
Abstract
Mycobacterium tuberculosis (Mtb) is the causative pathogen of tuberculosis, the most lethal infectious disease resulting in 1.3 million deaths annually. Treatments against Mtb are increasingly impaired by the growing prevalence of antimicrobial drug resistance, which necessitates the development of new antibiotics or alternative therapeutic approaches. Upon infecting host cells, predominantly macrophages, Mtb becomes critically dependent on lipids as a source of nutrients. Additionally, Mtb produces numerous lipid-based virulence factors that contribute to the pathogen’s ability to interfere with the host’s immune responses and to create a lipid rich environment for itself. As lipids, lipid metabolism and manipulating host lipid metabolism play an important role for the virulence of Mtb, this review provides a state-of-the-art overview of mycobacterial lipid metabolism and concomitant role of host metabolism and host-pathogen interaction therein. While doing so, we will emphasize unexploited bacteria-directed and host-directed drug targets, and highlight potential synergistic drug combinations that hold promise for the development of new therapeutic interventions.
1. Introduction
Tuberculosis, caused by Mycobacterium tuberculosis (Mtb), is the deadliest infectious disease worldwide, causing 1.3 million deaths every year (WHO Citation2023). Treatment of antibiotic-sensitive Mtb generally requires the use of 3-4 antibiotics for 4-6 months, which upon successful adherence cures 85% of the patients (WHO Citation2022). However, poor treatment adherence and the persistence of drug-tolerant dormant (i.e. non-replicating) bacteria impair treatment efficacy, potentially causing relapse, which in turn promotes the development of multi-drug (MDR) and extensively-drug resistant (XDR) Mtb strains (Pradipta et al. Citation2018; Goossens et al. Citation2020). Patients with MDR and XDR Mtb require more extensive and longer antibiotic regimens and still show higher morbidity and mortality rates rise up to 40% and 60%, respectively (Dheda et al. Citation2017). Currently, MDR and XDR strains already account for 5% of Mtb infections and their prevalence is rising rapidly, urging development of new antibiotics and alternative treatments, such as host-directed therapies (HDT) (Dheda et al. Citation2017).
Following aerosol transmission, Mtb infiltrates the lung and predominantly infects alveolar macrophages, where Mtb replicates and spreads for extended periods. Over time, infected lungs develop granulomas characterized by necrotic lipid-rich cores that are typically surrounded by macrophages, whom frequently are lipid-loaded (Peyron et al. Citation2008; Russell et al. Citation2009). To survive intracellularly, Mtb produces many virulence factors to manipulate the host cell via three major pathways: impairing (auto-)phagosomal degradation, dampening immune responses and acquiring nutrients to maintain metabolism. While Mtb is able to use both carbohydrates and lipids as energy sources, the bacterium becomes highly dependent on lipids in vivo (Pisu et al. Citation2020; Laval et al. Citation2021), which is supported by the fact that lipid-metabolising genes are abundantly present in the genome of Mtb (Cole et al. Citation1998). Surprisingly, however, only few mycobacterial antibiotics target Mtb’ lipid metabolism. Most notably, isoniazid and ethionamide inhibit the synthesis of the virulence factor and lipid-based cell wall component mycolic acid (Quemard et al. Citation1995). To promote the development of drugs targeting lipid metabolic pathways of Mtb, in vitro drug screen platforms, usually comprising planktonic cultures without any host immune cells, may have to be optimized to better reflect the conditions that Mtb encounters in vivo.
Our understanding of host and intracellular mycobacterial metabolism during human infections has expanded significantly during the last decades, which allows the design of more potent anti-tubercular drug regimens. Therefore, this review will first provide a state-of-the-art overview on mycobacterial lipid metabolism, in which lipid catabolism, ATP production, and lipid anabolism and potential drug targets within these systems will be described. Additionally, a partial description of carbohydrate metabolism is provided whenever relevant for production of lipid-based virulence factors. Next, the role of the host metabolism and concomitant host-pathogen interactions involved in the intracellular survival of Mtb will be discussed. Finally, possibilities for a combined bacteria-directed and host-directed compounds will be discussed in order to improve current treatment regimens for Mtb, including MDR and XDR Mtb.
2. Identification of essential proteins involved in lipid metabolism of Mycobacterium tuberculosis
2.1. Transporter proteins involved in uptake of lipids, especially cholesterol, are essential for Mtb survival in vivo
Mtb utilizes fatty acids (FAs) and cholesterol as nutrient sources, sequestered from host’ intracellular compartments (). Intracellular mycobacteria have been found to promote the formation of lipid droplets within host cells and stimulate access to these organelles rich in triacylglycerol (TAG), cholesterol and cholesteryl esters by mechanisms not completely understood (Daniel et al. Citation2011; Caire-Brändli et al. Citation2014; Santucci et al. Citation2018). Growth of Mtb on minimal media containing FA or cholesterol, combined with macrophage intracellular models and in vivo infection models, supports the identification of essential genes involved in lipid catabolism. Enzymes involved in key-steps of FA and cholesterol uptake, catabolism and storage may be feasible targets to treat mycobacterial infections ().
Figure 1. Overview lipid and carbohydrate uptake and catabolism. There are different routes for uptake and catabolism of lipids and carbohydrates within mtb. Starting molecules for each pathway are cholesterol, FAs, and glucose (red text). The different pathways contain proteins (italic) or metabolic intermediates (black text). Proteins were found to be essential for survival of mtb in vitro or in vivo (green text), while others are conditionally essential when grown on a specific nutrient source (orange text) (Sassetti and Rubin Citation2003; Griffin et al. Citation2011; DeJesus et al. Citation2017; Beites et al. Citation2021). The depicted pathways can be continuous (black arrow) or shown partially (discontinuous arrow). Pathways occur either in the macrophage phagosome or inside the bacteria. ‘?’ responsible protein not (yet) identified. Created with Bio-Render.com.
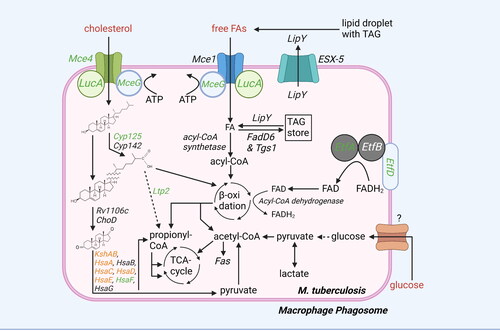
Mtb secretes lipase LipY through its ESX-5 efflux pump to release FA from host TAG (Deb et al. Citation2006; Daleke et al. Citation2011; Santucci et al. Citation2019; Barrientos et al. Citation2022). Host FAs and cholesterol are taken up by Mtb via the mycobacterial cell entry (Mce)1 and Mce4 transporter complexes, respectively (Pandey and Sassetti Citation2008; Nazarova et al. Citation2017), both of which require additional proteins for transport. MceG (commonly named mkl) uses ATP to provide Mce1 and Mce4 with energy for substrate uptake (Nazarova et al. Citation2017). In addition, lipid uptake coordinator A (LucA) acts as a scaffold protein to stabilize the protein complexes (Nazarova et al. Citation2017). Once FAs and cholesterol are imported into the cytoplasm of Mtb, they are either catabolised directly or stored as TAG and cholesteryl esters into intracytoplasmic lipid inclusions (ILIs) (Mallick et al. Citation2021), and in case of cholesterol potentially within the mycobacterial cell wall. LipY is also used to release FAs from TAG stored in the ILIs (Brzostek et al. Citation2009). Mce4 and MceG were found to be essential for Mtb growth on cholesterol (Griffin et al. Citation2011). In addition, several Mce4 subunits, such as MceG and LucA, are required for Mtb virulence in vivo (Griffin et al. Citation2011; Nazarova et al. Citation2017). On the other hand, for the FA transporter Mce1, studies have paradoxically demonstrated attenuated growth of mce1 mutants in mice one week after infection, as well as reduced granuloma formation, but such Mtb mutants have been reported to eventually continue replicating, becoming hypervirulent and resulting in earlier mortality in mice compared to wild type Mtb (Sassetti and Rubin Citation2003; Shimono et al. Citation2003; Gioffré et al. Citation2005; Lima et al. Citation2007).
2.2. Fatty acid-degrading enzymes expressed by mtb are redundant, while most cholesterol-degrading enzymes are essential for its survival
Catabolism of FAs and cholesterol within Mtb occurs via different processes (), but eventually produces similar end products including acetyl-CoA and, in case of odd-chain FAs and cholesterol, propionyl-CoA. FAs are first covalently attached to CoA by a large protein family of FadD acyl-CoA synthetases to form acyl-CoA (Cole et al. Citation1998). Acyl-CoA can be catabolised by β-oxidation (FadA, FadB, FadE and EchA enzymes) into acetyl-CoA for even-numbered FAs, or acetyl-CoA and propionyl-CoA for odd-numbered FAs. None of the enzymes specifically involved in the degradation of FAs through β-oxidation appear to be essential for Mtb survival in vivo, possible due to redundancy between members of these protein families (DeJesus et al. Citation2017). However, the action of all FadE acyl-CoA dehydrogenases depends on the oxidation of co-factor flavin adenine dinucleotide (FAD)H2 to FAD by electron transfer flavoproteins (Etf)A and EtfB, as well as their cognate oxidoreductase EtfD (i.e. Rv0338c) (Beites et al. Citation2021). Knocking down EtfA, EtfB or EtfD in Mtb resulted in abolished β-oxidation, impaired growth on FAs and, in case of EtfD, reduced Mtb survival in infected mice (Beites et al. Citation2021). Importantly, Mtb EtfD differs from its human counterpart and might therefore be an interesting target to intervene in mycobacterial lipid catabolism. Besides further degradation, acyl-CoA can also be used by acyltransferases for esterification with glycerol to create TAGs, which can be stored in ILIs as a nutrient reserve for Mtb in carbon-deprived environments (Daniel et al. Citation2004, Citation2011). The acyl-CoA synthetase FadD6 and acyltransferase triacylglycerol synthase (Tgs)1 were important for TAG storage by Mtb under dormant and intracellular conditions (Daniel et al. Citation2011, Citation2014).
For cholesterol, the P450 cytochrome enzymes Cyp125 or Cyp142 initiate the removal of the side chain from the sterol rings (Johnston et al. Citation2010; Griffin et al. Citation2011). Degradation of the side chain occurs through β-oxidation performed by cholesterol-specific enzymes, releasing acetyl-CoA and propionyl-CoA, and ends by the action of Ltp2, which cleaves of the final propionyl-CoA group from the sterol rings. Ring cleavage is initiated by oxidation performed by cholesterol oxidase ChoD and Rv1106c. Next, the rings are cleaved and degraded to pyruvate and propionyl-CoA by action of KstD, KshAB, HsaA-G, FadD3 and cholesterol-specific enzymes that perform β-oxidation. Most enzymes involved in cholesterol degradation are essential for Mtb growth on cholesterol, including Cyp125, Ltp2, KstD, KshAB, HsaA, HsaC-F, FadD3 and the enzymes involved in cholesterol β-oxidation (Yam et al. Citation2009; Hu et al. Citation2010; Griffin et al. Citation2011; VanderVen et al. Citation2015). Furthermore, Cyp125, HsaF, Ltp2 and β-oxidation enzymes FadE28/30/32 were found essential for Mtb to survive in mice (Sassetti and Rubin Citation2003). Of note, another study also found Rv1106c to be important for Mtb growth on cholesterol as well as its survival in mice, but not in infected macrophages or guinea pigs, making this protein an unlikely candidate to target therapeutically (Griffin et al. Citation2011; Yang et al. Citation2011).
Like in humans, in Mtb, acetyl-CoA derived from FA and cholesterol catabolism can be further degraded to produce ATP via the tricarboxylic acid (TCA) cycle, mitochondrial electron transport chain, and ATP synthase. Many proteins that are part of electron transport chain and ATP synthase are essential for Mtb survival and the latter is targeted by the antibiotic bedaquiline, used for MDR Mtb (Andries et al. Citation2005).
2.3. Propionyl-CoA is detoxified through the redundant methyl citrate cycle and vitamin B12-dependent methylmalonyl pathway
Accumulation of propionyl-CoA is highly toxic to Mtb. Propionyl-CoA derived from degradation of odd-chained fatty acids or cholesterol requires detoxification. Detoxification takes place either through the methyl citrate cycle (MCC) or the methylmalonyl pathway (MMP) (). The MCC converts propionyl-CoA to pyruvate and requires enzymes from the TCA cycle as well as three MCC-specific enzymes: methylcitrate synthase PrpC, methylcitrate dehydratase PrpD and, specifically in Mtb, isocitrate lyases (Icl)1/2 (Muñoz-Elías et al. Citation2006; Eoh and Rhee Citation2014). Pyruvate can be converted to acetyl-CoA and enter the TCA cycle. While Icl1/2, PrpC and PrpD are all required for growth on odd-chained FAs and their expression is upregulated in infected murine macrophages and mice, only the presence of either Icl1 or Icl2 is essential for Mtb survival in vivo, which might be explained by their additional role in gluconeogenesis as discussed below (Schnappinger Citation2003; Muñoz-Elías and Mckinney Citation2005; Mattow et al. Citation2006; Muñoz-Elías et al. Citation2006; Eoh and Rhee Citation2014). In the MMP, propionyl-CoA is first converted to (S)-methylmalonyl-CoA using the propionyl-CoA carboxylase complex ACCase 5 comprising α subunit AccA3, β subunit AccD5 and regulatory ε subunit AccE5 (Savvi et al. Citation2008; Bazet Lyonnet et al. Citation2014). All ACCase 5 subunits have been found required for optimal mycobacterial growth in vitro (Bazet Lyonnet et al. Citation2014; DeJesus et al. Citation2017). Next, a methylmalonyl-CoA epimerase, likely Rv1322A, converts (S)-methylmalonyl-CoA to (R)-methylmalonyl-CoA, which is turned into the TCA intermediate succinyl-CoA by methylmalonyl-CoA mutase (Mut)AB in a vitamin B12 (vitB12)-dependent manner (Savvi et al. Citation2008).
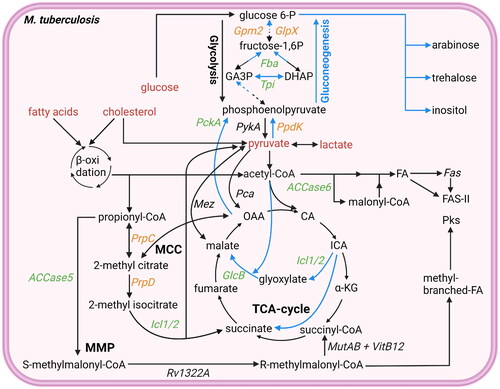
Figure 2. Overview glycolysis, gluconeogenesis, TCA cycle, MCC cycle, and MMC cycle. Mtb uses different pathways to generate ATP, produce molecules for lipid biosynthesis, or detoxify propionyl-CoA. Potential starting molecules for the different pathways are shown (red text). The different pathways contain proteins (italic) or metabolic intermediates (normal text). Proteins were found to be essential for survival of mtb in vitro or in vivo (green text), while others are conditionally essential when grown on a specific nutrient source (orange text) (Sassetti and Rubin Citation2003; Puckett et al. Citation2014Basu et al., 2018; Ganapathy et al. Citation2015; Gutka et al. Citation2015; DeJesus et al. Citation2017). Furthermore, the glyoxylate shunt, not present in mammals, which can be used for gluconeogenesis is shown (blue arrows). TCA cycle intermediates are also shown. Created with Bio-Render.com.
MCC and MMP-based detoxification of propionyl-CoA are redundant processes as supported by a study showing the rescue of Δicl1 Mtb growth by vitB12 supplementation (Lee et al. Citation2013). Furthermore, propionyl-CoA may be detoxified to a limited extent through the MMP without vitB12-dependent MutAB by incorporation of methylmalonyl-CoA into new methyl-branched lipids (Savvi et al. Citation2008), which are used for several important virulence factors as will be discussed further below. However, most likely, simultaneously blocking the MCC and MMP will be sufficient for propionyl-CoA intoxication and successful inhibition of Mtb.
2.4. Mtb requires the generation of carbohydrates from lipids in carbohydrate-deprived environments
Gluconeogenesis is the process of generating glucose-6-phosphate from non-carbohydrate carbon substrates. When Mtb is grown on lipids as a sole carbon source, gluconeogenesis is vital for its survival, likely because gluconeogenesis is necessary for the production of different virulence factors and cell wall components in the absence of carbohydrates (Puckett et al. Citation2014; Ganapathy et al. Citation2015; Gutka et al. Citation2015). In carbohydrate-deprived intracellular environments, Mtb exhibits the capability to generate carbohydrates by converting acetyl-CoA and propionyl-CoA into phosphoenolpyruvate, the starting molecule of gluconeogenesis.
Acetyl-CoA conversion into oxaloacetate is facilitated by a modified TCA cycle, not present in mammals, that contains a bypass known as the glyoxylate shunt, which requires the enzymes Icl1/2 as well as malate synthase G (GlcB) (Muñoz-Elías et al. Citation2006). Using this bypass, Mtb is able to convert acetyl-CoA into oxaloacetate without the typical release of CO2 (). As previously mentioned, having either isocitrate lyase (ICL) 1 or 2 is essential for Mtb virulence, while GlcB is generally required for Mtb growth, the essentiality of GlcB differs between strains (Muñoz-Elías et al. Citation2006; Eoh and Rhee Citation2014; DeJesus et al. Citation2017; Carey et al. Citation2018). The subsequent conversion of oxaloacetate to phosphoenolpyruvate is performed by phosphoenolpyruvate carboxykinase (PckA). By facilitating gluconeogenesis, PckA activity is essential for Mtb growth on FAs as sole carbon source and to replicate in mice or murine macrophages (Basu et al. Citation2018).
Propionyl-CoA can be converted to phosphoenolpyruvate through the two major routes of propionyl-CoA detoxification. The pyruvate obtained through propionyl-CoA detoxification via the MCC can be converted into phosphoenolpyruvate by pyruvate phosphate dikinase (PpdK), which is required for optimal growth of Mtb on cholesterol as a sole carbon source (Basu et al. Citation2018). In addition, the succinyl-CoA obtained through the MMP can be converted into oxaloacetate, through the final steps of the TCA cycle, and subsequently into phosphoenolpyruvate by PckA (Carey et al. Citation2018).
Many enzymes involved in gluconeogenesis, including triosephosphate isomerase (Tpi), fructose-1,6-bisphosphate aldolase (Fba) and fructose bisphosphatases Gpm2 and GlpX, have also been found essential for Mtb growth on FAs as sole carbon source and for replication in vivo (Puckett et al. Citation2014; Ganapathy et al. Citation2015; Gutka et al. Citation2015). While gluconeogenesis may be a potent target to treat Mtb during infection, most of the enzymes involved, except for PpdK, are all also expressed by humans, possibly leading to adverse effects (Wescott et al. Citation2018).
2.5. Virulence factors of the mycobacterial envelope
The mycobacterial envelope is very complex and predominantly consists of a plasma membrane, a periplasmic space comprising peptidoglycan and arabinogalactan, an outer membrane rich in mycolic acids and lipid-conjugates, and finally a loosely-bound polysaccharide-rich capsule. Mtb produces many lipid-based virulence factors that are embedded in the envelope, providing rigidity to the envelope, while also modulating host processes to create a more favorable environment for Mtb. The virulence factors described below are most essential for Mtb’ growth and virulence in vivo, including mycolic acids, trehalose dimycolates (TDMs), phthiocerol dimycocerosates (PDIMs) (), and phosphatidylinositol (PI)-based phosphatidylinositol mannosides (PIMs), lipomannan (LM), lipoarabinomannan (LAM), and mannose-capped LAM (ManLAM) ().
Figure 3. Overview lipid anabolism and production of lipid-based virulence factors: besides being used for ATP production, acetyl-CoA and propionyl-CoA are also used in lipid anabolism and virulence factor production. Potential starting molecules for the different pathways are shown (red text). The different pathways contain proteins (italic) or metabolic intermediates (normal text). Proteins were found to be essential for survival of mtb in vitro or in vivo (green text), while others are conditionally essential when grown on a specific nutrient source (orange text) (Sassetti and Rubin Citation2003; Kalscheuer et al. Citation2010; Varela et al. Citation2012; Bazet Lyonnet et al. Citation2014; DeJesus et al. Citation2017; Fay et al. Citation2019; Pohane et al. Citation2021). The depicted pathways can be continuous (black arrow) or shown partially (discontinuous arrow). Pathways occur either in the cytosol or on the outer membrane of the bacteria (outside the cytosol). Created with Bio-Render.com.
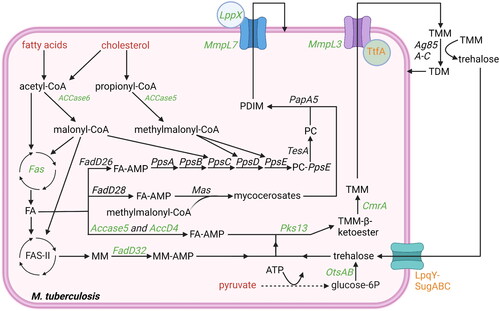
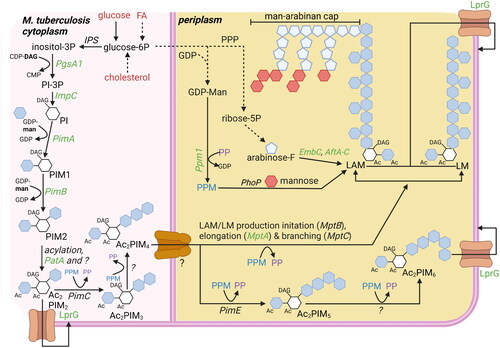
Figure 4. Overview carbohydrate pathways and production of carbohydrate-based virulence factors. To modulate host cells to deliver lipid nutrients, mtb requires carbohydrate-based virulence factors. Carbohydrate products can be derived from lipid nutrients in glucose-deprived conditions (). Potential starting molecules for the different pathways are shown (red text). The different pathways contain proteins (italic) or metabolic intermediates (normal text). Proteins were found to be essential for survival of mtb in vitro or in vivo (green text), while others are conditionally essential when grown on a specific nutrient source (orange text) (Jackson et al. Citation2000; Sassetti and Rubin Citation2003; Movahedzadeh et al. Citation2004; Kaur et al. Citation2007; Guerin et al. Citation2009; Movahedzadeh et al. Citation2010; Boldrin et al. Citation2014; DeJesus et al. Citation2017; Boldrin et al. Citation2021). The depicted pathways can be continuous (black arrow) or shown partially (discontinuous arrow). PPM (blue) and PP (purple) generated via the pentose-6P pathways are used both to elongate LAM and LM, as well as generate Ac2PIM6 from Ac2PIM6. The biosynthesis of PIMs starts on the cytosolic side of the plasma membrane, but from PIM4 onwards, the production occurs on the periplasmic side. ‘?’ responsible protein not identified. Created with Bio-Render.com.
2.5.1. FA biosynthesis in mtb
Mtb produces many virulence factors that are produced partially or fully from unbranched or branched FAs, including methyl-branched FAs (). Biosynthesis of unbranched FAs requires the carboxylation of acetyl-CoA to malonyl-CoA by ACCase 6, comprising α subunit AccA3 and β subunit AccD6 (Daniel et al. Citation2007; Pawelczyk et al. Citation2011). Subsequently, type I fatty acid synthase (Fas) is the main enzyme involved in the synthesis of unbranched FAs from acetyl-CoA using malonyl-CoA as the extender unit, releasing one carbon through CO2 at every elongation step, until a chain length of 16-26 carbons is reached (Cole et al. Citation1998; Zimhony et al. Citation2004). Further elongation, resulting in meromycolic acids, is performed by type II fatty acid synthase (FAS-II) complex, which involves InhA, the target of isoniazid and ethionamide (Quemard et al. Citation1995; Cole et al. Citation1998). Biosynthesis of methyl-branched FAs requires methylmalonyl-CoA, which, as discussed above, is produced from propionyl-CoA using ACCase5, harboring AccD5 as β subunit (Savvi et al. Citation2008; Bazet Lyonnet et al. Citation2014). Alternatively, methylmalonyl-CoA can theoretically be formed from succinyl-CoA by MutAB-mediated isomerization, the reverse of the reaction used for propionyl-CoA detoxification. Methyl-branched elongation of FAs by incorporating methyl-malonyl-CoA is performed by a number of multifunctional, Fas-like polyketide synthases (PKS) (Quadri Citation2014). One interesting example is Pks12, which performs alternating additions of malonyl-CoA and methylmalonyl-CoA to create mycoketide (Quadri Citation2014). The resulting methyl-branched FA is required for mannosyl-β-1-phosphomycoketides, a cell wall component of yet unknown function, but which is likely important as reflected by the requirement of Pks12 for Mtb survival in vivo and its recognition by human CD1c-restricted T cells (Sassetti and Rubin Citation2003; Matsunaga et al. Citation2004). The ACCase 5 and 6 subunits, Fas, and most proteins of the FAS-II complex are essential for Mtb growth in vitro (Sassetti and Rubin Citation2003; Bazet Lyonnet et al. Citation2014; DeJesus et al. Citation2017).
2.5.2. Mycolic acids and trehalose dimycolates (TDMs)
Mycolic acids are very long lipids (60-90 carbons) of which the majority becomes part of the mycolyl arabinogalactan-peptidoglycan (mAGP) complex that makes up most of the periplasmic space and outer membrane of Mtb. In addition, trehalose momomycolate (TMM) is produced by esterification of mycolic acid to trehalose ().
Mycolic acids are created by Pks13-mediated condensation of 24/26 carbon-long acyl-CoA, produced by Fas and activated by ACCase4 (i.e. AccA3 and AccD4), to a longer meromycolic acid, produced by FAS-II complex and activated by acyl-AMP synthetase FadD32 (Gavalda et al. Citation2009; Quadri Citation2014). This reaction is followed by CmrA (i.e. Rv2509)-mediated reduction of the β-keto ester bond, which subsequently enables Pks13 to ligate the mycolic acid to trehalose, resulting in TMM (Lea-Smith et al. Citation2007; Gavalda et al. Citation2009; Kim et al. Citation2023). The meromycolic acids used to produce the mycolic acids may undergo several modifications, including the addition of methyl, methoxy and ketone groups, as well as introducing cyclopropane rings, which are performed by MmaA1-4, CmaA2 and PcaA (Cole et al. Citation1998; Barkan et al. Citation2012). The trehalose of TMM can be produced from glucose, maltose or glycogen by several enzymes. Of these, OtsAB, which enables production of trehalose from glucose, is dominant and essential for Mtb growth (Murphy et al. Citation2005; DeJesus et al. Citation2017). Additionally, trehalose is recycled from trehalose-containing structures in the envelop upon internalization by the LpqY-SugA-C transporter, which is essential during intracellular infection when nutrients are limited (Sassetti and Rubin Citation2003; Kalscheuer et al. Citation2010; Pohane et al. Citation2021). The remaining enzymes involved in the production of TMM, including Fas, AccA3, AccD4, Pks13, FadD32 and CmrA are all essential for Mtb growth (DeJesus et al. Citation2017), and in vivo both Mma3 and PcaA are additionally required for Mtb survival (Sassetti and Rubin Citation2003; DeJesus et al. Citation2017). TMM is exported to the outer membrane by MmpL3/TtfA (i.e. Rv0383c) transporter complex, where its mycolic acid either becomes part of the mAGP complex or TMM is transferred to another TMM molecule to generate TDM (and a free trehalose molecule) (Fay et al. Citation2019; Su et al. Citation2019). TDM is a virulence factor suggested to impair lysosomal activity of the host and to prevent Mtb degradation (Puech et al. Citation2002; Fineran et al. Citation2016; Fay et al. Citation2019; Su et al. Citation2019). The export of TMM to the outer membrane by the MmpL3/TtfA transporter complex is essential for Mtb growth (Varela et al. Citation2012; DeJesus et al. Citation2017; Fay et al. Citation2019).
2.5.3. Phthiocerol dimycocerosates (PDIMs)
PDIMs comprise phthiocerol and mycocerosates (Passemar et al. Citation2014) (). For phthiocerol production, a long-chain FA is activated by the acyl-AMP synthetase FadD26 (Trivedi et al. Citation2004, Citation2005). The activated FA is transferred to PKS proteins phthiocerol polyketide synthase (Pps)A-E (and Rv2953 acting as enoyl reductase for PpsD), which elongate the acyl chain using three malonyl-CoA and two methylmalonyl-CoA substrates, resulting in two methyl branches (Trivedi et al. Citation2005; Siméone et al. Citation2007). PpsB and PpsC lack dehydratase and enoyl reductase domains, resulting in two hydroxyl-groups that will be used for ligation of two mycocerosates. In addition, PpsE performs acyltransferase activity, but leaves the resulting ketone group unreduced. For mycocerosate production, a long-chain FA is activated by the acyl-AMP synthetase FadD28 (Trivedi et al. Citation2004). The activated FA is transferred to the PKS mycocerosic acid synthase (Mas), which performs 3-4 elongation steps using of methylmalonyl-CoA as a substrate to generate methyl-branched mycocerosates. Next, phthiocerol is released from PpsE by the action of TesA and two mycocerosates are transferred from Mas to phthiocerol by PapA5 to generate PDIM (Trivedi et al. Citation2005). PDIM is exported to the outer membrane by the MmpL7/LppX transporter complex (Jain and Cox Citation2005; Sulzenbacher et al. Citation2006). Interestingly, the biosynthesis and export of PDIMs are likely physically linked, as MmpL7 was found to interact with PpsE (Jain and Cox Citation2005). Once incorporated in the outer membrane, PDIMs are thought to induce membrane damage in the host, which promotes phagosomal escape and can ultimately lead to pyrolytic host cell death, all of which are beneficial for Mtb (Augenstreich et al. Citation2020). In line with PDIMs function in host-pathogen interactions, the genes involved in PDIM biosynthesis were all found essential for M. bovis survival in cattle and mutagenesis of the MmpL7/LlpX transporter resulted in a loss of Mtb virulence in mice (Sassetti and Rubin Citation2003; DeJesus et al. Citation2017; Gibson et al. Citation2022).
2.5.4. Phosphatidyl inositol-based virulence factors
Besides FA based virulence factors, Mtb also produces several PI-based virulence factors, including PIMs, LM, LAM and ManLAM (). The different types of virulence factors are formed by the respective linkage of acyl-, mannosyl-, arabinosyl, and a further mannosyl-groups by different transferases. The PI core is created by converting glucose-6-phosphate into inositol-3-phosphate by inositol-3-phosphate synthase (IPS). Subsequently, inositol-3-phosphate is coupled to diacylglycerol by phosphosphatidylinositol synthase (PgsA1) and the 3-phosphate group is removed by phosphatase ImpC, of which the latter enzyme is essential (Movahedzadeh et al. Citation2004, Citation2010; Morii et al. Citation2013; DeJesus et al. Citation2017).
To create PIMs, PI is mannosylated and further acylated at several positions. The most abundant PIMs are di- (PIM2) or hexamannosylated (PIM6). Two mannosylations of PI are performed by PimA and PimB (i.e. Rv2188c) resulting in PIM2. The enzymes responsible for the mannosylation to create PIM6 are not fully deciphered yet but include PimC (in some Mtb strains, but not in H37Rv) and PimE (Korduláková et al. Citation2002; Kremer et al. Citation2002; Morita et al. Citation2006; Mishra et al. Citation2009). Acylation of the first added mannosyl group is performed by PatA, while additional acylation of the PI core is performed by a yet unknown acetyltransferase, resulting in AcPIM2/6 or Ac2PIM2/6 ().
Biosynthesis of both LM and LAM uses PIM4 as a starting molecule. Both require elongation by α-(1→6)-mannosyltransferase activity, likely first performed by MptB and subsequently by MptA, as well as branching by α-(1→2)-mannosyltransferase MptC (i.e. Rv2181) (Kaur et al. Citation2006, Citation2007; Mishra et al. Citation2007). The enzyme responsible for initiating the arabinosylation of the mannan backbone to create LAM is still unknown, but elongation is known to require α-(1→5)-arabinosyltransferase EmbC, one of the major drug targets of the antibiotic ethambutol, while branching requires α-(1→3)-arabinosyltransferase AftC and possibly AftD (Birch et al. Citation2008; Škovierová et al. Citation2009; Zhang et al. Citation2020). Arabinosylation of LAM is likely terminated by β-(1→2)-arabinosyltransferase AftB (Seidel et al. Citation2007). The arabinose required for the arabinosylation is generated from glucose-6-phosphate, which is first converted to ribose-5-phosphate in the pentose phosphate pathway and subsequently converted to arabinose by the actions of PrsA and UbiA (Roos et al. Citation2005). In pathogenic mycobacteria, LAM is often capped by the addition of mannose groups, the first of which is added by CapA and one or two more by MptC, resulting in ManLAM (Dinadayala et al. Citation2006; Kaur et al. Citation2008). Some studies have shown that ManLAM modulates phagocytes and causes early arrest of phagosome maturation, but the true impact of mannosylated caps on virulence and pathogenesis in vivo remains unclear (Fratti et al. Citation2003; Turner and Torrelles Citation2018).
The biosynthesis of PIMs starts on the cytosolic side of the plasma membrane, but the synthesis of PIM4, as well as LM and LAM, most likely takes place on the periplasmic side, thus requiring a flippase or translocase that remains to be identified (Jackson et al. Citation2021). Once PIMs, LM, and LAM have been produced, they can be transported to outer membrane by the lipoprotein transporter LprG, which recognizes the shared PI core sturcture (Drage et al. Citation2010; Gaur et al. Citation2014; Shukla et al. Citation2014). The enzymes responsible for PIM6 production (i.e. PimC/E) and mannose-capping of LAM (i.e. CapA and MptC) have been found dispensable for Mtb growth in vitro (Kremer et al. Citation2002; Morita et al. Citation2006; DeJesus et al. Citation2017). In contrast, most enzymes involved in biosynthesis of PI and LM are important for optimal growth in vitro, but their essentiality for Mtb survival in vivo, at least in immunocompetent mice, seems to be limited (Jackson et al. Citation2000; Sassetti and Rubin Citation2003; Movahedzadeh et al. Citation2004; Kaur et al. Citation2007; Movahedzadeh et al. Citation2010; DeJesus et al. Citation2017). Interestingly, the biosynthetic enzymes for PIM2 (i.e. PimA and PatA) and LAM (i.e. AftB and EmbC, the latter based on ethambutol treatment) production have been found essential for both Mtb growth in vitro and virulence in vivo, making these relevant targets for therapeutic interventions (Korduláková et al. Citation2002; Guerin et al. Citation2009; Boldrin et al. Citation2014; DeJesus et al. Citation2017; Boldrin et al. Citation2021). Finally, the transporter LprG was found dispensable for Mtb growth in vitro, but essential for Mtb survival in vivo, probably because its inhibition blocks the insertion of all described PI-based virulence factors into the outer cell wall (Sassetti and Rubin Citation2003; DeJesus et al. Citation2017).
3. Host-pathogen interactions
The main cellular tropism of Mtb in vivo is toward alveolar macrophages (AMs), cells located in the alveolar luminal space, a glucose-poor environment that is lined with pulmonary surfactant, consisting of 80% phospholipids, 10% cholesterol and 10% surfactant proteins (Milad and Morissette Citation2021; Pereverzeva et al. Citation2022). As AMs play a central role in degrading surfactant-derived lipids, AMs mainly utilize FA degradation and oxidative phosphorylation (OXPHOS) pathways for energy production, which fits ideally with the metabolic needs of Mtb, which predominantly uses lipids for intracellular metabolism (Pandey and Sassetti Citation2008; Daniel et al. Citation2011; Lee et al. Citation2013; Pisu et al. Citation2020). Furthermore, the inflammatory response in AMs seems relatively weak as compared to interstitial macrophages (IMs) (Pisu et al. Citation2020), further supporting their permissiveness toward Mtb infection.
3.1. Metabolic rewiring in Mtb-restrictive and Mtb-permissive macrophages
Upon recognition by macrophages, Mtb triggers toll-like receptor (TLR)2 ligation and, to a lesser extent, TLR4 ligation through interaction with its surface expressed lipid-based pathogen-associated molecular patterns (PAMPs), which promote bacterial internalization (). To carefully modulate the level of TLR ligation, Mtb expresses stimulatory PAMPs, including PIM2, PIM6, and LM, as well as immunomodulatory virulence factors like PDIM and ManLAM, that inhibit immune cell activation or stimulate anti-inflammatory responses (Quesniaux et al. Citation2004). Profound TLR ligation, which Mtb thus tries to prevent (Kilinç et al. Citation2021), induces the Warburg effect, i.e. the generation of ATP from glucose through aerobic glycolysis instead of more efficient OXPHOS despite the presence of sufficient oxygen, which facilitates rapid ATP generation and allows TCA intermediates to be used for the production of immunomodulatory and antibacterial effector molecules (Krawczyk et al. Citation2010; Tannahill et al. Citation2013; Weichhart et al. Citation2015; Lachmandas et al. Citation2016; Pisu et al. Citation2020). The utilization of TCA cycle intermediates is regulated by mammalian target of rapamycin complex 1 (mTORC1) and transcription factor hypoxia-inducible factor (HIF)-1α (Weichhart et al. Citation2015). Activation of mTORC1 stimulates HIF-1α translation, which is further enhanced in macrophages stimulated simultaneously with Mtb and interferon (IFN)-γ (Tannahill et al. Citation2013; Weichhart et al. Citation2015; Braverman et al. Citation2016; Lachmandas et al. Citation2016). Interestingly, gene expression of HIF-1α is also much higher in Mtb-restrictive interstitial macrophages compared to Mtb-permissive AMs (Pisu et al. Citation2020). In addition to stimulating aerobic glycolysis, HIF-1α also stimulates the expression of immune-responsive gene 1 (IRG1), which generates itaconate from TCA cycle intermediates (Shin et al. Citation2011; Li et al. Citation2022). Itaconate, a metabolite that is abundantly present in the lungs of Mtb-infected mice, has immunomodulatory effects by inhibiting production of mitochondrial reactive oxygen species and by providing negative feedback loop on type I interferon responses (Mills et al. Citation2018; Li et al. Citation2022; Wu et al. Citation2023). Furthermore, itaconate is directly antimicrobial for Mtb by targeting the MCC as well as the glyoxylate shunt through inhibition of Icl1/2, while itaconyl-CoA, the CoA derivative of itaconate, targets the MMP by inhibiting MutAB (Ruetz et al. Citation2019; Kwai et al. Citation2020). All these itaconate effects combined likely result in propionyl-CoA intoxication for Mtb when growing on a lipid-rich diet (Ruetz et al. Citation2019; Kwai et al. Citation2020).
Figure 5. Pathogen host interactions during mtb infections to sequester nutrients and promote intracellular survival. Upon recognition of mtb by TLRs, mtb is phagocytosed into macrophages. Once inside the macrophage, mtb exploits multiple mechanisms to survive intracellularly and to acquire nutrients. Mtb uses the virulence factors PDIM and ESAT6 to induce membrane permeabilisation of the phagosome to escape from the phagosome. Cytosolic mtb can migrate toward cytosolic LDs inside the macrophage. Alternatively, mtb can be recognized by the host and returned to a phagosome via xenophagy. However, potentially by stimulating TLR4 ligation, mtb can induce lipophagy of cytosolic LDs. As lipophagy and xenophagy use similar pathways, vacuoles containing both mtb and LDs can form, thereby representing a mechanism how mtb could also stimulate migration of nutrients toward itself. Once in a vacuole with LDs, mtb utilizes multiple lipid-based virulence factors to prevent phagosomal maturation and thereby protects itself from degradation. Additionally, mtb can stimulate the uptake of lipids by macrophages to increase the nutrient source available. To do so, mtb induces TLR2 ligation to stimulate mTORC1 signaling, which induces PPAR-γ expression. PPAR-γ is then thought to induce uptake of LDLs and FAs via endocytosis. The endocytosed lipids are then stored in cytosolic LDs in close proximity to mitochondria, thereby increasing the nutrient availability that mtb can acquire via the mechanisms described above. TLR2, blue; TLR4, green. Created with Bio-Render.com.
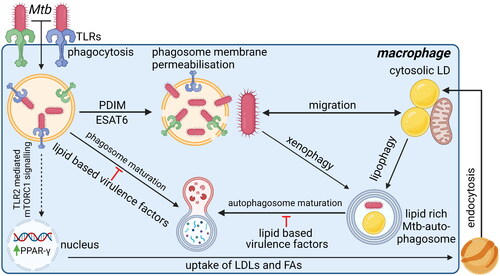
In contrast to the previously described host-beneficial responses induced after TLR-ligation, TLR2 activation also induces expression of transcription factor peroxisome proliferator-activated receptor (PPAR)-γ. PPAR-γ stimulates expression of genes involved in lipid uptake as well as lipid droplet formation, which was found to ultimately be host-detrimental (Rajaram et al. Citation2011) (). Expression of PPAR-γ is much higher in Mtb-permissive AMs as compared to Mtb-restrictive interstitial macrophages, which have a higher expression of HIF-1α instead (Pisu et al. Citation2020). Furthermore, upon Mtb infection, AMs remain more dependent on oxidative phosphorylation for energy consumption instead of aerobic glycolysis, which is dominant in Mtb-infected IMs. In addition, the TCA intermediate citrate can be exported into the cytosol and be used as a source of lipid biosynthesis, further contributing to the formation of lipid droplets and ultimately formation of foamy macrophages (Mehrotra et al. Citation2014). Also, in foamy macrophages mycobacteria depend predominantly on lipids, particularly cholesterol, for their metabolism (Hüsler et al. Citation2023). Cholesterol metabolism enhances mycobacterial expression of genes involved in persistence (Pawełczyk et al. Citation2021), and makes mycobacteria less prone to antibiotics (Laval et al. Citation2021; Samuels et al. Citation2022). Foamy macrophages are more prone to die by necrotic cell death instead of apoptosis, presumably due to overactive eicosanoid signalling (Russell et al. Citation2009; Pagán et al. Citation2022). Moreover, foamy macrophages are known to play a dominant role in disease pathophysiology (Russell et al. Citation2009).
3.2. Eicosanoid signaling during mycobacterial infections
During mycobacterial infections, lipid-based signaling molecules, especially eicosanoids, are pivotal to orchestrate an immune response that is sufficiently strong to eradicate mycobacteria, while limiting inflammation-associated tissue damage (Divangahi et al. Citation2010; Lalvani et al. Citation2012; Tobin et al. Citation2012; Sorgi et al. Citation2020). Hydrolysis of phospholipids gives rise to arachidonic acid, which can be stored in high concentrations within the hydrophobic core of lipid droplets (D’Avila et al. Citation2006; Bozza et al. Citation2011; Hüsler et al. Citation2023). Eicosanoids are synthesized mainly from arachidonic acid in a competitive fashion by multiple enzymes that form a complex signaling network (Sheppe and Edelmann Citation2021). Especially leukotriene B4 (LTB4), produced by 5-lipooxygenase (5-LOX) and leukotriene A4 hydrolase (LTA4H), as well as prostaglandin E2 (PGE2), formed by the action of cyclooxygenases (COX) and prostaglandin E synthase (PTGES) are both considered important during Mtb infections. Both pathways are antagonistic by competing for the same arachidonic acid, and an effective antimycobacterial response requires both pathways to be active (Divangahi et al. Citation2010; Peres-Buzalaf et al. Citation2011; Lalvani et al. Citation2012; Tobin et al. Citation2012; Srinivasan et al. Citation2014; Banks et al. Citation2019; Sorgi et al. Citation2020Jayaraman et al., 2013). LTB4 induces type I IFNs which promote inflammation and enhance anti-TB immunity (Banks et al. Citation2019), but overactive LTB4 signaling causes excessive tumor necrosis factor α (TNF-α) release and concomitant pyrolytic cell death, and is thereby detrimental to the host (Roca et al. Citation2019). In contrast, signaling by PGE2 stimulates the release of IL-1β, and this signaling axis promotes intracellular killing by macrophages (Peres-Buzalaf et al. Citation2011; Jayaraman et al. Citation2013). Additionally, PGE2 signaling limits systemic activation of the immune response by promoting apoptosis rather than pyrolytic forms of cell death and can counteract or limit the pro-inflammation induced by LTB4. A single nucleotide polymorphism (SNPs) has been identified in the promotor of LTA4H, leading to a disbalance in eicosanoid signaling due to low or high LTB4 levels, and both result in worse outcomes after TB, especially TB meningitis (Tobin et al. Citation2010, Citation2012; Yang et al. Citation2014). In short, eicosanoid signaling is of importance during Mtb infections wherein both LTB4 and PGE2 should both be active to induce sufficient inflammation, while preventing TNF-α overstimulation and concomitant pyrolytic cell death. TBCOX2, a clinical trial that used etoricoxib to inhibit COX-2 did not show any beneficial effects of this adjunctive therapy (Jenum et al. Citation2021), possibly due to the aforementioned SNP, and highlights the importance of personalized approach when targeting the eicosanoid pathway.
3.3. Battle for nutrients
As described above, TLR ligation promotes the uptake and synthesis of lipids that will be stored in lipid droplets (). These lipid droplets usually localize in proximity of mitochondria to regulate energy homeostasis of host cells (Olzmann and Carvalho Citation2019). The degradation of lipid droplets for energy production involves the mobilization and degradation of FAs, either via PPAR-α mediated lipolysis or via lysosomal degradation using a specialized form of autophagy, lipophagy (Hüsler et al. Citation2023). Mycobacteria escape from phagosomes using virulence factors ESX-1/ESAT-6 and PDIM by inducing phagosomal membrane permeabilisation and migrate toward cytosolic FAs derived from lipid droplets (van der Wel et al. Citation2007; Simeone et al. Citation2012; Quigley et al. Citation2017) (). Alternatively, mycobacteria actively promote migration of lipid droplets, which may involve TLR-mediated uncoupling of lipid droplets from mitochondria and redirection toward mycobacteria-containing vacuoles by mechanisms that have not been completely deciphered yet (Caire-Brändli et al. Citation2014; Daniel et al. Citation2011; Hüsler et al. Citation2023; Santucci et al. Citation2018). Lastly, host cells partially use the same machinery involved in autophagy to degrade lipids and cytosolic bacteria, depicted lipophagy and xenophagy, respectively, which may fuze in later stages of autophagosomal degradation (). Irrespective of the exact mechanism, vacuoles containing both mycobacteria and lipid droplets can form and/or associate in close proximity (Russell et al. Citation2009; Daniel et al. Citation2011; Almeida et al. Citation2023). Moreover, Mtb’ lipid-based virulence factor lipid sulfolipid-1 (SL-1) inhibits mTORC1 (Sachdeva et al. Citation2020), which leads to induction of autophagy, and certain Mtb strains are insensitive to autophagy-mediated degradation (Siregar et al. Citation2022; Kinsella et al. Citation2023), suggesting mycobacteria may also actively promote autophagy to gain access to host lipid droplets. Yet, vesicles dynamics are complicated and require further investigation in mycobacteria-infected macrophages.
In any case, mycobacteria within autophagosomes have to inhibit the normal process of autophagosomal maturation and subsequent fusion with lysosomes to prevent mycobacterial degradation (). Mtb has multiple lipid-based virulence factors, such as 1-tuberculosinyladenosine and SL-1, that impair lysosomal function of host cells (Bedard et al. Citation2023), likely by increasing the intraphagosomal pH, which impairs phagosomal maturation and thereby causes accumulation of (auto-)phagosomes in infected macrophages (Pisu et al. Citation2020; Caire-Brändli et al. Citation2014; Fineran et al. Citation2016; Sachdeva et al. Citation2020,The observed phenotype resembles lysosomal storage disorders to some extent, which host cells may try to overcome with lysosomal biogenesis, orchestrated by mTORC1 signaling and transcription factor EB (TFEB)-mediated gene expression (Fineran et al. Citation2016; Sachdeva et al. Citation2020). Remarkably, several host-directed compounds induced a similar phenotype of lysosomal storage disorders, while effectively reducing intracellular burden of Mtb (Heemskerk et al. Citation2021; Boland et al. Citation2023). These host-directed treatments indeed increased TFEB nuclear translocation (Heemskerk et al. Citation2021; Boland et al. Citation2023), and suggest mTORC1/TFEB signaling has a central role in overcoming impaired autophagosomal maturation. Thus, whether intracellular Mtb will be killed or survive within (auto)phagosomes, depends on whether host cells are able to overcome the impaired (auto)phagosomal maturation mediated by Mtb virulence factors. To maintain (auto)phagosomal degradation of host cells, TFEB-mediated lysosomal biogenesis seems to be crucial, likely orchestrated by signaling cues within lysosomes, but this process requires further investigation.
When in close proximity, ESAT-6 and PDIM, released by Mtb, also induce mitochondrial damage to impair host effector functions and to facilitate metabolic exchanges (Patrick and Watson Citation2021; Pagán et al. Citation2022). Host cells have multiple strategies to cope with mitochondrial damage with mTORC1 and leucine-rich repeat kinase 2 (LRRK2) emerging as important players during Mtb infection. mTORC1 is an important nutrient signaling hub that regulates the metabolic switch toward glycolysis, as discussed above. Moreover, mTORC1 suppresses autophagy, while stimulating mitochondrial biogenesis (Weichhart et al. Citation2015), the latter of which may help to overcome mycobacteria-induced mitochondrial damage and subsequent cell death (Pagán et al. Citation2022). LRRK2, an IFN-y induced gene, impairs phagosomal maturation of Mtb-containing phagosomes (Härtlova et al. Citation2018), but also maintains mitochondrial homeostasis and facilitates the repair of damaged phagosomal membranes during Mtb infection (Herbst et al. Citation2020; Weindel et al. Citation2020). Being a master regulator of specific forms of autophagy as well as phago-/endosomal trafficking by modulating activity of v-ATPase subunits (Gardet et al. Citation2010; Wallings et al. Citation2019; Patrick and Watson Citation2021), LRRK2 may prevent the degradation of mycobacteria-containing vacuoles or promote fusion with nutrient-rich endosomes. As LRRK2 is a known genetic risk factor for M. leprosy and was recently also identified as a risk factor for Mtb (Wang et al. Citation2018; Patrick and Watson Citation2021), better understanding of the kinetics and function of LRRK2 during Mtb infection is important and may identify novel potent therapeutic targets.
4. Therapeutic potential
Throughout the review, possibilities for intervention have been discussed for different aspects of bacterial metabolism. Importantly, routinely used in vitro drug testing models employ 7H9 broth or 7H10 agar plates, where Mtb has access to multiple carbon sources and which do not include any (immune) cells. Consequently, Mtb uses vastly different metabolic pathways in these in vitro models as compared to in vivo, and more representative models are therefore desired to enable the identification of bacteria-directed drugs that potently target the lipid metabolism of Mtb and act on intracellular bacteria. In addition, a combination of host- and bacteria-directed compounds likely act synergistic. Based on the targets described in this review, here we will discuss several examples of potential synergistic therapies that may act by: 1) limiting metabolism of lipids, 2) impairing cholesterol detoxification routes and 3) alleviate host-pathogen interactions to facilitate bacterial killing ().
Figure 6. Three suggestions for dual-acting therapies that target both mtb and host cells to facilitate synergistic therapies. (1) Impairing bacterial cholesterol metabolism by targeting bacteria-specific EtfD to impair β-oxidation or RV1625c to promote upregulation of bacterial cAMP, which impairs cholesterol metabolism. In addition, limit availability of cholesterol (and FAs) in host cells using statins and/or PPAR. (2) Promote intoxication by propionyl-CoA, which is produced as by-product during β-oxidation, by blocking detoxification pathways using ACCase5 and Icl1/2 inhibitors. Host itaconate will further inhibit Icl1/2, whose levels can be increased by promoting host macrophages polarization toward M1-like phenotype or administering endogenous itaconate. (3) Alleviating host-pathogen interaction will promote intracellular degradation of mtb. Inhibiting bacterial transporters MmpL7, MmpL3 and/or LprG will lead to reduced expression of virulence factors. In addition, stimulate host cells to mature their phagosomes and/or induce autophagy, both to promote (auto-)phagosomal degradation further impairs intracellular survival of mycobacteria. Drugs can either target mtb (blue/grey drug symbol) or the host (blue/red drug symbol). ‘?’ responsible protein not identified. Created with Bio-Render.com.
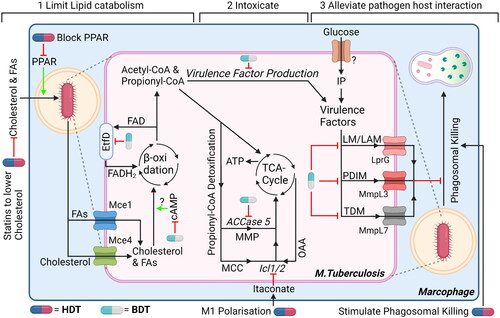
The first strategy aims to prevent Mtb from acquiring and catabolising lipids to impair energy production. Limiting production and/or delivery of FA and cholesterol to mycobacteria-containing phagosomes will likely impair mycobacterial metabolism. One approach that is currently being evaluated in clinical trials is to use statins to reduce circulating cholesterol levels. In addition, upregulation of bacterial cAMP levels by adenylyl cyclase Rv1625c agonist GSK2556286 was recently shown to block the ability of Mtb to use cholesterol for its metabolism (Brown et al. Citation2023). Although GSK2556286 alone did not reduce the Mtb burden in mice, GSK2556286 might still be interesting for combination therapies, for example by simultaneously targeting host’ cAMP, which is known to be modulated by mycobacteria (Knapp and McDonough Citation2014). Targeting host’ cAMP levels has been investigated using phosphodiesterase inhibitors in the context of TB, with phosphodiesterase-4 inhibitor CC-11050 having successfully completed phase II trials as adjunctive therapy (Wallis et al. Citation2021). Targeting PPAR-γ or PPAR-α to respectively limit lipid uptake or reduce lipolysis by host cells may also reduce intracellular availability of FA and cholesterol (Hüsler et al. Citation2023). The degradation of FAs and cholesterol through β-oxidation involves many redundant enzymes that are also expressed by mammalian host cells, thereby omitting β-oxidation itself from therapeutic intervention. However, Mtb EftD is pivotal to oxidize FADH2 to FAD, which is used as a co-factor both for FadE enzymatic activity during β-oxidation as well as succinate dehydrogenase activity in the TCA cycle. Lowering FAD levels by inhibition of EftD activity can thus interfere with both vital processes of Mtb. Moreover, Mtb EtfD is very different from its human counterpart and small molecule inhibitors against Mtb EtfD have already been identified (Székely et al. Citation2020).
The second strategy aims to kill Mtb by propionyl-CoA intoxication. During lipid metabolism, Mtb detoxifies propionyl-CoA mainly through the MMP and the MCC. If MMP and MCC are blocked using an ACCase 5 inhibitor (Lin et al. Citation2006) and an Icl1/2 inhibitor, respectively, toxic propionyl-CoA will accumulate within mycobacteria. An additional advantage of Icl1/2 inhibition is that the glyoxylate shunt of Mtb is also impaired, thereby blocking production of virulence factors arabinose, trehalose and inositol in glucose deprived conditions. Interestingly, M1-polarised macrophages produce a natural inhibitor of Icl1/2, namely itaconate, as well as itaconate-CoA, which can block MutAB from the MMP (Ruetz et al. Citation2019). Their production by macrophages can be stimulated using HDTs that promote macrophage activation by e.g. TLR agonist or adjuvants, as discussed elsewhere (Kilinç et al. Citation2021), or by administering endogenous itaconate (Coelho Citation2022). Finally, limiting availability of carbohydrates and especially even-chained fatty acids as described in the previous paragraph stimulates mycobacteria to metabolize cholesterol and likely further potentiates ACCase 5 and Icl1/2 inhibition.
The third strategy is to alleviate host-pathogen interaction by promoting lysosomal degradation by host cells and limiting the expression of mycobacterial virulence factors that counteract on this process. In addition to EmbC, which is already targeted by the antibiotic ethambutol, his review has highlighted multiple biosynthetic enzymes, including Fas, Pks13 and PatA (Sassetti and Rubin Citation2003; DeJesus et al. Citation2017; Boldrin et al. Citation2021), that may be targeted. Alternatively, inhibiting membrane transporters MmpL7, MmpL3 or LprG to prevent surface expression of PDIM, TDM or the PI-based virulence factors (PIM, LM and LAM), respectively, will also alleviate associated host-pathogen interactions. MmpL3 and MmpL7 are also important to maintain cell wall integrity and efflux activity of certain antibiotics, respectively (Kardan-Yamchi et al. Citation2019; Shao et al. Citation2020), further highlighting their feasibility and importance as therapeutic targets. Multiple MmpL3 inhibitors have already been developed (Shao et al. Citation2020), with SQ109 having successfully completed phase II studies and recently acquired approval by FDA as an orphan drug to treat MDR-TB (Singh et al. Citation2022). Interestingly, in mice, SQ109 was found to enhance M1 polarization of host macrophages, either due to a direct effect on host cells, or indirectly due to impaired MmpL3 activity. For MmpL7 and LprG, no clinically evaluated drugs are available to our knowledge, although some preclinical LprG inhibitors are available. Interestingly, mycobacterial resistance against pyrazinamide, a first-line drug with a yet unknown mechanism of action, associates with mutations in LprG (Shi et al. Citation2018), suggesting pyrazinamide may – directly or indirectly – target LprG. Combining therapies that inhibit virulence factor expression with host-directed compounds that promote lysosome degradation likely further impairs intracellular survival. Enhancing autophagy and phagosome maturation through host-directed therapy have been thoroughly investigated and is reviewed extensively elsewhere (Kilinç et al. Citation2021). Imatinib and metformin have both been investigated extensively in this regard and are both under clinical evaluation, but not yet in combination with inhibitors of Mtb virulence factor that impair lysosomal degradation.
5. Conclusion
This review summarized the main metabolic programs that are active in intracellular Mtb in which the many interconnected pathways highlight the versatility of Mtb. Mtb possesses many redundant enzymes and shares several enzymes involved in lipid metabolism with humans, which makes these unsuitable targets for therapeutic intervention. However, this review also highlights key enzymes that are pivotal in Mtb lipid metabolism and production of lipid-based virulence factors that may be interesting targets for drug development. Finally, as Mtb and host cells compete for the same nutrients and try to impair each other’s nutrient availability, designing anti-mycobacterial therapies that include both bacteria-directed and host-directed drugs will most likely mediate more effective therapies than those currently available. Such dual-acting therapies may have the potential to disrupt the current status quo in our battle to eradicate Mtb infections globally.
| List of abbreviations | ||
| (LppX) | = | 24kDa secreted lipoprotein |
| (5-LOX) | = | 5-lipooxygenase |
| (ACCase) | = | acyl-CoA carboxylase |
| (ATP) | = | adenosine tri-phosphate |
| (AMs) | = | alveolar macrophages |
| (Ag85) | = | antigen 85 |
| (AfTC) | = | arabinofuronasyltransferase |
| (AG) | = | arabinogalactan |
| (BDT) | = | bacteria-directed therapy |
| (ChoD) | = | cholesterol oxidase |
| (CA) | = | citric acid |
| (CoA) | = | coenzyme A |
| (cAMP) | = | cyclic adenosine monophosphate |
| (COX) | = | cyclooxygenases |
| (CDP) | = | cytidine diphosphate |
| (CMP) | = | cytidine monophosphate |
| (Cyp125) | = | cytochrome P450 125 |
| (Cyp142) | = | cytochrome P450 142 |
| (DAG) | = | diacylglycerol |
| (DHAP) | = | dihydroxyacetone phosphate |
| (ESAT6) | = | early secreted antigenic target 6 |
| (EtfD) | = | electron transfer flavoprotein D |
| (FA) | = | fatty acid |
| (Fas) | = | fatty acid synthase |
| (AMP) | = | fatty acyl adenylate |
| (FACL6) | = | fatty acyl-CoA ligase 6 |
| (FAD) | = | flavin adenine dinucleotide |
| (FADH2) | = | flavin adenine dinucleotide dihydrogen |
| (GlpX) | = | fructose-1,6-biphosphatase 1 class 2 |
| (Fba) | = | fructose-1,6-biphosphate aldolase |
| (Gpm2) | = | glucosephosphate mutase 2 |
| (GDP) | = | guanosine diphosphate |
| (HDT) | = | host-directed therapy |
| (HIF) | = | hypoxia-inducible factor |
| (IRG1) | = | immune-responsive gene 1 |
| (ImpC) | = | inositol monophosphate phosphatase C |
| (IP) | = | inositol phosphate |
| (IFN) | = | interferon |
| (IMs) | = | interstitial macrophages |
| (ILI) | = | intracytoplasmic lipid inclusions |
| (Icl1/2) | = | isocitrate lyase 1/2 |
| (ICA) | = | isocitric acid |
| (Ksh) | = | ketosteroid-9α-hydroxylase |
| (LRRK2) | = | leucine-rich repeat kinase 2 |
| (LTA4H) | = | leukotriene A4 hydrolase |
| (LTB4) | = | leukotriene B4 |
| (LipY) | = | lipaseY |
| (LD) | = | lipid droplet |
| (Ltp2) | = | lipid transfer protein 2 |
| (LucA) | = | lipid uptake coordinator A |
| (LAM) | = | lipoarabinomannan |
| (LM) | = | lipomannan |
| (LprG) | = | lipoproteinG |
| (LDL) | = | low-density lipoprotein |
| (GlcB) | = | malate synthase G |
| (MEZ) | = | malic enzyme |
| (Mce1) | = | mammalian cell entry 1 |
| (Mce4) | = | mammalian cell entry 4 |
| (MceG) | = | mammalian cell entry G |
| (mTORC1) | = | mammalian target of rapamycin complex 1 |
| (Man) | = | mannose |
| (PimA) | = | mannosyltransferase A |
| (PimB) | = | mannosyltransferase B |
| (PimE) | = | mannosyltransferase E |
| (MM) | = | meromycolate |
| (Rv1322A) | = | methyl malonyl-CoA epimerase |
| (MCC) | = | methylcitrate cycle |
| (MCC) | = | methylcitrate cycle |
| (PrpD) | = | methylcitrate dehydratase |
| (PrpC) | = | methylcitrate synthase |
| (MMP) | = | methyl-malonyl pathway |
| (MDR/XDR) | = | Multi-drug and extensive-drug resistant |
| (MutAB) | = | mutase AB |
| (MmpL3) | = | mycobacterium membrane protein large 3 |
| (MmpL7) | = | mycobacterium membrane protein large 7 |
| (Mtb) | = | Mycobacterium tuberculosis |
| (Mas) | = | mycocerosic acid synthase |
| (MAGP) | = | mycolate arabinogalactan peptidoglycan |
| (MAMT) | = | mycolic acid methyl transferase |
| (OOA) | = | oxaloacetate |
| (OXPHOS) | = | oxidative phosphorylation |
| (PAMPs) | = | pathogen-associated molecular patterns |
| (PPAR) | = | peroxisome proliferator activated receptor |
| (PI) | = | phosphatidylinositol |
| (PIM) | = | phosphatidylinositol mannoside |
| (PgsA1) | = | phosphatidylinositol phosphate synthase |
| (PckA) | = | phosphoenolpyruvate carboxykinase |
| (PC) | = | phthiocerol |
| (PDIM) | = | phthiocerol dimycocerosate |
| (Pps) | = | phthiocerol polyketide synthase |
| (Pks) | = | polyketide Synthase |
| (PapA5) | = | polyketide-associated protein A5 |
| (Ppm1) | = | polyprenyl Monophosphomannose Synthase |
| (PP) | = | polyprenylphosphate |
| (PPM) | = | polyprenylphosphate mannose |
| (PTGES) | = | prostaglandin E synthase |
| (PGE2) | = | prostaglandin E2 |
| (Pca) | = | pyruvate carboxylase |
| (PykA) | = | pyruvate kinase |
| (PpdK) | = | pyruvate phosphate dikinase |
| (SL-1) | = | sulfolipid-1 |
| (TtfA) | = | TMM transport factor A |
| (TLR) | = | toll-like receptor |
| (TFEB) | = | transcription factor EB |
| (TDM) | = | trehalose dimycolate |
| (TMM) | = | trehalose monomycolate |
| (TAG) | = | triacylglycerol |
| (Tgs1) | = | triacylglycerol synthase 1 |
| (TCA) | = | tricarboxylic acid |
| (Tpi) | = | triosephosphate isomerase |
| (TNF-α) | = | tumor necrosis factor α |
| (VitB12) | = | vitamin B12 |
| (α-KG) | = | α-ketoglutarate |
Disclosure statement
The authors declare no conflict of interest.
Additional information
Funding
References
- Almeida PEd, Pereira de Sousa NM, Rampinelli PG, Silva RVdS, Correa JR, D’Avila H. 2023. Lipid droplets as multifunctional organelles related to the mechanism of evasion during mycobacterial infection. Front Cell Infect Microbiol. 13:1102643. doi: 10.3389/fcimb.2023.1102643.
- Andries K, Verhasselt P, Guillemont J, Göhlmann HWH, Neefs J-M, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, et al. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science (1979). 307(5707):223–227. doi: 10.1126/science.1106753.
- Augenstreich J, Haanappel E, Sayes F, Simeone R, Guillet V, Mazeres S, Chalut C, Mourey L, Brosch R, Guilhot C, et al. 2020. phthiocerol dimycocerosates from Mycobacterium tuberculosis increase the membrane activity of bacterial effectors and host receptors. Front Cell Infect Microbiol. 10:420. doi: 10.3389/fcimb.2020.00420.
- Banks DA, Ahlbrand SE, Hughitt VK, Shah S, Mayer-Barber KD, Vogel SN, El-Sayed NM, Briken V. 2019. Mycobacterium tuberculosis inhibits autocrine type I IFN signaling to increase intracellular survival. J Immunol. 202(8):2348–2359. doi: 10.4049/jimmunol.1801303.
- Barkan D, Hedhli D, Yan HG, Huygen K, Glickman MS. 2012. Mycobacterium tuberculosis lacking all mycolic acid cyclopropanation is viable but highly attenuated and hyperinflammatory in mice. Infect Immun. 80(6):1958–1968. doi: 10.1128/IAI.00021-12.
- Barrientos OM, Langley E, González Y, Cabello C, Torres M, Guzmán-Beltrán S. 2022. Mycobacterium tuberculosis whiB3 and lipid metabolism genes are regulated by host induced oxidative stress. Microorganisms. 10(9):1821. doi: 10.3390/microorganisms10091821.
- Basu P, Sandhu N, Bhatt A, Singh A, Balhana R, Gobe I, Crowhurst NA, Mendum TA, Gao L, Ward JL, et al. 2018. The anaplerotic node is essential for the intracellular survival of Mycobacterium tuberculosis. J Biol Chem. 293(15):5695–5704. doi: 10.1074/jbc.RA118.001839.
- Bazet Lyonnet B, Diacovich L, Cabruja M, Bardou F, Quémard A, Gago G, Gramajo H. 2014. Pleiotropic effect of AccD5 and AccE5 depletion in acyl-coenzyme a carboxylase activity and in lipid biosynthesis in mycobacteria. PLoS One. 9(6):e99853. doi: 10.1371/journal.pone.0099853.
- Bedard M, van der Niet S, Bernard EM, Babunovic G, Cheng T-Y, Aylan B, Grootemaat AE, Raman S, Botella L, Ishikawa E, et al. 2023. A terpene nucleoside from M. tuberculosis induces lysosomal lipid storage in foamy macrophages. J Clin Investigat. 133(6):1–17. doi: 10.1172/JCI161944.
- Beites T, Jansen RS, Wang R, Jinich A, Rhee KY, Schnappinger D, Ehrt S. 2021. Multiple acyl-CoA dehydrogenase deficiency kills Mycobacterium tuberculosis in vitro and during infection. Nat Commun. 12(1):6593. doi: 10.1038/s41467-021-26941-1.
- Birch HL, Alderwick LJ, Bhatt A, Rittmann D, Krumbach K, Singh A, Bai Y, Lowary TL, Eggeling L, Besra GS, et al. 2008. Biosynthesis of mycobacterial arabinogalactan: identification of a novel α(1→3) arabinofuranosyltransferase. Mol Microbiol. 69(5):1191–1206. doi: 10.1111/j.1365-2958.2008.06354.x.
- Boland R, Heemskerk MT, Forn-Cuní G, Korbee CJ, Walburg KV, Esselink JJ, Carvalho dos Santos C, de Waal AM, van der Hoeven DCM, van der Sar E, et al. 2023. Repurposing Tamoxifen as Potential Host-Directed Therapeutic for Tuberculosis. mBio. 14(1):e0302422. doi: 10.1128/mbio.03024-22.
- Boldrin F, Anso I, Alebouyeh S, Sevilla IA, Geijo M, Garrido JM, Marina A, Cioetto Mazzabò L, Segafreddo G, Guerin ME, et al. 2021. The phosphatidyl-myo-inositol dimannoside acyltransferase PatA is essential for mycobacterium tuberculosis growth in vitro and in vivo. J Bacteriol. 203(7):e00439-20. doi: 10.1128/JB.00439-20.
- Boldrin F, Ventura M, Degiacomi G, Ravishankar S, Sala C, Svetlikova Z, Ambady A, Dhar N, Kordulakova J, Zhang M, et al. 2014. The phosphatidyl-myo-inositol mannosyltransferase PimA is essential for Mycobacterium tuberculosis growth in vitro and in vivo. J Bacteriol. 196(19):3441–3451. doi: 10.1128/JB.01346-13.
- Bozza PT, Bakker-Abreu I, Navarro-Xavier RA, Bandeira-Melo C. 2011. Lipid body function in eicosanoid synthesis: an update. Prostaglandins Leukot Essent Fatty Acids. 85(5):205–213. doi: 10.1016/j.plefa.2011.04.020.
- Braverman J, Sogi KM, Benjamin D, Nomura DK, Stanley SA. 2016. HIF-1α is an essential mediator of IFN-γ–dependent immunity to Mycobacterium tuberculosis. The Journal of Immunology. 197(4):1287–1297. doi: 10.4049/jimmunol.1600266.
- Brown KL, Wilburn KM, Montague CR, Grigg JC, Sanz O, Pérez-Herrán E, Barros D, Ballell L, VanderVen BC, Eltis LD, et al. 2023. Cyclic AMP-mediated inhibition of cholesterol catabolism in Mycobacterium tuberculosis by the novel drug candidate GSK2556286. Antimicrob Agents Chemother. 67(1):e0129422. doi: 10.1128/aac.01294-22.
- Brzostek A, Pawelczyk J, Rumijowska-Galewicz A, Dziadek B, Dziadek J. 2009. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J Bacteriol. 191(21):6584–6591. doi: 10.1128/JB.00488-09.
- Caire-Brändli I, Papadopoulos A, Malaga W, Marais D, Canaan S, Thilo L, de Chastellier C. 2014. Reversible lipid accumulation and associated division arrest of mycobacterium avium in lipoprotein-induced foamy macrophages may resemble key events during latency and reactivation of tuberculosis. Infect Immun. 82(2):476–490. doi: 10.1128/IAI.01196-13.
- Carey AF, Rock JM, Krieger IV, Chase MR, Fernandez-Suarez M, Gagneux S, Sacchettini JC, Ioerger TR, Fortune SM. 2018. TnSeq of Mycobacterium tuberculosis clinical isolates reveals strain-specific antibiotic liabilities. PLoS Pathog. 14(3):e1006939. doi: 10.1371/journal.ppat.1006939.
- Coelho C. 2022. Itaconate or how i learned to stop avoiding the study of immunometabolism. PLoS Pathog. 18(3):e1010361. doi: 10.1371/journal.ppat.1010361.
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE, III, et al. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 393(6685):537–544. & doi: 10.1038/31159.
- D’Avila H, Melo RCN, Parreira GG, Werneck-Barroso E, Castro-Faria-Neto HC, Bozza PT. 2006. Mycobacterium bovis bacillus calmette-guérin induces TLR2-mediated formation of lipid bodies: intracellular domains for eicosanoid synthesis in vivo. J Immunol. 176(5):3087–3097. doi: 10.4049/jimmunol.176.5.3087.
- Daleke MH, Cascioferro A, de Punder K, Ummels R, Abdallah AM, van der Wel N, Peters PJ, Luirink J, Manganelli R, Bitter W, et al. 2011. Conserved Pro-Glu (PE) and Pro-Pro-Glu (PPE) protein domains target LipY lipases of pathogenic mycobacteria to the cell surface via the ESX-5 pathway. Journal of Biological Chemistry. 286(21):19024–19034. doi: 10.1074/jbc.M110.204966.
- Daniel J, Deb C, Dubey VS, Sirakova TD, Abomoelak B, Morbidoni HR, Kolattukudy PE. 2004. Induction of a novel class of diacylglycerol acyltransferases and triacylglycerol accumulation in Mycobacterium tuberculosis as it goes into a dormancy-like state in culture. J Bacteriol. 186(15):5017–5030. doi: 10.1128/JB.186.15.5017-5030.2004.
- Daniel J, Maamar H, Deb C, Sirakova TD, Kolattukudy PE. 2011. Mycobacterium tuberculosis uses host triacylglycerol to accumulate lipid droplets and acquires a dormancy-like phenotype in lipid-loaded macrophages. PLoS Pathog. 7(6):e1002093. doi: 10.1371/journal.ppat.1002093.
- Daniel J, Oh TJ, Lee CM, Kolattukudy PE. 2007. AccD6, a member of the Fas II locus, is a functional carboxyltransferase subunit of the acyl-coenzyme A carboxylase in Mycobacterium tuberculosis. J Bacteriol. 189(3):911–917. doi: 10.1128/JB.01019-06.
- Daniel J, Sirakova T, Kolattukudy P. 2014. An Acyl-CoA synthetase in mycobacterium tuberculosis involved in triacylglycerol accumulation during dormancy. PLoS One. 9(12):e114877. doi: 10.1371/journal.pone.0114877.
- Deb C, Daniel J, Sirakova TD, Abomoelak B, Dubey VS, Kolattukudy PE. 2006. A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in mycobacterium tuberculosis. J Biol Chem. 281(7):3866–3875. doi: 10.1074/jbc.M505556200.
- DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, Boutte CC, Rubin EJ, Schnappinger D, Ehrt S, Fortune SM, et al. 2017. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio. 8(1):e02133–16. doi: 10.1128/mBio.02133-16.
- Dheda K, Gumbo T, Maartens G, Dooley KE, McNerney R, Murray M, Furin J, Nardell EA, London L, Lessem E, et al. 2017. The epidemiology, pathogenesis, transmission, diagnosis, and management of multidrug-resistant, extensively drug-resistant, and incurable tuberculosis. Lancet Respir Med. 5(4):291–360. doi: 10.1016/S2213-2600(17)30079-6.
- Dinadayala P, Kaur D, Berg S, Amin AG, Vissa VD, Chatterjee D, Brennan PJ, Crick DC. 2006. Genetic basis for the synthesis of the immunomodulatory mannose caps of lipoarabinomannan in Mycobacterium tuberculosis. J Biol Chem. 281(29):20027–20035. doi: 10.1074/jbc.M603395200.
- Divangahi M, Desjardins D, Nunes-Alves C, Remold HG, Behar SM. 2010. Eicosanoid pathways regulate adaptive immunity to Mycobacterium tuberculosis. Nat Immunol. 11(8):751–758. doi: 10.1038/ni.1904.
- Drage MG, Tsai H-C, Pecora ND, Cheng T-Y, Arida AR, Shukla S, Rojas RE, Seshadri C, Moody DB, Boom WH, et al. 2010. Mycobacterium tuberculosis lipoprotein LprG (Rv1411c) binds triacylated glycolipid agonists of Toll-like receptor 2. Nat Struct Mol Biol. 17(9):1088–1095. doi: 10.1038/nsmb.1869.
- Eoh H, Rhee KY. 2014. Methylcitrate cycle defines the bactericidal essentiality of isocitrate lyase for survival of mycobacterium tuberculosis on fatty acids. Proc Natl Acad Sci USA. 111(13):4976–4981. doi: 10.1073/pnas.1400390111.
- Fay A, Czudnochowski N, Rock JM, Johnson JR, Krogan NJ, Rosenberg O, Glickman MS. 2019. Two accessory proteins govern MmpL3 mycolic acid transport in mycobacteria. mBio. 10(3):e00850–19. doi: 10.1128/mBio.00850-19.
- Fineran P, Lloyd-Evans E, Lack NA, Platt N, Davis LC, Morgan AJ, Höglinger D, Tatituri RVV, Clark S, Williams IM, et al. 2016. Pathogenic mycobacteria achieve cellular persistence by inhibiting the Niemann-Pick Type C disease cellular pathway. Wellcome Open Res. 1:18. doi: 10.12688/wellcomeopenres.10036.1.
- Fratti RA, Chua J, Vergne I, Deretic V. 2003. Mycobacterium tuberculosis glycosylated phosphatidylinositol causes phagosome maturation arrest. Proc Natl Acad Sci USA. 100(9):5437–5442. doi: 10.1073/pnas.0737613100.
- Ganapathy U, Marrero J, Calhoun S, Eoh H, de Carvalho LPS, Rhee K, Ehrt S. 2015. Two enzymes with redundant fructose bisphosphatase activity sustain gluconeogenesis and virulence in Mycobacterium tuberculosis. Nat Commun. 6:7912. doi: 10.1038/ncomms8912.
- Gardet A, Benita Y, Li C, Sands BE, Ballester I, Stevens C, Korzenik JR, Rioux JD, Daly MJ, Xavier RJ, et al. 2010. LRRK2 is involved in the IFN-γ response and host response to pathogens. J Immunol. 185(9):5577–5585. doi: 10.4049/jimmunol.1000548.
- Gaur RL, Ren K, Blumenthal A, Bhamidi S, Gibbs S, Jackson M, Zare RN, Ehrt S, Ernst JD, Banaei N, et al. 2014. LprG-mediated surface expression of lipoarabinomannan is essential for virulence of mycobacterium tuberculosis. PLoS Pathog. 10(9):e1004376. doi: 10.1371/journal.ppat.1004376.
- Gavalda S, Léger M, van der Rest B, Stella A, Bardou F, Montrozier H, Chalut C, Burlet-Schiltz O, Marrakchi H, Daffé M, et al. 2009. The Pks13/FadD32 crosstalk for the biosynthesis of mycolic acids in Mycobacterium tuberculosis. J Biol Chem. 284(29):19255–19264. doi: 10.1074/jbc.M109.006940.
- Gibson AJ, Stiens J, Passmore IJ, Faulkner V, Miculob J, Willcocks S, Coad M, Berg S, Werling D, Wren BW, et al. 2022. Defining the Genes required for survival of Mycobacterium bovis in the bovine host offers novel insights into the genetic basis of survival of pathogenic mycobacteria. mBio. 13(4):e0067222. doi: 10.1128/mbio.00672-22.
- Gioffré A, Infante E, Aguilar D, Santangelo MDlP, Klepp L, Amadio A, Meikle V, Etchechoury I, Romano MI, Cataldi A, et al. 2005. Mutation in mce operons attenuates Mycobacterium tuberculosis virulence. Microbes Infect. 7(3):325–334. doi: 10.1016/j.micinf.2004.11.007.
- Goossens SN, Sampson SL, Rie AV. 2020. Mechanisms of drug-induced tolerance in mycobacterium tuberculosis. Clin Microbiol Rev. 34(1):e00141-20. doi: 10.1128/CMR.00141-20.
- Griffin JE, Gawronski JD, DeJesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 7(9):e1002251. doi: 10.1371/journal.ppat.1002251.
- Guerin ME, Kaur D, Somashekar BS, Gibbs S, Gest P, Chatterjee D, Brennan PJ, Jackson M. 2009. New insights into the early steps of phosphatidylinositol mannoside biosynthesis in mycobacteria: pimB′ is an essential enzyme of Mycobacterium smegmatis. J Biol Chem. 284(38):25687–25696. doi: 10.1074/jbc.M109.030593.
- Gutka HJ, Wang Y, Franzblau SG, Movahedzadeh F. 2015. Glpx gene in mycobacterium tuberculosis is required for in vitro gluconeogenic growth and in vivo survival. PLoS One. 10(9):e0138436. doi: 10.1371/journal.pone.0138436.
- Härtlova A, Herbst S, Peltier J, Rodgers A, Bilkei-Gorzo O, Fearns A, Dill BD, Lee H, Flynn R, Cowley SA, Davies P, Lewis PA, Ganley IG, Martinez J, Alessi DR, Reith AD, Trost M, Gutierrez MG. 2018. LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. Embo J. 37:1–17.
- Heemskerk MT, Korbee CJ, Esselink JJ, dos Santos CC, van Veen S, Gordijn IF, Vrieling F, Walburg KV, Engele CG, Dijkman K, et al. 2021. Repurposing diphenylbutylpiperidine-class antipsychotic drugs for host-directed therapy of Mycobacterium tuberculosis and Salmonella enterica infections. Sci Rep. 11(1):19634. doi: 10.1038/s41598-021-98980-z.
- Herbst S, Campbell P, Harvey J, Bernard EM, Papayannopoulos V, Wood NW, Morris HR, Gutierrez MG. 2020. LRRK 2 activation controls the repair of damaged endomembranes in macrophages. Embo J. 39(18):e104494.
- Hu Y, van der Geize R, Besra GS, Gurcha SS, Liu A, Rohde M, Singh M, Coates A. 2010. 3-Ketosteroid 9α-hydroxylase is an essential factor in the pathogenesis of Mycobacterium tuberculosis. Mol Microbiol. 75(1):107–121. doi: 10.1111/j.1365-2958.2009.06957.x.
- Hüsler D, Stauffer P, Hilbi H. 2023. Tapping lipid droplets: a rich fat diet of intracellular bacterial pathogens. Mol Microbiol. 120(2):194–209. doi: 10.1111/mmi.15120.
- Jackson M, Crick DC, Brennan PJ. 2000. Phosphatidylinositol is an essential phospholipid of mycobacteria. J Biol Chem. 275(39):30092–30099. doi: 10.1074/jbc.M004658200.
- Jackson M, Stevens CM, Zhang L, Zgurskaya HI, Niederweis M. 2021. Transporters involved in the biogenesis and functionalization of the mycobacterial cell envelope. Chem Rev. 121(9):5124–5157. doi: 10.1021/acs.chemrev.0c00869.
- Jain M, Cox JS. 2005. Interaction between polyketide synthase and transporter suggests coupled synthesis and export of virulence lipid in M. tuberculosis. PLoS Pathog. 1(1):e2. doi: 10.1371/journal.ppat.0010002.
- Jayaraman P, Sada-Ovalle I, Nishimura T, Anderson AC, Kuchroo VK, Remold HG, Behar SM. 2013. IL-1β promotes antimicrobial immunity in macrophages by regulating TNFR signaling and caspase-3 activation. J Immunol. 190(8):4196–4204. doi: 10.4049/jimmunol.1202688.
- Jenum S, Tonby K, Rueegg CS, Rühwald M, Kristiansen MP, Bang P, Olsen IC, Sellæg K, Røstad K, Mustafa T, et al. 2021. A phase I/II randomized trial of H56: IC31 vaccination and adjunctive cyclooxygenase-2-inhibitor treatment in tuberculosis patients. Nat Commun. 12(1):6774. doi: 10.1038/s41467-021-27029-6.
- Johnston JB, Ouellet H, Ortiz De Montellano PR. 2010. Functional redundancy of steroid C26-monooxygenase activity in Mycobacterium tuberculosis revealed by biochemical and genetic analyses. J Biol Chem. 285(47):36352–36360. doi: 10.1074/jbc.M110.161117.
- Kalscheuer R, Weinrick B, Veeraraghavan U, Besra GS, Jacobs WR. 2010. Trehalose-recycling ABC transporter LpqY-SugA-SugB-SugC is essential for virulence of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 107(50):21761–21766. doi: 10.1073/pnas.1014642108.
- Kardan-Yamchi J, Kazemian H, Haeili M, Harati AA, Amini S, Feizabadi MM. 2019. Expression analysis of 10 efflux pump genes in multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis clinical isolates. J Glob Antimicrob Resist. 17:201–208. doi: 10.1016/j.jgar.2019.01.003.
- Kaur D, Berg S, Dinadayala P, Gicquel B, Chatterjee D, McNeil MR, Vissa VD, Crick DC, Jackson M, Brennan PJ, et al. 2006. Biosynthesis of mycobacterial lipoarabinomannan: role of a branching mannosyltransferase. Proc Natl Acad Sci USA. 103(37):13664–13669. doi: 10.1073/pnas.0603049103.
- Kaur D, McNeil MR, Khoo K-H, Chatterjee D, Crick DC, Jackson M, Brennan PJ. 2007. New insights into the biosynthesis of mycobacterial lipomannan arising from deletion of a conserved gene. J Biol Chem. 282(37):27133–27140. doi: 10.1074/jbc.M703389200.
- Kaur D, Obregón-Henao A, Pham H, Chatterjee D, Brennan PJ, Jackson M. 2008. Lipoarabinomannan of Mycobacterium: mannose capping by a multifunctional terminal mannosyltransferase. Proc Natl Acad Sci USA. 105(46):17973–17977. doi: 10.1073/pnas.0807761105.
- Kilinç G, Saris A, Ottenhoff THM. 2021. Haks, M. C. Host-directed therapy to combat mycobacterial infections. Immunol Rev. 301:62–83.
- Kim SK, Dickinson MS, Finer-Moore J, Guan Z, Kaake RM, Echeverria I, Chen J, Pulido EH, Sali A, Krogan NJ, et al. 2023. Structure and dynamics of the essential endogenous mycobacterial polyketide synthase Pks13. Nat Struct Mol Biol. 30(3):296–308. doi: 10.1038/s41594-022-00918-0.
- Kinsella RL, Kimmey JM, Smirnov A, Woodson R, Gaggioli MR, Chavez SM, Kreamalmeyer D, Stallings CL. 2023. Autophagy prevents early proinflammatory responses and neutrophil recruitment during Mycobacterium tuberculosis infection without affecting pathogen burden in macrophages. PLoS Biol. 21(6):e3002159. doi: 10.1371/journal.pbio.3002159.
- Knapp GS, McDonough KA. 2014. Cyclic AMP signaling in mycobacteria. Microbiol Spectr. 2(2). doi: 10.1128/microbiolspec.MGM2-0011-2013.
- Korduláková J, Gilleron M, Mikus̃ová K, Puzo G, Brennan PJ, Gicquel B, Jackson M. 2002. Definition of the first mannosylation step in phosphatidylinositol mannoside synthesis: pimA is essential for growth of mycobacteria. J Biol Chem. 277(35):31335–31344. doi: 10.1074/jbc.M204060200.
- Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, et al. 2010. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 115(23):4742–4749. doi: 10.1182/blood-2009-10-249540.
- Kremer L, Gurcha SS, Bifani P, Hitchen PG, Baulard A, Morris HR, Dell A, Brennan PJ, Besra GS. 2002. Characterization of a putative α-mannosyltransferase involved in phosphatidylinositol trimannoside biosynthesis in Mycobacterium tuberculosis. Biochem. J. 363(3):437–447. doi: 10.1042/0264-6021:3630437.
- Kwai BXC, Collins AJ, Middleditch MJ, Sperry J, Bashiri G, Leung IKH. 2020. Itaconate is a covalent inhibitor of theMycobacterium tuberculosisisocitrate lyase. RSC Med Chem. 12:57–61.
- Lachmandas E, Beigier-Bompadre M, Cheng S, Kumar V, van Laarhoven A, Wang X, Ammerdorffer A, Boutens L, de Jong D, Kanneganti T, et al. 2016. Rewiring cellular metabolism via the AKT/mTOR pathway contributes to host defence against Mycobacterium tuberculosis in human and murine cells. Eur J Immunol. 46(11):2574–2586. doi: 10.1002/eji.201546259.
- Lalvani A, Behr MA, Sridhar S. 2012. Innate immunity to TB: a druggable balancing act. Cell. 148(3):389–391. doi: 10.1016/j.cell.2012.01.026.
- Laval T, Chaumont L, Demangel C. 2021. Not too fat to fight: the emerging role of macrophage fatty acid metabolism in immunity to Mycobacterium tuberculosis. Immunol Rev. 301(1):84–97. doi: 10.1111/imr.12952.
- Lea-Smith DJ, Pyke JS, Tull D, McConville MJ, Coppel RL, Crellin PK. 2007. The reductase that catalyzes mycolic motif synthesis is required for efficient attachment of mycolic acids to arabinogalactan. J Biol Chem. 282(15):11000–11008. doi: 10.1074/jbc.M608686200.
- Lee W, VanderVen BC, Fahey RJ, Russell DG. 2013. Intracellular Mycobacterium tuberculosis exploits host-derived fatty acids to limit metabolic stress. Journal of Biological Chemistry. 288(10):6788–6800. doi: 10.1074/jbc.M112.445056.
- Li Y, Li Y-C, Liu X-T, Zhang L, Chen Y-H, Zhao Q, Gao W, Liu B, Yang H, Li P, et al. 2022. Blockage of citrate export prevents TCA cycle fragmentation via Irg1 inactivation. Cell Rep. 38(7):110391. doi: 10.1016/j.celrep.2022.110391.
- Lima P, Sidders B, Morici L, Reader R, Senaratne R, Casali N, Riley LW. 2007. Enhanced mortality despite control of lung infection in mice aerogenically infected with a Mycobacterium tuberculosis mce1 operon mutant. Microbes Infect. 9(11):1285–1290. doi: 10.1016/j.micinf.2007.05.020.
- Lin T-W, Melgar MM, Kurth D, Swamidass SJ, Purdon J, Tseng T, Gago G, Baldi P, Gramajo H, Tsai S-C, et al. 2006. Structure-based inhibitor design of AccD5, an essential acyl-CoA carboxylase carboxyltransferase domain of Mycobacterium tuberculosis. Proc Natl Acad Sci USA. 103(9):3072–3077. doi: 10.1073/pnas.0510580103.
- Mallick I, Santucci P, Poncin I, Point V, Kremer L, Cavalier J-F, Canaan S. 2021. Intrabacterial lipid inclusions in mycobacteria: unexpected key players in survival and pathogenesis? FEMS Microbiology Reviews. 45(6):fuab029. doi: 10.1093/femsre/fuab029.
- Matsunaga I, Bhatt A, Young DC, Cheng T-Y, Eyles SJ, Besra GS, Briken V, Porcelli SA, Costello CE, Jacobs WR, Jr., et al. 2004. Mycobacterium tuberculosis pks12 produces a novel polyketide presented by CD1c to T cells. J Exp Med. 200(12):1559–1569. doi: 10.1084/jem.20041429.
- Mattow J, Siejak F, Hagens K, Becher D, Albrecht D, Krah A, Schmidt F, Jungblut PR, Kaufmann SHE, Schaible UE, et al. 2006. Proteins unique to intraphagosomally grown Mycobacterium tuberculosis. Proteomics. 6(8):2485–2494. doi: 10.1002/pmic.200500547.
- Mehrotra P, Jamwal SV, Saquib N, Sinha N, Siddiqui Z, Manivel V, Chatterjee S, Rao KVS. 2014. Pathogenicity of Mycobacterium tuberculosis Is Expressed by Regulating Metabolic Thresholds of the Host Macrophage. PLoS Pathog. 10(7):e1004265. doi: 10.1371/journal.ppat.1004265.
- Milad N, Morissette MC. 2021. Revisiting the role of pulmonary surfactant in chronic inflammatory lung diseases and environmental exposure. Eur Respir Rev. 30(162):210077. doi: 10.1183/16000617.0077-2021.
- Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, et al. 2018. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 556(7699):113–117. doi: 10.1038/nature25986.
- Mishra AK, Alderwick LJ, Rittmann D, Tatituri RVV, Nigou J, Gilleron M, Eggeling L, Besra GS. 2007. Identification of an α(1→6) mannopyranosyltransferase (MptA), involved in Corynebacterium glutamicum lipomanann biosynthesis, and identification of its orthologue in Mycobacterium tuberculosis. Mol Microbiol. 65(6):1503–1517. doi: 10.1111/j.1365-2958.2007.05884.x.
- Mishra AK, Batt S, Krumbach K, Eggeling L, Besra GS. 2009. Characterization of the Corynebacterium glutamicum ΔpimB′ ΔmgtA double deletion mutant and the role of Mycobacterium tuberculosis orthologues Rv2188c and Rv0557 in glycolipid biosynthesis. J Bacteriol. 191(13):4465–4472. doi: 10.1128/JB.01729-08.
- Morii H, Okauchi T, Nomiya H, Ogawa M, Fukuda K, Taniguchi H. 2013. Studies of inositol 1-phosphate analogues as inhibitors of the phosphatidylinositol phosphate synthase in mycobacteria. J Biochem. 153(3):257–266. doi: 10.1093/jb/mvs141.
- Morita YS, Sena CBC, Waller RF, Kurokawa K, Sernee MF, Nakatani F, Haites RE, Billman-Jacobe H, McConville MJ, Maeda Y, et al. 2006. PimE is a polyprenol-phosphate-mannose-dependent mannosyltransferase that transfers the fifth mannose of phosphatidylinositol mannoside in mycobacteria. J Biol Chem. 281(35):25143–25155. doi: 10.1074/jbc.M604214200.
- Movahedzadeh F, Smith DA, Norman RA, Dinadayala P, Murray-Rust J, Russell DG, Kendall SL, Rison SCG, McAlister MSB, Bancroft GJ, et al. 2004. The Mycobactenum tuberculosis ino1 gene is essential for growth and virulence. Mol Microbiol. 51(4):1003–1014. doi: 10.1046/j.1365-2958.2003.03900.x.
- Movahedzadeh F, Wheeler PR, Dinadayala P, Av-Gay Y, Parish T, Daffé M, Stoker NG. 2010. Inositol monophosphate phosphatase genes of Mycobacterium tuberculosis. BMC Microbiol. 10(1):50. doi: 10.1186/1471-2180-10-50.
- Muñoz-Elías EJ, Mckinney JDM. 2005. Tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 11(6):638–644. doi: 10.1038/nm1252.
- Muñoz-Elías EJ, Upton AM, Cherian J, McKinney JD. 2006. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol. 60(5):1109–1122. doi: 10.1111/j.1365-2958.2006.05155.x.
- Murphy HN, Stewart GR, Mischenko VV, Apt AS, Harris R, McAlister MSB, Driscoll PC, Young DB, Robertson BD. 2005. The OtsAB pathway is essential for trehalose biosynthesis in Mycobacterium tuberculosis. J Biol Chem. 280(15):14524–14529. doi: 10.1074/jbc.M414232200.
- Nazarova EV, Montague CR, La T, Wilburn KM, Sukumar N, Lee W, Caldwell S, Russell DG, VanderVen BC. 2017. Rv3723/LucA coordinates fatty acid and cholesterol uptake in Mycobacterium tuberculosis. Elife. 6:e26969. doi: 10.7554/eLife.26969.
- Olzmann JA, Carvalho P. 2019. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 20(3):137–155. doi: 10.1038/s41580-018-0085-z.
- Pagán AJ, Lee LJ, Edwards-Hicks J, Moens CB, Tobin DM, Busch-Nentwich EM, Pearce EL, Ramakrishnan L. 2022. mTOR-regulated mitochondrial metabolism limits mycobacterium-induced cytotoxicity. Cell. 185(20):3720–3738.e13. doi: 10.1016/j.cell.2022.08.018.
- Pandey AK, Sassetti CM. 2008. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci USA. 105(11):4376–4380. doi: 10.1073/pnas.0711159105.
- Passemar C, Arbués A, Malaga W, Mercier I, Moreau F, Lepourry L, Neyrolles O, Guilhot C, Astarie-Dequeker C. 2014. Multiple deletions in the polyketide synthase gene repertoire of mycobacterium tuberculosis reveal functional overlap of cell envelope lipids in host-pathogen interactions. Cell Microbiol. 16(2):195–213. doi: 10.1111/cmi.12214.
- Patrick KL, Watson RO. 2021. Mitochondria: powering the innate immune response to Mycobacterium tuberculosis infection. Infect Immun. 89(4):e00687-20. doi: 10.1128/IAI.00687-20.
- Pawelczyk J, Brzostek A, Kremer L, Dziadek B, Rumijowska-Galewicz A, Fiolka M, Dziadek J. 2011. Accd6, a key carboxyltransferase essential for mycolic acid synthesis in Mycobacterium tuberculosis, is dispensable in a nonpathogenic strain. J Bacteriol. 193(24):6960–6972. doi: 10.1128/JB.05638-11.
- Pawełczyk J, Brzostek A, Minias A, Płociński P, Rumijowska-Galewicz A, Strapagiel D, Zakrzewska-Czerwińska J, Dziadek J. 2021. Cholesterol-dependent transcriptome remodeling reveals new insight into the contribution of cholesterol to Mycobacterium tuberculosis pathogenesis. Sci Rep. 11(1):1–16. doi: 10.1038/s41598-021-91812-0.
- Peres-Buzalaf C, de Paula L, Frantz FG, Soares EM, Medeiros AI, Peters-Golden M, Silva CL, Faccioli LH. 2011. Control of experimental pulmonary tuberculosis depends more on immunostimulatory leukotrienes than on the absence of immunosuppressive prostaglandins. Prostaglandins Leukot Essent Fatty Acids. 85(2):75–81. doi: 10.1016/j.plefa.2011.04.024.
- Pereverzeva L, van Linge CCA, Schuurman AR, Klarenbeek AM, Ramirez Moral I, Otto NA, Peters-Sengers H, Butler JM, Schomakers BV, van Weeghel M, et al. 2022. Human alveolar macrophages do not rely on glucose metabolism upon activation by lipopolysaccharide. Biochim Biophys Acta Mol Basis Dis. 1868(10):166488. doi: 10.1016/j.bbadis.2022.166488.
- Peyron P, Vaubourgeix J, Poquet Y, Levillain F, Botanch C, Bardou F, Daffé M, Emile J-F, Marchou B, Cardona P-J, et al. 2008. Foamy macrophages from tuberculous patients’ granulomas constitute a nutrient-rich reservoir for M. tuberculosis persistence. PLoS Pathog. 4(11):e1000204. doi: 10.1371/journal.ppat.1000204.
- Pisu D, Huang L, Grenier JK, Russell DG. 2020. Dual RNA-seq of Mtb-infected macrophages in vivo reveals ontologically distinct host-pathogen interactions. Cell Rep. 30(2):335–350.e4. doi: 10.1016/j.celrep.2019.12.033.
- Pohane AA, Carr CR, Garhyan J, Swarts BM, Siegrist MS. 2021. Trehalose recycling promotes energy-efficient biosynthesis of the mycobacterial cell envelope. mBio. 12(1):1–19. doi: 10.1128/mBio.02801-20.
- Pradipta IS, Forsman LD, Bruchfeld J, Hak E, Alffenaar JW. 2018. Risk factors of multidrug-resistant tuberculosis: a global systematic review and meta-analysis. Journal of Infection. 77(6):469–478. doi: 10.1016/j.jinf.2018.10.004.
- Puckett S, Trujillo C, Eoh H, Marrero J, Spencer J, Jackson M, Schnappinger D, Rhee K, Ehrt S. 2014. Inactivation of fructose-1,6-bisphosphate aldolase prevents optimal co-catabolism of glycolytic and gluconeogenic carbon substrates in mycobacterium tuberculosis. PLoS Pathog. 10(5):e1004144. doi: 10.1371/journal.ppat.1004144.
- Puech V, Guilhot C, Perez E, Tropis M, Armitige LY, Gicquel B, Daffé M. 2002. Evidence for a partial redundancy of the fibronectin-binding proteins for the transfer of mycoloyl residues onto the cell wall arabinogalactan termini of Mycobacterium tuberculosis. Mol Microbiol. 44(4):1109–1122. doi: 10.1046/j.1365-2958.2002.02953.x.
- Quadri LEN. 2014. Biosynthesis of mycobacterial lipids by polyketide synthases and beyond. Crit Rev Biochem Mol Biol. 49(3):179–211. doi: 10.3109/10409238.2014.896859.
- Quemard A, Sacchettini JC, Dessen A, Vilcheze C, Bittman R, Jacobs WR, Jr., Blanchard JS. 1995. Enzymatic characterization of the target for isoniazid in mycobacterium tuberculosist. Biochemistry. 34(26):8235–8241. doi: 10.1021/bi00026a004.
- Quesniaux V, Fremond C, Jacobs M, Parida S, Nicolle D, Yeremeev V, Bihl F, Erard F, Botha T, Drennan M, et al. 2004. Toll-like receptor pathways in the immune responses to mycobacteria. Microbes Infect. 6(10):946–959. doi: 10.1016/j.micinf.2004.04.016.
- Quigley J, Hughitt VK, Velikovsky CA, Mariuzza RA, El-Sayed NM, Briken V. 2017. The cell wall lipid PDIM contributes to phagosomal escape and host cell exit of Mycobacterium tuberculosis. mBio. 8(2):e00148-17. doi: 10.1128/mBio.00148-17.
- Rajaram MVS, Brooks MN, Morris JD, Torrelles JB, Azad AK, Schlesinger LS. 2011. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor γ linking mannose receptor recognition to regulation of immune responses. J Immunol. 185(2):929–942. doi: 10.4049/jimmunol.1000866.
- Roca FJ, Whitworth LJ, Redmond S, Jones AA, Ramakrishnan L. 2019. TNF induces pathogenic programmed macrophage necrosis in tuberculosis through a mitochondrial-lysosomal-endoplasmic reticulum circuit. Cell. 178(6):1344–1361.e11. doi: 10.1016/j.cell.2019.08.004.
- Roos AK, Burgos E, Ericsson DJ, Salmon L, Mowbray SL. 2005. Competitive inhibitors of Mycobacterium tuberculosis ribose-5-phosphate isomerase B reveal new information about the reaction mechanism. J Biol Chem. 280(8):6416–6422. doi: 10.1074/jbc.M412018200.
- Ruetz M, Campanello GC, Purchal M, Shen H, McDevitt L, Gouda H, Wakabayashi S, Zhu J, Rubin EJ, Warncke K, et al. 2019. Itaconyl-CoA forms a stable biradical in methylmalonyl-CoA mutase and derails its activity and repair. Science (1979). 366(6465):589–593. doi: 10.1126/science.aay0934.
- Russell DG, Cardona PJ, Kim MJ, Allain S, Altare F. 2009. Foamy macrophages and the progression of the human tuberculosis granuloma. Nat Immunol. 10(9):943–948. doi: 10.1038/ni.1781.
- Sachdeva K, Goel M, Sudhakar M, Mehta M, Raju R, Raman K, Singh A, Sundaramurthy V. 2020. Mycobacterium tuberculosis (Mtb) lipid mediated lysosomal rewiring in infected macrophages modulates intracellular Mtb trafficking and survival. J Biol Chem. 295(27):9192–9210. doi: 10.1074/jbc.RA120.012809.
- Samuels AN, Wang ER, Harrison GA, Valenta JC, Stallings CL. 2022. Understanding the contribution of metabolism to Mycobacterium tuberculosis drug tolerance. Front Cell Infect Microbiol. 12:958555. doi: 10.3389/fcimb.2022.958555.
- Santucci P, Diomandé S, Poncin I, Alibaud L, Viljoen A, Kremer L, de Chastellier C, Canaan S. 2018. Delineating the physiological roles of the PE and catalytic domains of LipY in lipid consumption in mycobacterium-infected foamy macrophages. Infect Immun. 86(9):e00394-18. doi: 10.1128/IAI.00394-18.
- Santucci P, Smichi N, Diomandé S, Poncin I, Point V, Gaussier H, Cavalier J, Kremer L, Canaan S. 2019. Dissecting the membrane lipid binding properties and lipase activity of Mycobacterium tuberculosis LipY domains. FEBS Journal. 286(16):3164–3181. doi: 10.1111/febs.14864.
- Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci USA. 100(22):12989–12994. doi: 10.1073/pnas.2134250100.
- Savvi S, Warner DF, Kana BD, McKinney JD, Mizrahi V, Dawes SS. 2008. Functional characterization of a vitamin B12-dependent methylmalonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J Bacteriol. 190(11):3886–3895. doi: 10.1128/JB.01767-07.
- Schnappinger D. 2003. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: insights into the phagosomal environment. J Exp Med. 198:693–704.
- Seidel M, Alderwick LJ, Birch HL, Sahm H, Eggeling L, Besra GS. 2007. Identification of a novel arabinofuranosyltransferase AftB involved in a terminal step of cell wall arabinan biosynthesis in Corynebacterianeae, such as Corynebacterium glutamicum and Mycobacterium tuberculosis. J Biol Chem. 282(20):14729–14740. doi: 10.1074/jbc.M700271200.
- Shao M, McNeil M, Cook GM, Lu X. 2020. MmpL3 inhibitors as antituberculosis drugs. Eur J Med Chem. 200:112390.
- Sheppe AEF, Edelmann MJ. 2021. Roles of eicosanoids in regulating inflammation and neutrophil migration as an innate host response to bacterial infections. Infect Immun. 89(8):e0009521. doi: 10.1128/IAI.00095-21.
- Shi W, Chen J, Zhang S, Zhang W, Zhang Y. 2018. Identification of novel mutations in LprG (rv1411c), rv0521, rv3630, rv0010c, ppsC, and cyp128 associated with pyrazinoic acid/pyrazinamide resistance in Mycobacterium tuberculosis. Antimicrob Agent Chemotherap. 62(7):e00430–18.
- Shimono N, Morici L, Casali N, Cantrell S, Sidders B, Ehrt S, Riley LW. 2003. Hypervirulent mutant of Mycobacterium tuberculosis resulting from disruption of the mce1 operon. Proc Natl Acad Sci USA. 100(26):15918–15923. doi: 10.1073/pnas.2433882100.
- Shin J-H, Yang J-Y, Jeon B-Y, Yoon YJ, Cho S-N, Kang Y-H, Ryu DH, Hwang G-S. 2011. 1H NMR-based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J Proteome Res. 10(5):2238–2247. doi: 10.1021/pr101054m.
- Shukla S, Richardson ET, Athman JJ, Shi L, Wearsch PA, McDonald D, Banaei N, Boom WH, Jackson M, Harding CV, et al. 2014. Mycobacterium tuberculosis Lipoprotein LprG Binds lipoarabinomannan and determines its cell envelope localization to control phagolysosomal fusion. PLoS Pathog. 10(10):e1004471. doi: 10.1371/journal.ppat.1004471.
- Simeone R, Bobard A, Lippmann J, Bitter W, Majlessi L, Brosch R, Enninga J. 2012. Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PLoS Pathog. 8(2):e1002507. doi: 10.1371/journal.ppat.1002507.
- Siméone R, Constant P, Guilhot C, Daffé M, Chalut C. 2007. Identification of the missing trans-acting enoyl reductase required for phthiocerol dimycocerosate and phenolglycolipid biosynthesis in Mycobacterium tuberculosis. J Bacteriol. 189(13):4597–4602. doi: 10.1128/JB.00169-07.
- Singh M, Kumar S, Singh B, Jain P, Kumari A, Pahuja I, Chaturvedi S, Prasad DVR, Dwivedi VP, Das G, et al. 2022. The 1, 2-ethylenediamine SQ109 protects against tuberculosis by promoting M1 macrophage polarization through the p38 MAPK pathway. Commun Biol. 5(1). doi: 10.1038/s42003-022-03693-2.
- Siregar TAP, Prombutara P, Kanjanasirirat P, Kunkaew N, Tubsuwan A, Boonmee A, Palaga T, Khumpanied T, Borwornpinyo S, Chaiprasert A, Utaisincharoen P, Ponpuak M. 2022. The autophagy-resistant Mycobacterium tuberculosis Beijing strain upregulates KatG to evade starvation-induced autophagic restriction. Pathog Dis. 80(1):ftac004.
- Škovierová H, Larrouy-Maumus G, Zhang J, Kaur D, Barilone N, Korduláková J, Gilleron M, Guadagnini S, Belanová M, Prevost M-C, et al. 2009. AftD, a novel essential arabinofuranosyltransferase from mycobacteria. Glycobiology. 19(11):1235–1247. doi: 10.1093/glycob/cwp116.
- Sorgi CA, Soares EM, Rosada RS, Bitencourt CS, Zoccal KF, Pereira PAT, Fontanari C, Brandão I, Masson AP, Ramos SG, et al. 2020. Eicosanoid pathway on host resistance and inflammation during Mycobacterium tuberculosis infection is comprised by LTB4 reduction but not PGE2 increment. Biochim Biophys Acta Mol Basis Dis. 1866(3):165574. doi: 10.1016/j.bbadis.2019.165574.
- Srinivasan L, Ahlbrand S, Briken V. 2014. Interaction of mycobacterium tuberculosis with host cell death pathways. Cold Spring Harb Perspect Med. 4:1–15.
- Su CC, Klenotic PA, Bolla JR, Purdy GE, Robinson CV, Yu EW. 2019. MmpL3 is a lipid transporter that binds trehalose monomycolate and phosphatidylethanolamine. Proc Natl Acad Sci U S A. 166:11241–11246.
- Sulzenbacher G, Canaan S, Bordat Y, Neyrolles O, Stadthagen G, Roig-Zamboni V, Rauzier J, Maurin D, Laval F, Daffé M, et al. 2006. LppX is a lipoprotein required for the translocation of phthiocerol dimycocerosates to the surface of Mycobacterium tuberculosis. Embo J. 25(7):1436–1444. doi: 10.1038/sj.emboj.7601048.
- Székely R, Rengifo-Gonzalez M, Singh V, Riabova O, Benjak A, Piton J, Cimino M, Kornobis E, Mizrahi V, Johnsson K, et al. 2020. 6,11-Dioxobenzo [f]pyrido[1,2- a]indoles Kill Mycobacterium tuberculosis by Targeting Iron-Sulfur Protein Rv0338c (IspQ), A Putative Redox Sensor. ACS Infect Dis. 6(11):3015–3025. doi: 10.1021/acsinfecdis.0c00531.
- Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. 2013. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 496(7444):238–242. doi: 10.1038/nature11986.
- Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, Ko DC, Zou Y, Bang ND, Chau TTH, et al. 2012. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 148(3):434–446. doi: 10.1016/j.cell.2011.12.023.
- Tobin DM, Vary JC, Jr., Ray JP, Walsh GS, Dunstan SJ, Bang ND, Hagge DA, Khadge S, King M-C, Hawn TR, et al. 2010. The lta4h locus modulates susceptibility to mycobacterial infection in zebrafish and humans. Cell. 140(5):717–730. doi: 10.1016/j.cell.2010.02.013.
- Trivedi OA, Arora P, Sridharan V, Tickoo R, Mohanty D, Gokhale RS. 2004. Enzymic activation and transfer of fatty acids as acyl-adenylates in mycobacteria. Nature. 428(6981):441–445. doi: 10.1038/nature02384.
- Trivedi OA, Arora P, Vats A, Ansari MZ, Tickoo R, Sridharan V, Mohanty D, Gokhale RS. 2005. Dissecting the mechanism and assembly of a complex virulence mycobacterial lipid. Mol Cell. 17(5):631–643. doi: 10.1016/j.molcel.2005.02.009.
- Turner J, Torrelles JB. 2018. Mannose-capped lipoarabinomannan in Mycobacterium tuberculosis pathogenesis. Pathog Dis. 76(4):fty026.
- van der Wel N, Hava D, Houben D, Fluitsma D, van Zon M, Pierson J, Brenner M, Peters PJ. 2007. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. 129(7):1287–1298. doi: 10.1016/j.cell.2007.05.059.
- VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB, Memmott C, Crowe AM, Eltis LD, Perola E, Deininger DD, et al. 2015. Novel inhibitors of cholesterol degradation in mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog. 11(2):e1004679. doi: 10.1371/journal.ppat.1004679.
- Varela C, Rittmann D, Singh A, Krumbach K, Bhatt K, Eggeling L, Besra GS, Bhatt A. 2012. MmpL genes are associated with mycolic acid metabolism in mycobacteria and corynebacteria. Chem Biol. 19(4):498–506. doi: 10.1016/j.chembiol.2012.03.006.
- Wallings R, Connor-Robson N, Wade-Martins R. 2019. LRRK2 interacts with the vacuolar-type H+-ATPase pump a1 subunit to regulate lysosomal function. Hum Mol Genet. 28(16):2696–2710. doi: 10.1093/hmg/ddz088.
- Wallis RS, Ginindza S, Beattie T, Arjun N, Likoti M, Edward VA, Rassool M, Ahmed K, Fielding K, Ahidjo BA, et al. 2021. Adjunctive host-directed therapies for pulmonary tuberculosis: a prospective, open-label, phase 2, randomised controlled trial. Lancet Respir Med. 9(8):897–908. doi: 10.1016/S2213-2600(20)30448-3.
- Wang Z, Arat S, Magid-Slav M, Brown JR. 2018. Meta-analysis of human gene expression in response to Mycobacterium tuberculosis infection reveals potential therapeutic targets. BMC Syst Biol. 12(1):3. doi: 10.1186/s12918-017-0524-z.
- Weichhart T, Hengstschläger M, Linke M. 2015. Regulation of innate immune cell function by mTOR. Nat Rev Immunol. 15(10):599–614. doi: 10.1038/nri3901.
- Weindel CG, Bell SL, Vail KJ, West KO, Patrick KL, Watson RO. 2020. LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. Elife. 9:1–31. doi: 10.7554/eLife.51071.
- Wescott HH, Zuniga ES, Bajpai A, Trujillo C, Ehrt S, Schnappinger D, Roberts DM, Parish T. 2018. Identification of enolase as the target of 2-aminothiazoles in Mycobacterium Tuberculosis. Front Microbiol. 9:2542. doi: 10.3389/fmicb.2018.02542.
- WHO. 2022. Global tuberculosis report 2022. http://apps.who.int/bookorders.
- WHO. Tuberculosis fact sheet. 2023. https://www.who.int/news-room/fact-sheets/detail/tuberculosis.
- Wu Y. t, Xu WT, Zheng L, Wang S, Wei J, Liu MY, Zhou HP, Li QF, Shi X, Lv X. 2023. 4-octyl itaconate ameliorates alveolar macrophage pyroptosis against ARDS via rescuing mitochondrial dysfunction and suppressing the cGAS/STING pathway. Int Immunopharmacol. 118:110104.
- Yam KC, D’Angelo I, Kalscheuer R, Zhu H, Wang J-X, Snieckus V, Ly LH, Converse PJ, Jacobs WR, Strynadka N, et al. 2009. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 5(3):e1000344. doi: 10.1371/journal.ppat.1000344.
- Yang J, Chen J, Yue J, Liu L, Han M, Wang H. 2014. Relationship between human LTA4H polymorphisms and extra-pulmonary tuberculosis in an ethnic Han Chinese population in Eastern China. Tuberculosis. 94(6):657–663. doi: 10.1016/j.tube.2014.08.014.
- Yang X, Gao J, Smith I, Dubnau E, Sampson NS. 2011. Cholesterol is not an essential source of nutrition for Mycobacterium tuberculosis during infection. J Bacteriol. 193(6):1473–1476. doi: 10.1128/JB.01210-10.
- Zhang L, Zhao Y, Gao Y, Wu L, Gao R, Zhang Q, Wang Y, Wu C, Wu F, Gurcha SS, et al. 2020. Structures of cell wall arabinosyltransferases with the anti-tuberculosis drug ethambutol. Science (1979). 368(6496):1211–1219. doi: 10.1126/science.aba9102.
- Zimhony O, Vilchèze C, Jacobs WR.Jr. 2004. Characterization of Mycobacterium smegmatis expressing the Mycobacterium tuberculosis fatty acid synthase I (fas1) gene. J Bacteriol. 186(13):4051–4055. doi: 10.1128/JB.186.13.4051-4055.2004.