
Abstract
RNA methylation is a widespread regulatory mechanism that controls gene expression in physiological processes. In recent years, the mechanisms and functions of RNA methylation under diseased conditions have been increasingly unveiled by RNA sequencing technologies with large scale and high resolution. In this review, the fundamental concept of RNA methylation is introduced, and the common types of transcript methylation and their machineries are described. Then, the regulatory roles of RNA methylation, particularly N6-methyladenosine and 5-methylcytosine, in the vascular lesions of ocular and cardiopulmonary diseases are discussed and compared. The ocular diseases include corneal neovascularization, retinopathy of prematurity, diabetic retinopathy, and pathologic myopia; whereas the cardiopulmonary ailments involve atherosclerosis and pulmonary hypertension. This review hopes to shed light on the common regulatory mechanisms underlying the vascular lesions in these ocular and cardiopulmonary diseases, which may be conducive to developing therapeutic strategies in clinical practice.
Introduction
Epigenetic modifications, including chromatin remodeling, histone modification, DNA methylation, RNA modifications, and microRNA (miR)- and long noncoding RNA (lncRNA)-mediated regulation of gene expression, are involved in the initiation and progression of diseases. As one type of epigenetic modification, RNA modification regulates gene expression at the posttranscriptional level [Citation1], achieves controllable expression of genes without altering their sequences, and endows methylated RNAs with diverse biological functions. However, the low abundance of certain RNA species together with the difficulty in distinguishing modified nucleotides from unmodified nucleotides, used to impede studies of RNA methylation [Citation2]. However, with the development of high-throughput RNA sequencing technology, more than 170 types of modifications have been identified in a variety of RNAs, such as messenger RNA (mRNA), transfer RNA (tRNA), ribosomal RNA (rRNA), circular RNA (circRNA), and lncRNA [Citation3]. RNA methylation refers to the modification process of incorporating a methyl group into RNA bases under catalysis by RNA methyltransferases, with S-adenosyl-L-methionine (SAMe) as the donor [Citation4]. RNA methylation is closely associated with 3 types of proteins, including methyltransferase, demethylase, and RNA-binding protein (RBP). As the name indicates, methyltransferase and demethylase catalyze the processes of methylation and demethylation on RNA bases, respectively [Citation5]. RNA methylation regulates RNA processing, translation, transport, and degradation [Citation6], participating in the maintenance of physiological functions, whereas aberrant RNA methylation is involved in the pathogenesis and progression of diseases, such as cancer, metabolic disorders, and cardiovascular diseases [Citation7–10].
The mechanisms of RNA methylation in the eye have begun to be unveiled in recent years. Studies have shown that levels of RNA methylation ebb and flow under ocular pathological conditions, including corneal, retinal, and choroidal diseases [Citation11]. Moreover, as one of the major vascular systems in the body, cardiopulmonary vascular homeostasis is also regulated, to a considerable extent, by changes in RNA methylation levels under normal and diseased conditions. Although to our knowledge, there has been no translational or clinical study leveraging RNA methylation as interventional or therapeutic strategies to ocular and cardiopulmonary diseases, the pathogenesis and regulation of ocular blood vessels and cardiopulmonary vasculature may share common mechanisms and can be used for mutual reference. This review sought to introduce the fundamental conception and mechanisms of RNA methylation and discuss its regulatory roles in ocular and cardiopulmonary vascular diseases in the hope of inspiring pathogenic exploration and hinting at the interventional target search of these diseases.
RNA methylation
m6A methylation
N6-methyladenosine (m6A) is the most abundant base methylation in eukaryotic mRNA [Citation12] and occurs per 700-800 bp on average in polyadenylated RNA. M6A residues also exist at lower frequencies in other types of RNAs [Citation13]. More than 12,000 m6A sites have been identified in the transcripts of 7,000 genes in humans [Citation14]. The distribution of these sites is highly consistent, with the majority being in the coding region and 3′-untranslated regions (3′-UTRs) of genes. The consensus termination sequence of these sites is RRACH (R = G/A; H = A/C/U) [Citation3]. Certain target sequences of miRNAs and binding sites of RBPs partially overlap with m6A sites at the 3′-UTRs of transcripts, suggesting the associated mechanisms and physiological functions between these two types of RNA modifications [Citation15].
Three types of proteins, including m6A methyltransferases (writers), demethylases (erasers), and RBPs (readers), are responsible for performing, removing, and recognizing m6A modifications, respectively.
Writers
The main role of m6A methyltransferases is to catalyze RNA methylation. The core of the methyltransferase complex (MTC) consists of methyltransferase-like 3 (METTL3), methyltransferase-like 14 (METTL14), and Wilms tumor 1-associated protein (WTAP) () [Citation16]. METTL3 is the only subunit with catalytic activity and binds to the donor SAMe, whereas METTL14 is involved in the recognition and localization of RNA substrates and the stabilization of METTL3 conformation () [Citation3]. The METTL3/14 complex recognizes the GGACU consensus sequence and exhibits high catalytic activity toward m6A modification. In contrast, the transcript’s secondary structure exerts fewer effects on the catalytic activity [Citation17], indicating a sequence specific, but not a structure specific, manner in which the complex catalyzes m6A deposition. Additionally, WTAP serves as a regulatory subunit of MTC to mediate RNA targeting and splicing () [Citation18]. Despite the lack of methylation activity and a weak interaction with the METTL3/14 complex [Citation17], WTAP knockdown elicited 50% and 42% reductions in m6A modification in the transcriptomes of HeLa cells and 293FT cells, respectively [Citation17], implicating the crucial positive regulatory role of WTAP in RNA methylation.
Figure 1. The molecular mechanism underlying m6A RNA methylation in the cell. M6A is the most abundant base methylation in eukaryotic mRNA. In the nucleus, METTL3, METTL14, and WTAP form a methyltransferase complex, which recruits protein factors, such as HAKAI, ZC3H13, and RBM15/15B, to serve as “writers” and catalyze m6A methylation on nascent mRNAs. Conversely, FTO and ALKBH5 function as “erasers” to catalyze demethylation. Moreover, the m6A sites on mRNA are recognized by one of the “readers”, YTHDC1, mediating splicing and nuclear export. Following transport into the cytoplasm, the methylated mature mRNAs can be recognized by another “reader”, YTHDF2, and processed for degradation. Alternatively, YTHDF1 or elF3 may bind to the m6A sites at the 5’-UTR and promote translation of the transcript in a Cap-dependent manner. The third possibility is that the methylated mRNA is bound by a complex composed of YTHDF1, YTHDF2, and YTHDF3, fine tuning the balance between translation and degradation. M6A: N6-methyladenosine; METTL3: methyltransferase-like 3; METTL14: methyltransferase-like 14; WTAP: Wilms tumor 1-associated protein; FTO: Fat mass and obesity-associated protein; ALKBH5: AlkB homolog 5; YTHDC1: YT521-B homology domain-containing protein 1; YTHDF1: YT521-B homology domain-containing family protein 1; YTHDF2: YT521-B homology domain-containing family protein 2; YTHDF3: YT521-B homology domain-containing family protein 3; elF3: eukaryotic initiation factor 3.
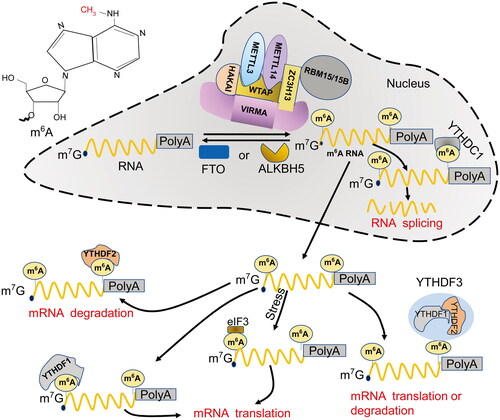
In addition to the catalytic core, MTC recruits VIRMA, HAKAI, KIAA1429, ZC3H13, RBM15/15B and other protein factors for substrate binding, regulation of catalytic activity, and maintenance of complex stability () [Citation19–24]. For instance, VIRMA acts as a scaffold for recruiting the core complex composed of METTL3/METTL14/WTAP and selectively catalyzes methylation at the 3′-UTR and near-stop codon regions of mRNAs; conversely, knocking down VIRMA results in a significant reduction in m6A abundance and downregulation of WTAP expression [Citation22]. In addition, HAKAI and KIAA1429 interact with WTAP and are required for the catalytic methylation of MTC; knocking down HAKAI or KIAA1429 substantially reduced the abundance of m6A [Citation19, Citation25]. RBM15/15B uses Zc3h13 as a linker to interact with WTAP and then binds to MELLT3 in a WTAP-dependent manner [Citation21], whereas Zc3h13 regulates nuclear retention of MTC [Citation16].
Moreover, histones contribute substantially to the accurate and dynamic deposition of m6A on RNA species. For example, histone H3K36me3 is recognized and bound by METTL14, which guides m6A modification near the H3K36me3 peak [Citation26]. Furthermore, the changes in the expression levels of H3K36me3-related methyltransferase and demethylase result in altered abundance of m6A in transcripts, indicating the involvement of histones and their activities in RNA methylation.
Erasers
Demethylases reverse the process of m6A modification on RNAs. Fat mass and obesity-associated protein (FTO) and AlkB homolog 5 (ALKBH5) are two RNA demethylases (). FTO interacts with splicing factors at nuclear speckles [Citation27] and is thought to demethylate m6A on nuclear RNAs [Citation28]. On the other hand, ALKBH5 is a nuclear protein capable of binding single-strand nucleotides and catalyzing m6A demethylation. It is colocalized with mRNA processing factors in nuclear speckles and profoundly influences the assembly, nuclear export, and metabolism of mRNAs [Citation29].
Readers
RBPs specifically recognize m6A sites in RNA and participate in splicing, nuclear export, and degradation of methylated transcripts. As one type of reader, YT521-B homology (YTH) domain-containing proteins recognize and bind to m6A sites with a special pocket-like structure [Citation30] with the assistance of low-complexity disordered regions [Citation31]. YTH domain-containing proteins are categorized into two types: YTH domain-containing protein (YTHDC) and YTHDF (YTH domain-containing family protein) [Citation31]. YTHDC is predominantly located in the nucleus, and YTHDF is a cytoplasmic protein () [Citation25]. The mammalian YTH domain-containing proteins include YTHDC1, YTHDC2, YTHDF1, YTHDF2, and YTHDF3 [Citation32,Citation33]. YTHDC1 regulates RNA splicing by recruiting the splicing factor SRSF3 [Citation34] and mediating the nuclear export of spliced mRNAs [Citation12]. YTHDC2 is mainly expressed in the testis [Citation25], binds to m6A through recognition of RRACH consensus sequence and accelerates mRNA degradation, actively regulating spermatogenesis [Citation32]. Moreover, YTHDF1 promotes translation in a cap-dependent manner, YTHDF2 accelerates mRNA decay, and YTHDF3 cooperates with the former two paralogs, delicately modulating the translation and degradation of m6A-containing mRNAs in the cytoplasm () [Citation35].
Other RBPs have also been reported to regulate RNA translation, metabolism, and splicing by recognizing and binding m6A sites on RNA. For example, insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs) are capable of recognizing the m6A-modified lncRNA DANCER and increasing its stability, thereby boosting the proliferation of pancreatic cancer cells and endowing them with stem cell-like characteristics [Citation36,Citation37]. Moreover, eukaryotic initiation factor 3 (eIF3) can directly bind to a single m6A at the 5′-UTR and initiate the cap-independent mRNA translation in the absence of the cap-binding factor eIF4E [Citation38], serving as an alternative to the YTHDF1-involved cap-dependent mRNA translation.
m5C methylation
5-Methylcytosine (m5C) exists in eukaryotic RNA at a much lower level than m6A [Citation39]. Similar to m6A, the m5C modification deposits a methyl group at specific cytosines on RNA via methyltransferase, regulating gene expression at the posttranscriptional level and influencing cell functions from versatile perspectives. In the past, research on m5C had been limited to tRNA and rRNA [Citation40], where only conservative m5C sites were identified [Citation41]. tRNA and rRNA are modified by methyltransferases of the NOL1/NOP2/SUN domain (NSUN) family containing two conserved cysteine residues [Citation42] as well as by the DNA methyltransferase DNMT2 [Citation43]. NSUN1, NSUN4, and NSUN5 catalyze rRNA m5C modification, whereas DNMT2, NSUN2, NSUN3, and NSUN6 recruit tRNAs as substrates [Citation44].
Recently, m5C modification has been found in mRNAs. The 3′-UTR and 5′-UTR of the messenger transcripts are the predominant m5C-enriched regions, in which cytosine-centered triplet ribonucleotides constitute the majority of the methylation sites [Citation45]. According to the literature reports, NSUN2 mediates m5C methylation at the 3′-UTRs of p53, p16, E2F3, BAK1, ERBB2, and CDK1 transcripts, leading to increased translational efficiency of these transcripts; in contrast, m5C methylation at the 5′-UTR of p27 mRNA suppresses its translational efficiency [Citation45]. These results indicate that the effects of NSUN2-mediated m5C on the translational efficiency of targeted transcripts are transcript- and site-dependent. In addition, m5C modification is also involved in the nuclear export of transcripts. Knocking down ALYREF, a reader protein of m5C, leads to abnormal RNA retention in the nuclei of HeLa cells [Citation46], indicating an m5C-dependent regulation of RNA nuclear export by ALYREF. Moreover, abundant m5C modifications were identified in the transcript genome of murine leukemia virus, and knocking down ALYREF markedly reduced the production of viral gag proteins and inhibited viral replication in murine NIH3T3 fibroblasts, revealing another biological function of m5C modification [Citation47].
m1A methylation
N1-methyladenosine (m1A), mainly distributed in tRNAs and a small number of mRNAs and noncoding RNAs, is formed by transferring a methyl group to the N1 site of adenine nucleoside on RNA. m1A modification is usually detected at nucleotide positions 9, 14, 22, 57, and 58 of both cytoplasmic and mitochondrial tRNAs [Citation48]. Methylation is catalyzed by TRMT10C, TRMT61B, and TRMT6A/61A [Citation49–51] and contributes to structural stability and facilitates correct folding of tRNAs. TRMT61B-mediated modification of m1A endows a positive charge to the elbow region of tRNA, putatively stabilizing its tertiary structure [Citation49]. On the other hand, ALKBH1 and ALKBH3 subserve demethylases of m1A modification, and the former primarily mediates m1A demethylation, inhibiting protein synthesis by curtailing the use of tRNAs in protein translation [Citation52]. In contrast, ALKBH3-mediated demethylation of m1A [Citation53] promoted protein synthesis, and knocking down ALKBH3 suppressed nascent protein synthesis in a human pancreatic cancer cell line [Citation54] and reduced the protein abundance of erb-b2 receptor tyrosine kinase 2 and proline-rich AKT1 substrate 1 in gastrointestinal cancers [Citation55–57].
Regulatory role of m6A in vascular lesions of ocular diseases
Corneal neovascularization
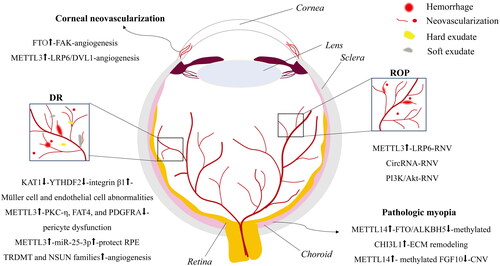
Corneal neovascularization refers to the hypoxia-induced invasion of limbal vessels from the peripheral to central cornea, leading to reduced corneal transparency, inflammatory response [Citation58], and severe vision impairment. The expression of FTO at the protein level was upregulated in a murine model of corneal neovascularization [Citation59]. FTO, a demethylase of m6A, is thought to reduce m6A levels in the transcript of focal adhesion kinase (Fak), a key proangiogenic gene, through an m6A reader YTHDF2, thereby increasing Fak transcript stability and bolstering the development of corneal neovascularization (). Moreover, in mice with corneal alkali burns [Citation60], corneal neovascularization was observed on day 2 and reached the maximum on day 8. At all the time points, the area of corneal neovascularization was smaller in endothelial cell-specific Mettl3 knockout mice than that in wild-type mice. Mechanistically, METTL3 increased m6A modification on the transcripts of low-density lipoprotein receptor related protein 6 (LRP6) and disheveled 1 (DVL1) genes, boosting the translation of these 2 mRNAs in a YTHDF1-dependent manner, augmenting the activity of Wnt signaling pathway and promoting angiogenesis () [Citation60]. Therefore, appropriate regulation of m6A modification could be an effective strategy for preventing or rescuing corneal neovascularization.
Figure 2. An illustration of RNA methylation regulatory mechanisms responsible for vascular lesions in ocular diseases. The sketched RNA methylation-mediated regulatory mechanisms underlying vascular lesions in corneal neovascularization, DR, ROP, and pathologic myopia are illustrated. DR: diabetic retinopathy; ROP: retinopathy of prematurity; RNV: retinal neovascularization; CNV: choroidal neovascularization.
Retinopathy of prematurity
Retinopathy of prematurity (ROP) is a retinal vascular disease that occurs in premature infants. ROP is hallmarked by aberrant proliferation of retinal blood vessels [Citation61], which may lead to retinal neovascularization (RNV), retinal detachment, and even irreversible visual impairment [Citation62]. ROP has become the leading cause of childhood blindness. On the other hand, the eyes of mice are not fully vascularized at birth and hence may serve as experimental models of ROP [Citation63]. The murine model of ROP, designated oxygen-induced retinopathy (OIR), is a useful and convenient platform where pathogenic investigation and therapeutic modality assessment of ROP can be performed. Under the experimental setting of OIR, newborn mice at postnatal day 7 (P7) are continuously exposed to hyperoxia (75% oxygen) for 5 days, resulting in regression and obliteration of developing retinal vasculature; the mice are subsequently removed from the hyperoxia at P12 and maintained under normoxia for another 5 days, leading to relative hypoxia and aberrant proliferation of retinal vasculature. Studies have found that neovascularization in the OIR retina culminates at P17 and is gradually attenuated thereafter [Citation64].
Accumulating evidence has shown that mRNA methylation plays an important regulatory role in RNV. For example, Yao and colleagues observed increased levels of m6A modification in OIR retinas compared to their normal counterparts; they also found a similar trend of m6A modification in human umbilical vein endothelial cells (HUVECs) cultured under hypoxic conditions [Citation60]. Furthermore, the augmented m6A modification in endothelial cells was caused by significant upregulation of METTL3 gene expression. Then, the translation of LRP6 was boosted in a YTHDF1-dependent manner, hence activating canonical Wnt signaling and promoting angiogenesis (). In contrast, METTL3 silencing not only abolished the hypoxia-induced enhancement in the viability, migration, proliferation, and tube formation of human vascular endothelial cells but also inhibited RNV in the OIR mouse model.
Moreover, methylation of noncoding RNAs has also been reported to regulate RNV in OIR retinas. Zhou and colleagues [Citation65] found that m6A-methylated circRNA is involved in the formation of RNV under ischemia. As compared to the retinas of the normal control group, the retinas of the OIR group harbored 88 circRNAs differentially modified by m6A, and the expression of these circRNAs was altered to varying degrees (). However, the specific role of m6A-modified circRNAs in the pathological process of ischemia-induced RNV remains unclear. In addition, Peng et al. [Citation66] found dramatic changes in the m6A levels of mRNAs and lncRNAs in OIR retinas as opposed to normal retinas. Further Kyoto Encyclopedia of Genes and Genomes pathway analysis suggested that hypermethylation and hypomethylation may regulate RNV in OIR mice through PI3K/Akt and autophagy signaling pathways, respectively ().
Diabetic retinopathy
Diabetic retinopathy (DR) refers to a series of retinal damages, such as macular edema and RNV, in the milieu of diabetes-induced metabolic dysregulation of glucose and lipids. DR has become the leading cause of blindness in the working-age population [Citation67]. RNV, as one of the hallmarks of DR, has drawn a great deal of attention, and its pathogenic and regulatory mechanism is one of the hotspots in retinal vascular research. RNV is elicited by abnormal proliferation, migration, and tubulogenesis of retinal microvascular endothelial cells [Citation68]. Moreover, retinal Müller cells, one type of glial cell in the retina, are also activated under DR conditions and secrete proinflammatory cytokines, such as interleukin-1β (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), and vascular endothelial growth factor (VEGF), leading to chronic and sterile inflammation, vascular leakage, and neovascularization in the retina [Citation69]. More recently, Qi et al. [Citation70] found that lysine acetyltransferase 1 (KAT1) was significantly downregulated in the retinas of a streptozotocin (STZ)-induced mouse model of diabetes. In the in vitro experiments, the proliferative and migratory capabilities of retinal vascular endothelial cells were significantly boosted upon high glucose (HG) stimulation, the Müller cells were activated, and the levels of proinflammatory factors were elevated; nevertheless, all these cellular abnormalities were suppressed by upregulation of KAT1 expression. Mechanistically, KAT1, a typical histone acetyltransferase, activates the transcription of the YTHDF2 gene by acetylating histones at the gene’s promoter region, leading to upregulation of YTHDF2 gene expression. Increased abundance of YTHDF2 induces the degradation of m6A-modified messenger transcripts, such as integrin β1 mRNA, causing its instability and downregulated expression (). Hence, the HG-induced diabetic behavior of both retinal vascular endothelial cells and Müller cells was rectified, thereby antagonizing RNV and slowing DR progression.
Pericytes are vascular wall cells embedded in the basement membrane of capillaries. They wrap around capillary endothelial cells and shoulder the role of regulating vascular tension and maintaining the functional integrity of capillaries [Citation71]. Pericyte dysfunction is one of the initiating factors of microvascular remodeling, and pericyte loss, microaneurysms, and acellular capillaries are vascular features of DR [Citation72]. Suo et al. [Citation73] found that m6A modification can also regulate the functions and characteristics of pericytes under stimulation with HG and oxidative stress. m6A modification was augmented in pericytes as a result of METTL3 upregulation, which elicited the degradation of PKC-η, FAT4, and PDGFRA transcripts in a YTHDF2-dependent manner and rendered a significant decrease in the protein levels of the corresponding targets (). The reduced abundance of these 3 proteins then affects the proliferation and differentiation of pericytes. Moreover, overexpression of the Mettl3 gene aggravated HG-induced pericyte dysfunction, manifesting as decreased cell viability, increased macromolecular permeability, and increased cell death (). In contrast, Mettl3 silencing in pericyte cultures or pericyte-specific knockout of the Mettl3 gene in mice protected pericytes from diabetes-induced injury, indicating the important regulatory role of m6A modification in vascular complications of DR.
Recent evidence shows that retinal pigment epithelial cells (RPE) contribute significantly to vascular lesions in the diabetic retina [Citation74]. For instance, the expression of METTL3 and miR-25-3p was downregulated in peripheral venous blood samples of diabetic patients in comparison to that of normal subjects. The downregulated patterns of both genes were recapitulated in HG-stimulated RPE cells. Overexpression of METTL3 and miR-25-3p protected RPE cells from the decreased proliferation and increased apoptosis and pyroptosis induced by HG (). Furthermore, METTL3 overexpression induced miR-25-3p expression, which subsequently suppressed PTEN expression and enhanced Akt phosphorylation [Citation75]. These findings suggest that METTL3 may exert protective effects on RPE cells under HG conditions, such as diabetes, by targeting the miR-25-3p/PTEN/Akt axis (). The abovementioned studies implicate METTL3 and m6A deposition on its downstream genes as emerging therapeutic targets of this debilitating ocular disease.
The level of m5C modification is also closely related to DR-induced angiogenesis. Wang et al. [Citation76] performed data mining on the GSE12610 and GSE111465 datasets related to DR in the Gene Expression Omnibus and found that the expression of the Lrpprc, Nsun4, Nsun6, and Trdmt1 genes was significantly upregulated, and the expression of the Cbll1, Hnrnpc, Mettl3, and Wtap genes was significantly downregulated under DR conditions. Moreover, the upregulation of the genes Trdmt1, Nsun4, and Nsun6 was validated in rat retinal vascular endothelial cell cultures treated with HG (). As mentioned above, the TRDMT and NSUN families are crucial regulators of m5C modification of transcripts. Therefore, the results of this study indicate a regulatory role of m5C modification in DR progression.
Pathologic myopia
With the incidence of myopia skyrocketing in this decade around the globe, studies on the pathogenic and regulatory mechanisms underlying pathologic myopia have become a focus in eye research. Dysfunctions of choroidal vascular endothelial cells, insufficient perfusion of choroidal circulation [Citation11, Citation77], and hypoxia-induced remodeling of extracellular matrix (ECM) in sclera [Citation20] have been considered important myopigenic factors. m6A modification, one of the major regulatory mechanisms of epigenetics, also plays a role in the progression of this prevalent eye disorder and is closely associated with environmental stimuli and lifestyle. Indeed, Wen et al. [Citation11] performed methylated RNA immunoprecipitation (MeRIP) sequencing and found that m6A levels were significantly increased in the transcriptome of lens anterior capsules from cataract patients with high myopia compared to those from cataract patients without myopia. Moreover, the authors identified that the m6A levels at Chitinase 3-like protein 1 (CHI3L1) mRNA were significantly elevated, while those at Fibroblast growth factor 10 (FGF10) mRNA were lowered in cataract patients with high myopia compared to nonmyopic cataract patients () [Citation11]. Furthermore, combined MeRIP and mRNA sequencing analyses demonstrated upregulated expression of the methyltransferase METTL14 and downregulated expression of the demethylases FTO and ALKBH5, all of which might lead to hypermethylation of CHI3L1 transcripts, presumably increasing the abundance of the encoded protein and contributing to ECM remodeling [Citation11]. FGF10 regulates RNA metabolism by binding to an angiogenesis-related RBP known as RNA binding motif protein 10 (RBM10) [Citation78], which implies that the hypomethylation of FGF10 transcripts in cataract patients with high myopia might result in a diminished abundance of FGF10 protein and a weakened interaction between FGF10 and RBM10 proteins, thus increasing the risk of choroidal neovascularization in patients with high myopia () [Citation11].
Regulatory roles of RNA methylation in vascular lesions of cardiopulmonary diseases
Atherosclerosis
Atherosclerosis is characterized by the formation of plaques on the vascular wall, limiting blood flow in the artery. Atherosclerosis is caused by multiple factors, and the pathogenic mechanism has not been elucidated. The main risk factors for atherosclerosis, however, include hypertension, hyperlipidemia, diabetes, and lipidosis. Locally activated endothelial cells and persistently boosted vascular permeability have been shown to stimulate inflammation and plaque formation [Citation79]. Moreover, increased m6A modification and upregulated METTL3 gene expression were identified in vascular endothelial cells as well as in mouse aorta segments exposed to atherogenic stimuli () [Citation80]. On the one hand, the increased expression of METTL3 resulted in blunted RelA/p65 phosphorylation and subsequently enhanced NF-κB activity, heightened inflammatory response, and plaque formation. On the other hand, the upregulation of METTL3 gene expression led to hypermethylation of transcripts, as expected. Indeed, m6A-enhanced cross-linking and immunoprecipitation (eCLIP) identified that NLR family pyrin domain containing 1 (NLRP1) and KLF transcription factor 4 (KLF4) mRNAs are 2 downstream targets of METTL3-mediated hypermethylation. NLRP1 is a protein involved in inflammasome formation and in switching vascular endothelial cells from a resting state to a proinflammatory state () [Citation81]. KLF4 is a transcription factor that regulates endothelial function and maintains vascular homeostasis, it also inhibits inflammation in human vascular endothelial cells and coronary arteries [Citation89]. The METTL3-mediated m6A modification of NLPR1 and KLF4 mRNAs resulted in upregulation of NLRP1 expression and downregulation of KLF4 expression. Specifically, METTL3 with increased abundance induced hypermethylation near the end of the coding region of NLRP1mRNA. m6A methylation was then recognized by the reader YTHDF1, and the translation of the NLRP1 protein was promoted. For the KLF4 transcript, METTL3 causes hypermethylation at the 3′-UTR, which is recognized by YTHDF2 and induces the degradation of KLF4 mRNA () [Citation80]. The differential regulation of downstream target genes leads to endothelial cell inflammation, compensatory arterial expansion, plaque formation, and eventually atherosclerotic lesions in the vasculature.
Table 1. The regulatory roles of RNA methylation in cardiopulmonary diseases.
In addition to vascular endothelial cells, macrophages at vessel lesions are also involved in the inflammatory response to atherosclerosis. Stimulation with oxidized low-density lipoprotein (oxLDL) upregulated the expression of METTL3 and METTL14 in macrophages, while the m6A methylation level decreased in these immune cells. The reason for this paradoxical observation was due to Matrin-3 (Matr3), an RBP that promotes the formation of the METTL3-METTL14 complex, which has been known to mediate the degradation of target transcripts in an m6A methylation-dependent manner (). Therefore, Matr3 can inhibit the oxLDL-induced inflammatory response in macrophages by downregulating the expression of proinflammatory genes and the genes encoding signaling molecules in MAPK pathway via the METTL3-METTL14 complex. Consistently, the expression of MATR3 was decreased in oxLDL-stimulated macrophages and in monocytes in peripheral blood from the patients with coronary artery disease. Additionally, Matr3 overexpression significantly alleviated atherosclerosis in vivo () [Citation82]. These results suggest that MATR3 is a potential target to combat atherosclerosis.
NSUN2-mediated m5C methylation also contributes to atherosclerosis. In a study conducted by Luo et al. [Citation83], leukocyte adhesion to vascular endothelia and the incidence of atherosclerosis were significantly reduced in Nsun2 knockout rats compared to the wild-type counterparts. Furthermore, the protein level of ICAM-1 was significantly diminished in Nsun2 knockout rats (). Mechanistically, the m5C methylation mediated by NSUN2 promoted ICAM-1 translation, increasing leukostasis in endothelial cells and leading to vascular endothelial inflammation and atherosclerosis. Moreover, the level of ICAM-1 is related to angiotensin II (Ang II)-induced hypertension and vascular remodeling. Blocking ICAM-1 ameliorates Ang II-induced arterial hypertension, vascular hypertrophy, fibrosis, macrophage infiltration, and reactive oxygen species (ROS) production and improves vasodilation () [Citation90]. Therefore, the m5C-ICAM-1 axis may not only be pathogenic to atherosclerosis but also be associated with a plethora of vascular diseases.
Pulmonary hypertension
Pulmonary hypertension (PH) is a heterogeneous disease with high morbidity and fatality. According to the current criteria, patients with a mean pulmonary artery pressure (mPAP) > 20 mmHg by right heart catheterization are diagnosed with PH [Citation91]. Moreover, the 6th World Symposium on PH categorized PH into 5 groups: pulmonary arterial hypertension (PAH); PH due to left heart disease; PH due to lung diseases and/or hypoxia; PH due to pulmonary artery obstructions (particularly thromboembolic syndromes); and PH with unclear and/or multifactorial mechanisms [Citation92]. Among these groups, the pathogenic and regulatory mechanisms underlying hypoxia-induced PH have been widely studied due to the availability and accessibility of rodent models. For instance, the expression of myeloid ecotropic viral integration site 1 (MEIS1) was found to be downregulated at both the mRNA and protein levels in the pulmonary arteries of rats exposed to hypoxia and in pulmonary artery smooth muscle cells (PASMCs) cultured under hypoxic conditions. Moreover, overexpression of the Meis1 gene inhibited the proliferation and migration of PASMCs cultured under hypoxia. Conversely, knockout of the Meis1 gene induced the proliferation and migration of PASMCs under normoxia. In addition, the expression of Mettle14 was significantly increased in pulmonary arteries and PASMCs under hypoxia; knockdown of Mettl14 precluded the hypoxia-induced downregulation of Meis1 expression (). Therefore, the results of this study suggested that hypoxia-induced PH promoted the proliferation and migration of PASMCs through the METTL14/MEIS1/p21 signaling pathway [Citation84], and METTL14 might be an interventional target in rescuing hypoxia-induced PH. In addition, it was reported that the m6A level of circRNA was significantly changed in a rat model of hypoxia-induced PH. m6A modification downregulated the expression of circRNAs under hypoxia and affected the circRNA-miRNA–mRNA network. Furthermore, m6A-circXpo6 and m6A-circTmtc3 were downregulated in hypoxic PASMCs (). This might be due to m6A demethylation, which is regulated by hypoxia-inducible factor and mediated by one of the m6A readers, ALKBH5. However, the clinical relevance of m6A-modified circRNAs to PH needs further investigation () [Citation85].
PAH is another type of devastating PH characterized by progressively aggravated pulmonary artery remodeling, which is mainly caused by excessive proliferation and migration of PASMCs [Citation93]. Monocrotaline (MCT)-induced rats are a commonly used animal model for PAH. Zeng et al. [Citation86] established the MCT-induced model and identified significantly increased levels of m6A in the lungs of PAH rats compared to those of normal rats using the MeRIP method. Consistent with this finding, the upregulation of METTL3 and YTHDF1 as well as the downregulation of FTO and ALKBH5 were detected by western blots and immunofluorescence. Furthermore, the mRNAs with elevated levels of m6A were enriched in the pathways mediating inflammation, glycolysis, and interaction between ECM and cell surface receptor, whereas the genes with reduced levels of m6A were enriched in transforming growth factor-beta (TGF-β) signaling pathways (). The m6A-associated alterations indicate the contributions of m6A to PAH at the scale of the whole transcriptome and at the scale of the enriched signaling pathways. In another study, Zheng and coworkers [Citation87] downloaded microarray data (GSE149713) and single-cell sequencing data from MCT-induced PAH rats, as well as RNA sequencing data (GSE144274) from the patients with idiopathic PAH (iPAH) and healthy controls. The authors reanalyzed the data and screened out a differentially expressed gene, heterogeneous nuclear ribonucleoprotein A2/B1 (HNRNPA2B1), which was enriched in the pathways of muscle differentiation and vascular development. Based on the analysis, HNRNPA2B1 might regulate PASMC differentiation and phenotypic transition through the axes of miRNAs and their targets. The upregulation of HNRNPA2B1 was confirmed by immunohistochemistry in lung samples from MCT-PAH rats and from patients with PAH ().
Moreover, the hypoxia-induced PH mouse model and the MCT-induced PAH rat model can both be employed in the study. For instance, elevated m6A levels and YTHDF1 protein levels were detected in lung samples from iPAH patients and from hypoxia-induced PH mice and MCT-induced PAH rats, as well as in hypoxic PASMCs. In both in vivo and in vitro experiments, YTHDF1 deficiency promoted PASMC proliferation, phenotypic transformation, and PH development. Additionally, immunoprecipitation analysis of m6A-modified transcripts showed that the m6A level in the mRNA of melanoma antigen gene family member D1 (Maged1), a potential target of YTHDF1, was significantly increased in hypoxic PH when compared with that in normal control animals. In human and rodent PH samples, MAGED1 protein abundance was also boosted (). Mechanistically, YTHDF1 bound to m6A on Maged1 mRNA and increased translation of this transcript, thereby promoting vascular remodeling and compromising cardiac functions under PH conditions. Consistently, Maged1 silencing inhibited hypoxia-induced PASMC proliferation by downregulating the expression of proliferating cell nuclear antigen () [Citation88]. This suggests that targeting YTHDF1 and MAGED1 may be an effective therapeutic intervention for PH.
The common pathogenic mechanism between ocular vessels and cardiopulmonary vessels
Dyslipidemia-elicited vascular damage
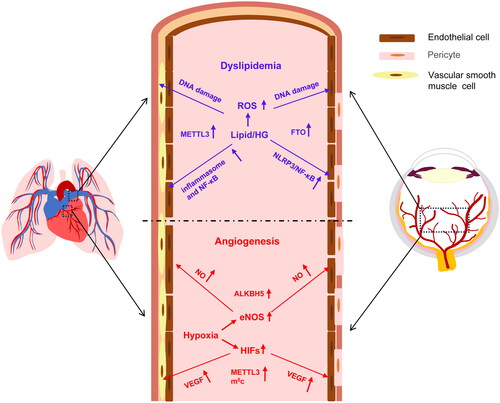
Dyslipidemia, characterized by elevated levels of cholesterol and triglycerides, causes endothelial damage and dysfunction through common mechanisms in eyes, lungs, and hearts. First, in HG-stimulated RPE cells, an increase in mitochondrial superoxide raises intracellular levels of ROS (), and the cells release IL-1β and IL-18, activating NLRP3 inflammasome and NF-κB signaling pathways, causing RPE cell damage under DR conditions [Citation94]. Moreover, HG-induced ROS production causes extensive DNA damage and remarkable impairment in endothelial cell repair system (). Therefore, ROS can trigger pyroptosis, parthanatos, and ferroptosis of endothelial cells, leading to vascular dysfunction, breakdown of the blood–retinal barrier, and neovascularization [Citation95]. Likewise, cardiopulmonary vascular smooth muscle cells also produce increased levels of ROS and aggravated DNA damage at the site of atherosclerotic plaques. Oxidative DNA damage accumulates to induce cell senescence and death; oxidized DNA also activates inflammasome and NF-κB signaling pathways and initiates inflammatory responses in vascular smooth muscle cells () [Citation96–100]. Among the vascular complications caused by dysregulated lipid metabolism, the oxidative stress, DNA damage, and inflammatory responses are the key pathological processes that are shared by DR and atherosclerosis and may be regulated by RNA methylation.
Figure 3. The molecular mechanisms shared by ocular and cardiopulmonary diseases. The dysregulated lipid and glucose metabolism generates an excessive amount of ROS, eliciting oxidative stress, DNA damage, and activation of NFκB signaling pathways and subsequent production of proinflammatory factors in both vascular endothelial cells and smooth muscle cells. Moreover, the expression of the transcription factors HIFs and the enzyme eNOS was upregulated under hypoxic conditions in vascular endothelial and smooth muscle cells, and their corresponding downstream factors, VEGF and NO, act through signaling pathways to promote cell proliferation, causing neovascularization and vascular remodeling in target tissues. HIFs: hypoxia-inducible factors; eNOS: endothelial nitric oxide synthase; VEGF: vascular endothelial growth factor; NO: nitric oxide; ROX: reactive oxygen species; NFκB: nuclear factor κ B; TGF-β: transforming growth factor-β.
Indeed, atherogenic stimuli in vascular endothelial cells upregulated METTL3 expression, elevated m6A RNA levels, and boosted phosphorylation of NF-κB family member RelA/p65, thereby augmenting monocyte adhesion and endothelial cell inflammation [Citation80]. Moreover, high glucose, a hallmark of dysregulated glucose metabolism, resulted in the upregulated expression of an m6A eraser FTO, reduced m6A levels on the transcript of TNFAIP3 Interacting Protein 1 (TNIP1), and the diminished abundance of TNIP1 mRNA. Subsequently, TNIP1 deficiency activated inflammatory factors such as NF-κB, and exacerbated retinal vascular endothelial cell injury and leakage [Citation101]. In addition, the endothelial cell-specific knockout of FTO protected mice from the high fat diet-induced glucose intolerance and insulin resistance and the cardiovascular disease-associated endothelial cell damage by enhancing AKT phosphorylation [Citation102].
Hypoxia-induced angiogenesis
Angiogenesis is a crucial process under physiological and pathological conditions, fulfilling the requirements of nutrients and oxygen for tissues. Both ocular and cardiopulmonary vessels entail the proliferation and migration of endothelial cells during hypoxia-induced angiogenesis. Hypoxia induces angiogenesis through signaling pathways mediated by hypoxia-inducible factors (HIFs), which in turn regulate the expression of downstream target genes.
One category of these target genes belongs to the machinery of RNA methylation, such as the methyltransferase family and YTHDF, the “writer” and “reader” of m6A methylation, respectively. For instance, hypoxia induces METTL3 expression through activation of both HIF-1α and HIF-2α, and the upregulated METTL3 increases the level of m6A RNA modification in endothelial cells. Therefore, overexpression of METTL3 promotes viability, proliferation, migration, and tube formation of HUVECs; whereas knockdown of METTL3 inhibits pathological angiogenesis [Citation60].
Another category of the hypoxia target genes includes VEGF and its receptor system, which constitutes a proangiogenic signaling pathway, promoting both ocular and cardiopulmonary angiogenesis by enhancing endothelial cell proliferation, migration, and tube formation [Citation103]. Anti-VEGF therapy has been widely used in DR, ROP, and age-related macular degeneration, rendering VEGF a crucial target for treating retinal vasculopathy [Citation104]. On the other hand, simultaneous inhibition of receptor signaling mediated by VEGF, platelet-derived growth factor, and FGF has been shown to prevent and reverse PAH as well as subsequent vascular remodeling [Citation105]. Furthermore, in hypoxia-induced PH, the VEGF signaling pathway not only participates in vascular remodeling characterized by arterial smooth muscle cell proliferation but also contributes to vascular leakage caused by enfeebled connectivity between endothelial cells () [Citation106]. Notably, there is evidence that the expression of VEGF can also be regulated by RNA methylation. In a swine model of cardiac failure with chronic kidney disease, cardiac-specific m5C-MeRIP and mRNA-sequencing identified a group of VEGF -related genes with elevated m5C levels but dampened mRNA levels as compared to the normal controls. This finding was validated by conventional qPCR at the transcript level and Western blot at the protein level [Citation107]. The RNA methylation-mediated downregulation and inhibition of VEGF signaling could be the mechanism underlying the compromised angiogenesis and microvascular rarefaction observed in the endocardia of the patients with heart failure.
Nitric oxide (NO) and endothelial nitric oxide synthase (eNOS) are the third category of the target genes involved in the hypoxia-induced angiogenesis. The eNOS-deficient OIR mice exhibited significant reduction in pathological neovascularization [Citation108]. Similarly, eNOS expression was upregulated and its activity was boosted in a PAH animal model under hypoxia. PI3K-AKT-eNOS pathway is activated in pulmonary artery endothelial cells, enhancing NO production and promoting endothelial cell proliferation, pulmonary microvascular muscularization, and pulmonary arterial remodeling () [Citation109,Citation110]. Interestingly, the eNOS signaling pathway can also be regulated by RNA methylation. Under proinflammatory and hypoxic conditions, the expression of m6A eraser ALKBH5 was upregulated in endothelial cells. Silencing ALKBH5 elevated the m6A levels on the sphingosine kinase-1 (SPHK1) transcript, resulting in degradation and instability of the SPHK1 transcript, diminishing phosphorylation of AKT and eNOS, and eventually supressing endothelial cell angiogenesis [Citation111].
Summary
Cardiopulmonary diseases are life-threatening and have been widely studied. The results of vessel studies in cardiopulmonary diseases can be a good reference for the study and management of vascular lesions in ocular diseases, whether in the exploration of pathogenesis or in the search for interventional targets. The mechanisms underlying major pathological processes, such as hypoxia-induced angiogenesis and dyslipidemia-elicited vascular endothelial cell damage, are common between ocular and cardiopulmonary vascular diseases.
RNA methylation, as a posttranscriptional epigenetic modification, navigates the disease course by regulating the key molecule expression in the signaling pathways orchestrating the major pathological processes. Specifically, m6A and m5C RNA modifications regulate the expression of disease-related genes through their writers, erasers, and readers. Generally speaking, if the disease-related transcript is hypermethylated, it would be more appropriate to select “writer” and “reader” as intervention targets; whereas the hypomethylated disease-related transcript would prefer an “eraser” as the intervention target [Citation112]. However, due to the complexity and specificity of the disease pathogenesis and regulation, the interventional target selection should also consider the uniqueness of RNA methylation at a specific target.
Author contributions
Y.Z. and Y.C. conceived, designed, and supervised the project. Y.Z. obtained fundings and revised versions of manuscript, and formatted and submitted manuscripts. S.L. searched and archived the literature, wrote the draft, and depicted the figures. All authors reviewed and approved the manuscript.
| Abbreviations | ||
| ALKBH5 | = | AlkB homolog 5 |
| CHI3L1 | = | Chitinase 3-like protein 1 |
| circRNA | = | circular RNA |
| CTD | = | C-terminal domain |
| DR | = | diabetic retinopathy |
| DVL1 | = | disheveled 1 |
| eCLIP | = | enhanced cross-linking and immunoprecipitation |
| ECM | = | extracellular matrix |
| eIF3 | = | eukaryotic initiation factor 3 |
| eNOS | = | endothelial nitric oxide synthase |
| FAK | = | focal adhesion kinase; FGF10: fibroblast growth factor 10 |
| FTO | = | fat mass and obesity-associated protein |
| Hexim1 | = | hexamethylene bisacetamide-induced protein 1 |
| HG | = | high glucose |
| HIFs | = | hypoxia-inducible factors |
| HUVECs | = | human umbilical vein endothelial cells |
| IGF2BPs | = | insulin-like growth factor 2 mRNA-binding proteins |
| IL-1β | = | interleukin-1β |
| KAT1 | = | lysine acetyltransferase 1 |
| KLF4 | = | KLF transcription factor 4 |
| lncRNA | = | long noncoding RNA |
| LRP6 | = | lipoprotein receptor related protein 6 |
| MAGED1 | = | melanoma antigen gene family member D1 |
| Matr3 | = | Matrin-3 |
| m1A | = | N1-methyladenosine |
| m6A | = | N6-methyladenosine |
| m5C | = | 5-Methylcytosine |
| MCT | = | Monocrotaline |
| MEIS1 | = | myeloid ecotropic viral integration site 1 |
| MeRIP | = | methylated RNA immunoprecipitation |
| METTL3 | = | methyltransferase-like 3 |
| METTL14 | = | methyltransferase-like 14 |
| miR | = | microRNA |
| mPAP | = | mean pulmonary artery pressure |
| mRNA | = | messenger RNA |
| MTC | = | methyltransferase complex |
| NLRP1 | = | NLR family pyrin domain containing 1 |
| NO | = | nitric oxide |
| NSUN | = | NOL1/NOP2/SUN domain |
| OIR | = | oxygen-induced retinopathy |
| oxLDL | = | oxidized low-density lipoprotein |
| PAH | = | pulmonary arterial hypertension |
| PASMCs | = | pulmonary artery smooth muscle cells |
| PH | = | pulmonary hypertension |
| RBM10 | = | RNA binding motif protein 10 |
| RBP | = | RNA-binding protein |
| RNV | = | retinal neovascularization |
| ROP | = | retinopathy of prematurity |
| ROS | = | reactive oxygen species |
| RPE | = | retinal pigment epithelial cells |
| rRNA | = | ribosomal RNA |
| SAMe | = | S-adenosyl-L-methionine |
| STZ | = | streptozotocin |
| TGF-β | = | transforming growth factor-beta |
| TNF-α | = | tumor necrosis factor-α |
| tRNA | = | transfer RNA |
| VEGF | = | vascular endothelial growth factor |
| WTAP | = | Wilms tumor 1-associated protein |
| YTH | = | YT521-B homology |
| YTHDC | = | YTH domain-containing protein |
| 3'-UTRs | = | 3'-untranslated regions. |
Disclosure statement
The authors declare no competing interests.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Chen X, Sun YZ, Liu H, et al. RNA methylation and diseases: experimental results, databases, Web servers and computational models. Brief Bioinform. 2019;20(3):896–917. doi: 10.1093/bib/bbx142.
- Li X, Xiong X, Yi C. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat Methods. 2016;14(1):23–31. doi: 10.1038/nmeth.4110.
- Huang H, Weng H, Chen J. The biogenesis and precise control of RNA m(6)A methylation. Trends Genet. 2020;36(1):44–52. doi: 10.1016/j.tig.2019.10.011.
- Traube FR, Carell T. The chemistries and consequences of DNA and RNA methylation and demethylation. RNA Biol. 2017;14(9):1099–1107. doi: 10.1080/15476286.2017.1318241.
- Xu H, Wang Z, Chen M, et al. YTHDF2 alleviates cardiac hypertrophy via regulating Myh7 mRNA decoy. Cell Biosci. 2021;11(1):132. doi: 10.1186/s13578-021-00649-7.
- Covelo-Molares H, Bartosovic M, Vanacova S. RNA methylation in nuclear pre-mRNA processing. Wiley Interdiscip Rev RNA. 2018;9(6):e1489. doi: 10.1002/wrna.1489.
- Wang T, Kong S, Tao M, et al. The potential role of RNA N6-methyladenosine in cancer progression. Mol Cancer. 2020;19(1):88. doi: 10.1186/s12943-020-01204-7.
- Han M, Liu Z, Xu Y, et al. Abnormality of m6A mRNA methylation is involved in Alzheimer’s disease. Front Neurosci. 2020;14:98. doi: 10.3389/fnins.2020.00098.
- Ling C, Rönn T. Epigenetics in human obesity and type 2 diabetes. Cell Metab. 2019;29(5):1028–1044. doi: 10.1016/j.cmet.2019.03.009.
- Qin Y, Li L, Luo E, et al. Role of m6A RNA methylation in cardiovascular disease (review). Int J Mol Med. 2020;46(6):1958–1972. doi: 10.3892/ijmm.2020.4746.
- Wen K, Zhang Y, Li Y, et al. Comprehensive analysis of transcriptome-wide m(6)A methylome in the anterior capsule of the lens of high myopia patients. Epigenetics. 2021;16(9):955–968. doi: 10.1080/15592294.2020.1834917.
- Roundtree IA, Evans ME, Pan T, et al. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187–1200. doi: 10.1016/j.cell.2017.05.045.
- Lavi U, Fernandez-Muñoz R, Darnell JE.Jr. Content of N-6 methyl adenylic acid in heterogeneous nuclear and messenger RNA of HeLa cells. Nucleic Acids Res. 1977;4(1):63–69. doi: 10.1093/nar/4.1.63.
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. doi: 10.1038/nature11112.
- Chen J, Fang X, Zhong P, et al. N6-methyladenosine modifications: interactions with novel RNA-binding proteins and roles in signal transduction. RNA Biol. 2019;16(8):991–1000. doi: 10.1080/15476286.2019.1620060.
- Garcias Morales D, Reyes JL. A birds’-eye view of the activity and specificity of the mRNA m(6) A methyltransferase complex. Wiley Interdiscip Rev RNA. 2021;12(1):e1618. doi: 10.1002/wrna.1618.
- Liu J, Yue Y, Han D, et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–95. doi: 10.1038/nchembio.1432.
- Wu R, Jiang D, Wang Y, et al. N (6)-methyladenosine (m(6)A) methylation in mRNA with A dynamic and reversible epigenetic modification. Mol Biotechnol. 2016;58(7):450–459. doi: 10.1007/s12033-016-9947-9.
- Schwartz S, Mumbach MR, Jovanovic M, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5’ sites. Cell Rep. 2014;8(1):284–296. doi: 10.1016/j.celrep.2014.05.048.
- Wu H, Chen W, Zhao F, et al. Scleral hypoxia is a target for myopia control. Proc Natl Acad Sci U S A. 2018;115(30):E7091–e7100. doi: 10.1073/pnas.1721443115.
- Knuckles P, Lence T, Haussmann IU, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes Dev. 2018;32(5-6):415–429. doi: 10.1101/gad.309146.117.
- Yue Y, Liu J, Cui X, et al. VIRMA mediates preferential m(6)A mRNA methylation in 3'UTR and near stop codon and associates with alternative polyadenylation. Cell Discov. 2018;4(1):10. doi: 10.1038/s41421-018-0019-0.
- Patil DP, Chen CK, Pickering BF, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537(7620):369–373. doi: 10.1038/nature19342.
- Wen J, Lv R, Ma H, et al. Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Mol Cell. 2018;69(6):1028–1038.e6. doi: 10.1016/j.molcel.2018.02.015.
- Balacco DL, Soller M. The m(6)A writer: rise of a machine for growing tasks. Biochemistry. 2019;58(5):363–378. doi: 10.1021/acs.biochem.8b01166.
- Huang H, Weng H, Zhou K, et al. Histone H3 trimethylation at lysine 36 guides m(6)A RNA modification co-transcriptionally. Nature. 2019;567(7748):414–419. doi: 10.1038/s41586-019-1016-7.
- Church C, Moir L, McMurray F, et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. 2010;42(12):1086–1092. doi: 10.1038/ng.713.
- Jia G, Fu Y, Zhao X, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–887. doi: 10.1038/nchembio.687.
- Zheng G, Dahl JA, Niu Y, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29. doi: 10.1016/j.molcel.2012.10.015.
- Luo S, Tong L. Molecular basis for the recognition of methylated adenines in RNA by the eukaryotic YTH domain. Proc Natl Acad Sci U S A. 2014;111(38):13834–13839. doi: 10.1073/pnas.1412742111.
- Patil DP, Pickering BF, Jaffrey SR. Reading m(6)A in the transcriptome: m(6)A-binding proteins. Trends Cell Biol. 2018;28(2):113–127. doi: 10.1016/j.tcb.2017.10.001.
- Hsu PJ, Zhu Y, Ma H, et al. Ythdc2 is an N(6)-methyladenosine binding protein that regulates mammalian spermatogenesis. Cell Res. 2017;27(9):1115–1127. doi: 10.1038/cr.2017.99.
- Wojtas MN, Pandey RR, Mendel M, et al. Regulation of m(6)A transcripts by the 3'-5’ RNA helicase YTHDC2 is essential for a successful meiotic program in the mammalian germline. Mol Cell. 2017;68(2):374–387.e12. doi: 10.1016/j.molcel.2017.09.021.
- Xiao W, Adhikari S, Dahal U, et al. Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol Cell. 2016;61(4):507–519. doi: 10.1016/j.molcel.2016.01.012.
- Shi H, Wang X, Lu Z, et al. YTHDF3 facilitates translation and decay of N(6)-methyladenosine-modified RNA. Cell Res. 2017;27(3):315–328. doi: 10.1038/cr.2017.15.
- Hu X, Peng WX, Zhou H, et al. IGF2BP2 regulates DANCR by serving as an N6-methyladenosine reader. Cell Death Differ. 2020;27(6):1782–1794. doi: 10.1038/s41418-019-0461-z.
- Walker MJ, Shortridge MD, Albin DD, et al. Structure of the RNA specialized translation initiation element that recruits eIF3 to the 5'-UTR of c-Jun. J Mol Biol. 2020;432(7):1841–1855. doi: 10.1016/j.jmb.2020.01.001.
- Meyer KD, Patil DP, Zhou J, et al. 5’ UTR m(6)A promotes cap-independent translation. Cell. 2015;163(4):999–1010. doi: 10.1016/j.cell.2015.10.012.
- Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42. doi: 10.1038/nrm.2016.132.
- Squires JE, Patel HR, Nousch M, et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012;40(11):5023–5033. doi: 10.1093/nar/gks144.
- Chen YS, Yang WL, Zhao YL, et al. Dynamic transcriptomic m(5) C and its regulatory role in RNA processing. Wiley Interdiscip Rev RNA. 2021;12(4):e1639. doi: 10.1002/wrna.1639.
- King MY, Redman KL. RNA methyltransferases utilize two cysteine residues in the formation of 5-methylcytosine. Biochemistry. 2002;41(37):11218–11225. doi: 10.1021/bi026055q.
- Chellamuthu A, Gray SG. The RNA methyltransferase NSUN2 and its potential roles in cancer. Cells. 2020;9(8):1758. doi: 10.3390/cells9081758.
- Trixl L, Lusser A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdiscip Rev RNA. 2019;10(1):e1510. doi: 10.1002/wrna.1510.
- Wei Z, Panneerdoss S, Timilsina S, et al. Topological characterization of human and mouse m(5)C epitranscriptome revealed by bisulfite sequencing. Int J Genomics. 2018;2018:1351964–1351919. doi: 10.1155/2018/1351964.
- Yang X, Yang Y, Sun BF, et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017;27(5):606–625. doi: 10.1038/cr.2017.55.
- Eckwahl M, Xu R, Michalkiewicz J, et al. 5-Methylcytosine RNA modifications promote retrovirus replication in an ALYREF reader protein-dependent manner. J Virol. 2020;94(13):e00544–00520. doi: 10.1128/jvi.00544-20.
- Oerum S, Dégut C, Barraud P, et al. m1A post-transcriptional modification in tRNAs. Biomolecules. 2017;7(1):20. doi: 10.3390/biom7010020.
- Vilardo E, Nachbagauer C, Buzet A, et al. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012;40(22):11583–11593. doi: 10.1093/nar/gks910.
- Chujo T, Suzuki T. Trmt61B is a methyltransferase responsible for 1-methyladenosine at position 58 of human mitochondrial tRNAs. RNA. 2012;18(12):2269–2276. doi: 10.1261/rna.035600.112.
- Li X, Xiong X, Zhang M, et al. Base-resolution mapping reveals distinct m(1)A methylome in nuclear- and mitochondrial-encoded transcripts. Mol Cell. 2017;68(5):993–1005.e9. doi: 10.1016/j.molcel.2017.10.019.
- Zhang C, Jia G. Reversible RNA Modification N(1)-methyladenosine (m(1)A) in mRNA and tRNA. Genomics Proteomics Bioinformatics. 2018;16(3):155–161. doi: 10.1016/j.gpb.2018.03.003.
- Chen Z, Qi M, Shen B, et al. Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA-derived small RNAs. Nucleic Acids Res. 2019;47(5):2533–2545. doi: 10.1093/nar/gky1250.
- Ueda Y, Ooshio I, Fusamae Y, et al. AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci Rep. 2017;7(1):42271. doi: 10.1038/srep42271.
- Zhao Y, Zhao Q, Kaboli PJ, et al. m1A regulated genes modulate PI3K/AKT/mTOR and ErbB pathways in gastrointestinal cancer. Transl Oncol. 2019;12(10):1323–1333. doi: 10.1016/j.tranon.2019.06.007.
- Ross JS, Fakih M, Ali SM, et al. Targeting HER2 in colorectal cancer: the landscape of amplification and short variant mutations in ERBB2 and ERBB3. Cancer. 2018;124(7):1358–1373. doi: 10.1002/cncr.31125.
- Han B, Cai H, Chen Y, et al. The role of TGFBI (βig-H3) in gastrointestinal tract tumorigenesis. Mol Cancer. 2015;14(1):64. doi: 10.1186/s12943-015-0335-z.
- Liu S, Romano V, Steger B, et al. Gene-based antiangiogenic applications for corneal neovascularization. Surv Ophthalmol. 2018;63(2):193–213. doi: 10.1016/j.survophthal.2017.10.006.
- Shan K, Zhou RM, Xiang J, et al. FTO regulates ocular angiogenesis via m(6)A-YTHDF2-dependent mechanism. Exp Eye Res. 2020;197:108107. doi: 10.1016/j.exer.2020.108107.
- Yao MD, Jiang Q, Ma Y, et al. Role of METTL3-dependent N(6)-methyladenosine mRNA modification in the promotion of angiogenesis. Mol Ther. 2020;28(10):2191–2202. doi: 10.1016/j.ymthe.2020.07.022.
- Hartnett ME. Pathophysiology of retinopathy of prematurity. Annu Rev Vis Sci. 2023;9(1):39–70. doi: 10.1146/annurev-vision-093022-021420.
- Bowl W, Lorenz B, Stieger K, et al. Fundus-controlled dark adaptometry in young children without and with spontaneously regressed retinopathy of prematurity. Transl Vis Sci Technol. 2019;8(3):62. doi: 10.1167/tvst.8.3.62.
- Ashton N, Ward B, Serpell G. Effect of oxygen on developing retinal vessels with particular reference to the problem of retrolental fibroplasia. Br J Ophthalmol. 1954;38(7):397–432. doi: 10.1136/bjo.38.7.397.
- Connor KM, Krah NM, Dennison RJ, et al. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat Protoc. 2009;4(11):1565–1573. doi: 10.1038/nprot.2009.187.
- Zhou Y, Li B, Wang Z, et al. m(6)A modifications of circular RNAs in ischemia-induced retinal neovascularization. Int J Med Sci. 2023;20(2):254–261. doi: 10.7150/ijms.79409.
- Peng Y, Wang Z, Li B, et al. N(6)-methyladenosine modifications of mRNAs and long noncoding RNAs in oxygen-induced retinopathy in mice. Exp Eye Res. 2022;220:109114. doi: 10.1016/j.exer.2022.109114.
- Cheung N, Mitchell P, Wong TY. Diabetic retinopathy. Lancet. 2010;376(9735):124–136. doi: 10.1016/s0140-6736(09)62124-3.
- Shao J, Fan G, Yin X, et al. A novel transthyretin/STAT4/miR-223-3p/FBXW7 signaling pathway affects neovascularization in diabetic retinopathy. Mol Cell Endocrinol. 2019;498:110541. doi: 10.1016/j.mce.2019.110541.
- Tu Y, Zhu M, Wang Z, et al. Melatonin inhibits Müller cell activation and pro-inflammatory cytokine production via upregulating the MEG3/miR-204/Sirt1 axis in experimental diabetic retinopathy. J Cell Physiol. 2020;235(11):8724–8735. doi: 10.1002/jcp.29716.
- Qi Y, Yao R, Zhang W, et al. KAT1 triggers YTHDF2-mediated ITGB1 mRNA instability to alleviate the progression of diabetic retinopathy. Pharmacol Res. 2021;170:105713. doi: 10.1016/j.phrs.2021.105713.
- Geevarghese A, Herman IM. Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Transl Res. 2014;163(4):296–306. doi: 10.1016/j.trsl.2014.01.011.
- Huang H. Pericyte-endothelial interactions in the retinal microvasculature. Int J Mol Sci. 2020;21(19):7413. doi: 10.3390/ijms21197413.
- Suo L, Liu C, Zhang QY, et al. METTL3-mediated N(6)-methyladenosine modification governs pericyte dysfunction during diabetes-induced retinal vascular complication. Theranostics. 2022;12(1):277–289. doi: 10.7150/thno.63441.
- Tonade D, Kern TS. Photoreceptor cells and RPE contribute to the development of diabetic retinopathy. Prog Retin Eye Res. 2021;83:100919. doi: 10.1016/j.preteyeres.2020.100919.
- Zha X, Xi X, Fan X, et al. Overexpression of METTL3 attenuates high-glucose induced RPE cell pyroptosis by regulating miR-25-3p/PTEN/Akt signaling cascade through DGCR8. Aging (Albany NY). 2020;12(9):8137–8150. doi: 10.18632/aging.103130.
- Wang X, Li X, Zong Y, et al. Identification and validation of genes related to RNA methylation modification in diabetic retinopathy. Curr Eye Res. 2023;48(11):1034–1049. doi: 10.1080/02713683.2023.2238144.
- Geng C, Liu S, Wang J, et al. Targeting the cochlin/SFRP1/CaMKII axis in the ocular posterior pole prevents the progression of nonpathologic myopia. Commun Biol. 2023;6(1):884. doi: 10.1038/s42003-023-05267-2.
- Loiselle JJ, Sutherland LC. RBM10: harmful or helpful-many factors to consider. J Cell Biochem. 2018;119(5):3809–3818. doi: 10.1002/jcb.26644.
- He M, Martin M, Marin T, et al. Endothelial mechanobiology. APL Bioeng. 2020;4(1):010904. doi: 10.1063/1.5129563.
- Chien CS, Li JY, Chien Y, et al. METTL3-dependent N(6)-methyladenosine RNA modification mediates the atherogenic inflammatory cascades in vascular endothelium. Proc Natl Acad Sci U S A. 2021;118(7):e2025070118. doi: 10.1073/pnas.2025070118.
- Bleda S, de Haro J, Varela C, et al. NLRP1 inflammasome, and not NLRP3, is the key in the shift to proinflammatory state on endothelial cells in peripheral arterial disease. Int J Cardiol. 2014;172(2):e282-284–e284. doi: 10.1016/j.ijcard.2013.12.201.
- Sun Z, Chen W, Wang Z, et al. Matr3 reshapes m6A modification complex to alleviate macrophage inflammation during atherosclerosis. Clin Immunol. 2022;245:109176. doi: 10.1016/j.clim.2022.109176.
- Luo Y, Feng J, Xu Q, et al. NSun2 deficiency protects endothelium from inflammation via mRNA methylation of ICAM-1. Circ Res. 2016;118(6):944–956. doi: 10.1161/circresaha.115.307674.
- Yao MZ, Ge XY, Liu T, et al. MEIS1 regulated proliferation and migration of pulmonary artery smooth muscle cells in hypoxia-induced pulmonary hypertension. Life Sci. 2020;255:117822. doi: 10.1016/j.lfs.2020.117822.
- Su H, Wang G, Wu L, et al. Transcriptome-wide map of m(6)A circRNAs identified in a rat model of hypoxia mediated pulmonary hypertension. BMC Genomics. 2020;21(1):39. doi: 10.1186/s12864-020-6462-y.
- Zeng Y, Huang T, Zuo W, et al. Integrated analysis of m(6)A mRNA methylation in rats with monocrotaline-induced pulmonary arterial hypertension. Aging (Albany NY). 2021;13(14):18238–18256. doi: 10.18632/aging.203230.
- Zheng H, Hua J, Li H, et al. Comprehensive analysis of the expression of N6-methyladenosine RNA methylation regulators in pulmonary artery hypertension. Front Genet. 2022;13:974740. doi: 10.3389/fgene.2022.974740.
- Hu L, Wang J, Huang H, et al. YTHDF1 regulates pulmonary hypertension through translational control of MAGED1. Am J Respir Crit Care Med. 2021;203(9):1158–1172. doi: 10.1164/rccm.202009-3419OC.
- Bai L, Shyy JYP. Shear stress regulation of endothelium: a double-edged sword. J Transl Int Med. 2018;6(2):58–61. doi: 10.2478/jtim-2018-0019.
- Lang PP, Bai J, Zhang YL, et al. Blockade of intercellular adhesion molecule-1 prevents angiotensin II-induced hypertension and vascular dysfunction. Lab Invest. 2020;100(3):378–386. doi: 10.1038/s41374-019-0320-z.
- Maron BA. Revised definition of pulmonary hypertension and approach to management: a clinical primer. J Am Heart Assoc. 2023;12(8):e029024. doi: 10.1161/jaha.122.029024.
- Galiè N, McLaughlin VV, Rubin LJ, et al. An overview of the 6th World Symposium on Pulmonary Hypertension. Eur Respir J. 2019;53(1):1802148. doi: 10.1183/13993003.02148-2018.
- Brock M, Schuoler C, Leuenberger C, et al. Analysis of hypoxia-induced noncoding RNAs reveals metastasis-associated lung adenocarcinoma transcript 1 as an important regulator of vascular smooth muscle cell proliferation. Exp Biol Med (Maywood). 2017;242(5):487–496. doi: 10.1177/1535370216685434.
- Bang E, Park C, Hwangbo H, et al. Spermidine attenuates high glucose-induced oxidative damage in retinal pigment epithelial cells by inhibiting production of ROS and NF-κB/NLRP3 inflammasome pathway. Int J Mol Sci. 2023;24(13):10550. doi: 10.3390/ijms241310550.
- Zheng D, Liu J, Piao H, et al. ROS-triggered endothelial cell death mechanisms: focus on pyroptosis, parthanatos, and ferroptosis. Front Immunol. 2022;13:1039241. doi: 10.3389/fimmu.2022.1039241.
- Shah A, Gray K, Figg N, et al. Defective base excision repair of oxidative DNA damage in vascular smooth muscle cells promotes atherosclerosis. Circulation. 2018;138(14):1446–1462. doi: 10.1161/circulationaha.117.033249.
- Zhao H, He Y. Lysophosphatidylcholine offsets the protective effects of bone marrow mesenchymal stem cells on inflammatory response and oxidative stress injury of retinal endothelial cells via TLR4/NF-κB signaling. J Immunol Res. 2021;2021:2389029–2389010. doi: 10.1155/2021/2389029.
- Qin X, Zhang Z, Xu H, et al. Notch signaling protects retina from nuclear factor-κB- and poly-ADP-ribose-polymerase-mediated apoptosis under high-glucose stimulation. Acta Biochim Biophys Sin (Shanghai). 2011;43(9):703–711. doi: 10.1093/abbs/gmr069.
- Qin M, Luo Y, Lu S, et al. Ginsenoside F1 ameliorates endothelial cell inflammatory injury and prevents atherosclerosis in mice through A20-mediated suppression of NF-kB signaling. Front Pharmacol. 2017;8:953. doi: 10.3389/fphar.2017.00953.
- Pan W, Yu H, Huang S, et al. Resveratrol protects against TNF-α-induced injury in human umbilical endothelial cells through promoting sirtuin-1-induced repression of NF-KB and p38 MAPK. PLoS One. 2016;11(1):e0147034. doi: 10.1371/journal.pone.0147034.
- Zhou C, She X, Gu C, et al. FTO fuels diabetes-induced vascular endothelial dysfunction associated with inflammation by erasing m6A methylation of TNIP1. J Clin Invest. 2023;133(19):e160517. doi: 10.1172/jci160517.
- Krüger N, Biwer LA, Good ME, et al. Loss of endothelial FTO antagonizes obesity-induced metabolic and vascular dysfunction. Circ Res. 2020;126(2):232–242. doi: 10.1161/circresaha.119.315531.
- Uemura A, Fruttiger M, D'Amore PA, et al. VEGFR1 signaling in retinal angiogenesis and microinflammation. Prog Retin Eye Res. 2021;84:100954. doi: 10.1016/j.preteyeres.2021.100954.
- Wu AL, Wu WC. Anti-VEGF for ROP and pediatric retinal diseases. Asia Pac J Ophthalmol (Phila). 2018;7(3):145–151. doi: 10.22608/apo.201837.
- Ambade AS, Jung B, Lee D, et al. Triple-tyrosine kinase inhibition attenuates pulmonary arterial hypertension and neointimal formation. Transl Res. 2019;203:15–30. doi: 10.1016/j.trsl.2018.07.010.
- Zhou W, Liu K, Zeng L, et al. Targeting VEGF-A/VEGFR2 Y949 signaling-mediated vascular permeability alleviates hypoxic pulmonary hypertension. Circulation. 2022;146(24):1855–1881. doi: 10.1161/circulationaha.122.061900.
- Eirin A, Chade AR. Cardiac epigenetic changes in VEGF signaling genes associate with myocardial microvascular rarefaction in experimental chronic kidney disease. Am J Physiol Heart Circ Physiol. 2023;324(1):H14–H25. doi: 10.1152/ajpheart.00522.2022.
- Smith TL, Oubaha M, Cagnone G, et al. eNOS controls angiogenic sprouting and retinal neovascularization through the regulation of endothelial cell polarity. Cell Mol Life Sci. 2021;79(1):37. doi: 10.1007/s00018-021-04042-y.
- Ji L, Su S, Xin M, et al. Luteolin ameliorates hypoxia-induced pulmonary hypertension via regulating HIF-2α-Arg-NO axis and PI3K-AKT-eNOS-NO signaling pathway. Phytomedicine. 2022;104:154329. doi: 10.1016/j.phymed.2022.154329.
- Erewele EO, Castellon M, Loya O, et al. Hypoxia-induced pulmonary hypertension upregulates eNOS and TGF-β contributing to sex-linked differences in BMPR2 (+/R899X) mutant mice. Pulm Circ. 2022;12(4):e12163. doi: 10.1002/pul2.12163.
- Kumari R, Dutta R, Ranjan P, et al. ALKBH5 regulates SPHK1-dependent endothelial cell angiogenesis following ischemic stress. Front Cardiovasc Med. 2021;8:817304. doi: 10.3389/fcvm.2021.817304.
- Zhu X, Zhou C, Zhao S, et al. Role of m6A methylation in retinal diseases. Exp Eye Res. 2023;231:109489. doi: 10.1016/j.exer.2023.109489.