
Abstract
The effects of long-term use of nicotine per se on cancer risk, in the absence of tobacco extract or smoke, are not clearly understood. This review evaluates the strength of published scientific evidence, in both epidemiological and animal studies, for the potential carcinogenic effects of nicotine per se; that is to act as a complete carcinogen or as a modulator of carcinogenesis. For human studies, there appears to be inadequate evidence for an association between nicotine exposure and the presence of or lack of a carcinogenic effect due to the limited information available. In animal studies, limited evidence suggests an association between long-term nicotine exposure and a lack of a complete carcinogenic effect. Conclusive studies using current bioassay guidelines, however, are missing. In studies using chemical/physical carcinogens or transgenic models, there appears to be inadequate evidence for an association between nicotine exposure and the presence of or lack of a modulating (stimulating) effect on carcinogenesis. This is primarily due to the large number of conflicting studies. In contrast, a majority of studies provides sufficient evidence for an association between nicotine exposure and enhanced carcinogenesis of cancer cells inoculated in mice. This modulating effect was especially prominent in immunocompromized mice. Overall, taking the human and animal studies into consideration, there appears to be inadequate evidence to conclude that nicotine per se does or does not cause or modulate carcinogenesis in humans. This conclusion is in agreement with the recent US Surgeon General’s 2014 report on the health consequences of nicotine exposure.
Introduction
Nicotine delivery systems (Shahab et al. Citation2013; Benowitz Citation2014) continue to evolve for nicotine replacement therapy (NRT) products and for electronic nicotine delivery systems (ENDS, e-cigarettes). As a result, millions of people are exposed to nicotine per se on a daily basis resulting in blood nicotine levels of approximately 5–40 ng/ml (). These products are often touted as “clean” nicotine delivery products as they contain pharmaceutical-grade nicotine as the only added active ingredient and do not contain tobacco (Schneider et al. Citation2001; Benowitz Citation2014; Flora et al. Citation2016). The recommended duration of use for approved NRT products is 8–12 weeks depending on the product type (US Food and Drug Administration Citation2013). The FDA, however, recently proposed the possibility of a 6-month extension of NRT use with healthcare provider consultation (US Food and Drug Administration Citation2013; Fucito et al. Citation2014). It seems that uncertainty regarding the potential adverse health effects (including cancer risk) of long-term use of nicotine per se may be, in part, responsible for the modest increase in the proposed duration of NRT use (Shields Citation2011; Grando Citation2014).
At present, nicotine is not considered a human carcinogen as noted in numerous statements from authoritative bodies (). For example, the latest report of the US Surgeon General concluded that “the evidence is inadequate to infer the presence or absence of a causal relationship between exposure to nicotine and risk for cancer” (US Department of Health and Human Services Citation2014, p. 8). In addition, the Tobacco Advisory Group of the UK Royal College of Physicians stated that there is no direct evidence that NRT is carcinogenic (UK Royal College of Physicians Citation2007). NRT product warning label statements (health-related) provide authoritative bodies with the opportunity to communicate their concern about the potential harmful effects of these products. For NRT products, the labels do not warn about potential cancer risks (US Food and Drug Administration Citation2015). Numerous review/opinion publications also suggest that the scientific evidence (animal and human) does not support a carcinogenic effect for long-term nicotine exposure (Benowitz Citation2011; Cardinale et al. Citation2012; Hecht Citation2012a, Citation2012b International Agency for Research on Cancer Citation2012, p. 134; Warren & Singh Citation2013; Schaal & Chellappan Citation2014; Sanner & Grimsrud Citation2015). A comprehensive evaluation of the published scientific literature on this topic (animal and human studies), however, is missing.
Table 1. Plasma and urinary concentrations of nicotine and cotinine in users of nicotine delivery systems (with comparison to conventional cigarettes).
Table 2. Statements by authoritative bodies on the potential carcinogenic effect of nicotine per se.
Carcinogenesis is a multi-staged process which operationally involves three stages: initiation, promotion and progression (Klaunig Citation2013). A complete carcinogen is a chemical that induces tumors, by itself, usually with initiating, promoting, and progressing properties. Genotoxicity is a required property of initiators. The available data on a genotoxic potential of nicotine are conflicting and have not been critically reviewed. Genotoxicity was not observed for nicotine or its four major metabolites at concentrations of up to 1 mg/ml in the Salmonella reverse mutation assay and in a sister chromatid exchange assay in Chinese hamster ovary cells (Doolittle et al. Citation1995). However, in recent in vitro genotoxicity studies examining strand-breaking activity assessed by the Comet assay, chromosome aberration or micronucleus formation, nicotine was found to be active in a concentration range between 160 ng/ml and 650 μg/ml (Argentin & Cicchetti Citation2004; Ginzkey et al. Citation2012; Citation2013; Bavarva et al. Citation2014; Ginzkey et al. Citation2014a, Citation2014b). This range is beyond the systemic nicotine levels achieved by using NRT products (), but at local sites of entry, such as at respiratory tract or oral epithelia, nicotine concentrations may indeed be higher than systemic concentrations (Jarvis et al. Citation1984). Genotoxic effects at systemically relevant nicotine concentrations (16 ng/ml) were reported in a few studies, such as in a cytokinesis-blocked micronucleus assay (Kleinsasser et al. Citation2005) and in a chromosomal aberration assay (Demirhan et al. Citation2011). Overall, definitive studies to determine the genotoxic potential of nicotine in users of nicotine delivery systems are missing.
Concern has been raised by authoritative bodies that nicotine might act as a promoter and/or progressor of an initiated carcinogenic process (). From a mechanistic standpoint, there is considerable evidence that nicotine exposure can affect many of the cellular processes that are considered important for the promotion or progression of the carcinogenic process. Numerous reviews have been published summarizing these mechanistic findings (Improgo et al. Citation2011; Cardinale et al. Citation2012; Jensen et al. Citation2012; Lee & Cooke Citation2012; Russo et al. Citation2012; Schuller Citation2012; Chu et al. Citation2013; Warren & Singh Citation2013; Grando Citation2014; Niu & Lu Citation2014; Schaal & Chellappan Citation2014; Schuller Citation2014). For example, nicotine has been reported to stimulate cell proliferation, inhibit apoptosis, induce cell migration and invasion, induce angiogenesis and inhibit immune functions. Such effects were often observed in vitro at systemically and/or locally relevant nicotine concentrations. In particular, the role of nAChRs in triggering intracellular signaling pathways that influence the carcinogenic process have been emphasized (Grando Citation2014).
Nicotine per se is a unique active ingredient for a consumer product in that the majority of nicotine’s effects are mediated by binding and activating nicotinic acetylcholine receptors (nAChRs) in a wide variety of neuronal (central and peripheral nervous system) and non-neuronal tissue. Consequently, nicotine exposure affects numerous systems, including neurologic, neuromuscular, cardiovascular, respiratory, immunological and gastrointestinal. The presence of different types of nAChRs, receptor upregulation and receptor desensitization influences these complex physiological effects. Numerous studies in experimental animals demonstrate that nicotine exposure results in a dramatic increase in both nAChR numbers and receptor desensitization in the brain resulting in tolerance to the central effects of nicotine (Marks et al. Citation1985; Renda & Nashmi Citation2014). In contrast, little is known about the response of peripheral nAChRs in regard to receptor upregulation and desensitization following nicotine exposure (Lam et al. Citation2016). Similarly, many types of cancer cells express a wide variety of nAChRs (Improgo et al. Citation2013), but few studies have characterized the effect of nicotine on receptor numbers and desensitization (Brown et al. Citation2013).
Based on the mechanistic studies, a case for biological plausibility has been proposed for a potential role of nicotine in carcinogenesis. Therefore, it seems appropriate for the present review to critically evaluate the strength of published scientific evidence, in both human and animal studies, for potential carcinogenic effects of nicotine per se. The potential of nicotine per se to act as a complete carcinogen or as a modulator of an initiated carcinogenic process are assessed in this review. Toxicokinetic considerations relevant for this evaluation are also briefly summarized. Mechanistic data on the potential carcinogenic effects of nicotine per se, however, will not be evaluated, as numerous mechanistic studies have recently been reviewed (see above). Finally, the words nicotine per se and nicotine are used synonymously in this review.
Methods
For the present review, evaluations of relevant published literature were carried out according to processes described in the sections below, which were adapted or modified from a wide variety of published frameworks (Hill Citation1965; International Agency for Research on Cancer Citation2007; Organisation for Economic Co-operation and Development Citation2009; Rhomberg et al. Citation2011; Goodman et al. Citation2013; Rhomberg et al. Citation2013; Prueitt et al. Citation2014; Willhite et al. Citation2014). These frameworks, including our own, have similar processes such as defining the study question, gathering relevant studies using inclusion and exclusion criteria, evaluating studies for quality, consistency and relevance, integrating evidence on related topics to draw conclusions and using these conclusions to determine a strength of evidence classification (Rhomberg et al. Citation2013).
Study question
The present review was conducted to answer the question: What is the potential carcinogenic effect of nicotine per se, at levels found in users of nicotine delivery systems? In this review, nicotine delivery systems refer to products that contain nicotine as the only added active ingredient, that contain pharmaceutical grade nicotine and do not contain tobacco. Due to the large number of in vitro and in vivo studies on this topic (), we chose to limit our answer to this question using published human and animal studies. In vitro studies and mechanistic data will not be critically evaluated in the present review. Thus, the objective of this review is to critically evaluate the strength of published scientific evidence, in both human and animal studies, for the potential carcinogenic effects of nicotine per se. In animal studies, the potential of nicotine to act as a complete carcinogen or as a modulator (stimulator) of carcinogenesis is evaluated. The goal for study evaluations is not to provide a yes or no answer to the question but to generate knowledge, communicate uncertainty about conclusions and enable an informed discussion about knowledge gaps and possible actions to be taken.
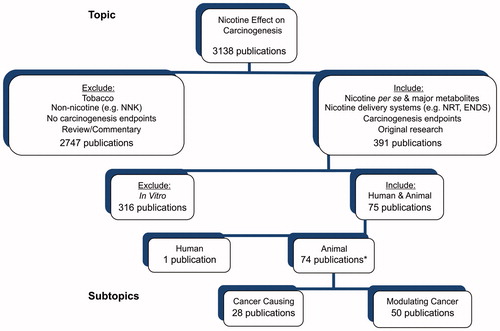
Figure 1. Overview of findings from literature searches and criteria used for the inclusion and exclusion of publications for critical evaluation. *The total number is only 74 because several publications include both complete and modulating cancer studies.
Literature search
An updated search of the relevant scientific literature was performed the final week of October 2015 in the Medline and Embase databases primarily relying on their hierarchical controlled-vocabulary thesauri. The concept of cancer (including carcinogenesis) was covered by selecting all records indexed to the most general cancer term, “Neoplasms” (“Neoplasm” in Embase), or to any of its narrower terms; 683 total Medline terms and 1002 in Embase. The cancer set was combined by Boolean AND with items indexed to either “Nicotine” or “Cotinine.” In Embase, allowance also was made for items indexed to either “Nicotine N Oxide” or “Nicotine N′ Oxide.” No such inclusion was necessary in Medline, since that database maps the oxides to “Nicotine,” modified by an “analogs & derivatives” sub-heading.
Since Medline makes titles and abstracts available before being fully indexed, a strategy such as the above must be supplemented by a “free-text” approach to pick up the mostly newer unindexed records. Thus, the “In-Process,” “Epub Ahead of Print” and “PubMed-Not-Medline” file segments were queried for either of nicotine or cotinine and any of cancer?, carcino?, tumor?, tumor?, cocarcinogen?, neoplas?, oncogenic or oncogenesis (where? represents zero or more characters).
Using the above search methodology, 3138 database records were identified. One of the authors (MWF) read the title and abstract (as available) for each of these records and applied the inclusion and exclusion criteria (as detailed in the Section “Inclusion/exclusion criteria”) to identify relevant studies for evaluation. This search strategy in combination with using secondary sources from publications and reviews (as described in the next paragraph) resulted in the identification of one publication for human studies and 75 publications for animal studies to be critically assessed and evaluated ().
In addition to the process described above, one of the authors (H. J. H.) identified recent reviews that addressed the potential of nicotine to act as a carcinogen or as a modulator of carcinogenesis (Benowitz Citation2011; Improgo et al. Citation2011; Lee et al. Citation2011; Singh et al. Citation2011; Cardinale et al. Citation2012; Jensen et al. Citation2012; Schuller Citation2012; Hecht Citation2012a; Warren & Singh Citation2013; Grando Citation2014; Schaal & Chellappan Citation2014; Schuller Citation2014). These reviews as well as original studies were found with PubMed searches performed in January 2015, using keywords “nicotine AND cancer,” and “nicotine AND carcinogen*”. Relatively old but relevant in vivo studies on nicotine and its metabolites were also discovered by checking earlier reviews (Larson et al. Citation1961; Schievelbein Citation1962; Levy & Martin Citation1989). All these reviews and original research publications were used as secondary sources to identify relevant published studies.
Inclusion/exclusion criteria
The articles identified as potentially eligible for inclusion in the present review were examined and confirmed as in scope by one of the authors (H. J. H.) based on inclusion/exclusion criteria described in detail below and illustrated in . In many cases, a single publication contained data from several relevant studies. For example, a single publication could contain results from using various strains of animals, various routes of nicotine administration or various kinds of cancer cells in xenograft experiments. The relevant studies present in a single publication are referred to as sub-studies. As a result, the 74 publications identified using animals contained 112 relevant animal studies or sub-studies (33 cancer causing and 79 modulating cancer). Thus, in the present review, 113 (sub-)studies (including one human study) were critically assessed for relevance and quality as well as evaluated for strength of evidence in categories such as human studies and animal studies including complete carcinogenesis and modulating carcinogenesis.
Studies included for evaluation are those in which nicotine, free base or salts, or the major metabolites of nicotine, cotinine or nicotine-N'-oxide (NNO), are administered. The half-life of nicotine is relatively short, especially in rodents, while the half-life of some of nicotine’s metabolites, such as cotinine and NNO, are longer (Matta et al. Citation2007). Therefore, it seems plausible that, findings that appear to be related to nicotine may instead result from one or more of its metabolites. Accordingly, studies that investigate the potential carcinogenicity of cotinine or NNO exposure were identified and are included in the current review.
Studies in which nicotine delivery systems are used for exposure are included. As previously mentioned, nicotine delivery systems refer to products that contain nicotine as the only added active ingredient, that contain pharmaceutical grade nicotine and do not contain tobacco. Examples would include the NRT products listed in as well as ENDS such as e-cigarettes. Accordingly, studies investigating the potential carcinogenicity of NRT products in humans and animals were identified and are included in the current review. In contrast, no relevant studies using ENDS were identified or evaluated.
Studies that use any product that contains tobacco for nicotine exposure are excluded. Epidemiological studies on Swedish snus (a smokeless tobacco) are often used as an indirect measure of the potential carcinogenic effect of long-term nicotine use in humans (Benowitz Citation2011). However, for this review, exposure to nicotine per se and snus are not considered equivalent. They differ in that a snus user is exposed not only to nicotine extracted from this tobacco product but also exposed to compounds that may mask a potential carcinogenic effect of nicotine (Hecht et al. Citation1986; Hoffmann et al. Citation1987; Prokopczyk et al. Citation1987).
Pesticide products that contain nicotine in combination with other tobacco alkaloids or contain nicotine of unknown purity are excluded. Studies that investigate the use of non-nicotine compounds or products are also excluded. Examples would include tobacco-specific nitrosamines such as 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) or N-nitrosonornicotine (NNN), and tobacco alkaloids other than nicotine.
Studies that describe the use of carcinogenesis endpoints or the possibility of detecting such are included. Examples of such endpoints include cancer diagnosis (human studies), gross pathology assessment, tissue histopathology analysis, tumor-related characteristics such as tumor size or volume, vascularization or metastasis. Studies identified in the literature search that do not include such endpoints are excluded from evaluation. Common non-carcinogenesis endpoints observed include those found in studies investigating metabolism, dependence and receptor activity (nAChRs).
Studies that are described in publications that are reviews, commentaries or opinions are not evaluated in this review. Only publications describing original research studies in some detail are included in this review.
Studies that are not conducted in humans or animals (e.g., in vitro) are excluded from evaluation in this review. Seventy-four relevant animal publications (whose studies met the inclusion criteria above) are assigned to one of the two subtopic categories, cancer causing or modulating cancer (). Cancer causing studies are those that investigate whether nicotine acts as a complete carcinogen. In these studies, carcinogenesis is followed in animals during long-term exposure to nicotine alone. The second subtopic category of animal studies comprises those evaluating the modulating (stimulating) effect of nicotine on carcinogenesis induced by other sources. Relevant modulating carcinogenesis studies are placed in one of the two subcategories depending on the source of carcinogenesis. These are a chemical, physical or transgenic source or the implantation of cancer cells (xenograft source).
Critical assessment of relevance and quality of studies
The in vivo studies reviewed differ in various aspects of study design, conduct and reporting. Thus, it is challenging to compare studies as well as to determine the strength of evidence for each of the studies that have passed the inclusion criteria in order to evaluate the potential carcinogenicity of nicotine. To facilitate this evaluation, certain criteria, based on current international guidelines for carcinogenicity testing (Organisation for Economic Co-operation and Development Citation2009), were used for judging relevance and quality of studies. Both criteria were combined to derive a score for the adequacy of a study for the purpose of this evaluation. Five design criteria on study relevance, including route of administration, group size, dose response, daily dose, duration of exposure as well as quality were scored separately as plus (+) or minus (−) and documented in the supplementary material (Supplementary Tables 1–3). If information was lacking, the study was assigned a minus score. Alternative approaches to judge study relevance and quality have been published. For instance, the Klimisch scores (Klimisch et al. Citation1997) were not considered discriminative enough for the current evaluation. This approach relies heavily on generally accepted international guidelines and/or Good Laboratory Practice and few studies in our review were conducted approaching such guidelines.
Our adequacy evaluation criteria are listed in and briefly explained as the following: adequate routes of administration were those that corresponded to the use of nicotine delivery systems in humans, i.e., inhalation, dermal and oral (Organisation for Economic Co-operation and Development Citation2009).
Table 3. Adequacy evaluation criteria for individual animal studies on the potential carcinogenicity of nicotine.
For the first category of studies, group sizes of approximately 100 or more (or 50 per sex) were considered adequate (Organisation for Economic Co-operation and Development Citation2009). Optimally, a dose–response relationship on the basis of at least three dose groups should be targeted (Organisation for Economic Co-operation and Development Citation2009), but each attempt to have more than one dose group was honored as adequate.
Average daily doses were calculated or estimated as a common denominator. Because this calculation is imprecise in view of the rapid nicotine metabolism in most laboratory animals, the actual dosing regimen was also provided in the Supplementary material, which lists the various studies in detail (Supplementary Tables 1–3). The highest dose level in the carcinogenicity studies should be selected to elicit some evidence of toxicity (Organisation for Economic Co-operation and Development Citation2009). Thus, any sign of nicotine-related toxicity, such as body weight or survival effects, was honored as indicative of a sufficiently high dosing and considered adequate. There might have been very acute and transient toxic reactions to nicotine injection, which were also honored here, although the same average daily dose given continuously, e.g., via the drinking water, might have avoided such toxic response. Another criterion of adequacy related to dosing or exposure was the assumption that the experimental nicotine exposure relative to body weight was similar or higher than in a user of nicotine delivery systems. For NRT, it is recommended that nicotine gum users consume no more than 24 units containing 4 mg nicotine each per day, while for nicotine inhaler users, not more than 16 units containing 10 mg nicotine each per day is recommended and sublingual tablet users can use up to 24 units containing 2 mg of nicotine each (Schneider et al. Citation2001). Thus, the upper limit of exposure to nicotine from NRTs can be estimated to be approximately 1 mg/(kg × d). Doses at this level or above were also considered adequate. The assessment of nicotine doses from experimental studies is hampered by the fact that quite often it was not stated whether doses were given in terms of pure nicotine or any salt and whether a racemic mixture or the pharmacologically active S(−)-enantiomer was used. If not stated otherwise, the data are interpreted as pure (−)-nicotine, which may lead to erroneous overestimates of nicotine doses of up to six-fold.
Study durations of ≥18 months were honored as adequate according to guidelines for assessing complete carcinogenesis (lower range of acceptable study durations selected for inclusion of studies with susceptible spontaneous or transgenic strains; Organisation for Economic Co-operation and Development Citation2009). For cancer-modulating studies, study duration was not a useful adequacy criterion. In many of these studies, the initial inducer of carcinogenicity was administered in a way that led to rapid tumor development. Therefore, animals in these studies were exposed to nicotine for relatively short periods of time, if these time periods were considered sufficient to induce cancer by the initial treatment alone. Because cancer growth was apparently easily observed in most of these studies, as a consequence of the initial treatment, any duration of nicotine exposure was honored as adequate for modulating carcinogenesis studies.
A final score was given based on a subjective evaluation of study quality. This included the availability of body weight or nicotine biomonitoring data, which were thought to at least improve the comparison of studies with similar design.
In order to achieve a comprehensive overview, no study was excluded from evaluation because it failed a certain study adequacy criterion. Rather, all studies that passed the inclusion criteria were evaluated. For assessing the strength of evidence for the studies evaluated, an overall adequacy score was determined by totaling the individual plus values for each individual sub-study. This approach gave all adequacy criteria the same weight. However, for practical reasons, only studies (or parts thereof, i.e., sub-studies) with high-adequacy scoring (with an overall score >2) were discussed in detail in the main body of the review, while narratives regarding studies or sub-studies with low-adequacy scoring (with an overall score ≤2) were placed in the Supplementary material (Supplementary Table 4). Scoring results were also used to roughly divide studies into two categories or tiers in other assessments (Goodman et al. Citation2014), but ranking of studies was avoided. Here, the overall results of studies with low-adequacy score were compared with those observed in high-adequacy score studies within the same subsection, to detect potential biases when stratifying by study adequacy.
Strength of evidence evaluation and classification
The evaluation and the integration of scientific evidence as well as strength of evidence classification (for human, animal, complete carcinogen, modulating carcinogenesis studies) are a matter of facilitated professional judgment, reflecting conclusions derived from evaluating relevant studies individually and collectively. The term “facilitated” refers to professional judgment that is guided by key factors or criteria for drawing conclusions from scientific studies (Rhomberg et al. Citation2013). The use of such criteria is described below. Key factors or criteria considered in the present review for evaluating and integrating scientific evidence were adapted from numerous sources and are described in brief below (Hill Citation1965; Goodman et al. Citation2013; Rhomberg et al. Citation2013). Criteria for evaluation and integrating evidence include consideration of the reproducibility, reliability and strength of the observed carcinogenic response, the presence of a dose (exposure)-carcinogenic response relationship, the timing (temporal relationship) and specificity of the carcinogenic response following nicotine exposure, and the dose and route of administration that is relevant to human nicotine exposure (nicotine delivery systems).
The framework described above uses an approach adapted from Bradford Hill and US Environmental Protection Agency (Hill Citation1965; Rhomberg et al. Citation2013). This approach was modified taking into account that conclusions in the present review did not consider mechanism of action data (biological plausibility) and limited information is available for long-term nicotine exposure in humans (lack of coherence). Therefore, it is important to remember that the strength of evidence conclusions and classifications in this review are a generalization. That is, we recognize the inherent difficulty in applying specific animal study results to arrive at conclusions for a more general study question (Rhomberg et al. Citation2013).
A thorough discussion of the strengths and weaknesses (based on the above criteria) of the relevant studies for each topic is provided in this review. Using our adequacy scoring system, less ideal studies (low-adequacy score) were not rejected outright but were summarized briefly as a narrative for each topic area (a more detailed evaluation of each study is found in the Supplementary material). The conclusions from the low-adequacy scored studies were then compared with the more detailed narrative and conclusions from the high-adequacy scored studies, again for each topic. Finally, the reasoning for the overall strength of evidence conclusion and classification was discussed in the present review for each topic (human, animal, cancer causing and cancer modulating studies).
The strength of evidence classifications used in the present review were adapted and modified from the Evaluation of Carcinogenic Risks outlined by the International Agency for Research on Cancer (IARC) (International Agency for Research on Cancer Citation2007). A major modification to the IARC strength of evidence assessment is the separation of the IARC classification “evidence suggesting lack of carcinogenicity” into two classifications: “limited evidence suggesting a lack of effect” and “sufficient evidence of a lack of effect.” With this revised classification in place, the strength of evidence conclusions are balanced. A conclusion and classification can be neutral (inadequate evidence) or can be deemed limited or sufficient in both directions, for a carcinogenic effect or for a lack of a carcinogenic effect. Strength of evidence classifications used for evaluating human and animal studies were as follows:
Sufficient evidence: Conclusive or highly suggestive studies are available for an association between nicotine exposure and either a lack of carcinogenic effect or a carcinogenic effect.
Limited evidence: The evidence from available studies is indicative of an association between nicotine exposure and either a lack of carcinogenic effect or a carcinogenic effect. Conclusive studies are missing.
Inadequate evidence: The available studies are of insufficient quality or consistency to permit a conclusion regarding an association between exposure to nicotine and carcinogenesis. Only conflicting or incomplete evidence is available.
We believe that one strength of the classification system described above is that it provides a balanced and symmetrical distribution for strength of evidence conclusions. On both sides of a neutral conclusion (inadequate evidence), there exists the same possible classification (sufficient or limited evidence) for either a carcinogenic effect or the lack of an effect. Similarly, Rhomberg et al. (Citation2011) supported a “two-pan balance” system for determining the weight of evidence as a more satisfactory means of drawing conclusions from an array of observations. These investigators suggested that many classification systems use “a single scale showing how much evidence in accord with a conclusion can be accumulated.” We believe that the IARC (International Agency for Research on Cancer Citation2007) and equipoise (Goodman et al. Citation2013) classification systems suffer from this limitation. Both systems favor observations demonstrating a positive effect, with only one availability category for a lack of an effect (sufficient evidence). Obviously, providing sufficient evidence for a lack of an effect is very difficult to accomplish especially in light of the fact that negative findings often go unpublished. Another strength is our classification system (sufficient, limited, and inadequate evidence) provides a clear communication that is easily understood, both from an overall conclusion point of view as well as the reasoning behind the message. Other frameworks using an equipoise classification system (e.g., equipoise and above or below equipoise), for example, are not easily understood.
As previously mentioned, a limitation of our strength of evidence conclusions is the absence of mechanistic data as well as little information in humans. Thus, our classification system is not meant to determine conclusions for a causal relationship between nicotine exposure and carcinogenesis.
Evidence from human studies
Epidemiological and clinical studies on the cancer risk associated with using nicotine delivery systems were identified and reviewed. One NRT study was identified as relevant and was critically evaluated. No other cancer-related studies with users of other nicotine delivery products (such as ENDS) were found.
Epidemiological studies with nicotine replacement therapy (NRT) use
The Lung Health Study investigated the cancer risk from using NRT products. This study prospectively investigated surveillance data on 3320 intervention participants who were enrolled in this study for 5 years and then followed up for 7.5 years (Murray et al. Citation2009). Nicotine gum use and smoking were determined by self-reporting, although the nicotine exposure from gum use may have been quite accurate as it was supplied for free to the participants of the study for the full 5 years. Most participants used either NRT or cigarettes, rather than using both concurrently. Using Cox proportional hazard regression analysis, the NRT alone was not a significant predictor of lung cancer [hazard ratio (HR) 1.02, 95% confidence intervals 0.95–1.09], while continued smoking was predictive [HR 1.08 (1.01–1.16)]. Neither NRT use nor continued smoking was significant predictors for all cancers and for gastrointestinal cancers in particular (including oral cancer). Survival from any diagnosis of cancer was the same between users of NRT and non-users. Most importantly, survival without any diagnosis of lung cancer was significantly higher for those participants below the median cigarette exposure during the study compared to those above the median level (determined in pack-years). There are a number of serious limitations regarding the interpretation of this study that were identified by the authors. First, the 5-year study duration and the follow-up period of 7.5 years are not considered long enough for lung cancer to develop. In addition, the total number of lung cancer cases in the study (morbidity and mortality) was only 75. Finally, the daily nicotine exposure in the NRT user group was approximately 2 mg (average of 2 gums per day with 2 mg nicotine per gum, assuming 50% extraction). Considering an appreciable first-pass effect upon oral nicotine exposure (see Section “Comparative toxicokinetics”), the estimated nicotine exposure in this study is approximately one order of magnitude below that observed for users of nicotine delivery products ().
In summary, this study provided no evidence for an effect of NRT use on cancers of the lung, the gastrointestinal tract or overall. This one study provides inadequate evidence for an association between nicotine exposure and the presence of or lack of a modulating effect on carcinogenesis following smoking cessation.
Conclusion on human studies
There is only one epidemiological study on the long-term use of NRT after smoking cessation, and this study provided no evidence for an effect of NRT use on cancers of the lung, gastrointestinal tract, or overall. This study, however, was relatively short given the reported 20–30 year latency period for lung cancer. Longer-term prospective epidemiological studies are required to support the hypothesis that nicotine does not cause cancer by itself or stimulate carcinogenesis. This would include surveillance studies after smokers switched to NRTs or ENDS as well as studies on users of these products without prior use of conventional tobacco products. Overall, for human studies (NRT use), there appears to be inadequate evidence for an association between nicotine exposure and the presence of or lack of a carcinogenic effect due to a limited number of studies.
Evidence from animal studies
Comparative toxicokinetics
Because there is little information from studies in humans, laboratory animal studies are important in evaluating the carcinogenic potential of nicotine, at levels found in users of nicotine delivery systems. The kinetics of nicotine absorption, distribution and metabolism are relevant for its pharmacological action (Benowitz et al. Citation2009), but also must be considered for the evaluation of its toxic activity, e.g., when comparing different routes of administration in various species and when extrapolating to the human nicotine user.
The time course for systemic nicotine distribution is fastest after inhalation (within seconds), intermediate for oral uptake and relatively delayed after dermal exposure (lag time of 1 h) (Benowitz et al. Citation2009). Once absorbed, nicotine is widely distributed in the body. As a consequence of the first-pass effect, only 30% of orally administered nicotine can reach systemic circulation (Matta et al. Citation2007). After inhalation or intravenous (i.v.) exposure, however, the first-pass effect of hepatic metabolism is avoided. The plasma half-life of nicotine in humans is approximately 2 h (Hukkanen et al. Citation2005). At an experimental dose of 1 mg/kg in rats and mice, half-lives of 0.75–1.6 h (Kyerematen et al. Citation1988; Schepers et al. Citation1993; Matta et al. Citation2007) and 6–9 min (Petersen et al. Citation1984; Siu & Tyndale Citation2007) were reported, respectively. These differences in elimination half-lives mirror differences in the rates and also patterns of nicotine metabolism among species. Thus, the route and mode of administration (e.g., injection versus continuous exposure) and the choice of species used for a study will determine differences in local and systemic exposures of nicotine and its metabolites that may lead to toxic and carcinogenic effects. Animal studies for the investigation of the potential carcinogenicity of nicotine were, therefore, sorted according to the route of administration used in the various studies reviewed herein.
Several nicotine metabolites have longer half-lives than the parent compound, such as cotinine in human smokers (16 h, Benowitz et al. Citation2009), rats (3 h, Schepers et al. Citation1993) and mice (25–50 min, Siu & Tyndale Citation2007), and have, therefore, been monitored as surrogates for nicotine exposure.
provides a schematic overview of the average range of levels for nicotine doses in users of nicotine delivery systems and the respective nicotine and cotinine levels in blood (based on the values from ). These data can be used to compare nicotine exposures in animal studies based on doses relative to body weight or on blood and urine levels, as available ().
Figure 2. Overview of nicotine biomonitoring data in mouse and rat studies (various shaped arrows). Data referring to human exposure and blood nicotine levels are presented as a black bar indicating the range of data presented in (users of nicotine delivery systems). For species comparison, mouse data are shown from studies with oral administration via the drinking water (red color: Pietilä et al. Citation1995; Sparks & Pauly Citation1999; Grabus et al. Citation2005; Arany et al. Citation2011; AlSharari et al. Citation2013), s.c. osmotic minipump (brown color: AlSharari et al. Citation2013) and feeding (violet color: Theophilus et al. Citation2012). Rat data are shown from nicotine inhalation (black color: Werley et al. Citation2014), continuous s.c. administration via minipump (dark blue color: Cheng et al. Citation2005), and feeding (blue color: Theophilus et al. Citation2012).
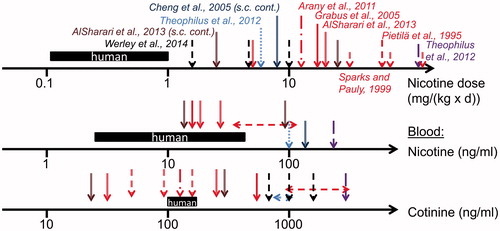
For instance, to achieve the same systemic nicotine concentration, higher nicotine doses relative to body weight have to be administered to rodents (especially mice) in comparison to humans. At drinking water doses of 20 mg/(kg × d) or higher, blood nicotine and cotinine levels can be achieved in mice similar to those in users of nicotine delivery systems ().
In animals, to maintain a nicotine blood level throughout the day that mimics the diurnal changes commonly seen in users of NRT or ENDS, inhalation or the oral administration of nicotine via the drinking water or the diet appear most suitable.
Potential of nicotine to cause cancer in animals
Studies aimed at evaluating the carcinogenic potential of nicotine can be divided into two categories: studies or study parts (sub-studies) with exposure to nicotine alone and studies or sub-studies with exposure to nicotine in combination with other exposures. The intention of reviewing the first category of studies is to evaluate the potential of nicotine to cause cancer, i.e., whether nicotine acts as a complete carcinogen. The intention of reviewing the latter category of studies is to evaluate the modulating effect of nicotine on the carcinogenic potency of these other exposures (Section “Potential of nicotine to modulate carcinogenesis”).
In the first category, several animal species were exposed to nicotine using different routes of administration in studies published over the past 100 years. A summary of reported conclusions from all reviewed studies on the potential of nicotine to act as a complete carcinogen is given in . A more detailed description of high-adequacy score studies is provided in the section below. A description of low-adequacy score studies as well as a more detailed overview of each study evaluated in this category is provided in the supplementary material (Supplementary Tables 4 and 1, respectively). At the end of the current section, both high and low-adequacy score studies are discussed in aggregate, and a conclusion on this part of the review is provided.
Table 4. Reported findings of relevant studies for evaluating the potential of nicotine to act as a complete carcinogen.
Inhalation exposure
Waldum et al. (Citation1996) conducted a 24-month inhalation study in Sprague–Dawley rats with nicotine exposure for 20 h/d, 5 d/week at a reported nicotine concentration of 0.5 mg/m3. A nicotine dose of 0.4 mg/(kg × d) can be estimated on the basis of an average body weight of 300 g/rat, a standard respiratory minute volume of 0.2 l (estimated according to Alexander et al. Citation2008) and assuming full retention of the inhaled nicotine (Feng et al. Citation2007). No food was available during the whole-body nicotine exposure. Plasma nicotine levels were found to exceed the range found in users of nicotine delivery products () by several folds. In this study, the determination of blood nicotine levels seems to be appropriate for comparison to users of nicotine delivery systems due to the long daily nicotine exposure, which probably resulted in a relatively stable steady-state concentration. The body weight of the nicotine-exposed rats was approximately 5% lower than that of the sham-exposed rats (Waldum et al. Citation1996) indicative of an effective nicotine exposure level. There was only one dose level, but this dose level was in an effective range. From the initial animals in the nicotine and sham control groups (68 and 34 rats, respectively), several were used for regular necropsies during the study. Thus, only 22 and 7 rats, respectively, were available for the final necropsy, which included examination of the brain, lungs, gastrointestinal tract, liver, kidneys and ovaries for tumors. For the rats undergoing necropsy during the course of the study, 36% and 24% of the nicotine and control groups, respectively, had tumors, such as age-related mammary tumors, which were found in both groups. Historical control data from this laboratory are not available, but mammary fibroadenomas in female Sprague–Dawley rats are spontaneous tumors frequently observed at the end of cancer studies (Dinse et al. Citation2010). Some tumor types were only found in nicotine-exposed rats, i.e., tumors of the anterior pituitary gland (8% incidence), ovary (5%) and skin (2%). In addition, two metastases of unknown origin were found in nicotine-exposed rats. None of these tumor incidences was statistically significantly different between nicotine and sham-exposure groups. Moreover, in historic controls, tumors have frequently been observed in various parts of the pituitary gland (between 1% and 39%) and occasionally also in the ovary (up to 0.6%), and skin (up to 1.7% incidence, Dinse et al. Citation2010). No lung tumors were detected in either group. Additionally, lung neuroendocrine hyperplasia was investigated, and no effect by nicotine was found. The authors of this study, which was a high-adequacy scoring study, concluded that they “did not find any tumorigenic effect of nicotine on any organ in the body.”
Oral administration
Wilson et al. exposed Albino rats to nicotine sulfate, nicotine tannate or nicotine bentonite via their food in order to investigate body weight and organ microscopic damage (Wilson & DeEds Citation1936; Wilson et al. Citation1938). For up to 10 months, the rats were exposed to four doses of nicotine sulfate at levels of 0.006–0.05% in the diet. These levels corresponded to doses of approximately 4 mg/(kg × d) to 33 mg/(kg × d). A no-observed adverse effect level of 4 mg/(kg × d) was reported based on the retarded body weight development and lower food consumption compared with control. Microscopic examination revealed that organs (liver, spleen, kidneys, lungs, adrenals, heart, testes, thyroid and pancreas) from nicotine-treated rats showed negligible structural difference from organs obtained from control animal (nicotine-free feed). This high-adequacy scored study is characterized by a suitable dose–response assessment, but it is hampered by the relatively short duration. Group sizes were not reported.
Toth (Citation1982) exposed Swiss mice in sufficiently large groups for their lifetime to 0.5 and 0.7 mg/ml nicotine via the drinking water. For the group exposed to the high concentration, the author reported daily doses of 4.3 and 5.3 mg of nicotine hydrochloride for female and male mice, respectively, which translates to average nicotine doses of approximately 150 mg/(kg × d) assuming a body weight of 25 g. Due to higher water consumption, the daily dose per mouse in the low-concentration group was only minimally smaller than in the high concentration group. Surprisingly, no toxicity, in particular, no impact on body weight development or survival, was reported at these doses, although the dose reported clearly exceeded that of other drinking water studies using mice. Histopathological examination of the liver, spleen, kidneys, bladder, thyroid, heart, testes, pancreas, ovaries, brain, nasal turbinates and lungs (at least four lobes) was performed but findings were not reported, except for tumor incidences. No increase in tumor incidence due to nicotine exposure was observed, in particular in lungs, which had a background tumor incidence of approximately 15%. The Swiss strain of mice is genetically predisposed to a relatively high lung cancer susceptibility (Manenti & Dragani Citation2005). The author of the study, which received the highest adequacy score in this review, concluded that nicotine was “not carcinogenic under the experimental conditions.”
Murphy et al. (Citation2011) exposed A/J mice to 0.2 mg/ml nicotine hydrogen tartrate (NHT) for 11 months via the drinking water. Water consumption was significantly lower in nicotine-exposed mice compared with sham-exposed mice, but there was no indication of dehydration. Body weights were not reported. Based on the reported weekly water consumption of 15 ml and an estimated body weight of 25 g, a daily nicotine dose of 6 mg/kg could be calculated. Plasma and urinary nicotine and cotinine levels were also reported. While the plasma cotinine level of mice was below that observed in humans, the urinary cotinine level found in the mice of this study exceeded that normally found in humans. No significant effect by nicotine on lung tumor multiplicity and size was observed, although there was an incidence of 15% (2/15 mice) adenocarcinomas in the nicotine-exposed group compared with the sham control with none. A similar pattern of numerically higher incidences and multiplicities of adenocarcinomas in the nicotine- versus sham-exposed mice was also observed in parallel groups that were pretreated with the tobacco-specific N-nitrosamine NNK. The A/J mouse is a strain susceptible to lung carcinogenesis, and thus, in this particular case, a study duration of 11 months should be sufficient for examining potential carcinogenic effects in this particular tissue (Stoner & Shimkin Citation1982); however, a group size of 19 is relatively small.
Hermann et al. (Citation2014) exposed C57/Bl6 mice for 18 months to nicotine via the drinking water at a nominal dose of 20 mg/(kg × d). The authors were particularly interested in mechanisms of pancreatic carcinogenesis. No effect on the area of pancreatic intra-epithelial neoplastic lesions by nicotine was observed. No other neoplastic findings were reported. This sub-study was conducted in parallel to a study using transgenic mice and reportedly was only intended to confirm earlier reports of no effects by nicotine on pancreatic tumorigenesis; on its own right, it suffered from a very small group size (n = 3) but otherwise received a high-adequacy score.
Nishikawa et al. (Citation1992) investigated the potential of nicotine to induce pancreatic carcinogenesis. Nicotine was administered to female Syrian Golden hamsters for 9 months at an estimated dose of 2.5 mg/(kg × d) via the drinking water (n = 30). The pancreas was carefully examined in serial sections of four anatomical lobes. In addition, the spleen and the duodenum were grossly examined. No neoplastic or preneoplastic lesions were detected in the pancreas.
Subcutaneous administration
Thompson et al. (Citation1973) administered nicotine (as a base) s.c. to male Fischer rats for up to 22 months at a daily dose of 1 mg/kg. Nicotine was administered in a gelatin matrix with the intention to prolong the absorption from the injection site and achieve a rather sustained nicotine distribution. The nicotine exposure was high enough to elicit a significant decrement in the body weight development of the rats (approximately 15% at maximum, which is generally considered acceptable for valid carcinogenicity studies). Starting group sizes were 38 for the nicotine treatment group and 10 for the vehicle control group, but mainly due to technical reasons, only 28 and six rats remained for the final dissection. The spleen, liver, adrenals, vertebra, lymph nodes, lungs, heart, kidneys, thymus, testes, anterior pituitary gland, skin, trachea, renal artery and aorta were routinely examined in the euthanized animals. No consistent differences in general pathology, which was described to be typical for aged rats of this strain, were found between the control and the nicotine-exposed groups. In the control group, two tumors were discovered, an adenocarcinoma of the lung and an adenoma of the anterior pituitary gland resulting in an incidence of 33%. Within the nicotine-exposed group, there were nine tumors present in eight rats resulting in an incidence of 29%. These tumors included three instances of pheochromocytoma, four cases of epidermoid carcinoma of the skin, one leukemia and one fibrosarcoma. The authors noted that all the tumors found are frequently observed in aged rats. In particular, pheochromocytomas indeed occur at rather high incidence in male rats (Greim et al. Citation2009). The only statistically significant difference in histopathology was the incidence of Leydig cell hyperplasia, which occurred in 89% of the nicotine-exposed and 66% of the control rats. The authors noted that the etiologic significance of the observation is unclear. Apparently, Leydig cell tumors are not commonly observed in other studies with this strain of rat and of similar duration. Overall, this negative study is characterized by its sufficient dosing and duration and the broad scope of organs and tissues examined, but it only had one dose level of nicotine and the group sizes were small.
Low-adequacy score studies
Relevant studies with low-adequacy scores using intratracheal installation (Yokohira et al. Citation2012), oral administration (Schoental & Head Citation1953; Truhaut & De Clercq Citation1961), dermal application (Schoental & Head Citation1953), s.c. injection (Staemmler Citation1935, Citation1936; Yun & Kim Citation1938; Hueper Citation1943; Eränkö et al. Citation1959a, Citation1959b; Thienes Citation1960; Schuller et al. Citation1995; Galitovskiy et al. Citation2012), i.v. injection (von Otto Citation1911; Kosdoba Citation1930) and i.p. injection (Schmähl & Habs Citation1976) were identified and evaluated. These studies are described in the Supplementary material as a narrative (Supplementary Table 4) and as an entry in the evidence table in the Supplementary material (Supplementary Table 1).
Discussion of aggregate evidence
High-adequacy score studies
Seven studies were identified with high-adequacy scoring and each varied widely in study design and quality. Overall, the greatest weaknesses often included the lack of dose–response analyses, a lack of sufficient group sizes, a lack of sufficient exposure duration and the lack of sufficient tissue histopathological analyses. However, all these studies were negative with regard to any potential carcinogenic effect of nicotine. These studies were conducted in numerous laboratories with three species (i.e., rats, mice and hamsters) and three different routes of administration, including the relevant inhalation and oral exposure routes. The highest adequacy scores were obtained by the two major dedicated carcinogenicity studies that were judged negative (lack of carcinogenic effect) by the authors (Toth Citation1982; Waldum et al. Citation1996). The negative study by Toth (Citation1982) came closest to a study design matching current bioassay guidelines.
Low-adequacy score studies
Sixteen low-scoring studies were reviewed in this section and were predominately considered negative (lack of carcinogenic effect) by their authors, with two exceptions using s.c. nicotine administration.
One exception relates to adrenal medulla adenocarcinomas or pheochromocytomas reported after s.c. nicotine administration to rats (Staemmler Citation1935). This effect was not dose dependent, and it was not observed again in other studies of similar design that were conducted in rats, mice, Guinea pigs and rabbits (Kosdoba Citation1930; Eränkö et al. Citation1959a, Citation1959b; Thienes Citation1960; Thompson et al. Citation1973), with one exception, i.e., a 22-month s.c. nicotine administration study in which three rats were observed in the nicotine group and none in the control (not statistically significant, Thompson et al. Citation1973). In particular, the Thienes (Citation1960) and Eränkö et al. (Citation1959a, Citation1959b) studies were performed in response to the findings reported by Staemmler (Citation1935), but the carcinogenic effect could not be reproduced. The occurrence of pheochromocytomas seems to be related to disturbances in catecholamine synthesis, which may indeed be the case in the nicotine exposure studies (Greim et al. Citation2009). A morphological effect of nicotine on adrenals might seem plausible, as nicotine can stimulate the release of corticosterone and catecholamines from the adrenal cortex and medulla, respectively, and hypertrophic adrenals were indeed described for i.v.-treated rabbits (Kosdoba Citation1930). However, an effect on adrenals was not reported in 90-d nicotine feeding studies with rats and mice at doses of 6 and 120 mg/(kg × d), respectively (Theophilus et al. Citation2012). In general, the relevance of pheochromocytomas in rat carcinogenicity studies for human risk assessment was questioned (Greim et al. Citation2009).
The second exception was the report that rhabdomyosarcomas and leiomyosarcomas were observed after s.c. nicotine administration to A/J mice for 5 d/week for 24 months (Galitovskiy et al. Citation2012). This study had small group sizes, and no statistical tests were performed. The development of spontaneous rhabdomyosarcomas was also observed in other studies using this mouse strain: Rhabdomyosarcomas at the hind legs and lower back were described to be rather frequent spontaneous tumors in A/J mice (34% incidence, Landau et al. Citation1998). In a chronic mainstream smoke inhalation study in A/J mice, rhabdomyosarcoma incidences of 27% and 43% in female and male control mice were observed, respectively, which tended to decrease with increasing mainstream smoke and thus nicotine exposure concentrations (Stinn et al. Citation2013).
Leiomyosarcomas have not been reported as a consequence of nicotine administration in other studies, and this finding would need to be reproduced in a more carefully designed study with a more appropriate route of administration. Interestingly, the authors reported only one mouse with pulmonary adenoma. The incidence of lung tumors after 24 months observed in this study is surprisingly low, as most other studies in A/J mice showed 100% incidence at this age (e.g., Stoner & Shimkin Citation1982).
In principle, the overall results of the low-adequacy studies agree with those of the high-adequacy studies, i.e., they do not suggest that nicotine is a complete carcinogen.
Comparative evaluation by dose
For a comparative evaluation, both high and low-adequacy studies were considered. The doses used in the various studies cover a relatively broad range for rats [0.3–33 mg/(kg × d)] and mice [1–150 mg/(kg × d)]. The doses used are similar or higher than those found for users of nicotine delivery systems. For example, in the lifetime mouse study conducted by Toth (Citation1982), the dose of nicotine administered via drinking water exceeded by two orders of magnitude that reported for human exposure following nicotine use (relative to body weight). The lifetime inhalation study (Waldum et al. Citation1996) reported blood nicotine levels beyond those reported for human users of nicotine. Both the Toth (Citation1982) and Waldum et al. (1996) studies reported the lack of nicotine-induced carcinogenicity. The most recent mouse study, which was reported as positive by their authors, used the lowest s.c. nicotine dose in this category (Galitovskiy et al. Citation2012), shedding additional doubt on the findings reported. A rat s.c. study with a similarly low apparent daily dose of nicotine for 22 months but with a pronounced nicotine-related body weight effect was negative for carcinogenesis (Thompson et al. Citation1973).
Nicotine concentration in body fluids may not be the best marker for comparing nicotine exposure from bolus injections due to the rather rapid and species-dependent clearance as well as the lack of standardizing sampling periods relative to nicotine administration. The only nicotine inhalation study available, however, exposed rats for 20 h/d, so the reported plasma nicotine value of 130 ng/ml most likely represents a stable, steady-state concentration (Waldum et al. Citation1996). However, the daily dose of 0.4 mg/(kg × d), estimated on the basis of the nicotine concentration in the aerosol and on certain assumptions of respiratory minute volume and body weight, does not fit the reported nicotine plasma level in comparison to other rat studies with sustained nicotine exposure (). In a 90-d mainstream smoke nose-only inhalation study (6 h/d) in rats with a nicotine concentration of 13 mg/m3 in the aerosol, a daily dose of 3.5 mg/kg can be estimated for the same strain and sex of rats using the same assumptions as above, which resulted in an average serum nicotine concentration of 280 ng/ml (Gaworski et al. Citation2008). In any case, the plasma nicotine value determined in the nicotine inhalation study (Waldum et al. Citation1996) most likely reflects a higher nicotine uptake than the estimated inhaled dose. As this study was conducted in a whole-body-exposure mode, nicotine deposited on the cage surfaces and the fur of the rats, which, as a consequence of self-grooming, can lead to several folds higher overall doses, as assumed from smoke inhalation studies (Mauderly et al. Citation1989; Haussmann et al. Citation1998). Based on the plasma nicotine values reported, the dose in this nicotine inhalation study (Waldum et al. Citation1996) appears to be sufficiently high to exceed human exposure from nicotine use and no carcinogenic potential was detected.
Figure 3. Overview of nicotine biomonitoring data in mouse and rat studies investigating potential nicotine-mediated carcinogenesis. Data (indicated by arrows) were generated in a rat inhalation study (Waldum et al. Citation1996) and in mouse studies with exposure to nicotine via drinking water (Wong et al. Citation2007; Maier et al. Citation2011; Murphy et al. Citation2011; Hermann et al. Citation2014; Li et al. Citation2015), via continuous s.c. administration (Hao et al. Citation2013), via i.v. administration (Li et al. Citation2015) and via patch and i.p. administration (Davis et al. Citation2009). Nicotine doses and nicotine and cotinine levels in blood and urine from users of nicotine delivery systems such as NRT products or ENDS are illustrated as black bars (blood) or diamonds and bars (urine) indicating the range of concentrations (from data presented in ). Positive (cancer stimulating) and negative studies are characterized by red and green colors, respectively.
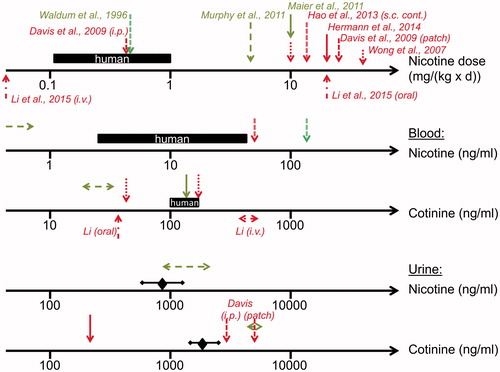
An average nicotine plasma level of 0.4 ng/ml was reported from a negative drinking water study in mice (Murphy et al. Citation2011), which is low, given the estimated dose of 6 mg/(kg × d) based on water consumption and body weight (). In plasma, 19 ng/ml cotinine level was determined on average (Murphy et al. Citation2011). Both the nicotine and cotinine plasma levels reported are relatively low compared to those in human nicotine users (). Urinary nicotine and cotinine concentrations of 1300 ng/ml and 4400 ng/ml, respectively, were also reported in this study, which are similar to and exceeding those found in users of nicotine delivery systems, respectively ( and ).
Summary of the evidence
The high-adequacy score studies were consistently negative (absence of a stimulating effect on carcinogenesis). The statistical power of only a few of the negative studies seemed to be sufficient, though. The finding of adrenal medulla adenocarcinoma in one low-adequacy study was not dose dependent and inconsistent with the results of other studies, although this type of effect might seem to be coherent with non-neoplastic effects of nicotine described in a few studies, i.e., adrenal hypertrophy. The findings of rhabdomyosarcomas and leiomyosarcomas in A/J mice injected with nicotine found in a low-adequacy score study was inconsistent with other studies, had a low strength of association (no statistical tests performed), and the low number of age-related spontaneous lung cancer cases was incoherent with historic controls of this strain of mice. As discussed in the Introduction, results on the potential genotoxicity of nicotine at relevant concentrations are conflicting and thus are not inconsistent with the relative absence of animal studies demonstrating nicotine as a complete carcinogen.
Conclusion
Overall, the animal studies on nicotine carcinogenicity available to date do not suggest that nicotine is a complete carcinogen. However, there has been no single study that would have passed the current criteria of a well-designed study according to generally agreed-upon guidelines, e.g., in terms of number and range of dose levels, statistical power, or biomonitoring of nicotine exposure by its metabolites in body fluids. Therefore, conclusive studies are missing. Nevertheless, two negative (lack of carcinogenic effect) studies were the highest adequacy scoring studies in this group. In conclusion, limited evidence suggests an association between long-term nicotine exposure and a lack of a complete carcinogenic effect.
Potential of nicotine to modulate carcinogenesis
This section reviews studies in which nicotine administration was tested for a potential modulating (stimulating or lack of stimulating) effect of tumorigenic processes induced by chemical and physical treatments and genetic manipulations (Section “Cancer induction by physical, chemical, and transgenic means”) as well as cellular treatments (xenograft studies, Section “Cancer xenograft studies”). At the end of each of these two sections, both high and low-adequacy score studies are discussed in aggregate and a conclusion is provided for each respective part of the review. Subsequently, in an attempt to further stratify results by study design variables, the results of these two sections are further discussed regarding the impact of various study design parameters used, e.g., route of administration, dose and dose rate, or the impact of immune competence.
Cancer induction by physical, chemical and transgenic means
A summary of reported conclusions from all reviewed studies in this category is given in . A description of high-adequacy studies is provided in the section below. A narrative description of low-adequacy studies as well as a more detailed overview of all studies evaluated in this category is provided in the Supplementary material (Supplementary Tables 4 and 2).
Table 5. Reported findings of relevant studies for evaluating the potential cancer modulating activity of nicotine in studies induced by physical, chemical, or transgenic means.
Oral administration
In a relatively early study, Freedlander et al. (Citation1956) investigated the potential of nicotine to modulate the tumorigenicity of UV light in mice (n = 100), which, under the conditions of the study, developed ear and eye tumors. The nicotine dose administered via the drinking water was increased from approximately 3 to 18 mg/(kg × d) over the course of the 7-month study. Tumor incidences were 42% in the control group and 35% in the group treated with nicotine via the drinking water. Thus, the authors concluded that there is no additive or co-carcinogenic effect of nicotine with UV light. There was no group exposed to nicotine alone.
In a study by Liu et al. (Citation2011), the bladder of Wistar rats was infused with N-methyl-nitrosurea (MNU) sufficient to induce bladder cancer within a few months. After the end of the MNU treatment, rats were randomized into four groups treated intragastrically with nicotine doses from 0 to 11 mg/(kg × d) for 2 months (n = 12). Although the groups were very small, animals were necropsized at given time points up to the conclusion of the study at 4 months. The authors reported a nicotine dose-dependent increase in tumor size (no data shown). In the high-dose nicotine group, two metastases were found. In addition, a nicotine dose-dependent increase in the frequency of mutated p53 genes was reported, which was apparently determined by immunohistochemistry. The authors concluded that nicotine may play an important role in the development of bladder cancer.
Murphy et al. (Citation2011) investigated the modulating activity of nicotine on NNK-induced carcinogenesis. Female A/J mice (n = 18) were initiated with a single i.p. injection of 80 mg/kg NNK and exposed to nicotine hydrogen tartrate via the drinking water for up to 11 months. In order to be able to study the potential impact of nicotine on various stages of the NNK-induced tumorigenesis, nicotine was administered either for 0.5 months before NNK administration, for 11 months after NNK administration, or throughout the study (with NNK administration after 0.5 months of nicotine exposure). Water consumption was lower than in sham-exposed mice, leading to an approximate nicotine dose of 6 mg/(kg × d). Plasma nicotine and cotinine levels were determined throughout the study and found to be relatively low, 0.66 and 31 ng/ml respectively, compared with levels reported for users of nicotine delivery products (). Lung tumor multiplicity was approximately 20, and there was no effect of nicotine on multiplicity, size and progression from benign to malignant lung tumors regardless of the nicotine exposure regimen. The advantages of this study were the targeted nicotine exposure at various stages of tumorigenesis, the chronic duration and the biomonitoring of nicotine exposure. Limitations were the intermediate group sizes relative to other studies in this category and the relatively low nicotine exposure as assessed by biomonitoring.
Maier et al. (Citation2011) conducted a relatively similar study to that above but with the use of AB6F1 mice. The nicotine dose delivered via the drinking water was estimated at 10 mg/(kg × d). No nicotine-related toxicity was observed. Nicotine exposure was for 3 months after i.p. treatment with 100 mg/kg NNK for three weeks (n = 10). A control group exposed to nicotine alone was included in this study. Although a relatively high NNK dose was used, the average tumor multiplicity was only approximately 1.5, which may be related to the relatively short duration and the loss of pulmonary tumor susceptibility by crossing of susceptible A/J with less susceptible C57Bl6 mice. Nicotine did not enhance lung carcinogenicity in terms of tumor multiplicity and volume. There was a numerical trend to a higher incidence of lung tumors in the nicotine-treated groups, which was not statistically significant. Serum cotinine levels were at 137 ng/ml, which according to the authors’ suggestion would be comparable with an NRT user with exposure to a 22 mg nicotine patch.
Maier et al. (Citation2011) also used a mouse model transgenic for a mutated human Kras gene, which is known to progress rapidly through pulmonary tumorigenesis, i.e., KrasLA2. In this model, tumors are apparent as early as 2 weeks of age, and they progress to adenocarcinomas within several months. Two weeks of nicotine exposure starting at an age of 3 weeks did not change tumor multiplicity or tumor burden. Six weeks of nicotine exposure starting at an age of 6 weeks did not affect tumor multiplicity, size or burden. A daily nicotine dose of 10 mg/(kg × d) was estimated (n = 5). If this treatment was continued until the death of the mice (approximately for 5 months), nicotine did not alter the overall life span either. It is unclear whether the life span was limited by the lung tumors, because data on tumor multiplicity or size were not reported for this latter sub-study. In the lung tumors found in this study, nicotine did not alter the activation status of a number of proteins associated with cellular growth signals, such as Akt, Erk, or the proliferation marker Ki-67. The group sizes in this sub-study are among the smallest in this category.
Hermann et al. (Citation2014) studied the effect of nicotine administration on pancreatic cancer development in various mouse models, apparently exposed to nicotine at a nominal concentration of 20 mg/(kg × d) via the drinking water. Kras+/LSLG12Vgeo;Elas-tTA/tetO-Cre and Kras+/LSLG12D; Trp53+/LSLR172H;Pdx-1-Cre (KPC) mice were exposed for 18 and 20 months, respectively, which resulted in a 10- and 4-fold increase in the area of pancreatic intraepithelial neoplasia lesions, respectively (n = 7). In addition, the grade or severity of the lesions was higher in the nicotine-treated groups. For the KPC mouse, an increased number of circulating pancreatic cells was also observed that was considered indicative of a metastatic phenotype. For the Kras+/LSLG12Vgeo mouse, a urinary cotinine level of 210 ng/ml was reported, which the authors suggested was similar to the level of intermediate smokers. This suggestion, however, was based on a reference that only reported blood cotinine levels, and in their abstract, the authors indeed discussed their result on the basis of blood levels. The reported value, as a urinary cotinine level, is much lower than what has been reported for users of nicotine delivery products (and ). A plethora of mechanistic investigations was included in this study pointing to a nicotine-induced acinar cell dedifferentiation via down-regulation of Gata6. For instance, nicotine seemed to increase tumor growth from murine acinar cells in nude mice, if these cells harbored a mutated Kras gene and were deficient of Gata6 (n = 2–3).
Nishikawa et al. (Citation1992) investigated the potential of nicotine to modulate the pancreatic carcinogenesis initiated by N-nitrosobis(2-oxopropyl)amine in female Syrian Golden hamsters. After completion of the initiation, nicotine was administered for 9 months at an estimated dose of 2.5 mg/(kg × d) via the drinking water (n = 30). The pancreas was carefully examined in serial sections of four anatomical lobes. In addition, the spleen and the duodenum were grossly examined. The authors claimed to find a tendency to enhanced pancreatic carcinogenesis in terms of adenocarcinoma and dysplasia incidence; however, no statistically significant effects for nicotine were reported.
Low-adequacy score studies
Relevant studies with low-adequacy scores using oral administration (Freedlander & French Citation1956; Ito et al. Citation1984; Nakada et al. Citation2012), cheek pouch application (Chen & Squier Citation1990; Chen et al. Citation1994), dermal application (Bock & Tso Citation1976; Bock Citation1980), s.c. injection (Rana & Bhagat Citation1970; Bhagat & Rana Citation1971; Gurkalo & Volfson Citation1982; Habs & Schmähl Citation1984; Schuller et al. Citation1995; Bersch et al. Citation2009; Hayashi et al. Citation2014) and i.p. injection (Habs & Schmähl Citation1976; Davis et al. Citation2009; Iskandar et al. Citation2013) were identified and evaluated. A study on the potential of nicotine to modulate chemotherapy was conducted using osmotic minipumps to administer nicotine (Berger & Zeller Citation1988), presumably via the s.c. route, although the actual route of administration was not specified. These studies are described in the Supplementary material as a narrative (Supplementary Table 4) and as an entry in the evidence table in the Supplementary material (Supplementary Table 2). No inhalation studies were identified.
Discussion of aggregate evidence
High-adequacy score studies
All high-adequacy score studies used oral nicotine administrations. Of the eight high-adequacy (sub-)studies identified and evaluated for a potential role of nicotine in modulating the carcinogenic effects of inducing treatments, four were negative (lack of a stimulating effect) and four were positive (stimulating effect). The positive rat study by Liu et al. (Citation2011) used an intragastric and thus most probably bolus administration of nicotine. The study is difficult to interpret because actual tumor data were not provided by the authors. Other studies in this category used sustained nicotine administration to mice or hamsters via the drinking water. The second set of two positive sub-studies in this context investigated the effect of nicotine administration on pancreatic neoplastic developments in particular models of Kras mutant mice (Hermann et al. Citation2014). However, in another Kras mutant mouse model, no effect of nicotine on pulmonary carcinogenesis was observed (Maier et al. Citation2011). Nominal nicotine doses of 10 (racemic mixture) and 20 mg/(kg × d) (unknown type of nicotine) were estimated for the negative and positive Kras mutant studies, respectively. With the uncertainty in the type of nicotine used in the positive study, a potential dose–response difference between the two studies as a reason for the differential outcome remains a possibility. A difference in the Kras biology of both models might also explain the difference in outcomes. Both studies, however, employed very small group sizes. Importantly, various sub-studies from two laboratories did not find a stimulating activity of oral nicotine for lung tumors induced by NNK, a tobacco-derived N-nitrosamine, regardless of the temporal relationship between the administrations of the two compounds (Maier et al. Citation2011; Murphy et al. Citation2011).
Low-adequacy score studies
Twenty-seven low-adequacy scoring (sub-)studies were identified and evaluated in this category. The three oral studies were all negative; included here are two studies with A/J mice that are susceptible to lung tumor formation (Freedlander & French Citation1956; Nakada et al. Citation2012). Combination with either urethane or NNK did not increase tumor risk. However, cheek pouch application studies with hamsters were reported to be positive in combination with chemical carcinogens (Chen & Squier Citation1990; Chen et al. Citation1994). Rather complex nicotine responses were obtained in studies that tried to identify whether nicotine would play a role in a rather common model for tobacco carcinogenesis, i.e., mouse skin painting (Bock & Tso Citation1976; Bock Citation1980). The main author concluded that the results of his experiments showed that nicotine per se can enhance carcinogenesis induced by the combination of benzo[a]pyrene and a promoter, although the mechanism of this presumed co-carcinogenesis and its relevance to humans remained unclear. Of the s.c. injection studies, a stimulating effect of nicotine was reported for pancreatic, pulmonary and gastric cancer models upon induction with dimethylbenzanthracene (Bersch et al. Citation2009), hyperoxia (Schuller et al. Citation1995) and methylnitronitrosoguanidine (Gurkalo & Volfson Citation1982), respectively. Other studies with s.c. nicotine injection were negative or apparently even showed a protective effect, e.g., in a colitis-associated cancer model (Hayashi et al. Citation2014). Upon i.p. injection, nicotine was reported to stimulate NNK-induced carcinogenesis in A/J mice (Davis et al. Citation2009; Iskandar et al. Citation2013), which apparently is in contrast to the results of NNK studies with A/J mice and nicotine administration via the drinking water (see “High-adequacy score studies” section). It would be interesting to understand from a mechanistic point of view, why bolus administrations of nicotine, as few as thrice per week, can result in a positive modulating effect.
Summary of the evidence
Across the many different routes of administration, which includes both sustained and bolus administrations, study results on carcinogenicity were not consistent, providing about half positive and half negative results, regardless of the adequacy score obtained. Except for the skin painting studies with mixed results, those studies with the highest statistical power in terms of group sizes tended to be negative. Most studies had very small group sizes. Dose–response studies were not performed. Dose–response analyses across studies were difficult due to the often missing description of the actual nicotine type used and the difficulty of assessing dose or dose rate with bolus injection studies administering only a few injections per week. However, a trend to a dose–responsive behavior across studies cannot be excluded. Dose-responsiveness would certainly need to be assessed in future studies using the biologically active nicotine enantiomer. Biological plausibility is difficult to assess, as the high-adequacy score studies lack sufficient mechanistic investigations. Of particular interest is to develop a better understanding of the toxicodynamic similarities and differences between sustained and bolus administrations. This is especially important in light of the well-known effect of nicotine dosing regimens on the induction and desensitization of nAChRs (Marks et al. Citation1985; Renda & Nashmi Citation2014; Lam et al. Citation2016). Interestingly, none of the studies in this section was conducted with immunocompromized animals, in contrast to the majority of studies conducted with xenografts (see “Cancer xenograft studies” section).
Conclusion
Conflicting results were reported for the effect of nicotine in cancer models with physical, chemical, or transgenic initiation. Additional mechanistic insight is required to allow an understanding of the negative and positive findings reported. Thus for animal studies using chemical, physical or transgenic models to initiate cancer, there is inadequate evidence for an association between nicotine exposure and the presence of or lack of a stimulating effect on carcinogenesis.
Cancer xenograft studies
In general, studies in this category investigated the ability of nicotine exposure to modulate (stimulate) tumor growth after cancer cells were inoculated in animals. Immunocompromized mice were commonly used in these studies. A summary of reported conclusions from all reviewed studies in this category is given in . A description of high-adequacy studies is provided in the section below. A description of low-adequacy studies as well as a more detailed overview of all studies evaluated in this category is provided in the Supplementary material (Supplementary Tables 4 and 2, respectively).
Table 6. Reported findings of relevant studies for evaluating the potential cancer modulating activity of nicotine in xenograft studies.
Oral administration
Jarzynka et al. (Citation2006) were interested in the combined effect of estradiol and nicotine on the growth of A549 cells subcutaneously (s.c.) implanted into nude ovariectomized mice. Nicotine was administered at two estimated doses of 20 and 40 mg/(kg × d) via the drinking water for 36 d (n = 8). Estradiol was administered via a pellet co-injected with the A549 cells. There is no mentioning of a sham operation with a pellet containing no estradiol as a control for potential physical effects. Nicotine by itself resulted in a numerical but statistically non-significant increase in tumor growth in the high dose group, which was characterized by the authors as positive (stimulates tumor growth). At the lower dose of nicotine, no effect was observed (data not shown). There was a statistically significant increase in cell proliferation due to nicotine in the tumor tissue, while there was only a numerical increase in vascularization. Estradiol by itself also resulted in a numerical but statistically non-significant increase in tumor growth. The combined administration of both substances resulted in an increased tumor volume that was significantly higher than in the untreated control or in the high-dose nicotine group. Given the moderate, if any, effect of nicotine on tumor growth in this study, the claimed combination effect could have been an estradiol effect and with no particular contribution by nicotine.
Shin et al. (Citation2004) investigated the role of nicotine in athymic nude mice inoculated with a gastric cancer cell line into the gastric wall. Nicotine was administered for 3 months at two doses of 11 and 62 mg/(kg × d) via the drinking water (n = 10). A decreased body weight effect was observed at the high nicotine dose. The area of the gastric wall covered by a tumor increased with increasing nicotine doses from 18 mm2 to 25 mm2 to 30 mm2 in parallel to an increasing cell proliferation index. Neovascularization determined as microvessel density as well as the expression of the vascular endothelial growth factor (VEGF) in the tumor tissue also increased in a parallel manner. These effects were dependent on the activity of cyclooxygenase-2 (COX-2). The authors concluded that their study revealed “a direct promoting action of nicotine on the growth of gastric tumor.”
Wong et al. (Citation2007) inoculated colon cancer cells s.c. into the flank of nude BALB/c mice. Nicotine was administered for 25 d at two doses of 10 and 40 mg/(kg × d) via the drinking water (unknown group size). No effects on drinking water consumption and body weight development were observed. No group size was reported. Higher tumor volumes were observed in a dose-dependent manner, and the tumor growth was attenuated by β1- and β2-adrenoceptor antagonists given by i.p. injections thrice per week for the course of the study. Plasma adrenaline and cotinine levels increased with increasing nicotine doses; the authors did not comment on the surprising finding of low levels of cotinine in the sham-treated group. In the tumor tissue, increased levels of both types of adrenoceptors, COX-2, prostaglandin E2 (PGE2), VEGF and microvessel density were observed. The authors concluded that the nicotine-dependent effect on tumor growth would be mediated by β-adrenergic activation and angiogenesis. The same group investigated an s.c. xenograft model with colon cancer cells that were pretreated in vitro with 10 nM, 100 nM and 1000 nM of nicotine for 5 h before inoculation (Ye et al. Citation2004). After 3 weeks, the volume of the tumors was increased in a nicotine concentration-dependent manner. The formation of these tumors could be inhibited by incubation of the cancer cells with inhibitors of the epidermal growth factor receptor or inhibitors of 5-lipoxygenase prior to treatment with nicotine.
Maier et al. (Citation2011) inoculated AB6F1 mice with three different cell lines derived from NNK-induced lung adenocarcinoma of the same strain and observed tumor development. The F1 generation of A/J and C57Bl6 mice, i.e., AB6F1 mice, was used in order to combine the A/J susceptibility to developing lung cancer and the C57B16 apparent preference for consuming nicotine-containing drinking water. Within the 2 weeks of nicotine exposure at an estimated dose of 10 mg/(kg × d), no modulating effect on tumor growth or the development of metastases was found. The group sizes in this sub-study are among the smallest in this category (n = 5).
Li et al. (Citation2015) investigated the potential antagonist effect of nicotine on the chemotherapeutic effect of the epidermal growth factor receptor (EGFR) inhibitor erlotinib. In one sub-study, nicotine was administered via the drinking water for 20 d after s.c. inoculation of PC9 NSCLC cells in nude BALB/c mice (group size unknown). A nicotine dose of 20 mg/(kg × d) was estimated, which resulted in serum cotinine levels of approximately 37 ng/ml. The authors did not explain why their control mice also had cotinine levels of up to 20 ng/ml. A small but statistically significant increase in tumor volume was observed. Interestingly, a parallel group of mice was exposed to i.v. nicotine injections for 5 d/week at doses of 0.06 mg/kg, and, in this group, a similar increase in tumor growth was observed as with the oral nicotine administration. Between days 21 and 36, erlotinib was also administered and inhibited the further growth of the xenograft tumors. Growth inhibition was less effective in the group with prior and concomitant oral nicotine exposure compared to that with i.v. injections. Nevertheless, in comparison with a control without nicotine, both types of nicotine treatment attenuated the growth-inhibitory effect of erlotinib.
Subcutaneous administration
Pratesi et al. (Citation1996) administered nicotine s.c. via osmotic minipumps to athymic nude BALB/c mice that were s.c. inoculated with tumor fragments developed from a small cell lung cancer cell line in the same mouse strain. Nicotine doses of 0.8 and 8 mg/(kg × d) were maintained for 2 weeks either shortly after inoculation or after tumors had grown to a certain size (n = 5–10). The time until a target tumor size was obtained was used to measure the potential modulating activity of nicotine on tumorigenesis. No effect was observed for nicotine, while in a parallel experiment a modulating activity for serotonin was claimed. The study suffered from very small group sizes.
Hao et al. (Citation2013) investigated the effect of nicotine on pulmonary metastasis formation from murine melanoma cells given i.v. to immunocompromized RAG2−/− mice. Nicotine was administered via s.c. osmotic minipumps at a dose rate of 13 mg/(kg × d), resulting in nicotine plasma levels of approximately 49 ng/ml (n = 10–13). Within 2 weeks, nicotine doubled the tumor volume determined via luminescence labeling of the melanoma cells. The nicotinic growth effect on metastases was largely diminished in RAG2−/− β2-nAChR−/− mice generating by crossing the RAG2−/− with mice knocked-out for this nAChR receptor. The authors interpreted these results to demonstrate that nicotine would exert an immunoinhibitory effect on TK cells via β2-nAChR, which would otherwise contain the growth of these melanoma metastases.
Berger and Zeller (Citation1988) investigated a potential interference with the chemotherapy of two types of rat cancer models. In a leukemia model driven by inoculation of rat leukemia cells and treated with cyclophosphamide for chemotherapy, nicotine had a borderline significant enhancing effect on the development of leukemia, i.e., nicotine was interpreted to impair the chemotherapy by cyclophosphamide, although only at the lowest of three cyclophosphamide doses. Nicotine was administered for 2 weeks via an osmotic minipump (no specification of the route of administration but presumably s.c., n = 8) at doses of 2.5 and 5 mg/(kg × d). No effect on the development of leukemia in this xenograft model was seen by nicotine alone.
Low-adequacy score studies
This section includes relevant studies with low-adequacy scores using i.v. injection (Paleari et al. Citation2008; Li et al. Citation2015), oral administration (Heeschen et al. Citation2001; Natori et al. Citation2003; Al-Wadei et al. Citation2009; Lee et al. Citation2010; Al-Wadei et al. Citation2012; Nakada et al. Citation2012; Banerjee et al. Citation2013; Khalil et al. Citation2013; Banerjee et al. Citation2014; Liu et al. Citation2015), dermal application (Davis et al. Citation2009), s.c. injection (Warren et al. Citation2012) and i.p. injection (Davis et al. Citation2009; Maier et al. Citation2011; Molfino et al. Citation2011; Treviño et al. Citation2012; Pillai et al. Citation2015; Yuge et al. Citation2015). In one study, nicotine was administered via an osmotic minipump; however, the actual route of delivery was not specified (Improgo et al. Citation2013). These studies are described in the Supplementary material as a narrative (Supplementary Table 4) and as an entry in the evidence table in the Supplementary material (Supplementary Table 2). No inhalation study was identified.
Discussion of aggregate evidence
High-adequacy score studies
Of the high-adequacy score (sub-)studies with oral nicotine administration, the four studies conducted with immunocompromized mice were considered positive by their authors (Shin et al. Citation2004; Jarzynka et al. Citation2006; Wong et al. Citation2007; Li et al. Citation2015), while the three sub-studies conducted with immunocompetent mice were negative (Maier et al. Citation2011). This differentiation is not as clear-cut as it might first seem, because the estimated nicotine dose used in the negative sub-studies were also among the lowest in this category. It is unclear whether other differences in study design could be responsible for the difference in outcome between these studies. The genetic set-up and thus the aggressiveness of the cancer cells used might also affect the responsiveness to nicotine.
Low-adequacy score studies
Twenty-four xenograft (sub-)studies with low-adequacy scores were identified. For those studies with oral nicotine administration, all were considered positive by their authors with the exception of those with intentionally low nicotine doses that were not expected to be positive and used for mechanistic investigations (Banerjee et al. Citation2013, Citation2014). Also, the negative study using s.c. nicotine administration was not intended to dose nicotine high enough to enhance tumor growth but rather to see whether lower doses might interfere with radio- and chemotherapy (Warren et al. Citation2012). The two studies with nicotine administration via s.c. minipumps used two different types of immunocompromized mice; the one with the higher estimated nicotine dose was positive (Hao et al. Citation2013), while the one with the lower estimated nicotine dose was negative (Pratesi et al. Citation1996). The sub-studies with i.p. nicotine injection were all positive except for two sub-studies, for which the nicotine administration frequency was not clearly identified (Maier et al. Citation2011). For the bolus types of nicotine administration, daily doses or dose rates are difficult to estimate and compare with other types of administrations. Both studies with sustained or bolus administration were considered positive, i.e., stimulating tumor growth from inoculated cancer cells. An interesting finding was the similar increase in tumor growth seen in one study after either oral or i.v. nicotine administration (Li et al. Citation2015). In contrast, in another study nicotine administration via both drinking water and i.p. injections were negative (Maier et al. Citation2011). In this category, studies using both immunocompetent (Heeschen et al. Citation2001; Natori et al. Citation2003; Davis et al. Citation2009; Nakada et al. Citation2012) and immunocompromized mice (Paleari et al. Citation2008; Al-Wadei et al. Citation2009; Lee et al. Citation2010; Al-Wadei et al. Citation2012; Treviño et al. Citation2012; Improgo et al. Citation2013; Khalil et al. Citation2013; Li et al. Citation2015; Liu et al. Citation2015; Pillai et al. Citation2015; Yuge et al. Citation2015) were considered positive by their authors. The negative studies by Maier et al. (Citation2011) and Molfino et al. (Citation2011) as well as the positive study by Davis et al. (Citation2009) used immunocompetent animals.
Summary of the evidence
There was a certain degree of consistency for the finding of cancer growth stimulating effects of nicotine across types of cancer cells used, sites of inoculation, routes of nicotine administration and laboratories conducting the studies in finding positive effects of nicotine in xenograft models. Such effects may also be biologically plausible in view of the in vitro findings on cell proliferation, anti-apoptosis, cell migration and invasion, and angiogenesis (see “Introduction” section), although a critical assessment of the degree of coherence in translating the in vitro findings to in vivo effects seem to be missing. The strengths of association between nicotine exposure and xenograft growth were variable from modest effects to severe differences as compared to controls. However, most studies used very small group sizes and dose–response analyses were rarely conducted.
Conclusion
The majority of studies in this category were positive across various routes of administration, various cancer cell types for inoculation, various organs and various types of hosts. In particular, positive findings were observed after both sustained and bolus nicotine administration. Some of the negative study results may be explained by the relatively low doses administered. Thus, a majority of studies provides sufficient evidence for an association between nicotine exposure and enhanced carcinogenesis of cancer cells inoculated in mice.
Discussion of cancer-modulating activity studies
Of the approximately 70 studies or sub-studies covered in this category, about 60% demonstrated the stimulation of a carcinogenic effect and about 40% showed a lack of such effect ( and ). To assess the relevance of these experimental animal studies to users of nicotine delivery systems (e.g., NRT products and ENDS), it would be helpful to understand the differences in study design parameters that may be responsible for the conflicting findings.
General insufficiency of available information
Common obstacles for the current assessment are the frequent absence of information on the type of nicotine (base, salt, and enantiomer) used in these studies as well as the common lack of information on nicotine exposure (e.g., accurate assessment of plasma nicotine or cotinine levels). Furthermore, practically all published studies did not provide sufficient information to fully judge their findings’ relevance and significance in a comparative manner. For instance, very few studies report body weight changes following nicotine treatment. At high doses, nicotine exposure in animals has been shown to result in a significant reduction in growth rate (body weight increase or maximally attained body weight), presumably due to a decrease in food intake (Wilson & DeEds Citation1936). Such an effect may or may not affect tumor growth in many of these relatively short-term studies. Such information would be useful for evaluating the actual nicotine exposure levels as well as the relevance of a carcinogenic effect (or lack of).
Most studies suffered from very small group sizes. Most studies in this category were conducted with mice, and consequently, no species-specific pattern for an effect or lack of an effect could be determined. The studies were conducted in three dozen different laboratories, and occasionally conflicting findings were derived in the same laboratory depending on the study conditions.
Segregation of studies by type of tumor initiation
Another approach to interpret the studies reviewed in this section is by the process in which the carcinogenic effect was established. Thirty-five studies or sub-studies used exposure of chemical or physical carcinogens or transgenic mice in combination with nicotine () while 35 studies or sub-studies, most of which were mouse studies, used the inoculation of cancer cells or tumor fragments (). Of the first group, 16/35 studies (46%) were positive, while the others were negative or, in few cases, even showed a protective effect. Of the second group, 24/35 were positive (69%). Among the three sub-studies that used mice transgenic with mutant Kras, two from one laboratory were positive (Hermann et al. Citation2014) and one was negative (Maier et al. Citation2011). In the study by Nakada et al. (Citation2012), sequential dosing with NNK and nicotine was negative, while nicotine administration after inoculation with cancer cells was positive. For studies investigating the cooperative effect of NNK and nicotine, most but not all were negative. Overall, nicotine’s effect on chemical/physical/transgenic-induced carcinogenesis does not appear to be dependent on the type of initiating agent. For xenograft studies, the evidence appears to be sufficient for a stimulating effect of nicotine on the growth of inoculated cancer cells.
Segregation of studies by route of administration
Both positive and negative results were found in studies investigating virtually all routes of nicotine administration. For determining study adequacy criteria, those routes of administration that a user of nicotine delivery systems may be exposed to, were weighed higher than others. This is justified based on the relevance for extrapolating the respective data to humans. However, the findings from animal studies evaluated in this section do not indicate that the influence of nicotine on carcinogenesis is dependent on a particular route of nicotine administration.
Comparative evaluation by dose or dose rate
Both a modulating effect and a lack of an effect on carcinogenesis were observed upon sustained nicotine administration or bolus nicotine administration. It is interesting to note that a similar increase in tumor growth was reported in a study using i.v. (bolus) or oral (sustained) nicotine administration side by side (Li et al. Citation2015). Of the studies using bolus administration of nicotine, there is no separation of positive and negative studies by dose stratification. Also, there is no stratification observed for the modulation of either physical/chemical- or cancer cell-induced carcinogenesis. If average daily doses are calculated for the positive bolus studies, these average doses are much lower than those that can be achieved by sustained exposure to nicotine, such as via drinking water, presumably due to rapid nicotine turnover. It remains to be investigated whether and how transient high nicotine exposures obtained by bolus administration might be sufficient to trigger events that may eventually stimulate carcinogenesis. Because of the rapid nicotine metabolism in the animal species investigated, it can be assumed that upon bolus administration, all nicotine is metabolized between administrations, even if performed on a daily basis. It may be concluded that the trigger obtained by a bolus administration seems to survive long enough to maintain a certain level of stimulating activity, such as proliferative or anti-apoptotic, between dosing intervals. This phenomenon may also be related to the observation that cancer cells were triggered to faster tumor growth upon inoculation by prior in vitro incubation with nicotine (Ye et al. Citation2004; Yu et al. Citation2012; Yu & Chang Citation2013). Otherwise, a repeated transient trigger, even with intermediate phases without nicotine present, may also be sufficient. A third and even more hypothetical possibility would be that the effects would not be mediated by nicotine itself but rather by its metabolites which have longer half-lives.
Of the drinking water studies in mice, as the largest example of sustained nicotine dosing, about half of the (sub-)studies were considered positive by their authors (cf. ). Most studies using chemical/physical carcinogens or transgenes in combination with nicotine were negative, while the majority of those using cancer cells to initiate the carcinogenic effect were positive. The negative sub-studies with inoculated cancer cells had relatively low estimated daily doses of nicotine administered. In particular, in the study by Jarzynka et al. (Citation2006), the low dose at 20 mg/(kg × d) was negative, while the high dose at 40 mg/(kg × d) was claimed positive by the authors (on the basis of only 20% increase in tumor volume). Among the drinking water studies, several used measures of tumor volume to assess the effect of nicotine. An attempt to establish a relationship across studies between the daily doses of nicotine received from the drinking water and tumor volume failed. Without demonstrating a dose–response relationship, the presence of a possible threshold for nicotine’s modulating effect could not be established. Interestingly, with chemical cancer induction, only comparatively low nicotine doses were used, which may or may not explain the negative results in this category. All these interpretations for possible trends in the available data or the lack thereof should be viewed cautiously due to the uncertainties in reporting the form of nicotine used. Future studies with chemical, transgene and cellular induction of cancer should aim at establishing nicotine dose–response relationships and potentially determine a nicotine threshold, if any, for these modulating effects.
Figure 4. Cancer modulating activity as judged positive or negative by respective authors for mouse studies using nicotine administration via drinking water (stratified by average estimated daily nicotine dose). Positive modulating activity refers to stimulating carcinogenesis. Negative activity is the lack of a stimulating effect. Symbols characterize chemical/physical/transgene-based (circles) and cancer cell-based (triangles) studies with high (full symbols) and low (open symbols) adequacy scores.
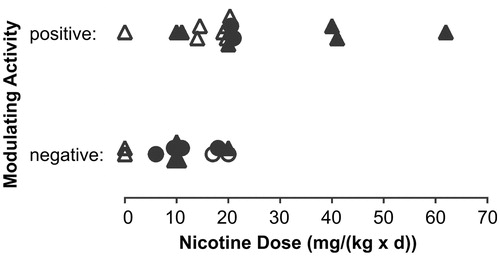
Mixed results were also observed with other sustained exposure methods. For example, using s.c. administration via osmotic minipumps at a dose of 8 mg/(kg × d), no stimulating effect on tumor growth was found in a study with mice (Pratesi et al. Citation1996). In contrast, a dermal patch delivering a nicotine dose of 25 mg/(kg × d) resulted in a 1.7-fold increase in tumor volume in mice (Davis et al. Citation2009).
Nicotine biomonitoring
If it is difficult to judge studies by their nominally reported doses, it may be more useful to stratify them by biomonitoring data that reflect tissue levels of nicotine or its metabolites. Systemic concentrations of nicotine and cotinine at a given per-kg dose are lower in mice than those in users of nicotine delivery systems () due to the rapid metabolism of both the parent compound and the primary metabolite. This rapid metabolism may not be reflected by their concentrations in urine, as these would integrate across the metabolic rate differences between species. The usefulness of all biomonitoring data depends on the knowledge of sampling periods relative to nicotine administration. However, sampling information is not available for the few studies that actually provided biomonitoring data.
Biomonitoring data are only available for seven recent studies, several of them conducted in sub-studies (). In the negative drinking water study performed using mice by Murphy and colleagues, plasma nicotine and cotinine levels were far below those found in users of nicotine delivery systems (), while urinary levels were similar (Murphy et al. Citation2011). In another drinking water study using a murine model, which administered a slightly higher estimated daily nicotine dose than in the previous study, a blood cotinine value similar to the average cotinine levels in users of nicotine delivery systems was reported (Maier et al. Citation2011). In this latter study, no modulation by nicotine of the carcinogenicity induced by NNK, transgenic mutant Kras, or inoculation with NNK-transformed cells was observed. A positive (stimulates tumor growth) study by Wong et al. (Citation2007) reported plasma cotinine values of 43 and 169 ng/ml after nicotine administration via the drinking water at estimated doses of 10 and 40 mg/(kg × d). The urinary cotinine levels in the positive (stimulates tumor growth) drinking water study by Hermann et al. (Citation2014) with mutant Kras mice were far below those of users of nicotine delivery systems (see “Oral administration” section).
Thus, based on the biomonitoring data available, a real stratification for negative and positive studies is still not possible. Biomonitoring data are critical for evaluating studies on the potential carcinogenesis of nicotine. Hopefully, future investigations will collect such data with consideration of species-specific toxicokinetics and transparent sampling methodology.
Applicability of the two-stage model of carcinogenesis
For the nicotine studies conducted in combination with chemical carcinogens, it might be informative to determine the amount of information available related to investigating the classic two-stage cancer model with initiation and promotion or investigating a co-carcinogenic effect (Moolgavkar & Knudson Citation1981; Cohen & Ellwein Citation1991). In fact, several published studies were indeed designed to unravel such relationships. Both cancer models (two-stage and co-carcinogenic) require specific timing for the dosing schedule of compounds under investigation. To facilitate the interpretation of data on this topic, the information whether any chemical compound given in combination with nicotine was given concomitantly (c) or sequentially (s) is available in the Supplementary material (Supplementary Table 2).
Murphy et al. (Citation2011) explicitly tested this question using nicotine administration in various combinations with NNK: nicotine was either given before, after or both before and after NNK administration. In all three cases, nicotine had no effect on NNK-induced tumorigenesis. In other studies, when nicotine was given after NNK, contrasting effects were observed: In two studies with nicotine administration via the drinking water, nicotine did not enhance tumorigenesis when NNK was given i.p. at 80–100 mg/kg either once or thrice (Maier et al. Citation2011; Nakada et al. Citation2012). In another study when NNK was i.p. injected twice at 100 mg/kg, an enhancing effect by nicotine was reported upon i.p. nicotine administration (Iskandar et al. Citation2013). Perhaps a critical difference between the studies is the nicotine dose rate.
Habs and Schmähl (Citation1984) investigated whether nicotine would affect tumor initiation or promotion by MNU by either administering nicotine during the week before MNU injection or for 3 months after MNU injection. Neither approach of nicotine treatment affected tumor incidence, size or histology.
Nicotine was also used in two studies with chemically-induced bladder cancer (Ito et al. Citation1984; Liu et al. Citation2011). The study by Liu and colleagues was considered positive after intragastric administration of nicotine, the other by Ito et al. was negative after administering nicotine via the food at a lower dose.
Skin painting studies have been the classic model to test for two-stage carcinogenesis and co-carcinogenesis (Rubin Citation2001; in particular for cigarette smoke, cf., Rubin Citation2002). In view of this, Bock et al. conducted a series of studies to determine the role of nicotine in tobacco condensate-related dermal carcinogenesis (Bock & Tso Citation1976; Bock Citation1980). Bock concluded that the results of his “experiments show that the enhancement of BaP-TPA carcinogenesis by nicotine is not due to a specific effect of the alkaloid on either initiation or promotion.” However these studies left him uncertain whether nicotine could be a co-carcinogen (Bock Citation1980).
It is obvious that insufficient information is available to classify nicotine in the context of the classic two-stage carcinogenesis model. This is not possible until we have a better understanding as to why some studies report tumorigenesis-stimulating effects from nicotine exposure while others report no effect. Cohen and Ellwein (Citation1991) suggested that the difficulty with this terminology (e.g., two-stage carcinogenesis model) is that it relies on specific experimental protocols for its definition. For instance, one feature of a promoter in the initiation-promotion model is reversibility, which has not been investigated for nicotine.
Impact of immune competence
Several immunocompromized mouse models were used in the evaluated studies, including nude, severe combined immunodeficient (scid) and non-obese diabetic (NOD)-scid mice. The development of immunodeficient mouse models (athymic nude mice) in the 1960s for in vivo investigation of human tumor growth and metastasis continued in the 1980s with the development of the scid mouse (RAG+/− or −/−) and into the 1990s with the NOD-scid mouse (Shultz et al. Citation2014). The adaptive immune system in these mouse models is severely compromised, but the innate immune system remains intact to varying degrees depending on the model (Shultz et al. Citation2014). Recent studies have demonstrated that the innate immune system, especially natural killer (NK) cells, plays a critical role in suppressing tumor growth in nude mice (Guerriero et al. Citation2011; Klier et al. Citation2011; Zhou et al. Citation2011).
Numerous published studies have clearly demonstrated that nicotine administration can inhibit both the innate and adaptive immune systems (Kalra et al. Citation2004; Han et al. Citation2014). In fact, nicotine administered for 6 weeks (s.c., minipump) to immunocompromized mice (RAG−/−) inhibited NK cell function and accelerated B16 tumor cell burden and metastasis in these animals (Hao et al. Citation2013). When dosed with sufficiently high nicotine doses using various routes of administration, 18 of 19 (sub-)studies with immunocompromized mice evaluated in this review were considered positive by the studies’ authors. It would be interesting to learn if these positive modulating effects of nicotine on tumor growth were dependent on immunosuppression (combination of animal model- and nicotine-derived).
Overall conclusion
There are numerous positive studies regarding the modulating activity of nicotine for the growth of inoculated cancer cells (1) using various kinds of cancer cells administered to various sites, (2) in various strains of mice, (3) employing various routes of nicotine administration and (4) conducted in various laboratories. Several of these studies investigated biological plausibility and suggest that angiogenesis might be involved as one mode of action by which nicotine may exert this growth-stimulating effect on cancer cells. Overall, there seems to be sufficient evidence to conclude that nicotine can stimulate carcinogenesis of inoculated cancer cells in laboratory animals, especially in immunocompromized mouse models.
The studies involving co-exposure with chemical carcinogens cannot be conclusively interpreted as showing either the presence or absence of a modulating effect of nicotine on the carcinogenesis of these compounds because of conflicting results and major qualitative or quantitative limitations. Thus, the existing evidence is inadequate to support the presence or absence of a carcinogenesis-modulating effect of nicotine when combined with chemical co-exposures.
For a risk assessment of nicotine, it would be helpful to understand why in some animal models, nicotine was indeed positive (stimulates carcinogenesis) while in others, negative (lack of stimulation). The current assessment did not produce any stratifying study design variable (e.g., dose) or biological principle that would allow a separation between positive and negative study outcomes with regard to a cancer-modulating effect of nicotine.
Studies with nicotine metabolites
Nicotine has a very short half-life in the laboratory rodent species commonly used for pharmacological and toxicological studies with nicotine (Matta et al. Citation2007) (cf., “Comparative toxicokinetics” section). Metabolites, such as cotinine and 3'-trans-hydroxycotinine, have a longer half-life than nicotine, though still shorter in rodents than in humans. In studies with mixed results with nicotine itself, e.g., some odd dose–response behavior in mouse skin painting studies (Bock Citation1980), it seemed possible that nicotine metabolites could either participate or interfere with the potential of nicotine to act as a carcinogen itself or to modulate the carcinogenesis of other materials. Several relevant studies were identified on these topics, and a summary of reported conclusions from all reviewed studies in this category is given in and . A more detailed description of high-adequacy score studies is provided in the section below. A description of low-adequacy score studies as well as a more detailed overview of each study evaluated in this category is provided in the Supplementary material (Supplementary Tables 4 and 3, respectively). At the end of the current section, both high and low-adequacy score studies are discussed in aggregate, and a conclusion on this part of the review is provided.
Table 7. Reported findings of relevant studies for evaluating the potential of nicotine metabolites to act as complete carcinogens.
Table 8. Reported findings of relevant studies for evaluating the potential cancer modulating activity of nicotine metabolites.
Oral administration
Truhaut et al. (Citation1964) exposed Wistar rats to cotinine via the drinking water at an estimated dose of 63 mg/(kg × d) (n ≥ 60 for cotinine group; n ≥ 15 for control). The rats showed signs of toxicity mainly during the first 6 months of the study. Between months 8 and 18, 12/15 rats that died in the cotinine group had malignant tumors, mainly lymphosarcomas in the digestive tract. Such lymphosarcomas were not observed in any of the 15 control rats euthanized after 18 months. Of the 45 rats euthanized after 18 months of cotinine exposure, several had benign lesions.
LaVoie et al. (Citation1985) wondered whether nicotine metabolites might be involved in smoking-related bladder cancer and, therefore, exposed Fischer 344 rats to urinary nicotine metabolites, i.e., cotinine, trans-NNO, and the mixture of cis- and trans-NNO, at toxic doses via the drinking water (n ≥ 33). No carcinogenic effect was seen after 18 months of exposure. To explore the potential promoting activity of the exposure to nicotine metabolites, parallel groups were initiated by dietary treatment with N-[4-(5-nitro-2-furyl)-2-thiazolyl]formamide (FANFT), a model carcinogen for the induction of bladder cancer in rats. In addition to bladder cancer, tumors at other sites also were induced by FANFT. The nicotine metabolites did not promote the FANFT-induced bladder cancer, rather if anything, NNO inhibited this effect. However, NNO induced an increase in forestomach cancer.
Freedlander et al. (Citation1956), in a relatively early study, investigated the potential of NNO to modulate the tumorigenicity of UV light in mice (n = 100), which under the conditions of this study developed ear and eye tumors. The NNO dose administered via the drinking water was increased from approximately 12–56 mg/(kg × d) over the course of the 7-month study. Tumor incidences were 42% in the control group and 41% in the group treated with NNO via the drinking water. The authors concluded that there is no additive or co-carcinogenic effect of NNO and nicotine (see above) with UV light. No treatment group was exposed to NNO alone.
Nakada et al. (Citation2012) applied two animal tumor models used for investigating the modulating potential of nicotine in parallel to cotinine (unclear group sizes). In one of these models, A/J mice were exposed to an initiating dose of NNK (80 mg/kg, i.p.), which was followed by cotinine administration via the drinking water. Two concentrations of cotinine were offered (with saccharine) resulting in estimated doses of 20 and 60 mg/(kg × d). After 4 months, adenocarcinomas had not yet developed, while the incidence of adenomas was significantly increased in the high cotinine dose group compared to the NNK-only group. The low dose of cotinine, as well as the same low dose of nicotine, did not lead to a significant effect on adenoma multiplicity.
Dermal application
Bock (Citation1980) investigated, in two sub-studies (n ≥ 45), whether the co-carcinogenic effect that he had seen with nicotine in mouse dermal carcinogenesis studies might be related to nicotine metabolites, which might have explained some of the odd dose/time–response behaviors observed with nicotine. Cotinine and NNO, respectively, were mixed with BaP and 12-O-tetradecanoylphorbol-13-acetate (TPA) in an experimental setup that showed co-carcinogenic activity with nicotine. With cotinine, there was a statistically significant increase in cancer incidence for the high dose tested. This increase was, however, not considered to be sufficiently high to suggest that metabolic formation of cotinine could account for the co-carcinogenic effect of nicotine in this assay system. With NNO, there was a statistically significant decrease in cancer incidence at both doses tested.
Low-adequacy score studies
This section includes relevant studies with low-adequacy scores using oral administration (Freedlander & French Citation1956; Schmähl & Osswald Citation1968; Nakada et al. Citation2012). Additionally, in view of the contrasting results seen in rats, Boyland (Citation1968) exposed mice to cotinine using various routes of administration. However, little information is available, as the data were only presented in a preliminary manner. Thus, this series of sub-studies was given a very low-adequacy score (for a detailed description, see Supplementary material, Supplementary Table 4). Boyland (Citation1968) concluded that cotinine might induce bladder cancer after implantation into the bladder within a pellet, but would be negative after dermal, s.c., and subscapular injections. Relevant concurrent controls were missing in these studies, which are necessary when using the bladder pellet implantation technique, which is known to induce bladder cancer by itself (Clayson Citation1974).
Discussion of aggregate evidence
High-adequacy score studies
In those studies investigating the effect of cotinine as a potential complete carcinogen, a positive effect observed with cotinine in the earliest study (Truhaut et al. Citation1964) could not be reproduced in a second study with the same strain of rats and the same concentration of cotinine in their drinking water (Schmähl & Osswald Citation1968). The authors of the second study even claimed to have administered a higher overall cumulative dose of cotinine, which is difficult to judge. The most recent study on this topic (LaVoie et al. Citation1985), with a higher adequacy score than the other studies in this section, did not reveal any increase in carcinogenicity by either cotinine or NNO.
Interestingly, the mouse skin painting study (Bock Citation1980) also suggested a trend towards higher tumor modulating activity of cotinine and a clear inhibition by NNO, as observed in the rat study in combination with FANFT (bladder carcinogen). The strongest positive cotinine study was in combination with NNK (Nakada et al. Citation2012). Mechanistically, the same anti-apoptotic effects were implicated in this study for cotinine that were also suggested for nicotine.
Low-adequacy score studies
The two oral low-adequacy score studies on cotinine in rats (Schmähl & Osswald Citation1968) and NNO in combination with urethane in mice (Freedlander & French Citation1956) were both negative. However, cotinine seemed to stimulate tumor growth in mice inoculated with Lewis carcinoma cells (Nakada et al. Citation2012). A complex study in mice using various routes of administration (Boyland Citation1968) was difficult to evaluate due to the lack of information.
Summary of the evidence
Studies with two nicotine metabolites were found, both testing their potential complete carcinogenicity or their potential to modulate carcinogenesis. The results from studies with cotinine were inconsistent, as were the study designs used therein. Biological plausibility, such as nAChR activation by cotinine was not assessed in the positive studies. There seemed to be a dose–response effect in one study, although only two dose levels were tested. For NNO, all available studies point to negative, if not protective effects, across various models. No biological plausibility was offered for this finding. It might be interesting to investigate the interaction of NNO with nAChRs.
Conclusion
Definitive carcinogenicity studies according to current standards are not available. The overall evidence is inadequate regarding a potential carcinogenic activity of cotinine, while there seems to be limited evidence for a lack of carcinogenicity of NNO.
Limitations of the current review
The current review has comprehensively and critically evaluated available evidence from epidemiological and animal carcinogenesis experiments. The review did not discuss the plethora of mechanistic information that is available on the potential of nicotine (and its metabolites), e.g., for epigenetic effects and in particular for modulating signal transduction pathways involved in various aspects of carcinogenesis, such as cell proliferation, inhibition of apoptosis, angiogenesis and invasion (Jensen et al. Citation2012; Russo et al. Citation2012; Chu et al. Citation2013; Improgo et al. Citation2013; Grando Citation2014; Niu & Lu Citation2014; Schaal & Chellappan Citation2014; Schuller Citation2014). There is also considerable published data on the interaction of nicotine and related compounds with nAChRs and its potential impact on carcinogenicity, but this information is not discussed in this review.
A surprisingly large number of animal studies were identified in this review with many different species, doses, routes and rates of administration, and co-exposures. Given the limited information provided on the dosing (e.g., the form of nicotine used in dosing) and the fast metabolism of nicotine in these species, it is extremely difficult to compare studies based on actual tissue levels of nicotine or its metabolites. Comparisons between studies are further complicated by the fact that biomonitoring was only rarely performed and methodological information was often missing.
The potential for nicotine to modulate the carcinogenic process induced by other materials may be relevant for humans in cases when carcinogenic processes are already present. However, all the studies reviewed that combine chemical carcinogens with nicotine exposure were conducted at very high doses of the initiating carcinogen, which may trigger other mechanisms of carcinogenesis not found at lower and more relevant doses. In addition, the cancer cells routinely used in xenograft models induced a very rapid and aggressive growth of tumors. It remains to be established to what degree such studies are relevant for human carcinogenesis and should serve as the basis for evaluating a modulating role of nicotine.
The potential of nicotine and its metabolites for endogenous nitrosation to yield carcinogenic N'-nitrosamines was not specifically addressed in this review. However, any potential risk stemming from such nitrosation should have been observed in the nicotine studies reviewed.
Knowledge gaps
Additional information on the long-term effect of nicotine exposure in humans is needed to determine the potential carcinogenic risk. This could be obtained in human studies by extending NRT use for longer periods than currently approved. Such studies should be conducted in former smokers as these users are presumably the most vulnerable population due to possible presence of already induced (pre-) cancerous processes. A second approach would be to follow up on studies with users of ENDS, again in former smokers and in ENDS users who had never smoked tobacco products.
Definitive laboratory animal studies are needed to assess the complete carcinogenic potential of nicotine. The design of an appropriate study or set of studies is challenging. For example, there are various routes of nicotine exposure for humans [inhalation (buccal/nasal), oral, dermal]. Doses and concentrations of nicotine and its metabolites in users of nicotine delivery systems need to be considered when designing and interpreting a carcinogenicity study of nicotine. Different allometric and biomonitoring parameters should be selected for a species comparison, of which systemic blood levels (with understood circadian changes) should be monitored.
For a better assessment of the modulating activity of nicotine, dose–response relationships would need to be established considering the same study design parameters as discussed above. Very recently, a nicotine threshold effect was considered possible based on in vitro and in vivo studies (Schuller Citation2014). Because the suggested mechanisms for this modulating effect are mainly non-genotoxic, a threshold of exposure to induce such effects may exist and requires further characterization for its potential application to users of nicotine delivery systems considering both toxico-kinetic and –dynamic species differences.
Nicotine exposure in an immunocompromized mouse model (nude, athymic) resulted in a positive modulating effect of carcinogenesis (tumor xenografts) in over 90% of the studies. This observation deserves further investigation, in light of nicotine’s ability to suppress the innate and adaptive immune response. It would be interesting to learn if there is an association between nicotine’s immunosuppressant activity and the observed cancer modulating effect in immunodeficient mice. This hypothesis could certainly serve as an alternate explanation for understanding the consistent cancer modulating effects of nicotine observed in this model system.
Overall conclusions
What are the potential carcinogenic effects of nicotine per se at levels found in users of nicotine delivery products? At present, public health statements on the subject indicate that “nicotine is not generally considered to be a carcinogen” (International Agency for Research on Cancer Citation2012). The latest report from the US Surgeon General concluded that “the evidence is inadequate to infer the presence or absence of a causal relationship between exposure to nicotine and risk for cancer” (US Department of Health and Human Services Citation2014). The purpose of the present review is to evaluate the strength of published scientific evidence, in both human and animal studies, for nicotine per se to act as a complete carcinogen or as a modulator of carcinogenesis. The findings from this comprehensive evaluation are summarized in .
Table 9. Strength of evidence classification for nicotine to act as a carcinogen or carcinogenesis modulator in human and animal studies.
For human studies, there appears to be inadequate evidence for an association between nicotine exposure and the presence of or lack of a carcinogenic effect. A limited number of studies are available on the subject with only one epidemiological study identified that investigated the cancer risk from using NRTs. The study provided no evidence for an effect of NRT use on cancers of the lung, the gastrointestinal tract, or overall. However, inadequate evidence was concluded due to the limited follow up time (12 years) for a chronic disease such as cancer and due to the low dose of nicotine (NRT) routinely used by participants of this study.
In animal studies, suggestive but still limited evidence suggests an association between long-term nicotine exposure and a lack of a complete carcinogenic effect. The two rodent studies with the highest adequacy scores reported the absence of a carcinogenic effect with lifetime exposure to nicotine (Toth Citation1982; Waldum et al. Citation1996). However, conclusive studies using current bioassay guidelines are missing.
In approximately 70 animal studies, nicotine was investigated for its ability to modulate (stimulate) the carcinogenic process induced by administration of chemical/physical carcinogens, inoculation with cancer cells, or in transgenic models. In 35 studies using chemical/physical carcinogens and transgenic models, there appears to be inadequate evidence for an association between nicotine exposure and the presence of or lack of a modulating (stimulate) effect on carcinogenesis. Evidence was deemed inadequate due to the large number of conflicting studies (approximately 50%). In contrast, a majority of studies (69%) provide sufficient evidence to conclude that nicotine can stimulate carcinogenesis of inoculated cancer cells in animals, especially in immunocompromized mouse models. Inconsistent findings on nicotine’s ability to modulate carcinogenesis may result from the use of numerous animal models and a wide variety of dosing regimens for nicotine administration. Comparisons between studies, however, are complicated by the fact that nicotine biomonitoring was rarely performed and methodological information was often missing.
Overall, taking both the human and animal studies into consideration, there appears to be inadequate evidence to conclude that nicotine per se does or does not cause or modulate carcinogenesis in humans. This conclusion agrees with the recent US Surgeon General’s 2014 report on the health consequences of nicotine exposure (US Department of Health and Human Services Citation2014).
Haussmann_Fariss_cancer_nicotine_review-Revised-SUPPLEMENTARY_MATERIAL.pdf
Download PDF (744.2 KB)Acknowledgements
The authors acknowledge the editorial assistance of Eileen Y. Ivasauskas of Accuwrit Inc. and the assistance of Alan I. Goldsmith of the Research Information Group, Altria Client Services LLC (ALCS), who performed the literature search in Medline and Embase databases. The authors thank Mohamadi Sarkar and Scott Appleton of ALCS for their scientific review of this paper. The authors also gratefully acknowledge the valuable comments offered by the editor and those offered by four external reviewers selected by the Editor and anonymous to the authors. These comments were very helpful in revising the manuscript.
Declaration of interest
The employment affiliation of the authors is shown on the cover page. M. W. F. is a current employee of ALCS and H. J. H. is a former employee of Philip Morris International. H. J. H. served as a paid consultant to ALCS for preparation of this review. H. J. H. is an independent toxicology consultant for commercial firms including companies that manufacture and sell tobacco products. ALCS is an affiliate of Philip Morris USA Inc., U.S. Smokeless Tobacco Company LLC and NuMark LLC which are manufacturers and marketers of various tobacco products in the United States including cigarettes, smokeless tobacco and e-vapor products, respectively. The authors have not testified in litigation or represented ALCS or affiliates in meetings with FDA regarding the topic of this review. The ALCS legal department reviewed this paper solely in connection with intellectual property protection. The opinions and conclusions of the authors are their own, and do not necessarily reflect the position of ALCS or its affiliates.
Related Research Data
References
- Al-Wadei HAN, Al-Wadei MH, Ullah MF, Schuller HM. 2012. Gamma-amino butyric acid inhibits the nicotine-imposed stimulatory challenge in xenograft models of non-small cell lung carcinoma. Curr Cancer Drug Targets. 12:97–106.
- Al-Wadei HAN, Plummer HK III, Schuller HM. 2009. Nicotine stimulates pancreatic cancer xenografts by systemic increase in stress neurotransmitters and suppression of the inhibitory neurotransmitter γ-aminobutyric acid. Carcinogenesis. 30:506–511.
- Alexander DJ, Collins CJ, Coombs DW, Gilkison IS, Hardy CJ, Healey G, Karantabias G, Johnson N, Karlsson A, Kilgour JD, et al. 2008. Association of Inhalation Toxicologists (AIT) working party recommendation for standard delivered dose calculation and expression in non-clinical aerosol inhalation toxicology studies with pharmaceuticals. Inhal Toxicol. 20:1179–1189.
- AlSharari SD, Akbarali HI, Abdullah RA, Shahab O, Auttachoat W, Ferreira GA, White KL, Lichtman AH, Cabral GA, Damaj MI. 2013. Novel insights on the effect of nicotine in a murine colitis model. J Pharmacol Exp Ther. 344:207–217.
- Arany I, Grifoni S, Clark JS, Csongradi E, Maric C, Juncos LA. 2011. Chronic nicotine exposure exacerbates acute renal ischemic injury. Am J Physiol Renal Physiol. 301:F125–F133.
- Argentin G, Cicchetti R. 2004 Genotoxic and antiapoptotic effect of nicotine on human gingival fibroblasts. Toxicol Sci. 79:75–81.
- Banerjee J, Al-Wadei HA, Al-Wadei MH, Dagnon K, Schuller HM. 2014. Differential modulation of nicotine-induced gemcitabine resistance by GABA receptor agonists in pancreatic cancer cell xenografts and in vitro. BMC Cancer. 14:725.
- Banerjee J, Al-Wadei HAN, Schuller HM. 2013. Chronic nicotine inhibits the therapeutic effects of gemcitabine on pancreatic cancer in vitro and in mouse xenografts. Eur J Cancer. 49:1152–1158.
- Bavarva JH, Tae H, McIver L, Garner HR. 2014. Nicotine and oxidative stress induced exomic variations are concordant and overrepresented in cancer-associated genes. Oncotarget. 5:4788–4798.
- Benowitz NL. 2011. Smokeless tobacco as a nicotine delivery device: harm or harm reduction? Clin Pharmacol Ther. 90:491–493.
- Benowitz NL. 2014. Emerging nicotine delivery products. Implications for public health. Ann Am Thorac Soc. 11:231–235.
- Benowitz NL, Hukkanen J, Jacob P III. 2009. Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol. 192:29–60.
- Berger MR, Zeller WJ. 1988. Interaction of nicotine with anticancer treatment. Klin Wochenschr. 66: 127–133.
- Bersch VP, Osvaldt AB, Edelweiss MI, Schumacher RC, Wendt LR, Abreu LP, Blom CB, Abreu GP, Costa L, Piccinini P, et al. 2009. Effect of nicotine and cigarette smoke on an experimental model of intraepithelial lesions and pancreatic adenocarcinoma induced by 7,12-dimethylbenzanthracene in mice. Pancreas. 38:65–70.
- Bhagat B, Rana MW. 1971. Antitumor activity of antiserum nerve growth factor (anti-NGF). Proc Soc Exp Biol Med. 138:983–984.
- Bock FG. 1980. Cocarcinogenic properties of nicotine. In: Gori GB, Bock FG, editors. A safe cigarette? Cold Spring Harbor (NY): Cold Spring Harbor Laboratory. p. 129–136.
- Bock FG, Tso TC. 1976. Chemical and biological identification of tumorigenic components of tobacco. In: Wynder EL, Hoffmann D, Gori GB, editors. Proceedings of the third world conference on smoking and health. Washington (DC): Government Printing Office. p. 161–174.
- Boyland E. 1968. The possible carcinogenic action of alkaloids of tobacco and betel nut. Planta Med. 1968:13–23.
- Brown KC, Perry HE, Lau JK, Jones DV, Pulliam JF, Thornhill BA, Crabtree CM, Luo H, Chen YC, Dasgupta P. 2013. Nicotine induces the up-regulation of the α7-nicotinic receptor (α7-nAChR) in human squamous cell lung cancer cells via the Sp1/GATA protein pathway. J Biol Chem. 288:33049–33059.
- Cardinale A, Nastrucci C, Cesario A, Russo P. 2012. Nicotine: specific role in angiogenesis, proliferation and apoptosis. Crit Rev Toxicol. 42:68–89.
- Chen YP, Johnson GK, Squier CA. 1994. Effects of nicotine and tobacco-specific nitrosamines on hamster cheek pouch and gastric mucosa. J Oral Pathol Med. 23:251–255.
- Chen YP, Squier CA. 1990. Effect of nicotine on 7,12-dimethylbenz[a]anthracene carcinogenesis in hamster cheek pouch. J Natl Cancer Inst. 82:861–864.
- Cheng SY, Glazkova D, Serova Sabban EL. 2005. Effect of prolonged nicotine infusion on response of rat catecholamine biosynthetic enzymes to restraint and cold stress. Pharmacol Biochem Behav. 82:559–568.
- Chu KM, Cho CH, Shin VY. 2013. Nicotine and gastrointestinal disorders: its role in ulceration and cancer development. Curr Pharm Des. 19:5–10.
- Clayson DB. 1974. Editorial: bladder carcinogenesis in rats and mice: possibility of artifacts. J Natl Cancer Inst. 52:1685–1689.
- Cohen SM, Ellwein LB. 1991. Genetic errors, cell proliferation, and carcinogenesis. Cancer Res. 51:6493–6505.
- Davis R, Rizwani W, Banerjee S, Kovacs M, Haura E, Coppola D, Chellappan S. 2009. Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS One. 4:e7524.
- Demirhan O, Demir C, Tunc E, nandiklioglu N, Sutcu E, Sadikoglu N, Ozcan B. 2011. The genotoxic effect of nicotine on chromosomes of human fetal cells: the first report described as an important study. Inhal Toxicol. 23:829–834.
- Dinse GE, Peddada SD, Harris SF, Elmore SA. 2010. Comparison of NTP historical control tumor incidence rates in female Harlan Sprague Dawley and Fischer 344/N Rats. Toxicol Pathol. 38:765–775.
- Doolittle DJ, Winegar R, Lee CK, Caldwell WS, Hayes AW, de Bethizy JD. 1995. The genotoxic potential of nicotine and its major metabolites. Mutat Res. 344:95–102.
- Eränkö O, Hopsu V, Räisäinen L. 1959a. Changes induced by prolonged administration of nicotine in the distributions of cholinesterases and acid phosphatase in the adrenal medulla of the rat. J Neurochem. 4:332–337.
- Eränkö O, Hopsu V, Räisäinen L. 1959b. Effect of prolonged administration nicotine on the medullary volume and the distribution of noradrenaline in the adrenals of the rat, the mouse and the guinea pig. Endocrinology. 65:293–297.
- Feng S, Kapur S, Sarkar M, Muhammad R, Mendes P, Newland K, Roethig HJ. 2007. Respiratory retention of nicotine and urinary excretion of nicotine and its five major metabolites in adult male smokers. Toxicol Lett. 173:101–106.
- Flora JW, Meruva N, Huang CB, Wilkinson CT, Ballentine R, Smith DC, Werley MS, McKinney WJ. 2016. Characterization of potential impurities and degradation products in electronic cigarette formulations and aerosols. Regul Toxicol Pharmacol. 74:1–11.
- Freedlander BL, French FA. 1956. Absence of co-carcinogenic action of oxidation products of nicotine in initiation of pulmonary adenomas in mice with urethane. Proc Am Assoc Cancer Res. 2:109.
- Freedlander BL, French FA, Furst A. 1956. The nonadditive effect of nicotine and nicotine N-oxide on the carcinogenicity of ultraviolet light. Proc Am Assoc Cancer Res. 2:109.
- Fucito LM, Bars MP, Forray A, Rojewski AM, Shiffman S, Selby P, West R, Foulds J, Toll BA. Writing Committee for the SRNT Policy and Treatment Networks. 2014. Addressing the evidence for FDA nicotine replacement therapy label changes: a policy statement of the Association for the Treatment of Tobacco Use and Dependence and the Society for Research on Nicotine and Tobacco. Nicotine Tob Res. 16:909–914.
- Galitovskiy V, Chernyavsky AI, Edwards RA, Grando SA. 2012. Muscle sarcomas and alopecia in A/J mice chronically treated with nicotine. Life Sci. 91:1109–1112.
- Gaworski CL, Lemus-Olalde R, Carmines EL. 2008. Toxicological evaluation of potassium sorbate added to cigarette tobacco. Food Chem Toxicol. 46:339–351.
- Ginzkey C, Steussloff G, Koehler C, Burghartz M, Scherzed A, Hackenberg S, Hagen R, Kleinsasser NH. 2014a. Nicotine derived genotoxic effects in human primary parotid gland cells as assessed in vitro by comet assay, cytokinesis-block micronucleus test and chromosome aberrations test. Toxicol in Vitro. 28:838–846.
- Ginzkey C, Steussloff G, Koehler C, Hackenberg S, Richter E, Hagen R, Kleinsasser NH. 2014b. Nicotine causes genotoxic damage but is not metabolized during long-term exposure of human nasal miniorgan cultures. Toxicol Lett. 229:303–310.
- Ginzkey C, Stueber T, Friehs G, Koehler C, Hackenberg S, Richter E, Hagen R, Kleinsasser NH. 2012. Analysis of nicotine-induced DNA damage in cells of the human respiratory tract. Toxicol Lett. 208:23–29.
- Ginzkey C, Friehs G, Koehler C, Hackenberg S, Hagen R, Kleinsasser NH. 2013. Assessment of nicotine-induced DNA damage in a genotoxicological test battery. Mutat Res Genet Toxicol Environ Mutagen. 751:34–39.
- Goodman JE, Prueitt RL, Sax SN, Bailey LA, Rhomberg LR. 2013. Evaluation of the causal framework used for setting national ambient air quality standards. Crit Rev Toxicol. 43:829–849.
- Goodman JE, Prueitt RL, Sax SN, Lynch HN, Zu K, Lemay JC, King JM, Venditti FJ. 2014. Weight-of-evidence evaluation of short-term ozone exposure and cardiovascular effects. Crit Rev Toxicol. 44:725–790.
- Grabus SD, Martin BR, Batman AM, Tyndale RF, Sellers E, Damaj MI. 2005. Nicotine physical dependence and tolerance in the mouse following chronic oral administration. Psychopharmacology (Berlin). 178:183–192.
- Grando SA. 2014. Connections of nicotine to cancer. Nat Rev Cancer. 14:419–429.
- Greim H, Hartwig A, Reuter U, Richter-Reichhelm HB, Thielmann HW. 2009. Chemically induced pheochromocytomas in rats: mechanisms and relevance for human risk assessment. Crit Rev Toxicol. 39:695–718.
- Guerriero JL, Ditsworth D, Catanzaro JM, Sabino G, Furie MB, Kew RR, Crawford HC, Zong W. 2011. DNA alkylating therapy induces tumor regression through an HMGB1-mediated activation of innate immunity. J Immunol. 186:3517–3526.
- Gurkalo VK, Volfson NI. 1982. Nicotine influence upon the development of experimental stomach tumors. Arch Geschwulstforsch. 52:259–265.
- Habs M, Schmähl D. 1976. Influence of five different postnatal lifelong treatments on the transplacental carcinogenicity of ethylnitrosourea in Sprague–Dawley rats. Cancer Lett. 2:93–100.
- Habs M, Schmähl D. 1984. Influence of nicotine on N-nitrosomethylurea-induced mammary tumors in rats. Klin Wochenschr. 62: 105–108.
- Han Y, Ling MT, Mao H, Zheng J, Liu M, Lam KT, Liu Y, Tu W, Lau YL. 2014. Influenza virus-induced lung inflammation was modulated by cigarette smoke exposure in mice. PLoS One. 9:e86166.
- Hao J, Shi FD, Abdelwahab M, Shi SX, Simard A, Whiteaker P, Lukas R, Zhou Q. 2013. Nicotinic receptorβ2 determines NK cell-dependent metastasis in a murine model of metastatic lung cancer. PLoS One. 8:e57495.
- Haussmann H-J, Gerstenberg B, Göcke W, Kuhl P, Schepers G, Stabbert R, Stinn W, Teredesai A, Tewes FJ. 1998. 12-Month inhalation study on room-aged cigarette sidestream smoke in rats. Inhal Toxicol. 10:663–697.
- Hayashi S, Hamada T, Zaidi SF, Oshiro M, Lee J, Yamamoto T, Ishii Y, Sasahara M, Kadowaki M. 2014. Nicotine suppresses acute colitis and colonic tumorigenesis associated with chronic colitis in mice. Am J Physiol Gastrointest Liver Physiol. 307:G968–G978.
- Hecht SS. 2012a. Research opportunities related to establishing standards for tobacco products under the Family Smoking Prevention and Tobacco Control Act. Nicotine Tob Res. 14:18–28.
- Hecht SS. 2012b. Lung carcinogenesis by tobacco smoke. Int J Cancer. 131:2724–2732.
- Hecht SS, Rivenson A, Braley J, Di Bello J, Adams JD, Hoffmann D. 1986. Induction of oral cavity tumors in F344 rats by tobacco-specific nitrosamines and snuff. Cancer Res. 46:4162–4166.
- Heeschen C, Jang JJ, Weis M, Pathak A, Kaji S, Hu RS, Tsao PS, Johnson FL, Cooke JP. 2001. Nicotine stimulates angiogenesis and promotes tumor growth and atherosclerosis. Nat Med. 7:833–839.
- Hermann PC, Sancho P, Cañamero M, Martinelli P, Madriles F, Michl P, Gress T, de Pascual R, Gandia L, Guerra C, et al. 2014. Nicotine promotes initiation and progression of KRAS-induced pancreatic cancer via Gata6-dependent dedifferentiation of acinar cells in mice. Gastroenterology. 147:1119–1133.
- Hill AB. 1965. The environment and disease: association or causation? Proc R Soc Med. 58:295–300.
- Hoffmann D, Adams JD, Lisk D, Fisenne I, Brunnemann KD. 1987. Toxic and carcinogenic agents in dry and moist snuff. J Natl Cancer Inst. 79:1281–1286.
- Hueper WC. 1943. Experimental studies in cardiovascular pathology. VII. Chronic nicotine poisoning in rats and dogs. Arch Pathol. 35:846–856.
- Hukkanen J, Jacob III P, Benowitz NL. 2005.Metabolism and disposition kinetics of nicotine. Pharmacol Rev. 57:79–115.
- Improgo MR, Soll LG, Tapper AR, Gardner PD. 2013. Nicotinic acetylcholine receptors mediate lung cancer growth. Front Physiol. 4:1–6. doi: 10.3389/fphys.2013.00251.
- Improgo M, Tapper AR, Gardner PD. 2011. Nicotinic acetylcholine receptor-mediated mechanisms in lung cancer. Biochem Pharmacol. 82:1015–1021.
- International Agency for Research on Cancer. 2007. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans, vol. 89, Smokeless Tobacco and Some Tobacco-specific N-Nitrosamines. Lyon, France: World Health Organization.
- International Agency for Research on Cancer. 2012. A Review of Human Carcinogens, Part E: Personal Habits and Indoor Combustions. [cited 2015 Mar 26]. Available from: http://monographs.iarc.fr/ENG/Monographs/vol100E/mono100E.pdf/.
- Iskandar AR, Liu C, Smith DE, Hu KQ, Choi SW, Ausman LM, Wang XD. 2013. β-Cryptoxanthin restores nicotine-reduced lung SIRT1 to normal levels and inhibits nicotine-promoted lung tumorigenesis and emphysema in A/J mice. Cancer Prev Res(Phila). 6:309–320.
- Ito N, Fukushima S, Shirai T, Hagiwara A, Imaida K. 1984. Drugs, food additives and natural products as promoters in rat urinary bladder carcinogenesis. IARC Sci Publ. 56:399–407.
- Jarvis M, Tunstall-Pedoe H, Feyerabend C, Vesey C, Salloojee Y. 1984. Biochemical markers of smoke absorption and self reported exposure to passive smoking. J Epidemiol Community Health. 38:335–339.
- Jarzynka MJ, Guo P, Bar-Joseph I, Hu B, Cheng SY. 2006. Estradiol and nicotine exposure enhances A549 bronchioloalveolar carcinoma xenograft growth in mice through the stimulation of angiogenesis. Int J Oncol. 28:337–344.
- Jensen K, Afroze S, Munshi MK, Guerrier M, Glaser SS. 2012. Mechanisms for nicotine in the development and progression of gastrointestinal cancers. Transl Gastrointest Cancer. 1:81–87.
- Kalra R, Singh SP, Pena-Philippides JC, Langley RJ, Razani-Boroujerdi S, Sopori ML. 2004. Immunosuppressive and anti-inflammatory effects of nicotine administered by patch in an animal model. Clin Diagn Lab Immunol. 11:563–568.
- Khalil AA, Jameson MJ, Broaddus WC, Lin PS, Chung TD. 2013. Nicotine enhances proliferation, migration, and radioresistance of human malignant glioma cells through EGFR activation. Brain Tumor Pathol. 30:73–83.
- Klaunig JE. 2013. Chapter 8: chemical carcinogenesis. In: Klaassen CD, editor. Casarett and Doull's toxicology: the basic science of poison. New York: McGraw-Hill.
- Kleinsasser NH, Sassen AW, Semmler MP, Harreus UA, Licht AK, Richter E. 2005. The tobacco alkaloid nicotine demonstrates genotoxicity in human tonsillar tissue and lymphocytes. Toxicol Sci. 86:309–317.
- Klier U, Maletzki C, Gottmann N, Kreikemeyer B, Linnebacher M. 2011. Avitalized bacteria mediate tumor growth control via activation of innate immunity. Cell Immunol. 269:120–127.
- Klimisch HJ, Andreae M, Tillmann U. 1997. A systematic approach for evaluating the quality of experimental toxicological and ecotoxicological data. Regul Toxicol Pharmacol. 25:1–5.
- Kosdoba AS. 1930. Zur Frage der experimentellen Pathologie der Nebennieren bei intravenöser Nicotineinverleibung [On the question of experimental pathology of the adrenals upon intravenous nicotine application]. Arch Klin Chir. 156:550–566.
- Kyerematen GA, Taylor LH, de Bethizy JD, Vesell ES. 1988. Pharmacokinetics of nicotine and 12 metabolites in the rat. Application of a new radiometric high performance liquid chromatography assay. Drug Metab Dispos. 16:125–129.
- Lam DC, Luo SY, Fu KH, Lui MM, Chan KH, Wistuba II, Gao B, Tsao SW, Ip MS, Minna JD. 2016. Nicotinic acetylcholine receptor expression in human airway correlates with lung function. Am J Physiol Lung Cell Mol Physiol. 310:L232–L239.
- Landau JM, Wang ZY, Yang GY, Ding W, Yang CS. 1998. Inhibition of spontaneous formation of lung tumors and rhabdomyosarcomas in A/J mice by black and green tea. Carcinogenesis. 19:501–507.
- Larson PS, Haag HB, Silvette H. 1961. Tobacco – experimental and clinical studies, a comprehensive account of the world literature. Baltimore (MD): The Williams & Wilkins Company.
- La Voie EJ, Shigematsu A, Rivenson A, Mu B, Hoffmann D. 1985. Evaluation of the effects of cotinine and nicotine-N'-oxides on the development of tumors in rats initiated with N-[4-(5-nitro-2-furyl)-2-thiazolyl]formamide. J Natl Cancer Inst. 75:1075–1081.
- Lee CH, Huang CS, Chen CS, Tu SH, Wang YJ, Chang YJ, Tam KW, Wei PL, Cheng TC, Chu JS, et al. 2010. Overexpression and activation of the alpha9-nicotinic receptor during tumorigenesis in human breast epithelial cells. J Natl Cancer Inst. 102:1322–1335.
- Lee CH, Wu CH, Ho YS. 2011. From smoking to cancers: novel targets to neuronal nicotinic acetylcholine receptors. J Oncol. 2011:693424.
- Lee J, Cooke JP. 2012. Nicotine and pathological angiogenesis. Life Sci. 91:1058–1064.
- Levy LS, Martin PA. 1989. Toxicology of nicotine – its role in the aetiology of cancer due to cigarette smoking and cardiovascular disease. In: Wald N, Froggatt P, editors. Nicotine, smoking, and the low tar programme. Oxford: Oxford University Press. p. 11–23.
- Li H, Wang S, Takayama K, Harada T, Okamoto I, Iwama E, Fujii A, Ota K, Hidaka N, Kawano Y, et al. 2015. Nicotine induces resistance to erlotinib via cross-talk between α 1 nAChR and EGFR in the non-small cell lung cancer xenograft model. Lung Cancer. 88:1–8.
- Liu D, Pan F, Li B, Han X, Li W, Shi Y, Pang Z, Zhang Q. 2011. Intervention of nicotine on MNU-induced bladder cancer in rats. J Huazhong Univ Sci Technol Med Sci. 31:103–106.
- Liu W, Yi DD, Guo JL, Xiang ZX, Deng LF, He L. 2015. Nuciferine, extracted from Nelumbo nucifera Gaertn, inhibits tumor-promoting effect of nicotine involving Wnt/β-catenin signaling in non-small cell lung cancer. J Ethnopharmacol. 165:83–93.
- Maier CR, Hollander MC, Hobbs EA, Dogan I, Linnoila RI, Dennis PA. 2011. Nicotine does not enhance tumorigenesis in mutant K-ras-driven mouse models of lung cancer. Cancer Prev Res(Phila). 4:1743–1751.
- Manenti G, Dragani TA. 2005. Pas1 haplotype-dependent genetic predisposition to lung tumorigenesis in rodents: a meta-analysis. Carcinogenesis. 26:875–882.
- Marks MJ, Stitzel JA, Collins AC. 1985. Time course study of the effects of chronic nicotine infusion on drug response and brain receptors. J Pharmacol Exp Ther. 235:619–628.
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, et al. 2007. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berlin). 190:269–319.
- Mauderly JL, Bechtold WE, Bond JA, Brooks AL, Chen BT, Cuddihy RG, Harkema JR, Henderson RF, Johnson NF, Rithidech K. 1989. Comparison of 3 methods of exposing rats to cigarette smoke. Exp Pathol. 37:194–197.
- Molfino A, Logorelli F, Citro G, Bertini G, Ramaccini C, Bollea MR, Rossi FF, Laviano A. 2011. Stimulation of the nicotine antiinflammatory pathway improves food intake and body composition in tumor-bearing rats. Nutr Cancer. 63:295–299.
- Moolgavkar SH, Knudson AGJ. 1981. Mutation and cancer: a model for human carcinogenesis. J Natl Cancer Inst. 66:1037–1052.
- Murphy SE, von Weymarn LB, Schutten MM, Kassie F, Modiano JF. 2011. Chronic nicotine consumption does not influence 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis. Cancer Prev Res (Phila). 4:1752–1760.
- Murray RP, Connett JE, Zapawa LM. 2009. Does nicotine replacement therapy cause cancer? Evidence from the Lung Health Study. Nicotine Tob Res. 11:1076–1082.
- Nakada T, Kiyotani K, Iwano S, Uno T, Yokohira M, Yamakawa K, Fujieda M, Saito T, Yamazaki H, Imaida K, et al. 2012. Lung tumorigenesis promoted by anti-apoptotic effects of cotinine, a nicotine metabolite through activation of PI3K/Akt pathway. J Toxicol Sci. 37:555–563.
- Natori T, Sata M, Washida M, Hirata Y, Nagai R, Makuuchi M. 2003. Nicotine enhances neovascularization and promotes tumor growth. Mol Cells. 16:143–146.
- Nishikawa A, Furukawa F, Imazawa T, Yoshimura H, Mitsumori K, Takahashi M. 1992. Effects of caffeine, nicotine, ethanol and sodium selenite on pancreatic carcinogenesis in hamsters after initiation with N-nitrosobis(2-oxopropyl)amine. Carcinogenesis. 13:1379–1382.
- Niu XM, Lu S. 2014. Acetylcholine receptor pathway in lung cancer: new twists to an old story. World J Clin Oncol. 5:667–676.
- Organisation for Economic Co-operation and Development. 2009. OECD guideline for the testing of chemicals. 451 Carcinogenicity studies. Paris: OECD Publishing.
- Paleari L, Catassi A, Ciarlo M, Cavalieri Z, Bruzzo C, Servent D, Cesario A, Chessa L, Cilli M, Piccardi F, et al. 2008. Role of alpha7-nicotinic acetylcholine receptor in human non-small cell lung cancer proliferation. Cell Prolif. 41:936–959.
- Petersen DR, Norris KJ, Thompson JA. 1984. A comparative study of the disposition of nicotine and its metabolites in three inbred strains of mice. Drug Metab Dispos. 12:725–731.
- Pietilä K, Laakso I, Ahtee L. 1995. Chronic oral nicotine administration affects the circadian rhythm of dopamine and 5-hydroxytryptamine metabolism in the striata of mice. Naunyn Schmiedebergs Arch Pharmacol. 353:110–115.
- Pillai S, Trevino J, Rawal B, Singh S, Kovacs M, Li X, Schell M, Haura E, Bepler G, Chellappan S. 2015. β-Arrestin-1 mediates nicotine-induced metastasis through E2F1 target genes that modulate epithelial–mesenchymal transition. Cancer Res. 75:1009–1020.
- Pratesi G, Cervi S, Balsari A, Bondiolotti G, Vicentini LM. 1996. Effect of serotonin and nicotine on the growth of a human small cell lung cancer xenograft. Anticancer Res. 16:3615–3619.
- Prokopczyk G, Adams JD, La Voie EJ, Hoffmann D. 1987. Effect of snuff and nicotine on DNA methylation by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Carcinogenesis. 8:1395–1397.
- Prueitt RL, Lynch HN, Zu K, Sax SN, Venditti FJ, Goodman JE. 2014. Weight-of-evidence evaluation of long-term ozone exposure and cardiovascular effects. Crit Rev Toxicol. 44:791–822.
- Rana MW, Bhagat B. 1970. Chemical carcinogenesis in immunosympathectomized mice. Anat Rec. 166:365.
- Renda A, Nashmi R. 2014. Chronic nicotine pretreatment is sufficient to upregulate α4 nicotinic receptors and increase oral nicotine self-administration in mice. BMC Neurosci. 15:89.
- Rhomberg LR, Bailey LA, Goodman JE, Hamade AK, Mayfield D. 2011. Is exposure to formaldehyde in air causally associated with leukemia? – a hypothesis-based weight-of-evidence analysis. Crit Rev Toxicol. 41:555–621.
- Rhomberg LR, Goodman JE, Bailey LA, Prueitt RL, Beck NB, Bevan C, Honeycutt M, Kaminski NE, Paoli G, Pottenger LH, et al. 2013. A survey of frameworks for best practices in weight-of-evidence analyses. Crit Rev Toxicol. 43:753–784.
- Rubin H. 2001. Synergistic mechanisms in carcinogenesis by polycyclic aromatic hydrocarbons and by tobacco smoke: a bio-historical perspective with updates. Carcinogenesis. 22:1903–1930.
- Rubin H. 2002. Selective clonal expansion and microenvironmental permissiveness in tobacco carcinogenesis. Oncogene. 21:7392–7411.
- Russo P, Cardinale A, Margaritora S, Cesario A. 2012. Nicotinic receptor and tobacco-related cancer. Life Sci. 91:1087–1092.
- Sanner T, Grimsrud TK. 2015. Nicotine: carcinogenicity and effects on response to cancer treatment-a review. Front Oncol. 5:196.
- Schaal C, Chellappan SP. 2014. Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol Cancer Res. 12:14–23.
- Schepers G, Rustemeier K, Walk RA, Hackenberg U. 1993. Metabolism of S-nicotine in noninduced and aroclor-induced rats. Eur J Drug Metab Pharmacokinet. 18:187–197.
- Schievelbein H. 1962. Nikotin, Rauchen und Organismus [Nicotine, smoking and organism]. Beitr Tabakforsch Int. 1:199–274.
- Schmähl D, Habs M. 1976. Life-span investigations for carcinogenicity of some immune-stimulating, immunodepressive and neurotropic substances in Sprague–Dawley rats. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol. 86:77–84.
- Schmähl D, Osswald H. 1968. Fehlen einer carcinogenen Wirkung von Cotinin bei Ratten [Lack of a carcinogenic effect of cotinine in rats]. Zeitschrift Für Krebsforschung. 71:198.
- Schneider NG, Olmstead RE, Franzon MA, Lunell E. 2001. The nicotine inhaler: clinical pharmacokinetics and comparison with other nicotine treatments. Clin Pharmacokinet. 40:661–684.
- Schoental R, Head MA. 1953. Tobacco alkaloids. In: 31st Annual Report. British Empire Cancer Campaign; p. 269.
- Schuller HM. 2012. Regulatory role of the α7nAChR in cancer. Curr Drug Targets. 13:680–687.
- Schuller HM. 2014. Impact of neuro-psychological factors on smoking-associated lung cancer. Cancers (Basel). 6:580–594.
- Schuller HM, McGavin MD, Orloff M, Riechert A, Porter B. 1995. Simultaneous exposure to nicotine and hyperoxia causes tumors in hamsters. Lab Invest. 73:448–456.
- Shahab L, Brose LS, West R. 2013. Novel delivery systems for nicotine replacement therapy as an aid to smoking cessation and for harm reduction: rationale, and evidence for advantages over existing systems. CNS Drugs. 27:1007–1019.
- Shields PG. 2011. Long-term nicotine replacement therapy: cancer risk in context. Cancer Prev Res(Phila). 4:1719–1723.
- Shin VY, Wu WK, Ye YN, So WH, Koo MW, Liu ES, Luo JC, Cho CH. 2004. Nicotine promotes gastric tumor growth and neovascularization by activating extracellular signal-regulated kinase and cyclooxygenase-2. Carcinogenesis. 25:2487–2495.
- Shultz LD, Goodwin N, Ishikawa F, Hosur V, Lyons BL, Greiner DL. 2014. Human cancer growth and therapy in immunodeficient mouse models. Cold Spring Harbor Protocol. 2014:694–708.
- Singh S, Pillai S, Chellappan S. 2011. Nicotinic acetylcholine receptor signaling in tumor growth and metastasis. J Oncol. 2011:456743.
- Siu ECK, Tyndale RF. 2007. Characterization and comparison of nicotine and cotinine metabolism in vitro and in vivo in DBA/2 and C57BL/6 mice. Mol Pharmacol. 71:826–834.
- Sparks JA, Pauly JR. 1999. Effects of continuous oral nicotine administration on brain nicotinic receptors and responsiveness to nicotine in C57Bl/6 mice. Psychopharmacology (Berl). 141:145–153.
- Staemmler M. 1935. Die chronische Vergiftung mit Nicotin. Ergebnisse experimenteller Untersuchungen an Ratten [Chronic poisoning with nicotine. Results of experimental studies in rats]. Vichows Archiv. 295:366–393.
- Staemmler M. 1936. Über geschwulstartige Bildungen im Nebennierenmark als Folge experimenteller Nicotinvergiftungen [Tumor-like developments in the adrenal medulla as a consequence of experimental nicotine poisoning]. Klin Wochenschr. 15:404–407.
- Stinn W, Berges A, Meurrens K, Buettner A, Gebel S, Lichtner RB, Janssens K, Veljkovic E, Xiang Y, Roemer E, et al. 2013. Towards the validation of a lung tumorigenesis model with mainstream cigarette smoke inhalation using the A/J mouse. Toxicology. 305:49–64.
- Stoner GD, Shimkin MB. 1982. Strain A mouse lung tumor assay. J Am Coll Toxicol. 1:145–169.
- Theophilus EH, Hayes JR, Potts RJ, Ayres PH, Williams CD, Garner CD. 2012. Toxicological evaluation of smokeless tobacco: 90-day rodent feeding studies. Exp Toxicol Pathol. 64:15–24.
- Thienes CH. 1960. Chronic nicotine poisoning. Ann N Y Acad Sci. 90:239–248.
- Thompson JG, Irwin FD, Kanematsu S, Seraydarian K, Suh M. 1973. Effects of chronic nicotine administration and age in male Fischer-344 rats. Toxicol Appl Pharmacol. 26:606–620.
- Toth B. 1982. Effects of long term administration of nicotine hydrochloride and nicotinic acid in mice. Anticancer Res. 2:71–73.
- Treviño JG, Pillai S, Kunigal S, Singh S, Fulp WJ, Centeno BA, Chellappan SP. 2012. Nicotine induces inhibitor of differentiation-1 in a Src-dependent pathway promoting metastasis and chemoresistance in pancreatic adenocarcinoma. Neoplasia. 14:1102–1114.
- Truhaut R, De Clercq M. 1961. Sur le rôle éventuel de la nicotine et de ses dérivés de pyrogénation dans les phénomènes de cancérisation [On the eventual role of nicotine and its derivatives of pyrogenation in the phenomena of canceration]. C R Hebd Seances Acad Sci. 253:1506–1508.
- Truhaut R, De Clercq M, Loisillier F. 1964. Sur les toxicitès aiguë et chronique de la cotinine, et sur son effet cancérigène chez le rat [On the acute and chronic toxicity of cotinine, and on its cancerogenic effect in the rat]. Pathol Biol (Paris). 12:39–42.
- UK Royal College of Physicians. 2007. Harm reduction in nicotine addiction. Helping people who can't quit. [cited 2015 Jun 16]. Available from: http://www.tobaccoprogram.org/pdf/4fc74817-64c5-4105-951e-38239b09c5db.pdf/.
- US Department of Health and Human Services. 2014. The health consequences of smoking – 50 years of progress. A report of the Surgeon General. [cited 2015 Jun 16]. Available from: http://www.surgeongeneral.gov/library/reports/50-years-of-progress/full-report.pdf/.
- US Food and Drug Administration. 2013. Modifications to labeling of nicotine replacement therapy products for over-the-counter human use. Fed Regist. 78:19718–19721.
- US Food and Drug Administration. 2015. Drugs@FDA, Nicorette. [cited 2016 Mar 7]. Available from: http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm/.
- von Otto C. 1911. Über anatomische Veränderungen des Herzens infolge von Nikotin (Experimentalstudie) [About anatomical changes of the heart due to nicotine (experimental study)]. Arch Path Anat. 205:384–397.
- Waldum HL, Nilsen OG, Nilsen T, Rørvik H, Syversen U, Sandvik AK, Haugen OA, Torp SH, Brenna E. 1996. Long-term effects of inhaled nicotine. Life Sci. 58:1339–1346.
- Warren GW, Singh AK. 2013. Nicotine and lung cancer. J Carcinog. 12:1.
- Warren GW, Romano MA, Kudrimoti MR, Randall ME, McGarry RC, Singh AK, Rangnekar VM. 2012. Nicotinic modulation of therapeutic response in vitro and in vivo. Int J Cancer. 131:2519–2527.
- Werley MS, Jerome AM, Oldham MJ. 2014. Toxicological evaluation of aerosols of a tobacco extract formulation and nicotine formulation in acute and short-term inhalation studies. Inhal Toxicol. 26:207–221.
- Willhite CC, Karyakina NA, Yokel RA, Yenugadhati N, Wisniewski TM, Arnold IM, Momoli F, Krewski D. 2014. Systematic review of potential health risks posed by pharmaceutical, occupational and consumer exposures to metallic and nanoscale aluminum, aluminum oxides, aluminum hydroxide and its soluble salts. Crit Rev Toxicol. 44:1–80.
- Wilson RH, De Eds F. 1936. Chronic nicotine toxicity: I. Feeding of nicotine sulfate, tannate, and bentonite. J Ind Hyg Toxicol. 18:553–564.
- Wilson RH, McNaught JB, De Eds F. 1938. Chronic nicotine toxicity: IV. Effect of nicotine-containing diets on histology and weights of organs of albino rats. J Ind Hyg Toxicol. 20:468–481.
- Wong HPS, Yu L, Lam EKY, Tai EKK, Wu WKK, Cho CH. 2007. Nicotine promotes colon tumor growth and angiogenesis through β-adrenergic activation. Toxicol Sci. 97:279–287.
- Ye YN, Liu ES, Shin VY, Wu WK, Luo JC, Cho CH. 2004. Nicotine promoted colon cancer growth via epidermal growth factor receptor, c-Src, and 5-lipoxygenase-mediated signal pathway. J Pharmacol Exp Ther. 308:66–72.
- Yokohira M, Nakano Y, Hashimoto N, Yamakawa K, Ninomiya F, Kishi S, Saoo K, Imaida K. 2012. Toxicity of nicotine by repeated intratracheal instillation to F344 rats. J Toxicol Pathol. 25:257–263.
- Yu CC, Chang YC. 2013. Enhancement of cancer stem-like and epithelial-mesenchymal transdifferentiation property in oral epithelial cells with long-term nicotine exposure: reversal by targeting SNAIL. Toxicol Appl Pharmacol. 266:459–469.
- Yu MA, Kiang A, Wang-Rodriguez J, Rahimy E, Haas M, Yu V, Ellies LG, Chen J, Fan JB, Brumund KT, et al. 2012. Nicotine promotes acquisition of stem cell and epithelial-to-mesenchymal properties in head and neck squamous cell carcinoma. PLoS One. 7:e51967.
- Yuge K, Kikuchi E, Hagiwara M, Yasumizu Y, Tanaka N, Kosaka T, Miyajima A, Oya M. 2015. Nicotine induces tumor growth and chemoresistance through activation of the PI3K/Akt/mTOR pathway in bladder cancer. Mol Cancer Ther. 14:2112–2120.
- Yun IS, Kim SS. 1938. Influence of continuous subcutaneous injection of nicotine on adrenal gland and anterior lobe of hypophysis. Trans Jap Pathol Soc. 28:426–428.
- Zhou P, Ma B, He W, Xu D, Wang X. 2011. CpG oligodeoxynucleotide stimulates protective innate immunity against human renal cell carcinoma xenografted in nude mice. J Immunother. 34:535–541.