding with testing platelets for activation-induced changes [Citation326,Citation343,Citation344]. P-selectin analyses have also been used to characterize platelets from patients with α-granule deficiency, although normal levels of P-selectin have been reported in some cases of GPS [Citation345–348]. P-selectin also mediates the interaction of activated platelets with neutrophils and monocytes [Citation349,Citation350], which can be evaluated in WB samples, analyzing for events that show dual-labeling for platelet (usually with CD41, CD61 or CD42a or CD42b antibodies) and leukocyte markers [Citation323,Citation341] that are not normally evaluated when performing flow cytometry for PFDs [Citation351–355].
Granulophysin is a membrane protein of the tetraspanin family that is normally sequestered in lysosomes and DG and becomes expressed on the outer surface of platelets after activation [Citation356,Citation357]. Granulophysin is reduced in platelets from persons with DGD [Citation356,Citation358,Citation359], which may affect platelet activation as it is postulated to modulate integrin-mediated signaling and other activation-induced changes [Citation360,Citation361]. Platelet granulophysin has been tested to evaluate platelet secretion and DGD presence [Citation338,Citation347].
Other probes of platelet activation have also been used for flow cytometry. PAC-1 is a monoclonal antibody that recognizes an epitope on αIIbβ3 associated with activation-induced conformational changes [Citation330,Citation362,Citation363]. Activation-induced changes in platelet αIIbβ3 can also be evaluated using fibrinogen antibodies [Citation246,Citation363–365], fluorescent fibrinogen, ligand-induced binding site (LIBS) antibodies that assess αIIbβ3 conformational change, and antibodies that detect conformational changes in platelet collagen receptors [Citation366–369]. Protocols for testing these markers have been described [Citation306,Citation313,Citation315,Citation316,Citation318,Citation338,Citation347,Citation370,Citation371].
Mepacrine uptake and release
Mepacrine is a fluorescent, antimalarial drug that mimics serotonin and accumulates in platelet DG that has been used as a reagent to assess platelet DG and platelet DG secretion defects [Citation372,Citation373]. Briefly, platelets are incubated at 37 °C with mepacrine (e.g. for 30 min), then its uptake (by test compared to control platelets) is measured by flow cytometry [Citation250,Citation374] and it can be reassessed after stimulation, to quantify DG secretion [Citation250,Citation375]. Mepacrine testing needs to be done within 6 h of blood collection, and while there can be some background staining of the platelet cytoplasm and RBC, mepacrine assessment shows a good negative predictive value and moderate diagnostic accuracy compared to ADP content measurement [Citation376]. Concomitant testing of CD63 expression can be helpful to evaluate for DGD, even if lumiaggregometry findings are normal, as it can help distinguish DGD from secretion defects [Citation377]. Nonetheless, few diagnostic laboratories use mepacrine and CD63 markers for PFD diagnosis.
Genetic investigations for platelet disorders
Genetic investigations have provided important insights on the causes of some inherited platelet disorders, and the genes that are mutated in many PFDs and ITs have now been identified [Citation2]. With the transition from Sanger sequencing to high-throughput sequencing (HTS), either with whole-exome sequencing (WES), whole-genome sequencing (WGS), or targeted gene sequencing (TGS), genetic testing for the diagnosis of PFDs continues to improve. The cost of some HTS, such as WGS, has fallen ∼1000-fold, with WGS now costing <1000 USD [Citation378]. These improvements have made HTS much more feasible for both research and clinical settings.
For the investigation of inherited PFDs, the yields are dependent on the HTS platforms, the genes and regions covered, and the phenotype and pretest probability of an inherited disorder (e.g. clinical suspicion of an inherited PFD based on symptoms and family history; the presence of thrombocytopenia; clinical or laboratory findings suggestive of a known disorder). One study that evaluated a 72 genes panel, comprised of genes associated with inherited PFDs or known to be critically important to platelet physiology, and interpreted the findings according to the American College of Medical Genetics (ACMG) guidelines, observing a diagnostic yield of ∼88% for patients with a clinical and laboratory phenotype that was suggestive of a particular PFD, whereas the yield was less (∼54%) when the features were not suggestive of specific etiology [Citation4]. Similarly, the UK-GAPP study that used a WES approach to study a cohort of patients with IT had a yield of 46%, while their 30 genes panel had a 26% yield in the same population [Citation379,Citation380]. Both studies found a large number of variants of unknown significance (VUS), in ∼22 and 51% of subjects, respectively. The ThromboGenomics has studied a panel of 96 genes by HTS, which included 66 platelet genes, showing a diagnostic yield of ∼48% in patients with an abnormal platelet count but a yield of only 26% in patients with platelet dysfunction (defined as abnormal aggregation, reduced membrane glycoprotein levels, and/or abnormal secretion). It also detected molecular abnormalities in about 3% of patients with completely normal coagulation and platelet studies [Citation2], some of which were VUS, suggesting that the true yield of the investigations is lower. Molecular studies of pediatric patients with platelet abnormalities have yielded a lower rate of molecular diagnosis, with about 16–24% showing pathologic variants [Citation3,Citation381]. Similar low yields have been seen for cohort studies of PFDs, which limit selection bias [Citation129]. The SSC for Genetics in Thrombosis and Haemostasis of the ISTH has developed a cloud-based clinical software interface to encourage the community to submit curated genetic variants for diagnostic-grade genes causing bleeding, thrombosis, and platelet disorders, to improve diagnosis, variant classification, and regular bulk data transfer to ClinVar [Citation382].
For some rare, severe PFDs, a molecular diagnosis can be helpful to prenatal counseling or patient care. For example, some MYH9 mutations are predictive of renal disease and of disease severity, which is helpful for planning follow-up and treatment [Citation383]. However, access to molecular investigations is limited in some regions of the world. Additionally, some molecular defects are best evaluated by specific approaches, as some HTS methods do not detect mutations outside of the coding regions. Copy number variation (CNV) can be missed by some molecular testing strategies [Citation384]. One example is QPD, in which the causative genetic mutation is a gain-of-copy number mutation due to tandem duplication of PLAU, for which specific diagnostic testing has been developed based on the unique breakpoint [Citation385].
While it is intellectually satisfying to identify the cause of an inherited PFD, there are ethical issues to consider. Some disorders have other health implications that the patient who is being tested for may not be expecting. For example, the diagnosis of a hereditary PFD with a predisposition to leukemia (e.g. from pathogenic ANKRD26, RUNX1, or ETV6 mutations) may cause unintended psychological distress and increase the need for medical monitoring. Questions have been raised about whether it is appropriate to test for such disorders in young children, particularly when they are too young to independently provide informed consent. These issues have been addressed in some recent guidelines [Citation386,Citation387]. Some suggest a stratified approach to genetic testing, with the first priority of assessing for gene mutations with management implications, and second, to evaluate for mutations in genes with a higher risk of harm (i.e. mutations associated with an increased risk of malignancy) after obtaining informed consent [Citation388].
It is too early to recommend molecular diagnosis as a first-line investigation, except for some rare disorders that are difficult to diagnose otherwise (e.g. QPD) or conditions for which there is a high clinical suspicion of a specific PFD. However, as knowledge on the pathogenesis of PFD improves, and database information on different variants expands, the yields of HTS may improve. Such improvements could change recommendations on which conditions to investigate for a molecular cause and the best molecular strategies to employ.
Conclusion
The investigation and diagnosis of an inherited PFD have evolved, from the recent validation of the ISTH-BAT score and its application for prediction of future bleeding to improved standardization of essential investigations, such as LTA, and improved guidance on the selection of other methods to consider, including specific tests for testing for DGD, expanded application of immunofluorescence staining for PFD diagnosis, and the use of flow cytometry panels and HTS, among others. However, some tests that are recommended for PFD diagnosis are performed in a minority of clinical laboratories and require further standardization and validation to verify utility beyond research investigations.
With the continued expansion of molecular databases that are helping to identify non-pathogenic variants and improvements in knowledge on the molecular pathogenesis of inherited PFD, HTS could have a growing role in PFD diagnosis, as long as ethical issues are properly considered.
Author contributions
AB led the manuscript preparation, with contributions from ST and CPH. CPMH supervised the manuscript preparations. ST prepared the manuscript figures.
| Abbreviations | ||
| 5-HT | = | serotonin |
| AA | = | arachidonic acid |
| ACD | = | acid-citrate-dextrose |
| ACMG | = | American College of Medical Genetics |
| ARC | = | arthrogryposis-renal dysfunction-cholestasis |
| BAT | = | bleeding assessment tool |
| BSS | = | Bernard-Soulier syndrome |
| BT | = | bleeding time |
| CalDAG-GEFI | = | calcium and diacylglycerol-regulated guanine nucleotide exchange factor I |
| CAMT | = | congenital amegakaryocytic thrombocytopenia |
| CAT | = | calibrated automated thrombogram |
| CHS | = | Chediak-Higashi syndrome |
| CLSI | = | Clinical and Laboratory Standards Institute |
| CNV | = | copy number variation |
| CRP-XL | = | cross linked collagen-related peptide |
| CT | = | closure time |
| CV | = | coefficient of variation |
| DGD | = | δ-granule deficiency |
| ECAT | = | External quality Control of diagnostic Assays and Tests |
| ELISA | = | enzyme-linked immunosorbent assay |
| EQA | = | external quality assessment |
| FERMT3 | = | Kindlin 3 |
| GPS | = | Gray platelet syndrome |
| GT | = | Glanzmann thrombasthenia |
| HIT | = | heparin-induced thrombocytopenia |
| HPLC | = | high-performance liquid chromatography |
| HPS | = | Hermansky-Pudlak syndrome |
| HTS | = | high throughput sequencing |
| ISTH | = | International Society of Thrombosis and Haemostasis |
| IT | = | inherited thrombocytopenia |
| ITP | = | immune thrombocytopenia |
| LIBS | = | ligand induced binding site |
| LTA | = | light transmittance aggregometry |
| MA | = | maximal aggregation |
| MPD | = | mean platelet diameter |
| MPL | = | thrombopoietin receptor |
| MPV | = | mean platelet volume |
| NASCOLA | = | North American Specialized Coagulation Laboratory Association |
| NSAID | = | non-steroidal anti-inflammatory drug |
| PE | = | phosphatidyl ethanolamine |
| PFD | = | platelet function disorder |
| PPP | = | platelet poor plasma |
| PRP | = | platelet rich plasma |
| PS | = | phosphatidyl serine |
| PTS | = | Jacobsen/Paris-Trousseau syndrome |
| QPD | = | Quebec platelet disorder |
| RI | = | reference interval |
| SERT | = | Serotonin transporter |
| SRA | = | serotonin release assay |
| TAR | = | thrombocytopenia with absent radius |
| TEM | = | transmission electron microscopy |
| TG | = | thrombin generation |
| TGS | = | target gene sequencing |
| THPO | = | thrombopoietin |
| TRAP | = | thrombin receptor-activating peptide |
| TS | = | thin section |
| uPA | = | urokinase plasminogen activator |
| VMAT2 | = | vesicular monoamine transporter 2 |
| VUS | = | variant of unknown significance |
| VWD | = | von Willebrand disease |
| VWF | = | von Willebrand factor |
| WAS | = | Wiskott-Aldrich syndrome |
| WB | = | whole blood |
| WBA | = | whole blood aggregometry |
| WES | = | whole-exome sequencing |
| WGS | = | whole-genome sequencing |
| WM | = | whole mount |
| XLT | = | X-linked thrombocytopenia |
Disclosure statement
The authors have no conflicts of interest to declare.
Additional information
Funding
References
- Megy K, Downes K, Simeoni I, et al. Curated disease-causing genes for bleeding, thrombotic, and platelet disorders: communication from the SSC of the ISTH. J Thromb Haemost. 2019;17(8):1253–1260.
- Downes K, Megy K, Duarte D, et al. Diagnostic high-throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood. 2019;134(23):2082–2091.
- Romasko EJ, Devkota B, Biswas S, et al. Utility and limitations of exome sequencing in the molecular diagnosis of pediatric inherited platelet disorders. Am J Hematol. 2018;93(1):8–16.
- Bastida JM, Lozano ML, Benito R, et al. Introducing high-throughput sequencing into mainstream genetic diagnosis practice in inherited platelet disorders. Haematologica. 2018;103(1):148–162.
- Simeoni I, Stephens JC, Hu F, et al. A high-throughput sequencing test for diagnosing inherited bleeding, thrombotic, and platelet disorders. Blood. 2016;127(23):2791–2803.
- Stritt S, Nurden P, Turro E, et al. A gain-of-function variant in DIAPH1 causes dominant macrothrombocytopenia and hearing loss. Blood. 2016;127(23):2903–2914.
- Noetzli L, Lo RW, Lee-Sherick AB, et al. Germline mutations in ETV6 are associated with thrombocytopenia, red cell macrocytosis and predisposition to lymphoblastic leukemia. Nat Genet. 2015;47(5):535–538.
- Hayward CPM, Moffat KA, Raby A, et al. Development of North American consensus guidelines for medical laboratories that perform and interpret platelet function testing using light transmission aggregometry. Am J Clin Pathol. 2010;134(6):955–963.
- Gresele P, Subcommittee on Platelet Physiology of the International Society on Thrombosis and Hemostasis. Diagnosis of inherited platelet function disorders: guidance from the SSC of the ISTH. J Thromb Haemost. 2015;13(2):314–322.
- Harrison P, Mackie I, Mumford A, et al. Guidelines for the laboratory investigation of heritable disorders of platelet function. Br J Haematol. 2011;155(1):30–44.
- Cattaneo M, Cerletti C, Harrison P, et al. Recommendations for the standardization of light transmission aggregometry: a consensus of the working party from the platelet physiology subcommittee of SSC/ISTH. J Thromb Haemost. 2013;11(6):1183–1189.
- Hayward CPM, Moffat KA, Plumhoff E, et al. External quality assessment of platelet disorder investigations: results of international surveys on diagnostic tests for dense granule deficiency and platelet aggregometry interpretation. Semin Thromb Hemost. 2012;38(6):622–631.
- Lentaigne C, Freson K, Laffan MA, et al. Inherited platelet disorders: toward DNA-based diagnosis. Blood. 2016;127(23):2814–2823.
- Rust S, Rosier M, Funke H, et al. Tangier disease is caused by mutations in the gene encoding ATP-binding cassette transporter 1. Nat Genet. 1999;22(4):352–355.
- Bastida JM, Benito R, Janusz K, et al. Two novel variants of the ABCG5 gene cause xanthelasmas and macrothrombocytopenia: a brief review of hematologic abnormalities of sitosterolemia. J Thromb Haemost. 2017;15(9):1859–1866.
- Wang Z, Cao L, Su Y, et al. Specific macrothrombocytopenia/hemolytic anemia associated with sitosterolemia. Am J Hematol. 2014;89(3):320–324.
- Latham SL, Ehmke N, Reinke PYA, et al. Variants in exons 5 and 6 of ACTB cause syndromic thrombocytopenia. Nat Commun. 2018;9(1):4250.
- Kunishima S, Okuno Y, Yoshida K, et al. ACTN1 mutations cause congenital macrothrombocytopenia. Am J Hum Genet. 2013;92(3):431–438.
- Bottega R, Marconi C, Faleschini M, et al. ACTN1-related thrombocytopenia: identification of novel families for phenotypic characterization. Blood. 2015;125(5):869–872.
- Noris P, Perrotta S, Seri M, et al. Mutations in ANKRD26 are responsible for a frequent form of inherited thrombocytopenia: analysis of 78 patients from 21 families. Blood. 2011;117(24):6673–6680.
- Pippucci T, Savoia A, Perrotta S, et al. Mutations in the 5′ UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am J Hum Genet. 2011;88(1):115–120.
- Kahr WHA, Pluthero FG, Elkadri A, et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun. 2017;8:14816.
- Takenouchi T, Kosaki R, Niizuma T, et al. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: yet another locus for thrombocytopenia and developmental delay. Am J Med Genet. 2015;167(11):2822–2825.
- Morison IM, Cramer Bordé EM, Cheesman EJ, et al. A mutation of human cytochrome c enhances the intrinsic apoptotic pathway but causes only thrombocytopenia. Nat Genet. 2008;40(4):387–389.
- Di Paola J, Porter CC. ETV6-related thrombocytopenia and leukemia predisposition. Blood. 2019;134(8):663–667.
- Stevenson WS, Rabbolini DJ, Beutler L, et al. Paris-Trousseau thrombocytopenia is phenocopied by the autosomal recessive inheritance of a DNA-binding domain mutation in FLI1. Blood. 2015;126(17):2027–2030.
- Favier R, Jondeau K, Boutard P, et al. Paris-Trousseau syndrome: clinical, hematological, molecular data of ten new cases. Thromb Haemost. 2003;90(5):893–897.
- Breton-Gorius J, Favier R, Guichard J, et al. A new congenital dysmegakaryopoietic thrombocytopenia (Paris-Trousseau) associated with giant platelet alpha-granules and chromosome 11 deletion at 11q23. Blood. 1995;85(7):1805–1814.
- Nurden P, Debili N, Coupry I, et al. Thrombocytopenia resulting from mutations in filamin a can be expressed as an isolated syndrome. Blood. 2011;118(22):5928–5937.
- Levin C, Koren A, Pretorius E, et al. Deleterious mutation in the FYB gene is associated with congenital autosomal recessive small-platelet thrombocytopenia. J Thromb Haemost. 2015;13(7):1285–1292.
- Seo A, Gulsuner S, Pierce S, et al. Inherited thrombocytopenia associated with mutation of UDP-galactose-4-epimerase (GALE). Hum Mol Genet. 2019;28(1):133–142.
- Nichols KE, Crispino JD, Poncz M, et al. Familial dyserythropoietic anaemia and thrombocytopenia due to an inherited mutation in GATA1. Nat Genet. 2000;24(3):266–270.
- Mehaffey MG, Newton AL, Gandhi MJ, et al. X-linked thrombocytopenia caused by a novel mutation of GATA-1. Blood. 2001;98(9):2681–2688.
- Monteferrario D, Bolar NA, Marneth AE, et al. A dominant-negative GFI1B mutation in the gray platelet syndrome. N Engl J Med. 2014;370(3):245–253.
- Izumi R, Niihori T, Suzuki N, et al. GNE myopathy associated with congenital thrombocytopenia: a report of two siblings. Neuromuscul Disord. 2014;24(12):1068–1072.
- Revel-Vilk S, Shai E, Turro E, et al. GNE variants causing autosomal recessive macrothrombocytopenia without associated muscle wasting. Blood. 2018;132(17):1851–1854.
- Savoia A, Kunishima S, De Rocco D, et al. Spectrum of the mutations in Bernard-Soulier syndrome. Hum Mutat. 2014;35(9):1033–1045.
- Othman M. Platelet-type von willebrand disease: a rare, often misdiagnosed and underdiagnosed bleeding disorder. Semin Thromb Hemost. 2011;37(5):464–469.
- Thompson AA, Nguyen LT. Amegakaryocytic thrombocytopenia and radio-ulnar synostosis are associated with HOXA11 mutation. Nat Genet. 2000;26(4):397–398.
- Niihori T, Ouchi-Uchiyama M, Sasahara Y, et al. Mutations in MECOM, encoding oncoprotein EVI1, cause radioulnar synostosis with amegakaryocytic thrombocytopenia. Am J Hum Genet. 2015;97(6):848–854.
- Lentaigne C, Greene D, Sivapalaratnam S, et al. Germline mutations in the transcription factor IKZF5 cause thrombocytopenia. Blood. 2019;134(23):2070–2081.
- Botero JP, Lee K, Branchford BR, et al. Glanzmann thrombasthenia: genetic basis and clinical correlates. Haematologica. 2020;105(4):888–894.
- Nurden AT, Pillois X. ITGA2B and ITGB3 gene mutations associated with Glanzmann thrombasthenia. Platelets. 2018;29(1):98–101.
- Takeichi T, Torrelo A, Lee JYW, et al. Biallelic mutations in KDSR disrupt ceramide synthesis and result in a spectrum of keratinization disorders associated with thrombocytopenia. J Invest Dermatol. 2017;137(11):2344–2353.
- Boyden LM, Vincent NG, Zhou J, et al. Mutations in KDSR cause recessive progressive symmetric erythrokeratoderma. Am J Hum Genet. 2017;100(6):978–984.
- Hurtado B, Trakala M, Ximénez-Embún P, et al. Thrombocytopenia-associated mutations in ser/thr kinase MASTL deregulate actin cytoskeletal dynamics in platelets. J Clin Invest. 2018;128(12):5351–5367.
- Melhem M, Abu-Farha M, Antony D, et al. Novel G6B gene variant causes familial autosomal recessive thrombocytopenia and anemia. Eur J Haematol. 2017;98(3):218–227.
- Hofmann I, Crispin A, Campagna D, et al. Congenital thrombocytopenia and myelofibrosis due to germline mutations in G6b-B—a megakaryocyte-specific immunoreceptor tyrosine-based inhibitory motif (ITIM) receptor. Blood. 2017;130:1630.
- Ihara K, Ishii E, Eguchi M, et al. Identification of mutations in the c-mpl gene in congenital amegakaryocytic thrombocytopenia. Proc Natl Acad Sci USA. 1999;96(6):3132–3136.
- Muraoka K, Ishii E, Tsuji K, et al. Defective response to thrombopoietin and impaired expression of c-mpl mRNA of bone marrow cells in congenital amegakaryocytic thrombocytopenia. Br J Haematol. 1997;96(2):287–292.
- Seri M, Pecci A, Bari F, et al. MYH9-Related disease: May-Hegglin anomaly, Sebastian syndrome, Fechtner syndrome, and Epstein syndrome are not distinct entities but represent a variable expression of a single illness. Medicine. 2003;82(3):203–215.
- Bottega R, Pecci A, De Candia E, et al. Correlation between platelet phenotype and NBEAL2 genotype in patients with congenital thrombocytopenia and α-granule deficiency. Haematologica. 2013;98(6):868–874.
- Nesin V, Wiley G, Kousi M, et al. Activating mutations in STIM1 and ORAI1 cause overlapping syndromes of tubular myopathy and congenital miosis. Proc Natl Acad Sci USA. 2014;111(11):4197–4202.
- Hayward CPM, Rivard GE. Quebec platelet disorder. Expert Rev Hematol. 2011;4(2):137–141.
- Manchev VT, Hilpert M, Berrou E, et al. A new form of macrothrombocytopenia induced by a germ-line mutation in the PRKACG gene. Blood. 2014;124(16):2554–2563.
- Tartaglia M, Mehler EL, Goldberg R, et al. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet. 2001;29(4):465–468.
- Marconi C, Di Buduo CA, LeVine K, et al. Loss-of-function mutations in PTPRJ cause a new form of inherited thrombocytopenia. Blood. 2019;133(12):1346–1357.
- Albers CA, Paul DS, Schulze H, et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet. 2012;44(4):435–439.
- Heremans J, Garcia-Perez JE, Turro E, et al. Abnormal differentiation of B cells and megakaryocytes in patients with Roifman syndrome. J Allergy Clin Immunol. 2018;142(2):630–646.
- Song WJ, Sullivan MG, Legare RD, et al. Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet. 1999;23(2):166–175.
- Kauskot A, Pascreau T, Adam F, et al. A mutation in the gene coding for the sialic acid transporter SLC35A1 is required for platelet life span but not proplatelet formation. Haematologica. 2018;103(12):e613–e617.
- Fletcher SJ, Johnson B, Lowe GC, et al. SLFN14 mutations underlie thrombocytopenia with excessive bleeding and platelet secretion defects. J Clin Invest. 2015;125(9):3600–3605.
- Turro E, Greene D, Wijgaerts A, et al. A dominant gain-of-function mutation in universal tyrosine kinase SRC causes thrombocytopenia, myelofibrosis, bleeding, and bone pathologies. Sci Transl Med. 2016;8(328):328ra30.
- Misceo D, Holmgren A, Louch WE, et al. A dominant STIM1 mutation causes stormorken syndrome. Hum Mutat. 2014;35(5):556–564.
- Markello T, Chen D, Kwan JY, et al. York platelet syndrome is a CRAC channelopathy due to gain-of-function mutations in STIM1. Mol Genet Metab. 2015;114(3):474–482.
- Noris P, Marconi C, De Rocco D, et al. A new form of inherited thrombocytopenia due to monoallelic loss of function mutation in the thrombopoietin gene. Br J Haematol. 2018;181(5):698–701.
- Pleines I, Woods J, Chappaz S, et al. Mutations in tropomyosin 4 underlie a rare form of human macrothrombocytopenia. J Clin Invest. 2017;127(3):814–829.
- Stritt S, Nurden P, Favier R, et al. Defects in TRPM7 channel function deregulate thrombopoiesis through altered cellular Mg(2+) homeostasis and cytoskeletal architecture. Nat Commun. 2016;7(1):11097.
- Kunishima S, Kobayashi R, Itoh TJ, et al. Mutation of the beta1-tubulin gene associated with congenital macrothrombocytopenia affecting microtubule assembly. Blood. 2009;113(2):458–461.
- Jin Y, Mazza C, Christie JR, et al. Mutations of the Wiskott-Aldrich syndrome protein (WASP): hotspots, effect on transcription, and translation and phenotype/genotype correlation. Blood. 2004;104(13):4010–4019.
- Lanzi G, Moratto D, Vairo D, et al. A novel primary human immunodeficiency due to deficiency in the WASP-interacting protein WIP. J Exp Med. 2012;209(1):29–34.
- Pecci A, Ragab I, Bozzi V, et al. Thrombopoietin mutation in congenital amegakaryocytic thrombocytopenia treatable with romiplostim. EMBO Mol Med. 2018;10(1):63–75.
- Wielders SJH, Broers J, ten Cate H, et al. Absence of platelet-dependent fibrin formation in a patient with Scott syndrome. Thromb Haemost. 2009;102(1):76–82.
- Millington-Burgess SL, Harper MT. Gene of the issue: ANO6 and Scott syndrome. Platelets. 2020;31(7):964–967.
- Suzuki J, Umeda M, Sims PJ, et al. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468(7325):834–838.
- Berrou E, Soukaseum C, Favier R, et al. A mutation of the human EPHB2 gene leads to a major platelet functional defect. Blood. 2018;132(19):2067–2077.
- Nagy M, Mastenbroek TG, Mattheij NJA, et al. Variable impairment of platelet functions in patients with severe, genetically linked immune deficiencies. Haematologica. 2018;103(3):540–549.
- Moser M, Nieswandt B, Ussar S, et al. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med. 2008;14(3):325–330.
- Jurk K, Schulz AS, Kehrel BE, et al. Novel integrin-dependent platelet malfunction in siblings with leukocyte adhesion deficiency-III (LAD-III) caused by a point mutation in FERMT3. Thromb Haemost. 2010;103(5):1053–1064.
- Moroi M, Jung SM, Okuma M, et al. A patient with platelets deficient in glycoprotein VI that lack both collagen-induced aggregation and adhesion. J Clin Invest. 1989;84(5):1440–1445.
- Jandrot-Perrus M, Hermans C, Mezzano D. Platelet glycoprotein VI genetic quantitative and qualitative defects. Platelets. 2019;30(6):708–713.
- Merideth MA, Introne WJ, Wang JA, et al. Genetic variants associated with Hermansky-Pudlak syndrome. Platelets. 2020;31(4):544–547.
- Huizing M, Helip-Wooley A, Westbroek W, et al. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet. 2008;9:359–386.
- Huizing M, Malicdan MCV, Wang JA, et al. Hermansky-Pudlak syndrome: mutation update. Hum Mutat. 2020;41(3):543–580.
- Nagle DL, Karim MA, Woolf EA, et al. Identification and mutation analysis of the complete gene for Chediak-Higashi syndrome. Nat Genet. 1996;14(3):307–311.
- Castermans D, Volders K, Crepel A, et al. SCAMP5, NBEA and AMISYN: three candidate genes for autism involved in secretion of large dense-core vesicles. Hum Mol Genet. 2010;19(7):1368–1378.
- Nurden P, Savi P, Heilmann E, et al. An inherited bleeding disorder linked to a defective interaction between ADP and its receptor on platelets. Its influence on glycoprotein IIb-IIIa complex function. J Clin Invest. 1995;95(4):1612–1622.
- Lecchi A, Femia EA, Paoletta S, et al. Inherited dysfunctional platelet P2Y12 receptor mutations associated with bleeding disorders. Hamostaseologie. 2016;36(4):279–283.
- Adler DH, Cogan JD, Phillips JA, et al. Inherited human cPLA(2alpha) deficiency is associated with impaired eicosanoid biosynthesis, small intestinal ulceration, and platelet dysfunction. J Clin Invest. 2008;118(6):2121–2131.
- Yagmur E, Weiskirchen R, Schedel A, et al. PTGS1 compound heterozygosity impairs gene expression and platelet aggregation and is associated with severe bleeding complications. Thromb Haemost. 2013;110(5):1083–1085.
- Palma-Barqueros V, Bohdan N, Revilla N, et al. PTGS1 gene variations associated with bleeding and platelet dysfunction. Platelets. 2021;32(5):710–716.
- Canault M, Ghalloussi D, Grosdidier C, et al. Human CalDAG-GEFI gene (RASGRP2) mutation affects platelet function and causes severe bleeding. J Exp Med. 2014;211(7):1349–1362.
- Desai A, Bergmeier W, Canault M, et al. Phenotype analysis and clinical management in a large family with a novel truncating mutation in RASGRP2, the CalDAG-GEFI encoding gene. Res Pract Thromb Haemost. 2017;1(1):128–133.
- Mundell SJ, Mumford A. TBXA2R gene variants associated with bleeding. Platelets. 2018;29(7):739–742.
- Hirata T, Kakizuka A, Ushikubi F, et al. Arg60 to leu mutation of the human thromboxane A2 receptor in a dominantly inherited bleeding disorder. J Clin Invest. 1994;94(4):1662–1667.
- Geneviève D, Proulle V, Isidor B, et al. Thromboxane synthase mutations in an increased bone density disorder (Ghosal syndrome). Nat Genet. 2008;40(3):284–286.
- Gissen P, Johnson CA, Morgan NV, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004;36(4):400–404.
- Cullinane AR, Straatman-Iwanowska A, Zaucker A, et al. Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat Genet. 2010;42(4):303–312.
- Palma-Barqueros V, Revilla N, Sánchez A, et al. Inherited platelet disorders: an updated overview. IJMS. 2021;22(9):4521.
- Pecci A, Balduini CL. Inherited thrombocytopenias: an updated guide for clinicians. Blood Rev. 2021;48:100784.
- Bury L, Falcinelli E, Gresele P. Learning the ropes of platelet count regulation: inherited thrombocytopenias. JCM. 2021;10(3):533.
- Nurden P, Stritt S, Favier R, et al. Inherited platelet diseases with normal platelet count: phenotypes, genotypes and diagnostic strategy. Haematologica. 2021;106(2):337–350.
- Barbosa MD, Nguyen QA, Tchernev VT, et al. Identification of the homologous beige and Chediak-Higashi syndrome genes. Nature. 1996;382(6588):262–265.
- Fager Ferrari M, Leinoe E, Rossing M, et al. Germline heterozygous variants in genes associated with familial hemophagocytic lymphohistiocytosis as a cause of increased bleeding. Platelets. 2018;29(1):56–64.
- Cramer Bordé E, Ouzegdouh Y, Ledgerwood EC, et al. Congenital thrombocytopenia and cytochrome C mutation: a matter of birth and death. Semin Thromb Hemost. 2011;37(6):664–672.
- Fellner M, Parakra R, McDonald KO, et al. Altered structure and dynamics of pathogenic cytochrome c variants correlate with increased apoptotic activity. Biochem J. 2021;478(3):669–684.
- Othman M, Kaur H, Emsley J. Platelet-type von Willebrand disease: new insights into the molecular pathophysiology of a unique platelet defect. Semin Thromb Hemost. 2013;39(6):663–673.
- Bury L, Falcinelli E, Chiasserini D, et al. Cytoskeletal perturbation leads to platelet dysfunction and thrombocytopenia in variant forms of Glanzmann thrombasthenia. Haematologica. 2016;101(1):46–56.
- Cattaneo M. The P2 receptors and congenital platelet function defects. Semin Thromb Hemost. 2005;31(2):168–173.
- Nieuwenhuis HK, Akkerman JW, Houdijk WP, et al. Human blood platelets showing no response to collagen fail to express surface glycoprotein Ia. Nature. 1985;318(6045):470–472.
- Noris P, Guidetti GF, Conti V, et al. Autosomal dominant thrombocytopenias with reduced expression of glycoprotein Ia. Thromb Haemost. 2006;95(3):483–489.
- Lacruz RS, Feske S. Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci. 2015;1356(1):45–79.
- Liang M, Soomro A, Tasneem S, et al. Enhancer-gene rewiring in the pathogenesis of Quebec platelet disorder. Blood. 2020;136(23):2679–2690.
- Diamandis M, Adam F, Kahr WHA, et al. Insights into abnormal hemostasis in the Quebec platelet disorder from analyses of clot lysis. J Thromb Haemost. 2006;4(5):1086–1094.
- Quiroga T, Goycoolea M, Matus V, et al. Diagnosis of mild platelet function disorders. Reliability and usefulness of light transmission platelet aggregation and serotonin secretion assays. Br J Haematol. 2009;147(5):729–736.
- Hayward CPM, Pai M, Liu Y, et al. Diagnostic utility of light transmission platelet aggregometry: results from a prospective study of individuals referred for bleeding disorder assessments. J Thromb Haemost. 2009;7(4):676–684.
- Quiroga T, Goycoolea M, Panes O, et al. High prevalence of bleeders of unknown cause among patients with inherited mucocutaneous bleeding. A prospective study of 280 patients and 299 controls. Haematologica. 2007;92(3):357–365.
- Philipp CS, Dilley A, Miller CH, et al. Platelet functional defects in women with unexplained menorrhagia. J Thromb Haemost. 2003;1(3):477–484.
- Quiroga T, Goycoolea M, Muñoz B, et al. Template bleeding time and PFA-100 have low sensitivity to screen patients with hereditary mucocutaneous hemorrhages: comparative study in 148 patients. J Thromb Haemost. 2004;2(6):892–898.
- Seravalli V, Linari S, Peruzzi E, et al. Prevalence of hemostatic disorders in adolescents with abnormal uterine bleeding. J Pediatr Adolesc Gynecol. 2013;26(5):285–289.
- Gupta PK, Charan VD, Saxena R. Spectrum of von Willebrand disease and inherited platelet function disorders amongst Indian bleeders. Ann Hematol. 2007;86(6):403–407.
- Vo KT, Grooms L, Klima J, et al. Menstrual bleeding patterns and prevalence of bleeding disorders in a multidisciplinary adolescent haematology clinic. Haemophilia. 2013;19(1):71–75.
- Hayward CPM, Moffat KA, Liu Y. Laboratory investigations for bleeding disorders. Semin Thromb Hemost. 2012;38(7):742–752.
- Israels SJ, McNicol A, Robertson C, et al. Platelet storage Pool deficiency: diagnosis in patients with prolonged bleeding times and normal platelet aggregation. Br J Haematol. 1990;75(1):118–121.
- Nieuwenhuis HK, Akkerman JW, Sixma JJ. Patients with a prolonged bleeding time and normal aggregation tests may have storage Pool deficiency: studies on one hundred six patients. Blood. 1987;70(3):620–623.
- Gresele P, Harrison P, Bury L, et al. Diagnosis of suspected inherited platelet function disorders: results of a worldwide survey. J Thromb Haemost. 2014;12(9):1562–1569.
- Oved JH, Lambert MP, Kowalska MA, et al. Population based frequency of naturally occurring loss-of-function variants in genes associated with platelet disorders. J Thromb Haemost. 2021;19(1):248–254.
- Šrámek A. Usefulness of patient interview in bleeding disorders. Arch Intern Med. 1995;155(13):1409.
- Brunet J, Badin M, Chong M, et al. Bleeding risks for uncharacterized platelet function disorders. Res Pract Thromb Haemost. 2020;4(5):799–806.
- McKay H, Derome F, Haq MA, et al. Bleeding risks associated with inheritance of the Quebec platelet disorder. Blood. 2004;104(1):159–165.
- Gresele P, Orsini S, Noris P, et al. Validation of the ISTH/SSC bleeding assessment tool for inherited platelet disorders: a communication from the platelet physiology SSC. J Thromb Haemost. 2020;18(3):732–739.
- Gresele P, Falcinelli E, Bury L, et al. The ISTH bleeding assessment tool as predictor of bleeding events in inherited platelet disorders: communication from the ISTH SSC Subcommittee on Platelet Physiology. J Thromb Haemost. 2021;19(5):1364–1371.
- Tosetto A, Rodeghiero F, Castaman G, et al. A quantitative analysis of bleeding symptoms in type 1 von Willebrand disease: results from a multicenter European study (MCMDM-1 VWD). J Thromb Haemost. 2006;4(4):766–773.
- Page LK, Psaila B, Provan D, et al. The immune thrombocytopenic purpura (ITP) bleeding score: assessment of bleeding in patients with ITP. Br J Haematol. 2007;138(2):245–248.
- Rodeghiero F, Tosetto A, Abshire T, et al. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8(9):2063–2065.
- Fogarty PF, Tarantino MD, Brainsky A, et al. Selective validation of the WHO bleeding scale in patients with chronic immune thrombocytopenia. Curr Med Res Opin. 2012;28(1):79–87.
- Mittal N, Naridze R, James P, et al. Utility of a paediatric bleeding questionnaire as a screening tool for von Willebrand disease in apparently healthy children. Haemophilia. 2015;21(6):806–811.
- Rodeghiero F, Michel M, Gernsheimer T, et al. Standardization of bleeding assessment in immune thrombocytopenia: report from the international working group. Blood. 2013;121(14):2596–2606.
- Mauer AC, Khazanov NA, Levenkova N, et al. Impact of sex, age, race, ethnicity and aspirin use on bleeding symptoms in healthy adults. J Thromb Haemost. 2011;9(1):100–108.
- Badin MS, Iyer JK, Chong M, et al. Molecular phenotype and bleeding risks of an inherited platelet disorder in a family with a RUNX1 frameshift mutation. Haemophilia. 2017;23(3):e204–e213.
- Brunet JG, Iyer JK, Badin MS, et al. Electron microscopy examination of platelet whole mount preparations to quantitate platelet dense granule numbers: implications for diagnosing suspected platelet function disorders due to dense granule deficiency. Int J Lab Hem. 2018;40(4):400–407.
- Sharma T, Brunet JG, Tasneem S, et al. Thrombin generation abnormalities in commonly encountered platelet function disorders. Int J Lab Hematol. 2021;43(6):1557–1565.
- Hayward CPM, Moffat KA, George TI, et al. Report on the international society for laboratory hematology survey on guidelines to support clinical hematology laboratory practice. Int J Lab Hematol. 2016;38(Suppl 1):133–138.
- Castilloux JF, Moffat KA, Liu Y, et al. A prospective cohort study of light transmission platelet aggregometry for bleeding disorders: is testing native platelet-rich plasma non-inferior to testing platelet count adjusted samples? Thromb Haemost. 2011;106(10):675–682.
- Briggs C, Longair I, Kumar P, et al. Performance evaluation of the Sysmex haematology XN modular system. J Clin Pathol. 2012;65(11):1024–1030.
- Schoorl M, Schoorl M, Oomes J, et al. New fluorescent method (PLT-F) on Sysmex XN2000 hematology analyzer achieved higher accuracy in low platelet counting. Am J Clin Pathol. 2013;140(4):495–499.
- Zandecki M, Genevieve F, Gerard J, et al. Spurious counts and spurious results on haematology analysers: a review. Part I: platelets. Int J Lab Hematol. 2007;29(1):4–20.
- Baccini V, Geneviève F, Jacqmin H, et al. Platelet counting: ugly traps and good advice. Proposals from the French-Speaking cellular hematology group (GFHC). JCM. 2020;9(3):808.
- Lardinois B, Favresse J, Chatelain B, et al. Pseudothrombocytopenia—a review on causes, occurrence and clinical implications. JCM. 2021;10(4):594.
- Payne BA, Pierre RV. Pseudothrombocytopenia: a laboratory artifact with potentially serious consequences. Mayo Clin Proc. 1984;59(2):123–125.
- Savage RA. Pseudoleukocytosis due to EDTA-induced platelet clumping. Am J Clin Pathol. 1984;81(3):317–322.
- Zhang L, Xu J, Gao L, et al. Spurious thrombocytopenia in automated platelet count. Lab Med. 2018;49(2):130–133.
- Cohen AM, Cycowitz Z, Mittelman M, et al. The incidence of pseudothrombocytopenia in automatic blood analyzers. Haematologia. 2000;30(2):117–121.
- Silvestri F, Virgolini L, Savignano C, et al. Incidence and diagnosis of EDTA-dependent pseudothrombocytopenia in a consecutive outpatient population referred for isolated thrombocytopenia. Vox Sang. 1995;68(1):35–39.
- Anchinmane VT, Sankhe SV. Utility of peripheral blood smear in platelet count estimation. Int J Res Med Sci. 2019;7(2):434–437.
- Noris P, Klersy C, Zecca M, et al. Platelet size distinguishes between inherited macrothrombocytopenias and immune thrombocytopenia. J Thromb Haemost. 2009;7(12):2131–2136.
- Noris P, Biino G, Pecci A, et al. Platelet diameters in inherited thrombocytopenias: analysis of 376 patients with all known disorders. Blood. 2014;124(6):e4–e10.
- Balduini CL, Pecci A, Loffredo G, et al. Effects of the R216Q mutation of GATA-1 on erythropoiesis and megakaryocytopoiesis. Thromb Haemost. 2004;91(1):129–140.
- Freson K, Devriendt K, Matthijs G, et al. Platelet characteristics in patients with X-linked macrothrombocytopenia because of a novel GATA1 mutation. Blood. 2001;98(1):85–92.
- Greinacher A, Pecci A, Kunishima S, et al. Diagnosis of inherited platelet disorders on a blood smear: a tool to facilitate worldwide diagnosis of platelet disorders. J Thromb Haemost. 2017;15(7):1511–1521.
- Zaninetti C, Greinacher A. Diagnosis of inherited platelet disorders on a blood smear. JCM. 2020;9(2):539.
- Rodgers RPC, Levin J. A critical reappraisal of the bleeding time. Semin Thromb Hemost. 1990;16(1):1–20.
- Gewirtz AS, Miller ML, Keys TF. The clinical usefulness of the preoperative bleeding time. Arch Pathol Lab Med. 1996;120(4):353–356.
- Favaloro EJ, Bonar R. An update on quality control for the PFA-100/PFA-200. Platelets. 2018;29(6):622–627.
- Favaloro EJ, Facey D, Henniker A. Use of a novel platelet function analyzer (PFA-100) with high sensitivity to disturbances in von Willebrand factor to screen for von Willebrand’s disease and other disorders. Am J Hematol. 1999;62(3):165–174.
- Kundu SK, Heilmann EJ, Sio R, et al. Description of an in vitro platelet function analyzer-PFA-100. Semin Thromb Hemost. 1995;21(Suppl 2):106–112.
- Harrison P, Robinson MS, Mackie IJ, et al. Performance of the platelet function analyser PFA-100 in testing abnormalities of primary haemostasis. Blood Coagul Fibrinolysis. 1999;10(1):25–31.
- Kundu SK, Heilmann EJ, Sio R, et al. Characterization of an in vitro platelet function analyzer, PFA-100™. Clin Appl Thromb Hemost. 1996;2(4):241–249.
- Poujol C, Nurden A, Paponneau A, et al. Ultrastructural analysis of the distribution of von willebrand factor and fibrinogen in platelet aggregates formed in the PFA-100. Platelets. 1998;9(6):381–389.
- Edwards A, Jakubowski JA, Rechner AR, et al. Evaluation of the INNOVANCE PFA P2Y test cartridge: sensitivity to P2Y(12) blockade and influence of anticoagulant. Platelets. 2012;23(2):106–115.
- Scavone M, Germanovich K, Femia EA, et al. Usefulness of the INNOVANCE PFA P2Y test cartridge for the detection of patients with congenital defects of the platelet P2Y₁₂ receptor for adenosine diphosphate. Thromb Res. 2014;133(2):254–256.
- Koessler J, Ehrenschwender M, Kobsar A, et al. Evaluation of the new INNOVANCE® PFA P2Y cartridge in patients with impaired primary haemostasis. Platelets. 2012;23(8):571–578.
- Tsantes A, Ikonomidis I, Papadakis I, et al. Evaluation of the role of the new INNOVANCE PFA P2Y test cartridge in detection of clopidogrel resistance. Platelets. 2012;23(6):481–489.
- Hayward CPM, Harrison P, Cattaneo M, et al. Platelet function analyzer (PFA)-100 closure time in the evaluation of platelet disorders and platelet function. J Thromb Haemost. 2006;4(2):312–319.
- Favaloro EJ. Clinical utility of closure times using the platelet function analyzer-100/200. Am J Hematol. 2017;92(4):398–404.
- Podda GM, Bucciarelli P, Lussana F, et al. Usefulness of PFA-100 testing in the diagnostic screening of patients with suspected abnormalities of hemostasis: comparison with the bleeding time. J Thromb Haemost. 2007;5(12):2393–2398.
- Favaloro EJ. Clinical utility of the PFA-100. Semin Thromb Hemost. 2008;34(8):709–733.
- Moenen FCJI, Vries MJA, Nelemans PJ, et al. Screening for platelet function disorders with multiplate and platelet function analyzer. Platelets. 2019;30(1):81–87.
- Born GVR. Aggregation of blood platelets by adenosine diphosphate and its reversal. Nature. 1962;194(4832):927–929.
- O’brien JR. Platelet aggregation: part I some effects of the adenosine phosphates, thrombin, and cocaine upon platelet adhesiveness. J Clin Pathol. 1962;15(5):446–452.
- Christie D, Avari T, Carrington L, et al. Platelet function testing by aggregometry: approved guidelines. Vol. 38. Wayne (PA): Clinical and Laboratory Standards Institute; 2008.
- Hayward CPM, Moffat KA, Brunet J, et al. Update on diagnostic testing for platelet function disorders: what is practical and useful? Int J Lab Hematol. 2019;41(S1):26–32.
- Althaus K, Zieger B, Bakchoul T, et al. Standardization of light transmission aggregometry for diagnosis of platelet disorders: an inter-laboratory external quality assessment. Thromb Haemost. 2019;119(7):1154–1161.
- Hendra TJ, Oughton J, Smith CC, et al. Exercise-induced changes in platelet aggregation; a comparison of whole blood and platelet rich plasma techniques. Thromb Res. 1988;52(5):443–451.
- Wang JS, Jen CJ, Chen HI. Effects of exercise training and deconditioning on platelet function in men. Arterioscler Thromb Vasc Biol. 1995;15(10):1668–1674.
- Aldemir H, Kiliç N. The effect of time of day and exercise on platelet functions and platelet-neutrophil aggregates in healthy male subjects. Mol Cell Biochem. 2005;280(1–2):119–124.
- Vicari AM, Margonato A, Macagni A, et al. Effects of acute smoking on the hemostatic system in humans. Clin Cardiol. 1988;11(8):538–540.
- Bolliger D, Seeberger MD, Tanaka KA, et al. Pre-analytical effects of pneumatic tube transport on impedance platelet aggregometry. Platelets. 2009;20(7):458–465.
- Lorenzen H, Frøstrup AB, Larsen AS, et al. Pneumatic tube transport of blood samples affects global hemostasis and platelet function assays. Int J Lab Hematol. 2021;43(5):1207–1215.
- Stegnar M, Knezevic A, Bozic-Mijovski M. The effect of pre-analytical variables on light transmittance aggregometry in citrated platelet-rich plasma from healthy subjects. Clin Chem Lab Med. 2010;48(10):1463–1465.
- Kaiser AFC, Neubauer H, Franken CC, et al. Which is the best anticoagulant for whole blood aggregometry platelet function testing? Comparison of six anticoagulants and diverse storage conditions. Platelets. 2012;23(5):359–367.
- Maurer-Spurej E, Pfeiler G, Maurer N, et al. Room temperature activates human blood platelets. Lab Invest. 2001;81(4):581–592.
- Merolla M, Nardi MA, Berger JS. Centrifugation speed affects light transmission aggregometry. Int J Lab Hematol. 2012;34(1):81–85.
- Femia EA, Pugliano M, Podda G, et al. Comparison of different procedures to prepare platelet-rich plasma for studies of platelet aggregation by light transmission aggregometry. Platelets. 2012;23(1):7–10.
- Mani H, Luxembourg B, Kläffling C, et al. Use of native or platelet count adjusted platelet rich plasma for platelet aggregation measurements. J Clin Pathol. 2005;58(7):747–750.
- Linnemann B, Schwonberg J, Mani H, et al. Standardization of light transmittance aggregometry for monitoring antiplatelet therapy: an adjustment for platelet count is not necessary. J Thromb Haemost. 2008;6(4):677–683.
- Cattaneo M, Lecchi A, Zighetti ML, et al. Platelet aggregation studies: autologous platelet-poor plasma inhibits platelet aggregation when added to platelet-rich plasma to normalize platelet count. Haematologica. 2007;92(5):694–697.
- Hayward CPM, Moffat KA, Pai M, et al. An evaluation of methods for determining reference intervals for light transmission platelet aggregation tests on samples with normal or reduced platelet counts. Thromb Haemost. 2008;100(1):134–145.
- Favaloro EJ, Mohammed S. Platelet function testing: auditing local practice and broader implications. Clin Lab Sci. 2010;23(1):21–31.
- Hayward CPM, Moffat KA, Castilloux JF, et al. Simultaneous measurement of adenosine triphosphate release and aggregation potentiates human platelet aggregation responses for some subjects, including persons with Quebec platelet disorder. Thromb Haemost. 2012;107(04):726–734.
- Blavignac J, Bunimov N, Rivard GE, et al. Quebec platelet disorder: update on pathogenesis, diagnosis, and treatment. Semin Thromb Hemost. 2011;37(6):713–720.
- Faraday N, Scharpf RB, Dodd-O JM, et al. Leukocytes can enhance platelet-mediated aggregation and thromboxane release via interaction of P-selectin glycoprotein ligand 1 with P-selectin. Anesthesiology. 2001;94(1):145–151.
- Praticò D, Iuliano L, Alessandri C, et al. Polymorphonuclear leukocyte-derived O2-reactive species activate primed platelets in human whole blood. Am J Physiol. 1993;264(5 Pt 2):H1582–1587.
- Sun P, McMillan-Ward E, Mian R, et al. Comparison of light transmission aggregometry and multiple electrode aggregometry for the evaluation of patients with mucocutaneous bleeding. Int J Lab Hematol. 2019;41(1):133–140.
- Al Ghaithi R, Drake S, Watson SP, et al. Comparison of multiple electrode aggregometry with lumi-aggregometry for the diagnosis of patients with mild bleeding disorders. J Thromb Haemost. 2017;15(10):2045–2052.
- Haas T, Cushing MM, Varga S, et al. Usefulness of multiple electrode aggregometry as a screening tool for bleeding disorders in a pediatric hospital. Platelets. 2019;30(4):498–505.
- Awidi A, Maqablah A, Dweik M, et al. Comparison of platelet aggregation using light transmission and multiple electrode aggregometry in Glanzmann thrombasthenia. Platelets. 2009;20(5):297–301.
- Albanyan A, Al-Musa A, AlNounou R, et al. Diagnosis of Glanzmann thrombasthenia by whole blood impedance analyzer (MEA) vs. light transmission aggregometry. Int J Lab Hematol. 2015;37(4):503–508.
- Karkouti K, Callum J, Wijeysundera DN, et al. Point-of-care hemostatic testing in cardiac surgery: a stepped-wedge clustered randomized controlled trial. Circulation. 2016;134(16):1152–1162.
- McGlasson D, Fritsma G. Whole blood platelet aggregometry and platelet function testing. Semin Thromb Hemost. 2009;35(2):168–180.
- Sweeney JD, Hoernig LA, Michnik A, et al. Whole blood aggregometry. Influence of sample collection and delay in study performance on test results. Am J Clin Pathol. 1989;92(5):676–679.
- Dyszkiewicz-Korpanty AM, Frenkel EP, Sarode R. Approach to the assessment of platelet function: comparison between optical-based platelet-rich plasma and impedance-based whole blood platelet aggregation methods. Clin Appl Thromb Hemost. 2005;11(1):25–35.
- Skipper MT, Rubak P, Stentoft J, et al. Evaluation of platelet function in thrombocytopenia. Platelets. 2018;29(3):270–276.
- Tiedemann Skipper M, Rubak P, Halfdan Larsen O, et al. Thrombocytopenia model with minimal manipulation of blood cells allowing whole blood assessment of platelet function. Platelets. 2016;27(4):295–300.
- Moffat K, Ledford-Kraemer M, Nichols W, et al. Variability in clinical laboratory practice in testing for disorders of platelet function: results of two surveys of the North American specialized coagulation laboratory association. Thromb Haemost. 2005;93(3):549–553.
- Cattaneo M. Light transmission aggregometry and ATP release for the diagnostic assessment of platelet function. Semin Thromb Hemost. 2009;35(2):158–167.
- Pai M, Wang G, Moffat KA, et al. Diagnostic usefulness of a lumi-aggregometer adenosine triphosphate release assay for the assessment of platelet function disorders. Am J Clin Pathol. 2011;136(3):350–358.
- Feinman RD, Lubowsky J, Charo I, et al. The lumi-aggregometer: a new instrument for simultaneous measurement of secretion and aggregation by platelets. J Lab Clin Med. 1977;90(1):125–129.
- Lotta LA, Maino A, Tuana G, et al. Prevalence of disease and relationships between laboratory phenotype and bleeding severity in platelet primary secretion defects. PLoS One. 2013;8(4):e60396.
- Badin MS, Graf L, Iyer JK, et al. Variability in platelet dense granule adenosine triphosphate release findings amongst patients tested multiple times as part of an assessment for a bleeding disorder. Int J Lab Hematol. 2016;38(6):648–657.
- De Robertis E. Electron microscope observations of the platelet-fibrin relationship in blood clotting. Blood. 1955;10(5):528–533.
- Chen D, Uhl CB, Bryant SC, et al. Diagnostic laboratory standardization and validation of platelet transmission electron microscopy. Platelets. 2018;29(6):574–582.
- Woods GM, Kudron EL, Davis K, et al. Light transmission aggregometry does not correlate with the severity of δ-granule platelet storage Pool deficiency. J Pediatr Hematol Oncol. 2016;38(7):525–528.
- Israels SJ, Robertson C, Mcnicol A. Identification of patients with storage Pool deficiency using ATP release and dense granule counts. Hematology. 1997;2(2):161–167.
- Gunning WT, Yoxtheimer L, Smith MR. Platelet aggregation assays do not reliably diagnose platelet delta granule storage Pool deficiency. J Hematol. 2020;10(4):196–201.
- White JG. The dense bodies of human platelets: inherent electron opacity of the serotonin storage particles. Blood. 1969;33(4):598–606.
- White JG. Use of the electron microscope for diagnosis of platelet disorders. Semin Thromb Hemost. 1998;24(2):163–168.
- Weiss HJ, Lages B, Vicic W, et al. Heterogeneous abnormalities of platelet dense granule ultrastructure in 20 patients with congenital storage Pool deficiency. Br J Haematol. 1993;83(2):282–295.
- Asher L, Hata J. Platelet electron microscopy: utilizing LEAN methodology to optimize laboratory workflow. Pediatr Dev Pathol. 2020;23(5):356–361.
- Hayward CPM, Moffat KA, Spitzer E, et al. Results of an external proficiency testing exercise on platelet dense-granule deficiency testing by whole mount electron microscopy. Am J Clin Pathol. 2009;131(5):671–675.
- Westmoreland D, Shaw M, Grimes W, et al. Super-resolution microscopy as a potential approach to diagnosis of platelet granule disorders. J Thromb Haemost. 2016;14(4):839–849.
- Gunning WT, Calomeni EP. A brief review of transmission electron microscopy and applications in pathology. J Histotechnol. 2000;23(3):237–246.
- White JG. Electron opaque structures in human platelets: which are or are not dense bodies? Platelets. 2008;19(6):455–466.
- White JG. Electron microscopy methods for studying platelet structure and function. Methods Mol Biol. 2004;272:47–63.
- Glauert AM, Lewis PR. Biological specimen preparation for transmission electron microscopy. Princeton, NJ: Princeton University Press; 2014.
- Sawatzke CL, Solomons CC. Fixation and embedding of small volumes of platelets for transmission electron microscopy. J Clin Pathol. 1980;33(6):600–602.
- White JG. Effects of ethylenediamine tetracetic acid (EDTA) on platelet structure. Scand J Haematol. 1968;5(4):241–254.
- White JG. Platelet microtubules and giant granules in the Chediak-Higashi syndrome. Am J Med Technol. 1978;44(4):273–278.
- White JG. Platelet storage Pool deficiency in Jacobsen syndrome. Platelets. 2007;18(7):522–527.
- White JG, de Alarcon PA. Platelet spherocytosis: a new bleeding disorder. Am J Hematol. 2002;70(2):158–166.
- Breton-Gorius J, Vainchenker W, Nurden A, et al. Defective alpha-granule production in megakaryocytes from gray platelet syndrome: ultrastructural studies of bone marrow cells and megakaryocytes growing in culture from blood precursors. Am J Pathol. 1981;102(1):10–19.
- Mezzano D, Aranda E, Foradori A. Comparative study of size, total protein, fibrinogen and 5-HT content of human and canine platelet density subpopulations. Thromb Haemost. 1986;56(3):288–292.
- Jedlitschky G, Greinacher A, Kroemer HK. Transporters in human platelets: physiologic function and impact for pharmacotherapy. Blood. 2012;119(15):3394–3402.
- Maurer-Spurej E, Pittendreigh C, Solomons K. The influence of selective serotonin reuptake inhibitors on human platelet serotonin. Thromb Haemost. 2004;91(01):119–128.
- Sheridan D, Carter C, Kelton JG. A diagnostic test for heparin-induced thrombocytopenia. Blood. 1986;67(1):27–30.
- Warkentin TE, Arnold DM, Nazi I, et al. The platelet serotonin-release assay. Am J Hematol. 2015;90(6):564–572.
- Holmsen H, Ostvold AC, Day HJ. Behaviour of endogenous and newly absorbed serotonin in the platelet release reaction. Biochem Pharmacol. 1973;22(20):2599–2608.
- Zhou L, Schmaier AH. Platelet aggregation testing in platelet-rich plasma: description of procedures with the aim to develop standards in the field. Am J Clin Pathol. 2005;123(2):172–183.
- Holmsen H, Dangelmaier CA. Measurement of secretion of serotonin. Methods Enzymol. 1989;169:205–210.
- Wall JE, Buijs-Wilts M, Arnold JT, et al. A flow cytometric assay using mepacrine for study of uptake and release of platelet dense granule contents. Br J Haematol. 1995;89(2):380–385.
- Kumar AM, Kumar M, Deepika K, et al. A modified HPLC technique for simultaneous measurement of 5-hydroxytryptamine and 5-hydroxyindoleacetic acid in cerebrospinal fluid, platelet and plasma. Life Sci. 1990;47(19):1751–1759.
- Pussard E, Guigueno N, Adam O, et al. Validation of HPLC-amperometric detection to measure serotonin in plasma, platelets, whole blood, and urine. Clin Chem. 1996;42(7):1086–1091.
- Ge S, Woo E, White JG, et al. Electrochemical measurement of endogenous serotonin release from human blood platelets. Anal Chem. 2011;83(7):2598–2604.
- Anderson GM, Hall LM, Yang JX, et al. Platelet dense granule release reaction monitored by high-performance liquid chromatography-fluorometric determination of endogenous serotonin. Anal Biochem. 1992;206(1):64–67.
- Bossant MJ, Ninio E, Delautier D, et al. Quantitation of paf-acether by release of endogenous platelet serotonin assessed by liquid chromatography with electrochemical detection. Anal Biochem. 1989;182(2):419–423.
- Torfs SC, Maes AA, Delesalle CJ, et al. Comparative analysis of serotonin in equine plasma with liquid chromatography-tandem mass spectrometry and enzyme-linked immunosorbent assay. J Vet Diagn Invest. 2012;24(6):1035–1042.
- Ge S, Wittenberg NJ, Haynes CL. Quantitative and real-time detection of secretion of chemical messengers from individual platelets. Biochemistry. 2008;47(27):7020–7024.
- Kluge H, Bolle M, Reuter R, et al. Serotonin in platelets: comparative analyses using new enzyme immunoassay and HPLC test kits and the traditional fluorimetric procedure. Lab J Lab Med. 1999;23(6):360–364.
- Sono-Koree NK, Crist RA, Frank EL, et al. A high-performance liquid chromatography method for the serotonin release assay is equivalent to the radioactive method. Int J Lab Hem. 2016;38(1):72–80.
- Chan SL, Yi X, Wysocki E, et al. Development of a nonradioactive platelet serotonin uptake and release assay by micro-liquid chromatography tandem mass spectrometry using minimal blood volume. Am J Clin Pathol. 2019;152(6):718–724.
- Chauveau J, Fert V, Morel AM, et al. Rapid and specific enzyme immunoassay of serotonin. Clin Chem. 1991;37(7):1178–1184.
- Fouassier M, Bourgerette E, Libert F, et al. Determination of serotonin release from platelets by HPLC and ELISA in the diagnosis of heparin-induced thrombocytopenia: comparison with reference method by [C]-serotonin release assay. J Thromb Haemost. 2006;4(5):1136–1139.
- Holmsen H, Storm E, Day HJ. Determination of ATP and ADP in blood platelets: a modification of the firefly luciferase assay for plasma. Anal Biochem. 1972;46(2):489–501.
- David JL, Herion F. Assay of platelet ATP and ADP by the luciferase method: Some theoretical and practical aspects. Adv Exp Med Biol. 1972;34:341–354.
- Weiss HJ, Witte LD, Kaplan KL, et al. Heterogeneity in storage Pool deficiency: studies on granule-bound substances in 18 patients including variants deficient in alpha-granules, platelet factor 4, beta-thromboglobulin, and platelet-derived growth factor. Blood. 1979;54(6):1296–1319.
- Lages B, Holmsen H, Weiss HJ, et al. Thrombin and ionophore A23187-induced dense granule secretion in storage Pool deficient platelets: evidence for impaired nucleotide storage as the primary dense granule defect. Blood. 1983;61(1):154–162.
- Cattaneo M, Canciani MT, Lecchi A, et al. Released adenosine diphosphate stabilizes thrombin-induced human platelet aggregates. Blood. 1990;75(5):1081–1086.
- D’Souza L, Glueck HI. Measurement of nucleotide pools in platelets using high pressure liquid chromatography. Thromb Haemost. 1977;38(04):0990–1001.
- Leoncini G, Buzzi E, Maresca M, et al. Alkaline extraction and reverse-phase high-performance liquid chromatography of adenine and pyridine nucleotides in human platelets. Anal Biochem. 1987;165(2):379–383.
- von PM, Gambaryan S, Schütz C, et al. Determination of ATP and ADP secretion from human and mouse platelets by an HPLC assay. Transfus Med Hemother. 2013;40(2):109–116.
- Blair P, Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. 2009;23(4):177–189.
- Ohkawa R, Hirowatari Y, Nakamura K, et al. Platelet release of beta-thromboglobulin and platelet factor 4 and serotonin in plasma samples. Clin Biochem. 2005;38(11):1023–1026.
- Kaplan KL, Nossel HL, Drillings M, et al. Radioimmunoassay of platelet factor 4 and beta-thromboglobulin: development and application to studies of platelet release in relation to fibrinopeptide a generation. Br J Haematol. 1978;39(1):129–146.
- Takahashi H, Yoshino N, Shibata A. Measurement of platelet factor 4 and beta-thromboglobulin by an enzyme-linked immunosorbent assay. Clin Chim Acta Int J Clin Chem. 1988;175(1):113–114.
- Schraw T, Whiteheart S. The development of a quantitative enzyme-linked immunosorbent assay to detect human platelet factor 4. Transfusion. 2005;45(5):717–724.
- Mumford AD, Frelinger AL, Gachet C, et al. A review of platelet secretion assays for the diagnosis of inherited platelet secretion disorders. Thromb Haemost. 2015;114(1):14–25.
- Wolfs JLN, Comfurius P, Rasmussen JT, et al. Activated scramblase and inhibited aminophospholipid translocase cause phosphatidylserine exposure in a distinct platelet fraction. Cell Mol Life Sci. 2005;62(13):1514–1525.
- Agbani EO, Poole AW. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood. 2017;130(20):2171–2179.
- Prodan CI, Stoner JA, Cowan LD, et al. Higher coated-platelet levels are associated with stroke recurrence following nonlacunar brain infarction. J Cereb Blood Flow Metab. 2013;33(2):287–292.
- Kirkpatrick AC, Stoner JA, Dale GL, et al. Elevated coated-platelets in symptomatic large-artery stenosis patients are associated with early stroke recurrence. Platelets. 2014;25(2):93–96.
- Kirkpatrick AC, Vincent AS, Dale GL, et al. Coated-platelets predict stroke at 30 days following TIA. Neurology. 2017;89(2):125–128.
- Zhao L, Bi Y, Kou J, et al. Phosphatidylserine exposing-platelets and microparticles promote procoagulant activity in Colon cancer patients. J Exp Clin Cancer Res. 2016;35:54.
- Guo L, Tong D, Yu M, et al. Phosphatidylserine-exposing cells contribute to the hypercoagulable state in patients with multiple myeloma. Int J Oncol. 2018;52(6):1981–1990.
- Panova-Noeva M, Marchetti M, Spronk HM, et al. Platelet-induced thrombin generation by the calibrated automated thrombogram assay is increased in patients with essential thrombocythemia and polycythemia vera. Am J Hematol. 2011;86(4):337–342.
- Colucci G, Stutz M, Rochat S, et al. The effect of desmopressin on platelet function: a selective enhancement of procoagulant COAT platelets in patients with primary platelet function defects. Blood. 2014;123(12):1905–1916.
- Wartiovaara-Kautto U, Joutsi-Korhonen L, Ilveskero S, et al. Platelets significantly modify procoagulant activities in haemophilia A. Haemophilia. 2011;17(5):743–751.
- Santagostino E, Mancuso ME, Tripodi A, et al. Severe hemophilia with mild bleeding phenotype: molecular characterization and global coagulation profile. J Thromb Haemost. 2010;8(4):737–743.
- Szanto T, Nummi V, Jouppila A, et al. Platelets compensate for poor thrombin generation in type 3 von Willebrand disease. Platelets. 2020;31(1):103–111.
- Castoldi E, Duckers C, Radu C, et al. Homozygous F5 deep-intronic splicing mutation resulting in severe factor V deficiency and undetectable thrombin generation in platelet-rich plasma. J Thromb Haemost. 2011;9(5):959–968.
- Duckers C, Simioni P, Spiezia L, et al. Residual platelet factor V ensures thrombin generation in patients with severe congenital factor V deficiency and mild bleeding symptoms. Blood. 2010;115(4):879–886.
- Rugeri L, Quélin F, Chatard B, et al. Thrombin generation in patients with factor XI deficiency and clinical bleeding risk. Haemophilia. 2010;16(5):771–777.
- Brunet JG, Sharma T, Tasneem S, et al. Thrombin generation abnormalities in Quebec platelet disorder. Int J Lab Hem. 2020;42(6):801–809.
- Chelle P, Montmartin A, Damien P, et al. Tissue factor pathway inhibitor is the main determinant of thrombin generation in haemophilic patients. Haemophilia. 2019;25(2):343–348.
- MacDonald S, White D, Langdown J, et al. Investigation of patients with unclassified bleeding disorder and abnormal thrombin generation for physiological coagulation inhibitors reveals multiple abnormalities and a subset of patients with increased tissue factor pathway inhibitor activity. Int J Lab Hem. 2020;42(3):246–255.
- Hemker HC, Giesen P, Al Dieri R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemos Thromb. 2003;33(1):4–15.
- Hemker HC, Giesen PL, Ramjee M, et al. The thrombogram: monitoring thrombin generation in platelet-rich plasma. Thromb Haemost. 2000;83(4):589–591.
- Rosing J, Bevers E, Comfurius P, et al. Impaired factor X and prothrombin activation associated with decreased phospholipid exposure in platelets from a patient with a bleeding disorder. Blood. 1985;65(6):1557–1561.
- Tohidi-Esfahani I, Lee CSM, Liang HPH, et al. Procoagulant platelets: laboratory detection and clinical significance. Int J Lab Hematol. 2020;42(S1):59–67.
- Jennings LK, Ashmun RA, Wang WC, et al. Analysis of human platelet glycoproteins IIb-IIIa and Glanzmann’s thrombasthenia in whole blood by flow cytometry. Blood. 1986;68(1):173–179.
- Giannini S, Cecchetti L, Mezzasoma AM, et al. Diagnosis of platelet-type von Willebrand disease by flow cytometry. Haematologica. 2010;95(6):1021–1024.
- Cohn RJ, Sherman GG, Glencross DK. Flow cytometric analysis of platelet surface glycoproteins in the diagnosis of Bernard-Soulier syndrome. Pediatr Hematol Oncol. 1997;14(1):43–50.
- Marti GE, Magruder L, Schuette WE, et al. Flow cytometric analysis of platelet surface antigens. Cytometry. 1988;9(5):448–455.
- Halliez M, Fouassier M, Robillard N, et al. Detection of phosphatidyl serine on activated platelets' surface by flow cytometry in whole blood: a simpler test for the diagnosis of Scott syndrome. Br J Haematol. 2015;171(2):290–292.
- Stenberg PE, McEver RP, Shuman MA, et al. A platelet alpha-granule membrane protein (GMP-140) is expressed on the plasma membrane after activation. J Cell Biol. 1985;101(3):880–886.
- De Cuyper IM, Meinders M, van de Vijver E, et al. A novel flow cytometry-based platelet aggregation assay. Blood. 2013;121(10):e70–e80.
- Navred K, Martin M, Ekdahl L, et al. A simplified flow cytometric method for detection of inherited platelet disorders–a comparison to the gold standard light transmission aggregometry. PLoS One. 2019;14(1):e0211130.
- Vinholt PJ, Frederiksen H, Hvas AM, et al. Measurement of platelet aggregation, independently of patient platelet count: a flow-cytometric approach. J Thromb Haemost. 2017;15(6):1191–1202.
- Boknäs N, Macwan AS, Södergren AL, et al. Platelet function testing at low platelet counts: when can you trust your analysis? Res Pract Thromb Haemost. 2019;3(2):285–290.
- Podda G, Scavone M, Femia EA, et al. Aggregometry in the settings of thrombocytopenia, thrombocytosis and antiplatelet therapy. Platelets. 2018;29(7):644–649.
- Jurk K, Shiravand Y. Platelet phenotyping and function testing in thrombocytopenia. J Clin Med. 2021;10(5):1114.
- Frelinger AL, Rivera J, Connor DE, et al. Consensus recommendations on flow cytometry for the assessment of inherited and acquired disorders of platelet number and function: communication from the ISTH SSC Subcommittee on Platelet Physiology. J Thromb Haemost. 2021;19(12):3193–3202.
- Busuttil-Crellin X, McCafferty C, Van Den Helm S, et al. Guidelines for panel design, optimization, and performance of whole blood multi-color flow cytometry of platelet surface markers. Platelets. 2020;31(7):845–852.
- Andres O, Henning K, Strauß G, et al. Diagnosis of platelet function disorders: a standardized, rational, and modular flow cytometric approach. Platelets. 2018;29(4):347–356.
- Frelinger AL, Grace RF, Gerrits AJ, et al. Platelet function tests, independent of platelet count, are associated with bleeding severity in ITP. Blood. 2015;126(7):873–879.
- Boknäs N, Ramström S, Faxälv L, et al. Flow cytometry-based platelet function testing is predictive of symptom burden in a cohort of bleeders. Platelets. 2018;29(5):512–519.
- van Asten I, Schutgens REG, Baaij M, et al. Validation of flow cytometric analysis of platelet function in patients with a suspected platelet function defect. J Thromb Haemost. 2018;16(4):689–698.
- Dovlatova N, Lordkipanidzé M, Lowe GC, et al. Evaluation of a whole blood remote platelet function test for the diagnosis of mild bleeding disorders. J Thromb Haemost. 2014;12(5):660–665.
- Huskens D, Li L, Florin L, et al. Flow cytometric analysis of platelet function to improve the recognition of thrombocytopathy. Thromb Res. 2020;194:183–189.
- Blair TA, Frelinger AL. Platelet surface marker analysis by mass cytometry. Platelets. 2020;31(5):633–640.
- Spurgeon BEJ, Naseem KM. High-throughput signaling profiling in blood platelets by multiplexed phosphoflow cytometry. Methods Mol Biol. 2018;1812:95–111.
- Spurgeon BEJ, Naseem KM. Phosphoflow cytometry and barcoding in blood platelets: Technical and analytical considerations. Cytometry B Clin Cytom. 2020;98(2):123–130.
- Pasalic L, Pennings GJ, Connor D, et al. Flow cytometry protocols for assessment of platelet function in whole blood. Methods Mol Biol. 2017;1646:369–389.
- Linden MD. Platelet flow cytometry. Methods Mol Biol. 2013;992:241–262.
- Michelson AD, Barnard MR, Krueger LA, et al. Evaluation of platelet function by flow cytometry. Methods. 2000;21(3):259–270.
- Pedersen OH, Nissen PH, Hvas AM. Platelet function investigation by flow cytometry: sample volume, needle size, and reference intervals. Platelets. 2018;29(2):199–202.
- Ramström S, Södergren AL, Tynngård N, et al. Platelet function determined by flow cytometry: new perspectives? Semin Thromb Hemost. 2016;42(3):268–281.
- Ritchie JL, Alexander HD, Rea IM. Flow cytometry analysis of platelet P-selectin expression in whole blood-methodological considerations. Clin Lab Haematol. 2000;22(6):359–363.
- Schmidt V, Hilberg T. ThromboFix platelet stabilizer: advances in clinical platelet analyses by flow cytometry? Platelets. 2006;17(4):266–273.
- Hu H, Daleskog M, Li N. Influences of fixatives on flow cytometric measurements of platelet P-selectin expression and fibrinogen binding. Thromb Res. 2000;100(3):161–166.
- Shattil SJ, Hoxie JA, Cunningham M, et al. Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. J Biol Chem. 1985;260(20):11107–11114.
- Dovlatova N, May JA, Fox SC. Remote platelet function testing-significant progress towards widespread testing in clinical practice. Platelets. 2015;26(5):399–401.
- Hagberg IA, Lyberg T. Blood platelet activation evaluated by flow cytometry: optimised methods for clinical studies. Platelets. 2000;11(3):137–150.
- Huskens D, Sang Y, Konings J, et al. Standardization and reference ranges for whole blood platelet function measurements using a flow cytometric platelet activation test. PLOS One. 2018;13(2):e0192079.
- Nurden P, Tandon N, Takizawa H, et al. An acquired inhibitor to the GPVI platelet collagen receptor in a patient with lupus nephritis. J Thromb Haemost. 2009;7(9):1541–1549.
- Giannini S, Mezzasoma AM, Guglielmini G, et al. A new case of acquired Glanzmann's thrombasthenia: diagnostic value of flow cytometry. Cytometry B Clin Cytom. 2008;74(3):194–199.
- McEver RP, Martin MN. A monoclonal antibody to a membrane glycoprotein binds only to activated platelets. J Biol Chem. 1984;259(15):9799–9804.
- Metzelaar MJ, Wijngaard PL, Peters PJ, et al. CD63 antigen. A novel lysosomal membrane glycoprotein, cloned by a screening procedure for intracellular antigens in eukaryotic cells. J Biol Chem. 1991;266(5):3239–3245.
- Rubak P, Nissen PH, Kristensen SD, et al. Investigation of platelet function and platelet disorders using flow cytometry. Platelets. 2016;27(1):66–74.
- Curvers J, de Wildt-Eggen J, Heeremans J, et al. Flow cytometric measurement of CD62P (P-selectin) expression on platelets: a multicenter optimization and standardization effort. Transfusion. 2008;48(7):1439–1446.
- Furie B, Furie B, Flaumenhaft R. A journey with platelet P-Selectin: the molecular basis of granule secretion, signalling and cell adhesion. Thromb Haemost. 2001;86(1):214–221.
- Pasalic L. Assessment of platelet function in whole blood by flow cytometry. Methods Mol Biol. 2017;1646:349–367.
- Berman CL, Yeo EL, Wencel-Drake JD, et al. A platelet alpha granule membrane protein that is associated with the plasma membrane after activation. Characterization and subcellular localization of platelet activation-dependent granule-external membrane protein. J Clin Invest. 1986;78(1):130–137.
- Ruf A, Patscheke H. Flow cytometric detection of activated platelets: comparison of determining shape change, fibrinogen binding, and P-selectin expression. Semin Thromb Hemost. 1995;21(2):146–151.
- Michelson AD, Benoit SE, Kroll MH, et al. The activation-induced decrease in the platelet surface expression of the glycoprotein Ib-IX complex is reversible. Blood. 1994;83(12):3562–3573.
- Lages B, Shattil SJ, Bainton DF, et al. Decreased content and surface expression of alpha-granule membrane protein GMP-140 in one of two types of platelet alpha Delta storage Pool deficiency. J Clin Invest. 1991;87(3):919–929.
- Lages B, Sussman II, Levine SP, et al. Platelet alpha granule deficiency associated with decreased P-selectin and selective impairment of thrombin-induced activation in a new patient with gray platelet syndrome (alpha-storage Pool deficiency). J Lab Clin Med. 1997;129(3):364–375.
- van Velzen JF, Laros-van Gorkom BAP, Pop GAM, et al. Multicolor flow cytometry for evaluation of platelet surface antigens and activation markers. Thromb Res. 2012;130(1):92–98.
- Gresele P, Falcinelli E, Bury L. Laboratory diagnosis of clinically relevant platelet function disorders. Int J Lab Hematol. 2018;40(Suppl 1):34–45.
- McEver RP, Cummings RD. Perspectives series: cell adhesion in vascular biology. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100(3):485–491.
- de Gaetano G, Cerletti C, Evangelista V. Recent advances in platelet-polymorphonuclear leukocyte interaction. Haemostasis. 1999;29(1):41–49.
- Maugeri N, Baldini M, Ramirez GA, et al. Platelet-leukocyte deregulated interactions foster sterile inflammation and tissue damage in immune-mediated vessel diseases. Thromb Res. 2012;129(3):267–273.
- Brill A, Fuchs TA, Savchenko AS, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. 2012;10(1):136–144.
- Asaduzzaman M, Lavasani S, Rahman M, et al. Platelets support pulmonary recruitment of neutrophils in abdominal sepsis. Crit Care Med. 2009;37(4):1389–1396.
- Peyton BD, Rohrer MJ, Furman MI, et al. Patients with venous stasis ulceration have increased monocyte-platelet aggregation. J Vasc Surg. 1998;27(6):1109–1115; discussion 1115–1116.
- Furman MI, Barnard MR, Krueger LA, et al. Circulating monocyte-platelet aggregates are an early marker of acute myocardial infarction. J Am Coll Cardiol. 2001;38(4):1002–1006.
- Gerrard JM, Lint D, Sims PJ, et al. Identification of a platelet dense granule membrane protein that is deficient in a patient with the Hermansky-Pudlak syndrome. Blood. 1991;77(1):101–112.
- Nieuwenhuis HK, van Oosterhout JJ, Rozemuller E, et al. Studies with a monoclonal antibody against activated platelets: evidence that a secreted 53,000-molecular weight lysosome-like granule protein is exposed on the surface of activated platelets in the circulation. Blood. 1987;70(3):838–845.
- Shalev A, Michaud G, Israels SJ, et al. Quantification of a novel dense granule protein (granulophysin) in platelets of patients with dense granule storage Pool deficiency. Blood. 1992;80(5):1231–1237.
- Nishibori M, Cham B, McNicol A, et al. The protein CD63 is in platelet dense granules, is deficient in a patient with Hermansky-Pudlak syndrome, and appears identical to granulophysin. J Clin Invest. 1993;91(4):1775–1782.
- Israels SJ, McMillan-Ward EM. CD63 modulates spreading and tyrosine phosphorylation of platelets on immobilized fibrinogen. Thromb Haemost. 2005;93(2):311–318.
- Pols MS, Klumperman J. Trafficking and function of the tetraspanin CD63. Exp Cell Res. 2009;315(9):1584–1592.
- Shattil SJ, Cunningham M, Hoxie JA. Detection of activated platelets in whole blood using activation-dependent monoclonal antibodies and flow cytometry. Blood. 1987;70(1):307–315.
- Abrams CS, Ellison N, Budzynski AZ, et al. Direct detection of activated platelets and platelet-derived microparticles in humans. Blood. 1990;75(1):128–138.
- Jackson CW, Jennings LK. Heterogeneity of fibrinogen receptor expression on platelets activated in normal plasma with ADP: analysis by flow cytometry. Br J Haematol. 1989;72(3):407–414.
- Kasahara K, Takagi J, Sekiya F, et al. Analysis of distribution of receptors among platelets by flow cytometry. Thromb Res. 1987;45(6):763–770.
- Schoolmeester A, Vanhoorelbeke K, Katsutani S, et al. Monoclonal antibody IAC-1 is specific for activated alpha2beta1 and binds to amino acids 199 to 201 of the integrin alpha2 I-domain. Blood. 2004;104(2):390–396.
- Heilmann E, Hynes LA, Burstein SA, et al. Fluorescein derivatization of fibrinogen for flow cytometric analysis of fibrinogen binding to platelets. Cytometry. 1994;17(4):287–293.
- Faraday N, Goldschmidt-Clermont P, Dise K, et al. Quantitation of soluble fibrinogen binding to platelets by fluorescence-activated flow cytometry. J Lab Clin Med. 1994;123(5):728–740.
- Frelinger AL, Cohen I, Plow EF, et al. Selective inhibition of integrin function by antibodies specific for ligand-occupied receptor conformers. J Biol Chem. 1990;265(11):6346–6352.
- Yaw HP, Van Den Helm S, Linden M, et al. Whole blood flow cytometry protocol for the assessment of platelet phenotype, function, and cellular interactions. Platelets. 2021;32(6):786–793.
- Ignatova AA, Ponomarenko EA, Polokhov DM, et al. Flow cytometry for pediatric platelets. Platelets. 2019;30(4):428–437.
- Rendu F, Nurden AT, Lebret M, et al. Relationship between mepacrine-labelled dense body number, platelet capacity to accumulate 14C-5-HT and platelet density in the Bernard-Soulier and Hermansky-Pudlak syndromes. Thromb Haemost. 1979;42(2):694–704.
- Lorez HP, Da Prada M, Rendu F, et al. Mepacrine, a tool for investigating the 5-hydroxytryptamine organelles of blood platelets by fluorescence microscopy. J Lab Clin Med. 1977;89(1):200–206.
- Gordon N, Thom J, Cole C, et al. Rapid detection of hereditary and acquired platelet storage Pool deficiency by flow cytometry. Br J Haematol. 1995;89(1):117–123.
- Ramström AS, Fagerberg IH, Lindahl TL. A flow cytometric assay for the study of dense granule storage and release in human platelets. Platelets. 1999;10(2–3):153–158.
- van Asten I, Blaauwgeers M, Granneman L, et al. Flow cytometric mepacrine fluorescence can be used for the exclusion of platelet dense granule deficiency. J Thromb Haemost. 2020;18(3):706–713.
- Cai H, Mullier F, Frotscher B, et al. Usefulness of flow cytometric mepacrine uptake/release combined with CD63 assay in diagnosis of patients with suspected platelet dense granule disorder. Semin Thromb Hemost. 2016;42(3):282–291.
- The cost of sequencing a human genome. Genome.gov [cited 2021 Nov 21]. Available from: https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost
- Johnson B, Lowe GC, Futterer J, et al. Whole exome sequencing identifies genetic variants in inherited thrombocytopenia with secondary qualitative function defects. Haematologica. 2016;101(10):1170–1179.
- Johnson B, Doak R, Allsup D, et al. A comprehensive targeted next-generation sequencing panel for genetic diagnosis of patients with suspected inherited thrombocytopenia. Res Pract Thromb Haemost. 2018;2(4):640–652.
- Andersson NG, Rossing M, Fager Ferrari M, et al. Genetic screening of children with suspected inherited bleeding disorders. Haemophilia. 2020;26(2):314–324.
- Megy K, Downes K, Morel-Kopp MC, et al. GoldVariants, a resource for sharing rare genetic variants detected in bleeding, thrombotic, and platelet disorders: communication from the ISTH SSC Subcommittee on Genomics in Thrombosis and Hemostasis. J Thromb Haemost. 2021;19(10):2612–2617.
- Pecci A, Ma X, Savoia A, et al. MYH9: Structure, functions and role of non-muscle myosin IIA in human disease. Gene. 2018;664:152–167.
- Kosugi S, Momozawa Y, Liu X, et al. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019;20(1):117.
- Paterson AD, Rommens JM, Bharaj B, et al. Persons with Quebec platelet disorder have a tandem duplication of PLAU, the urokinase plasminogen activator gene. Blood. 2010;115(6):1264–1266.
- Committee on Bioethics, Committee on Genetics, American College of Medical Genetics, et al. Ethical and policy issues in genetic testing and screening of children. Pediatrics. 2013;131(3):620–622.
- Downes K, Borry P, Ericson K, et al. Clinical management, ethics and informed consent related to multi-gene panel-based high throughput sequencing testing for platelet disorders: communication from the SSC of the ISTH. J Thromb Haemost. 2020;18(10):2751–2758.
- Greinacher A, Eekels JJM. Diagnosis of hereditary platelet disorders in the era of next-generation sequencing: “primum non nocere”. J Thromb Haemost. 2019;17(3):551–554.