
Abstract
To the multiple factors that may eventually result in colorectal cancer (CRC), strains of E. coli have now been added, in particular strains producing colibactin from their polyketide synthesis (pks) locus. The evidence and mechanistic explanations for this unfortunate effect of what is in most cases a harmless commensal are discussed in the first part of this review. In the second part, observations are presented and discussed that do not fit with the hypothesis that colibactin-producing E. coli produce CRC. The last part of this review is reserved for an alternative explanation of the function of this enigmatic colibactin, a toxin that has not yet been isolated. It is hypothesized that E. coli preferentially colonizes cancerous lesions as an effect rather than a cause and that colibactin production provides a selective advantage to compete with other bacteria.
Introduction
In the past two decades, insights into the multifactorial processes that eventually result in cancer have grown considerably. To the many factors that in combination contribute to tumour formation, bacterial components of the human’s microbiome have now been added. With the discovery that Helicobacter pylori carriage is a risk factor for gastric cancer (Caruso and Fucci Citation1990), other species colonizing the gastrointestinal tract have been investigated for potential cancer-inducing properties. Particular strains belonging to Escherichia coli have been identified as a potential risk factor for colorectal cancer (CRC).
The species E. coli can be divided into four phylotypes (A, B1, B2, and D). These were originally defined based on their multilocus enzyme electrophoresis patterns (MLEE) (Selander et al. Citation1986) and this grouping was later refined with DNA-based multilocus sequence typing (MLST) and confirmed by whole genome sequences [reviewed in Chaudhuri and Henderson (Citation2012)]. Commensal E. coli strains frequently belong to phylotype A, while phylotype B2 strains are more frequent carriers of virulence genes compared to the other phylotypes, and often cause extraintestinal infections (Johnson et al. Citation2008). Nevertheless, not all B2 strains are virulent: it is the predominant commensal phylotype in people living in temperate zones (Escobar-Páramo et al. Citation2004).
Cancer-inducing properties of E. coli strains belonging to B2 have been demonstrated, and with the recent accumulation of observations providing mechanistic evidence, there should be little reason for doubt that these strains can cause CRC. Nevertheless, there are a number of problems with the commonly presented interpretation of published data. The accumulative evidence for a link between E. coli and CRC is reviewed in the first part of this treatise. The interested reader is directed to more extensive reviews that deal with specific aspects of the mentioned processes in more detail, for instance Lax (Citation2005) and Gagnière et al. (Citation2016) as well as reviews cited below. The second part summarizes a number of observations that do not fit the published interpretations. In the third part, an alternative explanation is given that could account for the observations. In view of the evidence discussed here, this author is not convinced that E. coli is implicated in causing CRC, when considering these bacteria in the ecological environment of the human gut where they reside. In a holistic view, E. coli strains are not responsible for cancer that is sometimes observed together with their presence. However, the experimental evidence that has been collected so far is not suitable to test how valid this alternative hypothesis is.
Part one – the evidence
E. coli of phylotype B2 is frequently present in CRC patients
The proposed connection between E. coli and CRC started with repeated observations that E. coli are frequently found to colonize cancer lesions and neighbouring epithelium, often accumulating there at large numbers, so that sometimes they are the only cultivable organisms in close contact to the diseased site (Swidsinski et al. Citation1998; Martin et al. Citation2004; Buc et al. Citation2013; Bonnet et al. Citation2014; Raisch et al. Citation2014). The earliest study, from Germany, detected E. coli by PCR in 62% of adenoma and 77% of carcinoma biopsy specimens, compared to 12% positive biopsies from symptomatic, and 3% from asymptomatic controls (Swidsinski et al. Citation1998). Subsequent observations from the United Kingdom reported presence of invasive E. coli, by gentamycin protection assay, in biopsies from 71% patients with Crohn’s disease, 57% with CRC, 48% with ulcerative colitis, and 29% of controls (Martin et al. Citation2004). A number of studies related to a long-running French cohort of patients in which CRC patients were compared to diverticulosis cases, reporting slightly different numbers between the publications (Buc et al. Citation2013; Bonnet et al. Citation2014; Raisch et al. Citation2014). In the latter, 66% of biopsies from CRC were positive for E. coli strains by culture, compared to 19% in the diverticulosis control group.
Since many virulent E. coli strains belong to phylotype B2, it is no surprise that, of the virulent strains isolated from CRC cases, a higher fraction (43%) belonged to this phylotype than in the diverticulosis control group (32%); nevertheless, the majority of E. coli from CRC cases (57%) belonged to different phylotypes (Raisch et al. Citation2014).
An epidemiological link between phylotype B2 E. coli and CRC can also be postulated, as the frequency of these strains in the gut of people living on a Western diet has increased in the past decade (Escobar-Páramo et al. Citation2004), while the frequency of CRC also increased (Sandler Citation1996). Applying the mechanistic explanation that microorganisms causing chronic inflammation can result in cancer (Kipanyula et al. Citation2013), E. coli could thus result in CRC: people with chronic inflammation of gut tissue are also at higher risk of CRC (Herrinton et al. Citation2012). The complex interplay between gut microbiota, inflammation and cancer is reviewed in more detail elsewhere (Arthur and Jobin Citation2013). It is often stated that phylotype B2 E. coli are typically long-term colonizers of the human gut, based on a study that involved infants (Nowrouzian and Oswald Citation2012); in that case, the virulence factors these strains frequently produce would result in a prolonged inflammatory response, with eventually the malign outcome of CRC.
E. coli of phylotype B2 is cytotoxic in vitro
When particular E. coli strains of the B2 phylotype are incubated in vitro with various epithelial cell lines, they cause these cells to elongate, arrest their cell cycle and force them to enter senescence (Nougayrède et al. Citation2006; Maddocks et al. Citation2009; Secher et al. Citation2013; Cougnoux et al. Citation2014; Vizcaino et al. Citation2014). The effect is due to a group of compounds collectively named cyclomodulins (Nougayrède et al. Citation2005), which introduce double-strand DNA breaks in the target cells [reviewed in Gagnière et al. (Citation2016)]. Cyclomodulins produced by E. coli include cytolethal distending toxin (CDT) [reviewed in Grasso and Frisan (Citation2015)], cytotoxic necrotizing factor (Cnf) (Fabbri et al. Citation2013), cycle inhibiting factor (Cif) (Marchès et al. Citation2003), intimin-dependent attachment encoded by eae (Maddocks et al. Citation2009) and colibactin (Nougayrède et al. Citation2006), produced by the pks locus that will be introduced in detail in the next section (the term colibactin was also used in the past for a non-related siderophore). The cytotoxic phenotype is overrepresented in E. coli isolated from CRC patients. For instance, 26 cyclomodulin-positive E. coli strains (as defined by PCR detection of pks-specific genes) were isolated from 50 biopsies from CRC patients, compared to 17 cyclomodulin-negative stains (Bonnet et al. Citation2014). In a follow-up publication of the same study, 88 strains from CRC patients were compared to 46 strains obtained from a control group of diverticulosis patients. Split out for the different cyclomodulin factors, 23 of the 88 CRC-associated strains were positive for pks, 18 for cnf, 6 for cdt, and 3 for cif; most of these belonged to phylotype B2 (Raisch et al. Citation2014).
Double-strand DNA breaks can be induced by various mechanisms
The cyclomodulins that can be found in B2 E. coli strains can introduce DNA damage in mammalian cells via different mechanisms (). The mode of action of cytotoxin CDT, encoded by the cdtABC locus that can be found in a variety of species (Lara-Tejero and Galan Citation2000), has been elucidated. The secreted toxin consists of the three gene products, of which the active subunit CdtB is injected into the target cell; this is transported into the nucleus where it introduces DNA double-strand breaks [reviewed in Grasso and Frisan (Citation2015)]. DNAse activity of CdtB has been demonstrated, although it is much weaker than that of other DNAses (Lara-Tejero and Galan Citation2000).
Table 1. Cyclomodulins of E. coli.
Pathogenic E. coli strains containing the eae gene coding for intimin induce double-strand breaks using a different mechanism: they inject effector molecules into the cell by means of a Type III secretion system, which down-regulate the cell’s DNA mismatch repair system, with DNA strand breaks as a result (Maddocks et al. Citation2009).
Strains expressing Cnf1 employ yet another mechanism. This single-molecule protein binds to the target cell with its N-terminal binding domain, and translocates itself across the membrane to internalize into endosomes. The C-terminal part of the protein, released after splicing, has enzymatic activity and deaminates Rho-GTPases. The resulting actin cytoskeleton activation causes ruffling of the cell. The overall effect of this process induces tumour cell motility, invasiveness and metastasis, multinucleation, and amitotic cell division [reviewed in Fabbri et al. (Citation2013)].
The cyclomodulin that is the focus of this review is the most enigmatic. Colibactin was shown to induce double-strand DNA breaks in mammalian cells (Nougayrède et al. Citation2006; Cuevas-Ramos et al. Citation2010), but its mechanism of action is still hypothetical, as the toxin has never been isolated. In the first publication describing the cyclomodulin effect of colibactin, it was shown that direct contact between bacteria and the target cells was required and that the bacteria need to be alive for the toxic effect (Nougayrède et al. Citation2006). In this publication the locus name pks was introduced, short for polyketide synthesis (pks). Although the required contact between bacteria and cells was confirmed in a second publication (Buc et al. Citation2013), a year later it was demonstrated that the medium of cultured cells that were infected with colibactin-expressing E. coli could also induce proliferation of uninfected cells, suggesting that cell-mediated messengers enforced the tumour-inducing properties of pks (Dalmasso et al. Citation2014). However, these authors observed the same effect by incubation with 10% foetal calf serum.
Colibactin is most likely a combination of hybrid molecules containing both a peptide and a polyketide. The pks locus responsible for its biosynthesis was first characterized from probiotic E. coli Nissle 1917 (EcN) (Homburg et al. Citation2007). This pks locus is present on a 54-kb long genomic island that contains at least 18 genes (). These include three non-ribosomal peptide synthases (NRPS, enzymes building peptides without the assistance of ribosomes), three polyketide synthases (PKS), two NRPS/PKS hybrids, and nine accessory enzymes (Nougayrède et al. Citation2006; Homburg et al. Citation2007). Sixteen of the genes (clbB-clbQ) are transcribed from at least three polycistronic messengers, while clbA and clbR are transcribed from the other strand (Homburg et al. Citation2007; Putze et al. Citation2009). All genes except clbM are required for active expression of colibactin (Nougayrède et al. Citation2006). The presence of direct repeat flanks, its position in a tRNA gene (asnW), and presence of an integrase gene suggest the island was introduced into the chromosome by horizontal gene transfer (Putze et al. Citation2009). The genes clbB and clbN can be used as markers for presence of the complete island (Johnson et al. Citation2008).
Figure 1. Schematic representation of the pks locus of E. coli. Genes coding for polyketide synthases are shown in dark grey, non-ribosomal peptide synthase genes are shown in light grey, and the two fusion genes are shown bicoloured. The regulator clbR and the integrase (int) are shown in black. Protein functions for ClbA, ClbM, and ClbP are indicated. Modified after Nougayrède et al. (Citation2006) and Homburg et al. (Citation2007). Small accessory genes are not shown.
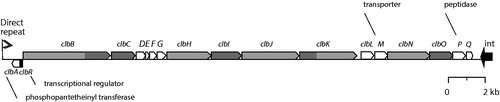
Before zooming in on the nature of colibactin, deduced from the pks genes in want of the isolated product, the in vivo evidence that this locus is involved in carcinogenesis is briefly reviewed.
In vivo models show that pks can induce cancer
A number of publications have described the cancer-inducing properties of B2 E. coli strains in vivo, determined using different mouse models (). One model makes use of IL-10 double knockout mice (IL-10−/−) to induce colitis in response to their gut microflora. These animals are treated with the carcinogen azoxymethane (AOM), and the animals are raised germ-free before they are mono-colonized with E. coli. Using this assay, the carcinogenic effect of E. coli strain NC101 was demonstrated (Arthur et al. Citation2012). This B2 strain was originally isolated from the feces of a wildtype (WT) mouse (Kim et al. Citation2005) and contains the pks locus. Colonization in WT mice does not result in colitis or tumours. In the literature, the strain is both described as “adherent-invasive” [as in Arthur et al. (Citation2012)] and “nonpathogenic” [e.g. in Patwa et al. (Citation2011)]; presence of potential cyclomodulin genes other than pks was not investigated. Arthur et al. (Citation2012) showed that inactivation of genes belonging to the pks locus reduced the cancer-inducing property of the strain: although animals mono-colonized with mutants still produced cancer, the disease was established later, with fewer or smaller tumours (Arthur et al. Citation2012).
Table 2. Mouse models demonstrating cancer-inducing properties of E. coli.
An alternative mouse model was used to test the carcinogenic properties of strain E. coli 11G5, a B2 strain obtained from a human CRC biopsy (Bonnet et al. Citation2014). This model compared 5- to 6-week-old C57B/6J-Apc−/+ mice (Min mice) with WT animals, raised under specific-pathogen-free conditions and treated with streptomycin prior to inoculation. After 50 d of colonization with either strain 11G5 or the K-12 strain MG1665 as a control, 50% of non-infected controls and 57% of K-12-colonized Min mice had developed colonic polyps, compared to 92% of animals colonized with strain 11G5. The WT mice did not develop neoplasia, despite being colonized with high levels of E. coli 11G5 (Bonnet et al. Citation2014).
A third murine model, that depends on colon loops, was used to demonstrate the rapid DNA-damaging effect of pks-bearing E. coli strain SP15 (Cuevas-Ramos et al. Citation2010). The strain, originally isolated from a newborn with meningitis by Johnson et al. (Citation2002), was used to introduce a marker gene (green fluorescence protein [GFP]) under regulation of clbA promoter. This mutant was inoculated into artificially closed colon loops in WT mice, and after 6 h colon tissue was analysed. This demonstrated that bacteria in close contact to the intestinal brush border actively expressed the marker gene (Cuevas-Ramos et al. Citation2010). Colonocytes already contained double-strand DNA breaks at this stage, as was concluded from immunohistology and detection of the S139-phosphorylation of histone H2AX. By means of isogenic pks− mutants, it was demonstrated that this effect was three times more frequent with E. coli containing active pks genes (Cuevas-Ramos et al. Citation2010).
A fourth murine model used subcutaneous injection of tumour cells infected with E. coli expressing colibactin from a pks-containing bacterial artificial chromosome (pBAC) (Cougnoux et al. Citation2014). As expected the animals developed tumours, both in the control group (E. coli without pks) and in the treatment group, but in the latter the tumours were larger. Since colibactin inhibited rather than stimulated cell growth (20% inhibition in vitro), it was tested if the observed in vivo tumour proliferation was due to indirect effects. It turned out to result from growth factors produced by cells after contact with colibactin (Cougnoux et al. Citation2014).
Confusingly, at high multiplicity of infection (MOI), E. coli strains expressing pks can actually suppress the proliferation of tumour cells, at least in a murine model using xenografts of E. coli-infected HC116 cells (Dalmasso et al. Citation2014). This tumour-suppressing effect was observed with an MOI of 100, while at an MOI of 20, tumour growth was accelerated by pks-positive E. coli (the strain used here was not specified, and without pks MOI had no effect on tumour growth) (Dalmasso et al. Citation2014). This resulted from a so-called “bystander effect” where infected cells could force neighbouring cells into senescence via cytokine signalling pathways (Secher et al. Citation2013). That cells treated with pks-positive E. coli produced a variety of cytokines or growth factors in vitro was demonstrated by intramuscular injection of cell culture supernatant in mice (Cougnoux et al. Citation2014).
Proposed colibactin structures as deduced from the pks locus genes
Several genes of the pks locus have been characterized in detail. Gene clbA, coded on the opposite strand compared to most other genes of this locus, encodes a phosphopantetheinyl transferase () whose activity is essential for cytotoxic activity (Nougayrède et al. Citation2006). Enzymes transferring phosphopantetheinyl groups typically modify PKS and NRPS enzymes; in this case ClbA modifies the three polypeptide synthases of the pks locus (Nougayrède et al. Citation2006). The gene clbP codes for a peptidase (Dubois et al. Citation2011) and its function is to splice a precursor of colibactin (pre-colibactin) into its active form (Dubois et al. Citation2011; Cougnoux et al. Citation2012). This peptidase contains an N-terminal cleavage site and is anchored in the inner membrane of the periplasmic space with its C-terminal domain (Cougnoux et al. Citation2012). The protein responsible for transporting pre-colibactin through the membrane is ClbM, a protein belonging to the group of multidrug and toxic compound extrusion proteins (MATE) (Mousa et al. Citation2016, Citation2017), although this gene was not found essential for colibactin production (Nougayrède et al. Citation2006).
A number of intermediates produced by the pks locus were identified by comparing artificial expression of the locus of strains IHE3034 or EcN in a pks-negative strain; a clbP-negative mutant was used as a control (Vizcaino et al. Citation2014). A total of 101 small molecule intermediates could be identified by mass spectrometry of ethyl-acetate extracts derived from bacterial cultures, but by comparison of the IHE3034-derived and the EcN-derived profiles only nine molecules were shared by both. The most abundant intermediate detected was N-myristoyl-D-Asn (an asparagine connected by a peptide bond to a 14-C saturated lipid). This compound was also identified by others (Bian et al. Citation2013; Brotherton and Balskus Citation2013). Related compounds that were predicted based on their mass contained a double bond in the lipid group (cis-7-tetradecenoyl-D-Asn), a longer lipid tail, or addition of various extra (amino acid) groups to the Asn end of the molecule (Vizcaino et al. Citation2014). Three molecules could be produced synthetically, but none had cyclomodulin activity. One of these could partly inhibit growth of Bacillus subtilis (Vizcaino et al. Citation2014).
A structure of pre-colibactin and the resulting final colibactin after splicing by ClbP peptidase was proposed a year later (Vizcaino and Crawford Citation2015). These predictions were based on pks gene activity and metabolic extrapolation combined with biosynthesis of colibactin using radio-labelled isotopes and NMR analysis. The use of a ClbP mutant allowed the analysis of pre-colibactin. This and the proposed structure for colibactin after its final cleavage by ClbP is shown in . Note that the part shown to the left of the peptidase cleavage site in the figure was also predicted by Brotherton and Balskus (Citation2013) and Bian et al. (Citation2013), though represented in a slightly different way. The presumed active site of the toxin is a cyclopropane group, a highly reactive group not without reason described as a “warhead” (Vizcaino and Crawford Citation2015). A minute amount of pre-colibactin could be isolated, enough to test its activity to DNA after peptide cleavage. Surprisingly, not DNA double-strand breaks, but DNA cross-linking was shown to result from co-incubation of colibactin with purified DNA (shown in ). The authors speculate that the double-strand breaks observed in cell assays were downstream effects resulting from the bystander effect (Vizcaino and Crawford Citation2015).
Figure 2. Proposed structures of colibactin and pre-colibactin. (A) The originally proposed structure of pre-colibactin (top), colibactin A after cleavage by ClbP (middle) and colibactin cross-linked with two sites to DNA (bottom), after (Vizcaino and Crawford Citation2015). (B) The revised structure of pre-colibactin A, and derivatives pre-colibactin B and C (insets) after (Healy et al. Citation2016). The part abbreviated with “R” remained unrevised and is thus identical to what is shown to the left. (C) The revised structure of colibactin A, with an imine group (the 5-ring to the left of the molecule) that presumably enhances DNA reactivity, after (Healy et al. Citation2016). (D) The structure of colibactin according to (Li et al. Citation2016).
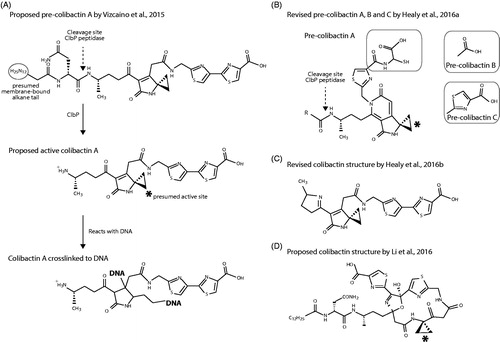
A slightly revised molecular structure was subsequently published (Healy et al. Citation2016), together with pre-colibactin B and pre-colibactin C. These intermediates contained a different side group attached to the S/N-containing thiazoline ring, as shown in the insets of . This proposed structure of pre-colibactin A was also suggested by Zha et al. (Citation2016), who used a clbP/clbG mutant to isolate larger quantities of the molecule. However, these intermediates were subsequently described as inactive by-products due to the clbP mutation (Healy et al. Citation2016). These authors proposed that active colibactin contained an imine group after processing by ClbP, as shown in , which would enhance its reactivity to DNA.
Not everybody agreed with the proposed colibactin structure and an alternative structure with a much larger cyclic backbone was put forward () (Li et al. Citation2016). How such a large macromolecule could leave the bacteria to pass the outer membrane was not addressed.
Part two – the doubt
Despite the seemingly overwhelming evidence that E. coli strains expressing cyclomodulins including colibactin can induce CRC, the argumentation can be countered with other observations or alternative interpretations that, when looking at the bigger picture, weakens the link between phylotype B2 E. coli and CRC considerably.
E. coli is not the only species preferentially colonizing the CRC gut
Culture- or PCR-dependent detection of E. coli selectively reports findings of this species only. Although multiple studies have thus described overrepresentation of E. coli in CRC biopsies, they have ignored a number of other genera that may also have been selectively present. The microbiome of CRC patients is quite different to that of a healthy gut. When rectal biopsies from healthy sites were compared between adenoma patients and controls, this time analysed by metagenomic analysis of 16S ribosomal DNA sequences, a wide variety of differences were observed (Sanapareddy et al. Citation2012), indicative of major changes in the microbiome of a healthy vs. a CRC gut. Thus, not only E. coli benefits from the presence of cancer lesions: the gut microbiome of CRC patients undergoes major shifts, in which the phyla Firmicutes, Verrucomicrobia, Proteobacteria (including members other than E. coli), and other phyla are increased. At the genus level at least 30 genera were significantly different between patients and healthy controls, and at OTU level there were at least 87 marked differences (Sanapareddy et al. Citation2012). Fewer shifts were observed in a Chinese cohort (Wang et al. Citation2012), and a French study reported noticeable shifts in Prevotella/Bacteroides in stool samples of CRC patients vs. controls (Sobhani et al. Citation2011). A study performed in the United States observed lower diversity in CRC cases, as was reported in most of the other studies, with enrichment for Fusobacterium and Porphyromonas (Ahn et al. Citation2013). Given the complexity of a gut microflora, the interplay amongst microbial species, and that between microbes and the host, it is probably a bit simplistic to concentrate on one species only, and to consider that species solely responsible for the onset of CRC.
Indeed, a number of other species may play a role in onset of CRC, summarized in a recent review (Gagnière et al. Citation2016). As CRC progresses, the bacterial diversity directly adhering to the lesions shifts with developmental stages, for instance resulting in expansion of Fusobacterium (Castellarin et al. Citation2012; Chen et al. Citation2012). That members of this genus have carcinogenic properties, for which its FadA adhesin is important, is reviewed elsewhere (Rubinstein et al. Citation2013; Allen-Vercoe and Jobin Citation2014).
By ignoring the vast changes occurring in the microbiome of CRC patients, the link between gut bacteria and CRC is frequently narrowed down to just E. coli, and then specifically to one phylotype of the species. Although it is understandable that a well-characterized species is used as the model organism to investigate a disease as complex as CRC, it also creates a bandwagon effect: the more evidence is collected, the more research will focus on this type of organisms only, although they are not the only and necessarily even the key players in the onset of CRC. The concentrated attention on one species only is a serious limitation of models using germ-free raised animals monoassociated with a single bacterial strain.
Is E. coli enrichment cause or effect of CRC?
The concentration of attention is even narrower than described above when it explicitly investigates the action of colibactin. This ignores the diversity of phylotype B2 E. coli strains that were found enriched and in association with cancer lesions: they can contain a variable number of different cyclomodulins or other virulence factors, resulting in either pro-inflammatory or anti-inflammatory reactions. Review articles (e.g. Gagnière et al. (Citation2016)) compare the pathovar adhesive-invasive E. coli (AIEC) with other E. coli types, but the pro-inflammatory activity of different E. coli strains is quite diverse, depending on their virulence mechanism.
The diversity within the B2 isolates under investigation is illustrated by the following example. Of the 38 B2 isolates originating from CRC-patients of the French cohort, 23 (60%) were pks positive and 15 were pks negative, while 11 did not contain any detectable cyclomodulin genes (Raisch et al. Citation2014). In a recent update of that French cohort study, still only 50% of E. coli isolated from CRC was pks positive (Gagnière et al. Citation2017). While pks was the most frequently reported gene locus in isolates from both the CRC and control group, cnf1 was present in 42% of the CRC isolates and in 4 of the 6 diverticulosis isolates. E. coli B2 phylotype strains may further produce cdt (Raisch et al. Citation2014), eae (Maddocks et al. Citation2009; Arthur et al. Citation2014), or afa-1 adhesin (Prorok-Hamon et al. Citation2014), while cnf and pks are often found in combination (Raisch et al. Citation2014). Each of these cyclomodulins may have carcinogenic properties.
Apart from their variable virulence genes content, there may be multiple other genes that are consistently conserved within B2 genomes that set them apart from the other phylotypes. The pks gene locus, that is present in approximately 80% of B2 strains, may be a marker of the majority of these strains, but that they are found enriched in CRC lesions may not be related to that property at all: alternatively it could be due to other shared genes that result in a selective advantage to live in this environment.
Indeed, the association of (high numbers of) E. coli B2 strains with cancerous lesions may be considered as evidence of a reversed causal relationship: these bacteria are attracted to lesions and specifically colonize these (Stritzker et al. Citation2010; Kocijancic et al. Citation2016). This characteristic is so strong, that the bacteria have actually been suggested as a potential diagnostic tool for early detection of CRC (Hill et al. Citation2011; Danino et al. Citation2015). It can be observed not only in the gut, but even with intravenously injected bacteria, as these move towards cancer tissue specifically (Brader et al. Citation2008; Kocijancic et al. Citation2016). The further CRC is developed (TNM status II or III instead of status I), the stronger the association with B2-type E. coli becomes (Bonnet et al. Citation2014). Such observations do not suggest E. coli presence is a cause but rather is the result of tumour development.
The preference for colonization of cancerous tissue is not the result of bacterial motility or motility-dependent chemotaxis (inactivating motility did not affect their preference to colonize tumours), but seems to be a passive process. Transportation by the host’s macrophages is not involved, as depletion of these actually increased tumour bacterial loads, but bacterial metabolism is required, as inactivation of aromatic amino acid biosynthesis (AroA mutations) inhibited tumour colonization (Stritzker et al. Citation2010). These authors argue that the bacteria, once growing inside tumours, would induce necrosis by their endotoxin and other factors, which might actually result in infiltration of macrophages or neutrophils that can then enclose the bacteria and clean up tumour tissue. Of note is that these experiments were done with pks-positive EcN, while none of the results indicated tumour growth as a result of colonization.
The affinity of E. coli for tumours is not reduced when the myristoylation pathway is blocked by mutation of msbB, which affects LPS biosynthesis and reduces its endotoxicity (Stritzker et al. Citation2010). This is an interesting observation in light of the proposed incorporation of myristic acid into colibactin (Brotherton et al. Citation2015).
The cancer-specific colonization by E. coli may depend on the interaction with a host receptor called carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6), which is over-expressed in cancer cells (Schölzel et al. Citation2000), and whose overexpression can also be induced by B2 E. coli strains (Raisch et al. Citation2014). Since the interaction works both ways (cancer cells express the host factor to which E. coli binds, whereas those bacteria also increase the factor’s expression) it is difficult to entangle cause and effect here. Nevertheless, selective colonization of cancer lesions could explain the overabundance of a variety of E. coli types. The bacteria might benefit from the changed conditions in these lesions, for instance a lower oxygenation level that is typical for tumours (Höckel and Vaupel Citation2001), as suggested by Kocijancic et al. (Citation2016).
In conclusion, the explanation that E. coli B2 bacteria cause lesions can be countered with the alternative explanation that existing lesions specifically select for E. coli B2 strains.
Is colibactin really inducing cancer?
The pks locus is most likely horizontally acquired by E. coli. It can also be found in other members of the Enterobacteriaceae, such as Klebsiella pneumoniae, Enterobacter aerogenes, and Citrobacter koseri (Putze et al. Citation2009). Cytotoxicity of these pks-positive strains was demonstrated in vitro, but whether any of these, which can all be present in a gut microbiome, are associated with CRC has not been reported. If colibactin were the causative of cancer, one would expect members of these other pks-positive species to be overrepresented as well. In the French cohort, 39% of E. coli strains obtained from CRC loci was pks negative (Raisch et al. Citation2014).
Variability in outcome of in vitro cell assays can be problematic, as one culture batch of E. coli may be cytotoxic, while the next batch, cultured under identical conditions, is not, as was observed with EcN (Dr. Birgit Klinkert, personal communication). The strongest evidence that colibactin can induce cancer comes from mouse experiments, but it must be appreciated that the applied models are extremely artificial. One model requires inactivation of an essential interleukin gene (IL10) before E. coli can colonize, with germ-free animals that are then colonized with one E. coli strain only, and also treated with a carcinogen. Other bacterial species are able to induce cancer under these conditions as well, for instance Streptococcus bovis (now known as S. gallolyticus) (Abdulamir et al. Citation2011), or Bacteroides vulgatus (Uronis et al. Citation2009). Even non-defined SPF-flora can produce cancer in 60–80% of animals by this model (Arthur et al. Citation2012). When these germ-free IL10 knockout mice are allowed to collect a microflora from their cage environment, this results in inflammation (Arthur et al. Citation2014), while the initial overrepresentation of Enterobacteriaceae compared to WT animals decreases over time (Arthur et al. Citation2014). This suggests that in the IL10 mouse model, E. coli is not very capable of long-term colonization when it has to compete with other organisms present. The animals did develop colitis but no cancer, presumably because the mutagen AOX was not used (Arthur et al. Citation2014).
In at least one mouse model a bystander effect was considered responsible for accelerated tumour growth ((Dalmasso et al. Citation2014) but how this can be the result of colibactin activity was not addressed.
In another mouse study, Enterococcus faecalis was used as a negative control, which can result in an inflammatory response in these animals but does not result in cancer (Arthur et al. Citation2012). However, this Gram-positive species does not produce LPS, and thus the cellular and immune responses will be different to that generated by E. coli, so the control experiment is not completely comparable.
In a murine cancer model inducing breast carcinomas by injection with a carcinoma cell line, it was demonstrated that EcN specifically colonizes the tumour cells, but the fate of the tumours was not investigated over time (Stritzker, Hill, et al. Citation2010). This work was not conducted to investigate carcinogenic properties of EcN. In an earlier study, the animals whose tumours were colonized by EcN were followed for at least 25 d but it was not reported that their tumours were growing faster than control animals (Stritzker et al. Citation2007), which would be expected for a pks-positive strain living inside these tumours.
The rat is probably a better model than the mouse, not only because it is more easily colonized by E. coli, but apc-mutated animals actually develop tumours in the colon rather than in the small intestine (Irving et al. Citation2014). It would be of interest to see if the carcinogenic properties of pks-positive E. coli can be reproduced in this alternative animal model. Those experiments have not yet been described, but newborn rats have recently been colonized with a (non-specified) pks-producing E. coli strain and its isogenic pks-negative mutants. This did not result in significant differences in histological scores or villus length in the rat’s jejuna at day 58 (the colon was not investigated); the fraction of phosphorylated histone (γH2AX)-positive cells, used as a marker for DNA double-strand breaks, did also not increase (Payros et al. Citation2017). One would think these results demonstrated lack of carcinogenic activity of colibactin, but the authors then combined pks-producing E. coli with the mycotoxin deoxynivalenol (DON), also known as “vomitoxin”, and with this combination histological scores increased, villi lengths decreased and the fraction of γH2AX-positive cells increased. DON by itself did not produce these effects (Payros et al. Citation2017). This is all the more surprising, since DON contains a cyclopropane group, just like colibactin does. Why two toxins, both with a similar, highly reactive cyclopropane group, administered separately do not have any effect on the health of a rat gut, but in combination cause DNA lesions (for which colibactin, but not DON, is held responsible) was not explained.
That B2 strains are able to colonize the human gut persistently is often stated with reference to Nowrouzian and Oswald (Citation2012); however, that study described colonization in infants. There are far fewer data that these strains colonize an adult gut persistently. For instance, EcN does not seem to colonize the adult gut for very long (Prilassnig et al. Citation2007; Joeres-Nguyen-Xuan et al. Citation2010). Although it is sometimes stated that long colonization correlates with presence of the pks locus, within phylotype B2 isolates, pks-positive strains did not colonize children longer than pks-negative strains (Nowrouzian and Oswald Citation2012).
The explanation that colibactin-producing E. coli B2 bacteria cause lesions can be questioned for a number of reasons. summarizes the main observations for and against the carcinogenic effects of colibactin.
Table 3. Observations pro and against the carcinogenic effect of E. coli colibactin.
Does colibactin cause DNA cross-linking in epithelial cells?
There remain a number of questions to be addressed in the colibactin-CRC story, as recently listed in another review (Balskus Citation2015), and that list is still incomplete. A number of small-molecule intermediates resulting from the pks genes have been detected (Vizcaino et al. Citation2014) but none of these are responsible for the mutagenic effect of colibactin. These products are released in the culture media, but colibactin can only act during direct contact between bacteria and target cell so it is not secreted (Nougayrède et al. Citation2006). Thus, colibactin does not pass the mucus layer all by itself, but how would cell contact initiate the activation of the final “warhead” product or force it to enter the target cell in order to produce DNA damage?
The active site of the toxin, the cyclopropane group, can cross-link purified DNA, but it is not clear why pre-colibactin is inactive, since its predicted structure also contains the highly labile cyclopropane group. There is no chemical explanation how cleavage by ClbP adds reactivity to the toxin, as the highly energetic ring strain of cyclopropane is not compensated in any of the proposed pre-colibactin precursors (). Possibly, the only reason why pre-colibactin does not react with cells is because it is present in the periplasmic space of the bacteria, presumably bound to the inner membrane with its lipid tail. This then raises the question why the operon would contain ClbS, a protein not essential for cytotoxicity but required to protect the E. coli cell against damage by its own colibactin (Bossuet-Greif et al. Citation2016). Since there is no DNA present in the periplasmic space, which is where colibactin is activated by ClbP, it is a mystery how this protection would work, if DNA were the only and exclusive substrate for this toxin.
After cleavage by ClbP, the active colibactin must leave the bacterial cell through the bacterial outer membrane and pass the cellular membrane somehow, though it is not described why direct contact between bacteria and target cells is required for toxin activity. It cannot be because its precursor is membrane-bound, since the outer membrane separates the precursor from the target cells.
The biggest question, however, remains this: How can colibactin pass the bacterial outer membrane and the cellular membrane, translocate through the cytoplasm where proteins and RNA are present, nevertheless remaining active until it has entered the nucleus, to then specifically react with DNA packed inside chromosomes without reacting to the proteins surrounding this DNA? Any other chemical substance capable of electrophilic attack (which would be true for most proteins and certainly for RNA) will force the cyclopropane ring structure to open (de Meijere Citation1979). It is not explained why only cellular DNA would be the target of colibactin and other biomolecules refrain from inactivating the toxin before it reached the nucleus, if colibactin were able to enter the cell in its active form at all. For the active component of cyclomodulin CDT, translocation of the enzymic CdtB protein has been demonstrated, and its enzymatic DNAse activity has been characterized [reviewed by Grasso and Frisan (Citation2015)]. Likewise, Intimin requires a type 3 secretion system to be injected into the target cell (). But how DNA-damaging colibactin as proposed by Vizcaino and Crawford (Citation2015) can enter the nucleus without losing its reactivity is hard to understand. It should be noted that, although DNA damage following incubation of cells with pks-positive E. coli has been demonstrated multiple times in vitro, so far nobody has been able to detect a DNA molecule cross-linked to colibactin in mouse tissue, or alkylated DNA that would result from the postulated toxin’s activity.
In conclusion, if the final product of the pks locus is both very reactive and very instable, it would lose its activity or react with substrates before reaching the chromosomes of the eukaryotic cell. If it is stable enough to do this, it is worrisome that it cannot be isolated or demonstrated in bacterial culture medium.
Part three – the alternative
In an attempt to incorporate the published observations into a better fitting hypothesis, an alternative relationship between phylotype B2 E. coli and CRC is proposed here. This last part of this review is rather short of citations, as the proposed hypothesis has not been widely studied. The alternative explanation builds on the hypothesis that phylotype B2 E. coli are not causative of cancer but their enhanced and prolonged colonization is the result of the disease.
The hypothesis is first of all based on the characteristic that these E. coli strains are attracted to CRC tissue and multiple near, on or inside cancer lesions. They may do so because of the abundance of iron, the less oxygenated environment, the presence of specific cellular adhesins, and possibly for other reasons. This preferred colonization explains why these bacteria are found in association with CRC, and why they can have a variety of pathogenic or non-pathogenic properties. The key property that they must all share, which dictates their presence on or near to cancer lesions, is their increased capacity to colonize such sites, and this is something that can be experimentally verified. A thorough bioinformatic analysis might even reveal what kind of gene products would be involved, provided a gene set can be identified that all CRC-colonizing E. coli strains have in common.
The alternative hypothesis furthermore proposes that colibactin does not act as a carcinogen, but as a bacteriocin. Colibactin is a very reactive compound that is likely to attack bacteria competing for a place to live in. This makes sense from an evolutionary perspective, since E. coli occupies a niche where it meets multiple competitors. If colibactin provides a selective advantage to the bacteria carrying the pks locus, it explains why up to 80% of the strains belonging to B2 phylotype contain the locus; it would also explain the longer colonization that has been proposed for these strains. One of the colibactin biosynthetic intermediates has been shown to inhibit Bacillus subtilis growth, and that colibactin has antibacterial activity was proposed as an evolutionary advantage (Vizcaino et al. Citation2014). Incorporating this may hint to its natural function. In the opinion of this author, the “warhead” colibactin is unlikely to reach mammalian epithelial cells, as it would rapidly be consumed by other bacteria. Support for this view could even be obtained in vivo, using the mouse model but now mixing pks-positive E. coli with a normal gut flora; it is predicted that this would abolish the induction of cancer. The cancer-inducing properties are possibly the result of highly artificial models.
In combination, the two parts of this alternative hypothesis explain why sometimes (near to) pure cultures of E. coli can be found on cancer lesions. If this observation is not resulting from E. coli being easier to cultivate compared to other organisms in the gut, it can be explained by increased colonization properties of B2 E. coli and from antibacterial activity of colibactin. The hypothesis that these E. coli bacteria are not a cause but a result of cancer is testable and should be pursued.
Epilogue
It may be hard to change the view on colibactin, or to redirect the emphasis of colibactin research from the hot topic of microbial carcinogenesis to the relatively boring subject of bacterial competition. But from an ecological perspective, competition with other bacteria is all that matters to bacterial proliferation, while inducing cancer in the host is not relevant for their survival. Bacterial evolution is mostly driven by competition. It is likely that the phylotype B2 E. coli that have increased in frequency over the past decades have employed their capacity to colonize an environment that has increased in numbers over the years: the gut of an individual living on a Western diet, with the diseased gut of unfortunate CRC patients as an ultimate consequence.
Why does it matter how we interpret the relationship between particular E. coli strains and CRC? The starting hypothesis of any research drives experimental design, which again forces the results into one direction or another, leading to scientific insights merging into a particular alley. As experimental evidence accumulates, it becomes harder to investigate alternative possibilities. Thus, a lot of research has already been conducted to demonstrate a carcinogenic effect of colibactin, but the obvious alternative, that the compound provides a selective advantage against the background microflora that E. coli constantly competes with, has hardly been studied.
There is one more aspect to consider. Patients suffering from CRC are not helped by work demonstrating that they can, at least in part, “blame” their bacteria for the disease. How would such insights change treatment or prevention strategies? CRC is a multifactorial disease, with a number of manipulable factors putting far more weight to the equation than E. coli does, and these are factors that individuals can actually influence to reduce their risk: for instance by a healthy diet and low alcohol intake. A proven risk factor for CRC is a diet rich in red meat and fat while low in fibre and vegetables, so that up to 80% of CRC cases are attributed to diet (Candela et al. Citation2011). These risk factors can be reduced by changes in diet and habits. But what to do about E. coli in the gut? It would be unwise to treat people at risk with antibiotics to eradicate those bacteria, or even start this treatment with infants to avoid they collect the “wrong type” of bacteria. Should we indeed regard B2 phylotype E. coli as “not wanted in the gut” as was recently discussed (Secher et al. Citation2016)? Even if antibiotic treatment were considered (which, from a microbiological viewpoint is not a clever strategy, to say the least), individuals would rapidly be recolonized. Thus, the insights in how colibactin can cause cancer in animal studies, or induces cell arrest in in vitro assays, may address highly interesting academical questions, but they do not have a lot of relevance to the prevention or treatment of CRC.
Disclosure statement
The author works as a consultant for companies producing probiotic products containing E. coli, including Ardeypharm GmbH. These companies had no influence on the content of the article.
References
- Abdulamir AS, Hafidh RR, Abu Bakar F. 2011. The association of Streptococcus bovis/gallolyticus with colorectal tumors: the nature and the underlying mechanisms of its etiological role. J Exp Clin Cancer Res. 30:11.
- Ahn J, Sinha R, Pei Z, Dominianni C, Wu J, Shi J, Goedert JJ, Hayes RB, Yang L. 2013. Human gut microbiome and risk for colorectal cancer. J Natl Cancer Inst. 105:1907–1911.
- Allen-Vercoe E, Jobin C. 2014. Fusobacterium and Enterobacteriaceae: important players for CRC? Immunol Lett. 162:54–61.
- Arthur JC, Gharaibeh RZ, Mühlbauer M, Perez-Chanona E, Uronis JM, McCafferty J, Fodor AA, Jobin C. 2014. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat Commun. 5:4724.
- Arthur JC, Jobin C. 2013. The complex interplay between inflammation, the microbiota and colorectal cancer. Gut Microbes. 4:253–258.
- Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan TJ, Campbell BJ, Abujamel T, Dogan B, Rogers AB. 2012. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science. 338:120–123.
- Balskus EP. 2015. Colibactin: understanding an elusive gut bacterial genotoxin. Nat Prod Rep. 32:1534–1540.
- Bian X, Fu J, Plaza A, Herrmann J, Pistorius D, Stewart AF, Zhang Y, Müller R. 2013. In vivo evidence for a prodrug activation mechanism during colibactin maturation. Chembiochem. 14:1194–1197.
- Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, Déchelotte P, Bonnet R, Pezet D, Darfeuille-Michaud A. 2014. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res. 20:859–886.
- Bossuet-Greif N, Dubois D, Petit C, Tronnet S, Martin P, Bonnet R, Oswald E, Nougayrède JP. 2016. Escherichia coli ClbS is a colibactin resistance protein. Mol Microbiol. 99:897–908.
- Brader P, Stritzker J, Riedl CC, Zanzonico P, Cai S, Burnazi EM, Ghani ER, Hricak H, Szalay AA, Fong Y, et al. 2008. Escherichia coli Nissle 1917 facilitates tumor detection by positron emission tomography and optical imaging. Clin Cancer Res. 14:2295–2302.
- Brotherton CA, Balskus EP. 2013. A prodrug resistance mechanism is involved in colibactin biosynthesis and cytotoxicity. J Am Chem Soc. 135:3359–3362.
- Brotherton CA, Wilson M, Byrd G, Balskus EP. 2015. Isolation of a metabolite from the pks island provides insights into colibactin biosynthesis and activity. Org Lett. 17:1545–1548.
- Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, Pezet D, Bonnet R. 2013. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS One. 8:e56964.
- Candela M, Guidotti M, Fabbri A, Brigidi P, Franceschi C, Fiorentini C. 2011. Human intestinal microbiota: cross-talk with the host and its potential role in colorectal cancer. Crit Rev Microbiol. 37:1–14.
- Caruso ML, Fucci L. 1990. Histological identification of Helicobacter pylori in early and advanced gastric cancer. J Clin Gastroenterol. 12:601–602.
- Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, et al. 2012. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22:299–306.
- Chaudhuri RR, Henderson IR. 2012. The evolution of the Escherichia coli phylogeny. Infect Genet Evol. 12:214–226.
- Chen W, Liu F, Ling Z, Tong X, Xiang C. 2012. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS One. 7:e39743.
- Cougnoux A, Dalmasso G, Martinez R, Buc E, Delmas J, Gibold L, Sauvanet P, Darcha C, Déchelotte P, Bonnet M, et al. 2014. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype . Gut. 63:1932–1942.
- Cougnoux A, Gibold L, Robin F, Dubois D, Pradel N, Darfeuille-Michaud A, Dalmasso G, Delmas J, Bonnet R. 2012. Analysis of structure-function relationships in the colibactin-maturating enzyme ClbP. J Mol Biol. 424:203–214.
- Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède JP. 2010. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA. 107:11537–11542.
- Dalmasso G, Cougnoux A, Delmas J, Darfeuille-Michaud A, Bonnet R. 2014. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes. 5:675–680.
- Danino T, Prindle A, Kwong GA, Skalak M, Li H, Allen K, Hasty J, Bhatia SN. 2015. Programmable probiotics for detection of cancer in urine. Sci Transl Med. 7:289ra84.
- de Meijere A. 1979. Bonding properties of cyclopropane and their chemical consequences. Angew Chem Int Ed Engl. 18:809–826.
- Dubois D, Baron O, Cougnoux A, Delmas J, Pradel N, Boury M, Bouchon B, Bringer MA, Nougayrède JP, Oswald E, et al. 2011. ClbP is a prototype of a peptidase subgroup involved in biosynthesis of nonribosomal peptides. J Biol Chem. 286:35562–35570.
- Escobar-Páramo P, Grenet K, Le Menac’h A, Rode L, Salgado E, Amorin C, Gouriou S, Picard B, Rahimy MC, Andremont A, et al. 2004. Large-scale population structure of human commensal Escherichia coli isolates. Appl Environ Microbiol.70:5698–5700.
- Fabbri A, Travaglione S, Ballan G, Loizzo S, Fiorentini CT. 2013. The cytotoxic necrotizing factor 1 from E. coli: a Janus toxin playing with cancer regulators. Toxins. 5:1462–1474.
- Gagnière J, Bonnin V, Jarrousse AS, Cardamone E, Agus A, Uhrhammer N, Sauvanet P, Déchelotte P, Barnich N, Bonnet R, et al. 2017. Interactions between microsatellite instability and human gut colonization by Escherichia coli in colorectal cancer. Clin Sci. 131:471–485.
- Gagnière J, Raisch J, Veziant J, Barnich N, Bonnet R, Buc E, Bringer MA, Pezet D, Bonnet M. 2016. Gut microbiota imbalance and colorectal cancer. World J Gastroenterol. 22:501–518.
- Grasso F, Frisan T. 2015. Bacterial genotoxins: merging the DNA damage response into infection biology. Biomolecules. 5:1762–1782.
- Healy AR, Nikolayevskiy H, Patel JR, Crawford JM, Herzon SB. 2016. A mechanistic model for colibactin-induced genotoxicity. J Am Chem Soc. 138:15563–15570.
- Healy AR, Vizcaino MI, Crawford JM, Herzon SB. 2016. Convergent and modular synthesis of candidate precolibactins. Structural revision of precolibactin A. J Am Chem Soc. 138:5426–5432.
- Herrinton LJ, Liu L, Levin TR, Allison JE, Lewis JD, Velayos F. 2012. Incidence and mortality of colorectal adenocarcinoma in persons with inflammatory bowel disease from 1998 to 2010. Gastroenterology. 143:382–389.
- Hill PJ, Stritzker J, Scadeng M, Geissinger U, Haddad D, Basse-Lüsebrink TC, Gbureck U, Jakob P, Szalay AA. 2011. Magnetic resonance imaging of tumors colonized with bacterial ferritin-expressing Escherichia coli. PLoS One. 6:e25409.
- Höckel M, Vaupel P. 2001. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J Natl Cancer Inst. 93:266–276.
- Homburg S, Oswald E, Hacker J, Dobrindt U. 2007. Expression analysis of the colibactin gene cluster coding for a novel polyketide in Escherichia coli. FEMS Microbiol Lett. 275:255–262.
- Irving AA, Yoshimi K, Hart ML, Parker T, Clipson L, Ford MR, Kuramoto T, Dove WF, Amos-Landgraf JM. 2014. The utility of Apc-mutant rats in modeling human colon cancer. Dis Model Mech. 7:1215–1225.
- Joeres-Nguyen-Xuan TH, Boehm SK, Joeres L, Schulze J, Kruis W. 2010. Survival of the probiotic Escherichia coli Nissle 1917 (EcN) in the gastrointestinal tract given in combination with oral mesalamine to healthy volunteers. Inflamm Bowel Dis. 16:256–262.
- Johnson JR, Johnston B, Kuskowski MA, Nougayrède JP, Oswald E. 2008. Molecular epidemiology and phylogenetic distribution of the Escherichia coli pks genomic island. J Clin Microbiol. 46:3906–3911.
- Johnson JR, Oswald E, O’Bryan TT, Kuskowski MA, Spanjaard L. 2002. Phylogenetic distribution of virulence-associated genes among Escherichia coli isolates associated with neonatal bacterial meningitis in the Netherlands. J Infect Dis. 185:774–784.
- Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, Huycke MM, Sartor RB. 2005. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology. 128:891–906.
- Kipanyula MJ, Seke Etet PF, Vecchio L, Farahna M, Nukenine EN, Nwabo Kamdje AH. 2013. Signaling pathways bridging microbial-triggered inflammation and cancer. Cell Signal. 25:403–416.
- Kocijancic D, Felgner S, Frahm M, Komoll RM, Iljazovic A, Pawar V, Rohde M, Heise U, Zimmermann K, Gunzer F, et al. 2016. Therapy of solid tumors using probiotic Symbioflor-2: restraints and potential. Oncotarget. 7:22605–22622.
- Lara-Tejero M, Galán JE. 2000. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science. 290:354–357.
- Lax AJ. 2005. Opinion: bacterial toxins and cancer-a case to answer? Nat Rev Microbiol. 3:343–349.
- Li ZR, Li J, Gu JP, Lai JYH, Duggan BM, Zhang WP, Li ZL, Li YX, Tong RB, Xu Y, et al. 2016. Divergent biosynthesis yields a cytotoxic aminomalonate-containing precolibactin. Nat Chem Biol. 12:773–775.
- Maddocks OD, Short AJ, Donnenberg MS, Bader S, Harrison DJ. 2009. Attaching and effacing Escherichia coli downregulate DNA mismatch repair protein in vitro and are associated with colorectal adenocarcinomas in humans. PLoS One. 4:e5517.
- Marchès O, Ledger TN, Boury M, Ohara M, Tu X, Goffaux F, Mainil J, Rosenshine I, Sugai M, De Rycke J, et al. 2003. Enteropathogenic and enterohaemorrhagic Escherichia coli deliver a novel effector called Cif, which blocks cell cycle G2/M transition. Mol Microbiol. 50:1553–1567.
- Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. 2004. Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology. 127:80–93.
- Mousa JJ, Newsome RC, Yang Y, Jobin C, Bruner SD. 2017. ClbM is a versatile, cation-promiscuous MATE transporter found in the colibactin biosynthetic gene cluster. Biochem Biophys Res Commun. 482:1233–1239.
- Mousa JJ, Yang Y, Tomkovich S, Shima A, Newsome RC, Tripathi P, Oswald E, Bruner SD, Jobin C. 2016. MATE transport of the E. coli-derived genotoxin colibactin. Nat Microbiol. 1:15009.
- Nougayrède JP, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, Buchrieser C, Hacker J, Dobrindt U, Oswald E, et al. 2006. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science. 313:848–851.
- Nougayrède JP, Taieb F, De Rycke J, Oswald E. 2005. Cyclomodulins: bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 13:103–110.
- Nowrouzian FL, Oswald E. 2012. Escherichia coli strains with the capacity for long-term persistence in the bowel microbiota carry the potentially genotoxic pks island. Microb Pathog. 53:180–182.
- Patwa LG, Fan TJ, Tchaptchet S, Liu Y, Lussier YA, Sartor RB, Hansen JJ. 2011. Chronic intestinal inflammation induces stress-response genes in commensal Escherichia coli. Gastroenterology.141:1842–1851.
- Payros D, Dobrindt U, Martin P, Secher T, Bracarense AP, Boury M, Laffitte J, Pinton P, Oswald E, Oswald IP. 2017. The food contaminant deoxynivalenol exacerbates the genotoxicity of gut microbiota. MBio. 8:e00007–00017.
- Prilassnig M, Wenisch C, Daxboeck F, Feierl G. 2007. Are probiotics detectable in human feces after oral uptake by healthy volunteers? Wien Klin Wochenschr. 119:456–462.
- Prorok-Hamon M, Friswell MK, Alswied A, Roberts CL, Song F, Flanagan PK, Knight P, Codling C, Marchesi JR, Winstanley C, et al. 2014. Colonic mucosa-associated diffusely adherent afaC + Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut. 63:761–770.
- Putze J, Hennequin C, Nougayrède JP, Zhang W, Homburg S, Karch H, Bringer MA, Fayolle C, Carniel E, Rabsch W, et al. 2009. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect Immun. 77:4696–4703.
- Raisch J, Buc E, Bonnet M, Sauvanet P, Vazeille E, de Vallée A, Déchelotte P, Darcha C, Pezet D, Bonnet R, et al. 2014. Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World J Gastroenterol. 20:6560–6572.
- Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. 2013. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/beta-catenin signaling via its FadA adhesin. Cell Host Microbe. 14:195–206.
- Sanapareddy N, Legge RM, Jovov B, McCoy A, Burcal L, Araujo-Perez F, Randall TA, Galanko J, Benson A, Sandler RS, et al. 2012. Increased rectal microbial richness is associated with the presence of colorectal adenomas in humans. ISME J. 6:1858–1868.
- Sandler RS. 1996. Epidemiology and risk factors for colorectal cancer. Gastroenterol Clin North Am. 25:717–735.
- Schölzel S, Zimmermann W, Schwarzkopf G, Grunert F, Rogaczewski B, Thompson J. 2000. Carcinoembryonic antigen family members CEACAM6 and CEACAM7 are differentially expressed in normal tissues and oppositely deregulated in hyperplastic colorectal polyps and early adenomas. Am J Pathol. 156:595–605.
- Secher T, Brehin C, Oswald E. 2016. Early settlers: which E. coli strains do you not want at birth? Am J Physiol Gastrointest Liver Physiol. 311:G123–G129.
- Secher T, Samba-Louaka A, Oswald E, Nougayrède JP. 2013. Escherichia coli producing colibactin triggers premature and transmissible senescence in mammalian cells. PLoS One. 8:e77157.
- Selander RK, Caugant DA, Ochman H, Mussser JM, Gilmour MN, Whittam TS. 1986. Methods of multilocus enzyme electrophoresis for bacterial population genetics and systematics. Appl Environ Microbiol. 51:873–884.
- Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, Corthier G, Tran Van Nhieu J, Furet JP. 2011. Microbial dysbiosis in colorectal cancer (CRC) patients. PLoS One. 6:e16393.
- Stritzker J, Hill PJ, Gentsche I, Szalay AA. 2010. Myristoylation negative msbB-mutants of probiotic E. coli Nissle 1917 retain tumor specific colonization properties but show less side effects in immunocompetent mice. Bioeng Bugs. 1:139–145.
- Stritzker J, Weibel S, Hill PJ, Oelschlaeger TA, Goebel W, Szalay AA. 2007. Tumor-specific colonization, tissue distribution, and gene induction by probiotic Escherichia coli Nissle 1917 in live mice. Int J Med Microbiol. 297:151–162.
- Stritzker J, Weibel S, Seubert C, Götz A, Tresch A, van Rooijen N, Oelschlaeger TA, Hill PJ, Gentschev I, Szalay AA. 2010. Enterobacterial tumor colonization in mice depends on bacterial metabolism and macrophages but is independent of chemotaxis and motility. Int J Med Microbiol. 300:449–456.
- Swidsinski A, Khilkin M, Kerjaschki D, Schreiber S, Ortner M, Weber J, Lochs H. 1998. Association between intraepithelial Escherichia coli and colorectal cancer. Gastroenterology.115:281–286.
- Uronis JM, Mühlbauer M, Herfarth HH, Rubinas TC, Jones GS, Jobin C. 2009. Modulation of the intestinal microbiota alters colitis-associated colorectal cancer susceptibility. PLoS One. 4:e6026.
- Vizcaino MI, Crawford JM. 2015. The colibactin warhead crosslinks DNA. Nat Chem. 7:411–417.
- Vizcaino MI, Engel P, Trautman E, Crawford JM. 2014. Comparative metabolomics and structural characterizations illuminate colibactin pathway-dependent small molecules. J Am Chem Soc. 136:9244–9247.
- Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, Jia W, Cai S, Zhao L. 2012. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 6:320–329.
- Zha L, Wilson MR, Brotherton CA, Balskus EP. 2016. Characterization of polyketide synthase machinery from the pks island facilitates isolation of a candidate precolibactin. ACS Chem Biol. 11:1287–1295.