
Abstract
Adverse outcome pathways (AOPs) are frameworks starting with a molecular initiating event (MIE), followed by key events (KEs) linked by KE relationships (KERs), ultimately resulting in a specific adverse outcome. Relevant data for the pathway and each KE/KER are evaluated to assess biological plausibility, weight-of-evidence, and confidence. We aimed to describe an AOP relevant to chemicals directly inducing mutation in cancer critical gene(s), via the formation of chemical-specific pro-mutagenic DNA adduct(s), as an early critical step in tumor etiology. Such chemicals have mutagenic modes-of-action (MOA) for tumor induction. To assist with developing this AOP, Aflatoxin B1 (AFB1) was selected as a case study because it has a rich database and is considered to have a mutagenic MOA. AFB1 information was used to define specific KEs, KERs, and to inform development of a generic AOP for mutagen-induced hepatocellular carcinoma (HCC). In assessing the AFB1 information, it became clear that existing data are, in fact, not optimal and for some KEs/KERs, the definitive data are not available. In particular, while there is substantial information that AFB1 can induce mutations (based on a number of mutation assays), the definitive evidence – the ability to induce mutation in the cancer critical gene(s) in the tumor target tissue – is not available. Thus, it is necessary to consider the patterns of results in the weight-of-evidence for KEs and KERs. It was important to determine whether there was sufficient evidence that AFB1 can induce the necessary critical mutations early in the carcinogenic process, which was the case.
Introduction
Adverse outcome pathways
Adverse outcome pathways (AOPs) are a means to organize and describe the steps (key events or KEs), from an initial interaction resulting from chemical (or other toxic agent) exposure, through a series of KEs, leading to an adverse health outcome (AO). AOPs array KEs in a temporal sequence and describe the relationships between them (key event relationships or KERs) (Ankley et al. Citation2010; Yauk et al. Citation2013; Meek et al. Citation2014a, Citation2014b; Becker et al. Citation2015a, Citation2015b; Burden et al. Citation2015; Edwards et al. Citation2015; Patlewicz et al. Citation2015; Yauk et al. Citation2015; Marchetti et al. Citation2016; OECD Citation2016). It is useful to think of an AOP as a description of the etiology of the events between initial chemical interaction with the biological targets and the AO. AOPs begin with a molecular initiating event (MIE) (Ankley et al. Citation2010) that is the event triggered by the first interaction with the chemical substance, which may then start the process that could lead to an apical outcome.
The Organization for Economic Cooperation and Development (OECD) established a program and a formal process for the development of AOPs under the guidance of the Extended Advisory Group on Molecular Screening and Toxicogenomics (EAGMST) (OECD Citation2013, Citation2014). To support this program, the United States Environmental Protection Agency (USEPA) has collaborated with OECD and other organizations to develop an AOP Wiki, which provides access to all information on the various AOPs available to date (https://aopkb.org/aopwiki/index.php). Individuals may submit an overview proposal for inclusion in the EAGMST work plan for their review and approval. AOP authors enter their detailed AOP description into the Wiki, and it is reviewed, first internally by the EAGMST and then externally by international experts. Once the AOP is deemed scientifically sound, it is submitted for endorsement by the OECD Working Group of National Coordinators (WNT) and the OECD Task Force on Hazard Assessment, and then posted as a reviewed AOP. These posted OECD AOPs are, however, viewed as “evergreen” with the goal that they be updated and revised as relevant new information becomes available.
A fundamental reason for establishing the OECD AOP program was to develop a large number of AOPs that would be publicly available. These could ultimately be used for various product stewardship and regulatory applications, including read-across, hazard identification, and risk assessment. As the program was developing and individuals were gaining experience in the construction of AOPs, case studies applicable to a single or to small numbers of chemicals with identical modes of action (MOAs), were approved by the EAGMST. Several are available on the Wiki (https://aopkb.org/aopwiki/index.php) or in the published literature (Russom et al. Citation2014; Becker et al. Citation2015b; Perkins et al. Citation2015; Bal-Price et al. Citation2017; Fay et al. Citation2017).
More recently, as experience was gained, AOPs have come to be viewed as toxicological pathways that are agnostic to specific chemicals or classes of chemicals but that are recognized as biologically plausible, based on understanding of the toxicology of specific chemicals or chemical classes and the biology of disease processes. Thus, chemically agnostic AOPs can be considered to have as their underpinning, chemical-/chemical class-specific knowledge and information. They are developed based on our general knowledge of biology but are (and should be) informed by chemical-specific information and any other stressors that might be involved in initiation or participating in the pathway.
Once developed, AOPs can be used, together with chemical-specific information on exposure, toxicokinetics, and toxicodynamics, to predict adverse outcomes for chemicals determined to induce the specific KEs that are a part of the AOP. We have developed an AOP that is relevant to those chemicals that have a mutagenic MOA for hepatocellular carcinoma (HCC) and a MIE that is the chemical induction of pro-mutagenic DNA adducts leading directly to the induction of a mutation in a cancer critical gene.
Hepatocellular carcinoma
HCC is one of the most common human cancers (Parkin et al. Citation1993; Grisham Citation1996; Staib et al. Citation2003). It is also the most frequently found cancer in laboratory rodents exposed to chemical carcinogens in 2-year bioassays; between one-third and one-half of all rodent carcinogens induce HCCs (Gold et al. Citation1991; Huff et al. Citation1991). There is extensive information on HCC etiology and pathology in rodents and other species, including humans (Grisham Citation1996). HCC is histologically similar in mice, rats, and humans (Frith et al. Citation1980; Goldfarb and Pugh Citation1986; Grisham Citation1996).
Extensive research has been conducted to understand the pathogenesis of HCC in rodents and humans, including both the pathology and molecular alterations as the liver cells progress from normal cells to metastatic tumors (Grisham Citation1996; Staib et al. Citation2003). Mutation and subsequent alterations in the expression of proto-oncogenes (such as myc and ras) and tumor suppression genes (p53), as well as alterations in growth factors, are all involved in HCC etiology (Grisham Citation1996; Staib et al. Citation2003). Sequencing of DNA isolated from tumor tissue shows that the proportion of tumors with mutations in the various oncogenes/tumor suppressor genes varies in different species. A 1996 review (Grisham Citation1996) provides a detailed summary of information from multiple species.
In humans, various exposures are associated with HCC; these include infection with hepatitis B virus (HBV) and/or hepatitis C virus (HCV), alcohol use, and exposure to specific chemicals (Sukowati et al. Citation2016). A large proportion (60–80%) of human tumors occur in cirrhotic livers (Grisham Citation1996), which can result from acute and chronic hepatitis. In addition, several epidemiological studies have indicated elevated risks for HCC from certain pesticides and from smoking tobacco (Grisham Citation1996). Studies from humans exposed to pharmaceuticals identified three that are associated with an increased risk of HCC: combined oral contraceptive steroid therapies, azathioprine, and anabolic steroids (Grisham Citation1996). The dietary consumption of aflatoxin (AFB1) from grains that have been contaminated with fungi is also a major cause of human HCC (Grisham Citation1996; Kew Citation2003; Staib et al. Citation2003; Kirk et al. Citation2005; Long et al. Citation2006; Szymañska et al. Citation2009; de Carvalho et al. Citation2013; Palliyaguru and Wu Citation2013).
HBV can play a large role in the development of human HCC, even in regions where AFB1 exposure is endemic. This interaction between AFB1 and viral infection has motivated much research (Farazi et al. Citation2003; Kew Citation2003; Farazi and DePinho Citation2006a, Citation2006b; Gouas et al. Citation2010; Chittmittrapap et al. Citation2013). HBV infection is thought to increase the possibility that individual cells will progress down the pathway to tumor formation. It seems reasonable to consider that this infection may contribute to increased cell proliferation, leading to increased clonal expansion of AFB1-induced critical cancer gene mutant cells. HBV infection might also contribute to inflammation and increased cell proliferation (Levrero and Zucman-Rossi Citation2016; Zhu et al. Citation2017) that could result in an increased number of spontaneously mutated cells containing mutations in cancer critical genes (such as p53), also resulting in HCC through a non-mutagenic MOA. HBV infections can also produce hepatic fibrosis and cirrhosis (Sukowati et al. Citation2016). It is clear and well documented, however, that although concurrent exposure to AFB1 and HBV greatly increases the incidence of HCC, AFB1 exposure alone is sufficient to cause HCC (Staib et al. Citation2003). That is, viral infection is not required for the formation of HCC in humans following exposure to AFB1.
To construct our AFB1 case study, it was necessary to integrate the large variety of information (from numerous different experimental designs, species, and endpoints), and to piece together the specific data that were useful to identify the KEs and to describe the various relationships (KERs) between the KEs. Note that the focus of the current manuscript was not a comprehensive evaluation of either the mutagenicity or carcinogenicity of AFB1, but rather, case study development as proof of concept for an AOP.
Development of an AOP for HCC (relevant to chemicals with a mutagenic MOA)
The fact that there is an extensive amount of published data for HCC, including experimental animal and human data on chemical-specific carcinogens, makes HCC a good case study for development of an AOP. It is known, based largely on both human and rodent studies of HCC, that chemical exposure can result in formation of pro-mutagenic DNA adducts. For other tumor types (e.g. kidney and skin), adducts have been shown to result in mutations and some of the mutant cells gain the ability to divide and proliferate into pre-neoplastic lesions that can progress to malignant tumors (Chakravarti et al. Citation2008; Wang et al. Citation2011).
There are a number of AOPs that lead to HCC, some of which rely on an early chemical-induced mutation in a cancer critical gene and others of which do not. One set of AOPs relies on pro-mutagenic DNA adducts or other MIEs interacting with the genome and leading directly to cancer critical gene mutation. Development of this AOP – Formation of pro-mutagenic DNA adducts leads to HCC – fits into this AOP family. In another set of AOPs, the MIE does not involve early chemical-induced mutation, but rather other non-mutagenic events such as compensatory proliferation of cells following liver injury, or receptor-mediated promotion of cell replication.
As a first step in the construction of this AOP, we developed a case study for the OECD EAGMST Program (accessed at https://aopkb.org/aopwiki/index.php/Aop:46). AFB1 was selected as the case study example because it is one of the most data-rich chemicals associated with HCC, with data available in both rodents and in humans. Based on these data, it is generally agreed that AFB1 has a mutagenic MOA. For chemicals with a mutagenic MOA for cancer, the critical, defining key event is the chemical induction of mutations in the cancer critical gene or gene(s) involved in the etiology of specific tumors as an early event in the overall carcinogenic process.
As a starting point for review of the literature, we used searches and results from a published paper (Pottenger et al. Citation2014) coauthored by two of the authors of the current manuscript (LHP and RSS). We updated and expanded this search to include information on later steps in HCC. Papers were evaluated for their utility in contributing to an AOP using criteria and definitions provided by the OECD in their guidance, user’s handbook and programs (see section OECD Format for Constructing AOPs below).
Comprehensive reviews on AFB1 have been published covering several decades of research and multiple species (Eaton and Gallagher Citation1994; Sudakin Citation2003; Kensler et al. Citation2004; Groopman and Kensler Citation2005; Bedard and Massey Citation2006; Kensler et al. Citation2011; Kew Citation2013). A large number of studies provide useful information; however, there are no comprehensive studies optimally designed for understanding the MOA for AFB1-induced HCC. That is, there are no rodent studies in which all the KEs from AFB1 exposure leading to HCC were evaluated. Investigators over the years have focused on individual parts of the process with most information concerning the following aspects: (1) AFB1 metabolism and subsequent DNA adduct formation; (2) AFB1 exposure and the induction of mutation in a variety of organisms and marker genes (not related to cancer etiology); (3) AFB1 exposure and pre-neoplastic liver lesions; and (4) AFB1 exposure and HCC. In addition, studies in humans have involved isolating DNA from tumor tissue and conducting DNA sequence analysis to determine the presence of specific oncogene/tumor suppressor gene mutations in the tumor DNA (p53, in particular). There is also a very large literature on chemoprevention that clearly and convincingly supports identification of the essential steps (the KEs) in the etiology of HCC.
A specific mutation in the p53 gene has been identified in human HCC tumors associated with AFB1 exposures. It is hypothesized that this specific codon 249 mutation is either the driver mutation or one of the important gene mutations that is an essential KE in AFB1-induced HCC. This mutation is present in a substantial proportion of human HCCs believed to be related, at least in part, to AFB1 exposure; for example, the codon 249 mutation is detected in up to 50% of liver cancers in Qidong, China (Hsu et al. Citation1991) and in Mozambique, both areas with high likelihood of AFB1 exposure. The codon 249 G:C to T:A mutation in the third base is seen in up to 75% of HCC in high-incidence areas of China and East Africa (Gouas and Shi Citation2009; Gouas et al. Citation2010). In contrast, this specific mutation is very rare in HCC from areas with no or low exposure to AFB1 (Bressac et al. Citation1991; Hsu et al. Citation1991). Furthermore, p53 mutations that are found in HCCs from areas without high AFB1 exposure (Europe and North America) occur in codons distributed throughout the p53 gene (Grisham Citation1996), rather than just in codon 249. This specific p53 codon 249 mutation is also very rare in HCCs that are associated with the use of contraceptive steroids and viral exposures (Grisham Citation1996) or in other types of tumors (Gouas and Shi Citation2009). Thus, this very specific mutation provides for the possibility of linking AFB1 exposure with HCC and identifying the KEs in the etiology of AFB1-induced HCC.
Another key piece of information comes from a variety of studies using surrogate genes (non-oncogene/tumor suppressor, genes), conducted both in vitro and in vivo; these have been designed to determine the range of possible mutations that can be induced by AFB1. The majority of mutations induced by AFB1 exposure in surrogate genes are also G:C to T:A transversions; this is consistent with the hypothesis that the codon p53 mutation (which is a G:C to T:A transversion mutation) found in human tumors is the AFB1-induced mutation involved in the initiation of the carcinogenic process (Shen and Ong Citation1996; Staib et al. Citation2003; Chawanthayatham et al. Citation2017). presents a summary of the various pieces of information that were used to develop and support the AFB1 case study AOP.
Table 1. AFB1 data supporting an AOP for “Formation of pro-mutagenic DNA adducts leads to HCC”.
We constructed an AOP based on the AFB1 case study, a chemical to which the scientific community has attributed a mutagenic MOA: that is, formation of pro-mutagenic AFB1 DNA adducts leads to HCC. There are other DNA-reactive substances believed to induce HCC via a similar AOP invoking a mutagenic MOA. These include substances such as 2-acetylaminofluorene (2-AAF), which like AFB1, is also capable of forming bulky DNA adducts following metabolic activation, with at least some data to support the KEs that we describe below (Cohen and Arnold Citation2011).
The current AOP effort focused mainly on AFB1 information, evaluated originally as a case study. We note that in addition to underpinning this particular AOP, utilizing available information for a data-rich chemical such as AFB1 helps demonstrate the types of information that are required to conduct an objective assessment of unknown chemicals for their ability to induce the KEs and ultimately the AO for any particular AOP.
AOP relevant to chemicals inducing HCC via a mutagenic MOA: formation of pro-mutagenic DNA adducts leads to HCC
There can be several plausible MOAs for the chemical induction of HCC and, thus, several different AOPs that end with HCC as the AO; the early KEs that distinguish these are usually dichotomized as mutagenic or non-mutagenic. An increase in cells with mutations in cancer critical genes is eventually part of every cancer etiology. A mutagenic MOA is distinguished from a non-mutagenic MOA by an early, influential KE that involves chemically induced mutations in cancer critical gene(s) (Preston and Williams Citation2005). There are several initiating events that can lead to chemically induced mutation. Our AOP is based on the formation of chemical-specific pro-mutagenic DNA adducts as the MIE. Other examples of MIEs that lead to mutation in cancer critical genes could be intercalation or the formation of DNA cross-links. The MIE or early KE for chemicals that have a non-mutagenic MOA may be the compensatory proliferation of cells following liver injury leading to increased formation of tumors, or receptor-mediated promotion of cell replication (Jarabek et al. Citation2009; Pottenger et al. Citation2014). Becker et al. (Citation2015b) have constructed an AOP for dioxin-like chemicals that bind with the aryl hydrocarbon receptor (AHR) and cause sustained activation of the AHR receptor (the MIE), ultimately leading to HCC and bile duct tumors in rodents. This AOP is an example of the set of AOPs with MIEs like the induction of sustained activation of a receptor, toxicity, or chronic hepatitis. They all have increased cell proliferation (and not mutation in a critical gene) as an early influential KE; they all represent chemicals that act by non-mutagenic MOAs.
An overarching goal of AOP development is the elaboration of a network of AOPs that may differ in one or more of the MIE, KE, and/or AO events. That is, AOPs should be viewed as families of pathways describing the individual various etiologies that start with the different but specific MIEs, include the specific KEs and the specific AO; these will intersect at key nodes (i.e. the same MIEs/KEs/AOs), and lead to a network of AOPs (Villeneuve et al. Citation2014).
It is clear that the major difference between AOPs that either do or do not involve chemically induced mutation leading to HCC (and, in fact, for all tumor types) is in the MIE and the first few KEs. Once mutations occur in the cancer critical genes, and the cells containing these mutations proliferate, the steps or KEs are the same; the later KEs for HCC converge, irrespective of the original MIE, leading to the same AO.
Drawing upon the information learned in the construction of the AFB1 case study, we can construct an AOP that reflects a mutagenic MOA for chemically induced HCC that includes as its MIE the formation of a pro-mutagenic DNA adduct and, as the influential KE2, the early induction of mutation in cancer critical gene(s). The MIE and KEs for this AOP are these ():
Figure 1. Key events for the AOP on mutagenic MOA for HCC: formation of pro-mutagenic DNA adducts leads to HCC.
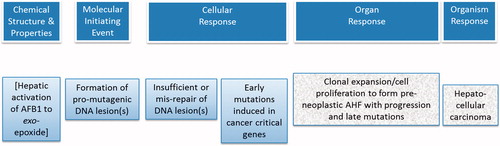
MIE: Formation of pro-mutagenic DNA adducts;
KE1: Insufficient repair or mis-repair of pro-mutagenic DNA adducts;
KE2: Early induced mutation in cancer critical genes;
KE3: Cellular proliferation, clonal expansion of mutant cells, and progression;
AO: Hepatocellular carcinoma.
OECD format for constructing AOPs
The OECD has provided a template for outlining AOPs and for describing and evaluating data in support of its KEs and KERs. For example, the KE descriptions include discussion of the level of biological organization, evidence for taxonomic applicability, techniques for measuring or detecting the KE, and evidence of essentiality of the identified KEs. Descriptions of KERs include application of evolved Bradford Hill considerations in evaluating weight of supporting evidence, focusing on biological plausibility, essentiality, empirical evidence, and uncertainties (Meek et al. Citation2014a, Citation2014b; Becker et al. Citation2015a). The terminology for levels of evidence for KEs and KERs were defined by OECD guidance and user’s handbook in 2014 and updated in 2016 (OECD Citation2014, Citation2016) and are shown in .
Table 2. AOP evidence levels evaluated based on OECD Guidance (OECD Citation2014) and user’s handbook (OECD Citation2016).
For an AOP, it is necessary to assess the evidence as to whether the MIE or early KEs are essential to the AOP. Only MIEs and KEs that are necessary steps in the pathway should be used as key events that will be useful for assessing whether unknown chemicals might cause a particular AO. Thus, it is important for future regulatory applications that the necessity of the KEs and the essentiality of the MIE and early KEs be adequately detailed as part of the AOP development process.
Description of the MIE and KEs
This section describes the MIE and the KEs for this AOP and provides a summary of the information related to each event based on available data for AFB1. In addition, we discuss available evidence supporting the essentiality of the MIE and KEs for the AOP.
Many chemicals must undergo absorption, metabolism, and distribution to the target tissues before they can interact with DNA and form adducts, including those that are pro-mutagenic. A chemical-specific MOA would be incomplete without such information. AOPs, however, do not include a discussion of this aspect of the overall process from exposure to AO. Our AFB1 case study was dependent on understanding how and where AFB1 is metabolized and which metabolites are responsible for forming the DNA adducts that are ultimately involved in the mutation. There are several publications that provide an extensive summary of the metabolism of AFB1 (Degen and Neumann Citation1981; Ueng et al. Citation1995; Guengerich et al. Citation1996; Groopman and Kensler Citation2005; Pottenger et al. Citation2014). While AFB1 produces several metabolites, the important metabolite for the mutagenic MOA for HCC is the reactive exo-8,9-epoxide of AFB1 (exo-epoxide). This is formed in hepatocytes and extra-hepatically via metabolism of the parent AFB1 by CYP450 (Larsson et al. Citation1990; Larsson and Tjälve Citation1993), and it is the reactive metabolite that binds to nuclear DNA forming pro-mutagenic DNA adducts.
A number of mammalian species have been investigated for their AFB1 metabolic pathways, and the same pathways are applicable to many (perhaps all) mammalian species; it has even been demonstrated in certain birds such as turkeys (Gregory et al. Citation1983; IARC Citation1993). Humans, non-human primates, rats, mice, poultry, and fish have all demonstrated susceptibility to AFB1-induced liver tumors, albeit in some cases to varying degrees; thus, the necessary metabolic activation is inferred (Asplin and Carnaghan Citation1961; Eaton and Gallagher Citation1994). Species that preferentially metabolize AFB1 to the exo-8,9-epoxide are more susceptible to AFB1 carcinogenicity. Conjugation with glutathione by glutathione-S-transferase (GST) enzymes or other enzymatic detoxification of AFB1 metabolites can reduce available levels of the exo-epoxide and impede subsequent steps and the progression of KEs. For example, although mice demonstrate increased AFB1 oxidation by CYP450 to the exo-epoxide, they are less susceptible to AFB1-induced liver cancer than rats (Degen and Neumann Citation1981). This difference is believed to be due to the constitutive presence of GST-alpha conjugative detoxification activity in mice vs. rats, where this activity is not found (Monroe and Eaton Citation1987). This higher detoxification capability can also be caused by dietary exposures to compounds that modulate CYP450 expression or modify detoxification activities (Roebuck et al. Citation1991; Primiano et al. Citation1995; Kensler et al. Citation1998a; Wang et al. Citation1999; Elegbede and Gould Citation2002; Yates et al. Citation2006). In short, although outside of the formal realm of AOPs, studies in many species consistently point to AFB1 metabolism to reactive forms as an essential step in the sequence of events for AFB1-induced HCC.
MIE: formation of pro-mutagenic DNA adducts
The MIE is the formation of chemically induced pro-mutagenic DNA adduct(s). For every chemical that causes cancer through a mutagenic MOA and which interacts with DNA to form adducts, one or more of these adducts will be a pro-mutagenic adduct; that is, an adduct that, when not repaired properly, can lead to the induction of mutations in daughter cells, which can initiate the tumor process. AFB1 exposure initially results in only one type of pro-mutagenic DNA adduct, the 8,9-dihydro-8-(N7-guanyl)-9-hydroxyaflatoxin B1 adduct [N7-AFB1-G] (Croy et al. Citation1978). Following insufficient or mis-repair, this adduct can result in a G:C to T:A transversion mutation.
This initial AFB1-induced pro-mutagenic DNA adduct comes from the exo-epoxide, which intercalates into DNA and then binds to the nucleophilic N7-G residue via an SN2 reaction. Very few data were identified that provide dose–response context for adduct formation in the mammalian liver target tissue. Appleton et al. (Citation1982) measured AFB1-related radioactivity in hepatic DNA following i.p. injections of from 10 to 1000 ng/kg bw of radiolabelled AFB1. Buss et al. (Citation1990) quantified radioactivity bound to DNA and demonstrated a linear dose–response for AFB1-related radioactivity in rat liver with administered dose using a log-scale range of single doses of [3 H]-AFB1 administered via oral gavage.
The N7-AFB1-G adduct has a short half-life; it can spontaneously depurinate, leaving an apurinic (AP) site, which typically is rapidly repaired (Denissenko et al. Citation1998). AP sites, formed by spontaneous depurination, are pro-mutagenic lesions, and are the predominant background or endogenous lesion identified to date in DNA from untreated controls, with about 30,000 AP sites/cell present ubiquitously and continuously (Swenberg et al. Citation2010). Alternatively, the N7-AFB1-G can be repaired (described below) or can ring-open to form the formamidopyrimidine adduct, called the AFB1-FAPy adduct. Thus, although the N7-AFB1-G is considered to be a pro-mutagenic lesion due to its capability to intercalate in DNA and its bulkiness (Bailey et al. Citation1996), it may not be the most important DNA adduct in the process of AFB1-induced tumorigenesis.
Once the exo-epoxide is bound to the N7-guanine (N7-AFB1-G), it can then ring-open to form the 8,9-dihydro-8-(2,6-diamino-4-oxo-3,4-pyrimid-9-yl-foramido)-9-hydroxyaflatoxin B1, or AFB1-FAPy adduct (Brown et al. Citation2006). The AFB1-FAPy adduct has a longer half-life than the N7-AFB1-G adduct, and it demonstrates higher mutagenic efficiency or potency than the N7-AFB1-G (Brown et al. Citation2006). Data indicate that about 20% of the N7-AFB1-G adducts undergo spontaneous ring-opening to become AFB1-FAPy adducts (Croy and Wogan Citation1981; Bedard et al. Citation2005); others report that by about 24 h post-exposure, AFB1-FAPy adducts predominate (Croy and Wogan Citation1981; Boysen et al. Citation2009). These pro-mutagenic adducts do not spontaneously depurinate, but accumulate over time, which likely contributes to their increased mutagenic efficacy (Smela et al. Citation2002).
The pro-mutagenicity of these two adducts was demonstrated by assessing their mutagenic efficiency; that is, number of mutants formed/adduct, such that if all adducts result in mutations, then the mutagenic efficiency =100%. This was reported for the non-human primate-derived kidney cell line COS-7, in a site-specific mutation assay using shuttle vectors. By transfecting an adducted single strand shuttle vector under stringent conditions, no repair can take place there is (no intact strand to serve as a template), so one can accurately measure the mutagenic efficiency. The N7-AFB1-G adducts demonstrated a mutagenic efficiency of 45% in COS-7 kidney cells (Lin et al. Citation2014a); thus, 45% of adducts present resulted in a measureable mutation in the transfected plasmid. The predominant mutation observed was G:C to T:A transversion. In contrast, in the same in vitro system, the N7-AFB1-FAPy adduct had a mutagenic efficiency of 97% (Lin et al. Citation2014b), an extremely potent mutagenic efficiency in this system.
Sensitive analytical techniques (Himmelstein et al. Citation2009) are available for structural quantitation of specific DNA adducts, including the AFB1-specific DNA adducts. These methods include high pressure liquid chromatography (HPLC) or liquid chromatography (LC) separation, coupled with tandem mass spectrometry (HPLC-MS/MS or LC-MS/MS). These techniques allow for definitive identification of the AFB1-related adducts using authentic standards. These capabilities can be further enhanced by use of stable isotope-labeled test materials, e.g. with 13 C, 15 N, or deuterium. Increased sensitivity is reported with accelerated mass spectrometry (AMS) approaches; these require the use of radiolabelled (14 C) test material but can detect adducts down into the attomolar range (10−18).
Codon 249 of the p53 gene has been identified as a particular target of AFB1-induced adduction and subsequent mutation. Puisieux et al. (Citation1991) provide evidence that the AFB1 epoxide binds preferentially to codon 249 of the p53 gene. Using a plasmid containing full-length human p53 DNA, adduct formation was observed in p53 exons 5, 6, 7, and 8 (equaling a total of 1086 bases), and 20% of the bases were targeted by AFB1 with a preference for guanine residues. Binding of AFB1 to p53 sequences was restricted to fewer residues and was more specific for guanine than was the binding of the carcinogen benzo[a]pyrene (B[a]P) (Puisieux et al. Citation1991). Binding of AFB1 in the region around codon 249 of p53 was reported to be “stronger” than that of B[a]P. The last nucleotide of codon 249 is a guanine and was targeted by AFB1 but not by B[a]P. As already noted, this guanine residue is the mutational hotspot in human liver cancers from patients in high AFB1 exposure regions. Denissenko et al. (Citation1998) applied PCR techniques to DNA from human HepG2 cells exposed to AFB1 metabolically activated by rat liver microsomes. They observed base changes in p53 coding regions in addition to codon 249 mutations at this site.
For AFB1, the essentiality of this MIE is demonstrated by the reduction of DNA adducts and the subsequent demonstrated reduction and/or elimination of AHF (KE3) and of liver tumors (AO) when chemopreventive agents such as oltipraz (Kensler et al. Citation1987, Citation1998a) and CDDO-Im (Johnson et al. Citation2014) are administered along with AFB1. The effects of the chemopreventive agents are due at least in part to modulation of AFB1 metabolism to reactive forms. For example, inhibition of activation results in reduced formation of the critical exo-epoxide, which then results in a demonstrated decrease in the MIE, reduced adduct formation. Likewise, increased GST activity results in increased conjugative detoxification of the exo-epoxide. In both cases, less reactive metabolite is available to form the critical pro-mutagenic DNA adducts, resulting in fewer adducts, thus reduced MIE and subsequent reduction in AHF and liver tumors (Guengerich et al. Citation1996). Recent work with Nrf2 knock-out rats has provided more mechanistic evidence to support the essentiality of the formation of AFB1-N7-G adducts. AFB1-treated Nrf2 knock-out rats had increased DNA adducts, and decreased gene expression of GST and anti-oxidative enzymes, even with CDDO-Im treatment; wild-type rats treated with CDDO-Im and AFB1 demonstrated significantly reduced AFB1-DNA adducts as described earlier (Taguchi et al. Citation2016). The Nrf2-Keap1 signaling system is discussed under chemoprevention below.
The evidence supporting the formation of a pro-mutagenic DNA adduct as the MIE is strong, and it stems from many datasets in different biological systems. Supporting data include the formation of N7-AFB1-G and N7-AFB1-FAPy DNA adducts after AFB1 exposure, which have been demonstrated across phyla, from bacteria through yeast, fish, birds, and including many mammalian systems up through non-human primates and humans (Croy et al. Citation1978; IARC 1993; Cupid et al. Citation2004).
KE1: insufficient repair or mis-repair of pro-mutagenic DNA adduct(s)
The first KE following the MIE is the insufficient or mis-repair of the pro-mutagenic DNA adduct(s). For AFB1, the induced pro-mutagenic DNA adducts, N7-AFB1-G and/or AFB1-FAPy, are recognized by various DNA repair proteins or systems, which then initiate the repair processes (Bedard and Massey Citation2006).
The DNA repair system that recognizes the damage can depend in part upon which stage of the cell cycle is in progress when the pro-mutagenic DNA adducts are detected and on the type of DNA damage present. Initial steps depend on the repair system, and can include recruitment of a series of proteins specific to that repair system and can also include blocking of the progression of the DNA replication fork. This latter step (blocking the replication fork) ensures that DNA replication waits for repair to occur before proceeding, preventing replication of the damaged DNA, thus avoiding a mutation (Hanawalt Citation2003; Hanawalt et al. Citation2003; Kunkel Citation2004; Brown et al. Citation2011; Dianov and Hübscher Citation2013; Kozmin and Jinks-Robertson Citation2013). For many of the DNA repair systems, the repair process is quite faithful; that is, damage is correctly repaired with the original base substituted for the adducted base, resulting in no change in the DNA sequence. For example, with excision repair (ER), the damaged base (BER) or oligomer (NER) is excised and the opposite, undamaged strand is copied to make a usually faithful repair. When an incorrect base is inserted, then it is a mis-repair and the change in primary DNA sequence can result in a mutation. When on-going DNA replication is not stopped, and DNA replication continues across an adducted base (typically a non-instructional lesion), there is no opportunity for repair. Depending on the process involved (e.g. error-free or error-prone translesion synthesis, TLS, or even post-replication repair), this can result in the incorporation of an incorrect base into the nascent DNA strand. When the cell divides, this error is “fixed” as a permanent change in the primary DNA sequence, resulting in a mutation.
Data from diverse cell types and systems demonstrate that AFB1-induced DNA adducts are repaired by a variety of processes, including SOS repair, NER, homologous recombination (HR), and post-replication repair (Bedard and Massey Citation2006). Following depurination of the AFB1-N7-G adduct, the remaining AP sites are repaired mainly by BER (Bedard and Massey Citation2006). Guo et al. (Citation2005) conducted in studies in transgenic yeast modified to express human CYP1A2, and evaluated several DNA repair systems for efficacy towards the AFB1-induced DNA adducts. Mutations were more likely to be induced in strains deficient in certain repair systems and were also induced in strains with active secondary repair pathways; these include pathways for error-prone post-replication repair and those relying on apurinic endonucleases. However, NER appears to be most important for repair of AFB1-induced adducts in mammalian systems (Bedard and Massey Citation2006).
There is no simple method for direct measurement of DNA repair of specific adducts. One approach to measuring the level of insufficient or mis-repair is the quantitation of chemical-specific DNA adducts, such as the N7-AFB1-G and AFB1-FAPy DNA adducts, before and after DNA repair occurs. This can be combined with DNA sequence data to determine whether adducts were correctly repaired or not. While such a dataset would provide a quantitative assessment of DNA repair/mis-repair, this is not a very practical approach. Measurement of mutations following AFB1 treatment of yeast with a variety of DNA repair deficiencies has been used to elucidate the role of DNA repair (Guo et al. Citation2005) by comparing mutant frequency (MF) in wild type versus DNA repair-deficient strains. In this instance, MF serves as a surrogate marker for insufficient or mis-repair of the AFB1 DNA adducts, and it can be used in situations with specific DNA repair deficiencies, such as NER.
A different approach to obtaining supporting data would be to use the exquisitely sensitive analytical techniques available for structural quantification of these chemical-specific DNA adducts. This requires specialized analytical chemistry techniques conducted on DNA isolated from tissues or cells and subjected to neutral thermal or enzyme or acid hydrolysis to release the N7-adducted bases, which are then further analyzed (Himmelstein et al. Citation2009). In this case, the strategy would be to measure adducts before and after repair has had time to occur. However, it should be noted that this approach would not provide information as to whether the repair did or did not result in a mutation.
Another approach that could address measuring DNA repair is one using transgenic animals, such as the Fluorescent Yellow Direct Repeat (FYDR) mice that react to DNA damage-initiation of HR with a fluorescent response (Hendricks et al. Citation2003). Recent work has shown that the HR response is linked to BER glycosylase activity (Kiraly et al. Citation2014) and induced by inflammation-related DNA damage and cell proliferation (Kiraly et al. Citation2015). This can be measured in liver albeit not as robustly as pancreas and some other tissues (Wiktor-Brown et al. Citation2006a, Citation2006b). Some data from alkylating agents, methyl methane sulfonate (MMS) and ethyl nitrosourea (ENU), indicate that such an approach might also work for AFB1. A similar fluorescent-linked probe approach has been described for an in vitro assay using transgenic mouse embryonic stem cells (mES) with two reporters, one aimed at revealing induction of DNA repair and the other at oxidative stress, called ToxTracker (Hendriks et al. Citation2012). This model cannot address the specific questions of AFB1 effects in hepatocytes but could measure activation of genes related to DNA repair.
Measurements of repair of AFB1-induced DNA adducts have focused mainly on in vitro systems with bacteria, yeast, and mammalian cells, including cell lines derived from rats and non-human primates (Sarasin et al. Citation1977; Leadon et al. Citation1981; Levy et al. Citation1992; Oleykowski et al. Citation1993; Guo et al. Citation2005; Gross-Steinmeyer and Eaton Citation2012). Relevant recent studies evaluated mutation induction of AFB1 in Caenorhabditis elegans (Leung et al. Citation2010; Meier et al. Citation2014). It is likely that all species capable of DNA repair following the MIE – formation of pro-mutagenic DNA adduct (e.g. binding of AFB1 exo-epoxide to DNA) – are subject to insufficient or mis-repair; for AFB1, these species include bacteria, yeast, birds, mammals, and fish.
Evidence supporting the insufficient repair or mis-repair of pro-mutagenic DNA adducts is strong, albeit mainly indirect. This evidence comes from different biological systems and datasets, mostly mammalian, and it is based in large part on the biological understanding of a DNA-reactive mode of action requiring a permanent change in primary DNA sequence (mutation). As DNA repair pathways are highly conserved in eukaryotic species, taxonomic applicability can be assumed for all phyla, including rats, mice, woodchucks, humans, monkeys, tree shrews, birds, trout, and lower eukaryotes.
KE2: early induced mutation in cancer critical gene(s)
The second KE is mutation formation early in the etiology of a tumor in a cancer critical gene; this is the influential early step that would apply to chemicals that induce cancer via mutagenic MOA. A mutation is generally considered to be a permanent, transmissible (to daughter cells) change in the amount or structure of the genetic material; this is by contrast to transitory or non-transmissible changes, and alterations leading to cell death. There is no substantial biological impact when the mutation results in no change in the coded amino acid due to the redundant nature of the genetic code for amino acids. Even a change in amino acid sequence may not result in any change in the activity or function of the expressed protein product. This can occur if an amino acid is exchanged for a chemically similar amino acid or is located in the protein where there is no important function. Thus, depending on the specific altered function, the biological impact may be none, or slight, or substantial. A mutation in a cancer relevant gene is required to initiate the process of dysregulated cell proliferation, which ultimately leads to the development of HCC. For example, if the mutation is induced in codon 249 of the p53 gene (as it appears often to be for AFB1), there is potential for substantial biological impact, including induction of a mutation leading towards tumor formation.
Generally, for chemicals that induce tumors by chemically induced mutation, there is no information on which oncogenes/tumor suppressor genes are involved in the tumor etiology, or which specific mutations within those genes are the critical mutations. For AFB1, however, there is information that helps to identify a candidate for that critical mutation. Human tumor DNA has been sequenced, and a codon 249 mutation in the p53 gene can be found in a substantial proportion of human HCCs in regions with AFB1 exposure. This specific mutation is not prevalent in other types of tumors (Gouas and Shi Citation2009). Because the p53 gene is known to be important in the etiology of various tumors, it has been hypothesized that this mutation is the critical one or one of the important gene mutations that is represented by KE2 in AFB1-induced HCC.
However, finding a particular mutation in tumor DNA (even in a high fraction of the tumors) does not provide definitive proof it was that specific mutation which was the initial influential mutation (the KE), starting the progression to tumor formation. As will be discussed further, there are a number of steps that take place in the etiology of tumor development, and the mutations that are seen in DNA from tumor tissue may have occurred at any stage in that process. The key question is whether a particular mutation occurs as a direct result of adduct formation at this site early in the process, or by some different mechanism. As a general strategy to evaluate whether a chemical can induce the type(s) of mutations that are observed in tumors, the set of possible mutations induced by the chemical of interest can be compared with the mutations that are observed in the DNA from tumors. By linking these two pieces of information, it is possible to draw conclusions on whether a chemical is capable of inducing the specific mutations through the induction of specific oncogene/tumor suppressor gene mutations. It is important to note that chemicals cause a variety of mutations across the genome. It is only those specific mutations that occur in the cancer critical gene that would be the key event in this AOP.
Using AFB1 as an example, there is a large body of literature concerning the types of mutations that can be induced by AFB1 with several studies involving the p53 gene. This provides a pattern for a weight of the evidence evaluation. Several published studies indicated that codon 249 of the p53 gene adopts an unusually mutagenic adduct conformation based on the local DNA sequence; the authors concluded that a higher mutation rate may be observed at this location by comparison to other sites because of increased DNA polymerase bypass (Puisieux et al. Citation1991; Lin et al. Citation2014a, Citation2014b). In addition, using restriction fragment length polymorphism/polymerase chain reaction genotypic analysis, Aguilar et al. (Citation1993), showed that AFB1 preferentially induced a G:C to T:A transversion at the third position of p53 codon 249 in human-derived HepG2 cells exposed to AFB1. Denissenko et al. (Citation1998) observed adduct formation at codons other than 249 in in vitro studies of human-derived HepG2 hepatocytes exposed to microsomally activated AFB1. They cite this finding as well as what they describe as incomplete correspondence between sites of persistent AFB1 damage and specific codon 249 mutation as evidence suggesting that AFB1 may not be involved in the mutation of this site, or that additional mechanisms may be required for selection of codon 249 mutants in HCC.
There is substantial evidence in a variety of gene mutation assays from bacteria to mammals, evaluating a range of target genes, that AFB1 induces G:C to T:A transversions. A G:C to T:A transversion mutation is expected for both AFB1-related pro-mutagenic DNA adducts, N7-AFB1-G and AFB1-FAPy, and it is found in the p53 mutations from human tumor DNA. Various mutation assays that assess the types of mutations chemically induced in surrogate genes can be used to evaluate the full spectrum of mutational events potentially induced by a chemical. In bacteria and in mammalian cells (both in vitro and in vivo) the primary mutation associated with AFB1 exposure is a G:C to T:A transversion (Foster et al. Citation1983; Dycaico et al. Citation1996). For example, studies in the AS52 assay, an in vitro mammalian transgenic mutation assay that measures mutations in the guanine phosphoribosyl transferase (gpt) gene, find that exposure to AFB1 leads predominantly to G:C to T:A transversions; a number of other types of mutations were also seen (Wattanawaraporn et al. Citation2012).
There are no AFB1 data that directly demonstrate essentiality of KE2. Some indirect evidence of the essentiality of mutation for tumor development is provided by the clear species difference between adult mice and adult rats as well as clear differences between neonatal mice and adult mice. These species and age differences provide evidence that the induction of mutation by AFB1 (in surrogate genes) and the formation of liver tumors are linked. Adult mice have higher constitutive alpha-GST, and so much lower amounts of active AFB1 exo-epoxide metabolite are available for adduct formation (IARC 1993). Consistent with this, adult mice are almost refractory to AFB1-induced liver tumor formation, and no increase in MF is seen in the in vivo Big Blue™ mutation assay for adult mice exposed to AFB1. Lac I mutants isolated from the AFB1-exposed adult mice showed a spontaneous mutational spectrum. Rats, however, showed a large increase in MF and, more specifically, a large increase in G:C to T:A transversions (Dycaico et al. Citation1996), in concordance with increased liver tumors. Thus, with few AFB1-induced mutations in liver, adult mice have very low rates of HCC following AFB1 treatment; in contrast, rats show substantial increases in AFB1-induced MF as well as increased rates of HCC following AFB1 treatment, providing indirect evidence of essentiality of KE2.
In addition, for mice, there is a difference in AFB1 mutation effects and liver tumor induction between neonatal and adult mice. Neonatal wild-type mice form ∼200 times the level of carcinogen-DNA adducts and have >100-fold lower levels of GST than do adult wild-type mice. These results indicate that neonatal mice may be more susceptible to AFB1 because of an inability of proliferating hepatocytes to detoxify the carcinogen at the same rate as adult mice (Sell Citation2003). Big Blue™ neonatal mice treated with AFB1 (at 6 mg/kg – a dose that results in tumors) showed an increase in mutations in an indicator gene (cII mutation) with G:C to T:A transversion as the major mutation; this is a hallmark of AFB1 mutagenicity. Big Blue™ adult mice treated at 6 and 60 mg/kg (doses that do not produce tumors) did not have a significant increase in cII mutations. This study comparing neonatal and adult mice provides another example of increased AFB1-induced mutations correlating with increased liver tumors, while no increases in AFB1-induced mutation correlated with no increase in tumors. The adult mice did display a different mutational spectrum than untreated controls (Chen et al. Citation2010); such changes in mutational spectrum without significant increases in transgenic MF may indicate that the chemical exposure is actually inducing mutations, but that the particular experiment did not have enough statistical power to detect the increase in MF. In any case, the difference in tumorigenicity between neonatal and adult mice provides support for the essentiality of the influential KE2: early mutation in cancer critical genes.
Techniques to evaluate whether a chemical can induce a particular (or even any) cancer gene mutation in target tissues are not readily available. As already indicated, it is possible to identify specific mutations in oncogene/tumor suppressor genes in tumor DNA using sequencing. Such an approach will reveal the presence of mutations when almost every cell contains the mutation. Historically, however, it has not been technically feasible to detect of critical cancer-gene-specific mutations occurring at low frequency and particularly as very rare (1 per 100,000 cells) events in target tissue(s). A recently developed method, allele-specific competitive blocker-polymerase chain reaction (known as ACB-PCR), has proved useful in providing such information, and data on specific chemical-induced mutations (some in cancer target tissues) following exposure to specific chemical are now available (Parsons et al. Citation2005, Citation2010). There are no such data for AFB1.
Evidence supporting the induction of mutations in cancer critical genes is strong, but it is mainly indirect. A high percentage of human tumors from AFB1 endemic regions have a specific p53 gene mutation, while those from regions without endemic AFB1 do not. While there is no direct evidence that AFB1 can induce this specific p53 gene mutation in tumor target tissue, there is substantial evidence that AFB1 induces the same class of mutation (G:C to T:A transversion). Thus, the pattern of mutations that can be induced by AFB1 is consistent with the type of mutation seen in human tumors.
KE3: cellular proliferation, clonal expansion of mutant cells, and progression
KE3 in the AOP is, in fact, a series of events common to various AOPs for HCC, including those AOPs that do not involve the induction of mutation in a cancer critical gene that occurs early in the etiology of the tumor. KE3 is, thus, a node where other AOPs may intersect on the path to the AO of HCC. These steps may be associated with the observation of additional new mutations late in carcinogenesis, and in fact, additional mutations (beyond the initiating mutation) are generally involved in the development of tumors.
The process by which mutations in tumor-critical genes lead to alterations in cellular growth homeostasis and clonal expansion of mutated cells is unknown. It is possible that the presence of DNA damage acts to alter biological pathways controlling cellular growth homeostasis by alteration in the expression of genes. The specific mechanisms by which cells become transformed are not fully known.
The initiated cells, mutated in the cancer critical gene(s), begin to undergo clonal expansion, resulting in the formation of altered hepatic foci (AHF) and further dysregulated cellular proliferation. It appears that single transformed cells, in which apoptosis is blocked by tumor-critical mutations, will grow into AHF (Grasl-Kraupp et al. Citation1997). AHF are clonal in origin and are identified with immunohistochemical techniques, typically due to their synthesis/expression of GST-P.
In mammals, AHF as a pre-neoplastic lesion has been recognized for many years as part of the general etiology of HCC (Goldsworthy et al. Citation1986; Beer and Pitot Citation1987; Sargent et al. Citation1989; Pitot Citation1990; Pitot et al. Citation1990, Citation1991; Dragan et al. Citation1995). In rats, the presence of AHF with altered growth characteristics has been observed in a number of studies (Newberne Citation1976; Nishizumi et al. Citation1977; Manson et al. Citation1984; Bannasch et al. Citation1985; Godlewski et al. Citation1985; Fischer Citation1986; Fischer et al. Citation1987; Gil et al. Citation1988; Harada et al. Citation1989b; Roebuck et al. Citation1991; Youngman and Campbell Citation1992a, Citation1992b). The mechanisms involved in the formation of AHF appear to be generalizable across species and likely apply to fish and birds as well as to mammals including humans (Kirby et al. Citation1990; Thoolen et al. Citation2012; Ribback et al. Citation2013).
Originally, these foci were observed as histologically different from the surrounding parenchyma (Bannasch et al. Citation1985; Gil et al. Citation1988; Harada et al. Citation1989a, Citation1989b). In addition, immunohistochemical techniques to demonstrate enzyme alterations were used to identify AHF foci, most notably, the occurrence of a placental form of GST (GST-P) (Godlewski et al. Citation1985; Kirby et al. Citation1990; Dragan et al. Citation1994, Citation1995). Both measurements reflect focal growth because single cells with the altered phenotype cannot be detected using the immunohistochemical staining technique. There has been extensive research to document the growth and occurrence of foci; the data are generally expressed as the number of AHF per volume of liver and the volume fraction of the liver occupied by AHF (Dragan et al. Citation1997). Quantitative stereology has also been used to quantify the growth of AHF (Xu et al. Citation1990a, Citation1990b; Dragan et al. Citation1995; Pitot et al. Citation1996). Growth of foci appears to follow the Moolgavkar–Venzon–Knudson model of initiation and promotion (Dewanji et al. Citation1991; Dragan et al. Citation1995). A number of agents regarded as tumor promoters appear to enhance the growth of foci, acting to inhibit apoptosis and also to create an overall proliferative stimulus (Angsubhakorn et al. Citation2002; Wyde et al. Citation2002).
Evidence supporting cell proliferation and the formation of pre-neoplastic lesions is strong, and these effects have been directly observed following exposure to chemicals including AFB1. Furthermore, AHF have been observed universally in livers from AFB1-treated mammals, birds, and fish (Cullen et al. Citation1990; Kirby et al. Citation1990; Kimura et al. Citation2004; Kensler et al. Citation2011; Pottenger et al. Citation2014).
Cells in the pre-neoplastic lesions can progress toward a malignant phenotype. However, not all AHF progress to HCC. Although not definitively described, it is during this common KE that the cells within the foci can undergo additional mutations; it has been observed that tumor cells can be diploid, near diploid, or aneuploid (Grisham Citation1996). Presumably, these additional mutations result in changes that permit unregulated growth, and introduce other tumor-related characteristics or hallmarks of cancer (Hanahan and Weinberg Citation2000, Citation2011). Many processes likely contribute to this KE, including inflammation, oxidative stress, and gene expression changes; for example, induction or repression of transcription factors such as Nrf-2/KEAP1 or NF-κsz, may regulate the activities and responses to this environment.
Nodular aggregates of hepatocytes varying in size are seen in human cirrhotic livers (Grisham Citation1996). These nodules, which are termed adenomatous hyperplastic (AHN) or macroregenerative nodules (MRN) in cirrhotic livers, and hyperplastic nodules (HN) in non-cirrhotic livers, are understood to be the immediate pre-neoplastic precursors of HCC (Grisham Citation1996). In rodents, the HN nodules are commonly seen in animals exposed to chemicals that ultimately cause HCC. Some research has been conducted in humans, and substantial research has been conducted in rodents to characterize the phenotype of these various nodules (Godlewski et al. Citation1985; Kirby et al. Citation1990; Pitot et al. Citation1990; Dragan et al. Citation1994, Citation1995; Grisham Citation1996). Cell proliferation appears to be 6- to 7-fold greater in HN than in surrounding liver parenchyma (Dragan et al. Citation1994).
While these additional changes are not identified in detail, they contribute to the overall tumor milieu that is described as hallmarks of cancer by Hanahan and Weinberg (Citation2000, Citation2011). Thus, the evidence for progression of pre-neoplastic lesions and the accumulation of additional mutations involved ultimately in the malignant tumor HCC is strong. These mutations occur later in the etiology of the tumor, for example, as a result of increased compensatory cell proliferation following exceedance of a toxicity (or homeostatic) threshold which then enhances the probability of fixation of a spontaneous mutation in a cancer critical gene. It should be noted that multiple mutational events likely can occur between the initiation of the first biological event in tumorigenesis and the tumor AO.
AO: hepatocellular carcinoma (HCC)
The AO is HCC, which can vary in size along a continuum from a few, microscopically visible, malignant cells to nodules to large masses. It is clear that mutational events are associated with HCCs and that they have a broad range of cytological and histological features of HCC. The cells in these tumors can be diploid, near-diploid, or aneuploid. They can appear relatively morphologically normal or have markedly abnormal ratios of nuclear to cytoplasmic areas and/or show bizarre spindle shapes (Grisham Citation1996). The fact that the neoplastic cells within a tumor can be clonal or quasi-clonal has been determined using a number of techniques, including analyzing X-linked genes, mosaic livers of chimeric animals, and – for viral-induced tumors – the pattern of genomic integration of the virus into DNA (Grisham Citation1996). The observation of HCCs with neoplastic cells that are clonal (or even quasi-clonal) is consistent with an initial chemically induced mutation that starts the process of tumor development and encompasses stages with additional mutations and dysregulated cell proliferation/growth (Luo et al. Citation2013).
The evidence for this AOP (i.e. the chemically induced formation of pro-mutagenic DNA adducts leading to HCC) is strong. The weight of the evidence indicates that the mutation induced by the chemical (starting with a specific chemical-induced pro-mutagenic DNA adduct – the MIE) is responsible for initiating the process that ultimately led to the formation of the tumor. Depending upon the events occurring during the KE3 stage, that mutation would be expected to be still present in the majority of tumor cells. Additional mutational events during these later stages may also result in alterations in some of the cells involved in the pre-neoplastic lesions, thus also affecting the presence of the initiating mutation.
HCC has been observed in the vast majority of studies of AFB1-treated mammals, birds, and fish (Cullen et al. Citation1990; Kirby et al. Citation1990; Kimura et al. Citation2004; Kensler et al. Citation2011; Pottenger et al. Citation2014). An exception is a small study in woodchucks, wherein it was noted that liver tumors occurred only in AFB1-treated animals also exposed to woodchuck hepatitis virus (Bannasch et al. Citation1995).
It should be noted that while exposure to AFB1 does result in the formation of HCC in a large variety of species, some investigations of codon 249 mutation in the tumors of certain nonhuman primates, ducks, and squirrels do not show a high frequency of this mutation (Denissenko et al. Citation1998).
Chemoprevention studies with AFB1 demonstrate essentiality of the MIE and KEs
Essentiality of KEs and support for the AOP
Essentiality is assessed for the overall AOP, rather than KE by KE. The OECD guidance and user’s handbook (OECD Citation2016) provide the defining question for essentiality as “are downstream KEs and/or the AO prevented if an upstream key event is blocked?” Examples of the types of studies that might address this issue include (1) stop-exposure, (2) reversibility/antagonism, and (3) knock-out models. Evidence can be direct (e.g. knock-out of KE1 or early KEs leads to blockage of all downstream KEs) or indirect (e.g. impact on a modulating factor for early KEs leads to expected pattern of effects on later KEs). Weight of evidence for essentiality is based on the types of available evidence. High (strong) evidence is defined as “direct evidence from specifically designed experimental studies illustrating prevention or corresponding impact on downstream KEs and/or the AO if upstream KEs are blocked or modified”. Moderate evidence is defined as “indirect evidence that modification of one or more upstream KEs is associated with a corresponding (increase or decrease) in the magnitude or frequency of downstream KEs.” Low evidence is defined as “no or contradictory experimental evidence that blocking or modulating/attenuating any of the KEs influences the KEs downstream or AO.”
A determination of strong evidence for essentially can be made by showing that downstream KEs and/or the AO do not occur in experiments that stop/interrupt exposure, or use chemicals that either block or reverse the formation of the MIE or one or more of the upstream KEs. These experiments must, by their nature, be conducted for specific chemicals. For AFB1, there is a large chemoprotectant/chemopreventive literature that provides support for the essentiality of the MIE and KEs proposed for this AOP.
Chemoprotectant/chemopreventive actions and essentiality
A number of substances have been shown to act as chemoprotectant or chemopreventive agents against AFB1-induced liver tumors (Kensler et al. Citation2004; Yates et al. Citation2007; Johnson et al. Citation2014). They may act at several points in the AOP beginning with exposure to AFB1 and the formation of AFB1-specific pro-mutagenic DNA adducts (MIE), and ending with the occurrence of tumors (AO). The actions of selected chemoprotectants are briefly described below and linked to the KEs in the AOP, including some discussion of various cellular signaling pathways that likely underlie their actions (Ma and He Citation2012; Jaramillo and Zhang Citation2013; Taguchi et al. Citation2016).
Chemoprotective agents may affect three aspects of this AOP:
protection against the formation of pro-mutagenic adducts;
protection against the formation of mutations in tumor-critical genes;
protection against the formation of pre-neoplastic lesions (AHF) and progression to tumors through appropriate cell cycle checkpoints or cytotoxicity.
A chemoprotective agent may affect more than one KE. At the metabolism stage of the pathway (which occurs prior to the MIE), chemoprotectants could enhance the AFB1 detoxification metabolic pathways (e.g. GST conjugation). This can decrease formation or alter availability of the pro-mutagenic exo-epoxide metabolite, and thereby block or reduce all the subsequent KEs. Treatment of rats with oltipraz before exposure to AFB1 provides a specific example for this case study, in which the MIE, AFB1-binding to DNA, was specifically reduced and KE3, the GGT-positive hepatic foci (markers for eventual liver tumors), was also reduced (Kensler et al. Citation1987). Other studies evaluated both adduct formation and impact on later KEs; they demonstrate that a 65–70% reduction in AFB1-induced DNA adducts (MIE), attributed to increased GST activity, corresponded with up to 100% reduction in both AHF and in liver tumors in rats (Roebuck et al. Citation1991; Kensler et al. Citation1998b; Elegbede and Gould Citation2002; Yates et al. Citation2006; Johnson et al. Citation2014). These data support the essentiality of the MIE. In addition, given that AFB1 adducts are only pro-mutagenic ones, as a substantial portion (30–35%) of adducts remain in rats with basically no AHF and no AO/HCC, the concept of “essential but not sufficient” is also demonstrated for the MIE.
Data from studies in human populations show that treatment with known modulators of AFB1 metabolism (e.g. chlorophyllin) resulted in reduced urinary levels of N7-AFB1-G (derived from AFB1-induced DNA adducts) (Egner et al. Citation2006), indicating reduced levels of the MIE. Some studies suggest that KE1 (Insufficient/Mis-repair of DNA) such as eukaryotic NER expression, is induced by exposure to phytochemicals (Gross-Steinmeyer et al. Citation2010), and alteration of DNA repair has been suggested as a pathway of chemoprevention for AFB1 carcinogenesis (Gross-Steinmeyer and Eaton Citation2012). This would then affect the AOP at KE1/KE2, with increased faithful and adequate repair inhibiting or reducing subsequent KEs and the AO; this is, then, a demonstration of essentiality.
At the KE3 stage, chemoprotectants may act to block or lessen the progression of cells containing AFB1-induced critical mutations to pre-neoplastic lesions and ultimately to tumors. For example, some chemoprevention studies suggest some of these agents may block cell proliferation and various aspects of progression, such as inflammation, oxidative stress, and apoptosis. Activation of the Nrf2-Keap1 pathway may intervene at this level (Kensler et al. Citation2011; Taguchi et al. Citation2016). Using the recently developed Nrf2 knock-out rats, Taguchi and colleagues demonstrated several targets for Nrf2 impact on AFB1-related effects resulting in a reduced level of AHF. These include the binding to the Antioxidant Regulatory Element (ARE) of many ARE-regulated genes; these include those affecting xenobiotic metabolism (GSTs, UDPGT, etc.) and genes affecting oxidative stress and inflammation, which may play a role in the later KE3 AHF formation (GST-P, SOD, NQO, HO, etc.). Thus, the chemoprotective agents that affect Nrf2 can act between AHF/progression and HCC tumor formation, between KE3 and the AO (Kensler et al. Citation2004; Yates et al. Citation2007; Liby et al. Citation2008; Olden and Vulimiri Citation2014; Taguchi et al. Citation2016).
Actions of some specific chemoprotectants
A number of chemopreventive/chemoprotectants have proved useful in protecting rodents and/or humans from the AFB1-induced KEs and/or AO; these include selenium and many phytochemicals such as chlorophyllin, tea polyphenols, and red ginseng. Oltipraz and a triterpenoid – 1-[2-Cyano-3-,12-dioxoleana-1,9(11)-dien-28-oyl] imidazole (CDDO-Im) – has been used in studies of AFB1 and HCC induction. We discuss some additional details below, as they provide substantial significant support for the essentiality of the KEs in this AOP.
The 1,2-dithiol-3-thione, oltipraz, appears to be chemoprotective by affecting several processes involved in AFB1-induced HCC. Oltipraz can increase GST activity along with other Phase II enzymes (Roebuck et al. Citation1991; Primiano et al. Citation1995; Kensler et al. Citation1998b; Wang et al. Citation1999; Elegbede and Gould Citation2002; Yates et al. Citation2006). It also likely acts by activating the Nrf2-Keap1 pathway, mentioned above (Kensler et al. Citation2011). It may also increase DNA repair; however, there is only limited evidence for this hypothesis (O'Dwyer et al. Citation1997). In a series of experiments conducted in rats and in hepatic tissues in vitro, dietary administration of oltipraz prior to and then concurrent with AFB1 dosing resulted in remarkable alterations in many key markers including the following: decreases in AFB1 binding to DNA (decreased MIE); increased induction of Phase I and Phase II hepatic enzymes (including CYP450 and GST); and decreased volume of GGT+ hepatic foci (decreased AHF, or KE3). All these changes were biologically and statistically significant (Kensler et al. Citation1987).
Johnson et al. (Citation2014) demonstrated that administration of the triterpenoid CDDO-Im prior to and during 28 days of dosing rats with AFB1 provided complete protection against the formation of both pre-neoplastic lesions (KE3) and liver tumors. This protection occurred despite the fact that the rats maintained a substantial (although reduced) burden of AFB1 DNA adducts (MIE) in their liver. Triterpenoids are also potent inducers of the Nrf2-Keap1 ARE pathway, which may also affect KE3 as discussed previously (Taguchi et al. Citation2016). Commentaries (Eaton and Schaupp Citation2014; Olden and Vulimiri Citation2014) on the Johnson paper both noted that Nrf2 activation introduced a nonlinearity into the AFB1 adduct-to-tumor relationship and might also provide significant protection in human populations exposed to AFB1.
Key event relationships (KERs)
A KER describes the relationships between an upstream and a downstream event. They can be adjacent (i.e. between sequential events) or non-adjacent (i.e. between events that are not contiguous). The OECD guidance and user’s handbook indicates that development of the AOP requires description and assessment of the evidence supporting the biological plausibility, the empirical support for, and uncertainties and/or conflicting evidence for the AOP and associated KERs.
Biological plausibility of KERs
The OECD guidance (OECD Citation2014, Citation2016) provides a table that summarizes the information to be considered for each of the adjacent and non-adjacent KERs as to their biological plausibility; these are noted in of this manuscript. The defining question for this assessment is “is there a relationship (i.e. structural or functional) between KEup and KEdown consistent with the established biological knowledge?”
Empirical support of KERs
In , we also provide the OECD guidance on empirical support for the adjacent and non-adjacent KERs. The defining question is “does the empirical evidence support that a change in KEup leads to an appropriate change in KEdown?” For assessing the empirical support, it is important to consider the evolved Bradford Hill considerations (Meek et al. Citation2014a, Citation2014b; Becker et al. Citation2015a) including dose–response concordance, temporality, and incidence concordance. The evidence and an assessment of its strength for our AFB1 case study are included in the KER discussion section below. Following current practice and guidance we have arrayed supporting empirical information in a dose- and time-concordance table (). In assessing the appropriateness of the selected MIE and KEs, it is necessary to demonstrate that the events occur in a temporal sequence. It is also important to demonstrate that, for the MIE and each of the KEs, there is a dose–response relationship between chemical exposure and the magnitude of the response of the MIE or KE. provides an example for an optimal data set, based in part on available published information; the “+” and “−” symbols indicate predicted responses for an ideal dataset, upon which we have layered the available supporting data. KEs for which there are reliable in vivo dose–response data with AFB1 are shaded; KEs for which there are some in vivo data for AFB1 are hashed; KEs with only limited relevant data/information for AFB1 are not shaded or hashed. This direct juxtaposition emphasizes what data are and what are not available for AFB1.
AFB1 is a data-rich chemical, and it clearly induces HCC in many species. Much of the research for AFB1 was conducted over the last several decades; however, rather than having a goal of understanding all the key steps between exposure and HCC, most of the published studies were designed to investigate specific effects (often in vitro) that represent only a portion of the AOP. Moreover, there are very few studies designed to provide dose–response data for the KEs. Thus, attempting to array the available useful information on AFB1 in a dose- and time-concordant manner is an extremely challenging and unsatisfactory experience, emphasizing the scarcity of appropriate dose–response data for the identified KEs as indicated in . Data are largely inadequate to develop the quantitative relationships for the KERs. The induction of pre-neoplastic lesions (KE3; AHF) and HCC might be an exception. However, quantitative analysis of the later KEs/KERs is not the focus of this effort.
Table 3. Hypothetical dose- and time-concordance table for AFB1 data on key events (KEs), with some qualitative data.
There are sufficient data to indicate that at least one hot-spot for mutation induction occurs in codon 249 (G:C to T:A transversion) of the p53 gene. Because of a lack of technical methods, there are no studies conclusively demonstrating that AFB1 exposure induces these specific mutations in vivo and in the target tissue and critical gene (oncogene/tumor suppressor gene). To be optimal, demonstration of a specific cancer gene mutation would have to be shown following soon after a relatively short exposure to AFB1, and long before the observation of any pre-neoplastic AHF lesions or HCCs.
There have, however, been studies in which rodents have been exposed to AFB1 and either pro-mutagenic DNA adducts, or increased mutant frequency in a surrogate gene (Lac I or cII) of a transgenic rodent have been observed (Autrup et al. Citation1996; Macé et al. Citation1997). Some of these data are shown in . There is some dose–response information for the hepatic formation of AFB1 DNA adducts in rodents, but the in vivo rodent mutagenesis studies were conducted with only a single administration of a single mutagenic dose level; therefore, no dose–response data on in vivo mutation induction are available.
Data from studies with chemoprevention agents add considerably to the WOE and understanding of the KEs as necessary, but not sufficient. That is, demonstration of early KEs (e.g. DNA adduct formation) does not ensure that later KEs or the adverse outcome are a given. In particular, two chemoprevention datasets are included in : Roebuck et al. (Citation1991) with oltipraz, and Johnson et al. (Citation2014) with CDDO-Im. Both datasets demonstrated a substantial reduction – even elimination – in tumor burden, with a concomitant decrease in AHF, that was correlated with a reduction in, but not total elimination of, the pro-mutagenic DNA adducts (Johnson et al. Citation2014). Both datasets demonstrate that the presence of pro-mutagenic adducts, alone, does not result in AHF or HCC; that is, pro-mutagenic DNA adduct formation is viewed as necessary but not sufficient.
Uncertainties and conflicting evidence for KERs and AOP
Finally, the OECD guidance on AOP development provides for an overall assessment as to the degree of uncertainty and conflicting evidence for the KERs and the AOP. The defining question is “are there inconsistencies in the empirical support across taxa, species which don’t align with the appropriate pattern for the hypothesized KERs and AOP? Are there significant knowledge gaps or uncertainties with regard to the relationship between the KEs and overall AOP?” The language in the OECD guidance (as provided in ) is somewhat counterintuitive; the descriptor “strong” for gaps and inconsistencies means that there are few data gaps and little inconsistent data. We have used somewhat different descriptors in our evaluations below and in the summary .
Both adjacent KERs and non-adjacent KERs are described below and illustrated in and , respectively. summarizes the conclusions of our assessments of the weight of the evidence that underpins these KERs, based on the definitions provided in the OECD guidance.
Figure 2. Adjacent KERs for the AOP on mutagenic MOA for HCC: formation of pro-mutagenic DNA adducts leads to HCC.
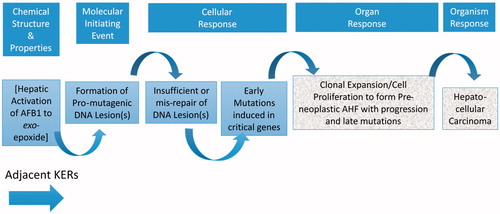
Figure 3. Non-adjacent KERs for the AOP on mutagenic MOA for HCC: formation of pro-mutagenic DNA adducts leads to HCC.
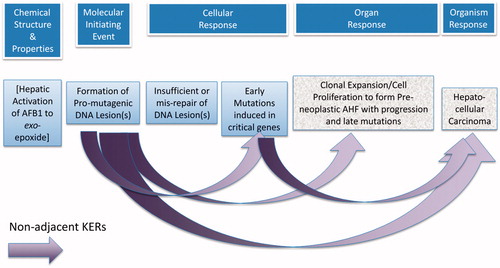
Table 4. AFB1: formation of pro-mutagenic DNA adducts leads to hepatocellular carcinoma: determination of strength of evidence levels.
Description of the adjacent key event relationships
KER1: MIE to KE1 (formation of pro-mutagenic adduct directly to insufficient/mis-repair of pro-mutagenic adducts)
DNA repair systems in multiple species can recognize and attempt to repair damage caused by the presence of bulky adducts. For AFB1, the two pro-mutagenic adducts formed from the exo-epoxide are N7AFB1-G and AFB1-FAPy; both these adducts can induce DNA repair (Guo et al. Citation2005). When DNA repair is faulty (mis-repair) or when it does not occur ahead of DNA replication, then the incorrect/inadequate repair can result in a mutation when the DNA replicates. The biological plausibility for the KER1: MIE → KE1 is strong.
Empirical support for KER1 is moderate and indirect. At this time, techniques to provide a direct measurement of insufficient or mis-repair are not available, and there are no quantitative data applicable to this KER. The only feasible techniques for indirect assessment of this KER include assessing DNA adducts and the mutations which result from potential insufficient or mis-repair of them, or using the relatively recent FYDR transgenic mouse, as described previously. There are data from in vitro systems and in surrogate genes that provide support for this KER.
Species and tissue differences in repair capacities provide indirect empirical support for KER1. For instance, nuclear extracts from the livers of mice administered AFB1 in vivo repair N7-AFB1-G adducts at a greater rate than AFB1-FAPy adducts; in rat liver, repair rates of both types of adducts were similar (Bedard et al. Citation2005).
KER2: KE1 to KE2 (insufficient repair or mis-repair of pro-mutagenic DNA adducts directly to early (in the etiology of the tumor) induced mutation in cancer critical genes)
Insufficient or incorrect DNA repair followed by DNA replication results in permanently fixing a mutation once replication of that erroneous base has occurred. This is well established in the field of mutagenesis for a wide variety of genes. However, obtaining direct evidence that insufficient repair or mis-repair leads to the induction of mutations in tumor-critical genes is technically challenging.
Based on data from the surrogate gene mutation assays, AFB1 is able to induce the type of mutation (G:C to T:A transversion) that is seen in codon 249 of the p53 gene in human HCCs from regions with high AFB1 exposure. The fact that this class of mutations can be induced by AFB1 indicates insufficient or mis-repair of the pro-mutagenic DNA adducts is biologically plausible. Thus, biological plausibility is rated strong for KER2: KE1 → KE2.
Empirical data for KER2 are mostly indirect. The majority of both N7-AFB1-G and AFB1-FAPy adducts are replaced following depurination (and subsequent BER) or repair by NER (Bedard and Massey Citation2006) but mis-repair occurs occasionally (Alekseyev et al. Citation2004; Kew Citation2013; Mulder et al. Citation2014). If BER/NER capacity is insufficient, mutations may result through other repair pathways such as post-replication repair, or if replication continues across the unrepaired lesion. The evidence supporting the role of NER in humans is strong. This is evident when evaluating populations in The Gambia and China, which indicate an increased risk factor for HCC in cases with high AFB1 exposure and populations with polymorphisms in the DNA repair gene X-ray repair cross complementary group (XRCC) (Kirk et al. Citation2005; Long et al. Citation2006, Citation2008). In addition, these polymorphisms are associated with both increased levels of AFB1-DNA adducts and with mutations at codon 249 of p53 (Long et al. Citation2008, Citation2009).
Yeast strains with different DNA repair deficiencies show increased mutant frequency in a surrogate gene mutation assay following AFB1 treatment compared to those with wild type DNA repair capability (Guo et al. Citation2005). NER, HR repair, post-replication repair, and several cell cycle checkpoint proteins all function in the repair of AFB1 adducts in yeast. Error-prone post-replication repair and apurinic endonucleases are secondary repair pathways that are used by cells when the major pathways are not functional or are overwhelmed. These secondary pathways tend to produce mutations. The mechanism of DNA repair in Archaea is similar to that in eukaryotes. Banarjee et al. (Citation2011) showed the P2 DNA polymerase in S. solfataricus bypasses the N7-AFB1-G adducts in an error-free manner, but conducts error-prone replication past the AFB1-FAPy adduct and mis-inserts dATP, consistent with G:C to T:A transversion (Banerjee et al. Citation2011). Although very few data are available on dose–response, the induction of surrogate mutations occurs in a dose-responsive manner in in vivo assays; surrogate mutations (Big Blue® transgene) were measured shortly after exposure to AFB1 (Dycaico et al. Citation1996). Because empirical support for KER2: KE1 → K2 is limited to indirect evidence, it is rated as moderate.
The evidence for KER2 has few inconsistencies/uncertainties. Clearly, AFB1 can induce the type of gene mutations required to initiate a mutation in a cancer critical gene such as p53. However, there is only indirect evidence that AFB1 actually does induce the specific cancer critical gene mutation in the liver of exposed animals (or humans); however, such indirect data are available from several sources. There are no specific data on whether cancer critical gene mutation(s) happens early in the etiology of the tumor.
KER3: KE2 to KE3 [early induced mutation in cancer critical genes directly to cellular proliferation, clonal expansion of mutant cells, and progression]
The biological plausibility for KE2 to KE3 is strong; however, there are no data for AFB1 demonstrating that cells containing critical gene mutations specifically lead to cellular proliferation and clonal expansion, and tumor progression; this is also true for the majority of, if not all, chemicals that act via this AOP. This KER must occur both for AFB1 and other chemicals. This conclusion is based on chemoprevention studies and also on a plethora of initiation–promotion studies for chemically induced tumors in rodents – which primarily tie DNA adducts to pre-neoplastic lesions. Thus, biological plausibility is strong for KER3: KE2 → KE3.
Empirical support for KER3: KE2 to KE3 is weak. While studies have been conducted to quantitate clonal expansion and pre-neoplastic foci following exposure to several chemicals including AFB1, there are no data with which to evaluate the quantitative relationship between cells carrying critical cancer gene mutations and clonal expansion/pre-neoplastic foci.
In vitro data collected from two human-derived cell lines suggest that doses of AFB1 sufficient to induce DNA damage also induce an incomplete cell cycle checkpoint response, likely involving p53; this suggests a potential mechanism that might ultimately lead to tumor formation in vivo (Gursoy-Yuzugullu et al. Citation2011).
For AFB1, the product of the p53 gene mutated at codon 249 may itself impair the apoptotic process. In Hep3B human-derived hepatoma cells, transfection with mutant p53 (p53mt249) increased transcription of insulin-like growth factor II (IGF-II), which could then induce cell proliferation. Wild-type p53 inhibited binding of the TATA-box binding protein (TBP) and the Sp1 transcription factors to the IGF-II promoter, possibly by binding to this promoter. However, p53mt249 increased the formation of transcription complexes, possibly due to lack of binding to the promoter (Lee et al. Citation2000).
Overall, no important inconsistencies or data gaps were identified in the available data.
KER4: KE3 to AO (AHF with progression and late mutations directly to HCC)
The biological plausibility for KER4: KE3 → AO is strong. Initiation–promotion studies provide substantial evidence of this link. Multiple biological processes are involved in tumor development/progression, including apoptosis, inflammation, the development of a tumor microenvironment, interference with the anti-oxidant response, and others.
There are six general mechanisms that could be operating in this overall progression: (1) decrease in the activity of the Keap1/Nrf2/ARE pathway; (2) decrease in p53 function leading to increased survival in the presence of genomic instability; (3) changes in the tumor microenvironment within cells; (4) alterations in apoptosis; (5) changes in Wnt/β-catenin signaling; and (6) gene expression changes that may be related to cancer promotion/progression (Caballero et al. Citation2004; Haridas et al. Citation2004; Ikeda et al. Citation2004; Yates et al. Citation2007; Liby et al. Citation2008; Gross-Steinmeyer and Eaton Citation2012; Liby and Sporn Citation2012; Ma and He Citation2012; Gong et al. Citation2013; He et al. Citation2013; Jia et al. Citation2013; Dang et al. Citation2014; Miyajima et al. Citation2014). In addition to these six possible mechanisms, epigenetic changes in DNA methylation may occur and may be related to the progression of cancer (Kwak et al. Citation2001; Zhang et al. Citation2003; Fabregat Citation2009; Honda et al. Citation2011; Shelton and Jaiswal Citation2013).
Empirical evidence is available for early stages of KER4. The progression of AHF to liver cancer has been well studied with several hepatotoxins (Pitot et al. Citation1990; Sargent et al. Citation1996; Dragan et al. Citation1994, Citation1995, Citation1997). There is a plethora of initiation–promotion studies that provide substantial evidence and some quantitative information concerning the link between clonal expansions of foci to HCC. There is a seemingly strong relationship between AHF and tumors, and AHF have long been considered as pre-neoplastic lesions for HCC (Bannasch et al. Citation1985; Ikeda et al. Citation2004; Ribback et al. Citation2013); however, there are no specific endpoints and no direct measures for post-AHF stages. One might predict increasing volume of AHF (Dragan et al. Citation1997) and could quantify the growth of AHF (Xu et al. Citation1990a, Citation1990b; Dragan et al. Citation1995; Pitot et al. Citation1996). Likely acquisition of at least some hallmarks of cancer, such as invasiveness and unregulated growth, would be expected as potential markers that might be used to develop quantitative data for this KER (Hanahan and Weinberg Citation2000, Citation2011).
Chemoprevention studies suggest a strong relationship between AHF and HCC tumor formation (Kensler et al. Citation2004; Liby et al. Citation2008; Yates et al. Citation2007; Olden and Vulimiri Citation2014), that would also encompass KER4. Hence, the empirical support for early AHF-related events in KER4 is strong.
Empirical evidence for later stages of KER4 is sparse. For chemicals such as AFB1, in which the critical (or one of the critical) mutation(s) appears to involve p53, a defective p53 gene may contribute to telomere dysfunction. Continuous hepatocyte renewal may be associated with telomere shortening and chromosomal instability (Miura et al. Citation1997; Golubovskaya et al. Citation1999; Farazi et al. Citation2003). In transgenic mice, telomere dysfunction enhanced tumor initiation but interfered with tumor progression/promotion. However, p53 mutation leading to inactivation of the gene, in combination with telomere dysfunction and increased hepatocyte turnover, may increase the risk of HCC (Farazi et al. Citation2003; Zhang et al. Citation2006; Pogribny et al. Citation2011; Zhang et al. Citation2012; Wu et al. Citation2013).
Intracellular signaling mechanisms are involved in the creation of the tumor microenvironment (Hanahan and Weinberg Citation2000, Citation2011). These include alterations in cellular circuits governing proliferation, differentiation, motility, and viability, all affecting the hallmarks of cancer.
TGF-β has a dual effect on hepatocytes – inhibiting proliferation and inducing apoptosis, and it may be affected by AFB1. Experiments using TGF-β null mutant mice show that this signal is required for normal development and appears to be involved in destruction of damaged cells in the liver. TGF-β also mediates interferon-induced apoptosis in AHF in rats. In rats, AFB1 exposure altered biological processes in hepatic stem-like cells related to cell motion, cell adhesion, immune response, and signal transduction (Yang and Ji Citation2014). Reduced levels of TGF-β produce an increase in E-cadherin and reduce cellular motility, one of the intracellular networks associated with the cancer hallmark of invasion and metastasis (Fransvea et al. Citation2008). Empirical evidence for these later events in KER4 is not as strong as for the earlier events.
Overall, no substantial inconsistencies were identified in the available database; there are gaps, however, as there are few direct data for aspects other than AHF, and quantitative data are very limited.
Non-adjacent key event relationships
We found that adjacent KERs for our AFB1 case study were difficult to measure and were not well represented in the reviewed literature. For instance, the KE “insufficient and/or mis-repair of adducted DNA,” is technically challenging to measure directly. With limited data available for adjacent KERs, we found it is particularly useful to consider the non-adjacent KERs (naKERs), for which it may be easier to obtain quantitative information. The same factors (biological plausibility, empirical evidence, and inconsistencies/uncertainties) are considered for the naKERs and the assessments are summarized in .
naKER1: MIE to KE2 (formation of pro-mutagenic DNA adducts indirectly to early induced mutation in cancer critical genes)
The biological plausibility for naKER1: MIE → KE2 is strong. Pro-mutagenic adducts formed by a number of chemicals (including AFB1 metabolites) may be repaired/removed, may result in cell death, or may result in mutations. The fidelity of the repair processes and probability of mis-repair determine whether mutations arise in tumor-critical genes. Both of the AFB1-induced adducts, N7-AFB1-G and AFB1-FAPy, can induce mutations (Brown et al. Citation2006; Lin et al. Citation2014a, Citation2014b). Published studies, including several in human populations, identified codon 249 of the p53 gene as a site with a higher than normal mutation rate (Puisieux et al. Citation1991; Jackson et al. Citation2003; Gouas and Shi Citation2009; Lin et al. Citation2014a, Citation2014b).
The empirical support for naKER1, MIE → KE2 and AFB1 is limited and indirect; therefore, we assessed it as moderate. There are no available data in either rodents or humans, which directly quantify the relationship between the numbers of pro-mutagenic AFB1 adducts and the frequency of mutation, either in surrogate genes or in the critical cancer genes in tumor target tissue. However, there are both in vitro and in vivo examples from other direct-acting alkylating agents which also induce HCC. These DNA-reactive mutagens demonstrate non-linear dose–responses for mutations measured in surrogate (non-cancer related) genes versus administered dose. DNA adduct data are also available for some of these agents (Doak et al. Citation2007; Gocke and Müller Citation2009; Pottenger et al. Citation2009; Bryce et al. Citation2010; Dobo et al. Citation2011), which demonstrate non-linear responses for adducts versus mutations, the response–response relationship. The induction of mutations can occur shortly after exposure to various chemicals, including AFB1. Thus, based on the overall pattern supported by data other than for AFB1, the empirical support for naKER1 can be assessed as moderate.
Our conclusion was that the consistency of the data and lack of uncertainties for naKER1 were moderate. There were very few inconsistencies in the data base, but a lack of direct evidence/data that AFB1 can actually induce the specific, cancer critical, gene mutation. However it is clear that AFB1 can induce the type of mutation, a G:C to T:A transversion, that is found in p53 – a cancer critical gene in a high percentage of HCC tumors in humans.
naKER2: MIE to KE3 [formation of pro-mutagenic DNA adducts indirectly to proliferation and clonal expansion of the critical gene mutated cells (pre-neoplastic lesions)]
The biological plausibility for naKER2 MIE→KE3 is strong. The theory that pro-mutagenic DNA adducts will lead to the proliferation and clonal expansion of critical gene mutated cells (and form pre-neoplastic lesions) is well accepted. AHF are believed to result from mutations expressed in cells that demonstrate reduced apoptosis and increased proliferation, likely linked to the induced mutations (Bailey et al. Citation1996; Giri et al. Citation2002; Hang et al. Citation2003; Alekseyev et al. Citation2004; Lin et al. Citation2014b).
AFB1 chemoprevention data indicate a strong relationship between adducts and AHF and/or tumor formation, a relationship which is not a simple linear dose–response (Kensler et al. Citation2004; Yates et al. Citation2007; Liby et al. Citation2008; Johnson et al. Citation2014; Olden and Vulimiri Citation2014).
The empirical support for this naKER2 is mostly indirect and judged to be moderate. Unfortunately, there are few data to support a quantitative relationship between adducts and AHF, largely because experiments with exposure to AFB1 did not include quantification of AFB1-induced adducts. There are, however, some indirect data. Chemoprevention data show that a reduction in adducts results in a decrease in and even ablation of foci. Some evaluations propose linearity in the dose–response for adduct formation as a function of administered dose in both rats and trout (Appleton et al. Citation1982; Bechtel Citation1989; Buss et al. Citation1990). Recent work of Johnson et al. (Citation2014), however, showed that linearity does not seem to apply to the relationship between pro-mutagenic DNA adduct formation and observation of AHF (or HCC tumors, the AO). AHF were reduced to background levels by the chemoprotectant CDDO-Im, while around 30% of the original level of N7-AFB1-G adducts were still present. These authors note that there remained ∼15,000 AFB1 DNA adducts per cell following treatment with the chemoprotectant, while AHF were at background levels, and no HCC tumors were induced (100% ablation).
Overall this naKER2 was determined to have no substantive inconsistencies, but there were gaps. The universality of both DNA adducts and AHF in the pathogenesis of liver cancer suggests that the wide taxonomic applicability noted elsewhere in this AOP is likely also true for this naKER.
naKER3: MIE to AO (formation of pro-mutagenic DNA adducts indirectly to HCC)
The biological plausibility for naKER3, MIE→AO is strong, and the theory is well accepted. The relationship of adducts to HCC depends upon two well-established relationships: between adducts and AHF, and between AHF and HCC. Indirect support also comes from chemoprevention data demonstrating that decreases in pro-mutagenic adducts resulted in decreased downstream events (AHF and HCC incidences) (Roebuck et al. Citation1991; Johnson et al. Citation2014). Indeed, these data also provide strong support for the essentiality of this naKER3: a reduction in MIE (reduced adducts) results in decreased or eliminated AO response (no tumors).
Empirical support for naKER3 is moderate. Formation of AFB1-induced DNA adducts clearly precedes tumor formation; thus, the necessary temporal pattern exists. The plethora of published chemoprevention studies support a relationship between AFB1-induced DNA adducts and HCC. Although data are not available for a quantitative analysis, there are data to indicate that low-dose linearity does not apply to this naKER3 between adducts and HCC. Recent work of Johnson et al. (Citation2014) showed that HCC incidence in AFB1-treated rats was reduced to zero by the administration of chemoprotectant CDDO-Im, while adduct burden remained substantial, albeit significantly reduced (∼30%), with ∼15,000 adducts per cell following treatment with the chemoprotectant. Other chemoprevention data also support the reduction or elimination of HCC, accompanied by a reduction in – but not a complete elimination of – AFB1-induced DNA adducts. This was, for example, demonstrated for oltipraz prevention data from humans demonstrating that administration of 500 mg oltipraz/week resulted in a decreased burden of AFB1-induced serum albumin adducts, which could serve as a biomarker for AFB1-induced DNA adducts (Kensler et al. Citation1987; Roebuck et al. Citation1991; Maxuitenko et al. Citation1993). The reduction in AFB1-albumin adducts presumably stems from a reduced availability of the exo-epoxide, and presumably parallels a reduced burden of AFB1-induced adducts generally; it is expected to result in a concomitant reduction in tumor burden eventually (Kensler et al. Citation1998a). Thus, while formation of AFB1 adducts may appear to be dose-linear at low dose, it is clear there is not a one-to-one correspondence between numbers of adducts and tumors. That is, only some small subset of these pro-mutagenic DNA adducts will result in mutations (non-linear dose–response) or ultimately non-linear development of HCC.
While there are gaps, we considered that information on naKER3 was strong on data consistency and lack of uncertainty. There are no definitive data on causality for the AFB1-specific pro-mutagenic adducts and HCC. Nevertheless, there is a strong relationship between pro-mutagenic DNA adducts and tumors. Furthermore, when chemopreventives are used to decrease this DNA adduct burden, the HCCs are either eliminated or markedly reduced.
naKER4: KE2 to AO (early induced mutation in cancer critical genes indirectly to HCC)
The biological plausibility for naKER4: KE2→AO is strong. The relationship of mutations to cancer is well-established. However, there are no specific studies addressing chemically induced (including AFB1-induced) critical gene mutations and the subsequent progression through AHF to HCC. The cells that are mutant for the critical cancer gene undergo a change in phenotype and clonally expand into pre-neoplastic lesions, some of which go on to form HCC. This specific naKER has not been directly measured; however, there is indirect evidence from studies in rats, humans, and trout. The same is true for essentiality, with no direct data linking KE2 to the AO, although the demonstrated presence of the p53 codon 249 GC → AT transversion mutation in HCC tumor tissue is a biomarker for AFB1-induced mutation.
Empirical support for naKER4: KE2 → AO is weak. Data adequate to establish quantitative dose–response relationships are not extant. A correlation between AFB1 exposure, codon 249 mutations, and HCC has been established in humans (Besaratinia et al. Citation2009). There is no quantitative information for the induction of these mutations and, thus, no quantitative understanding with data to support the relationship between the AFB1-induced critical cancer gene mutations and HCC. Hence, our assessment of overall empirical evidence of this naKER4 as weak.
The Dycaico et al. (Citation1996) Big Blue™ rat study and the Chen et al. (Citation2010) Big Blue™ neonatal mouse study show that AFB1 can induce mutations in a surrogate gene following a relatively short in vivo exposure time; however, these studies used a single dose only. In a review of p53 mutations at codon 249 (R249S), Gouas and colleagues (Gouas and Shi Citation2009; Gouas et al. Citation2010) report that cancer patients have significantly higher levels of circulating mutated DNA compared with healthy individuals. Using restriction enzyme digest methods (RFLP-PCR) R249S has been detected in circulating DNA from patients in The Gambia, and this was strongly associated with HCC. The presence of this mutation in DNA from human plasma was linked to the codon 249 p53 mutation in the tumors of individual patients. In addition, using a sensitive and quantitative mass spectrometric method, combined with other molecular techniques, Jackson et al. (Citation2003) were able to detect the R249S mutation in DNA from the plasma of patients from The Gambia both before and after clinical diagnosis of HCC in the same patient. These data provide good evidence of an appropriate temporal sequence of events: induction of the cancer-specific mutation precedes tumor formation.
This naKER4 was judged to have a low level of inconsistencies, but there is a moderate level of gaps or uncertainties. There are no data directly addressing this naKER4; however, there is a substantial amount of indirect information. There is a high association between exposure to AFB1 and codon 249 p53 gene mutations in human HCC cells, but there are not adequate data to provide any quantitative assessment.
Discussion
We used a case study on a data-rich chemical considered to have a mutagenic MOA for hepatocellular carcinoma as the basis to develop an AOP according to the guidelines established by the OECD program. Assembling and evaluating the available data for AFB1, in turn, prompted analysis of data requirements and limitations in establishing a mutagenic MOA for AFB1 and other mutagenic or genotoxic chemicals. A conclusion of our case study preparation is that demonstration of genotoxicity or mutagenicity does not equate to demonstration of a mutagenic MOA for tumor formation for a particular agent.
The critical, influential key event that defines a chemical’s mutagenic MOA for cancer is the chemical induction of mutation(s) in the cancer critical gene(s) that are involved in and occur early in the etiology of specific tumors. It is important to understand that mutagenic chemical exposure results in mutations across the genome. Only those specific mutations in cancer critical genes are reflected in KE2 of this AOP. Often there are multiple mutations that occur in the etiology of tumors. It is, however, the initial, early chemical induction of mutation in the cancer critical gene(s) that defines the mutagenic MOA. Additional mutations that occur later in tumorigenesis, during the process of increased cell proliferation and clonal expansion of mutated cells (in some cases due to genomic instability), are not the events that define a mutagenic MOA.
Our AOP is reflective of chemicals, such as AFB1, that either can directly form, or are metabolized to chemicals that can form pro-mutagenic DNA adducts which lead to the specific mutations in cancer relevant genes and ultimately lead to HCC. That is, the mutation induction is initiated by the formation of pro-mutagenic DNA adducts. There are additional, different, initiating events for the induction of mutations, such as intercalation or cross-linking. These should all be considered as separate AOPs with different MIEs that could converge with the AOP for HCC described in this paper. Thus, each of these putative AOPs would have an MIE and early KEs reflective of a specific type of DNA interaction, but the later KEs would not differ from the AOP for induction of pro-mutagenic DNA adducts leading to HCC. In addition, AOPs involving the chemical induction of mutation in a cancer critical gene can be developed for tumors other than HCC. This approach would make good use of the concept of an AOP network, with KEs that converge and perhaps diverge for separate target tissues.
Likewise, a set of AOPs could be developed for the various MIEs resulting in tumors but which do not involve the chemical induction of mutation. The AOP for HCC as a consequence of aryl hydrocarbon receptor binding, developed by Becker and colleagues, is one such AOP (Becker et al. Citation2015b). Thus, it is possible to create a family of AOPs dealing with various MIEs and KEs for tumor induction. Based on our understanding of the AOP wiki, some of these AOPs are already in development or are proposed/planned.
Although the current thinking about AOPs considers them to be chemically agnostic, we found that first developing a case study based on data from a specific chemical was invaluable to understanding the MIE and the KEs that constitute the AOP. While developing this AOP we identified a number of lessons that can generally be applied to the development and use of AOPs.
Among these was the importance of identifying the specific types of data that provide information supporting the AOP. The most desirable, and often least available, data are those that assess a chemical’s ability to induce a mutation in the gene(s) involved in the etiology of the tumor. The techniques to determine the ability of the chemical to induce specific cancer gene mutations have not been generally available in the past. This situation is changing, however, as new molecular technologies become available (Parsons et al. Citation2005, Citation2010). To establish a mutagenic MOA for a particular chemical it is critical to demonstrate its ability to induce the same type(s) of mutation(s) as those identified in the tumor. Thus, we specifically formulated the wording for the KEs in this AOP. In establishing the MIE and KEs for this AOP, we were careful to select specific terminology (i.e. “pro-mutagenic DNA adduct formation and “early induced mutation in critical genes”) that would precisely identify the specific events that are involved in the AOP, and not to use terminology that we felt would be too broad or inclusive. That is, we did not use the more general terms of “DNA adduct formation” and “induced gene mutation,” so as not to characterize inappropriately all chemicals that induce one or both of these types of events as potential hepatocellular carcinogens. It is well-recognized that not all DNA adducts are pro-mutagenic (Jarabek et al. Citation2009; Swenberg et al. Citation2010; Pottenger et al. Citation2014); moreover, considering the multiple steps required between the formation of the pro-mutagenic adduct and fixation of a heritable mutation (Pottenger and Gollapudi Citation2010), those that are pro-mutagenic do not always result in mutations.
One of the stated uses of AOPs is to provide quantitative information to inform the “real” dose–response for specific chemically induced AOs. This may be particularly important for cancer, as the current practice for chemicals that are determined to have mutagenic MOAs is to use linear low-dose extrapolation below an experimentally established point of departure. This default process is thought to establish upper bounds for effects, may lead to overestimate of risk, and is unlikely to reflect the biology of tumor formation. To use quantitative KER data to assess the overall dose–response for a chemical that fits a particular AOP, it is important that adequate and appropriate data be available. While there is a growing body of evidence to demonstrate that simple low-dose linear extrapolation is not supported by the available quantitative datasets across KERs (adjacent and non-adjacent), there are not adequate, high quality dose–response data on AFB1-related endpoints for quantitative definition of the dose–response relationships for these KERs (with the possible exception of KER4: AHF → AO). It is also important to recognize that the specific quantitative relationships for KERs will almost certainly vary by chemical. For instance, the rates of conversion of pro-mutagenic adducts to critical gene mutations will vary by the specific adduct and by the efficiency of DNA repair of that adduct in the target tissue.
Based on our overall evaluation of the data, we concluded that AFB1 causes HCC by inducing mutations in cancer critical genes. The consistent pattern of information strongly supports this judgment (which is also a judgment that AFB1 has a mutagenic MOA) although the database for AFB1 is not adequate to address the rigorous requirement of AOP development. The majority of studies, even those done to elucidate AFB1 mechanisms or MOA, were performed years prior to considerations of MOAs, or AOPs and recommendations for integrated approaches to testing and assessment (IATA) in this context.
Based on the early KEs that also define a mutagenic MOA, we have developed an AOP that is applicable not only to AFB1 but also to chemicals that share the same MIE and KEs. A goal of the OECD AOP program is to create a series of AOPs that can ultimately be used for a variety of applications, such as hazard identification, risk assessment, read-across, and the development and/or refinement of test guidelines. For hazard identification and read-across one could determine what types of biological effects the chemical being studied induces and then, using the set of AOPs, predict potential adverse outcomes that might be caused by that chemical. For such uses, it is important that the KEs be carefully defined and merit high confidence, as is discussed in Becker et al. (Citation2015a). In particular, reliable prediction is a key goal for AOPs, driven in part by the high-throughput data collection that provides an enormous source of information, but which is currently without an agreed-upon framework for reliable interpretation.
In the process of assessing the AFB1 information for the case study underpinning the AOP, it became clear that the AFB1 database available to define its MOA for HCC and to also develop an AOP is far from optimal. We realize both the need and the opportunity to articulate a hierarchical list for the utility of specific types of data to define a mutagenic MOA, as noted in Moore et al. (Citation2008). The influential KE that distinguishes a mutagenic AOP from a non-mutagenic AOP is the ability of the chemical to induce gene mutations in a tumor-critical gene and to induce it early in the etiology of the tumor; it is not sufficient merely to determine that a chemical can induce mutations in any one of a number of mutation assays. Furthermore, while one potential use of AOPs is to apply data from the KERs to develop quantitative relationships, it also became clear that it is necessary to establish the appropriate kinds of data and to use only such data for an overall quantitative assessment of the relationship between the MIE and the AO. While the MIE and KEs of this AOP are well established, and there are extensive data for AFB1, there were surprisingly very few data that might be appropriate to establish quantitative relationships for most of the KERs (either adjacent or non-adjacent).
Perhaps one of the most important outcomes from the development of this AOP is the insight on optimal design of research and the kind of data needed to establish and use AOPs. In evaluating the utility of this AOP, it is clear that there are significant knowledge gaps, despite the rich database. While, for AFB1 it appears that a specific p53 gene mutation is involved in the etiology of HCC, it is clear that not all HCC tumors have this genotype. Obviously, there are other, some as yet unidentified, cancer gene mutations that can be involved. Furthermore, the specific types of pro-mutagenic DNA adducts that can lead to the various cancer gene mutations need to be elucidated. For AFB1, it is the AFB1-N7-G and the AFB1-FAPy adducts, but clearly not all N7-alkyl-G adducts are pro-mutagenic; for low molecular weight molecules, such as ethylene/ethylene oxide and propylene/propylene oxide, the N7-alkoxy-G adducts (N7-hydroxyethyl-G and N7-hydroxypropyl-G), which predominate, have been shown to be not mutagenic in site-specific mutagenesis studies (Philippin et al. Citation2014). The construction of this AOP informs the need for test method development and IATA. There are clearly opportunities for a more systematic and rational approach to data collection for such an AOP. These issues continue to be the subject of further discussion by the risk assessment and genetic toxicology communities.
Declaration of interest
The authors' current affiliations are as shown on the cover page. RS Schoeny was formerly employed by the U.S. Environmental Protection Agency (U.S. EPA). MM Moore was formerly employed by the U.S. Food and Drug Administration, Department of Health and Human Services. LH Pottenger was formerly employed by The Dow Chemical Company, also a chemical manufacturer (like Olin Corporation). M. M. Moore’s participation in developing the OECD AOP and in drafting this manuscript was supported by both her current employer (Ramboll Environ) and by funds from the Center for Advancing Risk Assessment Science and Policy (ARASP) (https://arasp.americanchemistry.com/) of the American Chemistry Council. ARASP is a coalition of independent groups and associations that promotes the development and application of up-to-date, scientifically sound methods for conducting chemical assessments. While employed by the U.S. EPA, R. S. Schoeny participated in the development of the OECD AOP and the early drafts of this manuscript as a part of her official duties. Following her retirement from the U.S. EPA, her participation in this manuscript was supported by funds from ARASP. L. H. Pottenger, K. White, and R. A. Becker worked on this paper as part of their normal job responsibilities. L. H. Pottenger, K. White, and R. A. Becker participate in ARASP as part of their normal job responsibilities; they did not receive any additional compensation for their efforts on this paper. None of the authors have appeared in any legal or administrative proceedings related to the contents of the paper with the exceptions that (1) M. M. Moore and R. S. Schoeny (as US Government employees) participated in the development of the EPA’s External Review Draft Framework for Determining a Mutagenic Mode of Action for Carcinogenicity (2007 https://archive.epa.gov/osa/mmoaframework/web/pdf/mmoa-erd-final-83007.pdf) and (2) R. A. Becker has served as a member of the Organization for Economic Cooperation and Development (OECD) Extended Advisory Group on Molecular Screening and Toxicogenomics (EAGMST) and participated in review of the OECD AOP Handbook, served as a co-reviewer of early drafts of a number of AOPs under development as part of the EAGMST AOP program, and coauthored papers on an AOP of rat liver tumor promotion, assessing weight of evidence of AOPs and applying a scientific confidence framework to AOPs. The American Chemistry Council (ACC https://www.americanchemistry.com/) is an industry trade association that represents a diverse set of companies engaged in the business of chemistry. ACC works to improve environmental, health, and safety performance through Responsible Care®, common sense advocacy designed to address major public policy issues, and health and environmental research and product testing. For a list of ACC members see https://www.americanchemistry.com/Membership/MemberCompanies/. For a list of ARASP members, see https://arasp.americanchemistry.com/arasp/Membership-Information/ARASP-Members. Drafts of the Adverse Outcome Pathway described in this paper have been reviewed under the Organization for Economic Cooperation and Development (OECD) Extended Advisory Group on Molecular Screening and Toxicogenomics (EAGMST http://www.oecd.org/chemicalsafety/testing/adverse-outcome-pathways-molecular-screening-and-toxicogenomics.htm) and are also part of the AOP wiki (https://aopwiki.org/wiki/index.php/Main_Page). The authors had sole responsibility for the writing and content of the paper. The views and opinions expressed in the paper are those of the authors, and do not necessarily reflect the views or policies of the authors' current or former employers.
Acknowledgements
The authors gratefully acknowledge the comments offered by four reviewers selected by the editor and whose identity was not made known to the authors. These comments were very helpful in improving the manuscript. We wish to thank Ted W. Simon for his efforts on this paper, including helpful discussions, quantitative insights, and contributions to early drafts.
References
- Aguilar F, Hussain SP, Cerutti P. 1993. Aflatoxin B1 induces the transversion of G->T in codon 249 of the p53 tumor suppressor gene in human hepatocytes. Proc Natl Acad Sci USA. 90:8586–8590.
- Alekseyev YO, Hamm ML, Essignmann JM. 2004. Aflatoxin B1 formamidopyrimidine adducts are preferentially repaired by the nucleotide excision repair pathway in vivo. Carcinogenesis. 25:1045–1051.
- Angsubhakorn S, Pradermwong A, Phanwichien K, Nguansangiam S. 2002. Promotion of aflatoxin B1-induced hepatocarcinogenesis by dichlorodiphenyl trichloroethane (DDT). Southeast Asian J Trop Med Public Health. 33:613–623.
- Ankley GT, Bennett RS, Erickson RJ, Hoff DJ, Hornung MW, Johnson RD, Mount DR, Nichols JW, Russom CL, Schmieder PK, et al. 2010. Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem. 29:730–741.
- Appleton BS, Goetchius MP, Campbell TC. 1982. Linear dose-response curve for the hepatic macromolecular binding of aflatoxin B1 in rats at very low exposures. Cancer Res. 42:3659–3662.
- Asplin FD, Carnaghan RBA. 1961. The toxicity of certain groundnut meals for poultry with special reference to their effect on ducklings and chickens. Vet Rec. 73:1215–1219.
- Autrup H, Jorgensen EC, Jensen O. 1996. Aflatoxin B1 induced lacI mutation in liver and kidney of transgenic mice C57BL/6N: effect of phorone. Mutagenesis. 11:69–73.
- Bailey EA, Iyer RS, Stone MP, Harris TM, Essigmann JM. 1996. Mutational properties of the primary aflatoxin B1-DNA adduct. Proc Natl Acad Sci USA. 93:1535–1539.
- Bal-Price A, Lein PJ, Keil KP, Sethi S, Shafer T, Barenys M, Fritsche E, Sachana M, Meek ME. 2017. Developing and applying the adverse outcome pathway concept for understanding and predicting neurotoxicity. Neurotoxicology. 59:240–255.
- Banarjee S, Brown KL, Egli M, Stone MP. 2011. Bypass of aflatoxin B1 adducts by the sulfolobus solfataricus DNA polymerase IV. J Am Chem Soc. 133:12556–12568.
- Bannasch P, Benner U, Enzmann H, Hacker HJ. 1985. Tigroid cell foci and neoplastic nodules in the liver of rats treated with a single dose of aflatoxin B1. Carcinogenesis. 6:1641–1648.
- Bannasch P, Khoshkhou NI, Hacker HJ, Radaeva S, Mrozek M, Zillmann U, Kopp-Schneider A, Haberkorn U, Eglas M, Tolle T. 1995. Synergistic heptaocarcinogenic effect of hepadnaviral infection and dietary aflatoxin B1 woodchucks. Cancer Res. 55:3318–3330.
- Bechtel DH. 1989. Molecular dosimetry of hepatic aflatoxin B1-DNA adducts: linear correlation with hepatic cancer risk. Regul Toxicol Pharmacol. 10:74–81.
- Becker RA, Ankley GT, Edwards SW, Kennedy SW, Linkov I, Meek B, Sachana M, Segner H, Van Der Burg B, Villeneuve DL, et al. 2015a. Increasing scientific confidence in adverse outcome pathways: application of tailored Bradford Hill considerations for evaluating weight of evidence. Regul Toxicol Pharmacol. 72:514–537.
- Becker RA, Patelwicz G, Simon TW, Rowlands JC, Budinsky RA. 2015b. The adverse outcome pathway for rodent liver tumor promotion by sustained activation of the aryl hydrocarbon receptor. Regul Toxicol Pharmacol. 73:172–190.
- Bedard LL, Massey TE. 2006. Aflatoxin B1-induced DNA damage and its repair. Cancer Lett. 241:174–183.
- Bedard LL, Alessi M, Davey S, Massey TE. 2005. Susceptibility to aflatoxin B1-induced carcinogenesis correlates with tissue-specific differences in DNA repair activity in mouse and in rat. Cancer Res. 65:1265–1270.
- Beer DG, Pitot HC. 1987. Biological markers characterizing the development of preneoplastic and neoplastic lesions in rodent liver. Arch Toxicol Suppl. 10:68–80.
- Besaratinia A, Kim S, Hainaut P, Pfeifer GP. 2009. In vitro recapitulating of TP53 mutagenesis in hepatocellular carcinoma associated with dietary aflatoxin B1 exposure. Gastroenterology. 137:1127–1137.
- Boysen G, Pachkowski BF, Nakamura J, Swenberg JA. 2009. The formation and biological significance of N7-guanine adducts. Mutat Res. 678:76–94.
- Bressac B, Kew M, Wands J, Ozturk M. 1991. Selective G to T mutations of p53 gene in hepatocellular carcinoma from southern Africa. Nature. 350:429–431.
- Brown JA, Pack LR, Sanman LE, Suo Z. 2011. Efficiency and fidelity of human DNA polymerases λ and β during gap-filling DNA synthesis. DNA Repair (Amst). 10:24–33.
- Brown KL, Deng JZ, Iyer RS, Iyer LG, Voehler MW, Stone MP, Harris CM, Harris TM. 2006. Unraveling the aflatoxin-FAPY conundrum: structural basis for differential replicative processing of isomeric forms of the formamidopyrimidine-type DNA adduct of aflatoxin B1. J Am Chem Soc. 128:15188–15199.
- Bryce SM, Avlasevich SL, Bemis JC, Phonethepswath S, Dertinger SD. 2010. Miniaturized flow cytometric in vitro micronucleus assay represents an efficient tool for comprehensively characterizing genotoxicity dose-response relationships. Mutat Res. 703:191–199.
- Burden N, Sewell F, Andersen ME, Boobis A, Chipman JK, Cronin MTD, Hutchinson TH, Kimber I, Whelan M. 2015. Adverse outcome pathways can drive non-animal approaches for safety assessment. J Appl Toxicol. 35:971–975.
- Buss P, Caviezel M, Lutz WK. 1990. Linear dose-response relationship for DNA adducts in rat liver from chronic exposure to aflatoxin B1. Carcinogenesis. 11:2133–2135.
- Caballero F, Meiss R, Gimenez A, Batlle A, Vasquez E. 2004. Immunohistochemical analysis of heme oxygenase-1 in preneoplastic and neoplastic lesions during chemical heptacarcinogenesis. Int J Exp Pathol. 85:213–222.
- Chakravarti D, Venugopal D, Mailander P. 2008. The role of polycyclic aromatic hydrocarbon-DNA adducts in inducing mutations in mouse skin. Mutat Res. 649:161–178.
- Chawanthayatham S, Valentine IIIC, Fedeles B, Fox E, Loeb L, Levine S, Slocum S, Wogan G, Croy R, Essigmann J. 2017. Mutational spectra of aflatoxin B1 in vivo establish biomarkers of exposure for human hepatocellular carcinoma. Proc Natl Acad Sci USA. 114:E3101–E3109.
- Chen T, Heflich RH, Moore MM, Mei N. 2010. Differential mutagenicity of aflatoxin B1 in the liver of neonatal and adult mice. Environ Mol Mutagen. 51:156–163.
- Chittmittrapap S, Chieochansin T, Chaiteerakij R, Treeprasertuk S, Klaikaew N, Tangkijvanich P, Komolmit P, Poovorawan Y. 2013. Prevalence of aflatoxin induced P53 mutation at codon 249 (R249s) in heptacellular carcinoma patients with and without hepatitis B surface antigen (HBsAg). Asian Pacific J Cancer Prev. 14:7675–7679.
- Cohen SM, Arnold LL. 2011. Chemical carcinogenesis. Toxicol Sci. 120(S1): S76–S92.
- Croy RG, Wogan GN. 1981. Temporal patterns of covalent DNA adducts in rat liver after single and multiple doses of aflatoxin B1. Cancer Res. 41:197–203.
- Croy RG, Essigmann JM, Reinhold VN, Wogan GN. 1978. Identification of the principal aflatoxin B1-DNA adduct formed in vivo in rat liver. Proc Natl Acad Sci USA. 75:1745–1749.
- Cullen JM, Marion PL, Sherman GJ, Hong X, Newbold JE. 1990. Hepatic neoplasms in aflatoxin B1-treated, congenital duck hepatitis B virus-infected, and virus-free Pekin ducks. Cancer Res. 50:4072–4080.
- Cupid BC, Lightfoot TJ, Russell D, Grant SJ, Turner PC, Dingley KH, Curtis KD, Leveson SH, Turteltaub KW, Garner RC. 2004. The formation of AFB(1)-macromolecular adducts in rats and humans at dietary levels of exposure. Food Chem Toxicol. 42:559–569.
- Dang HT, Budhu A, Wang XW. 2014. The origin of cancer stem cells. J Hepatol. 60:1304–1305.
- de Carvalho FM, de Almeida Pereira T, Gonçalves PL, Jarske RD, Pereira FEL, Luoro ID. 2013. Heptacellular carcinoma and liver cirrhosis TP53 mutation analysis reflects a moderate dietary exposure to aflatoxins in Espírito Santo State, Brazil. Mol Biol Rep. 40:4883–4887.
- Degen GH, Neumann HG. 1981. Differences in aflatoxin B1-susceptibility of rat and mouse are correlated with the capability in vitro to inactivate aflatoxin B1-epoxide. Carcinogenesis. 2:299–306.
- Denissenko MF, Koudriakova TB, Smith L, O’Connor TR, Riggs AD, Pfeifer GP. 1998. The p53 codon 249 mutational hotspot in hepatocellular carcinoma is not related to selective formation or persistence of aflatoxin B1 adducts. Oncogene. 17:3007–3014.
- Dewanji A, Moolgavkar SH, Luebeck EG. 1991. Two-mutation model for carcinogenesis: Joint analysis of premalignant and malignant lesions. Math Biosci. 104:97–109.
- Dianov GL, Hübscher U. 2013. Mammalian base excision repair: the forgotten archangel. Nucleic Acids Res. 41:3483–3490.
- Doak SH, Jenkins GLS, Johnson GE, Quick E, Parry EM, Parry JM. 2007. Mechanistic influences for mutation induction curves after exposure to DNA-reactive carcinogens. Cancer Res. 67:3904–3911.
- Dobo KL, Fiedler RD, Gunther WC, Thiffeault CJ, Cammerer Z, Coffing SL, Shutsky T, Schuler M. 2011. Defining EMS ad ENU dose-response relationships using the Pig-a mutation essay in rats. Mutat Res. 725:13–21.
- Dragan YP, Hully J, Crow R, Mass M, Pitot HC. 1994. Incorporation of bromodeoxyuridine in glutathione S-transferase-positive hepatocytes during rat multistage hepatocarcinogenesis. Carcinogenesis. 15:1939–1947.
- Dragan YP, Hully J, Baker K, Crow R, Mass MJ, Pitot HC. 1995. Comparison of experimental and theoretical parameters of the moolgavkar-venzon-knudson incidence function for the stages of initiation and promotion in rat hepatocarcinogenesis. Toxicology. 102:161–175.
- Dragan YP, Campbell HA, Xu XH, Pitot HC. 1997. Quantitative stereological studies of a “'selection' protocol of hepatocarcinogenesis following initiation in neonatal male and female rats”. Carcinogenesis. 18:149–158.
- Dycaico MJJ, Stuart GRR, Tobal GMM, de Boer JGG, Glickman BWW, Provost GSS. 1996. Species-specific differences in hepatic mutant frequency and mutational spectrum among lambda/lacI transgenic rats and mice following exposure to aflatoxin B1. Carcinogenesis. 17:2347–2356.
- Eaton DL, Gallagher EP. 1994. Mechanisms of aflatoxin carcinogenesis. Annu Rev Pharmacol Toxicol. 34:135–172.
- Eaton DL, Schaupp CM. 2014. Of mice, rats, and men: could Nrf2 activation protect against aflatoxin heptocarcinogenesis in humans? Cancer Prev Res (Phila). 7:653–657.
- Edwards SW, Tan YM, Villeneuve DL, Meek ME, McQueen CA. 2015. Adverse outcome pathways-organizing toxicological information to improve decision making. J Pharmacol Exp Ther. 356:170–181.
- Egner P, Groopman J, Wang JS, Kensler T, Friesen M. 2006. Quantification of aflatoxin B1 N7 guanine in human urine by LC and isotope dilution tandem mass spectrometry. Chem Res Toxicol. 19:1191–1195.
- Elegbede AJ, Gould MN. 2002. Monoterpenes reduced adducts formation in rats exposed to aflatoxin B1. Afr J Biotechnol. 1:46–49.
- Fabregat I. 2009. Dysregulation of apoptosis in hepatocellular carcinoma cells. World J Gastroenterol. 15:513–520.
- Farazi PA, DePinho RA. 2006a. The genetic and environmental basis of hepatocellular carcinoma. Discov Med. 6:182–186.
- Farazi PA, DePinho RA. 2006b. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 6:674–687.
- Farazi PA, Glickman J, Jiang S, Yu A, Rudolph KL, DePinho RA. 2003. Differential impact of telomere dysfunction on initiation and progression of hepatocellular carcinoma. Cancer Res. 63:5021–5027.
- Fay KA, Villeneuve DL, LaLone CA, Song Y, Tollefsen KE, Ankley GT. 2017. Practical approaches to adverse outcome pathway development and weight-of-evidence evaluation as illustrated by ecotoxicological case studies. Environ Toxicol Chem. 36:1429–1449.
- Fischer G. 1986. Increased UDP-glucuronyltransferase and gamma-glutamyltranspeptidase in enzyme-altered rat liver lesions produced by low doses of aflatoxin B1. Virchows Arch B Cell Pathol. 51:443–460.
- Fischer G, Domingo M, Lodder D, Katz N, Reinacher M, Eigenbrodt E. 1987. Immunohistochemical demonstration of decreased L-pyruvate kinase in enzyme altered rat liver lesions produced by different carcinogens. Virchows Arch B Cell Pathol. 53:359–364.
- Foster PL, Eisenstadt E, Miller JH. 1983. Base substitution mutations induced by metabolically activated aflatoxin B1. Proc Natl Acad Sci USA. 80:2695–3698.
- Fransvea E, Angelotti U, Antonaci S, Giannelli G. 2008. Blocking transforming growth factor-beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology. 47:1557–1566.
- Frith CH, Kodell RL, Littlefield NA. 1980. Biologic and morphologic characteristics of hepatocellular lesions in BALB/c female mice fed 2-acetylaminofluorene. J Environ Pathol Toxicol. 3:121–138.
- Gil R, Callaghan R, Boix J, Pellin A, Llombart-Bosch A. 1988. Morphometric and cytophotometric nuclear analysis of altered hepatocyte foci induced by N-nitrosomorpholine (NNM) and aflatoxin B1 (AFB1) in liver of Wistar rats. Virchows Arch B Cell Pathol. 54:341–349.
- Giri I, Johnston DS, Stone MP. 2002. Mispairing of the 8,9-dihydro-8-(N7-guanyl)-9-hydroxy-aflatoxin B1 adduct with deoxyadenosine results in extrusion of the mismatched dA toward the major groove. Biochemistry. 41:5462–5472.
- Gocke E, Müller L. 2009. In vivo studies in the mouse to define a threshold for the genotoxicity of EMS and ENU. Mutat Res. 678:101–107.
- Godlewski CE, Boyd JN, Sherman WK, Anderson JL, Stoewsand GS. 1985. Hepatic glutathione S-transferase activity and aflatoxin B1-induced enzyme altered foci in rats fed fractions of Brussels sprouts. Cancer Lett. 28:151–157.
- Gold LS, Slone TH, Manley NB, Bernstein L. 1991. Target organs in chronic bioassays of 533 chemical carcinogens. Environ Health Perspect. 93:233–246.
- Goldfarb S, Pugh TD. 1986. Multistage rodent hepatocarcinogenesis. Prog Liver Dis. 8:597–620.
- Goldsworthy TL, Hanigan MH, Pitot HC. 1986. Models of hepatocarcinogenesis in the rat-contrasts and comparisons. Crit Rev Toxicol. 17:61–89.
- Golubovskaya VM, Filatov LV, Behe CI, Presnell SC, Hooth MJ, Smith GJ, Kaufmann WK. 1999. Telomere shortening, telomerase expression, and chromosome instability in rat hepatic epithelial stem-like cells. Mol Carcinog. 24:209–217.
- Gong P, Wang Y, Zhang J, Wang Z. 2013. Differential hepatic stem cell proliferation and differentiation after partial hepatectomy in rats. Mol Med Rep. 8:1005–1010.
- Gouas D, Shi H. 2009. The aflatoxin-induced TP53 mutation at codon 249 (R249S): biomarker of exposure, early detection and target for therapy. Cancer Lett. 286:29–37.
- Gouas D, Shi H, Hautefeuille A. 2010. Effects of the TP53 p.R249S mutant on proliferation and clonogenic properties in human hepatocellular carcinoma cell lines: interaction with hepatitis B virus X protein. Carcinogenesis. 31:1475–1482.
- Grasl-Kraupp B, Ruttkay-Nedecky B, Mullauer L, Taper H, Huber W, Bursch W, Schulte-Hermann R. 1997. Inherent increase of apoptosis in liver tumors: implications for carcinogenesis and tumor regression. Hepatology. 25:906–912.
- Gregory J, Goldstein S, Edds G. 1983. Metabolite distribution and rate of residue clearance in turkeys fed a diet containing aflatoxin B1. Food Chem Toxicol. 21:463–467.
- Grisham JW. 1996. Interspecies comparison of liver carcinogenesis: implications for cancer risk assessment. Carcinogenesis. 18:59–81.
- Groopman J, Kensler T. 2005. Role of metabolism and viruses in aflatoxin-induced liver cancer. Toxicol Appl Pharmacol. 206:131–137.
- Gross-Steinmeyer K, Eaton D. 2012. Dietary modulation of the biotransformation and genotoxicity of aflatoxin B(1). Toxicology. 299:69–79.
- Gross-Steinmeyer K, Stapleton P, Tracy J. 2010. Sulforaphane- and phenethyl isothiocyanate-induced inhibition of aflatoxin B1-mediated genotoxicity in human hepatocytes: role of GSTM1 genotype and CYP3A4 gene expression. Toxicol Sci. 116:422–432.
- Guengerich F, Johnson W, Ueng Y. 1996. Involvement of cytochrome P450, glutathione S-transferase, and epoxide hydrolase in the metabolism of aflatoxin B1 and relevance to risk of human liver cancer. Environ Health Perspect. 104:557–562.
- Guo Y, Breeden L, Zarbl H, Preston B. 2005. Expression of a human cytochrome p450 in yeast permits analysis of pathways for response to and repair of aflatoxin-induced DNA damage. Mol Cell Biol. 25:5823–5833.
- Gursoy-Yuzugullu O, Yuzugullu H, Yilmaz M, Ozturk M. 2011. Aflatoxin genotoxicity is associated with a defective DNA damage response bypassing p53 activation. Liver Int. 31:561–571.
- Hanahan D, Weinberg R. 2000. The hallmarks of cancer. Cell. 100:57–70.
- Hanahan D, Weinberg R. 2011. Hallmarks of cancer: the next generation. Cell. 144:646–674.
- Hanawalt PC. 2003. Four decades of DNA repair: from early insights to current perspectives. Biochimie. 85:1043–1052.
- Hanawalt PC, Ford JM, Lloyd DR. 2003. Functional characterization of global genomic DNA repair and its implications for cancer. Mutat Res. 544:107–114.
- Hang YZ, Hen YC, Hsan HA, Unn RML, Ee PL, Hen CC, Antella RMS. 2003. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation and its relationship to aflatoxin B1-DNA adducts and p53 mutation in hepatocellular carcinoma. Int J Cancer. 103:440–444.
- Harada T, Maronpot RR, Morris RW, Boorman GA. 1989a. Observations on altered hepatocellular foci in National Toxicology Program two-year carcinogenicity studies in rats. Toxicol Pathol. 17:690–706.
- Harada T, Maronpot RR, Morris RW, Stitzel KA, Boorman GA. 1989b. Morphological and stereological characterization of hepatic foci of cellular alteration in control Fischer 344 rats. Toxicol Pathol. 17:579–593.
- Haridas V, Hanausek M, Nishimura G. 2004. Triterpenoid electrophiles (avicins) activate the innate stress response by redox regulation of a gene battery. J Clin Invest. 113:65–73.
- He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, Shalapour S, Seki E, Yost SE, Jepsen K, et al. 2013. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell. 155:384–396.
- Hendricks CA, Almeida KH, Stitt MS, Jonnalagadda VS, Rugo RE, Kerrison GF, Engelward BP. 2003. Spontaneous mitotic homologous recombination at an enhanced yellow fluorescent protein (EYFP) cDNA direct repeat in transgenic mice. Proc Natl Acad Sci USA. 100:6325–6330.
- Hendriks G, Atallah M, Morolli B, Calleja F, Ras-Verloop B, Huijskens I, Raamsman M, van de Water B, Vrieling H. 2012. The ToxTracker assay: novel GFP reporter systems that provide mechanistic insight into the genotoxic properties of chemicals. Toxicol Sci. 125:285–298.
- Himmelstein M, Boogaard P, Cadet J, Farmer PB, Kim JH, Martin EA, Persaud R, Shuker DE. 2009. Creating context for the use of DNA adduct data in cancer risk assessment: II. Overview of methods of identification and quantitation of DNA damage. Crit Rev Toxicol. 39:679–694.
- Honda T, Yoshizawa H, Sundararajan C. 2011. Tricyclic compounds containing nonenolizable cyano enones. A novel class of highly potent anti-inflammatory and cytoprotective agents. J Med Chem. 54:1762–1778.
- Hsu IC, Metcalf RA, Sun T, Welsh JA, Wang NJ, Harris CC. 1991. Mutational hotspot in the p53 gene in human hepatocellular carcinomas. Nature. 350:427–428.
- Huff J, Cirvello J, Haseman J. 1991. Chemicals associated with site-specific neoplasia in 1394 long-term carcinogenesis experiments in laboratory rodents. Environ Health Perspect. 93:247–270.
- IARC. 1993. IARC monographs on the evaluation of carcinogenic risks to humans. Volume 56: Some naturally occurring substances: food items and constituents, heterocyclic aromatic amines and mycotoxins. [IARC] IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, France: IARC.
- Ikeda H, Nishi S, Sakai M. 2004. Transcription factor Nrf2/MafK regulates rat placental glutathione S-transferase gene during hepatocarcinogenesis. Biochem J. 380:515–521.
- Jackson PE, Kuang SY, Wang JB, Strickland PT, Muñoz A, Kensler TW, Qian GS, Groopman JD. 2003. Prospective detection of codon 249 mutations in plasma of hepatocellular carcinoma patients. Carcinogenesis. 24:1657–1663.
- Jarabek AM, Pottenger LH, Andrews LS, Casciano D, Embry MR, Kim JH, Preston RJ, Reddy MV, Schoeny R, Shuker D, et al. 2009. Creating context for the use of DNA adduct data in cancer risk assessment: I. Data organization. Crit Rev Toxicol. 39:659–678.
- Jaramillo M, Zhang D. 2013. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 27:2179–2191.
- Jia S, Ren J, Dong P, Meng X. 2013. Probing the hepatic progenitor cell in human hepatocellular carcinoma. Gastroenterol Res Pract. 2013:145253.
- Johnson N, Egner P, Baxter V, Sporn M. 2014. Complete protection against aflatoxin B1-induced liver cancer with a triterpenoid: DNA adduct dosimetry, molecular signature, and genotoxicity threshold. Cancer Prev Res (Phila). 7: 658–665.
- Kensler TW, Egner PA, Dolan PM, Groopman JD, Roebuck BD. 1987. Mechanism of protection against aflatoxin tumorigenicity in rats fed 5-(2-pyrazinyl)-4-methyl-1,2-dithiol-3-thione (oltipraz) and related 1,2-dithiol-3-thiones and 1,2-dithiol-3-ones. Cancer Res. 47:4271–4277.
- Kensler TW, He X, Otieno M, Egner PA, Jacobson LP, Chen B, Wang JS, Zhu YR, Zhang BC, Wang JB, et al. 1998a. Oltipraz chemoprevention trial in Qidong, People’s Republic of China: modulation of serum aflatoxin albumin adduct biomarkers. Cancer Epidemiol Biomarkers Prev. 7:127–134.
- Kensler T, Groopman J, Roebuck B. 1998b. Use of aflatoxin adducts as intermediate endpoints to assess the efficacy of chemopreventive interventions in animals and man. Mutat Res. 402:165–172.
- Kensler TW, Egner PA, Wang JB, Zhu YR, Zhang BC, Lu PX, Chen JG, Qian GS, Kuang SY, Jackson PE, et al. 2004. Chemoprevention of hepatocellular carcinoma in aflatoxin endemic areas. Gastroenterology. 127:S310–S318.
- Kensler TW, Roebuck BD, Wogan GN, Groopman JD. 2011. Aflatoxin: a 50-year Odyssey of mechanistic and translational toxicology. Toxicol Sci. 120:S28–S48.
- Kew MC. 2003. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 23:405–409.
- Kew MC. 2013. Aflatoxins as a cause of hepatocellular carcinoma. J Gastrointestin Liver Dis. 22:305–310.
- Kimura M, Lehmann K, Gopalan-Kriczky P, Lotlikar PD. 2004. Effect of diet on aflatoxin B1-DNA binding and aflatoxin B1-induced glutathione S-transferase placental form positive hepatic foci in the rat. Exp Mol Med. 36:351–357.
- Kiraly O, Gong G, Roytman MD, Yamada Y, Samson LD, Engelward BP. 2014. DNA glycosylase activity and cell proliferation are key factors in modulating homologous recombination in vivo. Carcinogenesis. 35:2495–2502.
- Kiraly O, Gong G, Olipitz W, Muthupalani S, Engelward BP. 2015. Inflammation-induced cell proliferation potentiates DNA damage-induced mutations in vivo. PLoS Genet. 11:e1004901
- Kirby GM, Stalker M, Metcalfe C, Kocal T, Ferguson H, Hayes MA. 1990. Expression of immunoreactive glutathione S-transferases in hepatic neoplasms induced by aflatoxin B1 or 1,2-dimethylbenzanthracene in rainbow trout (Oncorhynchus mykiss). Carcinogenesis. 11:2255–2257.
- Kirk GD, Turner PC, Gong Y, Lesi OA, Mendy M, Goedert JJ, Hall AJ, Whittle H, Hainaut P, Montesano R, Wild CP. 2005. Hepatocellular carcinoma and polymorphisms in carcinogen-metabolizing and DNA repair enzymes in a population with aflatoxin exposure and hepatitis B virus endemicity. Cancer Epidemiol Biomarkers Prev. 14:373–379.
- Kozmin SG, Jinks-Robertson S. 2013. The mechanism of nucleotide excision repair-mediated UV-induced mutagenesis in nonproliferating cells. Genetics. 193:803–817.
- Kunkel TA. 2004. DNA replication fidelity. J Biol Chem. 279:16895–16898.
- Kwak MK, Itoh K, Yamamoto M, Sutter TR, Kensler TW. 2001. Role of transcription factor Nrf2 in the induction of hepatic phase 2 and antioxidative enzymes in vivo by the cancer chemoprotective agent, 3H-1, 2-dimethiole-3-thione. Mol Med. 7:135–145.
- Larsson P, Hoedaya WI, Tjälve H. 1990. Disposition of 3H-aflatoxin B1 in mice: formation and retention of tissue bound metabolites in nasal glands. Pharmacol Toxicol. 67:162–171.
- Larsson P, Tjälve H. 1993. Distribution and metabolism of aflatoxin B1 in the marmoset monkey (Callithrix jacchus). Carcinogenesis. 14:1–6.
- Leadon SA, Tyrrell RM, Cerutti PA. 1981. Excision repair of aflatoxin B1-DNA adducts in human fibroblasts. Cancer Res. 41:5125–5129.
- Lee YI, Lee S, Das GC, Park US, Park SM, Lee YI. 2000. Activation of the insulin-like growth factor II transcription by aflatoxin B1 induced p53 mutant 249 is caused by activation of transcription complexes; implications for a gain-of-function during the formation of hepatocellular carcinoma. Oncogene. 19:3717–3726.
- Leung MCK, Goldstone JV, Boyd WA, Freedman JH, Meyer JN. 2010. Caenorhabditis elegans generates biologically relevant levels of genotoxic metabolites from aflatoxin B1 but not benzo[a]pyrene in vivo. Toxicol Sci. 118:444–453.
- Levrero M, Zucman-Rossi J. 2016. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. 64: S84–S101.
- Levy DD, Groopman JD, Lim SE, Seidman MM, Kraemer KH. 1992. Sequence specificity of aflatoxin B1-induced mutations in a plasmid replicated in xeroderma pigmentosum and DNA repair proficient human cells. Cancer Res. 52:5668–5673.
- Liby K, Yore MM, Roebuck BD, Baumgartner KJ, Honda T, Sundararajan C, Yoshizawa H, Gribble GW, Williams CR, Risingsong R, et al. 2008. A novel acetylenic tricyclic bis-(cyano enone) potently induces phase 2 cytoprotective pathways and blocks liver carcinogenesis induced by aflatoxin. Cancer Res. 68:6727–6733.
- Liby KT, Sporn MB. 2012. Synthetic oleanane triterpenoids: multifunctional drugs with a broad range of applications for prevention and treatment of chronic disease. Pharmacol Rev. 64:972–1003.
- Lin YC, Li L, Makarova AV, Burgers PM, Stone MP, Lloyd RS. 2014a. Error-prone replication bypass of the primary aflatoxin B1 DNA adduct, AFB1-N7-Gua. J Biol Chem. 289:18497–18506.
- Lin YC, Li L, Makarova AV, Burgers PM, Stone MP, Lloyd RS. 2014b. Molecular basis of aflatoxin-induced mutagenesis-role of the aflatoxin B1-formamidopyrimidine adduct. Carcinogenesis. 35:1461–1468.
- Long X, Ma Y, Deng Z. 2009. GSTM1 and XRCC3 polymorphisms: Effects on levels of aflatoxin B1-DNA adducts. Chin J Cancer Res. 21:177–184.
- Long XD, Ma Y, Qu DY, Liu YG, Huang ZQ, Huang YZ, Lin ZH, Wei NB, Zhou SC. 2008. The polymorphism of XRCC3 codon 241 and AFB1-related hepatocellular carcinoma in Guangxi Population, China. Ann Epidemiol. 18:572–578.
- Long XD, Ma Y, Wei YP, Deng ZL. 2006. The polymorphisms of GSTM1, GSTT1, HYL1*2, and XRCC1, and aflatoxin B1-related hepatocellular carcinoma in Guangxi population, China. Hepatol Res. 36:48–55.
- Luo M, Yang F, Huang SX, Kuang ZP, Luo XL, Li YD, Wu JN, Xie YA. 2013. Two-stage model of chemically induced hepatocellular carcinoma in mouse. Oncol Res. 20:517–528.
- Ma Q, He X. 2012. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol Rev. 64:1055–1081.
- Macé K, Aguilar F, Wang JS, Vautravers P, Gómez-Lechón M, Gonzalez FJ, Groopman J, Harris CC, Pfeifer AM. 1997. Aflatoxin B1-induced DNA adduct formation and p53 mutations in CYP450-expressing human liver cell lines. Carcinogenesis. 18:1291–1297.
- Manson MM, Green JA, Neal GE. 1984. Monoclonal antibody against tumour-derived cell line shows heterogeneity of altered hepatocytes during AFB1-induced carcinogenesis. Int J Cancer. 34:869–874.
- Marchetti F, Massarotti A, Yauk CL, Pacchierotti F, Russo A. 2016. The adverse outcome pathway (AOP) for chemical binding to tubulin in oocytes leading to aneuploid offspring. Environ Mol Mutagen. 57:87–113.
- Maronpot RR, Harada T, Murthy AS, Boorman GA. 1989. Documenting foci of hepatocellular alteration in two-year carcinogenicity studies: current practices of the National Toxicology Program. Toxicol Pathol. 17:675–683.
- Maxuitenko YY, MacMillan DL, Kensler TW, Roebuck BD. 1993. Evaluation of the post-initiation effects of oltipraz on aflatoxin B1-induced preneoplastic foci in a rat model of hepatic tumorigenesis. Carcinogenesis. 14:2423–2425.
- Meek ME, Boobis A, Cote I, Dellarco V, Fotakis G, Munn S, Seed J, Vickers C. 2014a. New developments in the evolution and application of the WHO/IPCS framework on mode of action/species concordance analysis. J Appl Toxicol. 34:1–18.
- Meek MEB, Palermo CM, Bachman AN, North CM, Jeffrey Lewis R. 2014b. Mode of action human relevance (species concordance) framework: Evolution of the Bradford Hill considerations and comparative analysis of weight of evidence. J Appl Toxicol. 34:595–606.
- Meier B, Cooke S, Weiss J, Bailly A. 2014. C. elegans whole-genome sequencing reveals mutational signatures related to carcinogens and DNA repair deficiency. Genome. 24:1624–1636.
- Miura N, Horikawa I, Nishimoto A, Ohmura H. 1997. Progressive telomere shortening and telomerase reactivation during hepatocellular carcinogenesis. Cancer Genet Cytogenet. 93:56–62.
- Miyajima A, Tanaka M, Itoh T. 2014. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell. 14:561–574.
- Monroe D, Eaton D. 1987. Comparative effects of butylated hydroxyanisole on hepatic in vivo DNA binding and in vitro biotransformation of aflatoxin B 1 in the rat and mouse. Toxicol Appl Pharmacol. 14:561–574.
- Moore MM, Manjanatha M, Heflich R. 2008. Research strategy to better inform the decision as to whether a carcinogen is a “mutagenic carcinogen,” in: 2008 Meeting of the Society of Toxicology, Baltimore, MD.
- Mulder J, Bondy G, Mehta R, Massey T. 2014. Up-regulation of nucleotide excision repair in mouse lung and liver following chronic exposure to aflatoxin B1 and its dependence on p53 genotype. Toxicol Appl Pharmacol. 275:96–103.
- Newberne PM. 1976. Experimental hepatocellular carcinogenesis. Cancer Res. 36:2573–2578.
- Nishizumi M, Albert RE, Burns FJ, Bilger L. 1977. Hepatic cell loss and proliferation induced by N-2-fluorenylacetamide, diethylnitrosamine, and aflatoxin B1 in relation to hepatoma induction. Br J Cancer. 36:192–197.
- O'Dwyer PJ, Johnson SW, Khater C, Krueger A, Matsumoto Y, Hamilton TC, Yao KS. 1997. The chemopreventive agent oltipraz stimulates repair of damaged DNA. Cancer Res. 57:1050–1053.
- OECD. 2013. Guidance document on developing and assessing adverse outcome pathways. Series on testing and assessment. No. 184. Paris, France: [OECD] Organisation for Economic Co-operation and Development.
- OECD. 2014. User’s handbook supplement to the guidance document for developing and assessing AOPs. Paris, France: [OECD] Organisation for Economic Co-operation and Development.
- OECD. 2016. Users' Handbook supplement to the Guidance Document for developing and assessing Adverse Outcome Pathways. Paris, France: [OECD] Organisation for Economic Co-operation and Development. Available at: http://www.oecd-ilibrary.org/environment/oecd-series-on-adverse-outcome-pathways_2415170x;jsessionid=20oo8q54271qt.x-oecd-live-03
- Olden K, Vulimiri S. 2014. Laboratory to community: chemoprevention is the answer. Cancer Prev Res (Phila). 7:648–652.
- Oleykowski C, Mayernik J, Lim S. 1993. Repair of aflatoxin B1 DNA adducts by the UvrABC endonuclease of Escherichia coli. J Biol Chem. 268:7990–8002.
- Palliyaguru D, Wu F. 2013. Global geographical overlap of aflatoxin and hepatitis C: controlling risk factors for liver cancer worldwide. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 30:534–540.
- Parkin DM, Pisani P, Ferlay J. 1993. Estimates of the worldwide incidence of eighteen major cancers in 1985. Int J Cancer. 54:594–606.
- Parsons B, McKinzie P, Heflich R. 2005. Allele-specific competitive blocker-PCR detection of rare base substitution. Mol Toxicol Protoc. 291:235–245.
- Parsons B, Myers M, Meng F, Wang Y. 2010. Oncomutations as biomarkers of cancer risk. Environ Mol Mutagen. 51:836–850.
- Patlewicz G, Simon T, Rowlands J. 2015. Proposing a scientific confidence framework to help support the application of adverse outcome pathways for regulatory purposes. Regul Toxicol Pharmacol. 71:463–477.
- Perkins EJ, Antczak P, Burgoon L, Falciani F, Garcia-Reyero N, Gutsell S, Hodges G, Kienzler A, Knapen D, McBride M, et al. 2015. Adverse outcome pathways for regulatory applications: examination of four case studies with different degrees of completeness and scientific confidence. Toxicol Sci. 148:14–25.
- Philippin G, Cadet J, Gasparutto D, Mazon G, Fuchs RP. 2014. Ethylene oxide and propylene oxide derived N7-alkylguanine adducts are bypassed accurately in vivo. DNA Repair (Amst). 22:133–136.
- Pitot H. 1990. Altered hepatic foci: their role in murine hepatocarcinogenesis. Annu Rev Pharmacol Toxicol. 30:465–500.
- Pitot HC, Dragan Y, Sargent L, Xu YH. 1991. Biochemical markers associated with the stages of promotion and progression during hepatocarcinogenesis in the rat. Environ Health Perspect. 93:181–189.
- Pitot HC, Dragan Y, Xu YH, Pyron M, Laufer C, Rizvi T. 1990. Role of altered hepatic foci in the stages of carcinogenesis. Prog Clin Biol Res. 340D:81–95.
- Pitot HC, Dragan YP, Teeguarden J, Hsia S, Campbell H. 1996. Quantitation of multistage carcinogenesis in rat liver. Toxicol Pathol. 24:119–128.
- Pogribny IP, Muskhelishvili L, Tryndyak VP, Beland FA. 2011. The role of epigenetic events in genotoxic hepatocarcinogenesis induced by 2-acetylaminofluorene. Mutat Res Toxicol Environ Mutagen. 722:106–113.
- Pottenger LH, Andrews LS, Bachman AN, Boogaard PJ, Cadet J, Embry MR, Farmer PB, Himmelstein MW, Jarabek AM, Martin EA, et al. 2014. An organizational approach for the assessment of DNA adduct data in risk assessment: case studies for aflatoxin B1, tamoxifen and vinyl chloride. Crit Rev Toxicol. 44:348–391.
- Pottenger LH, Gollapudi BB. 2010. Genotoxicity testing: moving beyond qualitative “screen and bin” approach towards characterization of dose-response and thresholds. Environ Mol Mutagen. 51:792–799.
- Pottenger LH, Schisler MR, Zhang F, Bartels MJ, Fontaine DD, McFadden LG, Bhaskar Gollapudi B. 2009. Dose-response and operational thresholds/NOAELs for in vitro mutagenic effects from DNA-reactive mutagens, MMS and MNU. Mutat Res. 678:138–147.
- Preston R, Williams G. 2005. DNA-reactive carcinogens: mode of action and human cancer hazard. Crit Rev Toxicol. 35:673–683.
- Primiano T, Egner P, Sutter T, Kelloff G. 1995. Intermittent dosing with oltipraz: relationship between chemoprevention of aflatoxin-induced tumorigenesis and induction of glutathione S-transferases. Cancer Res. 55:4319–4324.
- Puisieux A, Lim S, Groopman J, Ozturk M. 1991. Selective targeting of p53 gene mutational hotspots in human cancers by etiologically defined carcinogens. Cancer Res. 51:6185–6189.
- Ribback S, Calvisi D, Cigliano A, Sailer V, Peters M. 2013. Molecular and metabolic changes in human liver clear cell foci resemble the alterations occurring in rat hepatocarcinogenesis. J Hepatol. 58:1147–1156.
- Roebuck B, Liu Y, Rogers A, Groopman J. 1991. Protection against aflatoxin B1-induced hepatocarcinogenesis in F344 rats by 5-(2-pyrazinyl)-4-methyl-1,2-dithiole-3-thione (oltipraz): predictive role for short-term molecular dosimetry. Cancer Res. 51:5501–5506.
- Russom CL, LaLone CA, Villeneuve DL, Ankley GT. 2014. Development of an adverse outcome pathway for acetylcholinesterase inhibition leading to acute mortality. Environ Toxicol Chem. 33:2157–2169.
- Sarasin A, Smith C, Hanawalt P. 1977. Repair of DNA in human cells after treatment with activated aflatoxin B1. Cancer Res. 37:1786–1793.
- Sargent L, Dragan Y, Xu YH, Sattler G, Wiley J, Pitot HC. 1996. Karyotypic changes in a multistage model of chemical hepatocarcinogenesis in the rat. Cancer Res. 56:2985–2991.
- Sargent L, Xu YH, Sattler GL, Meisner L, Pitot HC. 1989. Ploidy and karyotype of hepatocytes isolated from enzyme-altered foci in two different protocols of multistage hepatocarcinogenesis in the rat. Carcinogenesis. 10:387–391.
- Sell S. 2003. Mouse models to study the interaction of risk factors for human liver cancer. Cancer Res. 63:7553–7562.
- Shelton P, Jaiswal A. 2013. The transcription factor NF-E2-related factor 2 (Nrf2): a protooncogene? FASEB J. 27:414–423.
- Shen H, Ong C. 1996. Mutations of the p53 tumor suppressor gene and ras oncogenes in aflatoxin hepatocarcinogenesis. Mutat Res. 366:23–44.
- Smela M, Hamm M, Henderson P, Harris CM, Harris TM, Essignmann JM. 2002. The aflatoxin B1 formamidopyrimidine adduct plays a major role in causing the types of mutations observed in human hepatocellular carcinoma. Proc Natl Acad Sci USA. 99:6655–6660.
- Staib F, Hussain SP, Hofseth L. 2003. TP53 and liver carcinogenesis. Hum Mutat. 21:201–216.
- Sudakin D. 2003. Dietary aflatoxin exposure and chemoprevention of cancer: a clinical review. J Toxicol Clin Toxicol. 41:195–204.
- Sukowati CHC, El-Khobar KE, Ie SI, Anfuso B, Muljono DH, Tiribelli C. 2016. Significance of hepatitis virus infection in the oncogenic initiation of hepatocellular carcinoma. World J Gastroenterol. 22:1497–1512.
- Swenberg J, Lu K, Moeller B, Gao L. 2010. Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology and risk assessment. Toxicol Sci. 120:S130–S145.
- Szymañska K, Chen JG, Cui Y, Gong YY, Turner PC, Villar S, Wild CP, Parkin DM, Hainaut P. 2009. TP53 R249S mutations, exposure to aflatoxin, and occurrence of hepatocellular carcinoma in a cohort of chronic hepatitis B virus carriers from Qidong, China. Cancer Epidemiol Biomarkers Prev. 18:1638–1643.
- Taguchi K, Takaku M, Egner PA, Morita M, Kaneko T, Mashimo T, Kensler TW, Yamamoto M. 2016. Generation of a new model rat: Nrf2 knockout rats are sensitive to aflatoxin B1 toxicity. Toxicol Sci. 152:40–52.
- Thoolen B, Ten Kate FJ, van Diest PJ, Malarkey DE, Elmore SA, Maronpot RR. 2012. Comparative histomorphological review of rat and human hepatocellular proliferative lesions. J Toxicol Pathol. 25:189–199.
- Ueng YF, Shimada T, Yamazaki H, Guengerich FP. 1995. Oxidation of aflatoxin B1 by bacterial recombinant human cytochrome P450 enzymes. Chem Res Toxicol. 8:218–225.
- Villeneuve DL, Crump D, Garcia-Reyero N, Hecker M, Hutchinson TH, LaLone CA, Landesmann B, Lettieri T, Munn S, Nepelska M, et al. 2014. Adverse outcome pathway (AOP) development I: strategies and principles. Toxicol Sci. 142:312–320.
- Wang JS, Shen X, He X, Zhu YR, Zhang BC, Wang JB, Qian GS, Kuang SY, Zarba A, Egner PA, et al. 1999. Protective alterations in phase 1 and 2 metabolism of aflatoxin B1 by oltipraz in residents of qidong, People's Republic of China. J Natl Cancer Inst. 91:347–354.
- Wang Y, Meng F, Arlt VM, Mei N, Chen T, Parsons BL. 2011. Aristolochic acid-induced carcinogenesis examined by ACB-PCR quantification of H-Ras and K-Ras mutant fraction. Mutagenesis. 26:619–628.
- Wattanawaraporn R, Kim MY, Adams J, Trudel LJ, Woo LL, Croy RG, Essigmann JM, Wogan GN. 2012. AFB(1)-induced mutagenesis of the gpt gene in AS52 cells. Environ Mol Mutagen. 53:567–573.
- Wiktor-Brown DM, Hendricks CA, Olipitz W, Engelwood BP. 2006a. Age-dependent accumulation of recombinant cells in the mouse pancreas revealed by in situ fluorescence imaging. Proc Natl Acad Sci USA. 103:11862–11867.
- Wiktor-Brown DM, Hendricks CA, Olipitz W, Rogers A, Engelward B. 2006b. Applications of fluorescence for detecting rare sequence rearrangements. In Vivo. Cell Cycle. 5:2715–2719.
- Wogan GN, Paglialunga S, Newberne PM. 1974. Carcinogenic effects of low dietary levels of aflatoxin B1 in rats. Food Cosmet Toxicol. 12:681–685.
- Wu H, Wang Q, Yang H, Tsai W, Chen C. 2013. Global DNA methylation in a population with aflatoxin B1 exposure. Epigenetics. 8:962–969.
- Wyde M, Cambre T, Lebetkin M, Eldridge S. 2002. Promotion of altered hepatic foci by 2,3,7,8-tetrachlorodibenzo-p-dioxin and 17beta-estradiol in male Sprague-Dawley rats. Toxicol Sci. 68:295–303.
- Xu YH, Campbell HA, Sattler GL, Hendrich S, Maronpot R, Sato K, Pitot HC. 1990a. Quantitative stereological analysis of the effects of age and sex on multistage hepatocarcinogenesis in the rat by use of four cytochemical markers. Cancer Res. 50:472–479.
- Xu YH, Maronpot R, Pitot HC. 1990b. Quantitative stereologic study of the effects of varying the time between initiation and promotion on four histochemical markers in rat liver during hepatocarcinogenesis. Carcinogenesis. 11:267–272.
- Yang L, Ji J. 2014. Transcriptome profiling of malignant transformed rat hepatic stem-like cells by aflatoxin B1. Neoplasma. 61:193–204.
- Yates MS, Kwak MK, Egner PA, Groopman JD, Bodreddigari S, Sutter TR, Baumgartner KJ, Roebuck BD, Liby KT, Yore MM, et al. 2006. Potent protection against aflatoxin-induced tumorigenesis through induction of Nrf2-regulated pathways by the triterpenoid 1-[2-cyano-3-,12-dioxooleana-1,9(11)-dien-28-oyl]imidazole. Cancer Res. 66:2488–2494.
- Yates MS, Tauchi M, Katsuoka F, Flanders KC, Liby KT, Honda T, Gribble GW, Johnson DA, Johnson JA, Burton NC, et al. 2007. Pharmacodynamic characterization of chemopreventive triterpenoids as exceptionally potent inducers of Nrf2-regulated genes. Mol Cancer Ther. 6:154–162.
- Yauk CL, Bishop J, Dearfield KL, Douglas GR, Hales BF, Luijten M, O’Brien JM, Robaire B, Sram R, van Benthem J, et al. 2013. The development of adverse outcome pathways for mutagenic effects for the organization for economic co-operation and development. Environ Mol Mutagen. 54:79–81.
- Yauk CL, Lambert IB, Meek MEB, Douglas GR, Marchetti F. 2015. Development of the adverse outcome pathway “alkylation of DNA in male premeiotic germ cells leading to heritable mutations” using the OECD’s users’ handbook supplement. Environ Mol Mutagen. 56:724–750.
- Youngman LD, Campbell TC. 1992a. Inhibition of aflatoxin B1-induced gamma-glutamyltranspeptidase positive (GGT+) hepatic preneoplastic foci and tumors by low protein diets: evidence that altered GGT + foci indicate neoplastic potential. Carcinogenesis. 13:1607–1613.
- Youngman LD, Campbell TC. 1992b. Attenuation of preneoplastic lesion development by dietary protein intervention: apparent persistence and regression. Cancer Lett. 66:165–174.
- Zhang Y, Chen Y, Ahsan H, Lunn R. 2003. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation and its relationship to aflatoxin B1-DNA adducts and p53 mutation in hepatocellular carcinoma. Int J Cancer. 103:440–444.
- Zhang Y, Rossner P, Chen Y. 2006. Aflatoxin B1 and polycyclic aromatic hydrocarbon adducts, p53 mutations and p16 methylation in liver tissue and plasma of hepatocellular carcinoma patients. Int J Cancer. 119:985–991.
- Zhang Y, Wu H, Yazici H, Yu M, Lee P, Santella RM. 2012. Global hypomethylation in hepatocellular carcinoma and its relationship to aflatoxin B(1) exposure. World J Hepatol. 4:169–175.
- Zhu H, Wu J, Shen X. 2017. Genome-wide association study: new genetic insights into HBV/HCV-related hepatocellular carcinoma genomes. Scand J Gastroenterol. 52:209–215.