
Abstract
Transthyretin (TTR), a homotetrameric protein found in plasma, cerebrospinal fluid, and the eye, plays a pivotal role in the onset of several amyloid diseases with high morbidity and mortality. Protein aggregation and fibril formation by wild-type TTR and its natural more amyloidogenic variants are hallmarks of ATTRwt and ATTRv amyloidosis, respectively. The formation of soluble amyloid aggregates and the accumulation of insoluble amyloid fibrils and deposits in multiple tissues can lead to organ dysfunction and cell death. The most frequent manifestations of ATTR are polyneuropathies and cardiomyopathies. However, clinical manifestations such as carpal tunnel syndrome, leptomeningeal, and ocular amyloidosis, among several others may also occur. This review provides an up-to-date listing of all single amino-acid mutations in TTR known to date. Of approximately 220 single-point mutations, 93% are considered pathogenic. Aspartic acid is the residue mutated with the highest frequency, whereas tryptophan is highly conserved. “Hot spot” mutation regions are mainly assigned to β-strands B, C, and D. This manuscript also reviews the protein aggregation models that have been proposed for TTR amyloid fibril formation and the transient conformational states that convert native TTR into aggregation-prone molecular species. Finally, it compiles the various in vitro TTR aggregation protocols currently in use for research and drug development purposes. In short, this article reviews and discusses TTR mutagenesis and amyloidogenesis, and their implications in disease onset.
1. Introduction
Protein aggregation and amyloid formation contribute to several debilitating diseases collectively known as Amyloidosis [Citation1]. To date, more than fifty amyloid diseases have been identified, including localized amyloidosis found in neurodegenerative conditions like Alzheimer’s and Parkinson’s diseases, and systemic amyloidosis such as transthyretin amyloidosis and lysozyme amyloidosis [Citation1]. These pathologies result from mutations, post-translational modifications, or partial proteolysis, and by abnormal folding or unfolding events affecting approximately fifty different peptides/proteins. These end up adopting non-native, misfolded conformations prone to aggregate into highly ordered soluble oligomers and insoluble fibrils with a characteristic cross-β structure – the amyloid substance. Generally, amyloid diseases are not a consequence of the loss of function of the native protein but result from the cytotoxic nature of the amyloid aggregates and/or the action of amyloid fibrils on inter-cellular communication and tissue physiology. Although most amyloids are found extracellularly, amyloid-like deposits are also found inside cells [Citation1].
Transthyretin amyloid disorders include sporadic age-related wild-type TTR amyloidosis (ATTRwt), hereditary TTR amyloidosis polyneuropathy (ATTRv-PN), hereditary TTR amyloidosis cardiomyopathy (ATTRv-CM), hereditary leptomeningeal TTR amyloidosis (ATTRv-LM) and hereditary ocular TTR amyloidosis (ATTRv-OC). Although some of the ATTR clinical manifestations have unmet medical needs, in the last decade several disease-modifying therapies have contributed to slowing down disease progression and, in some cases, have ameliorated disease symptoms. The continuing efforts to better understand the molecular mechanisms of disease progression and tissue specificity are critical for the rational development of new and improved therapies for the treatment of TTR amyloidoses.
2. Transthyretin structure and function
2.1. From prealbumin to transthyretin (TTR)
In 1942, a new protein was identified and named “prealbumin” due to its electrophoretic mobility just ahead of serum albumin, both in plasma [Citation2] and cerebrospinal fluid (CSF) [Citation3]. Later, in 1958, prealbumin was found to bind thyroid hormones, being therefore renamed as “thyroxine-binding prealbumin” [Citation4]. In 1969, additional studies showed that thyroxine-binding prealbumin could also bind the retinol-binding protein (RBP) [Citation5]. The International Union of Biochemists then coined the name “transthyretin” (TTR) in 1981 [Citation6] due to its ability to transport thyroid hormones (THs), especially thyroxine (T4), as well as retinol (vitamin A) in association with RBP.
2.2. Transthyretin biosynthesis, tissue concentration, and metabolism
TTR is a globular homotetrameric protein, with a total molecular mass of approximately 55 kDa, consisting of four monomers with an identical sequence of 127 amino acids (). In humans, TTR is mainly found in plasma [Citation7], CSF [Citation8–11], and the eye [Citation12–16]. Serum TTR accounts for nearly 90% of TTR in the human organism and is secreted by the liver to concentrations (as tetramers) varying from 0.17 to 0.42 mg/mL (3.1 to 7.6 μM) [Citation7].
Figure 1. Three-dimensional structure representation of the native TTRwt homotetramer. The structure is composed of four identical subunits (each represented in a different color) forming a central channel able to accommodate two thyroxine (T4) molecules (depicted in a ball-and-stick representation). The image was produced with UCSF Chimera [Citation19] and coordinates of the crystallographic structure of human TTRwt in a complex with T4 (PDB code: 1ICT).
![Figure 1. Three-dimensional structure representation of the native TTRwt homotetramer. The structure is composed of four identical subunits (each represented in a different color) forming a central channel able to accommodate two thyroxine (T4) molecules (depicted in a ball-and-stick representation). The image was produced with UCSF Chimera [Citation19] and coordinates of the crystallographic structure of human TTRwt in a complex with T4 (PDB code: 1ICT).](/cms/asset/fa6f2807-0d07-41bf-8acc-2ba3e66bc8e1/ilab_a_2350379_f0001_c.jpg)
In CSF, TTR has been reported as one of the most abundant proteins, along with serum albumin, prostaglandin-D synthase, and immunoglobulins [Citation8]. Since serum TTR does not cross the blood–brain barrier (BBB) to any significant extent, a different source of production apart from the liver must account for the protein in the CSF at a concentration range between 0.005 to 0.02 mg/mL (0.09 to 0.36 μM) [Citation11]. Indeed, TTR synthesis has been reported to occur in the epithelial cells of the choroid plexus [Citation9,Citation10]. Expression of the TTR gene was also found in Schwann cells, dorsal root ganglia [Citation17], and cortical and hippocampal neurons in response to amyloid-β (Aβ)-induced stress, in patients with Alzheimer’s disease (AD) [Citation18].
TTR is also found in the eye [Citation12–14]. Since the Bruch membrane that encircles more than half of the eye is a barrier to protein crossing, intraocular TTR synthesis is required and has been reported in the retinal pigment epithelium (RPE), the pigmented epithelium of the ciliary body, corneal endothelium, and the optic nerve fiber layer of the retina [Citation12–14]. The concentration of TTR in the aqueous humor is approximately 1.3 µg/mL (0.024 μM) [Citation15], while in the vitreous humor is nearly 18 µg/mL (0.327 μM) [Citation16].
TTR biosynthesis has also been described in several other tissues to a smaller extent, namely in the heart, skeletal muscle, spleen [Citation20], visceral yolk sac endoderm [Citation21], trophoblasts of placenta [Citation22], gastric ghrelin cells [Citation23], pineal gland [Citation24], meninges [Citation25] and pancreatic α-cells [Citation26,Citation27].
Furthermore, TTR serum levels are slightly higher in adulthood than in childhood, higher in men than in women, and start to decline after 40 years of age [Citation28]. The half-life of serum TTR is approximately 2 to 3 days in humans [Citation29,Citation30]. The main sites for TTR degradation are the liver (36–38%), muscle (12–15%), and skin (8–10%), along with other minor sites, such as the kidney, adipose tissue, testicles, and gastrointestinal tract (1–8%), and other tissues in less than 1% [Citation31]. Serum levels of TTR are routinely measured as an indicator of health status, since TTR is a typical negative acute-phase serum protein [Citation32]. TTR serum levels are reduced in conditions such as trauma, surgery, inflammation, bacterial infection, protein malnutrition, coronary artery disease, depression, Alzheimer’s disease, gestational diabetes mellitus, and Down’s syndrome [Citation33–39].
2.3. Transport of thyroid hormones and retinol
The term “transthyretin” describes the dual physiological role of the protein in the transport of thyroid hormones (THs) and retinol [Citation6]. Although binding with higher affinity to T4 (thyroxine or 3,5,3′,5′-tetraiodo-L-thyronine) than to T3 (triiodothyronine or 3,5,3′-triodo-L-thyronine), TTR is able to transport both THs. Thyroid hormones show a common structure consisting of a hydrophobic thyronine nucleus, which accounts for their poor solubility in water, a hydrophilic hydroxyl group attached to the phenolic ring, and three iodine atoms in positions 3, 5, and 3′ in the case of T3, and four iodine atoms in positions 3, 5, 3′ and 5′ in the case of T4. In human plasma, 99.97% of T4 and 99.70% of T3 are bound to TH distributor proteins (THDPs), namely human serum albumin (HSA), TTR, and thyroxine-binding globulin (TBG) [Citation40,Citation41]. THDPs circulate at different concentrations and show distinct dissociation constants (Kd) and affinity for THs [Citation42]. TBG, a monomeric 54 kDa protein with a single binding site, has the highest affinity for T4 and T3 with Kd of 0.1 and 2 nM, respectively. Due to its high binding affinity and despite its low plasma concentration of 0.015 mg/mL, TBG binds approximately 75% of both T4 and T3 in plasma. TTR has two binding sites with the highest affinity binding event with Kd values of 14 nM for T4 and 57 nM for T3 [Citation43]. Binding to a second thyronine molecule occurs with a significantly lower affinity, through negative cooperativity [Citation44]. Present in plasma at a concentration between 0.17 and 0.42 mg/mL, TTR binds nearly 15% of T4 and less than 5% of T3. Conversely, HSA, a monomeric 66.5 kDa protein with various binding sites, has the lowest affinity for THs with dissociation constants of 1.4 µM for T4 and 100 µM for T3. Due to its low binding affinity and despite its high serum concentration of 35-50 mg/mL, HSA binds less than 5% of T4 and less than 20% of T3. Additionally, a small fraction of THs is also distributed in the bloodstream by lipoproteins, including ApoB100, via interaction with its cell surface receptor, i.e. the low-density lipoprotein (LDL) receptor [Citation45,Citation46]. TTR is the only TH transport protein synthesized in the choroid plexus and, therefore, plays a significant role in transporting THs in the CSF and distributing THs in the brain [Citation47].
The transport of retinol is mediated by RBP [Citation48]. RBP circulates in the plasma bound to TTR. Both apo- (without) and holo- (with retinol) RBPs form the RBP-TTR complex with a stoichiometry of 2:1, with approximately 97 kDa [Citation49,Citation50]. However, the Kd of holo-RBP with TTR is significantly lower than with apo-RBP [Citation51], which is consistent with a retinol delivery mechanism where the stable holo-RBP-TTR complex is retained in the plasma, while the unbound apo-RBP, with lower molecular weight (21 kDa) is cleared by glomerular filtration [Citation52]. The binding sites for RBP are located at the surface of TTR, and each TTR molecule has four RBP-binding sites, two in each dimer. However, due to steric hindrance, only two RBP molecules bind simultaneously to TTR [Citation49]. RBP and TTR contribute 21 amino acids each to the protein–protein recognition interface, with most of these residues located in the C-terminal regions of the two proteins [Citation50]. Affinity measurements of TTR to RBP estimate a Kd of 150 to 190 nM for the first RBP molecule and 35 µM for the second RBP, indicating negative cooperativity [Citation53,Citation54]. Ratios of RBP:TTR in plasma are around 0.3 in healthy individuals [Citation55,Citation56] indicating that most of the circulating TTR remains free of RBP.
3. Transthyretin amyloidoses
3.1. Transthyretin amyloid diseases, symptoms, and geographic distribution
Transthyretin amyloidosis (ATTR) is manifested in sporadic and hereditary forms: wild-type TTR amyloidosis (ATTRwt) and hereditary TTR amyloidosis (ATTRv). In the case of hereditary TTR amyloidosis, polyneuropathy (ATTRv-PN) and cardiomyopathy (ATTRv-CM) are the most common clinical manifestations. While ATTRv-PN mostly affects the peripheral nervous system (PNS), ATTRv-CM mainly targets the heart. Additionally, other forms of ATTRv such as leptomeningeal amyloidosis (ATTRv-LM) and ocular amyloidosis (ATTRv-OC) affect the central nervous system (CNS) and the vitreous body of the eye, respectively. Nonetheless, some patients simultaneously exhibit multiple symptoms associated with distinct manifestations of ATTRv. That is the case of hereditary TTR amyloidosis polyneuropathy with muscle, vitreous, leptomeningeal, and cardiac involvement [Citation57–59].
In contrast, ATTRwt, formerly known as senile systemic amyloidosis, is a non-hereditary age-related systemic amyloidosis caused by TTRwt. The main features of each type of ATTR are described in .
Table 1. Summary of human transthyretin-associated amyloidoses.
In an effort to better understand the natural history and phenotypic heterogeneity of TTR amyloidosis, as well as improving diagnosis and treatment, the Transthyretin Amyloidosis Outcomes Survey (THAOS), the largest global, longitudinal, observational survey of patients with ATTR amyloidosis (inherited and wild-type), as well as of asymptomatic TTR gene carriers, has been established and formally ongoing since 2007 [Citation60].
3.2. Transthyretin amyloidogenic and non-amyloidogenic variants
Hereditary TTR amyloidoses (ATTRv) are fatal diseases triggered by TTR variants that have been classified as “amyloidogenic” TTR mutants. ATTRv amyloidoses are autosomal-dominant diseases, where only one amyloidogenic variant TTR allele is required to develop pathology. Most affected individuals are heterozygous for a pathogenic mutation and express both normal (TTRwt) and variant TTR (TTRv), but some homozygous patients have also been identified [Citation86,Citation87]. The majority of the mutations result from a single nucleotide substitution in the TTR gene [Citation88]. Transthyretin has been identified as one of the most mutated human proteins leading to the development of amyloid diseases [Citation89].
In 1952, the pioneering work of Corino de Andrade produced the first medical reports on the diagnosis of ATTRVal30Met amyloidosis among a group of families, from the north of Portugal [Citation90], conducing to the discovery of other foci in Japan (1968) and Sweden (1976) [Citation91, Citation92]. Until two decades ago, ATTRv-PN was thought to be an endemic disease restricted to those areas, but today is known to occur worldwide.
The human TTR gene is localized in chromosome 18 at position 12.1 (18q12.1) [Citation93], spanning approximately 6.9 kb (GenBank accession number: NG_009490.1, and Gene ID: 7276). The TTR gene is divided into four exons and three introns. Exon 1 encodes 23 amino acid residues, that is translated into 20 residues of a signal peptide and the first three residues of the mature protein. Exon 2 codes for amino acid residues 4 to 47, exon 3 for residues 48 to 92, and exon 4 for residues 93 to 127 [Citation94]. Hence, the amino acid numbering used throughout this manuscript to identify the various mutations in the protein sequence refers to the 127 residues that compose the mature TTR monomer (e.g.: Val30Met or V30M). Nonetheless, another counting system that includes the 20-amino acid peptide signal (labeled as p.) is also used elsewhere (e.g.: p.Val50Met or p.V50M).
lists the 216 point mutations identified in the TTR gene so far, by depicting their position in the 127 amino acid polypeptide chain of the TTR monomer [Citation80,Citation95–119]. includes all TTR mutations (amyloidogenic and non-amyloidogenic) reported in the open access page “Mutations in Hereditary Amyloidosis” (amyloidosismutations.com) [Citation95], in the Human Genetic Mutation database (hgmd.cf.ac.uk) ([Citation118]), in the ClinVar public archive of reports of human genetic variants (ncbi.nlm.nih.gov/clinvar) [Citation119], along with other TTR mutations that have been documented in the literature through scientific papers and reports available at PubMed Central (pubmed.com). Commonly identified variants are well documented and have proper information on their clinical phenotype and natural history. However, regarding rare variants, it is difficult to obtain information on their amyloidogenic potential and clinical significance due to a lack of family history, reduced penetrance, and other factors. Most mutations result in single amino acid substitutions distributed across the polypeptide chain, except for the Val122_del deletion mutation [Citation120], the Met13_dup and Glu51-Ser52_dup duplication mutations [Citation121,Citation122], and the synonymous mutations Ala108Ala and Thr119Thr [Citation123]. Double mutations have also been reported over time. lists some of these TTR double mutations found in ATTRv patients.
Figure 2. Amino-acid sequence derived from the human 127-residue TTR mature protein showing the position of 216 mutations formally identified. Non-amyloidogenic mutations are displayed in green, while aggressive amyloidogenic mutations are colored in red. Duplication mutations are in orange, and a deletion mutation is in blue. Elements of secondary structure are displayed according to the TTRwt crystallographic structure, at 1.15 Å resolution (PDB code: 8AWI), with the β-strands highlighted in light blue, the α-helix in light red, and loops and turns in black.
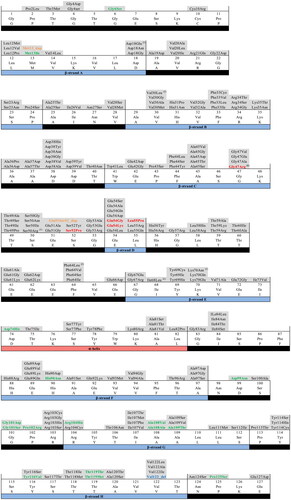
Table 2. TTR double mutations found in ATTRv patients and their protein sequence localization, according to the 127-residue numbering of the mature protein. These may occur in the same or different gene alleles.
A 7-residue length “window analysis” was employed here to scan the more than 200 TTR variants identified, as applied previously by other authors in 1996 to less than 50 TTR mutations [Citation130]. This 7-window “view” helps to highlight “hot spot” regions, that would otherwise be masked when using the simple “mutations per residue” view (also shown in by the solid grey graph). A sliding window of 7-residues in length was moved from the N-terminus to the C-terminus in steps of one residue. The total number of mutations found in each window was plotted against the number of the midpoint-residue of each interval (). All variants were treated independently so that multiple variants at a single site were considered separately when determining the number of variants within the scanning window. A random distribution of TTR mutations would be expected to produce an approximately linear horizontal plot along the polypeptide chain, with 11.7 residues at each window position with a standard deviation of ± 5.1. However, the observed distribution is quite different from a random distribution.
Figure 3. Naturally occurring mutation "hot spot" zones of the 127-amino acid human transthyretin subunit mature sequence. (A) Plot of the TTR mutation frequency along the polypeptide chain. A graphical record of the total number of mutations per amino acid is shown in solid grey. In addition, a graph representing a sliding window of 7-residues in length that moves along the sequence plotting the total number of mutations (solid circles and black trace) of each interval against the midpoint residue of the interval is also shown [Citation130]. Most aggressive amyloidogenic (red circles) and non-amyloidogenic (green circles) variants are identified. Regions with a high frequency of mutations are numbered from 1 to 7 (in blue). The duplication mutations (Met13_dup and Glu51-Ser52_dup) and the deletion mutation (Val122_del) were not considered in the analysis. Elements of secondary structure are displayed at the top of the figure: β-strands (blue) and α-helix (light red). (B) Three-dimensional structure representation of the TTR subunit backbone, with the elements of secondary structure (β-strands A to H) colored according to the number of mutations known per sequence position: “no substitutions” (grey); one (yellow); two (orange); three (red); four (dark red); and five or more (black). The image was produced with UCSF Chimera [Citation19], using the coordinates of the TTRwt crystallographic structure at 1.15 Å resolution (PDB code: 8AWI).
![Figure 3. Naturally occurring mutation "hot spot" zones of the 127-amino acid human transthyretin subunit mature sequence. (A) Plot of the TTR mutation frequency along the polypeptide chain. A graphical record of the total number of mutations per amino acid is shown in solid grey. In addition, a graph representing a sliding window of 7-residues in length that moves along the sequence plotting the total number of mutations (solid circles and black trace) of each interval against the midpoint residue of the interval is also shown [Citation130]. Most aggressive amyloidogenic (red circles) and non-amyloidogenic (green circles) variants are identified. Regions with a high frequency of mutations are numbered from 1 to 7 (in blue). The duplication mutations (Met13_dup and Glu51-Ser52_dup) and the deletion mutation (Val122_del) were not considered in the analysis. Elements of secondary structure are displayed at the top of the figure: β-strands (blue) and α-helix (light red). (B) Three-dimensional structure representation of the TTR subunit backbone, with the elements of secondary structure (β-strands A to H) colored according to the number of mutations known per sequence position: “no substitutions” (grey); one (yellow); two (orange); three (red); four (dark red); and five or more (black). The image was produced with UCSF Chimera [Citation19], using the coordinates of the TTRwt crystallographic structure at 1.15 Å resolution (PDB code: 8AWI).](/cms/asset/9a645ffc-342e-4883-8d03-601676ca8dff/ilab_a_2350379_f0003_c.jpg)
According to , there are two major peaks of mutation frequency, one centered at residue 52 and another centered around residues 33-36, that also define two major mutation “hot spot” regions, Region 1 and 2, formed by residues 44-59 and 26-43, respectively. Region 1 computed 27 substitutions in the 7-residue window, with a height of 3.0 standard deviations above the mean. Both of these regions map the mutation “hot spot” zone of TTR, mainly assigned to β-strand D and CD and DE loops in the case of Region 1, and to β-strand B, which is parallel and hydrogen bonded to β-strand C, the second half of β-strand C and the BC loop, for Region 2. Five more subsidiary regions can also be observed in : Region 3, composed of residues 60-76 assigned to the DE loop, β-strand E, and to the α-helix; Region 4, represented by residues 97-112 located in β-strand G, GH, and FG loops; Region 5, formed by residues 77-96 which corresponds to β-strand F and the αF loop; Region 6, with residues 113-125, corresponding to β-strand H and the C-terminus; and Region 7, allocated to residues 14-25 of the AB loop. The protein regions involved in dimer-dimer interactions include loops AB and GH which are part of Regions 4 and 7, and the segments involved in monomer-monomer interactions like β-strands H and F, located in Regions 5 and 6, can be seen in . Most of the mutations found in Region 1 were the first mutations to be identified in the TTR gene [Citation131]. summarizes the Regions identified, their amino-acid residues, localization in the polypeptide chain, exons involved, and number of mutations.
Table 3. Regions of TTR high mutation frequency, and their characterization concerning residue number, structure localization, exon localization, and number of mutations per region along the 127-amino acid mature sequence of human TTR. Duplication mutations (Met13_dup and Glu51-Ser52_dup) and the deletion mutation (Val122_del) were not considered in the analysis.
Another interesting observation, that can also be inferred from , is that the mutations classified as the most aggressive amyloidogenic TTR mutations (causing disease with an early age of onset, with multiple organ involvement and severe impairment), namely Gly47Arg [Citation132], Ser52Pro [Citation133], Glu54Gly [Citation134], Glu54Lys [Citation135] and Leu55Pro [Citation136] are all located in the Region 1 mutation “hot spot” (residues 44-59); whereas the TTR non-amyloidogenic mutations [Citation96,Citation99] (Gly6Ser, Met13Ile, Asp74His, His90Asn, Asp99Asn, Gly101Ser, Gly101Asp, Pro102Arg, Arg104His, Ala108Ala, Ala108Val, Ala109Thr, Tyr116Val, Thr119Thr, Thr119Met, and Pro125Ser) are preferentially located in Regions 4 and 6 (residues 97 to 125). In addition, some parts of the polypeptide chain present a low frequency of mutations, as is the case of the N-terminus (11 residues and 6 mutations), and the C-terminus (5 residues and 3 mutations). Exon 3 (residue 48 to 92) is the most mutated exon with 87 mutations, followed by exon 2 (residue 4 to 47) with 77 mutations, exon 4 (residue 93 to 127) with 50 mutations, and exon 1 (residue 1 to 3) with 2 mutations. The regions with the highest and the lowest mutation frequencies stand out when the 3D backbone structure of the TTR subunit is color-mapped (), revealing major “hot spots” at β-strands B, C, and D, as shown in red, dark red, and black.
A complete understanding of the effects of each point mutation on the overall structure and stability of the TTR monomers and tetramer is essential to unravel sequence–structure relationships. The group of mutations found to either stabilize or destabilize the protein varies according to the nature of the substituted amino acid. Some mutations affect crucial interactions within the protein, which in turn lead to misfolding and aggregation (amyloidogenic variants), while others can be innocuous (non-amyloidogenic or protective variants).
According to , aspartic acid is the most frequently replaced/mutated residue of the protein sequence, followed by arginine, isoleucine, phenylalanine, and alanine. TTRwt contains 5 aspartic acids (Asp) in its sequence (Asp18, Asp38, Asp39, Asp74, and Asp99) which have all been associated with one or more mutations per residue (Asp18Asn, Asp18Glu (2×), Asp18Gly, Asp38His, Asp38Tyr, Asp38Asn, Asp38Gly, Asp38Val, Asp38Ala, Asp39Tyr, Asp39Val, Asp74His, and Asp99Asn − 14 in total). In opposition, other residues show high conservation propensities, like tryptophan, asparagine, proline, and lysine. On the other hand, the residues that most frequently replaces the original residues present in TTRwt are serine, arginine, and alanine; a fact that directly correlates with the high number of RNA codons coding for these residues in the genetic code. Serine substitutes several different residues along the protein sequence and is present in 25 TTR mutants (Gly4Ser, Gly6Ser, Pro24Ser, Ala25Ser, Asn27Ser, Val28Ser, Arg34Ser, Pro43Ser, Phe44Ser, Ala45Ser, Thr49Ser, Glu54Ser, Phe64Ser, Ala81Ser, Ile84Ser, Ala91Ser, Ala97Ser, Gly101Ser, Arg103Ser, Ala109Ser, Tyr114Ser, Tyr116Ser, Ala120Ser, Asn124Ser, and Pro125Ser) which have largely been identified as amyloidogenic (exceptions are Gly6Ser, Gly101Ser, and Pro125Ser). Inversely, the residue which less frequently replace mutated residues is tryptophan (one single RNA codon), followed by phenylalanine, cysteine, and glutamine, with only two RNA codons available in the genetic code [Citation137]. TTRwt contains 2 tryptophan residues (Trp) in its sequence (Trp41 and Trp79), but only one mutation associated with Trp41 has been identified (the amyloidogenic mutation Trp41Leu).
Figure 4. TTR mutation map. TTRwt residues are represented in green (wt) and mutations in red (mut). The scheme depicts the type of amino acid (AA), number and location of known naturally occurring mutations in the mature 127-residue sequence of hTTR, as well as: i) AA prevalence in hTTRwt (A); ii) the number of mutations a given AA suffers (B) (e.g. Asp is the amino acid that suffers most mutations, in fact, all Asp residues in the sequence have suffered mutations); iii) number of times a certain AA is introduced by mutation (C) (e.g. Ser is the AA most often introduced by mutation, being involved in a total of 25 mutations, so far, mostly amyloidogenic); iv) respective mutation ratio relative to the prevalence of each AA in hTTRwt (D = B/A); and v) mutation ratio of the introduction of a given AA per 20 AA (E = C/20). The duplication mutations (Met13_dup and Glu51-Ser52_dup) and the deletion mutation (Val122_del) were not included in the diagram. N/A – non-applicable.

Additionally, even if all amyloidogenic, different pathogenic TTR mutations may produce different phenotypes, with different clinical manifestations and targeted organs, either leading to polyneuropathy, cardiomyopathy, vitreous opacities, carpal tunnel syndrome, CNS dysfunction, or leptomeningeal involvement. That is the case of residue in position 114, with 3 mutations identified (). While Tyr114Cys conduces to ATTRv-PN [Citation138], Tyr114Ser causes ATTRv-CM [Citation139], and Tyr114His is implicated in hereditary carpal tunnel syndrome ATTRv [Citation140]. On the other hand, in the case of residue in position 45 all five known amyloidogenic variants specifically target the heart: Ala45Val, Ala45Ser, Ala45Thr, Ala45Gly, and Ala45Asp [Citation111,Citation141–144]. Similarly, mutation of residue in position 47 also leads to a unique phenotype, in this case ATTRv-PN, as observed in Gly47Arg, Gly47Ala, Gly47Glu, and Gly47Val [Citation145–148].
Mutations of the TTR gene have already been associated with more than 200 pathogenic variants (), and the reason why different mutations produce distinct clinical manifestations and different onset ages is still under investigation. Even when multiple symptoms are present in patients carrying the same mutation, clinical phenotypes do not always coincide, and the same mutation may be associated with different symptoms, their severity, and age of onset, even within the same kindred [Citation149]. A clear example is the distinct pathological phenotype observed in Val30Met TTR carriers, with three main endemic foci in Portugal, Sweden, and Japan. Val30Met Portuguese and Japanese kindreds generally experience disease early onsets (below age 50, normally 30-40), severe disease, and high disease penetrance [Citation150,Citation151], while Val30Met Swedish families often present disease late onsets (above age 50, usually 60), intermediate disease severity, and low disease penetrance [Citation152]. Nevertheless, beyond these endemic foci, other unrelated Val30Met TTR carriers have also been identified worldwide, where late-onset cases have found to be more prevalent and widely spread [Citation153]. This clinical heterogeneity seems to be strongly correlated with the amyloid fibril composition found in the amyloid deposits of ATTRv patients. The early onset of the disease is associated with deposits containing only full-length TTR (type B fibrils), whereas the late onset of the disease is related to deposits with a mixture of full-length TTR and large amounts of C-terminal TTR fragments (type A fibrils) [Citation154, Citation155] and composed by a combination of amyloidogenic TTRv and TTRwt [Citation156]. Another example of heterogeneous disease onset is the case of the Thr60Ala TTR variant that has been associated with early and late disease onsets (23 and 70 years old women, respectively) [Citation157,Citation158], causing simultaneously severe restrictive ATTRv-CM and ATTRv-PN. These observations indicate that the basis of the amyloid phenotype expression is not only due to a certain disease-triggering mutation, but also the amyloid fibril composition and type and eventually other genetic and environmental factors [Citation73,Citation159–161]. Recent studies showed that non-coding regions and the variation of the mutations in the TTR gene may also influence the phenotypic heterogeneity of ATTRv [Citation162–169].
3.3. Transthyretin structural fluctuations that promote fibrillogenesis
TTR amyloidogenesis is initiated by the dissociation of the native tetramer into monomers that undergo conformational fluctuations to partially unfolded intermediates, which self-assemble into soluble oligomers and amyloid fibrils [Citation170]. Analysis of X-ray structures of tetrameric TTRwt and several of its natural variants revealed that both amyloidogenic and non-amyloidogenic mutations induce only minor changes in the overall tridimensional structure of the protein [Citation171,Citation172]. Hence, single amino-acid substitutions must affect the conformational stability and folding/unfolding transitions of the protein rather than its overall structure [Citation173]. The characterization of transient states populating the conformational ensemble between the natively folded functional TTR and aggregation-prone forms is of extreme relevance for understanding fibrillogenesis. The conformational changes identified as “amyloidogenic”, both by experimental and computational studies, point to structural perturbations occurring at different locations: the α-helix and adjacent EF loop [Citation174–177]; displacement of β-strands F, G, and H [Citation178]; displacement of β-strands C and D from the β-sheet [Citation179–184]; destabilization of the CBEF β-sheet [Citation180]; destabilization of the CD loop [Citation185] and FG loop [Citation186]; proteolytic cleavage of Lys48 − Thr49 peptide bond in the CD loop [Citation187]; destabilization of the DAGH β-sheet [Citation179,Citation188–190]; and cooperative conformational transitions to α-sheet of both TTR β-sheets [Citation191,Citation192]. Depending on the localization in the polypeptide chain and eventually the type of amino acid involved in the TTR amyloidogenic mutation, each one of these conformational changes may be related to local structural perturbations or partial unfolding, decreasing the conformational stability of the overall molecule, and accelerating tetramer dissociation and subsequent monomer aggregation into oligomers and amyloid fibrils.
4. Transthyretin aggregation
4.1. Transthyretin aggregation models
Understanding the mechanism of TTR fibrillogenesis is critical for the rational design of therapeutic approaches aimed at retarding, preventing, and/or reverting amyloid formation and fibril deposition. Attempts to unravel the molecular mechanisms of TTR amyloidogenesis led to many experimental and theoretical studies over the years, and several models have emerged. The various models focused on the explanation of different aspects of the aggregation pathway, given the experimental conditions used, and contributed to the overall understanding of the process, complementing each other. describes the main features underlying some of the models used to describe TTR aggregation in the past decades. Most models proposed the existence of a conformationally unstable intermediate that acquires conformational stability through aggregation, in the pathway to amyloid fibril formation [Citation193,Citation194]. Different starting oligomeric states have also been proposed over the years, from tetramers to dimers and monomers.
Table 4. Transthyretin aggregation models proposed over the years and their limitations.
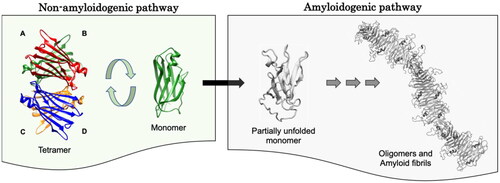
In the last two decades, a consensus has developed in the field that, in most instances, the molecular species that initiates the aggregation cascade is monomeric in nature ( and ). This implies tetramer dissociation to a monomeric intermediate as the first step of TTR fibril formation, followed by partial unfolding of the monomeric species and aggregation into amyloid. Since tetramer dissociation is the first step toward TTR amyloid formation and the rate-limiting step for fibrillogenesis [Citation208], it is paramount to characterize the dissociation pathway. The quaternary structure of TTR contains two distinct dimer-dimer interfaces – AB/CD and AC/BD, interfaces between transversal and longitudinal dimers, respectively ( and ). Several pathways may be envisaged for the dissociation of a homotetrameric protein. However, the dissociation mechanism reported for TTR consists of an initial AB/CD dimer-dimer scission followed by a rapid dissociation of dimers into monomers [Citation208]. This dissociation mechanism also rationalizes and points toward the relevance of stabilizing the tetrameric conformation by small molecule binding to the natural thyroxine binding sites which are located in the AB/CD dimer-dimer interface, according to one of the therapeutic approaches currently available to ATTR patients [Citation209]. Conversely, the refolding mechanism from unfolded monomers to native tetramers comprises a single intermediate with monomeric characteristics [Citation210].
Figure 5. Transthyretin amyloid cascade. The tetrameric native form of TTR undergoes dissociation to a non-native monomer which upon partial unfolding and self-aggregation forms prefibrillar species, such as soluble oligomers and, eventually, mature amyloid fibrils.
4.2. Transthyretin aggregation protocols
Several studies on the aggregation pathway of TTR have demonstrated that amyloid fibril formation is preceded by the dissociation of the native tetramer into non-native monomers that self-assemble into non-fibrillar cytotoxic oligomers, which are prone to form protofibrils and elongate into mature amyloid fibrils. lists the various TTR aggregation protocols reported in the literature. These are not only useful to study aggregation mechanisms but also to screen for potential aggregation inhibitors [Citation211,Citation212] and modulators [Citation213,Citation214]. The experimental conditions of these aggregation protocols vary widely with some making use of proteolytic cleavage, organic solvents, and temperature, while others submit the protein to sample aging, high pressure, low pH, changes in ionic strength, or use a combination of these destabilizing effects. TTR aggregation species formed in vitro have many morphological and tinctorial properties common to amyloid material found in vivo, and may therefore be used to study TTR fibrillation leading to ATTR amyloidosis [Citation215]. Depending on the goals of a particular study, different experimental conditions and protocols may be used, but it must be kept in mind that the kinetics of the processes and the structural and oligomeric features of the intermediate and final species vary. In particular, it must be stressed that the coexistence of different relative amounts of well-structured amyloid aggregates and amorphous aggregates varies significantly from condition to condition and protocol to protocol.
Table 5. Transthyretin aggregation protocols.
5. Conclusions
Transthyretin (TTR) is a transport protein for thyroid hormones (THs) and retinol in association with the retinol-binding protein (RBP). Nevertheless, TTR may exhibit additional biological functions albeit less known. More than 200 TTR natural variants have already been identified and most of them are pathogenic (∼93%). Hereditary transthyretin amyloidosis polyneuropathy (ATTRv-PN) and hereditary transthyretin amyloidosis cardiomyopathy (ATTRv-CM) are the most common clinical manifestations of hereditary ATTR. Additionally, wild-type TTR also forms amyloid leading to ATTRwt, a mainly age-related progressive cardiomyopathy with poor prognosis and characterized by heart failure. Among human proteins, TTR has a high rate of mutations leading to amyloid fibril formation. An average of 1.7 point mutations (216 mutations in total) have been identified per residue of the TTR polypeptide chain subunit (with 127 residues). TTR amyloid fibrils are highly stable β-structured protein aggregates. Different pathogenic TTR mutations can produce different phenotypes, with different clinical manifestations and targeted organs, either leading to polyneuropathy, cardiomyopathy, vitreous opacities, carpal tunnel syndrome, CNS dysfunction, or leptomeningeal involvement. Genotype–phenotype correlations are not only due to specific triggering mutations but distinct pathological phenotypes within different populations are observed for the same TTR mutated genotype. Most mutations result in single amino acid substitutions. “Hot spot” mutation regions are largely assigned to β-strands B, C, and D. The most aggressive amyloidogenic mutations are mainly localized in the 44-59 segment of the protein sequence, whereas non-amyloidogenic mutations are mostly located in the 97-125 segment. Aspartic acid is the most mutated residue, followed by arginine, isoleucine, phenylalanine, and alanine. Tryptophan, asparagine, proline, and lysine residues show high conservation propensities along the protein sequence.
TTR fibrillogenesis occurs upon tetramer dissociation and partial unfolding to non-native monomers that undergo self-assembly into cytotoxic soluble oligomers and then into mature amyloid fibril deposits. Monomeric TTR species have been identified as crucial intermediates in amyloid fibril formation, either by wild-type TTR (TTRwt) or by other amyloidogenic TTR variants (TTRv). TTR fibrillogenesis can be rapidly and efficiently reproduced in vitro through various experimental protocols taking advantage of different experimental conditions using proteolytic cleavage, organic solvents, high temperature, sample aging, high pressure, low pH, changes in ionic strength, or a combination of these protein destabilizing effects. These different experimental conditions, however, produce different oligomeric species, and in particular, different proportions of amyloid and amorphous aggregates, which must be considered when studying fundamental processes in protein aggregation and amyloidogenesis and when screening for amyloid inhibitors and modulators in drug discovery and development.
| Abbreviations | ||
| AD | = | Alzheimer’s disease |
| ATTR | = | TTR amyloidosis |
| ATTRwt | = | wild-type TTR amyloidosis |
| ATTRv | = | hereditary TTR amyloidosis |
| ATTRv-PN | = | hereditary TTR amyloidosis with polyneuropathy |
| ATTRv-CM | = | hereditary TTR amyloidosis with cardiomyopathy |
| ATTRv-LM | = | hereditary leptomeningeal TTR amyloidosis |
| ATTRv-OC | = | hereditary ocular TTR amyloidosis |
| BBB | = | blood–brain barrier |
| CNS | = | central nervous system |
| CR | = | Congo red |
| CSF | = | cerebrospinal fluid |
| DCVJ | = | (9-(2,2-dicyanovinyl)julolidine) |
| DMSO | = | dimethyl sulfoxide |
| EDTA | = | ethylenediaminetetraacetic acid |
| HSA | = | human serum albumin |
| hATTR | = | hereditary TTR amyloidosis |
| HTS | = | high throughput screening |
| hTTR | = | human TTR |
| LDL | = | low-density lipoprotein |
| PBS | = | phosphate-buffered saline |
| PDB | = | Protein Data Bank |
| PNS | = | peripheral nervous system |
| RBP | = | retinol-binding protein |
| RPE | = | retinal pigment epithelium |
| SDS | = | sodium dodecyl sulfate |
| T3 | = | triiodothyronine |
| T4 | = | thyroxine |
| TBG | = | thyroxine-binding globulin |
| TFE | = | 2,2,2-trifluoroethanol |
| THAOS | = | Transthyretin Amyloidosis Outcomes Survey |
| THDPs | = | thyroid hormone distributor proteins |
| THs | = | thyroid hormones |
| ThT | = | thioflavin-T |
| TTR | = | transthyretin |
| wt | = | wild-type |
Disclosure statement
The authors report there are no competing interests to declare.
Additional information
Funding
References
- Almeida ZL, Brito RMM. Structure and aggregation mechanisms in amyloids. Molecules. 2020;25(5):1195. doi:10.3390/molecules25051195.
- Seibert FB, Nelson JW. Electrophoretic study of the blood protein response in tuberculosis. J Biol Chem. 1942;143(1):29–38. doi:10.1016/s0021-9258(18)72655-0.
- Kabat EA, Moore DH, Landow H. An electrophoretic study of the protein components in cerebrospinal fluid and their relationship to the serum proteins 1. J Clin Invest. 1942;21(5):571–577. doi:10.1172/jci101335.
- Ingbar SH. Pre-albumin: a thyroxine-binding protein of human plasma. Endocrinology. 1958;63(2):256–259. doi:10.1210/endo-63-2-256.
- Raz A, Goodman DS. The interaction of thyroxine with human plasma prealbumin and with the prealbumin-retinol-binding protein complex. J Biol Chem. 1969;244(12):3230–3237. doi:10.1016/s0021-9258(18)93118-2.
- Nomenclature Committee of the International Union of Biochemistry (NC-IUB). Enzyme nomenclature. Recommendations 1978. Supplement 2: corrections and additions. Eur J Biochem. 1981;116(3):423–435.
- Smith FR, Goodman DS. The effects of diseases of the liver, thyroid, and kidneys on the transport of vitamin a in human plasma. J Clin Invest. 1971;50(11):2426–2436. doi:10.1172/JCI106741.
- Ramström M, Bergquist J. Proteomics of human cerebrospinal fluid. In Proteomics of human body fluids: principles, methods, and applications. Totowa (NJ): humana Press Inc.; 2007:269–284. doi:10.1007/978-1-59745-432-2_12.
- Aleshire SL, Bradley CA, Richardson LD, et al. Localization of human prealbumin in choroid plexus epithelium. J Histochem Cytochem. 1983;31(5):608–612. doi:10.1177/31.5.6341455.
- Dickson PW, Schreiber G. High levels of messenger RNA for transthyretin (prealbumin) in human choroid plexus. Neurosci Lett. 1986;66(3):311–315. doi:10.1016/0304-3940(86)90037-6.
- Vatassery GT, Quach HT, Smith WE, et al. A sensitive assay of transthyretin (prealbumin) in human cerebrospinal fluid in nanogram amounts by ELISA. Clin Chim Acta. 1991;197(1):19–25. doi:10.1016/0009-8981(91)90344-C.
- Martone RL, Herbert J, Dwork A, et al. Transthyretin is synthesized in the mammalian eye. Biochem Biophys Res Commun. 1988;151(2):905–912. doi:10.1016/S0006-291X(88)80367-X.
- Getz RK, Kennedy BG, Mangini NJ. Transthyretin localization in cultured and native human retinal pigment epithelium. Exp Eye Res. 1999;68(5):629–636. doi:10.1006/exer.1998.0646.
- Dwork AJ, Cavallaro T, Martone RL, et al. Distribution of transthyretin in the rat eye. Invest Ophthalmol Vis Sci. 1990;31(3):489–496.
- Grus FH, Joachim SC, Sandmann S, et al. Transthyretin and complex protein pattern in aqueous humor of patients with primary open-angle glaucoma. Mol Vis. 2008;14:1437–1445.
- Van Aken E, De Letter EA, Veckeneer M, et al. Transthyretin levels in the vitreous correlate with change in visual acuity after vitrectomy. Br J Ophthalmol. 2009;93(11):1539–1545. doi:10.1136/bjo.2009.158048.
- Murakami T, Ohsawa Y, Zhenghua L, et al. The transthyretin gene is expressed in Schwann cells of peripheral nerves. Brain Res. 2010;1348:222–225. doi:10.1016/j.brainres.2010.06.017.
- Li X, Masliah E, Reixach N, et al. Neuronal production of transthyretin in human and murine Alzheimer’s disease: is it protective? J Neurosci. 2011;31(35):12483–12490. doi:10.1523/jneurosci.2417-11.2011.
- Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera - a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605–1612. doi:10.1002/jcc.20084.
- Soprano DR, Herbert J, Soprano KJ, et al. Demonstration of transthyretin mRNA in the brain and other extrahepatic tissues in the rat. J Biol Chem. 1985;260(21):11793–11798. doi:10.1016/s0021-9258(17)39100-7.
- Soprano DR, Soprano KJ, Goodman DS. Retinol-binding protein and transthyretin mRNA levels in visceral yolk sac and liver during fetal development in the rat. Proc Natl Acad Sci U S A. 1986;83(19):7330–7334. doi:10.1073/pnas.83.19.7330.
- McKinnon B, Li H, Richard K, et al. Synthesis of thyroid hormone binding proteins transthyretin and albumin by human trophoblast. J Clin Endocrinol Metab. 2005;90(12):6714–6720. doi:10.1210/jc.2005-0696.
- Walker AK, Gong Z, Park WM, et al. Expression of serum retinol binding protein and transthyretin within mouse gastric ghrelin cells. Luque RM, ed. PLoS One. 2013;8(6):e64882. doi:10.1371/journal.pone.0064882.
- Martone RL, Mizuno R, Herbert J. The mammalian pineal gland is a synthetic site for TTR and RBP. J Rheumatol. 1993;20:175.
- Blay P, Nilsson C, Hansson S, et al. An in vivo study of the effect of 5-HT and sympathetic nerves on transferrin and transthyretin mRNA expression in rat choroid plexus and meninges. Brain Res. 1994;662(1-2):148–154. doi:10.1016/0006-8993(94)90807-9.
- Jacobsson B, Collins VP, Grimelius L, et al. Transthyretin immunoreactivity in human and porcine liver, choroid plexus, and pancreatic islets. J Histochem Cytochem. 1989;37(1):31–37. doi:10.1177/37.1.2642294.
- Jacobsson B. In situ localization of transthyretin-mRNA in the adult human liver, choroid plexus and pancreatic islets and in endocrine tumours of the pancreas and gut. Histochemistry. 1989;91(4):299–304. doi:10.1007/BF00493004.
- Ingenbleek Y, De Visscher M. Hormonal and nutritional status: critical conditions for endemic goiter epidemiology? Metabolism. 1979;28(1):9–19. doi:10.1016/0026-0495(79)90162-8.
- Socolow EL, Woeber KA, Purdy RH, et al. Preparation of I-131-labeled human serum prealbumin and its metabolism in normal and sick patients. J Clin Invest. 1965;44(10):1600–1609. doi:10.1172/JCI105266.
- Ando Y, Tanaka Y, Nakazato M, et al. Change in variant transthyretin levels in patients with familial amyloidotic polyneuropathy type I following liver transplantation. Biochem Biophys Res Commun. 1995;211(2):354–358. doi:10.1006/bbrc.1995.1820.
- Makover A, Moriwaki H, Ramakrishnan R, et al. Tissue sites of degradation and turnover in the rat. J Biol Chem. 1988;263(18):8598–8603. doi:10.1016/s0021-9258(18)68346-2.
- Ritchie RF, Palomaki GE, Neveux LM, et al. Reference distributions for the negative acute-phase proteins, albumin, transferrin, and transthyretin: a comparison of a large cohort to the world’s literature. J Clin Lab Anal. 1999;13(6):280–286. doi:10.1002/(SICI)1098-2825(1999)13:6 < 280::AID-JCLA5 > 3.0.CO;2-U.
- Johnson AM, Merlini G, Sheldon J, et al. Clinical indications for plasma protein assays: transthyretin (prealbumin) in inflammation and malnutrition. Clin Chem and Lab Med. 2007;45(3):419–426. doi:10.1515/CCLM.2007.051.
- Férard G, Gaudias J, Bourguignat A, et al. C-reactive protein to transthyretin ratio for the early diagnosis and follow-up of postoperative infection. Clin Chem Lab Med. 2002;40(12):1334–1338. doi:10.1515/CCLM.2002.230.
- Ingenbleek Y, Young VR. Significance of transthyretin in protein metabolism. Clin Chem Lab Med. 2002;40(12):1281–1291. doi:10.1515/CCLM.2002.222.
- Gloeckner SF, Meyne F, Wagner F, et al. Quantitative analysis of transthyretin, tau and amyloid-β in patients with dementia. J Alzheimer’s Dis. 2008;14(1):17–25. doi:10.3233/JAD-2008-14102.
- Liu M, Chen Y, Chen D. Association between transthyretin concentrations and gestational diabetes mellitus in Chinese women. Arch Gynecol Obstet. 2020;302(2):329–335. doi:10.1007/s00404-020-05599-y.
- Heywood W, Mills K, Wang D, et al. Identification of new biomarkers for Down’s syndrome in maternal plasma. J Proteomics. 2012;75(9):2621–2628. doi:10.1016/j.jprot.2012.03.007.
- Kharb, Rupsi, Sharma, Ankita, Chaddar, Monu Kumar, et al. Plasma proteome profiling of coronary artery disease patients: downregulation of transthyretin—an important event. Mediators Inflamm. 2020;2020:3429541. doi:10.1155/2020/3429541.
- Oppenheimer JH. Role of plasma proteins in the binding, distribution and metabolism of the thyroid hormones. N Engl J Med. 1968;278(21):1153–1162. doi:10.1056/nejm196805232782107.
- Mendel CM. The free hormone hypothesis: a physiologically based mathematical model. Endocr Rev. 1989;10(3):232–274. doi:10.1210/edrv-10-3-232.
- Janssen ST, Janssen OE. Directional thyroid hormone distribution via the blood stream to target sites. Mol Cell Endocrinol. 2017;458:16–21. doi:10.1016/j.mce.2017.02.037.
- Chang L, Munro SLA, Richardson SJ, et al. Evolution of thyroid hormone binding by transthyretins in birds and mammals. Eur J Biochem. 1999;259(1-2):534–542. doi:10.1046/j.1432-1327.1999.00076.x.
- Irace G, Edelhoch H. Thyroxine-Induced conformational changes in prealbumin. Biochemistry. 1978;17(26):5729–5733. doi:10.1021/bi00619a020.
- Benvenga S, Robbins J. Enhancement of thyroxine entry into low density lipoprotein (LDL) receptor-Competent fibroblasts by LDL: an additional mode of entry of thyroxine into cells. Endocrinology. 1990;126(2):933–941. doi:10.1210/endo-126-2-933.
- Benvenga S, Cahnmann H, Gregg R, et al. Binding of thyroxine to human plasma low density lipoprotein through specific interaction with apolipoprotein B (apoB-100). Biochimie. 1989;71(2):263–268. doi:10.1016/0300-9084(89)90063-1.
- Richardson SJ, Wijayagunaratne RC, D'Souza DG, et al. Transport of thyroid hormones via the choroid plexus into the brain: the roles of transthyretin and thyroid hormone transmembrane transporters. Front Neurosci. 2015;9(MAR):66. doi:10.3389/fnins.2015.00066.
- Ronne H, Ocklind C, Wiman K, et al. Ligand-dependent regulation of intracellular protein transport: effect of vitamin a on the secretion of the retinol-binding protein. J Cell Biol. 1983;96(3):907–910. doi:10.1083/jcb.96.3.907.
- Monaco HL, Rizzi M, Coda A. Structure of a complex of two plasma proteins: transthyretin and retinol-binding protein. Science. 1995;268(5213):1039–1041. doi:10.1126/science.7754382.
- Naylor HM, Newcomer ME. The structure of human retinol-binding protein (RBP) with its carrier protein transthyretin reveals an interaction with the carboxy terminus of RBP. Biochemistry. 1999;38(9):2647–2653. doi:10.1021/bi982291i.
- Fex G, Albertsson PA, Hansson B. Interaction between prealbumin and retinol-binding protein studied by affinity chromatography, gel filtration and two-Phase partition. Eur J Biochem. 1979;99(2):353–360. doi:10.1111/j.1432-1033.1979.tb13263.x.
- Goodman DS. Vitamin a transport and retinol-binding protein metabolism. Vitam Horm. 1975;32(C):167–180. doi:10.1016/S0083-6729(08)60011-4.
- Kopelman M, Cogan U, Mokady S, et al. The interaction between retinol-binding proteins and prealbumins studied by fluorescence polarization. Biochim Biophys Acta. 1976;439(2):449–460. doi:10.1016/0005-2795(76)90082-9.
- Rostom AA, Sunde M, Richardson SJ, et al. Dissection of multi-protein complexes using mass spectrometry: subunit interactions in transthyretin and retinol-binding protein complexes. Proteins. 1998;33(S2):3–11. doi:10.1002/(SICI)1097-0134(1998)33:2+ <3::AID-PROT2 > 3.0.CO;2-H.
- Shoji S, Nakagawa S. Serum prealbumin and retinol-binding protein concentrations in Japanese-Type familial amyloid polyneuropathy. Eur Neurol. 1988;28(4):191–193. doi:10.1159/000116264.
- Filteau SM, Willumsen JF, Sullivan K, et al. Use of the retinol-binding protein: transthyretin ratio for assessment of vitamin a status during the acute-phase response. Br J Nutr. 2000;83(5):513–520. doi:10.1017/S0007114500000659.
- Prashantha DK, Taly AB, Sinha S, et al. Familial amyloidotic polyneuropathy with muscle, vitreous, leptomeningeal, and cardiac involvement: phenotypic, pathological, and MRI description. Ann Indian Acad Neurol. 2010;13(2):142–144. doi:10.4103/0972-2327.64642.
- Brett M, Persey MR, Reilly MM, et al. Transthyretin Leu12Pro is associated with systemic, neuropathic and leptomeningeal amyloidosis. Brain. 1999;122 (Pt 2)(2):183–190. doi:10.1093/brain/122.2.183.
- McColgan P, Viegas S, Gandhi S, et al. Oculoleptomeningeal amyloidosis associated with transthyretin Leu12Pro in an African patient. J Neurol. 2015;262(1):228–234. doi:10.1007/s00415-014-7594-2.
- Dispenzieri A, Coelho T, Conceição I, et al. A consolidated overview of 14 years of global data from the transthyretin amyloidosis outcomes survey. J Card Fail. 2022;28(5):S111. doi:10.1016/j.cardfail.2022.03.284.
- Dungu JN, Anderson LJ, Whelan CJ, et al. Cardiac transthyretin amyloidosis. Heart. 2012;98(21):1546–1554. doi:10.1136/heartjnl-2012-301924.
- Ruberg FL, Grogan M, Hanna M, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-Art review. J Am Coll Cardiol. 2019;73(22):2872–2891. doi:10.1016/j.jacc.2019.04.003.
- Maurer MS, Elliott P, Comenzo R, et al. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation. 2017;135(14):1357–1377. doi:10.1161/circulationaha.116.024438.
- Ueda M, Horibata Y, Shono M, et al. Clinicopathological features of senile systemic amyloidosis: an ante-and post-mortem study. Mod Pathol. 2011;24(12):1533–1544. doi:10.1038/modpathol.2011.117.
- González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585–2594. doi:10.1093/eurheartj/ehv338.
- Ternacle J, Krapf L, Mohty D, et al. Aortic stenosis and cardiac amyloidosis. J Am Coll Cardiol. 2019;74(21):2638–2651. doi:10.1016/j.jacc.2019.09.056.
- Hafeez AS, Bavry AA. Diagnosis of transthyretin amyloid cardiomyopathy. Cardiol Ther. 2020;9(1):85–95. doi:10.1007/s40119-020-00169-4.
- Grogan M, Scott CG, Kyle RA, et al. Natural history of wild-type transthyretin cardiac amyloidosis and risk stratification using a novel staging system. J Am Coll Cardiol. 2016;68(10):1014–1020. doi:10.1016/j.jacc.2016.06.033.
- Ghosh S, Khanra D, Krishna V, et al. Wild type transthyretin cardiac amyloidosis in a young individual: a case report. Medicine (Baltimore). 2021;100(17):e25462. doi:10.1097/MD.0000000000025462.
- Connors LH, Sam F, Skinner M, et al. Heart failure resulting from age-related cardiac amyloid disease associated with wild-type transthyretin: a prospective, observational cohort study. Circulation. 2016;133(3):282–290. doi:10.1161/circulationaha.115.018852.
- Schmidt HH, Waddington-Cruz M, Botteman MF, et al. Estimating the global prevalence of transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2018;57(5):829–837. doi:10.1002/mus.26034.
- Zhen DB, Swiecicki PL, Zeldenrust SR, et al. Frequencies and geographic distributions of genetic mutations in transthyretin- and non-transthyretin-related familial amyloidosis. Clin Genet. 2015;88(4):396–400. doi:10.1111/cge.12500.
- Soares ML, Coelho T, Sousa A, et al. Haplotypes and DNA sequence variation within and surrounding the transthyretin gene: genotype-phenotype correlations in familial amyloid polyneuropathy (V30M) in Portugal and Sweden. Eur J Hum Genet. 2004;12(3):225–237. doi:10.1038/sj.ejhg.5201095.
- Jacobson DR, Alexander AA, Tagoe C, et al. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African-Americans. Amyloid. 2015;22(3):171–174. doi:10.3109/13506129.2015.1051219.
- Jacobson DR, Alexander AA, Tagoe C, et al. The prevalence and distribution of the amyloidogenic transthyretin (TTR) V122I allele in Africa. Mol Genet Genomic Med. 2016;4(5):548–556. doi:10.1002/mgg3.231.
- Dungu JN, Papadopoulou SA, Wykes K, et al. Afro-Caribbean heart failure in the United Kingdom. Circ: heart Failure. 2016;9(9):e003352. doi:10.1161/CIRCHEARTFAILURE.116.003352.
- Arruda-Olson AM, Zeldenrust SR, Dispenzieri A, et al. Genotype, echocardiography, and survival in familial transthyretin amyloidosis. Amyloid. 2013;20(4):263–268. doi:10.3109/13506129.2013.845745.
- Dharmarajan K, Maurer MS. Transthyretin cardiac amyloidoses in older North Americans. J Am Geriatr Soc. 2012;60(4):765–774. doi:10.1111/j.1532-5415.2011.03868.x.
- Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–2412. doi:10.1161/circulationaha.116.021612.
- Fan K, Zhu H, Xu H, et al. The identification of a transthyretin variant p.D38G in a Chinese family with early-onset leptomeningeal amyloidosis. J Neurol. 2019;266(1):232–241. doi:10.1007/s00415-018-9125-z.
- Mitsuhashi S, Yazaki M, Tokuda T, et al. Biochemical characteristics of variant transthyretins causing hereditary leptomeningeal amyloidosis. Amyloid. 2005;12(4):216–225. doi:10.1080/13506120500352404.
- Martins AC, Rosa AM, Costa E, et al. Ocular manifestations and therapeutic options in patients with familial amyloid polyneuropathy: a systematic review. Biomed Res Int. 2015;2015:282405–282409. doi:10.1155/2015/282405.
- Dammacco R, Merlini G, Lisch W, et al. Amyloidosis and ocular involvement: an overview. Semin Ophthalmol. 2020;35(1):7–26. doi:10.1080/08820538.2019.1687738.
- Beirão JM, Malheiro J, Lemos C, et al. Impact of liver transplantation on the natural history of oculopathy in Portuguese patients with transthyretin (V30M) amyloidosis. Amyloid. 2015;22(1):31–35. doi:10.3109/13506129.2014.989318.
- Hara R, Kawaji T, Ando E, et al. Impact of liver transplantation on transthyretin-related ocular amyloidosis in Japanese patients. Arch Ophthalmol. 2010;128(2):206–210. doi:10.1001/archophthalmol.2009.390.
- Reddi HV, Jenkins S, Theis J, et al. Homozygosity for the V122I mutation in transthyretin is associated with earlier onset of cardiac amyloidosis in the African American population in the seventh decade of life. J Mol Diagn. 2014;16(1):68–74. doi:10.1016/j.jmoldx.2013.08.001.
- Maia LF, Magalhães R, Freitas J, et al. CNS involvement in V30M transthyretin amyloidosis: clinical, neuropathological and biochemical findings. J Neurol Neurosurg Psychiatry. 2015;86(2):159–167. doi:10.1136/JNNP-2014-308107.
- Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36(4):411–423. doi:10.1002/mus.20821.
- Wang Y, Wang Q, Huang H, et al. A crowdsourcing open platform for literature curation in UniProt. PLoS Biol. 2021;19(12):e3001464. doi:10.1371/journal.pbio.3001464.
- Andrade C. A peculiar form of peripheral neuropathy: familiar atypical generalized amyloidosis with special involvement of the peripheral nerves. Brain. 1952;75(3):408–427. doi:10.1093/brain/75.3.408.
- Araki S, Mawatari S, Ohta M, et al. Polyneuritic amyloidosis in a Japanese family. Arch Neurol. 1968;18(6):593–602. doi:10.1001/archneur.1968.00470360015001.
- Andersson R. Familial amyloidosis with polyneuropathy. A clinical study based on patients living in northern Sweden [disseration]. Umeå (SE): Umeå Universitet; 1976.
- Jinno Y, Matsumoto T, Kamel T, et al. Localization of the human prealbumin gene to 18p11.1-q12.3 by gene dose effect study of Southern blot hybridization. Jinrui Idengaku Zasshi. 1986;31(3):243–248. doi:10.1007/BF01870754.
- Hiroyuki S, Naoko Y, Yasuyuki T, et al. Structure of the chromosomal gene for human serum prealbumin. Gene. 1985;37(1-3):191–197. doi:10.1016/0378-1119(85)90272-0.
- Rowczenio DM, Noor I, Gillmore JD, et al. Online registry for mutations in hereditary amyloidosis including nomenclature recommendations. Hum Mutat. 2014;35(9):E2403–E2412. doi:10.1002/humu.22619.
- Ando Y, Ueda M. Novel methods for detecting amyloidogenic proteins in transthyretin related amyloidosis. Front Biosci. 2008;13(14):5548–5558. doi:10.2741/3098.
- He S, Jin Y, Tian Z, et al. Establishment of an induced pluripotent stem cell line PUMCHi004-a from a hereditary transthyretin amyloid cardiomyopathy patient with transthyretin (TTR) p.Asp38Asn mutation. Stem Cell Res. 2020;49:102022. doi:10.1016/j.scr.2020.102022.
- Aono Y, Hamatani Y, Katoh N, et al. Late-onset hereditary ATTR amyloidosis with a novel p.P63S (P43S) Transthyretin variant. Intern Med. 2021;60(4):557–561. doi:10.2169/internalmedicine.5615-20.
- Groenning M, Campos RI, Fagerberg C, et al. Thermodynamic stability and denaturation kinetics of a benign natural transthyretin mutant identified in a danish kindred. Amyloid. 2011;18(2):35–46. doi:10.3109/13506129.2011.560215.
- Cuddy SAM, Dorbala S, Falk RH. Complexities and pitfalls in cardiac amyloidosis. Circulation. 2020;142(4):409–415. doi:10.1161/circulationaha.120.046680.
- Chen Z, Koh JS, Saini M, et al. Hereditary transthyretin amyloidosis- clinical and genetic characteristics of a multiracial South-East asian cohort in Singapore. J Neuromuscul Dis. 2021;8(4):723–733. doi:10.3233/JND-210656.
- Reznik EV, Nguyen TL, Borisovskaya SV, et al. A clinical case of the hereditary transthyretin amyloidosis. Russ Arch Intern Med. 2021;11(3):229–240. doi:10.20514/2226-6704-2021-11-3-229-240.
- Sant’Anna R, Almeida MR, Varejāo N, et al. Cavity filling mutations at the thyroxine-binding site dramatically increase transthyretin stability and prevent its aggregation. Sci Rep. 2017;7(1):44709. doi:10.1038/srep44709.
- Raivio VE, Jonasson J, Myllykangas L, et al. A novel transthyretin Lys70Glu (p.Lys90Glu) mutation presenting with vitreous amyloidosis and carpal tunnel syndrome. Amyloid. 2016;23(1):46–50. doi:10.3109/13506129.2015.1126574.
- Bergström J, Patrosso MC, Colussi G, et al. A novel type of familial transthyretin amyloidosis, ATTR Asn124Ser, with co-localization of κ light chains. Amyloid. 2007;14(2):141–145. doi:10.1080/13506120701259895.
- Choi K, Seok JM, Kim BJ, et al. Characteristics of South korean patients with hereditary transthyretin amyloidosis. J Clin Neurol. 2018;14(4):537–541. doi:10.3988/jcn.2018.14.4.537.
- Strahler JR, Rosenblum BB, Hanash SM. Identification and characterization of a human transthyretin variant. Biochem Biophys Res Commun. 1987;148(1):471–477. doi:10.1016/0006-291X(87)91135-1.
- Lavigne Moreira C, Marques VD, Lourenço CM, et al. Transthyretin Asp38Tyr: a new mutation associated to a late onset neuropathy. J Peripher Nerv Syst. 2015;20(1):60–62. doi:10.1111/jns.12112.
- Lavigne-Moreira C, Marques VD, Gonçalves MVM, et al. The genetic heterogeneity of hereditary transthyretin amyloidosis in a sample of the Brazilian population. J Peripher Nerv Syst. 2018;23(2):134–137. doi:10.1111/jns.12259.
- Patel JK, Rosen AM, Chamberlin A, et al. Three newly recognized likely pathogenic gene variants associated with hereditary transthyretin amyloidosis. Neurol Ther. 2022;11(4):1595–1607. doi:10.1007/s40120-022-00385-1.
- Thimm A, Oubari S, Hoffmann J, et al. A novel TTR mutation (p.Ala65Val) underlying late-onset hereditary transthyretin (ATTRv) amyloidosis with mixed cardiac and neuropathic phenotype: a case report. BMC Neurol. 2022;22(1):469. doi:10.1186/S12883-022-02952-3/FIGURES/4.
- Ikeda K, Yamamoto D, Usui K, et al. A case of transthyretin variant amyloidosis with a TTR A97D (p.A117D) Mutation manifesting remarkable asymmetric neuropathy. Intern Med. 2022;62(15):2261–2266. Published online doi:10.2169/internalmedicine.0798-22.
- Jiang M, Wang M, Tao Z, et al. Biochemical and biophysical properties of an unreported T96R mutation causing transthyretin cardiac amyloidosis. Amyloid. 2022;30(2):188–198. doi:10.1080/13506129.2022.2142109.
- Ma Q, Wang M, Huang Y, et al. Identification of a novel transthyretin mutation D39Y in a cardiac amyloidosis patient and its biochemical characterizations. Front Cardiovasc Med. 2023;10:1091183. doi:10.3389/FCVM.2023.1091183.
- Lyng CS, Gude E, Hodt A, et al. First Norwegian case of hereditary ATTR amyloidosis with a novel transthyretin variant. Scand Cardiovasc J. 2023;57(1):2174269. doi:10.1080/14017431.2023.2174269.
- Waddington-Cruz M, Ackermann EJ, Polydefkis M, et al. Hereditary transthyretin amyloidosis: baseline characteristics of patients in the NEURO-TTR trial. Amyloid. 2018;25(3):180–188. doi:10.1080/13506129.2018.1503593.
- Neculae G, Adam R, Beyer R, et al. Novel transthyretin variant linked to cardiac amyloidosis in the Romanian population. Arch Cardiovasc Dis. 2024;117(1):S38–S39. doi:10.1016/j.acvd.2023.10.068.
- Stenson PD, Mort M, Ball EV, et al. The human gene mutation database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139(10):1197–1207. doi:10.1007/S00439-020-02199-3/FIGURES/5.
- Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(Database issue):D980–D985. doi:10.1093/NAR/GKT1113.
- Uemichi T, Liepnieks JJ, Benson MD. A trinucleotide deletion in the transthyretin gene (ΔV122) in a kindred with familial amyloidotic polyneuropathy. Neurology. 1997;48(6):1667–1670. doi:10.1212/WNL.48.6.1667.
- Klimtchuk ES, Prokaeva T, Frame NM, et al. Unusual duplication mutation in a surface loop of human transthyretin leads to an aggressive drug-resistant amyloid disease. Proc Natl Acad Sci U S A. 2018;115(28):E6428–E6436. doi:10.1073/pnas.1802977115.
- Waddington-Cruz M, Ando Y, Amass L, et al. Feasibility of assessing progression of transthyretin amyloid polyneuropathy using nerve conduction studies: findings from the transthyretin amyloidosis outcomes survey (THAOS). J Peripher Nerv Syst. 2021;26(2):160–166. doi:10.1111/JNS.12444.
- Sharp N. Mutations matter even if proteins stay the same. Nature. 2022;606(7915):657–659. doi:10.1038/d41586-022-01091-6.
- Skare J, Jones LA, Myles N, et al. Two transthyretin mutations (glu42gly, his90asn) in an Italian family with amyloidosis. Clin Genet. 1994;45(6):281–284. doi:10.1111/J.1399-0004.1994.TB04030.X.
- da Silva-Batista JA, Marques W, Oliveira MTdJS, et al. Presence of val30Met and val122ile mutations in a patient with hereditary amyloidosis. J Hum Genet. 2020;65(8):711–713. doi:10.1038/S10038-020-0749-3.
- Lim A, Prokaeva T, Connors LH, et al. Identification of a novel transthyretin Thr59Lys/Arg104His. A case of compound heterozygosity in a Chinese patient diagnosed with familial transthyretin amyloidosis. Amyloid. 2002;9(2):134–140. doi:10.3109/13506120208995246.
- Jacobson DR, Buxbaum JN. A double-variant transthyretin allele (SER 6, ILE 33) in the Israeli patient “SKO” with familial amyloidotic polyneuropathy. Hum Mutat. 1994;3(3):254–260. doi:10.1002/HUMU.1380030313.
- Saraiva MJM. Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum Mutat. 2001;17(6):493–503. doi:10.1002/HUMU.1132.
- González-Duarte A, Cárdenas-Soto K, Bañuelos CE, et al. Amyloidosis due to TTR mutations in Mexico with 4 distincts genotypes in the index cases. Orphanet J Rare Dis. 2018;13(1):107. doi:10.1186/S13023-018-0801-Y/FIGURES/2.
- Serpell LC, Goldsteins G, Dacklin I, et al. The “edge strand” hypothesis: prediction and test of a mutational “hot-spot” on the transthyretin molecule associated with FAP amyloidogenesis. Amyloid. 1996;3(2):75–85. doi:10.3109/13506129609014359.
- Saraiva MJM. Transthyretin mutations in health and disease. Hum Mutat. 1995;5(3):191–196. doi:10.1002/humu.1380050302.
- Salvi F, Pastorelli F, Plasmati R, et al. Early onset aggressive hereditary amyloidosis: report of an Italian family with TTR Arg47 mutation. Neurol Sci. 2005;26(2):140–142. doi:10.1007/s10072-005-0449-y.
- Mangione PP, Porcari R, Gillmore JD, et al. Proteolytic cleavage of Ser52Pro variant transthyretin triggers its amyloid fibrillogenesis. Proc Natl Acad Sci U S A. 2014;111(4):1539–1544. doi:10.1073/pnas.1317488111.
- Kim HS, Kim S-M, Kang S-W, et al. An aggressive form of familial amyloid polyneuropathy caused by a Glu54Gly mutation in the transthyretin gene. Eur J Neurol. 2005;12(8):657–659. doi:10.1111/j.1468-1331.2005.01005.x.
- Togashi S, Watanabe H, Nagasaka T, et al. An aggressive familial amyloidotic polyneuropathy caused by a new variant transthyretin lys 54. Neurology. 1999;53(3):637–639. doi:10.1212/wnl.53.3.637.
- Jacobson DR, McFarlin DE, Kane I, et al. Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet. 1992;89(3):353–356. doi:10.1007/BF00220559.
- Gardini S, Cheli S, Baroni S, et al. On nature’s strategy for assigning genetic code multiplicity. Dupuy D, ed. PLoS ONE. 2016;11(2):e0148174. doi:10.1371/journal.pone.0148174.
- Ueno S, Uemichi T, Yorifuji S, et al. A novel variant of transthyretin (Tyr114 to cys) deduced from the nucleotide sequences of gene fragments from familial amyloidotic polyneuropathy in Japanese sibling cases. Biochem Biophys Res Commun. 1990;169(1):143–147. doi:10.1016/0006-291X(90)91445-X.
- Nakase T, Yamashita T, Matsuo Y, et al. Hereditary ATTR amyloidosis with cardiomyopathy caused by the novel variant transthyretin Y114S (p.Y134S). Intern Med. 2019;58(18):2695–2698. doi:10.2169/internalmedicine.2456-18.
- Murakami T, Tachibana S, Endo Y, et al. Familial carpal tunnel syndrome due to amyloidogenic transthyretin his 114 variant. Neurology. 1994;44(2):315–318. doi:10.1212/wnl.44.2.315.
- Janunger T, Anan I, Holmgren G, et al. Heart failure caused by a novel amyloidogenic mutation of the transthyretin gene: ATTR Ala45Ser. Amyloid. 2000;7(2):137–140. doi:10.3109/13506120009146252.
- Saraiva MJ, Almeida MdR, Sherman W, et al. A new transthyretin mutation associated with amyloid cardiomyopathy. Am J Hum Genet. 1992;50(5):1027–1030.
- Klaassen SHC, Lemmink HH, Bijzet J, et al. Late onset cardiomyopathy as presenting sign of ATTR A45G amyloidosis caused by a novel TTR mutation (p.A65G). Cardiovasc Pathol. 2017;29:19–22. doi:10.1016/j.carpath.2017.04.002.
- Misumi Y, Doki T, Ueda M, et al. Myopathic phenotype of familial amyloid polyneuropathy with a rare transthyretin variant: ATTR Ala45Asp. Amyloid. 2014;21(3):216–217. doi:10.3109/13506129.2014.932277.
- Murakami T, Maeda S, Yi S, et al. A novel transthyretin mutation associated with familial amyloidotic polyneuropathy. Biochem Biophys Res Commun. 1992;182(2):520–526. doi:10.1016/0006-291X(92)91763-G.
- Ferlini A, Patrosso MC, Repetto M, et al. A new mutation (TTR ala-47) in the transthyretin gene associated with hereditary amyloidosis. Hum Mutat. 1994;4(1):61–64. doi:10.1002/humu.1380040110.
- Pelo E, Da Prato L, Ciaccheri M, et al. Familial amyloid polyneuropathy with genetic anticipation associated to a gly47glu transthyretin variant in an Italian kindred. Amyloid. 2002;9(1):35–41. doi:10.3109/13506120209072443.
- Booth DR, Soutar AK, Hawkins PN, et al. Three new amyloidogenic transthyretin gene mutations: advantages of direct sequencing. In: kisikevsky R, Benson MD, Frangione B, Gauldie J, Muckle TJ, Young ID, eds. Amyloid and amyloidosis 1993. 1st ed. Kingston (ON): Parthenon Publishing Group Ltd; 1994:456–458.
- Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8(1):31. doi:10.1186/1750-1172-8-31.
- Lemos C, Coelho T, Alves-Ferreira M, et al. Overcoming artefact: Anticipation in 284 portuguese kindreds with familial amyloid polyneuropathy (FAP) ATTRV30M. J Neurol Neurosurg Psychiatry. 2014;85(3):326–330. doi:10.1136/jnnp-2013-305383.
- Ueda M, Yamashita T, Misumi Y, et al. Origin of sporadic late-onset hereditary ATTR Val30Met amyloidosis in Japan. Amyloid. 2018;25(3):143–147. doi:10.1080/13506129.2018.1531842.
- Hellman U, Suhr O. Regional differences and similarities of FAP in Sweden. Amyloid. 2012;19 (sup1). 53–54.
- Koike H, Misu K-I, Ikeda S-I, et al. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset form. Arch Neurol. 2002;59(11):1771–1776. doi:10.1001/archneur.59.11.1771.
- Ihse E, Ybo A, Suhr OB, et al. Amyloid fibril composition is related to the phenotype of hereditary transthyretin V30M amyloidosis. J Pathol. 2008;216(2):253–261. doi:10.1002/PATH.2411.
- Suhr OB, Lundgren E, Westermark P. One mutation, two distinct disease variants: unravelling the impact of transthyretin amyloid fibril composition. J Intern Med. 2017;281(4):337–347. doi:10.1111/joim.12585.
- Koike H, Ando Y, Ueda M, et al. Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J Neurol Sci. 2009;287(1-2):178–184. doi:10.1016/J.JNS.2009.07.028.
- Keppel SC, Brannagan TH, Helmke S, et al. Early-Onset of transthyretin amyloidosis in a young Afro-Caribbean woman with Thr60Ala mutation. JACC Case Rep. 2020;2(13):2063–2067. doi:10.1016/j.jaccas.2020.08.030.
- Kotani N, Hattori T, Yamagata S, et al. Transthyretin Thr60Ala appalachian-type mutation in a Japanese family with familial amyloidotic polyneuropathy. Amyloid. 2002;9(1):31–34. doi:10.3109/13506120209072442.
- Soares ML, Coelho T, Sousa A, et al. Susceptibility and modifier genes in portuguese transthyretin V30M amyloid polyneuropathy: complexity in a single-gene disease. Hum Mol Genet. 2005;14(4):543–553. doi:10.1093/hmg/ddi051.
- Santos D, Coelho T, Alves-Ferreira M, et al. Variants in RBP4 and AR genes modulate age at onset in familial amyloid polyneuropathy (FAP ATTRV30M). Eur J Hum Genet. 2016;24(5):756–760. doi:10.1038/ejhg.2015.180.
- Olsson M, Hellman U, Planté-Bordeneuve V, et al. Mitochondrial haplogroup is associated with the phenotype of familial amyloidosis with polyneuropathy in Swedish and French patients. Clin Genet. 2009;75(2):163–168. doi:10.1111/J.1399-0004.2008.01097.X.
- Alves-Ferreira M, Coelho T, Santos D, et al. A trans-acting factor may modify age at onset in familial amyloid polyneuropathy ATTRV30M in Portugal. Mol Neurobiol. 2018;55(5):3676–3683. doi:10.1007/s12035-017-0593-4.
- Polimanti R, Di Girolamo M, Manfellotto D, et al. In silico analysis of TTR gene (coding and non-coding regions, and interactive network) and its implications in transthyretin-related amyloidosis. Amyloid. 2014;21(3):154–162. doi:10.3109/13506129.2014.900487.
- Iorio A, De Angelis F, Di Girolamo M, et al. Most recent common ancestor of TTR Val30Met mutation in Italian population and its potential role in genotype-phenotype correlation. Amyloid. 2015;22(2):73–78. doi:10.3109/13506129.2014.994597.
- Polimanti R, Di Girolamo M, Manfellotto D, et al. Functional variation of the transthyretin gene among human populations and its correlation with amyloidosis phenotypes. Amyloid. 2013;20(4):256–262. doi:10.3109/13506129.2013.844689.
- Iorio A, De Lillo A, De Angelis F, et al. Non-coding variants contribute to the clinical heterogeneity of TTR amyloidosis. Eur J Hum Genet. 2017;25(9):1055–1060. doi:10.1038/ejhg.2017.95.
- Iorio A, De Angelis F, Di Girolamo M, et al. Population diversity of the genetically determined TTR expression in human tissues and its implications in TTR amyloidosis. BMC Genomics. 2017;18(1):254. doi:10.1186/s12864-017-3646-1.
- Polimanti R, Nuñez Y, Gelernter J. Increased risk of multiple outpatient surgeries in African-American carriers of transthyretin Val122Ile mutation is modulated by non-coding variants. J Clin Med. 2019;8(2):269. doi:10.3390/jcm8020269.
- Sikora JL, Logue MW, Chan GG, et al. Genetic variation of the transthyretin gene in wild-type transthyretin amyloidosis (ATTRwt). Hum Genet. 2015;134(1):111–121. doi:10.1007/s00439-014-1499-0.
- Quintas A, Vaz DC, Cardoso I, et al. Tetramer dissociation and monomer partial unfolding precedes protofibril formation in amyloidogenic transthyretin variants. J Biol Chem. 2001;276(29):27207–27213. doi:10.1074/jbc.M101024200.
- Hörnberg A, Eneqvist T, Olofsson A, et al. A comparative analysis of 23 structures of the amyloidogenic protein transthyretin. J Mol Biol. 2000;302(3):649–669. doi:10.1006/jmbi.2000.4078.
- Palaninathan SK. Nearly 200 X-Ray crystal structures of transthyretin: what do they tell Us about this protein and the design of drugs for TTR amyloidoses? Curr Med Chem. 2012;19(15):2324–2342. doi:10.2174/092986712800269335.
- Brito RMM, Damas AM, Saraiva MJ. Amyloid formation by transthyretin: from protein stability to protein aggregation. Curr Med Chem Endocr Metab Agents. 2003;3(4):349–360. doi:10.2174/1568013033483230.
- Palaninathan SK, Mohamedmohaideen NN, Snee WC, et al. Structural insight into pH-Induced conformational changes within the native human transthyretin tetramer. J Mol Biol. 2008;382(5):1157–1167. doi:10.1016/j.jmb.2008.07.029.
- Dasari AKR, Hung I, Michael B, et al. Structural characterization of cardiac ex vivo transthyretin amyloid: insight into the transthyretin misfolding pathway in vivo. Biochemistry. 2020;59(19):1800–1803. doi:10.1021/acs.biochem.0c00091.
- Schmidt M, Wiese S, Adak V, et al. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat Commun. 2019;10(1):5008. doi:10.1038/s41467-019-13038-z.
- Ferguson JA, Sun X, Dyson HJ, et al. Thermodynamic stability and aggregation kinetics of EF helix and EF loop variants of transthyretin. Biochemistry. 2021;60(10):756–764. doi:10.1021/acs.biochem.1c00073.
- Yang M, Lei M, Bruschweiler R, et al. Initial conformational changes of human transthyretin under partially denaturing conditions. Biophys J. 2005;89(1):433–443. doi:10.1529/biophysj.105.059642.
- Yang M, Yordanov B, Levy Y, et al. The sequence-dependent unfolding pathway plays a critical role in the amyloidogenicity of transthyretin. Biochemistry. 2006;45(39):11992–12002. doi:10.1021/bi0609927.
- Rodrigues JR, Simões CJV, Silva CG, et al. Potentially amyloidogenic conformational intermediates populate the unfolding landscape of transthyretin: insights from molecular dynamics simulations. Protein Sci. 2010;19(2):202–219. doi:10.1002/pro.289.
- Lai Z, Colón W, Kelly JW. The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry. 1996;35(20):6470–6482. doi:10.1021/bi952501g.
- Olofsson A, Ippel JH, Wijmenga SS, et al. Probing solvent accessibility of transthyretin amyloid by solution NMR spectroscopy. J Biol Chem. 2004;279(7):5699–5707. doi:10.1074/jbc.M310605200.
- Serag AA, Altenbach C, Gingery M, et al. Arrangement of subunits and ordering of β-strands in an amyloid sheet. Nat Struct Biol. 2002;9(10):734–739. doi:10.1038/nsb838.
- Goldsteins G, Persson H, Andersson K, et al. Exposure of cryptic epitopes on transthyretin only in amyloid and in amyloidogenic mutants. Proc Natl Acad Sci U S A. 1999;96(6):3108–3113. doi:10.1073/pnas.96.6.3108.
- Yee AW, Aldeghi M, Blakeley MP, et al. A molecular mechanism for transthyretin amyloidogenesis. Nat Commun. 2019;10(1):925. doi:10.1038/s41467-019-08609-z.
- Wang Y-S, Huang C-H, Liou G-G, et al. A molecular basis for tetramer destabilization and aggregation of transthyretin Ala97Ser. Protein Sci. 2023;32(4):e4610. doi:10.1002/PRO.4610.
- Dasari AKR, Arreola J, Michael B, et al. Disruption of the CD loop by enzymatic cleavage promotes the formation of toxic transthyretin oligomers through a common transthyretin misfolding pathway. Biochemistry. 2020;59(25):2319–2327. doi:10.1021/acs.biochem.0c00079.
- Zhou S, Cheng J, Yang T, et al. Exploration of the misfolding mechanism of transthyretin monomer: insights from Hybrid-Resolution simulations and markov state model analysis. Biomolecules. 2019;9(12):889. doi:10.3390/biom9120889.
- Lim KH, Dyson HJ, Kelly JW, et al. Localized structural fluctuations promote amyloidogenic conformations in transthyretin. J Mol Biol. 2013;425(6):977–988. doi:10.1016/j.jmb.2013.01.008.
- Lim KH, Dasari AKR, Hung I, et al. Structural changes associated with transthyretin misfolding and amyloid formation revealed by solution and solid-State NMR. Biochemistry. 2016;55(13):1941–1944. doi:10.1021/acs.biochem.6b00164.
- Armen RS, Alonso DOV, Daggett V. Anatomy of an amyloidogenic intermediate: conversion of β-sheet to α-sheet structure in transthyretin at acidic pH. Structure. 2004;12(10):1847–1863. doi:10.1016/j.str.2004.08.005.
- Steward RE, Armen RS, Daggett V. Different disease-causing mutations in transthyretin trigger the same conformational conversion. Protein Eng Des Sel. 2008;21(3):187–195. doi:10.1093/protein/gzm086.
- Kelly JW. Alternative conformations of amyloidogenic proteins govern their behavior. Curr Opin Struct Biol. 1996;6(1):11–17. doi:10.1016/S0959-440X(96)80089-3.
- Uversky VN, Segel DJ, Doniach S, et al. Association-induced folding of globular proteins. Proc Natl Acad Sci U S A. 1998;95(10):5480–5483. doi:10.1073/pnas.95.10.5480.
- Terry CJ, Damas AM, Oliveira P, et al. Structure of Met30 variant of transthyretin and its amyloidogenic implications. Embo J. 1993;12(2):735–741. doi:10.1002/j.1460-2075.1993.tb05707.x.
- Eneqvist T, Andersson K, Olofsson A, et al. The β-slip: a novel concept in transthyretin amyloidosis. Mol Cell. 2000;6(5):1207–1218. doi:10.1016/s1097-2765(00)00117-9.
- Ferrão-Gonzales AO, Souto SO, Silva JL, et al. The preaggregated state of an amyloidogenic protein: Hydrostatic pressure converts native transthyretin into the amyloidogenic state. Proc Natl Acad Sci U S A. 2000;97(12):6445–6450. doi:10.1073/pnas.97.12.6445.
- Schormann N, Murrell JR, Benson MD. Tertiary structures of amyloidogenic and non-amyloidogenic transthyretin variants: new model for amyloid fibril formation. Amyloid. 1998;5(3):175–187. doi:10.3109/13506129809003843.
- Olofsson A, Ippel HJ, Baranov V, et al. Capture of a dimeric intermediate during transthyretin amyloid formation. J Biol Chem. 2001;276(43):39592–39599. doi:10.1074/jbc.M103599200.
- Serag AA, Altenbach C, Gingery M, et al. Identification of a subunit interface in transthyretin amyloid fibrils: evidence for self-assembly from oligomeric building blocks. Biochemistry. 2001;40(31):9089–9096. doi:10.1021/bi010655s.
- Lashuel HA, Lai Z, Kelly JW. Characterization of the transthyretin acid denaturation pathways by analytical ultracentrifugation: implications for wild-type, V30M, and L55P amyloid fibril formation. †Biochemistry. 1998;37(51):17851–17864. doi:10.1021/BI981876+.
- Nettleton EJ, Sunde M, Lai Z, et al. Protein subunit interactions and structural integrity of amyloidogenic transthyretins: evidence from electrospray mass spectrometry. J Mol Biol. 1998;281(3):553–564. doi:10.1006/jmbi.1998.1937.
- Lindgren M, Sörgjerd K, Hammarström P. Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy. Biophys J. 2005;88(6):4200–4212. doi:10.1529/biophysj.104.049700.
- Lindgren M, Hammarström P. Amyloid oligomers: spectroscopic characterization of amyloidogenic protein states. Febs J. 2010;277(6):1380–1388. doi:10.1111/j.1742-4658.2010.07571.x.
- Inouye H, Domingues FS, Damas AM, et al. Analysis of x-ray diffraction patterns from amyloid of biopsied vitreous humor and kidney of transthyretin (TTR) Met30 familial amyloidotic polyneuropathy (FAP) patients: Axially arrayed TTR monomers constitute the protofilament. Amyloid. 1998;5(3):163–174. doi:10.3109/13506129809003842.
- Saelices L, Nguyen BA, Chung K, et al. A pair of peptides inhibits seeding of the hormone transporter transthyretin into amyloid fibrils. J Biol Chem. 2019;294(15):6130–6141. doi:10.1074/jbc.RA118.005257.
- Quintas A, Saraiva MJM, Brito RMM. The tetrameric protein transthyretin dissociates to a non-native monomer in solution. A novel model for amyloidogenesis. J Biol Chem. 1999;274(46):32943–32949. doi:10.1074/jbc.274.46.32943.
- Foss TR, Wiseman RL, Kelly JW. The pathway by which the tetrameric protein transthyretin dissociates. Biochemistry. 2005;44(47):15525–15533. doi:10.1021/bi051608t.
- Coelho T, Merlini G, Bulawa CE, et al. Mechanism of action and clinical application of tafamidis in hereditary transthyretin amyloidosis. Neurol Ther. 2016;5(1):1–25. doi:10.1007/s40120-016-0040-x.
- Jesus C, Almeida Z, Vaz D, et al. A new folding kinetic mechanism for human transthyretin and the influence of the amyloidogenic V30M mutation. Int J Mol Sci. 2016;17(9):1428. doi:10.3390/ijms17091428.
- Simões CJV, Almeida ZL, Costa D, et al. A novel bis-furan scaffold for transthyretin stabilization and amyloid inhibition. Eur J Med Chem. 2016;121:823–840. doi:10.1016/j.ejmech.2016.02.074.
- Simões CJV, Almeida ZL, Cardoso AL, et al. Lead optimization of resilient next-generation transthyretin stabilizers for multiple target-product profiles: approaching the CNS. Amyloid. 2019;26(sup1):77–78. doi:10.1080/13506129.2019.1583195.
- Ferreira E, Almeida ZL, Cruz PF, et al. Searching for the best transthyretin aggregation protocol to study amyloid fibril disruption. Int J Mol Sci. 2022;23(1):391. doi:10.3390/ijms23010391.
- Almeida ZL, Brito RMM. Amyloid disassembly: what can We learn from chaperones? Biomedicines. 2022;10(12):3276. doi:10.3390/biomedicines10123276.
- Cardoso I, Goldsbury CS, Müller SA, et al. Transthyretin fibrillogenesis entails the assembly of monomers: a molecular model for in vitro assembled transthyretin amyloid-like fibrils. J Mol Biol. 2002;317(5):683–695. doi:10.1006/jmbi.2002.5441.
- Sant’Anna R, Braga C, Varejão N, et al. The importance of a gatekeeper residue on the aggregation of transthyretin implications for transthyretin-related amyloidoses. J Biol Chem. 2014;289(41):28324–28337. doi:10.1074/jbc.M114.563981.
- Faria TQ, Almeida ZL, Cruz PF, et al. A look into amyloid formation by transthyretin: Aggregation pathway and a novel kinetic model. Phys Chem Chem Phys. 2015;17(11):7255–7263. doi:10.1039/c4cp04549a.
- Jarvis JA, Craik DJ, Wilce MCJ. X-ray-diffraction studies of fibrils formed from peptide fragments of transthyretin. Biochem Biophys Res Commun. 1993;192(3):991–998. doi:10.1006/bbrc.1993.1514.
- Dirix C, Meersman F, MacPhee CE, et al. High hydrostatic pressure dissociates early aggregates of TTR 105-115, but not the mature amyloid fibrils. J Mol Biol. 2005;347(5):903–909. doi:10.1016/j.jmb.2005.01.073.
- Gustavsson Å, Engström U, Westermark P. Normal transthyretin and synthetic transthyretin fragments from amyloid-like fibrils in vitro. Biochem Biophys Res Commun. 1991;175(3):1159–1164. doi:10.1016/0006-291X(91)91687-8.
- Bergström J, Engström U, Yamashita T, et al. Surface exposed epitopes and structural heterogeneity of in vivo formed transthyretin amyloid fibrils. Biochem Biophys Res Commun. 2006;348(2):532–539. doi:10.1016/j.bbrc.2006.07.140.
- Colon W, Kelly JW. Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry. 1992;31(36):8654–8660. doi:10.1021/bi00151a036.
- Groenning M, Campos RI, Hirschberg D, et al. Considerably unfolded transthyretin monomers preceed and exchange with dynamically structured amyloid protofibrils. Sci Rep. 2015;5(1):11443. doi:10.1038/srep11443.
- Pires RH, Karsai Á, Saraiva MJ, et al. Distinct annular oligomers captured along the assembly and disassembly pathways of transthyretin amyloid protofibrils. Muller DJ, ed. PLoS One. 2012;7(9):e44992. doi:10.1371/journal.pone.0044992.
- Frangolho A, Correia BE, Vaz DC, et al. Oligomerization profile of human transthyretin variants with distinct amyloidogenicity. Molecules. 2020;25(23):5698. doi:10.3390/molecules25235698.
- Dolado I, Nieto J, Saraiva MJM, et al. Kinetic assay for high-throughput screening of in vitro transthyretin amyloid fibrillogenesis inhibitors. J Comb Chem. 2005;7(2):246–252. doi:10.1021/cc049849s.
- Saelices L, Chung K, Lee JH, et al. Amyloid seeding of transthyretin by ex vivo cardiac fibrils and its inhibition. Proc Natl Acad Sci U S A. 2018;115(29):E6741–E6750. doi:10.1073/pnas.1805131115.
- Foguel D, Suarez MC, Ferrão-Gonzales AD, et al. Dissociation of amyloid fibrils of α-synuclein and transthyretin by pressure reveals their reversible nature and the formation of water-excluded cavities. Proc Natl Acad Sci U S A. 2003;100(17):9831–9836. doi:10.1073/pnas.1734009100.
- Cardoso I, Merlini G, Saraiva MJ. 4 ′‐Iodo‐4′‐deoxydoxorubicin and tetracyclines disrupt transthyretin amyloid fibrils in vitro producing noncytotoxic species: Screening for TTR fibril disrupters. Faseb J. 2003;17(8):803–809. doi:10.1096/fj.02-0764com.
- Bonifácio MJ, Sakaki Y, Saraiva MJ. “In vitro” amyloid fibril formation from transthyretin: the influence of ions and the amyloidogenicity of TTR variants. Biochim Biophys Acta. 1996;1316(1):35–42. doi:10.1016/0925-4439(96)00014-2.
- Lundberg E, Olofsson A, Westermark GT, et al. Stability and fibril formation properties of human and fish transthyretin, and of the Escherichia coli transthyretin-related protein. Febs J. 2009;276(7):1999–2011. doi:10.1111/j.1742-4658.2009.06936.x.
- Marcoux J, Mangione PP, Porcari R, et al. A novel mechano‐enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med. 2015;7(10):1337–1349. doi:10.15252/emmm.201505357.
- Mangione PP, Verona G, Corazza A, et al. Plasminogen activation triggers transthyretin amyloidogenesis in vitro. J Biol Chem. 2018;293(37):14192–14199. doi:10.1074/jbc.RA118.003990.
- Saelices L, Johnson LM, Liang WY, et al. Uncovering the mechanism of aggregation of human transthyretin. J Biol Chem. 2015;290(48):28932–28943. doi:10.1074/jbc.M115.659912.
- Dasari AKR, Yi S, Coats MF, et al. Toxic misfolded transthyretin oligomers with different molecular conformations formed through distinct oligomerization pathways. Biochemistry. 2022;61(21):2358–2365. doi:10.1021/acs.biochem.2c00390.
- Moreira L, Beirão JM, Beirão I, et al. Oligomeric TTR V30M aggregates compromise cell viability, erythropoietin gene expression and promoter activity in the human hepatoma cell line Hep3B. Amyloid. 2015;22(2):93–99. doi:10.3109/13506129.2015.1007497.