
Abstract
The mercapturic acid pathway is a major route for the biotransformation of xenobiotic and endobiotic electrophilic compounds and their metabolites. Mercapturic acids (N-acetyl-l-cysteine S-conjugates) are formed by the sequential action of the glutathione transferases, γ-glutamyltransferases, dipeptidases, and cysteine S-conjugate N-acetyltransferase to yield glutathione S-conjugates, l-cysteinylglycine S-conjugates, l-cysteine S-conjugates, and mercapturic acids; these metabolites constitute a “mercapturomic” profile. Aminoacylases catalyze the hydrolysis of mercapturic acids to form cysteine S-conjugates. Several renal transport systems facilitate the urinary elimination of mercapturic acids; urinary mercapturic acids may serve as biomarkers for exposure to chemicals. Although mercapturic acid formation and elimination is a detoxication reaction, l-cysteine S-conjugates may undergo bioactivation by cysteine S-conjugate β-lyase. Moreover, some l-cysteine S-conjugates, particularly l-cysteinyl-leukotrienes, exert significant pathophysiological effects. Finally, some enzymes of the mercapturic acid pathway are described as the so-called “moonlighting proteins,” catalytic proteins that exert multiple biochemical or biophysical functions apart from catalysis.
1. Introduction
The mercapturic acid pathway plays a significant role in the detoxication of electrophiles formed by the biotransformation of xenobiotics. Indeed, the elaboration of the mercapturic acid pathway was interwoven with investigations into the metabolic fate of chemicals. Although the detoxication of xenobiotic-derived electrophiles has received the most emphasis, the mercapturic acid pathway also has a significant role in the enzymatic processing of endobiotics, particularly leukotrienes and steroids. Some enzymes of the mercapturic acid pathway are the so-called moonlighting proteins, i.e. polypeptide chains with multiple biochemical or biophysical functions (Jeffery Citation1999, Citation2018). For example, the dipeptidase aminopeptidase N is also known as CD13 and functions not only as a hydrolase but also as a viral receptor and a signaling molecule (Mina-Osorio Citation2008).
Mercapturic acids (S-substituted N-acetyl-l-Cys) arise from the glutathione transferase (GST)-catalyzed reaction of glutathione (GSH) with electrophilic species to give GSH S-conjugates that undergo sequential hydrolysis to l-Cys-Gly S-conjugates and thence to l-Cys S-conjugates, which undergo N-acetylation to yield the mercapturic acid (). The metabolites formed by the mercapturic acid pathway have been proposed to constitute a “mercapturomic” profile (Gonçalves-Dias et al. Citation2019a, Citation2019b). Mercapturic acids are polar (carboxylates at physiological pH) and are readily excreted by the kidney.
Figure 1. The mercapturic acid pathway, as illustrated with the biotransformation of monochlorobimane. GSH: glutathione; GST: glutathione transferase; GGT: γ-glutamyltransferase; NAT8: Cys S-conjugate N-acetyltransferase; AcCoA: acetyl-CoA; 1, monochlorobimane; 2, l-γ-glutamyl-S-[(2,5,6-trimethyl-1,7-dioxo-1H,7H-pyrazolo[1,2-a]pyrazol-3-yl)methyl]-l-Cys-Gly; 3, S-[(2,5,6-trimethyl-1,7-dioxo-1H,7H-pyrazolo[1,2-a]pyrazol-3-yl)methyl]-l-Cys-Gly; 4, S-[(2,5,6-trimethyl-1,7-dioxo-1H,7H-pyrazolo[1,2-a]pyrazol-3-yl)methyl]-l-Cys; 5, N-acetyl-S-[(2,5,6-trimethyl-1,7-dioxo-1H,7H-pyrazolo[1,2-a]pyrazol-3-yl)methyl]-l-Cys.
![Figure 1. The mercapturic acid pathway, as illustrated with the biotransformation of monochlorobimane. GSH: glutathione; GST: glutathione transferase; GGT: γ-glutamyltransferase; NAT8: Cys S-conjugate N-acetyltransferase; AcCoA: acetyl-CoA; 1, monochlorobimane; 2, l-γ-glutamyl-S-[(2,5,6-trimethyl-1,7-dioxo-1H,7H-pyrazolo[1,2-a]pyrazol-3-yl)methyl]-l-Cys-Gly; 3, S-[(2,5,6-trimethyl-1,7-dioxo-1H,7H-pyrazolo[1,2-a]pyrazol-3-yl)methyl]-l-Cys-Gly; 4, S-[(2,5,6-trimethyl-1,7-dioxo-1H,7H-pyrazolo[1,2-a]pyrazol-3-yl)methyl]-l-Cys; 5, N-acetyl-S-[(2,5,6-trimethyl-1,7-dioxo-1H,7H-pyrazolo[1,2-a]pyrazol-3-yl)methyl]-l-Cys.](/cms/asset/dbbf9445-aebf-45fb-87ce-d1ae51d55756/itxc_a_1692191_f0001_b.jpg)
Mercapturic acid formation was first reported in 1879 (Baumann and Preusse Citation1879; Jaffé Citation1879): dogs given bromobenzene or chlorobenzene excreted S-(bromophenyl)- or S-(chlorophenyl)mercapturic acid. Since these early observations, the several steps in the mercapturic acid pathway have been elucidated. The identification and characterization of GSH and the later finding that GSH is the source of the sulfur atom in mercapturic acids was intertwined with the clarification of the mercapturic acid pathway (Barnes et al. Citation1959; Boyland and Chasseaud Citation1969; Meister Citation1988). These observations led to the discovery of the GSTs and their central role in mercapturic acid formation (Booth et al. Citation1961; Habig et al. Citation1974b).
Baumann and Preusse (Baumann and Preusse Citation1879) and Jaffé (Jaffé Citation1879) isolated mercapturic acids from acidified urine of dogs given bromobenzene or chlorobenzene. Studies on the metabolic fate of naphthalene and several aromatic hydrocarbons in rats and rabbits showed that mercapturic acids were found in urine only after acidification (Boyland et al. Citation1957; Knight and Young Citation1957); these observations led to the concept that precursors of mercapturic acids, so-called premercapturic acids, were formed and excreted in urine. Boyland and Sims (Boyland and Sims Citation1958) identified the acid-labile precursor of N-acetyl-S-(naphthalen-1-yl)cysteine as N-acetyl-S-(2-hydroxy-1,2-dihydronaphthalen-1-yl)-l-cysteine; similarly, Knight and Young (Knight and Young Citation1958) also demonstrated the formation of premercapturic acids from benzene, naphthalene, anthracene, and halobenzenes.
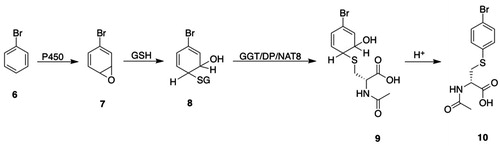
The formation of an intermediate premercapturic acid is characteristic of aromatic ring conjugation by GSH, and can be illustrated by the metabolism of bromobenzene (6, ) to bromobenzene oxide 7 (), which may react with GSH to give GSH S-conjugate 8 (); hydrolysis and N-acetylation of the GSH S-conjugate 8 () gives the premercapturic acid 9 (); acid-catalyzed dehydration of intermediate 9 () yields the observed S-(4-bromophenyl)mercapturic acid 10 ().
Figure 2. Premercapturic acids as intermediates in the formation of mercapturic acids. P450: cytochromes P450; GSH: glutathione; GGT: γ-glutamyltransferase; DP: dipeptidases; NAT8: Cys S-conjugate N-acetyltransferase; 6, bromobenzene; 7, bromobenzene oxide; 8, S-(4-bromo-6-hydroxycyclohexa-2,4-dien-1-yl)GSH; 9, N-acetyl-S-(4-bromo-6-hydroxycyclohexa-2,4-dien-1-yl)-l-Cys; 10, S-(4-bromophenyl)mercapturic acid.
Structural assignments of mercapturic acids formed by the acid-catalyzed dehydration of premercapturic acids of aromatic compounds may require revision because of rearrangements of premercapturic acids after acidification (Jeffery and Jerina Citation1975); these authors also questioned whether premercapturic acids were the products of enzymatic reactions or whether they were formed by the nonenzymatic reaction of arene oxides with GSH.
For most compounds, mercapturic acid formation is a detoxication pathway, but some mercapturic acids or precursor Cys S-conjugates undergo bioactivation to reactive intermediates (Hashmi et al. Citation1992; Anders and Dekant Citation1998). A discussion of the bioactivation and toxicity of mercapturic acids and Cys S-conjugates is not, however, within the scope of this review. The metabolites of endogenous substrates formed by the mercapturic acid pathway may play a role in respiratory diseases, cancer, neurological disorders, and cardiometabolic diseases that contribute to the pathophysiological development of chronic inflammatory and metabolic disorders (Gonçalves-Dias et al. Citation2019a, Citation2019b).
The objective of this review is to provide a broad, detailed presentation of the formation of mercapturic acids. Descriptions of the several contributing enzymes, their discovery and characterization, substrate selectivities, inhibitors, species and tissue distribution, and biological functions are included. The review is intended to be useful to experienced investigators, students, and new investigators seeking an informative guide to the mercapturic acid pathway. Previous reviews of the mercapturic acid pathway have been published (Boyland and Chasseaud Citation1969; Wood Citation1970; Chasseaud Citation1973, Citation1976; Tate Citation1980; Bakke Citation1990; Commandeur et al. Citation1995; Cooper and Hanigan Citation2010, Citation2018).
2. Glutathione transferases1,2
2.1. Introduction
GSTs catalyze the reaction of electrophilic substrates with GSH (), the first step in the mercapturic acid pathway. The GSTs play a major role in the detoxication of toxic electrophiles: the GSH S-conjugates that are formed are typically less toxic, more water soluble, and more readily excreted than the precursor compounds.
There are three distinct mammalian GST superfamilies: (1) soluble or cytosolic GSTs, (2) mitochondrial Kappa-class GST, and (3) microsomal MAPEG (Membrane-Associated Proteins in Eicosanoid and Glutathione metabolism) (Pearson Citation2005; Zimniak and Singh Citation2007; Oakley Citation2011; Brown and Babbitt Citation2012). The cytosolic GSTs and the Kappa-class GSTs are evolutionarily derived from common ancestral proteins (Martin Citation1995; Frova Citation2006; Atkinson and Babbitt Citation2009). The structural differences and similarities between the cytosolic and Kappa-class GSTs led Armstrong and coworkers (Ladner et al. Citation2004) to propose parallel (convergent) evolution pathways for each class with thioredoxin/glutaredoxin proteins as the suggested starting points (Frova Citation2006). The bacterial fosfomycin resistance proteins (frp) catalyze a GST reaction and are members of the vicinal oxygen chelate (VOC) superfamily (Armstrong Citation2000, Citation2010; He and Moran Citation2011; Armstrong et al. Citation2018); these metalloenzymes are unrelated to other members of the GST superfamilies.
The GSTs were discovered during the search for the source of the Cys moiety in mercapturic acids (for a review, see Boyland and Chasseaud Citation1969). Two findings established GSH as the source of the Cys moiety in mercapturic acids: one, GSH S-conjugates given to animals are excreted as mercapturic acids (Barnes et al. Citation1959; Bray et al. Citation1959a). Two, liver cytosolic enzymes catalyze the formation of GSH S-conjugates (Booth et al. Citation1961; Combes and Stakelum Citation1961). Subsequent studies showed that liver enzymes catalyze the reaction of GSH with a diverse group of compounds (Booth et al. Citation1961; Al-Kassab et al. Citation1963; Boyland and Williams Citation1965; Johnson Citation1966; Suga et al. Citation1967; Gillham Citation1973; Armstrong Citation2010; Cooper and Hanigan Citation2010; Armstrong et al. Citation2018).
The cytosolic GSTs were the first GSTs to be identified and have been extensively studied (for reviews, see Mannervik and Danielson Citation1988; Coles et al. Citation1990; Awasthi et al. Citation1994; Hayes and Pulford Citation1995; Sheehan et al. Citation2001; Sherratt and Hayes Citation2002; Hayes et al. Citation2005; Josephy and Mannervik Citation2006; Wu and Dong Citation2012; Mohana and Achary Citation2017; Allocati et al. Citation2018). Later studies showed that GST activity was also present in hepatic microsomal, mitochondrial, peroxisomal, and nuclear fractions (Suga et al. Citation1967; Glatt and Oesch Citation1977; Kraus and Gross Citation1979; Morgenstern et al. Citation1979; Wahllander et al. Citation1979; Nishino and Ito Citation1990b).
This section will focus on structure and properties of GSTs along with their role in the mercapturic acid pathway. A discussion of the genetics and phylogeny of GSTs is beyond the scope of this review. Several reviews are, however, available (Nebert and Vasiliou Citation2004; Pearson Citation2005; Dourado et al. Citation2008a; Josephy Citation2010; Board and Menon Citation2013). The polymorphic variants in human GSTs have been summarized, and the functional effects of the polymorphisms, as well as their clinical and toxicological relevance, have been discussed (Strange et al. Citation2001; Holley et al. Citation2007; Board and Menon Citation2013).
2.2. Cytosolic GST superfamily (EC 2.5.1.18)3
2.2.1. Discovery and nomenclature
Early studies in which GST activities were measured with a range of substrates indicated that several GSTs were present in rat liver tissue and were assigned names based on the assay substrate used, i.e. GSH S-aryltransferase, GSH S-alkyltransferase, GSH S-aralkyltransferase, GSH S-epoxidetransferase, and GSH S-alkenetransferase (Boyland and Chasseaud Citation1969; Chasseaud Citation1974).
Gillham (Citation1971, Citation1973) reported the partial purification of GSTs that catalyze the conjugation of 1-araalkyl sulfates with GSH. Jakoby and coworkers described the column chromatographic purification and preliminary characterization of rat liver GSTs (Fjellstedt et al. Citation1973; Pabst et al. Citation1973, Citation1974; Habig et al. Citation1974b, Citation1976); all are dimeric proteins with subunit molecular masses of approximately 25 kDa and holoenzyme molecular masses of approximately 45 kDa. When activities were measured with a panel of substrates, the purified GSTs showed broad and overlapping substrate selectivities, although some isozymes showed clear substrate preferences.
Later studies demonstrated the utility of immobilized GSH or S-hexylGSH affinity chromatography for purification of GSTs (Simons and Vander Jagt Citation1977; Guthenberg and Mannervik Citation1979). Affinity chromatography with high-pressure liquid chromatography (HPLC) and mass spectrometry for the characterization of GSTs was also reported (Rouimi et al. Citation1995; Listowsky et al. Citation1998).
Earlier, Levi et al. (Citation1969) identified two hepatic cytosolic proteins, designated “Y” and “Z,” that bound large hydrophobic nonsubstrate compounds, e.g. sulfobromophthalein, bilirubin, indocyanine green, and other organic anions, and were termed ligandins (Habig et al. Citation1974a; Ketley et al. Citation1975). The rat liver protein “Y” was later demonstrated to be identical with GST B (Litwack et al. Citation1971; Habig et al. Citation1974a). There is considerable heterogeneity in the properties of ligandin binding sites among GSTs (Oakley Citation2011): with hGSTP1-1, the ligandin binding site occupies part of the second substrate binding site (“H-site”) (Oakley et al. Citation1999), whereas with hGSTA1-1, a large site that spans both subunits serves as a ligandin binding site (Le Trong et al. Citation2002). The ligandin binding site in hGSTO1-1 is found deep within the dimer interface (Brock et al. Citation2013).
GST Theta (GSTT) (Meyer et al. Citation1991), GST Zeta (GSTZ) (Board et al. Citation1997, Citation2001), and GST Omega (GSTO) (Board et al. Citation2000) were identified with BLAST searches of the human expressed sequence tag (EST) database. Sigma-class GSTs (GSTS) were discovered by sequence alignment and phylogenetic analysis of GST-like proteins in cephalopod tissues (Tomarev and Zinovieva Citation1988).
The multiplicity of GSTs and the several substrates used to measure activity led to inconsistent and confusing nomenclature. Rat GSTs can be differentiated based on column chromatographic behavior, substrate selectivity, isoelectric point, SDS-polyacrylamide gel electrophoresis, amino acid composition, and sensitivity to inhibitors (Bass et al. Citation1977; Jakoby Citation1978; Mannervik and Jensson Citation1982; Yälçin et al. Citation1983; Jakoby et al. Citation1984).
A standardized nomenclature for the human GSTs based on amino acid and gene sequences was described by Mannervik et al. (Citation1992, Citation2005) and has been extended to other species (Hayes and Pulford Citation1995). The criteria for classifying mammalian GSTs have been summarized (Sheehan et al. Citation2001). With the standardized nomenclature, each subunit encoded by a discrete gene has its own designation. Subunits are grouped into classes or gene families of GSTs and are numbered sequentially with Arabic numerals. The recognized classes of cytosolic GSTs include: Alpha, Mu, Pi, Theta, Zeta, Omega, and Sigma, which are identified by upper-case letters, e.g. A, M. P, T, Z, O, and S. Species designations are indicated by a lower-case letter preceding the GST abbreviation, e.g. h, human; r, rat; m, murine. GSTs are dimeric proteins, and subfamily homo- and heterodimers are denoted with Arabic numerals separated by a hyphen. For example, a homodimeric rat Alpha-class GST from subfamily A would be written as rGSTA1-1. Several designations have been used in the literature for allelic variants, although currently most authors use the “star” designation, e.g. GSTM1*A-1*A.
Subunits are identified by primary DNA sequence homology. Members of the same class share 40–50% sequence identity but less than approximately 25–30% sequence identity with GSTs in other classes. The nomenclature for human, rat, and mouse GSTs is summarized in . A mouse GST database is available (http://www.people.virginia.edu/∼wrp/gst_mouse/gst_mouse.html; accessed 31 October 2019).
Table 1. Human cytosolic glutathione transferases.
Table 2. Mouse cytosolic glutathione transferasesTable Footnote*.
Table 3. Rat cytosolic glutathione transferasesTable Footnote*.
2.2.2. Structure and properties common to cytosolic GSTs
The determination of the amino acid and DNA sequences for cytosolic GSTs was followed by crystallographic analysis, which provided insight into the relationship between structure and catalytic activity. Indeed, three-dimensional structures are available for all human cytosolic GSTs, except hGSTA5-5, for many rat and mouse cytosolic GSTs, and for Kappa-class GST (Oakley Citation2011; Wu and Dong Citation2012; Rose et al. Citation2017). Reviews about the structure of GSTs are available (Armstrong Citation1997; Salinas and Wong Citation1999; Sheehan et al. Citation2001; Pearson Citation2005; Frova Citation2006; Dourado et al. Citation2008a; Atkinson and Babbitt Citation2009; Armstrong Citation2010; Oakley Citation2011; Wu and Dong Citation2012; Mohana and Achary Citation2017; Armstrong et al. Citation2018).
Canonical cytosolic GSTs are homo- or heterodimeric proteins; although homodimers apparently predominate in vivo, heterodimeric GSTs, e.g. GSTA1-2, have been purified from human, rat, and hamster tissues (Mannervik and Jensson Citation1982; Tu and Reddy Citation1985; Bogaards et al. Citation1992; Czerwinski et al. Citation1996). The subunit masses are approximately 25 kDa (Eaton and Bammler Citation1999; Mohana and Achary Citation2017).
Early studies recognized that the requirement for GSH and a second electrophilic substrate likely indicated the presence of distinct substrate and electrophile binding sites (Jakobson et al. Citation1977). Mannervik et al. (Citation1978, Citation1985) proposed the terms “G-site” and “H-site” for the binding sites for GSH and the second substrate, respectively.
Each subunit consists of distinct N- and C-terminal domains (also termed Domains 1 and 2, respectively) that are connected by a short linker sequence between helices α3 and α4 (Ji et al. Citation1992; Reinemer et al. Citation1992; Dirr et al. Citation1994b) (). Among the cytosolic GSTs, amino acid sequence similarities are highest (∼70%) for the N-terminal domains but are lower (∼30%) and vary considerably in the C-terminal domains (Salinas and Wong Citation1999; Armstrong Citation2010); in spite of the differences in sequence similarities, the spatial structures of, for example, GSTA, -M, and -P are very similar (Dirr et al. Citation1994b).
Figure 3. (A) Structural representation of cytosolic GSTs. The thioredoxin fold of the N-terminal domain is represented by β1α1β2α2β3β4α3 (filled arrows and rectangles). The α-helices of the C-terminal domain are shown as stippled rectangles; the number of α-helices in the C-terminal domain varies among the GST subfamilies. The broken line represents the linker between the N- and C-terminal domains. Modified from Frova (Citation2006); (B) crystal structure of hGSTA1 in complex with S-(benzyl)GSH; (C) crystal structure of hGSTA1-1 in complex with S-(benzyl)GSH (PDB 1GUH). Panels (B) and (C) are reproduced with permission from the publisher and from Wu and Dong (Citation2012); the authors thank Prof. Wu for providing (B) and (C).
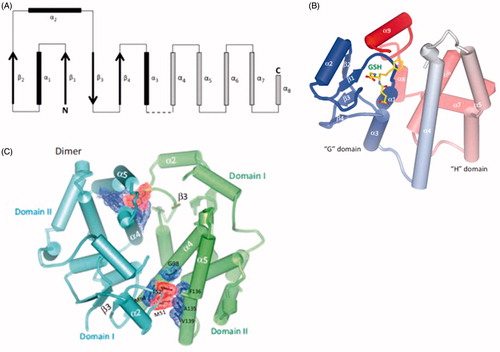
The N-terminal domain adopts the thioredoxin fold represented by a β1α1β2 motif at the N-terminus and is connected to a β3β4α3 motif at the C-terminus by a loop containing the α2 helix (). The C-terminal domain contains five or more α-helices, depending on the subfamily, e.g. GSTA, -T and -O have an α9 helix (Dirr et al. Citation1994b; Sheehan et al. Citation2001; Frova Citation2006; Dourado et al. Citation2008a; Oakley Citation2011; Wu and Dong Citation2012).
The H-site is typically located in the cleft between the N- and C-domains, and amino acid residues from both domains contribute to the interaction of substrates with the H-site (Mohana and Achary Citation2017). Crystallographic analyses of GSTs showed that amino acid residues of the G- and H-sites are found in both the N-terminal and C-terminal domains (Ji et al. Citation1992; Sinning et al. Citation1993; Ji et al. Citation1994; Wilce and Parker Citation1994; Martin Citation1995; Sheehan et al. Citation2001; Patskovsky et al. Citation2006; Deponte Citation2013). Mohana and Achary (Citation2017) indicate that the H-site for cytosolic GSTs, except GSTK, can be divided into three regions: for GSTA, the loop connecting α1 and β2 is designated as H1; residues from the C-terminal portion of α4 constitute the H2 region; and variable residues from the C-terminal α-helix constitute the H3 region. Details about the composition of the H-sites for each of the cytosolic GSTs are available (Mohana and Achary Citation2017). The size, topography, and amino acid residues of the H-site play a critical role in the electrophilic substrate selectivity of GSTs (Kurtovic et al. Citation2008; Shokeer and Mannervik Citation2010b; Wu and Dong Citation2012; Modén and Mannervik Citation2014; Mohana and Achary Citation2017). Activity-based probes that target the H- and G-sites of cytosolic GSTs have been reported (Stoddard et al. Citation2017).
Although the highly conserved structure and sequence of the G-site is well documented, GSTs vary in the identity and location of catalytically important amino acid residues in the G-site (for a description of amino-acid residues involved in GSH binding, see Mohana and Achary Citation2017). Binding of GSH to the G-site is dependent on polar interactions between GSH and the β3β4α3 motif, and the thiol of GSH forms hydrogen bonds with the wall of the G-site.
A critical Tyr residue in the G-site, which serves to stabilize the GSH thiolate, is replaced in several GSTs by a Ser or Cys. Accordingly, Atkinson and Babbitt (Atkinson and Babbitt Citation2009) designated GSTA, GSTM, GSTP, and GSTS as “Y-GSTs” and GSTT, GSTZ, and GSTO as “S/C-GSTs.” The highly conserved G-site shows high selectivity for GSH, although some GSH analogs are substrates (Chen et al. Citation1988; Adang et al. Citation1989, Citation1990); the γ-Glu residue of GSH is a major binding determinant (Adang et al. Citation1990).
The quaternary structure of cytosolic GST dimers is characterized by a two-fold axis of symmetry that extends through the interface. The nature of the interactions between the dimers differs among the cytosolic GST subfamilies. In the Alpha-, Mu-, and Pi-class GSTs, subunit interactions are attributed primarily to a lock-and-key motif consisting of hydrophobic interactions between an aromatic amino acid (Phe52 in hGSTA, Phe56 in hGSTM, and Tyr49 in hGSTP) that protrudes from the loop connecting α2-helix with β3-strand in the N-terminal domain and interacts with a hydrophobic pocket formed by α4 and α5 helices on the C-terminal domain of the second monomer (Sinning et al. Citation1993; Sayed et al. Citation2000; Sheehan et al. Citation2001; Hegazy et al. Citation2004; Alves et al. Citation2006; Thompson et al. Citation2006). Electrostatic interactions between polar residues on the β4 strand and the α3 helix with residues on the α4 helix of the second monomer also contribute to stabilization of GSTA, GSTM, and GSTP dimers (Thompson et al. Citation2006; Wu and Dong Citation2012). With Sigma-class GSTs, polar interactions between subunits serve to maintain their dimeric structure (Stevens et al. Citation1998), whereas with Omega-class GSTs, nonpolar interactions between subunits dominate (Board et al. Citation2000). With hGSTZ, nonpolar interactions between monomers prevail, but a polar interaction between Glu81 and Arg96 is also observed (Polekhina et al. Citation2001).
The properties of the subunit interface may exert significant effects on catalytic activity. Binding of the cosubstrate in GSTP1-1 increases the affinity for GSH by several folds (Caccuri et al. Citation1996). With hGSTA1-1, the side-chain of Trp21 in the N-terminal domain interacts with helices α6 and α8 in the C-terminal domain (Balchin et al. Citation2010); the W21A mutant affects the H-site (decreased kcatCDNB) but has little effect on the G-site (little change in kcatGSH).
Although cytosolic GSTs are functional dimers, the question has been raised whether catalytically competent GST monomers exist in cells. Conflicting conclusions about the existence of stable, catalytically active GSTP1 monomers have been reported, but the preponderance of the data indicate that catalytically active GSTP1 monomers are likely not present in cells (for a summary, see Fabrini et al. Citation2009). It was also reported that regulation of JNK signaling was attributable to a GSTP1 monomer (Adler et al. Citation1999); subsequent studies show, however, that dimeric GSTP1-1 rather than the monomer GSTP1 most likely serves to regulate JNK signaling (Gildenhuys et al. Citation2010b).
GSTs catalyze the reaction of GSH with a range of carbon-, sulfur-, and oxygen containing electrophilic substrates (Deponte Citation2013). In addition to catalyzing transferase reactions (SNAr, Michael additions, epoxide ring opening), where the GSH moiety is incorporated in the product GSH S-conjugate, GSTs also catalyze isomerization reactions and the reduction of peroxides and disulfides. The catalytic mechanisms of transferase, isomerization, and reductive reactions show considerable mechanistic diversity. The mechanism of GST-catalyzed reactions has been the subject of intense investigation (for reviews, see Sheehan et al. Citation2001; Dourado et al. Citation2008a; Atkinson and Babbitt Citation2009; Wu and Dong Citation2012). Deponte (Citation2013) has described several models of GST-catalyzed reactions and illustrated how they differ among the GST classes and substrates.
A major event in the catalytic cycle is the formation and stabilization of the GSH thiol as a thiolate (Armstrong Citation1997; Armstrong Citation2010). It was originally proposed that the Tyr7 residue of hGSTP1-1 acted as a general base in promoting the formation of the GSH thiolate (GS-) (Karshikoff et al. Citation1993). Later studies showed, however, that at the pH optimum for hGSTA1-1 the Tyr residue was largely present as the conjugate acid (Björnestedt et al. Citation1995); moreover, the Y9F mutant retains partial catalytic activity. Rather than serving as a general base, the catalytically important Tyr is now believed to play a key role in stabilizing the reactive, negatively charged GSH thiolate. With C/S-GSTs, a hydrogen-bonding interaction between the active-site Ser or Cys plays a major role in stabilizing the GSH thiolate (e.g. Liu et al. Citation1992). In GSTA1-1-, GSTM1-1-, and GSTP1-1-catalyzed reactions, the binding of GSH to the G-site is accompanied by a decrease in the pKa of the thiol group from 9.1 to 6.2–6.6 (Caccuri et al. Citation1999; Dourado et al. Citation2008a). Other studies on the binding and activation of GSH and the fate of the thiol proton in cytosolic GSTs indicate that a multistep process is involved and that a water molecule may assist in catalysis (Xiao et al. Citation1996; Parraga et al. Citation1998; Caccuri et al. Citation1999; Dourado et al. Citation2008b, Citation2009, Citation2010b). Regardless of the mechanistic details of deprotonation of GSH to form the highly nucleophilic thiolate, the most significant features of the overall GSH activation process include the rapid formation and stabilization of the thiolate upon binding of GSH to the G-site, where it is poised to react with electrophilic substrates.
In hGSTM1-1 the direct (first-sphere) interaction of the GSH thiolate with Tyr6 is supplemented by a range of second-sphere electrostatic interactions (Xiao et al. Citation1996). Arg15 also stabilizes the GSH thiolate in hGSTA1-1 (Björnestedt et al. Citation1995). The differences in catalytic rates and regio- and stereoselectivity observed among cytosolic GSTs are attributed to differences in stabilization of transition states of the specific reactions (Armstrong Citation2010; Deponte Citation2013).
The presence of H- and G-sites raised the question of whether both subunits were catalytically active (all-of-the-sites reactivity) in the holoenzyme or whether only one site was active (half of the sites reactivity). The two active sites present in each subunit are kinetically independent (Danielson and Mannervik Citation1985). GSTA1-1 exhibits all-of-the-sites reactivity in SN2Ar reactions but half-of the-sites reactivity in addition reactions (Lien et al. Citation2001).
As with many other xenobiotic-metabolizing enzymes, the cytosolic GSTs are induced by a range of chemicals (for reviews, see Higgins and Hayes Citation2011; Boušová and Skálová Citation2012; Ma and He Citation2012). A detailed discussion of the induction of GSTs by chemicals is beyond the scope of this review, but some salient points merit mention. Two xenobiotic-responsive elements (XREs) are implicated in the induction of GSTA1-1: one requires the aryl-hydrocarbon receptor (AhR) for the induction of GSTA1-1 by planar aromatic compounds, e.g. β-naphthoflavone (Rushmore et al. Citation1990); these compounds also induce Phase-I enzymes, e.g. cytochromes P450, and are termed bifunctional inducers. A second group of compounds, e.g. phenolic antioxidants, induce the transcriptional activation of GSTA1-1 independent of the AhR and are termed monofunctional inducers; the XRE involved was named the antioxidant-responsive element (ARE) (Rushmore and Pickett Citation1990). It was also demonstrated that bifunctional inducers require biotransformation by cytochromes P450 to induce GSTs. The bZIP transcription factor Nrf2 is required for the ARE-dependent transcriptional activation of GSTs (Itoh et al. Citation1997). In addition to Nrf2, other transcription factors regulate the activity of ARE (Motohashi et al. Citation2002; Hayes et al. Citation2005). The activity of the microsomal GSTs is also induced by xenobiotics (Higgins and Hayes Citation2011).
Activity-based protein profiling (ABPP) of murine cytosolic GSTs has been used to explore the effect of benzo[a]pyrene (B[a]P) induction on protein expression and on G- and H-site activities (Stoddard et al. Citation2019). The approach involves the use of two activity-based probes, a GSH-based photoaffinity probe that targets the G-site and a reactive GST inhibitor that binds to H-site residues, which are incubated with tissue cytosols followed by protein and proteomic analysis (Stoddard et al. Citation2017). In mice given B[a]P, both expression and G- and H-site activities of GSTA2, -M4, and -P1 were increased, whereas an expression-independent increase in H-site activity was found with GSTM1, -2, and -6. In contrast, an expression-independent inhibition of G- and H-site activities of GSTA4 was observed. The mechanisms underlying these observed changes have not been elucidated but the possible involvement of post-translational modification-induced conformational changes was suggested.
2.2.3. Substrates
Cytosolic GSTs exhibit broad substrate selectivity for xenobiotic substrates, although some GST classes show significant selectivity. Substrates for the several classes of cytosolic GSTs are discussed below. A summary of the catalytic activities of rat, mouse, and human GSTs has been published (Hayes and Pulford Citation1995). Deponte (Citation2013) has reviewed how structural features of the cytosolic GSTs determine substrate selectivity. GSTs also catalyze reactions with physiological substrates, including steroid isomerization, maleylacetoacetic acid isomerization, prostaglandin reduction and synthesis, and organic peroxide reduction (Sharma et al. Citation2007). Prodrugs of cancer chemotherapeutic agents that undergo GST-catalyzed activation have also been reported (Morgan et al. Citation1998; Zhao and Wang Citation2006; Axarli et al. Citation2009; Johansson et al. Citation2011; Ruzza and Calderan Citation2013; Ramsay and Dilda Citation2014).
2.2.4. Inhibitors
Considerable effort has been expended in the development of GST (particularly GSTP) inhibitors, largely to overcome GST-dependent drug resistance (for reviews, see van Bladeren and van Ommen Citation1991; Tew et al. Citation1997; Burg and Mulder Citation2002; Townsend and Tew Citation2003; Mahajan and Atkins Citation2005; Mathew et al. Citation2006; Zhao and Wang Citation2006; Li et al. Citation2010; Thurairatnam Citation2012; Wu and Batist Citation2013; Mohana and Achary Citation2017). Inhibitors of the several classes of cytosolic GSTs are discussed below. Few GST inhibitors that show activity in vitro appear to have been tested for activity in vivo but compounds effective in animal models have been reported (Ouwerkerk-Mahadevan et al. Citation1996; Mulder and Ouwerkerk-Mahadevan Citation1997; Ouwerkerk-Mahadevan and Mulder Citation1998; Gale and Tew Citation2001).
2.2.5. Tissue and species distribution
The cytosolic GSTs are widely distributed in mammals, and individual GSTs often differ in tissue distribution. For example, Alpha- and Mu-class GSTs are intensely expressed in the liver, whereas Pi-class GST is scarcely expressed in the liver but is highly expressed in various nonhepatic tissues. For compilations of the tissue distribution of GSTs, see Baars et al. (Citation1981), Mannervik (Citation1985), Mannervik and Widersten (Citation1995), Rowe et al. (Citation1997), Lantum et al. (Citation2002a), Dhanani and Awasthi (Citation2007), Holley et al. (Citation2007) and Mohana and Achary (Citation2017). Activity-based probes that are targeted at the H- and G-sites of GSTs have been described (Stoddard et al. Citation2017); these probes allow site-specific profiling of cytosolic GST activities in multiple tissues.
2.2.6. Cytosolic GST classes
2.2.6.1. Alpha-class (GSTA)
2.2.6.1.1. Structure and properties
Alpha-class GSTs have been found in all mammalian species studied. Links to the human, mouse, and rat genes encoding Alpha-class GSTs are listed in . The expression of hGSTA5 has been proposed to be limited to unidentified physiological or pathological conditions (Singh et al. Citation2010).
The crystal structure of hGSTA1-1 with the bound inhibitor S-(benzyl)GSH showed two domains in each monomer and identified the key amino acid residues in the G- and H-sites that form hydrogen bonds and salt links with the inhibitor (Sinning et al. Citation1993). Arg15, a G-site residue that is located near the juncture with the H-site, was proposed to be within hydrogen bonding distance of the GSH sulfur atom of the bound inhibitor; a more recent crystal structure of the GSH-hGSTA1-1 complex does not, however, show hydrogen bonding between Arg15 and GSH (Grahn et al. Citation2006, PDB: 1PKW). Arg 15 is conserved in all GSTA class members except GSTA5-5 (Singh et al. Citation2010); mutating Arg15 to Leu, Ala, or His in GSTA1-1 causes a substantial reduction in catalytic activity (Björnestedt et al. Citation1995). Arg15 has a modest influence on the affinity of GSTA1-1 for GSH, but mutation of Arg15 to Leu raises the pKa of bound GSH from 6.5 to 7.6, indicating that Arg15 contributes to the stabilization of the GSH thiolate in the active site. In the absence of hydrogen bonding between Arg15 and the GSH thiolate, the stabilization of the thiolate by Arg15 was attributed to electrostatic forces that may involve its participation in an electron-sharing network that contributes to the deprotonation of GSH and stabilization of GSH thiolate (Winayanuwattikun and Ketterman Citation2005; Gildenhuys et al. Citation2010a). A computational analysis of the activation of GSH by GSTA1-1 indicates that Arg15 forms an ion-dipole and a hydrogen bond with the Cys carbonyl of GSH that persists through the transition state and in the GSH thiolate complex (Dourado et al. Citation2010a).
Alpha-class GSTs, especially GSTA3-3, play an important role in steroidogenesis by catalyzing the isomerization of the C5–C6 double bond in Δ5-androstene-3,17-dione (Δ5-AD) (13, ) to form the testosterone precursor Δ4-androstene-3,17-dione (14, ) (Johansson and Mannervik Citation2001). The hGSTA3-3 H-site differs in three residues from the H-site of hGSTA1-1 and differs in five H-site residues from hGSTA2-2. Compared with hGSTA3-3, the catalytic efficiency, as represented by kcat/Km, for isomerization of the C5-C6 double bond in Δ5-AD (13, ) to the C4–C5 position is 10-fold lower for hGSTA1-1 and 5000-fold lower for hGSTA2-2. Site-directed mutagenesis in which five H-site residues in hGSTA2-2 were replaced by the corresponding five variant H-site residues (Phe10, Gly12, Leu111, Ala208, and Ala216) of hGSTA3-3 resulted in a 3500-fold increase in the hGSTA2-2 kcat/Km for isomerization of Δ5-AD, making it comparable to that of hGSTA3-3 (Pettersson et al. Citation2002).
Figure 4. GSTA substrates. 11, 1-chloro-2,4-dinitrobenzene; 12, S-(2,4-dinitrophenyl)glutathione; 13, Δ5-androstene-3,17-dione (Δ5-AD, carbon atoms 4, 5, and 6 are numbered); 14, Δ4-androstene-3,17-dione (carbon atoms 4, 5, and 6 are numbered); 15, cumene hydroperoxide; 16, 2-phenyl-2-propanol; 17, 4-hydroxy-2-nonenal (HNE); 18, l-γ-Glu-S-[2-hydroxy-1-(2-oxoethyl)heptyl]-l-Cys-Gly (HNE-GSH); 19, aflatoxin B1-8,9-epoxide; 20, 8,9-dihydro-8-(S-glutathionyl)-9-hydroxyaflatoxin B1; 21, busulfan; 22, γ-Glu-β-(S-tetrahydrothiophenium)-Ala-Gly; 23, azathioprine; 24, 6-mercaptopurine; 25, 1-methyl-4-nitro-5-(glutathion-S-yl)-1H-imidazole; 26, melphalan; 27, N-(2-glutathion-S-ylethyl)-N-(2-chloroethyl)-l-phenylalanine; 28, chlorambucil; 29, N-(2-glutathion-S-ylethyl)-N-(2-chloroethyl)benzenebutanoic acid; 30, (7 R,8S,9R,10S)-(-)-syn-benzo[a]pyrene-7,8-diol 9,10-epoxide ((-)-syn-BPDE); 31, (7 R,8S,9R,10R)-10-(glutathion-S-yl)-7,8,9,10-tetrahydrobenzo[a]pyrene-7,8,9-triol; 32, (8S,10S)-7,8,9,10-tetrahydro-6,8,11-trihydroxy-8-(2-hydroxyacetyl)-1-methoxy-10-[[2,3,6-trideoxy-3-[[(2,4-dinitrophenyl)sulfonyl]amino]-α-l-lyxo-hexopyranosyl]oxy]-5,12-naphthacenedione (DNS-DOX); 33, doxorubicin; 34, N-(3′-((2,4-dinitrophenyl)sulfonamido)-3-oxo-3H-spiro[isobenzofuran-1,9′-xanthen]-6′-yl)acetamide (DNs-AcRh); 35, 2-(3-(acetylimino)-6-amino-3H-xanthen-9-yl)benzoic acid (N-acetylrhodamine); 36, 2-(7-((2,4-dinitrophenyl)sulfonamido)-4-methyl-2-oxochroman-3-yl)acetic acid (DNs-Coum); 37, 2-(7-amino-4-methyl-2-oxochroman-3-yl)acetic acid; 38, (Z)-N-(9-amino-5H-benzo[a]phenoxazin-5-ylidene)-2,4-dinitrobenzenesulfonamide (DNs-CV). 39, cresyl violet; 40, 4-acetyl-2-nitro-N-(2-oxo-4-(trifluoromethyl)-2H-chromen-7-yl)benzenesulfonamide (ANs-AFC); 41, S-(4-acetyl-2-nitrophenyl)GSH; 42, 7-amino-4-(trifluoromethyl)coumarin (AFC); 43, 4-[3-(4-acetyl-2-nitrobenzenesulfonyl)phenyl]-4-methoxyspiro[1,2-dioxetane-3,2′-adamantane] (ANs-AMPD); 44, 3-(2-spiroadamantane)-4-methoxy-4-phenyl-1,2-dioxetane (AMPD).
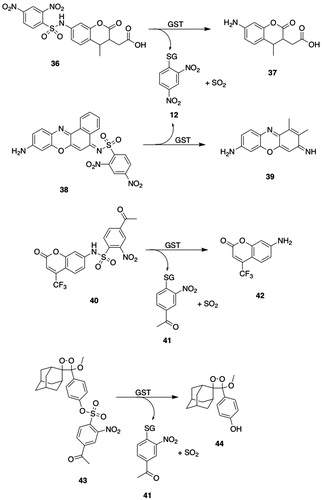
The crystal structure of the complex of hGSTA2-2 with Δ5-AD (13, ) shows that the steroid substrate cannot achieve an orientation in the active site similar to the one seen in hGSTA3-3, due in part to unfavorable steric interactions with Phe111 and Met208, which do not occur with Leu111 and Ala208 in hGSTA3-3 (Tars et al. Citation2010). Examination of the crystal structure of hGSTA3-3 in a complex with both GSH and Δ5-AD revealed that Δ5-AD (13, ) is bound with a C4 hydrogen atom proximal to the GSH thiolate and the phenolic hydroxyl of Tyr9 is located near C6. A model was suggested in which Tyr9 plays an “auxiliary” role in catalysis by forming a hydrogen bond with the sulfur of GSH to facilitate its conversion to the thiolate, which functions as an acid/base catalyst in the proton transfer from C4 to C6 during isomerization of the double bond to the C4–C5 position (Tars et al. Citation2010).
A quantum mechanics/molecular mechanics computational analysis found that the isomerization reaction is a concerted, but asynchronous, reaction in which GSH serves as an acid/base catalyst that requires Tyr9 to act as a proton shuttle (Calvaresi et al. Citation2012). In this proposed mechanism, Arg15 forms a hydrogen bond with the sulfur of GSH thiolate. A similar proton donor function for Tyr9 had been suggested earlier by Gu et al. (Citation2004). The proposal that the GSH thiolate effects both proton abstraction from C4 of Δ5-AD (13, ) and transfer of the proton to C6 (Tars et al. Citation2010) was modified as the result of a DFT computational analysis of the reaction (Dourado et al. Citation2014). The modified proposal retains the initial proton abstraction from C4 by the GSH thiolate to form GSH and the negatively charged enolate of Δ5-AD (13, ), which is followed by breaking of the weakened hydrogen bond between GSH and Tyr9 and a conformational rearrangement of Tyr9 that allows it serve as the proton donor to C6 of the enolate to form Δ4-androstene-3,17-dione (14, ). Following a second conformational change, Tyr9 is protonated by GSH to return the enzyme to its original state. Other studies indicate that Arg15 is as important as Tyr9 in the isomerization reaction and plays an important role in decreasing the pKa of GSH (Robertson et al. Citation2017).
4-Hydroxy-2-nonenal (17, ), formed by the peroxidative degradation of arachadonic acid, is a substrate for GSTs (Ålin et al. Citation1985). hGSTA4-4 shows far higher catalytic activities than hGSTA1-1 with 4-hydroxy-2-nonenal (17, ) and related toxic alkenal products of lipid peroxidation (Hubatsch et al. Citation1998). The high catalytic activity of hGSTA4-4 for 4-hydroxy-2-nonenal (17, ) is attributed to the H-site Tyr212 residue, which enhances the electrophilicity of the aldehyde by forming a specific hydrogen bond with the carbonyl oxygen, causing increased polarization of the carbonyl group (Bruns et al. Citation1999).
SNPs have been identified in hGSTA1, hGSTA2, and hGSTA3 (for a summary of GSTA allelic polymorphisms and their racial distribution, see Coles and Kadlubar Citation2005).
Alpha-class GSTs are induced by many agents (for compilations of GSTA inducers, see Morel et al. Citation1994; Hayes and Pulford Citation1995). hGSTA1/A2 activities are increased in human subjects with high vegetable intake (Sreerama et al. Citation1995; Hoensch et al. Citation2002). Ethoxyquin and coumarin are inducers of rat GSTA5-5 (Hayes et al. Citation1998). 3 H-1,2-Dithiole-3-thione is an inducer of Alpha-class GSTs (Li et al. Citation2005b). In mice given B[a]P, activity-based protein profiling (see Section 2.2.2 above) revealed an expression-independent decrease in both G- and H-site activities in GSTA4 (Stoddard et al. Citation2019).
2.2.6.1.2. Substrates
Although the Alpha-class GSTs show broad substrate selectivity, some Alpha-class GSTs show substrate preferences (for compilations of Alpha-class GST substrates, see Hayes and Pulford Citation1995; Coles and Kadlubar Citation2005).
CDNB (11, ) is frequently used as an assay substrate for cytosolic GSTs; significant differences are, however, found in its reaction rates with purified human GSTs (Czerwinski et al. Citation1996): the reaction rates for GSTA1-2 and GSTA2-2 are much less (<10%) than the rate for GSTA1-1. With recombinant hGSTs and with CDNB (11, ) as the substrate, the reaction rates for GSTA1-1 and GSTA2-2 were similar but were larger than those found with GSTA3-3 or GSTA4-4 (Johansson and Mannervik Citation2001).
GSTA3-3 preferentially catalyzes the isomerization of Δ5-androstene-3,17-dione (13, ) to Δ4-androstene-3,17-dione (14, ) (a testosterone precursor) and of Δ5-pregnene-3,20-dione to Δ4-pregnene-3,20-dione (progesterone) (Benson et al. Citation1977; Johansson and Mannervik Citation2001; Raffalli-Mathieu and Mannervik Citation2005; Dourado et al. Citation2014). Δ5-Androstene-3,17-dione (13, ) and cumene hydroperoxide (15, ) have been proposed as class-distinguishing substrates for GSTA (Mannervik et al. Citation1985). hGSTA3-3-catalyzed steroid isomerase activity is 7- and 1657-folds greater than that of hGSTA1-1 and hGSTA2-2, respectively (Johansson and Mannervik Citation2002). hGSTA4-4 shows selectivity for 4-hydroxy-2-nonenal (HNE) (17, ) and related α,β-unsaturated aldehydes (Hubatsch et al. Citation1998; Balogh and Atkins Citation2011). Rat GSTA5-5 and the GSTA5 heterodimers GSTA1-5 and GSTA3-5 show high selectivity for aflatoxin B1-8,9-epoxide (19, ) (Hayes et al. Citation1998). The resistance of mice to aflatoxin B1 (19, ) has been attributed to the high expression of GSTA3-3 (YcYc) in mouse liver (Hayes et al. Citation1992).
hGSTA catalyzes the reduction of cumene hydroperoxide (15, ) and phospholipid hydroperoxides (Mannervik et al. Citation1985; Hurst et al. Citation1998; Zhao et al. Citation1999a). Purified human GSTA1-1 selectively catalyzes the conjugation of busulfan (21, ) (Czerwinski et al. Citation1996); lower (<10%) activity is also observed with GSTA1-2 and GSTA2-2.
Azathioprine (23, ) is a widely used immunosuppressant that is a prodrug of 6-mercaptopurine (24, ); the drug is administered orally and is activated primarily by GST-catalyzed conjugation with GSH (Modén and Mannervik Citation2014). The reaction involves attack of the GSH thiolate on the imidazole ring to produce stoichiometric quantities of 6-mercaptopurine (24, ) and the imidazole–GSH conjugate (25, ) (Eklund et al. Citation2006). GSTA1-1, GSTM1-1, and GSTA2-2 are the principal GSTs responsible for activation of azathiopurine in human liver and intestine, with GSTA2-2 having more than twice the specific activity and twice the catalytic efficiency (kcat/Km) of either GSTA1-1 or GSTM1-1. GSTA2*E is an allelic variant of GSTA2 (GSTA2*A) that has 60–70% of the specific activity of GSTA2*A and a somewhat lower catalytic efficiency than GSTA2*A with CDNB (11, ) as substrate, but exhibits four-fold greater specific activity and catalytic efficiency than GSTA2*A with azathiopurine (23, ) as substrate (Ning et al. Citation2004; Zhang et al. Citation2010). Pro110 is conserved in all GSTA2 variants except GSTA2*E, which contains Ser110; this residue is near the C-terminus of α-helix 4, where it could potentially function as an H-site residue (Modén and Mannervik Citation2014).
Melphalan (26, ), a chemotherapeutic alkylating agent containing a reactive nitrogen mustard moiety, undergoes hGSTA1-1-catalyzed biotransformation to the monoglutatathione S-conjugate (27, ) and the diglutathione S-conjugate (Dulik et al. Citation1986; Hall et al. Citation1994a; Paumi et al. Citation2001). The glutathionylation reaction takes place between the GSH thiolate and the aziridinium ion intermediate formed from the nitrogen mustard (Bolton et al. Citation1993). The structurally similar chemotherapeutic agent chlorambucil (28, ) is a substrate for hGSTA1-1 and 2-2, and is biotransformed to the analogous monoglutathione (29, ) and diglutathione S-conjugates; monoglutathione conjugate 29 is quantitatively the most significant product (Ciaccio et al. Citation1990; Meyer et al. Citation1992). GSTA1-1 catalyzes the conversion of chlorambucil (28, ) to conjugate 29 () with four-fold greater efficiency than the conversion of mephalan (26, ) to its monoglutathione S-conjugate (27, ) (Paumi et al. Citation2001). The formation of the chlorambucil monoglutathione conjugate (29, ) results in product inhibition of GSTA1-1, whereas the mephalan conjugate 27 () is a less effective inhibitor of the complex (Meyer et al. Citation1992; Paumi et al. Citation2001). The bonding interactions of the monoglutathione conjugate of chlorambucil (29, ) with the active-site residues of GSTA1-1 were deduced from a crystal structure of the complex (Karpusas et al. Citation2013). Expression of GSTs has been associated with development of resistance to various anticancer agents. The expression of GSTA1-1 activity in MCF7 and HepG2 cells is insufficient to cause maximum resistance to chlorambucil (28, ) in the absence of efflux transporters MRP1 (ABCC1) or MRP2 (ABCC2) (Morrow et al. Citation1998; Paumi et al. Citation2001; Smitherman et al. Citation2004). Both transporters efficiently eliminate the chlorambucil monoglutathione conjugate (29, ) from cells, which prevents the conjugate from inhibiting the catalytic activity of GSTA1-1.
GSTs play a major role in detoxifying bay- and fjord-region diol epoxides, which are the ultimate, DNA-modifying metabolites of polycyclic aromatic hydrocarbon (PAH) carcinogens (Thakker et al. Citation1985). Several classes of cytosolic GSTs, including hGSTA, hGSTM, and hGSTP, catalyze the reaction of GSH with diol epoxide metabolites of PAHs (Robertson and Jernström Citation1986; Sundberg et al. Citation1997). GSTA1-1 catalyzes the conjugation of all PAH-derived diol epoxide isomers studied, although the catalytic efficiencies (Vmax/Km) differ significantly with the absolute configuration of the diol epoxide isomers (Jernström et al. Citation1992, Citation1996); the preferred site of attack is at the benzylic oxiranyl carbon with the R configuration. The catalytic efficiencies of hGSTA1-1-catalyzed conjugation of the four isomers of benzo[a]pyrene diol epoxide (BPDE) follow the order: (−)-syn (30, ) > (+)-syn > (+)-anti > (-)-anti (Jernström et al. Citation1996). The catalytic efficiencies for conjugation of (+)-anti-BPDE are, however, five-fold higher with hGSTA1-1 than with hGSTA2-2, even though the two isozymes differ by only four residues in the H-site (Singh et al. Citation2004). Mutation of H-site Ile11 to Ala11 in hGSTA2-2 results in a seven-fold increase in catalytic efficiency, which, according to molecular modeling studies, is attributable to relief of unfavorable steric interactions between the substrate and the bulky Ile11 residue.
The catalytic efficiency of murine GSTA1-1 with (+)-anti-BPDE is significantly higher than that of mGSTA2-2, -3-3, and -4-4, mGSTP1-1, and mGSTM1-1 (Xia et al. Citation1998); also, murine GSTA1-2 catalyzes the conjugation of (+)-anti-BPDE at rates significantly higher than other murine GSTAs, except GSTA1-1 (Hu et al. Citation1996; Xia et al. Citation1999). The mGSTA1 and mGSTA-2 subunits differ by only 10 amino acid residues; by use of chimeric enzymes, molecular modeling, and site-directed mutagenesis, it was shown that the high catalytic activity of mGSTA1-2 and, thus, the even higher activity of GSTA1-1, can be attributed to Met207 and Ile221 in the mGSTA1 subunit.
The overexpression of certain GSTs in cancer cells has led to the design of GSTA-activated prodrugs as potential chemotherapeutic agents. Sulfonamides having the general structure R-HN-SO2-Ar, where Ar is an electrophilic aromatic ring, can be cleaved by GSTs, including GSTA1-1, to yield RNH2, SO2, and GS-Ar (Koeplinger et al. Citation1999; Zhao et al. Citation1999b). A sulfonamide-based prodrug of metformin (N1,N1-dimethyl-N4-(2-nitro-4-(trifluoromethyl)benzenesulfonamide)-bisguanidine) is cleaved by rat liver GSTs and by hGSTA1-1, -M2-2, and -P1-1 (Rautio et al. Citation2014). van Gisbergen et al. (van Gisbergen et al. Citation2016) reported that a sulfonamide prodrug in which R is doxorubicin and Ar is 2,4-dinitrobenzene (32, ) is metabolized to doxorubicin (33, ) at a much higher rate by GSTA1-1 than by either GSTP1-1 or by rat liver MGST1; the cytotoxicity of the prodrug in cells overexpressing GSTA1-1, however, was substantially lower than that of an analog in which Ar (2-nitro-4-acetylbenzene) was less electrophilic than 2,4-dinitrobenzene and which was converted to free doxorubicin at a much lower rate by GSTA1-1. GSTA1-1-activated, sulfonamide-based prodrugs in which R is an analog of bombesin, a peptide that is recognized and bound by tumor cell receptors, have been prepared for use in targeted drug delivery (Axarli et al. Citation2009).
The GSTA-catalyzed cleavage of sulfonamides was exploited to develop fluorogenic substrates (Zhang et al. Citation2011a). The nonfluorescent compounds DNs-AcRh (34, ), DNs-Coum (36, ), and DNs-CV (38, ) release fluorescent derivatives of N-acetylrhodamine (35, ), 2-(7-amino-4-methyl-2-oxochroman-3-yl)acetic acid (37, ), and cresyl violet (39, ), respectively, after the GSTA-catalyzed cleavage of the 2,4-dinitrophenylsulfonyl moiety. The activities of fluorogenic substrates 34, 36, and 38 () were at least twofold higher for GSTA1-1 than for GSTA2-2, -3-3, and -4-4, GSTM1-1 and -2-2, GSTP1-1, GSTT1-1, and MGST1.
Further studies with sulfonamide- and sulfonate ester-based GSTA substrates led to the development of 19F-NMR and bioluminescent probes (Ito et al. Citation2012; Shibata et al. Citation2013; Ito et al. Citation2014). Benzenesulfonamide 40 () was cleaved by GSTA1-1 at much higher rates than with GSTM1-1, GSTP1-1, or MGST1 to release 7-amino-4-(trifluoromethyl)coumarin (42, ); the cleavage of benzenesulfonamide 40 () in GSTA1-expressing Escherichia coli cells was demonstrated by 19F NMR and by fluorescent intensity. 1,2-Dioxetane 43 (), which was designed as a bioluminescent probe for GSTA, is cleaved by GSTA to release 1,2-dioxetane 44 (); luminescence in E. coli cells transfected with GSTA1 was readily detected.
GSTA shows a strong preference for GSH as the co-substrate. Studies with several GSH analogs and Alpha-class GSTs showed that with rat GSTA1-1 and GSTA2-2 all analogs except α-l-Glu-l-Cys-Gly were inactive (Adang et al. Citation1988a, Citation1988b).
2.2.6.1.3. Inhibitors
Indomethacin (Hall et al. Citation1989), curcumin and analogs (Appiah-Opong et al. Citation2009), and hypericin (Lu and Atkins Citation2004) have been reported to inhibit GSTA. Triethyllead chloride inhibits the GSTA-catalyzed conjugation of BSP, both in vitro and in vivo (Byington and Hansbrough Citation1979). Tributyltin acetate and triphenyltin chloride are potent inhibitors of GSTA class enzymes (Tipping et al. Citation1979; Warholm et al. Citation1986).
Increased expression of GSTs is associated with antitumor drug resistance; accordingly, there has been a search for GST inhibitors that might be used clinically to enhance the effectiveness of chemotherapeutic agents (Townsend and Tew Citation2003; Sau et al. Citation2010).
GSH analogs have been prepared and tested as GSTA inhibitors (for a review, see Burg and Mulder Citation2002). A series of GSH analogs based on a l-γ-Glu-D-2-aminoadipic acid backbone, e.g. (R)-5-ethyloxycarbonyl-2-γ-(S)-glutamylamino-N-2-heptylpentamide (45, ) are selective inhibitors of GSTA1-1 and -2-2, both in vitro and in vivo (Ouwerkerk-Mahadevan et al. Citation1995; Ouwerkerk-Mahadevan and Mulder Citation1998). l-γ-Glu-(S-9-fluorenylmethyl)-l-Cys-Gly and its carbamate congener (46, ), which is resistant to hydrolysis by GGT, inhibit hGSTA1-1 (also GSTM1-1 and GSTP1-1) (Cacciatore et al. Citation2005). In a series of 2-(pyrrolesulfonylmethyl)-N-arylimines, compound 47 () was the most potent inhibitor of hGSTA1-1 (Ki = 71 μM) (Koutsoumpli et al. Citation2012).
Figure 5. GSTA inhibitors. 45, (R)-5-ethyloxycarbonyl-2-γ-(S)-glutamylamino-N-2-heptylpentamide; 46, L-γ-(γ-oxa)glutamyl-(S-9-fluorenylmethyl)-l-Cys-Gly; 47, 2-{[(1-methyl-1H-pyrrol-2-yl)sulfonyl]methyl}-N-[(1E)-(4-nitrophenyl)methylene]aniline; 48, 2-hydroxy-4-bromo-2′-hydroxybenzophenone N-acetylhydrazone; 49, 2,2′-dihydroxy-5-phenylbenzophenone; 50, 9-oxo-9H-xanthene-4-carbaldehyde; 51, diaminobis(2-(2,3-dichloro-4-(2-methylenebutanoyl)phenoxy)acetoxy)platinum(VI) chloride (ethacraplatin); 52, ethacrynic acid.
![Figure 5. GSTA inhibitors. 45, (R)-5-ethyloxycarbonyl-2-γ-(S)-glutamylamino-N-2-heptylpentamide; 46, L-γ-(γ-oxa)glutamyl-(S-9-fluorenylmethyl)-l-Cys-Gly; 47, 2-{[(1-methyl-1H-pyrrol-2-yl)sulfonyl]methyl}-N-[(1E)-(4-nitrophenyl)methylene]aniline; 48, 2-hydroxy-4-bromo-2′-hydroxybenzophenone N-acetylhydrazone; 49, 2,2′-dihydroxy-5-phenylbenzophenone; 50, 9-oxo-9H-xanthene-4-carbaldehyde; 51, diaminobis(2-(2,3-dichloro-4-(2-methylenebutanoyl)phenoxy)acetoxy)platinum(VI) chloride (ethacraplatin); 52, ethacrynic acid.](/cms/asset/74d40ad2-1e8a-41c7-a618-6e36b58d730c/itxc_a_1692191_f0005_b.jpg)
Halogen-substituted 1,4-benzoquinones and 1,4-naphthoquinones were identified as irreversible inhibitors of rat Alpha-class GSTs (Vos et al. Citation1989). Further studies showed that the GSH S-conjugate of tetrachloro-1,4-benzoquinone (2-S-glutathionyl-3,5,6-trichloro-1,4-benzoquinone) and analogs are active site-directed inhibitors of rat Alpha-class GSTs that covalently modify Cys residues (van Ommen et al. Citation1988, Citation1989, Citation1991).
2,2′-Dihydroxybenzophenones and analogs have been investigated as GSTA inhibitors. 2-Hydroxy-4-bromo-2′-hydroxybenzophenone N-acetylhydrazone (48, ) is a potent inhibitor (IC50 = 0.18 µM) of hGSTA1-1 (Perperopoulou et al. Citation2014). Further studies on 2,2′-dihydroxybenzophenones showed that benzophenone 49 () inhibited hGSTA1-1 (IC50 = 1.77 µM) but was a weak inhibitor of hGSTP1A-1A (Pouliou et al. Citation2015). Both benzophenones 48 and 49 are weak inhibitors of GSTP1-1, although analog 48 () inhibits GSTM1-1 (Ki = 22.3 µM) (Georgakis et al. Citation2017). Xanthone 50 () inhibits hGSTA1-1 in colon cancer cell lysates and is weakly cytotoxic in Caco-2 cells (Zoi et al. Citation2013).
Ethacraplatin (51, ), which represents the GST inhibitor ethacrynic acid (52, ) tethered to the anticancer drug cisplatin, was designed to deliver ethacrynic acid (52, ) to inhibit GSTs and a Pt(IV) therapeutic agent (Ang et al. Citation2005). Ethacraplatin (51, ) is a potent inhibitor of both GSTA1-1 and GSTP1-1 and is a more potent cytotoxic agent than cisplatin in several cancer cell lines. Mass spectral analysis of GSTA1-1 incubated with ethacraplatin (51, ) indicated the formation of a GSTA1-1-ethacrynic acid (52, ) adduct. An analysis of the molecular interactions of ethacraplatin (51, ) with hGSTP1-1 has been reported (Parker et al. Citation2011).
2.2.6.1.4. Tissue and species distribution
Alpha-class GSTs are found in most tissues, although the distribution of isozymes varies with the tissue (Davies et al. Citation1993; Rozell et al. Citation1993; Sundberg et al. Citation1993; Forkert et al. Citation1999; Desmots et al. Citation2001; Morel et al. Citation2002; Coles and Kadlubar Citation2005; Mohana and Achary Citation2017). The subcellular localization of Alpha-class GSTs differs among isozymes. Activity with CDNB (11, ) as the substrate was found in cytosolic, microsomal, and mitochondrial fractions of rat liver and was ascribed to Alpha- and Mu-class GSTs (Bhagwat et al. Citation1998). Later studies confirmed the cytosolic and mitochondrial location of GSTA4 in mouse hepatocytes (Desmots et al. Citation2001). Other studies reported that GSTA4-4 was found solely in human liver mitochondria (Gardner and Gallagher Citation2001), but the presence of GSTA1-1 and GSTA4-4 in mouse liver cytosol and mitochondria has also been observed (Raza et al. Citation2002; Robin et al. Citation2003). Mitochondrial GSTs are encoded by nuclear DNA, synthesized in the cytosol, and apparently targeted to mitochondria by constitutive signal sequences (Raza Citation2011). With GSTA4-4, however, phosphorylation and binding to Hsp70 are required for efficient translocation into mitochondria (Robin et al. Citation2003). Western blot analysis shows that GSTA (Ya–Ya) is present in the matrix and membrane fractions of rat liver mitochondria and catalyzes the reaction of GSH with HNE (17, ) (Chen et al. Citation2002). GSTA4-4 is also found in sheep and human liver microsomes (Prabhu et al. Citation2001, Citation2004).
2.2.6.2. Mu-class (GSTM)
2.2.6.2.1. Structure and properties
As with other cytosolic GSTs, Mu-class GSTs are homodimeric proteins with molecular masses of approximately 50 kDa. Five human, six rat, and seven mouse Mu-class GSTM isozymes have been identified (); the genes encoding hGSTM1 to hGSTM5 are located on chromosome band 1p13.
hGSTM1 has been well characterized as having two allelic variants, hGSTM1a and hGSTM1b (Widersten et al. Citation1991). GSTM2-2 was originally purified from skeletal muscle and the cDNA sequence reported (Board et al. Citation1988; Vorachek et al. Citation1991). hGSTM3-3 was identified in testis and brain (Campbell et al. Citation1990b). hGSTM4-4 is present in several organs, and its specific activity with CDNB (11, ) is lower than that of other Mu-class GSTs (Comstock et al. Citation1994).
GSTM1-1, along with GSTA1-1 and GSTP1-1, has an active site Tyr residue. Crystallographic and computational analysis show, however, that the binding interactions of GSH at the G-site of GSTM1-1 differ from that of other cytosolic GSTs (Dourado et al. Citation2010b). In contrast to GSTA1-1, Trp7 and Trp45 in GSTM1-1 form hydrogen bonds with GSH. Moreover, in GSTM1-1, proton transfer from GSH to His107 to form the GSH thiolate is assisted by two water molecules that serve as a bridge, which contrasts with the proposal that a single water molecule acts as a bridge in the transfer of a proton from the thiol group of GSH to the GSH glutamyl α-carboxylate during GSH activation by GSTA1-1 and GSTP1-1.
The most studied hGSTM1 polymorphism is a gene deletion whose frequency ranges from 50 to 78% among ethnic groups (Moyer et al. Citation2007). Other studies show functionally significant polymorphisms in hGSTM1 that may be associated with responses to chemotherapy (Moyer et al. Citation2007; Agúndez and Ladero Citation2008; Piacentini et al. Citation2013). The hGSTM1/hGSTT1 double-null genotype is associated with poor treatment outcome in acute myeloid leukemia and with increased risk of drug-induced liver injury (Lucena et al. Citation2008; Xiao et al. Citation2014).
Mice lacking the entire Gstm locus (GstmΔ/Δ) were generated by deleting all coding sequences and regulatory elements encoding GSTMs (Xiang et al. Citation2014). No expression of GSTM1, -2, -3, -4, -5, -6, and -7 was detected in liver, testes, heart, jejunum, and brain of GstmΔ/Δ mice, although decreased conjugation of CDNB (11, ) was observed in kidney and lung cytosol. A Gstm1−/− mouse line was also generated; the deletion of Gstm1 failed to alter the expression of the neighboring Gstm2 and Gstm4 genes.
The expression of Mu-class GSTs in rodents is induced by butylated hydroxyanisole, phenobarbital, flavonoids, and dexamethasone (McLellan and Hayes Citation1989; Bhagwat et al. Citation1998; Boušová and Skálová Citation2012). Mice fed an “egg white-based diet,” an “amino acid-based diet,” or lab chow containing 2% flavone for three days showed a marked increase in Ggst3 expression and a lesser increase in Gstm2 and Gstm4 (Rudolf et al. Citation2008); similarly, total hepatic GSTM activity in mice and rats with bromosulfophthalein as the substrate was significantly increased.
2.2.6.2.2. Substrates
Human GSTM1-1 catalyzes the conjugation of CDNB (11, ) with GSH at rates higher than those observed with GSTA1-1 or GSTP1-1 (Czerwinski et al. Citation1996).
Trans-4-Phenyl-3-butene-2-one (53, ) and bromosulfophthalein have been proposed as class-distinguishing substrates for GSTM (Mannervik et al. Citation1985). Rat GSTM1-1 and -2-2 catalyze both the addition of GSH to trans-4-phenyl-3-buten-2-one (53, ) and the elimination of GSH from 4-(glutathion-S-yl)-phenyl-2-butanone (54, ) (Chen and Armstrong Citation1995). Purified human GSTM1-1 catalyzes the conjugation of busulfan (21, ) with GSH but at a lower (∼50%) rate than that found with GSTA1-1 (Czerwinski et al. Citation1996).
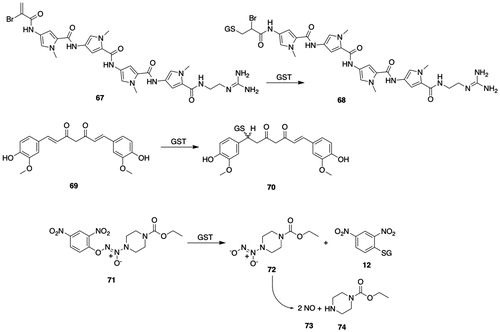
Figure 6. GSTM substrates. 53, trans-4-phenyl-3-buten-2-one; 54, 4-(glutathion-S-yl)-phenyl-2-butanone; 55, 5-cyano-N-[3-[(1R)-1-[(6R)-5,6-dihydro-4-hydroxy-2-oxo-6-(2-phenylethyl)-6-propyl-2H-pyran-3-yl]propyl]phenyl]- 2-pyridinesulfonamide (PNU-109112); 56, (6R)-3-[(1R)-1-(3-aminophenyl)propyl]-5,6-dihydro-4-hydroxy-6-(2-phenylethyl)-6-propyl-2H-pyran-2-one (PNU-143070); 57, 3-cyano-1,2-dimethyl-1-nitrosoguanidine; 58, N-cyano-N′-methylpropanimidamide; 59, S-nitroso-GSH; 60, γ-Glu-α-amino-β-[[2-ethyl-N,N,N′,N′-tetrakis(2-chloroethyl)-phosphorodiamidate]-sulfonyl]propionyl-Gly; 61, tetrakis(2-chloroethyl)phosphorodiamidate; 62, N5-((R)-1-((carboxymethyl)amino)-1-oxo-3-(vinylsulfonyl)propan-2-yl)-l-Gln; 63, 5-(pentafluorobenzoylamino)fluorescein; 64, 2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)-5-(2,3,5,6-tetrafluoro-4-(glutathion-S-yl)benzamido)benzoic acid; 65, 5-(3,4-dinitrobenzoylamino)-2,7-dichlorofluorescein; 66, 5-(3-nitro-4-glutathion-S-ylbenzoylamino)-2,7-dichlorofluorescein; 67, brostallicin; 68, brostallicin-GSH adduct; 69, curcumin; 70, (E)-7-(glutathion-S-yl)-5-hydroxy-1,7-bis(4-hydroxy-3-methoxyphenyl)hept-1-en-3-one; 71, O2-(2,4-dinitrophenyl)-1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate (JS-K); 72, 1-[(4-ethoxycarbonyl)piperazin-1-yl]diazen-1-ium-1,2-diolate; 73, nitric oxide; 74, ethyl piperazine-1-carboxylate.
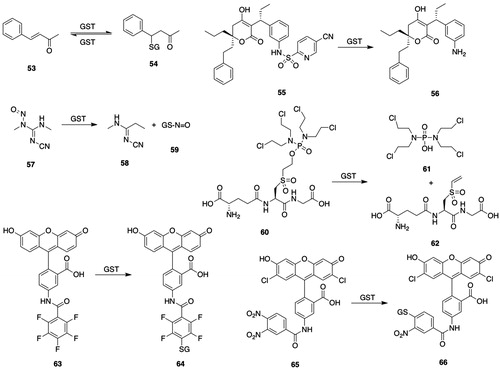
Although sulfonamides were formerly considered to be metabolically inert, GSTs catalyze the cleavage of some sulfonamides. HIV-protease inhibitor 56 () is released upon cleavage of the sulfonamide group by reaction of GSH with the prodrug (PNU-109112) (55, ), which is a substrate for hGSTM1-1 and to a lesser extent hGSTA1-1 and GSTP1-1 (Koeplinger et al. Citation1999; Zhao et al. Citation1999b).
hGSTM1-1 and hGSTM2-2 show 84% sequence identity but specific activities with aminochrome and 3-cyano-1,2-dimethyl-1-nitrosoguanidine (57, ) as substrates are 100-fold greater with hGSTM2-2 than with hGSTM1-1 (Hansson et al. Citation1999a, Citation1999b).
hGSTM1a-1a catalyzes the release of the cytotoxic tetrakis(2-chloroethyl)phosporodiamidate (61, ) from the GSH-based prodrug (60, ) by a proposed elimination mechanism at a rate that is much faster than that observed with hGSTA1-1 or hGSTP1-1 (Lyttle et al. Citation1994b). Replacement of Gly in compound 60 () with phenylGly yields an analog that is a poor substrate for hGSTM1a-1a, but is readily cleaved by hGSTA1-1 and hGSTP1-1.
Rat liver GSTM1-1 catalyzes the conjugation of 5-(pentafluorobenzoylamino)fluorescein (63, ) to GSH S-conjugate 64 () (Arttamangkul et al. Citation1999); although both the substrate and product are fluorescent, they can be readily separated by HPLC or TLC. The reaction is also catalyzed by hGSTA1-1 and hGSTP1-1 but at rates lower than those by GSTM1-1. 5-(3,4-Dinitrobenzoylamino)-2,7-dichlorofluorescein (65, ) is a substrate for hGSTM1-1 and also hGSTM2-2, hGSTM3-3, hGSTA2-2, and hGSTP1-1 (Fujikawa et al. Citation2015).
Brostallicin (67, ) is an anticancer prodrug that is highly cytotoxic to cells that overexpress GSTM2-2 or GSTP1-1 or both. hGSTM1-1, hGSTA1-1, and hGSTP1-1 catalyze the reaction of GSH with the α-bromoacrylamido group of brostallicin (67, ) to yield the GSH S-conjugate (68, ), which is a DNA alkylating agent (Geroni et al. Citation2002; Pezzola et al. Citation2010). Brostallicin (67, ) is a potent inhibitor of GSTM2-2-catalyzed conjugation of CDNB (11, ) (Pezzola et al. Citation2010).
hGSTM1a-1a along with hGSTA1-1, hGSTA2-2, and hGSTP1-1, but not hGSTT1-1, catalyzes the reaction of curcumin (69, ) with GSH to form diastereomeric GSH conjugates (70, ) (Usta et al. Citation2007); the adducts are, however, unstable and decompose to give curcumin (69, ) and other products.
hGSTM1-1 catalyzes the activation of the diazenium diolate prodrug (JS-K, 71, ) of nitric oxide (73, ) at a much greater rate than either hGSTA1-1 or hGSTP1-1 (Shami et al. Citation2003); accordingly, JS-K is cytotoxic in multiple myeloma cell lines and in patient derived multiple myeloma cells (Kiziltepe et al. Citation2007).
2.2.6.2.3. Inhibitors
Given its putative role in the development of resistance to chemotherapeutic agents, there has been considerable interest in developing GSTM inhibitors to overcome resistance to chemotherapeutic agents (Lo and Ali-Osman Citation2007). 4-O-Decyl-gabosine D (75, ) inhibits hGSTM1-1 (KiGSH = 3.2 μM) and synergizes cisplatin cytotoxicity (IC50 = 12 μM) in A549 lung cancer cells (Wang et al. Citation2011b).
Figure 7. GSTM inhibitors. 75, 4-O-decyl-gabosine D; 76, (S)-γ-Glu-(2RS)-(±)-2-amino-(di-n-butoxyphosphinyl)-acetyl-Gly; 77, γ-l-Glu-d-aminoadipic acid; 78, (R)-5-carboxy-2-γ-(S)-glutamylamino-N-2-heptylpentamide; 79, 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX); 68, N-(benzyloxy)-4-(((7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)thio)methyl)benzamide; 80, N-(benzyloxy)-4-(((7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)thio)methyl)benzamide; 81, 4-(glutathion-S-yl)-7-nitrobenzo[c][1,2,5]oxadiazole; 82, S-(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)-γ-(l-γ-oxaglutamyl)-l-Cys-Gly; 83, 1,5-bis(4-hydroxy-3-methoxyphenyl)-1,4-pentadiene-3-one; 84, 1-chloro-2,4-dinitronaphthalene.
![Figure 7. GSTM inhibitors. 75, 4-O-decyl-gabosine D; 76, (S)-γ-Glu-(2RS)-(±)-2-amino-(di-n-butoxyphosphinyl)-acetyl-Gly; 77, γ-l-Glu-d-aminoadipic acid; 78, (R)-5-carboxy-2-γ-(S)-glutamylamino-N-2-heptylpentamide; 79, 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX); 68, N-(benzyloxy)-4-(((7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)thio)methyl)benzamide; 80, N-(benzyloxy)-4-(((7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)thio)methyl)benzamide; 81, 4-(glutathion-S-yl)-7-nitrobenzo[c][1,2,5]oxadiazole; 82, S-(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)-γ-(l-γ-oxaglutamyl)-l-Cys-Gly; 83, 1,5-bis(4-hydroxy-3-methoxyphenyl)-1,4-pentadiene-3-one; 84, 1-chloro-2,4-dinitronaphthalene.](/cms/asset/f4cd68c4-d93b-4a39-817a-9a7092a461f9/itxc_a_1692191_f0007_b.jpg)
Kunze and coworkers (Kunze and Heps Citation2000) prepared a series of phosphono analogs of GSH as potential GST inhibitors. One member of the series (76, ) is a potent inhibitor of hGSTM1-1 (IC50 = 4.7 μM); GSH analog 76 () also inhibited porcine liver GSTA and GSTM as well as porcine lung GSTP1-1. Although compound 76 () was resistant to GGT-catalyzed hydrolysis and was stable in cell media, cellular uptake was poor; the methyl glycinate analog was a much weaker inhibitor of GSTM1-1 (IC50 = 67 μM) but was taken up by cells and underwent intracellular hydrolysis to form 76 (). The GSH analog γ-l-Glu-d-aminoadipic acid (77, ), which is resistant to GGT-catalyzed hydrolysis, potently inhibits rat GSTM1-1- and GSTM2-2-catalyzed reactions (Ki = 8–34 μM) (Adang et al. Citation1991). Related compounds, e.g. (R)-5-carboxy-2-γ-(S)-glutamylamino-N-2-heptylpentamide (78, ), are modest inhibitors of rat GSTM1-1 and -2-2 (Ouwerkerk-Mahadevan and Mulder Citation1998).
6-(7-Nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol (NBDHEX, 79, ) is a potent inhibitor of hGSTM2-2 (IC50 = ≤0.01 μM), but also inhibits hGSTP1-1 and activates JNK and apoptosis in human osteosarcoma cell lines (Ricci et al. Citation2005; Sau et al. Citation2012). The crystal structure of 7-nitrobenzoxadiazole 79 () bound to hGSTM2-2 shows that it forms a σ-complex with GSH and that the complex is stabilized by bonding interactions between the 7-nitro group and H-site Arg residues (Federici et al. Citation2009). Structure–activity studies aimed at identifying analogs of 7-nitrobenzodiazolyl 79 () with improved pharmacological and toxicological properties yielded additional potent inhibitors of GSTM2-2, such as compound 80 (). Analog 80 (IC50 = 0.003 uM) was a 67-fold more potent inhibitor of GSTM2-2 than of GSTP1-1 and a 3-fold more potent inhibitor of GSTM2-2 than compound 79 () (Rotili et al. Citation2015). The 7-nitrobenzoxadiazolyl (NBD) S-conjugate of GSH (81, ) and the corresponding γ-oxaglutamyl isostere (82, ) are more potent inhibitors of hGSTM2-2 compared with hGSTP1-1 (Luisi et al. Citation2016).
Curcumin analogs inhibit hGSTM1-1, hGSTA1-1 and hGSTP1-1 (Appiah-Opong et al. Citation2009); of the several compounds studied, compound 83 () is a potent inhibitor of hGSTM1-1 (IC50 = 0.3 μM). Several 2,4-dinitronaphthalene derivatives inhibit hGSTM selectively (Groom et al. Citation2014): 1-chloro-2,4-dinitronaphthalene (84, ) is a much more potent inhibitor of hGSTM2-2 than of hGSTM1-1.
2.2.6.2.4. Tissue and species distribution
Mu-class GSTs are widely distributed in human tissues but are absent or weakly expressed in some tissues (Campbell et al. Citation1991; Awasthi et al. Citation1994; Sherratt and Hayes Citation2002; Dhanani and Awasthi Citation2007). The tissue distribution of GSTM1 to -5 has been summarized (Mohana and Achary Citation2017).
hGSTM2-2 is highly expressed in skeletal and cardiac muscle and regulates calcium signaling via ryanodine receptors (Abdellatif et al. Citation2007). A distinct GSTM subclass, human GSTM3 and rodent GSTM5, is selectively expressed in brain and testis (Rowe et al. Citation1998; Listowsky Citation2005). Human GSTM5 is selectively expressed in brain, lung, and testes and is not detectable in liver (Takahashi et al. Citation1993; Rowe et al. Citation1997); GSTM5 in Cynomolgus macaques, however, is expressed at higher levels in liver and adrenal gland than in testes (Uno et al. Citation2013).
Immunohistochemical analysis showed that Mu-class GSTs are widely expressed in rat tissues (Otieno et al. Citation1997). The distribution of GSTM isozymes differs among tissues: in the hamster, GSTM1 is highly expressed in liver but is absent in kidney, GSTM1–GSTM4 are expressed in testis, and GSTM4 is found in kidney (Bogaards et al. Citation1992). In rat kidney, Mu-class GSTs are strongly expressed in the distal tubules but are also found at other sites in the kidney (Davies et al. Citation1993; Rozell et al. Citation1993).
Catalytically active GSTM1-1 has been identified in mouse liver mitochondria (Raza et al. Citation2002; Sun et al. Citation2012).
2.2.6.2.5. Other biological functions
GSTM2-2, along with GSTA1-1 and GSTO1-1, are ryanodine receptor (RyR1, -2) modulators (Dulhunty et al. Citation2011). Ryanodine receptors are intracellular calcium channels; RyR1 is primarily expressed in skeletal muscle, and RyR2 is the principal RyR expressed in cardiac muscle. GSTM2-2 decreases RyR2 activity in cardiac muscle but activates RyR1 in skeletal muscle (Dulhunty et al. Citation2011). GSTM2-2 exerts mechanistically different effects on RyR in cardiac and skeletal muscle, which led to the proposal that the C- and N-terminal portions of GSTM2-2 interact with RyR2 and RyR1, respectively. The C-terminal domain (GSTM2C) of the GSTM2 subunit, which contains α-helices 4, 5, 6, 7, and 8 (GSTM2-2C H4-8), inhibits cardiac muscle RyR2 but fails to bind to skeletal muscle RyR1 (Liu et al. Citation2009). Further studies of GSTM2C H5-8 variants showed enhanced efficacy in inhibiting cardiac muscle RyR2 (Samarasinghe et al. Citation2015).
2.2.6.3. Pi-class (GSTP)
2.2.6.3.1. Structure and properties
As with other cytosolic GSTs, Pi-class GSTs are homodimeric proteins with molecular masses of approximately 50 kDa. One human, one rat, and two mouse Pi-class GSTM isozymes have been identified (); the gene encoding hGSTP1 is located on chromosome band 11q13. As with other so-called Y-GSTs, GSTP has a Tyr residue in its active site (Atkinson and Babbitt Citation2009).
GSTP shares many structural features in common with GSTA and GSTM although their amino-acid sequences show little identity (20–32%) (Dirr et al. Citation1994a, Citation1994b). Crystal structures of GSTP1-1 in complex with a range of ligands have been summarized (Wu and Dong Citation2012).
Two amino acid substitutions (I105V/GSTP*B and A114V/GSTP*C) are known to occur in hGSTP1 (Ali-Osman et al. Citation1997a; Board et al. Citation1998; Harris et al. Citation1998); the I105V variant is common across racial groups, whereas the A114V variant is rare. These hGSTP1 allelic variants may determine responsiveness to N,N′N′-triethylenethiophosphoramide (thioTEPA) therapy (Srivastava et al. Citation1999b).
2.2.6.3.2. Substrates
The substrate selectivity of human, rat, and mouse GSTP has been summarized (Hayes and Pulford Citation1995; Mohana and Achary Citation2017); hence, only the salient characteristics of substrates or newer findings will be discussed. To test the hypothesis that relatively few amino acid residues determine substrate selectivity, the Y108V mutant of GSTP was studied (Nuccetelli et al. Citation1998); interestingly, the substrate selectivity of the GSTP Y108V mutant resembles that of GSTA and shows marked increases in specific activities with 4-chloro-7-nitro-2,1,3-benzoxadiazole (85, ) and cumene hydroperoxide (15, ) as substrates. The specific activities for the biotransformation of benzoxodiazole 85 () vary among the cytosolic GSTs (Ricci et al. Citation1994).
Figure 8. GSTP substrates. 85, 4-chloro-7-nitro-2,1,3-benzoxadiazole (NBD-Cl); 86, 4-(glutathion-S-yl)-7-nitro-2,1,3-benzoxadiazole; 87, O2-[2,4-dinitro-5-(N-methyl-N-4-carboxyphenylamino)phenyl]-1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate (PABA/NO); 88, 2,4-dinitro-5-(glutathion-S-yl)-phenyl 4-methylaminobenzoate; 89, O2-(2,4-dinitrophenyl)diazeniumdiolate-2-(β-d-galactopyranosyl)-oleanolic acid hybrid; 90, 1-(glutathion-S-yl)-2,4-dinitro-5-{[2-(β-d-galactopyranosyl olean-12-en-28-oate-3-yl)-oxy-2-oxoethyl] piperazine-1-yl}benzene; 91, O2-(2,4-dinitrophenyl)diazeniumdiolate-2-PARP-1 inhibitor hybrid; 92, 4-[[3-(piperazine-1-carbonyl)phenyl]methyl]-2H-phthalzain-1-one; 93, 1-[(3,3-dimethyl-2-oxido-1-triazen-1-yl)oxy]-3-(glutathion-S-yl)-4,6-dinitrobenzene; 94, 1,3-(diglutathion-S-yl)-4,6-dinitrobenzene; 95, 6-chloroacetyl-2-dimethylaminonaphthalene (Cadan); 96, 6-(glutathion-S-yl)acetyl-2-dimethylaminonaphthalene; 97, 6-(5-methyl-2-nitrophenylsulfonyl)-luciferin; 98, 3-(glutathion-S-yl)-4-nitrotoluene; 99, luciferin; 100, 5-(3,4-dinitrobenzamido)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)benzoic acid (DNAF1); 101, 5-(3-nitro-4-(glutathion-S-yl)benzamido)-2-(6-hydroxy-3-oxo-3H-xanthen-9-yl)benzoic acid.
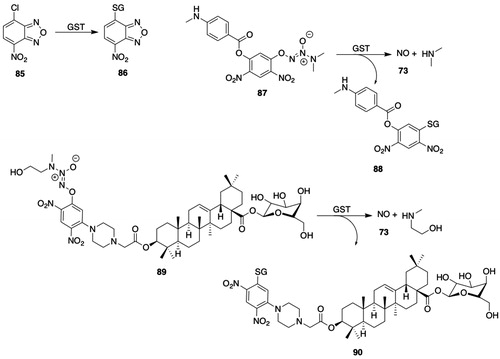
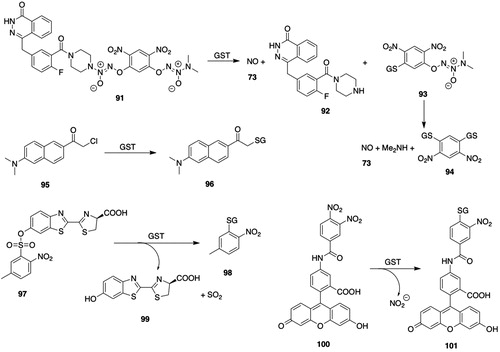
Chlorambucil (28, ) is also a substrate for hGSTP1-1 (Ciaccio et al. Citation1990). The rates of biotransformation of chlorambucil (28, ) differ among the several allelic variants of hGSTP1-1 (Pandya et al. Citation2000): for example, the rate of biotransformation is 15-fold higher with the I104/A113 variant than with the V104/V113 variant. The crystal structure of the chlorambucil–hGSTP1-1 complex shows that the substrate binds to the H-site and that the aromatic group forms stacking pi-pi interactions with Phe8 (Parker et al. Citation2008); moreover, rates of biotransformation of CDNB (11, ), ethacrynic acid (52, ) and benzoxadiazole 85 () vary among hGSTP1-1 allelic variants.
Ethacrynic acid (52, ) has been proposed as a class-distinguishing substrate for GSTP (Mannervik et al. Citation1985). The kcat/Km is the highest for hGSTP-catalyzed conjugation of acrolein compared with the hGSTA- and hGSTM-catalyzed reactions (Berhane and Mannervik Citation1990); accordingly, mGstP1/P2 null mice show enhanced susceptibility to cyclophosphamide-induced urotoxicity, which is associated with acrolein formation (Conklin et al. Citation2009).
hGSTP1-1 catalyzes the bioactivation of the prodrug brostallicin (67, ) (Pezzola et al. Citation2010). The enzyme catalyzes a Michael addition of GSH to brostallicin to yield an intermediate (68, ) that alkylates DNA in a sequence-specific manner.
Several diazeniumdiolate-based nitric oxide (73, ) prodrugs have been prepared as potential cancer chemotherapeutic agents. hGSTP1-1 catalyzes the release of nitric oxide (73, ) from prodrug 87 () (Findlay et al. Citation2004; Kim et al. Citation2015). Nitric oxide prodrug 87 () shows antiproliferative effects in both in vitro and in vivo models (Kogias et al. Citation2012). An analog of prodrug 87 () that contains a 4-cyano, rather than 4-nitro, substituent is more resistant to non-enzymatic hydrolysis of the ester linkage and undergoes less non-enzymatic activation by GSH than 87 (). Because of its antiproliferative and apoptotic potency and its potentially enhanced selectivity for hGSTP1-overexpressing cancer cells, the 4-cyano analog was proposed to be a more promising prodrug than compound 87 () (Kim et al. Citation2015). The hybrid diazeniumdiolate-oleanolic acid-based nitric oxide prodrug 89 () was prepared to impart hepatic selectivity and stability against non-enzymatic reaction with GSH (Fu et al. Citation2013). The rate of reaction of prodrug 89 () with GSTP was 7.5-fold greater than with GSTA. Prodrug 89 () released higher levels of nitric oxide (73, ) in human hepatocarcinoma cell lines than in normal liver cell lines. The hybrid diazeniumdiolate-based nitric oxide (73, ) prodrug 91 () was designed to release poly-ADP-ribose polymerase 1 (PARP-1) inhibitor 92 () (Maciag et al. Citation2014); PARP-1 inhibitor 92 () is an analog of the FDA-approved PARP-1 inhibitor olaparib. The design objective was to combine inhibition of PARP-1-dependent DNA repair with the cytotoxic effects of nitric oxide (73, ). The biotransformation of prodrug 91 () in A549 cells showed that the first step released nitric oxide (73, ) and PARP-1 inhibitor 92 (); reaction of GSH S-conjugate 93 () with GSH gave nitric oxide (73, ) and diGSH S-conjugate 94 (). hGSTP1-1 also catalyzed the release of PARP-1 inhibitor 92 () from prodrug 91 ().
Prodrugs of alkylating agents based on a GSH backbone that undergo GSTP-catalyzed activation have been described (Lyttle et al. Citation1994b; Satyam et al. Citation1996). For example, the phenylGly analog of prodrug 60 () undergoes a hGSTP1-1-catalyzed reaction to release a nitrogen-mustard alkylating agent (61, ) and is cytotoxic to MCF-7 breast cancer cells that overexpress hGSTP1-1 (Satyam et al. Citation1996; Townsend et al. Citation2002); the reaction is also catalyzed by hGSTA1-1 and hGSTM1-1. GSH S-conjugate 62 (), which is also formed upon GST-catalyzed activation of prodrug 61, is proposed to contribute to tumor cell cytotoxicity by inhibition of DNA-dependent protein kinase (Townsend et al. Citation2002).
Prodrugs of the cytostatic agent doxorubicin (33, ) have been developed as a strategy to overcome resistance to chemotherapeutic agents (Johansson et al. Citation2011). 2,4-Dinitrobenzenesulfonyl-doxorubicin (32, ) and 4-nitrobenzenesulfonyl-doxorubicin are substrates for hGSTP1-1 (and rMGST1). Doxorubicin prodrug 32 () is cytotoxic to V79 cells overexpressing hGSTP1-1.
There has been considerable interest in developing fluorogenic or bioluminescent substrates for cytosolic GSTs. Based on a 6-acetyl-2-dimethylaminonaphthalene scaffold, 6-chloroacetyl-2-dimethylaminonaphthalene (95, ) was an excellent substrate for GSTP1-1, although activity was also observed with GSTA and GSTM isozymes (Svensson et al. Citation2002). Luciferin sulfonate ester 97 () is biotransformed by GSTA, GSTM, and GSTP (Zhou et al. Citation2006); related quinolinyl luciferin derivatives are also substrates for GSTs. Fluorogenic substrates for GSTs that undergo glutathionylation and denitration have recently been reported (Fujikawa et al. Citation2008); fluorescein derivative 100 () undergoes an hGSTP1-1-catalyzed reaction to yield a fluorescent GSH S-conjugate (101, ). hGSTA1-1 and hGSTM1-1 also catalyze the reaction but at much lower rates. An analog of fluorescein derivative 100 () in which the carboxylic acid group was replaced with a methyl group was used to visualize GST activity in living HuCCT1 cells. Further studies with fluorescein derivative 100 () showed that in addition to GSTP1-1, the compound was also a substrate for GSTA2-2, GSTM1-1, and GSTM2-2 (Fujikawa et al. Citation2015). 3′,6′-Diacetyl fluorescein and 3′-methoxy-6′-acetylfluorescein undergo GSTP-catalyzed thiolysis to afford fluorescent products (Fujikawa et al. Citation2018); the reaction shows remarkable selectivity for hGSTP1-1: scant activity was observed with 12 other cytosolic hGSTs. GST-catalyzed thiolytic cleavage of esters has been previously reported (Keen and Jakoby Citation1978; Shyam et al. Citation1999). 3′,6′-Diacetyl fluorescein is also a substrate for porcine liver esterase, indicating that esterase-catalyzed hydrolysis could confound its use in imaging cellular GSTs.
2.2.6.3.3. Inhibitors
GSTP1-1 expression is increased in a range of human tumors (Mannervik et al. Citation1987; Hayes and Pulford Citation1995; O'Brien and Tew Citation1996). The overexpression of hGSTP1-1 is associated with resistance to chemotherapy with anticancer drugs that are substrates for hGSTP1-1 (Kodera et al. Citation1994; O'Brien and Tew Citation1996), which has led to a search for hGSTP1-1 inhibitors (Mahajan and Atkins Citation2005; Johansson et al. Citation2011).
The development and testing of hGSTP1-1 inhibitors as potential cancer chemotherapeutic agents has been reviewed (Kodera et al. Citation1994; Morales and Laborde Citation2007; Laborde Citation2010); three types of inhibitors have been identified: suicide inhibitors, competitive inhibitors, and hGSTP1-1-activated prodrugs. Given the potential clinical utility of hGSTP1-1 inhibitors, there has been much interest in preparing and testing compounds for their inhibitory properties.
Tributyltin acetate, triethyltin bromide, triphenyltin chloride, Rose Bengal, and Cibacron Blue are potent but nonselective inhibitors of mouse Pi-class enzymes (Warholm et al. Citation1986). Several 2,2′-dihydroxybenzophenones inhibit GSTP but are in general more potent inhibitors of GSTA (Pouliou et al. Citation2015). Curcumin (69, ) and related compounds inhibit GSTP1-1 (and GSTA1-1, GSTPA2-2, GSTM1-1, and GSTM2-2) (van Iersel et al. Citation1997; Hayeshi et al. Citation2007); of 34 curcumin analogs prepared as potential inhibitors of GSTP1-1, six were more potent than curcumin (69, ) (IC50 = 15 µM) with IC50 values of 0.4–1.6 µM (Appiah-Opong et al. Citation2009). Several compounds from the US Drug Collection (MicroSource Discovery Systems, Inc.), which includes drugs tested in clinical trials, including chlorophyllide, merbromin, ethacrynic acid (52, ), and hexachlorophene, are effective inhibitors of hGSTP1-1 (IC50 = 2.3–9.7 μM), but are even more potent inhibitors of GSTA3-3 (Musdal et al. Citation2013). 8-Methoxypsoralen is a competitive inhibitor of hGSTP1-1 (de Oliveira et al. Citation2014).
Ethacraplatin (51, ), in which ethacrynic acid (52, ) is tethered to Pt(IV), inhibits hGSTP1-1 and delivers Pt to cells (Ang et al. Citation2005). The results of X-ray crystallographic and molecular modeling analyses led to the proposal that ethacraplatin (51, ) initially forms a complex at the dimer interface of hGSTP1-1 and undergoes reduction resulting in the release of both Pt and the two ethacrynic acid ligands, which diffuse to the H-site of each subunit where they become covalently bound (Parker et al. Citation2011). Ethacrynic acid tethered to p-cymene ruthenium(II) or osmium(II) complexes inhibits hGSTP1-1 and is cytotoxic to two cancer cell lines (Păunescu et al. Citation2017).
Several groups of investigators have prepared and tested compounds based on the GSH backbone as GSTP1-1 inhibitors. Moreover, the observations that Alpha-, Mu-, and Pi-class GSTs are subject to product inhibition led to investigation of GSH S-conjugates as GST inhibitors (Meyer Citation1993; Quesada-Soriano et al. Citation2012). A cross-docking approach has been used to investigate the binding of GSH S-conjugates to GSTP (Lannutti et al. Citation2012). Preparation and testing of a panel of GSH analogs as potential isozyme-selective GST inhibitors showed that compound 102 () is a potent inhibitor of hGSTP1-1 (Ki = 0.42 μM) and is 58-fold or 438-fold more potent as an inhibitor of hGSTP1-1 than of hGSTA1-1 and hGSTM1a-1a or hGSTM2-2 (Flatgaard et al. Citation1993; Lyttle et al. Citation1994a; Oakley et al. Citation1997). The S-(4-chlorobenzyl) analog of compound 102 () was a highly selective inhibitor of hGSTP1-1 compared with hGSTPA1-1, hGSTM1a-1a, or hGSTM2-2. Moreover, the diethyl ester (ezatiostat, TLK199) analog of compound 102 () potentiated chlorambucil (28, ) cytotoxicity in several cell lines, whereas the less lipophilic dicarboxylic acid 102 () caused negligible potentiation (Morgan et al. Citation1996). Phosphono analogs of GSH, in which the Cys moiety of GSH is replaced by phosphonic acid esters, inhibit the human placental GSTP-catalyzed conjugation of CDNB (11, ) (Kunze Citation1996).
Figure 9. GSTP inhibitors. 102, γ-glutamyl-S-(benzyl)-cysteinyl-R-(−)-phenylglycine; 103, piperlongumine (1-[(E)-3-(3,4,5-trimethoxyphenyl)prop-2-enoyl]-2,3-dihydropyridin-6-one); 104, 3-(glutathion-S-yl)-3-(3,4,5-trimethoxyphenyl)propanoic acid; 105, (4-peptidyl- or 4-glutathion-S-yl)piperidin-2-one (R = FGLCSGPADTGR or GSH); 106, 3-cinnamyl-5(E)-bromomethylidenetetrahydro-2-furanone; 107, methyl 3-({2-[3-(4-nitrophenyl)-1,2,4-trioxan-6-yl]prop-2-enoyl}-oxy)benzoate; 108, 5,8-dihydroxy-1-(hydroxymethyl)naphtho[2,3-c]furan-4,9-dione; 109, 6,7-dihydroxy-3-(3′,4′-dihydroxyphenyl)coumarin; 110, 5-[3-chloro-4-(2-methylene-1-oxopropyl)phenoxymethyl]-3-phenyl-1,2,4-oxadiazole; 111, 6-((7 nitrobenzo[c][1,2,5]oxadiazol-4-yl)thio)hexyl benzoate (MC2753); 112, methyl N-(4,6-dichloro-1,3,5-triazin-2-yl)-N-(pent-4-yn-1-yl)-l-leucinate (LAS17); 113, S-glutathionyl ethylenesulfonyl fluoride; 114, phenylarsine oxide (PAO); 115, dinitrosyl-diglutathionyl iron complex (DNDGIC).
![Figure 9. GSTP inhibitors. 102, γ-glutamyl-S-(benzyl)-cysteinyl-R-(−)-phenylglycine; 103, piperlongumine (1-[(E)-3-(3,4,5-trimethoxyphenyl)prop-2-enoyl]-2,3-dihydropyridin-6-one); 104, 3-(glutathion-S-yl)-3-(3,4,5-trimethoxyphenyl)propanoic acid; 105, (4-peptidyl- or 4-glutathion-S-yl)piperidin-2-one (R = FGLCSGPADTGR or GSH); 106, 3-cinnamyl-5(E)-bromomethylidenetetrahydro-2-furanone; 107, methyl 3-({2-[3-(4-nitrophenyl)-1,2,4-trioxan-6-yl]prop-2-enoyl}-oxy)benzoate; 108, 5,8-dihydroxy-1-(hydroxymethyl)naphtho[2,3-c]furan-4,9-dione; 109, 6,7-dihydroxy-3-(3′,4′-dihydroxyphenyl)coumarin; 110, 5-[3-chloro-4-(2-methylene-1-oxopropyl)phenoxymethyl]-3-phenyl-1,2,4-oxadiazole; 111, 6-((7 nitrobenzo[c][1,2,5]oxadiazol-4-yl)thio)hexyl benzoate (MC2753); 112, methyl N-(4,6-dichloro-1,3,5-triazin-2-yl)-N-(pent-4-yn-1-yl)-l-leucinate (LAS17); 113, S-glutathionyl ethylenesulfonyl fluoride; 114, phenylarsine oxide (PAO); 115, dinitrosyl-diglutathionyl iron complex (DNDGIC).](/cms/asset/76fb84cf-37b1-4e5b-94b4-663134284530/itxc_a_1692191_f0009_b.jpg)
Zompra et al. (Citation2016) synthesized twenty-five GSH analogs and investigated their ability to serve as substrates or inhibitors of the hGSTP1-1 allozymes hGSTP1*A (Ile104/Ala113), hGSTP1*B (Val104/Ala113), and hGSTP1*C (Val104/Val113). One analog, l-γ-Glu-l-Cys-Sar, supported the conjugation of CDNB (11, ) with all allozymes. l-γ-Glu-Thi-Gly inhibited hGSTP1*A and hGSTP1*C but not hGSTP1*B, whereas l-γ-Glu-(S-(acetamidomethyl)-l-Cys-Gly was a better inhibitor of allozymes hGSTP1*B and hGSTP1*C than hGSTP1*A.
Piperlongumine (103, ) is a natural product that interacts with GSH-binding proteins, including GSTP1-1 (Adams et al. Citation2012). The interaction with GSH-binding proteins was first attributed to reaction of the C2–C3 olefin of the piperidin-2-one moiety with GSH followed by covalent reaction of the C7–C8 olefin of the GSH adduct with the proteins (Adams et al. Citation2012). Subsequent studies failed, however, to show adduction of GSTP1-1 (Harshbarger et al. Citation2017). Rather studies on the mechanism of inhibition of GSTP1-1 indicate that piperlongumine (103, ) is a prodrug and that GSH S-conjugate 104 (), formed by a reaction sequence that has not been unequivocally elucidated, binds to the H-site and, thereby, inhibits GSTP1-1 (Harshbarger et al. Citation2017).
Haloenol lactone 106 () is a highly selective inactivator of mouse liver GSTP and shows only minor inactivation of mGSTA or mGSTM (Zheng et al. Citation1996). Haloenol lactone 106 inactivates hGSTP1-1 by alkylation of Cys47 (Mitchell et al. Citation1998; Zheng et al. Citation2005). Several 1,2,4-trioxanes were prepared as potential inhibitors of hGSTP1-1 (Bräutigam et al. Citation2015); trioxane 107 () is a potent inhibitor of human GSTP1-1 (IC50 = 1.65–2.90 μM) but a weak inhibitor of hGSTA1-1 and hGSTM2-2 (IC50 = 74–98 μM).
Evaluation of a panel of natural plant products as inhibitors of recombinant hGSTP1-1 showed that isofuranonaphthoquinone 108 () was the most potent (Mukanganyama et al. Citation2011). 3-Arylcoumarin analogs, e.g. 109 () are strong inhibitors of human placental GSTP1-1, but their isozyme selectivity was not determined (Alparslan and Daniş Citation2015).
Oxadiazole analogs of ethacrynic acid, e.g. 110 () inhibit hGSTP1-1 and exhibit antiproliferative activity (Yang et al. Citation2010); interestingly, one analog was a poor inhibitor of hGSTP1-1 but still showed potent antiproliferative activity. Nitrobenzoxadiazole 79 () inhibits GSTP1-1 (IC50 = 0.80 μM) and activates JNK and induces apoptosis in human osteosarcoma cell lines (Ricci et al. Citation2005; Sau et al. Citation2012); preparation and testing of a range of nitrobenzoxadiazole derivatives showed that all were more potent inhibitors of GSTM2-2 than of GSTP1-1 (De Luca et al. Citation2015; Rotili et al. Citation2015). Nitrobenzoxadiazole 111 (), the benzoate ester of 79 (), partially inhibits GSTP1-1, perhaps because of negative cooperativity (Fulci et al. Citation2017); benzoate ester 111 () undergoes little non-enzymatic reaction with GSH, and docking results indicate that it binds near the dimer interface of GSTP1-1. Benzoate ester 111 () undergoes rapid microsomal carboxylesterase-catalyzed hydrolysis, which limits its utility as a drug candidate (Di Paolo et al. Citation2019). Compared with benzoate ester 111 (), the benzamide analog is hydrolytically stable, shows improved stability in the presence of GSH, inhibits GSTP1-1, disrupts the interaction between GSTP1-1 and TRAF2, and is cytotoxic in A375 cells.
Tyrosine-directed, irreversible inhibitors of GSTP have been reported. Leucinate ester 112 () inhibits hGSTP1-1 by modifying selectively Tyr108 (Crawford and Weerapana Citation2016). Covalent modification is attributed to the reaction of the dichlorotriazine moiety with Tyr108; no reaction was observed with the Y108F mutant. Proteomic analysis of HeLa cells incubated with ester 112 () confirmed the selective labeling of Tyr108 in hGSTP1-1. Binding of sulfonyl fluoride 113 () to the G-site of hGSTP1-1 positions an electrophilic group adjacent to the active site Tyr-108 (Shishido et al. Citation2017); the formation of a sulfonate ester of Tyr108 in hGSTP1-1 was confirmed by crystallographic analysis. The IC50 values for the inhibition of hGSTP1-1, hGSTM2-2, and hGSTA1-1 are 12, 50, and 12 μM, respectively.
Phenylarsine oxide (114, ) reacts rapidly with GSH to form a diGSH complex that is a potent inhibitor of GSTP1-1 (Ki = 90 nM) (Parker et al. Citation2017). In the absence of GSH, phenylarsine oxide (114, ) forms an intermonomer crosslink between Cys47 and Cys101; accordingly, phenylarsine oxide fails to inhibit the C47S/C101S mutant. The sequestration of phenylarsine oxide (114, ) by GSTP1-1 may represent a general detoxification and inactivation mechanism for arsenic-containing drugs and environmental agents.
Negative cooperativity in hGSTP1-1 is observed with dinitrosyl-diglutathionyl iron complex (DNDGIC, 115, ), which is a potent inhibitor (Ki = <1.5 nM) of hGSTP1-1 (Cesareo et al. Citation2005; Bocedi et al. Citation2016). In the reaction of DNDGIC (115, ) with hGSTP1-1, the active-site Tyr7 displaces one of the GSH moieties to bind the complex. The sequestration of nitric oxide (73, ) by iron and GSH to give DNDGIC (115, ) followed by its reaction with GSTP1-1 may protect against nitrosylative and oxidative stress, but DNDGIC (115, ) inactivates GSH reductase, which is important in maintaining the redox state of the cell. It has been proposed that negative cooperativity serves to protect cells by maintaining residual GST activity in the presence of DNDGIC (115, ) to protect against toxins (Bocedi et al. Citation2013); negative cooperativity is much more prominent in Tyr- and Ser-GSTs than in Cys-GSTs.
2.2.6.3.4. Tissue and species distribution
Although GSTP is widely distributed in human tissues, it is noteworthy that expression is low in hepatocytes (Sundberg et al. Citation1993; Awasthi et al. Citation1994; Dhanani and Awasthi Citation2007; Mohana and Achary Citation2017). Indeed, immunohistochemical studies showed that hepatocytes were unstained by antibodies to GSTP but that staining was observed in the bile duct (Campbell et al. Citation1991); hGSTP was, however, detected in most other organs, although the intensity varied widely. GSTP is highly expressed in human placenta (Guthenberg et al. Citation1979; Zusterzeel et al. Citation1999). As with hGSTP, rat GSTP is widely expressed but its expression is low in liver (Meyer et al. Citation1985; Hayes and Mantle Citation1986; Pemble et al. Citation1986; Davies et al. Citation1993; Rozell et al. Citation1993). GSTP is highly expressed in mouse kidney liver and urinary bladder (Conklin et al. Citation2009). GSTP1-1 is highly expressed in tumor tissue (Peters et al. Citation1989; Tsuchida et al. Citation1989; Tew Citation1994; Jardim et al. Citation2012; Tew Citation2016).
The expression of GSTP in initiated hepatocytes after treatment with carcinogens is a biomarker of hepatocarcinogenesis (Pitot et al. Citation1996). GSTP1-1 is the predominant GST found in many cell lines, including those used in the National Cancer Institute Drug Screening Program (Batist et al. Citation1986; Tew et al. Citation1996). The expression of GSTP mRNA in human tissue-derived cell lines varies widely (Bauer et al. Citation2006): GSTP mRNA is abundant in several lung cell lines but is absent in the HepG2 cell line. In addition to the enhanced expression of genes encoding hGSTP1, epigenetic factors also influence hGSTP1 gene expression (Zhang et al. Citation2014b).
GSTP is also present in nuclei and mitochondria in mammalian cell lines and tissues (Hall et al. Citation1994b; Ali-Osman et al. Citation1997b; Soh et al. Citation2005; Allen et al. Citation2007; Goto et al. Citation2009). The nuclear localization of hGSTP is significant because its expression is associated with drug resistance and poor treatment outcomes (Huang et al. Citation2003; Soh et al. Citation2005; Allen et al. Citation2007; Rolland et al. Citation2010).
2.2.6.3.5. Other biological functions
Although a thorough treatment of the many biological functions of GSTP, particularly those involved in cancer drug resistance, is beyond the scope of this discussion, several excellent reviews are available (Tew Citation1994; Hayes and Pulford Citation1995; Lage Citation2008; Tew et al. Citation2011; Vasieva Citation2011; Tew and Townsend Citation2012; Zhang et al. Citation2014a; Singh Citation2015; Tew Citation2016). For example, GSTP regulates JNK signaling (Adler et al. Citation1999; Sau et al. Citation2012) and controls the S-glutathionylation of a range of proteins (Townsend et al. Citation2009; Tew et al. Citation2011). GSTP also regulates estrogen response element (ERE)-dependent estrogen receptor alpha (ERα) signaling events (Liu et al. Citation2014b) and is an inhibitor of TRAF2 (De Luca et al. Citation2014).
2.2.6.4. Theta-class (GSTT)
Three Theta-class GSTs have been identified (Landi Citation2000; Coggan et al. Citation2002) (): GSTT1-1, GSTT2-2, and GSTT3-3. Theta-class GSTs are considered to be the progenitor of other GST classes and to be the most ancient of the GST classes (Pemble and Taylor Citation1992; Landi Citation2000). Theta-class GSTs are widely distributed in nature in contrast to Alpha-, Mu-, and Pi-class GSTs, which are found only in animals and fungi. Theta-class GSTs are characterized by their low activity with CDNB (11, ) as the substrate and by their failure to bind to immobilized GSH affinity matrices (Meyer et al. Citation1991; Hussey and Hayes Citation1992).
2.2.6.4.1. Structure and properties
Theta-class GSTs are homodimeric proteins with subunit molecular masses of approximately 27 kDa (Meyer et al. Citation1991; Tan and Board Citation1996). In contrast to Alpha-, Mu-, Pi-, and Sigma-class GSTs, in which Tyr is a catalytically critical active-site residue, Ser is the corresponding active-site residue in Theta-class GSTs (Wilce et al. Citation1995). Accordingly, site-directed mutation of the active-site Ser yields an inactive enzyme (Board et al. Citation1995; Caccuri et al. Citation1997).
Hiratsuka et al. (Citation1990) and Ogura et al. (Citation1991) described a rat liver GST that showed little amino-acid sequence homology (∼20%) to Alpha-, Mu-, and Pi-class GSTs, that showed high activity with reactive sulfate esters, and that was termed Yrs–Yrs. A protein purified from rat liver that showed high GSH peroxidase activity and activity with alkyl epoxides and alkyl halides was designated GST E (Meyer et al. Citation1984). Two proteins purified from rat liver that were designated GST 5–5 and GST 12–12 and one from human liver that was designated as human liver GST Theta (Meyer et al. Citation1991) were identified as Theta-class GSTs; subsequently, rat liver GST 5–5 and GST 12–12 were termed GSTT1-1 and GSTT2-2, respectively. GST Yrs–Yrs was further characterized as belonging to the Theta-class GSTs and was termed GSTT2-2 (Hussey and Hayes Citation1992; Tan et al. Citation1995). hGSTT1 and hGSTT2 subunits share approximately 54% amino-acid sequence identity (Flanagan et al. Citation1998). Genes encoding a third Theta-class GST, Gstt3, have been identified in mice and rats (Coggan et al. Citation2002) (Rat Genome Database 1562732) ( and ).
The crystal structures of hGSTT1-1 and hGSTT2-2 have been solved (McKinstry et al. Citation1998; Rossjohn et al. Citation1998; Tars et al. Citation2006). The crystal structures of hGSTT1-1 and -2-2 are similar (Tars et al. Citation2006); both show the canonical thioredoxin fold, but major differences are found in the C-terminus and in the loop connecting helices α4 and α5. In hGSTT1-1, the bulky side-chain of H-site residue Trp234 in C-terminus helix α9 hampers binding to both the H- and G-sites (Shokeer et al. Citation2005; Tars et al. Citation2006); in GSTT2-2 a 40-residue C-terminal extension, which does not prevent catalysis, blocks access to both the H- and G-sites, and a sulfate-binding site was identified (Rossjohn et al. Citation1998; Flanagan et al. Citation1999).
The substrate selectivities of hGSTT1-1 and -2-2 differ (Jemth and Mannervik Citation1997); H-site residues are poorly conserved between hGSTT1-1 and -2-2, which contributes substantially to the differences in substrate selectivity (Rossjohn et al. Citation1998; Tars et al. Citation2006). Preferred substrates for GSTT1-1 include halogenated hydrocarbons, e.g. dichloromethane (120, ) and 4-nitrobenzyl chloride (125, ), but the specific activities of the human enzyme are far lower than that of those of the rodent enzyme (Jemth and Mannervik Citation1997). Mutational studies that compared murine and hGSTT1-1 indicated that residue 234 (Arg and Trp in the wild-type murine and human proteins, respectively) is associated with the observed differences in specific activities (Larsson et al. Citation2004; Shokeer et al. Citation2005; Shokeer and Mannervik Citation2010a, Citation2010b); the finding that the specific activity of the W234R mutant of hGSTT1-1 with 4-nitrobenzyl chloride (125, ) as the substrate is approximately 50-fold higher than the wild-type enzyme highlights the importance of residue Trp234 (Shokeer et al. Citation2005). In addition, mutation of Met232 in the C-terminal helix of mGSTT1-1 l alters the specific activities for several substrates (Shokeer and Mannervik Citation2010a); with the M232A mutant, the specific activities with EPNP (116, ) or 4-nitrobenzyl chloride (125, ) as substrates are decreased by approximately 70% whereas the specific activity with cumene hydroperoxide (15, ) as the substrate is increased 30-fold.
Figure 10. GSTT substrates. 116, 1,2-epoxy-3-(4-nitrophenoxy)propane (EPNP); 117, S-(2-hydroxy-3-[4-nitrophenoxy]propyl)GSH; 118, phenethyl isothiocyanate; 119, S-(N-phenethylthiocarbamoyl)GSH; 120, dichloromethane; 121, S-(chloromethyl)GSH; 122, formaldehyde; 123, 1-menaphthyl sulfate; 124, S-(1-menaphthyl)GSH; 125, 4-nitrobenzyl chloride; 126, S-(4-nitrobenzyl)GSH; 127, 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU); 128, 1-(2[-glutathion-S-yl]ethyl-3-(2-chloroethyl)urea; 129, methyl parathion; 130, O-methyl O-(4-nitrophenyl) phosphorothioate; 131, S-(methyl)GSH.
![Figure 10. GSTT substrates. 116, 1,2-epoxy-3-(4-nitrophenoxy)propane (EPNP); 117, S-(2-hydroxy-3-[4-nitrophenoxy]propyl)GSH; 118, phenethyl isothiocyanate; 119, S-(N-phenethylthiocarbamoyl)GSH; 120, dichloromethane; 121, S-(chloromethyl)GSH; 122, formaldehyde; 123, 1-menaphthyl sulfate; 124, S-(1-menaphthyl)GSH; 125, 4-nitrobenzyl chloride; 126, S-(4-nitrobenzyl)GSH; 127, 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU); 128, 1-(2[-glutathion-S-yl]ethyl-3-(2-chloroethyl)urea; 129, methyl parathion; 130, O-methyl O-(4-nitrophenyl) phosphorothioate; 131, S-(methyl)GSH.](/cms/asset/7e45b4b6-7e76-4f8a-9d58-0d7a40d03fe9/itxc_a_1692191_f0010_b.jpg)
In contrast to hGSTT1-1, hGSTT2-2 shows activity with cumene hydroperoxide (15, ) and 1-menaphthyl sulfate (123, ) but no detectable activity with EPNP (116, ) or dichloromethane (120, ) as substrates (Hussey and Hayes Citation1992; Tan and Board Citation1996). hGSTT2-2 catalyzes the biotransformation of 1-menaphthyl sulfate (123, ) in the absence of GSH but in the presence of S-(methyl)GSH plus 2-mercaptoethanol or L-Cys (Tan et al. Citation1996). Moreover, the S11A mutant shows increased specific activity with 1-menaphthyl sulfate (123, ) as the substrate (Tan et al. Citation1996). A sulfatase reaction mechanism was proposed in which the binding of GSH to the G-site favors the entry of the second substrate, i.e. 1-menaphthyl sulfate (123, ), which loses sulfate to form the 1-menaphthyl carbonium ion that reacts with GSH to give S-(1-menaphthyl)GSH (123, ) (Tan et al. Citation1996). The proposal that GSTT2-2 acts as a sulfatase rather than a GST has been challenged based on data obtained with rGSTT2-2, which exhibits highly similar residue identity with hGSTT2-2 in the sulfate-binding site (Jemth and Mannervik Citation2000; Moyer et al. Citation2007; Agúndez and Ladero Citation2008). The authors found that Ser11 stabilizes the GST thiolate: the lower kcat values found for the S11A mutant of rGSTT2-2 with substrates 123 and 125 () compared with the wild-type enzyme are attributed to slower product release.
hGSTT1 is highly polymorphic (Thorn et al. Citation2012). The genes encoding hGSTT1 and hGST2 are located on band 22q11.2 (Tan et al. Citation1995; Webb et al. Citation1996). GSTT activity is found in human erythrocytes (Peter et al. Citation1989; Schröder et al. Citation1992); with methyl chloride as the substrate, approximately 40% of human subjects fail to conjugate methyl chloride with GSH and are termed “non-conjugators.” The non-conjugator phenotype is attributed to deletion of GSTT1 gene and the conjugator phenotype and GSTT1 genotype are comparable (Pemble et al. Citation1994). The gene-deletion genotype varies widely among ethnic groups and amounts to a low of 33.5% in Caucasian-Americans to a high of 73.5% in Han Chinese-Americans (Moyer et al. Citation2007; Agúndez and Ladero Citation2008).
Although null alleles have been the focus of most studies on GSTT polymorphisms, single-nucleotide polymorphisms (SNPs) are also known. Given the potential for GSTT-dependent bioactivation of carcinogens, the observed SNPs in GSTT1 merit investigation (Landi Citation2000; Agúndez and Ladero Citation2008). SNPs in GSTT1 affect the catalytic activity of GSTT1-1 by altering the stability and physical properties of the expressed protein (Josephy et al. Citation2009, Citation2011). Investigation of the genetic variants of GSTT1 and GSTT2 in rhesus and cynomolgus macaques identified a range of alleles (Uno et al. Citation2018). Activities of the D125G and R76C/D125G variants of GSTT1 were much lower than the wild-type enzyme or the R76C variant with 1-iodohexane and dibromomethane as substrates.
As with other GSTs, Theta-class GSTs are induced by a range of agents (for a review, see Higgins and Hayes Citation2011). In rats fed NSAIDs, including indomethacin, sulindac, ibuprofen, piroxicam, and aspirin, the expression of colonic GSTT1-1 is increased (van Lieshout et al. Citation1998). Among several potential chemopreventive agents studied, indole-3-carbinol is the most potent inducer of hepatic GSTT1-1 in male rats (Sherratt et al. Citation1998); in female rats, however, coumarin is the most potent inducer of hepatic GSTT1-1. In mice, treatment with clofibrate, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene, and phenobarbital induced expression of Gstt1, whereas clofibrate and ciprofibrate induced expression of Gstt2 (Knight et al. Citation2008); of 15 microsomal enzyme inducers studied, none induced expression of Gstt3. Apple extracts rich in polyphenols prevent DNA damage induced by hydroperoxide 15 () in colon cell lines by enhancing GSTT2-2 promoter activity (Miene et al. Citation2009; Petermann et al. Citation2009); the cytoprotective effects of the extracts may be attributed to flavan-3-ols, including procyanidins, and flavonols.
2.2.6.4.2. Substrates
The substrate selectivity of Theta-class GSTs differs among species and among the identified isoenzymes (Meyer et al. Citation1984; Thier et al. Citation1998; Landi Citation2000). As indicated above, although the crystal structures of hGSTT1-1 and hGSTT2-2 are similar, their substrate selectivities differ. For example, hGSTT1-1 shows high activity with 1,2-epoxy-3-(4-nitrophenoxy)propane (116, ) and phenethyl isothiocyanate (118, ) as substrates, but little or no activity with dichloromethane (120, ) or 1-menaphthyl sulfate (123, ) as substrates (Jemth and Mannervik Citation1997). In contrast, hGSTT2-2 shows high activity with cumene hydroperoxide (15, ) and 1-menaphthyl sulfate (123, ) as substrates (Meyer et al. Citation1991; Jemth and Mannervik Citation1997). Rat GSTT1-1 shows high activity with dichloromethane (120, ), 4-nitrobenzyl chloride (125, ), or cumene hydroperoxide (15, ) as substrates (Meyer et al. Citation1991). hGSTT1-1, hGST2-2, and erythrocytic hGSTT1-1 show scant or no activity with CDNB (11, ) as the substrate (Meyer et al. Citation1991; Hussey and Hayes Citation1992; Schröder et al. Citation1996). In contrast to GSTT1-1 expressed in solid tissues, human erythrocytic GSTT1-1 fails to accept EPNP (116, ) and 4-nitrobenzyl chloride (125, ) as substrates (Schröder et al. Citation1996).
hGSTT1-1 is an efficient catalyst for the denitrosation of BCNU (127, ) (Lien et al. Citation2002); little activity was observed with hGSTM2-2 and -3-3, and no activity was found with GSTA1-1, -2-2, and -4-4; GSTM1-1, -4-4, and 5-5; GSTP1-1; and rGSTT2-2. hGSTT1-1 is the major catalyst for the O-demethylation of methyl parathion (129, ), although lower activity was observed with hGSTA1-1, rGSTA5-5, and mGSTA3-3 (Abel et al. Citation2004). CDNB (11, ) is not a substrate for hGSTT2-2 and is a poor substrate for rGSTT1-1, but is a substrate for rGSTT2-2 and mGSTT3-3 (Coggan et al. Citation2002). Murine GSTT1-1 shows high activity with dichloromethane (120, ), EPNP (116, ) and cumene hydroperoxide (15, ), low activity with CDNB (11, ) and 1-menaphthyl sulfate (123, ), and no activity with ethacrynic acid (52, ) (Whittington et al. Citation1999; Coggan et al. Citation2002). With dichloromethane (120, ) as the substrate, the catalytic efficiency (kcat/Km) of murine GSTT1-1 is five-fold greater than that of human GSTT1-1 (Sherratt et al. Citation2002). In the biotransformation of dibromomethane to formaldehyde (122, ), GSH is required, but not consumed (Ahmed and Anders Citation1978).
GSTT2-2 isoenzymes show preference for reactive sulfate esters, e.g. 1-menaphthyl sulfate (123, ) and 5-sulfoxymethylchrysene, for lipid-derived alkenals, and for cumene hydroperoxide (15, ) (Hiratsuka et al. Citation1990, Citation1994; Tan and Board Citation1996). Rat and human GSTT2-2 show different substrate selectivities (Coggan et al. Citation2002): CDNB (11, ) is a substrate for rGSTT2-2 but not for hGSTT2-2, whereas ethacrynic acid (52, ) is a substrate for hGSTT2-2 but not for rGSTT2-2. Murine GSTT3-3 shows high selectivity for 4-nitrobenzyl chloride (125, ) and, similar to mGSTT1-1 and hGSTT2-2, exhibits low specific activity with 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole (85, ); unlike mGSTT1-1 and hGSTT2-2, mGSTT3-3 fails to catalyze the conjugation of 1-menaphthyl sulfate (123, ) (Coggan et al. Citation2002).
As expected, GSTT1-1 catalyzed reactions are decreased in Gstt1-null mice (Fujimoto et al. Citation2007); with dichloromethane (120, ) as the substrate, activity in liver and kidney cytosols from Gstt1-null mice was only 2.2% of that observed in wild-type mice, whereas with EPNP (116, ) or BCNU (127, ) as substrates, activity in Gstt1-null mice ranged from 8.7% to 42.1% or 13.2% to 23.9%, respectively, of wild-type controls. Similarly, the biotransformation of dichloromethane (120, ) in human liver samples with the GSTT1-null genotype is significantly reduced (Arakawa et al. Citation2012).
2.2.6.4.3. Inhibitors
Apart from the weak inhibition of tissue and erythrocyte GSTT1-1 by GSH S-conjugates, no ground-state or active-site directed inhibitors of Theta-class GSTs have apparently been reported (Ong and Clark Citation1986; Meyer Citation1993; Schröder et al. Citation1996). Sulfate and S-(1-menaphthyl)GSH (124, ) inhibit the GSTT2-2-catalyzed biotransformation of 1-menaphthyl sulfate (123, ) (Gillham Citation1973).
2.2.6.4.4. Tissue and species distribution
The tissue-specific distribution of hGSTT2-2 has been reported (Fagerberg et al. Citation2014; Mohana and Achary Citation2017).
Immunoblot analysis shows that hGSTT1-1 is highly expressed in erythrocytes and liver and is also expressed in lung, kidney, brain, skeletal muscle, heart, small intestine, and spleen, but not in lymphocytes (Juronen et al. Citation1996); N-terminal amino-acid sequence analysis and similarities in immunochemical properties indicate that liver and erythrocyte GSTT1-1 are identical. Immunohistochemical analysis shows that only trace amounts of GSTT1-1 are present in human lung (Sherratt et al. Citation1997). Western analysis shows that hepatic expression of GSTT1-1 is greater in the mouse than in the rat (Mainwaring et al. Citation1998); in the lung, mGSTT1-1 is expressed in bronchiolar epithelium and in type-II alveolar cells.
With EPNP (116, ) as the substrate, GSTT activities are high in rat epididymis and adrenal glands; intermediate in liver, testis, epididymal fluid, and seminal vesicles, and not detectable in mammary gland and skeletal muscle (Meyer et al. Citation1984).
In the mouse, Northern-blot analysis shows that all Theta-class GSTs are highly expressed in the liver and to a lesser extent in the kidney (Coggan et al. Citation2002); mGSTT3, but neither mGSTT1 nor mGSTT2, expression is observed in the testes. Immunohistochemical analysis and in situ hybridization showed differences in distribution patterns and enzyme activities of GSTT1-1 and GSTT2-2 in liver and lung tissues of the mouse, rat, and human; the human tissue levels are lower than those of either the mouse or rat (Mainwaring et al. Citation1996).
The low expression of GSTT1-1 in human and rat lung tissue compared with mouse lung tissue, as well as the comparatively low catalytic efficiency of hGSTT1-1 with dichloromethane (120, ) as substrate, indicates little bioactivation of dichloromethane in human lung tissue (Sherratt et al. Citation1997, Citation2002).
A protein isolated from rat liver mitochondria that shows N-terminal sequence identity with Theta-class GSTs and a possible mitochondrial import signal was designated GST 13-13 (Harris et al. Citation1991). There apparently have been no other reports of the characterization of GSTT 13-13, although “13-13” has been used as a designation for GSTT3-3 ().
2.2.6.5. Zeta-class (GSTZ/MAAI)
2.2.6.5.1. Structure and properties
GST Zeta (GSTZ) was identified with a BLAST search of the human expressed sequence tag (EST) database with the amino-terminal sequence of a GST-like protein identified in carnation petals (Board et al. Citation1997, Citation2001). As with GSTT (Meyer et al. Citation1991), GSTZ binds weakly to GSH-agarose affinity columns, which may have prevented its earlier discovery (Board et al. Citation1997). GSTZ is identical with maleylacetoacetate isomerase (MAAI), which catalyzes the penultimate step in the tyrosine degradation pathway (Fernández-Cañón and Peñalva Citation1998). Several reviews about Zeta-class GSTs and MAAI are available (Seltzer Citation1989a, Citation1989b; Anders et al. Citation2001; Board and Anders Citation2005, Citation2011).
Tong et al. (Citation1998a) purified a protein from rat liver that catalyzed the biotransformation of dichloroacetic acid (DCA) (132, ) to glyoxylic acid (133, ).
Figure 11. GSTZ substrates. 132, dichloroacetic acid (DCA); 133, glyoxylic acid; 134, maleylacetone; 135, fumarylacetone; 136, maleylacetoacetic acid (MAA); 137, fumarylacetoacetic acid; 138, (±)-2-bromo-3-(4-nitrophenyl)propanoic acid; 139, 2-(glutathion-S-yl)-3-(4-nitrophenyl)propanoic acid; 140, maleylpyruvic acid; 141, fumarylpyruvic acid; 142, cis-β-acetylacrylic acid ((Z)-4-oxopent-2-enoic acid); 143, (E)-4-oxopent-2-enoic acid.
Portions of the sequence of the purified protein matched that of GSTZ1-1 reported by Board et al. (Citation1997), and antibodies raised against hGSTZ1-1 reacted with the rat liver protein. Accordingly, the GST that catalyzes the biotransformation of DCA was identified as a Zeta-class GST.
hGSTZ1 encodes a 216-residue peptide with a molecular mass of 24 212 Da (Board et al. Citation1997). The gene encoding hGSTZ1 maps to a single locus on chromosome 14q24.3 (Blackburn et al. Citation1998).
Although hGSTZ1-1 fails to catalyze the conjugation of typical GST substrates, its crystal structure indicates that it is a member of the GST superfamily (Polekhina et al. Citation2001; Boone et al. Citation2014). hGSTZ1-1 shows the N-terminal canonical thioredoxin fold characteristic of other cytosolic GSTs and a C-terminal all α-helical domain. The G-site is found in a crevice between the domains, which may explain the low affinity of GSTZ1-1 for GSH-affinity columns. The dimer interface is dominated by hydrophobic interactions. α-Haloacids, but not α-haloacid esters and amides or β-haloacids, are substrates for GSTZ1-1 (Tong et al. Citation1998b); the crystal structure of hGSTZ1-1 indicates that the interaction of α-haloacids with Arg175 positions them for attack by GSH, whereas binding of β-haloacids fails to orient them for attack by GSH.
Kinetic studies with hGSTZ1c-1c indicate that the active-site Cys16 is not a catalytic residue, although Cys16 is has an important role in binding of both GSH and electrophilic co-substrates (Board et al. Citation2003; Ricci et al. Citation2004). The enzyme also shows half-site reactivity: binding of GSH to one subunit effectively silences the second subunit as it is unable to bind and activate GSH (Ricci et al. Citation2004).
Four hGSTZ1 allelic variants have been identified (Blackburn et al. Citation1998, Citation2000, Citation2001; Board and Anders Citation2005); these are designated hGSTZ1a-1a, hGSTZ1b-1b, hGSTZ1c-1c, and hGSTZ1d-1d. (The four allelic variants have also been termed KRT, KGT, EGT, and EGM, respectively (Stacpoole et al. Citation2008; Shroads et al. Citation2012).) The frequency of hGSTZ1 variants in a normal European population is: hGSTZ1a, 0.086; hGSTZ1b, 0.285; hGSTZ1c, 0.473; hGSTZ1d, 0.156 (Blackburn et al. Citation2001); similar distributions have been reported in other populations (Shroads et al. Citation2012). Although 10 SNPs have been identified upstream of the GSTZ1 start of transcription, only two were associated with changes in promoter activity (Fang et al. Citation2006).
The relationship between hGSTZ1 allelic variants and rates of biotransformation has been investigated. The specific activities for biotransformation of DCA (132, ) and (R)-2-chloropropionic acid are highest with hGSTZ1a-1a, whereas the rates of biotransformation of fluoroacetic acid and (S)-2-chloropropionic acid are highest with hGSTZ1b-1b, -1c-1c, and 1d-1d (Blackburn et al. Citation2000, Citation2001). The rate constants (kinact) for the GSH-dependent, DCA-induced inactivation of hGSTZ1-1 variants are similar for hGSTZ1b-1b, -1c-1c, and -1d-1d (∼7 × 10−2 min−1), which are higher than that of hGSTZ1a-1a (3 × 10−2 min−1) (Tzeng et al. Citation2000). With recombinant hGSTZ1-1, the rate constants (kcat/Km) for the biotransformation of maleylacetone (134, ) followed the order: 1d-1d = 1 b-1b > 1c-1c > 1a-1a, whereas the rate constants for the biotransformation of chlorofluoroacetic acid were similar among the four allelic variants (Lantum et al. Citation2002b). In children given DCA (132, ) for management of congenital mitochondrial disorders, the plasma drug t½ was shorter in hGSTZ1c-1c (EGT, see above) carriers compared with hGSTZ1c-1c noncarriers (Shroads et al. Citation2015).
2.2.6.5.2. Substrates
As indicated above, GSTZ1-1 fails to catalyze the conjugation of typical GST substrates or catalyzes the reactions at low rates (Board et al. Citation1997). It was later shown that substrates for GSTZ1-1 include DCA (132, ) and a range of α-haloalkanoic acids as well as maleylacetone (134, ) (Tong et al. Citation1998b; Lantum et al. Citation2002b). hGSTZ1-1 catalyzes the reduction of organic hydroperoxides, e.g. tert-butyl hydroperoxide, and the conjugation of ethacrynic acid (52, ) and NBD-Cl (85, ), albeit at relatively low rates, but shows no activity with CDNB (11, ) and several other known GST substrates (Board et al. Citation1997). The rate of hGSTZ1-1 or rat GSTZ1-1-catalzyed biotransformation of DCA is, however, much greater than that of tert-butyl hydroperoxide (Tong et al. Citation1998a).
GSTZ catalyzes the biotransformation of DCA (132, ) to glyoxylic acid (133, ) (Tong et al. Citation1998a, Citation1998b); in this reaction, GSH is required but not consumed. In addition to DCA, GSTZ catalyzes the biotransformation of fluoroacetic acid, bromochoro-, bromofluoro-, chlorofluoro, and dibromoacetic acid, 2-bromo-, 2-chloro-, and 2-iodopropionic acid, and 2,2-dichloropropionic acid, but not difluoroacetic acid, 2-fluoropropionic acid, or 3,3-dichloropropionic acid (Tong et al. Citation1998b).
GSTs have been implicated in the defluorination of fluoroacetic acid (Soiefer and Kostyniak Citation1983, Citation1984; Wang et al. Citation1986; Tecle and Casida Citation1989). Tu et al. (Citation2005) showed, however, that at least two enzymes catalyze the defluorination of fluoroacetic acid and that rat liver GSTZ1-1 is a minor catalyst. Other studies showed that recombinant hGSTZ1a-1a, -1 b-1b, -1c-1c, and 1d-1d all catalyze the defluorination of fluoroacetic acid but at rates that are much lower than those for the biotransformation of DCA (132, ) (Blackburn et al. Citation2000, Citation2001; Tu et al. Citation2006).
The selectivity of GSTZ for α-haloalkanoic acids was exploited to develop an assay substrate for GSTZ (Board et al. Citation2003): (±)-2-bromo-3-(4-nitrophenyl)propanoic acid (138, ) undergoes a GSTZ1-1-catalyzed reaction to give 2-(glutathion-S-yl)-3-(4-nitrophenyl)propanoic acid (139, ), which shows a spectral shift that allows quantification of reaction rates.
The isomerization of MAA (136, ) to fumarylacetoacetic acid (137, ) is catalyzed by GSH-requiring hepatic enzymes (Edwards and Knox Citation1956). Although the unstable MAA can be prepared by the homogentistate dioxygenase-catalyzed oxidation of homogentistic acid, the surrogate substrates maleylacetone (134, ), maleylpyruvic acid (140, ), and cis-β-acetylacrylic acid (142, ), which are biotransformed to the corresponding E-isomers, are most frequently used (Lack Citation1961; Fowler and Seltzer Citation1970; Lee and Seltzer Citation1989; Lantum et al. Citation2002b). The consensus mechanism of the GSTZ-catalyzed isomerization of MAA (136, ) involves the Michael addition of GSH to give a dienediolate intermediate, which allows rotation around the newly formed single bond, followed by elimination of GSH to form the trans-isomer, i.e. furmarylacetoacetic acid (137, ) (Seltzer and Lin Citation1979; Deponte Citation2013). Hence, GSH is required, but not consumed, in the isomerization reaction.
GSTZ is not detectable in Gstz1−/− mice and accordingly the biotransformation of chlorofluoroacetic acid and maleylacetone (134, ) is not detectable (Lim et al. Citation2004); GSTA-, GSTM-, and GSTP-class GSTs are, however, overexpressed in female Gstz−/− mice.
2.2.6.5.3. Inhibitors
No ground-state, competitive inhibitors of GSTZ1-1 have apparently been described. DCA (88, ) is, however, an irreversible inhibitor of GSTZ1-1: in human subjects given DCA, the t½ is prolonged after repeated dosing (Curry et al. Citation1985, Citation1991). Hepatic levels of GSTZ1-1 are decreased in rats given DCA (Anderson et al. Citation1999; Guo et al. Citation2006).
Kinetic studies showed that DCA (88, ) inactivated GSTZ in rat liver cytosol (Anderson et al. Citation1999); moreover, in rats given DCA, losses in enzyme activity and in the amount of immunoreactive GSTZ1-1 are observed. From these data, the turnover of GSTZ1-1 was found to be 0.21−1/day, which amounts to a t½ of 3.3 days. In addition to DCA, bromochloroacetic acid, dibromoacetic acid, and 2,2-dichloropropionic acid also inactivate GSTZ1-1, whereas fluorine-containing dihaloacetic acids and 2-chloropropionic acid fail to inactivate GSTZ1-1. These data, along with studies with [2H]dichloroacetic acid and [2H]chlorofluoroacetic acid (Wempe et al. Citation1999), allowed the proposal of a mechanism for the DCA-induced inactivation of GSTZ1-1: the formation of the S-(α-chlorocarboxymethyl)GSH intermediate results in the covalent modification of a critical residue in GSTZ1-1. Indeed, mass-spectral analysis of hGSTZ1c-1c inactivated by DCA showed that Cys16 is covalently modified by the S-(α-chlorocarboxymethyl)GSH intermediate (Anderson et al. Citation2002). The partition ratio for the DCA inactivation of hGSTZ1c-1c was 5.7 × 102, and the kcat for the biotransformation of DCA was 39 min−1.
2.2.6.5.4. Tissue and species distribution
hGSTZ1-1 is most intensely expressed in liver, but is also found in kidney, skeletal muscle, and brain (Board et al. Citation1997). In mice, GSTZ-catalyzed biotransformation of DCA (88, ) is highest in liver followed by lung and kidney (Lipscomb et al. Citation1995). Immunohistochemical studies show that GSTZ1-1 is present in most rat tissues but that expression is highest in liver, testes, and prostate (Lantum et al. Citation2002a; Mohana and Achary Citation2017). GSTZ1-1 is also found in human and rat liver mitochondrial matrix and, as with cytosolic GSTZ1-1, is inactivated by DCA (Li et al. Citation2011).
2.2.6.5.5. Other biological functions
Maleylacetoacetic acid (MAA) (136, ), fumarylacetoacetic acid (137, ), and succinylacetoacetic acid are electrophilic metabolites of tyrosine, and their formation contributes to the deleterious symptoms of hereditary tyrosinemia type 1 (Lindblad et al. Citation1977). Gstz−/− mice given Phe show hepatic necrosis, macrovesicular steatosis, splenic atrophy, decreased total white blood cell counts, and lowered hepatic GSH concentrations as well as increased serum succinylacetone concentrations, indicating that GSTZ1-1 plays a critical role in detoxifying Phe metabolites (Lim et al. Citation2004; Blackburn et al. Citation2006); treatment of mice with DCA (132, ), which inactivates GSTZ1-1, followed by feeding of Phe does not, however, result in decreased white blood cell counts (Theodoratos et al. Citation2009). The hepatotoxicity of acetaminophen is increased in Gstz−/− mice, perhaps because of the lowered hepatic GSH concentrations seen in Gstz-/- mice (Blackburn et al. Citation2006).
C/EBPα-dependent induction of GSTZ1-1 mRNA and protein is observed during adipogenesis in 3T3-L1 preadipocytes, indicating a role for GSTZ1-1 in adipocytes (Qiang and Farmer Citation2006).
Associations between hGSTZ1 polymorphisms and a range of human disorders have been investigated. In human subjects, no overall association between Parkinson’s Disease and hGSTZ1 polymorphisms was observed (Taylor et al. Citation2001); the Z1*C allele, which is the most common variant in Caucasian populations, was, however, less common in Parkinsonian subjects when stratified according to pesticide exposure. A SNP (rs2111699 located in intron 1 of GSTZ1) is associated with longevity in long-living subjects in Calabria, Italy (Di Cianni et al. Citation2013), but no association between hGSTZ1 polymorphisms and age-related macular degeneration was found (Othman et al. Citation2012). No association between hGSTZ1 allelic variants and susceptibility to breast cancer or to schizophrenia has been found (Nafissi et al. Citation2011; Saadat et al. Citation2012).
2.2.6.6. Omega-class (GSTO)
Omega-class GSTs are distinguished from most other cytosolic GSTs in that they fail to catalyze canonical conjugative reactions but catalyze reductive reactions and regulate cell-signaling pathways. Reviews about GSTO are available (Whitbread et al. Citation2003, Citation2005; Board Citation2011; Board and Menon Citation2016; Xie et al. Citation2018).
2.2.6.6.1. Structure and properties
Omega-class GSTs were identified by a BLAST search of the human EST database with hGSTT and hGSTZ sequences as query sequences (Board et al. Citation2000). These studies identified a human cDNA that was expressed, characterized, and designated as hGSTO1-1. Further studies on the genomic organization of GSTO1 revealed a second Omega-class gene, GSTO2 (Whitbread et al. Citation2003). The genes encoding hGSTO1 and hGSTO2 are located on chromosome band 10q25.1 (Board et al. Citation2000; Whitbread et al. Citation2003; Wang et al. Citation2005). Rat and mouse orthologues of hGsto2 have been described ( and ).
Omega-class GSTs differ from other cytosolic GSTs in that they have a Cys residue in the active-site rather than a Tyr or a Ser residue (Board et al. Citation2000; Board Citation2011). Moreover, human Omega-class GSTs show only about 20% amino-acid sequence identity with other cytosolic GSTs. hGSTO2-2 shows 64% amino-acid sequence identity with hGSTO1-1 (Whitbread et al. Citation2003, Citation2005).
The crystal structures of hGSTO1-1 and -2-2 complexed with substrates and inhibitors have been solved (Board Citation2011; Zhou et al. Citation2012). The thioredoxin fold and C-terminal domain with two helices are present in both hGSTO1-1 and -2-2, and both show an extended N-terminus loop. Although the G-sites are similar in hGSTO1-1 and -2-2, the H-site is more occluded in hGSTO2-2 due to substitution of Lys128 for Gly128.
Before the Omega-class GSTs were fully characterized, four proteins were described that were later shown to be Omega-class GSTs: S-phenacylglutathione reductase (Kitada et al. Citation1985); dehydroascorbic acid (144, ) reductase (Maellaro et al. Citation1994; Ishikawa et al. Citation1998); “p28,” a 28-kDa stress response protein (Kodym et al. Citation1999); and MMAV (146, ) reductase (Zakharyan and Aposhian Citation1999).
Figure 12. GSTO substrates. 144, dehydroascorbic acid; 145, ascorbic acid; 146, methylarsonic acid (MMAV); 147, methylarsonous acid (MMAIII); 148, dimethylarsinic acid (DMAV); 149, dimethylarsinous acid (DMAIII); 150, 2,4-dichlorophenacyl chloride; 151, 2,4-dichloroacetophenone; 152, S-(4-nitrophenacyl)GSH; 153, 4-nitroacetophenone; 154, 2-hydroxyethane disulfide; 155, 2-hydroxyethanethiol.
hGSTO1 and hGSTO2 are polymorphic (Mukherjee et al. Citation2006; Board Citation2011). In hGSTO1, the E155/E208 variant predominates in Australian-European and Chinese population (Whitbread et al. Citation2003; Schmuck et al. Citation2005). Interestingly, thioltransferase, dehydroascorbic acid reductase, methylarsonic acid reductase, and dimethylarsinic acid reductase activities of GSTO1-1 are increased in the A140/ΔE155/E208 and A140/ΔE155/K208 allelic variants compared with similar variants that retain E155; an exception is the A140/E155/K208 variant, which exhibits dimethylarsinic acid reductase activity similar to the corresponding ΔE155 variant (Whitbread et al. Citation2003; Schmuck et al. Citation2005). The E155 deletion in hGSTO1-1 causes not only an increase in specific activity but also a substantial decrease in stability (Whitbread et al. Citation2003; Schmuck et al. Citation2005, Citation2008).
The association between hGSTO1 and hGSTO2 polymorphisms has been investigated and attempts to identify at-risk populations and differences in arsenic metabolism in arsenic-exposed populations have been made (Marnell et al. Citation2003; Tanaka-Kagawa et al. Citation2003; Yu et al. Citation2003; Schmuck et al. Citation2005; Whitbread et al. Citation2005; Chowdhury et al. Citation2006; Mukherjee et al. Citation2006; Paiva et al. Citation2008; Schmuck et al. Citation2008; Escobar-García et al. Citation2012). For example, in arsenic-exposed subjects in the Mexican state of Coahuila, the A140D, but not the E208K, polymorphism is associated with increased excretion of arsenic metabolites (Escobar-García et al. Citation2012); in addition, the A140D and E208K polymorphisms are associated with expression of genes encoding IL-8, Apaf-1, and TGF-β.
hGSTO1 polymorphisms have been associated, however inconsistently, with age-of-onset of Alzheimer’s disease and Parkinson’s disease (Kolsch et al. Citation2004; Whitbread et al. Citation2004; Li et al. Citation2006; Allen et al. Citation2012; Piacentini et al. Citation2012).
Of 15 microsomal enzyme inducers studied in mice, only butylated hydroxylanisole significantly increased the expression of Gsto1 RNA (Knight et al. Citation2008). The expression of GSTO1-1 is increased or decreased in a range of treatments or disorders, including cancer, cataract, neurodegenerative disease, pulmonary disease, and inflammation (Board and Menon Citation2016).
2.2.6.6.2. Substrates
GSTO catalyzes several reactions: S-phenacylglutathione reductase, thioltransferase, deglutathionylation, dehydroascorbic acid reductase, α-haloketone reductase, methylarsonic acid reductase, and dimethylarsinic acid reductase (Board et al. Citation2000; Zakharyan et al. Citation2001; Board and Anders Citation2007; Board et al. Citation2008; Board Citation2011; Board and Menon Citation2016). GSTO1-1 shows low activity with CDNB (11, ) and little or no activity with many canonical GST substrates (Board et al. Citation2000). hGSTO2-2 shows high dehydroascorbic acid (144, ) reductase activity compared with hGSTO1-1 (Schmuck et al. Citation2005; Zhou et al. Citation2012). In contrast to hGSTO1-1, recombinant hGSTO2-2 has been reported to show high activity with CDNB (11, ) as the substrate (Wang et al. Citation2005); the dehydroascorbic acid (144, ) reductase and thioltransferase activities for hGSTO2-2 reported by Wang et al. (Citation2005), are much lower than those reported in other studies (Board et al. Citation2000; Menon and Board Citation2013).
2,4-Dichlorophenacyl chloride (150, ), a metabolite of chlorfenvinphos, undergoes GSH-dependent biotransformation to 2,4-dichloroacetophenone (151, ) (Hutson et al. Citation1976; Brundin et al. Citation1982; Kitada et al. Citation1985); the intermediate GSH S-conjugate S-(2,4-dichlorophenacyl)GSH, which is formed non-enzymatically, rather than α-haloketone 150 (), is the actual substrate for the enzyme, and several thiols, e.g. GSH, 2-mercaptoethanol, L-Cys, cysteamine, can serve as second substrates. The reduction of phenacyl halides is catalyzed by hGSTO1-1, but not by hGSTO2-2 (Board and Anders Citation2007).
The finding that GSTO1-1 catalyzes the GSH-dependent reduction of phenacyl halides led to the development of a specific assay substrate for hGSTO1-1 (Board and Anders Citation2007; Board et al. Citation2008). hGSTO1-1 catalyzes the reduction of S-(4-nitrophenacyl)GSH (152, ) in the presence of 2-mercaptoethanol to 4-nitroacetophenone (153, ) (Board et al. Citation2008); the reaction is selective for hGSTO1-1: little or no reaction was observed with hGSTO2-2, hGSTP1-1, hGSTA1-1, hGSTA2-2, hGSTA4-4, hGSTM1-1, hGSTM2-2, hGSTM4-4, hGSTT1-1, hGSTT2-2, or hGSTZ1-1. Németi et al. (Németi et al. Citation2015b) modified the assay to increase its sensitivity by using HPLC to quantify 4-nitroacetophenone (153, ) formation and by the use of tris(2-carboxyethyl)phosphine (TCEP) as the reductant.
GSTO plays a critical role in the reduction of weakly toxic pentavalent arsenic to highly toxic trivalent arsenic (Watanabe and Hirano Citation2013). The reduction of methylarsonic acid (MMAV, 146, ) to methylarsonous acid (MMAIII, 147, ) is catalyzed by MMAV (146, ) reductase, which requires GSH as a second substrate (Zakharyan and Aposhian Citation1999). Later studies showed that MMAV (146, ) reductase is identical with hGSTO1-1 (Zakharyan et al. Citation2001) and that both hGSTO1-1 and hGSTO2-2 catalyze the reduction of methylarsonic acid (146, ), as well as the reduction of dimethylarsinic acid (148, ), but that rates of reduction of dimethylarsinic acid (148, ) found with hGSTO2-2 are four-fold lower than those found with hGSTO1-1 (Schmuck et al. Citation2005). Investigation of the reduction of dimethylarsinic acid (148, ) in vivo in rats and in rat liver cytosol showed that GSH is required, but, on the basis of purification studies, the authors concluded that GSTO1-1 was not involved (Németi and Gregus Citation2013). Further investigation showed, however, that reduction of dimethylarsinic acid (146, ) by rat liver cytosol was inhibited by the GSTO1-1-selective substrate S-(4-nitrophenacyl)GSH (152, ) (Németi et al. Citation2015a); additional purification studies confirmed the role of GSTO1-1 in the reduction of dimethylarsinic acid (148, ).
In studies with Gsto1-null mice, the hepatic GSTO1-1-catalyzed reduction of methylarsonic acid (146, ) amounted to 20% that of wild-type mice, indicating that pathways in addition to GSTO may be involved (Chowdhury et al. Citation2006); in Gsto1-null mice given sodium arsenate, high concentrations of methylarsonous acid (147, ) were found in the kidneys. (The reduction of arsenate to arsinite is catalyzed by a range of phosphorolytic-arsenolytic enzymes, including purine nucleoside phosphorylase-a, glyceraldehyde-3-phosphate dehydrogenase, and glycogen phosphorylase (Yu et al. Citation2003; Németi and Gregus Citation2009).)
Rates for the thioltransferase reaction with 2-hydroxyethyl disulfide (154, ) as the substrate are similar with both hGSTO1-1- and hGSTO2-2 (Schmuck et al. Citation2005). The glutathionylation and deglutathionylation of proteins is a redox detoxification process and also acts as a regulatory switch (Board and Menon Citation2016). hGSTO1-1, but not hGSTO2-2, catalyzes the deglutathionylation of the glutathionylated synthetic peptide SQLWC-SGLSN (Menon and Board Citation2013); expression of hGSTO1-1 in T47-D cells, which show reduced GSTO1-1 activities, decreased the total cellular glutathionylated protein concentration.
Early studies showed the presence of a GSH-dependent dehydroascorbic acid reductase in a range of rat tissues (Maellaro et al. Citation1994; Paolicchi et al. Citation1996; Ishikawa et al. Citation1998), an activity now attributed to GSTO. Both hGSTO1-1 and hGSTO2-2 catalyze the reduction of dehydroascorbic acid (144, ) to ascorbic acid (145, ), but rates with hGSTO2-2 are approximately100-fold higher than those with hGSTO1-1 (Schmuck et al. Citation2005) or with glutaredoxin-dependent dehydroascorbic acid reductase (Wells et al. Citation1990; Park and Levine Citation1996). Interestingly, the crystal structure of hGSTO1-1-C32S mutant in complex with ascorbic acid (145, ) shows that ascorbic acid is bound to the G-site (Zhou et al. Citation2012); this is the first report of a GST substrate or reaction product that does not contain GSH as a structural component being bound in the G-site of a GST.
2.2.6.6.3. Inhibitors
Given the putative role of Omega-class GSTs in arsenic bioactivation, cancer, and neurodegeneration, there has been considerable interest in identifying GSTO inhibitors (Board and Menon Citation2016). Xie et al. (Citation2018) have reviewed the development of small-molecule inhibitors as an example of hit discovery for difficult targets, e.g. GSTO1. The role of GSTO1-1 in lipopolysaccharide-stimulated inflammation and ROS production also indicates that inhibitors may find therapeutic uses (Menon et al. Citation2014, Citation2015; Board and Menon Citation2016).
The cytokine release inhibitory drug CRID 2 (156, ) covalently binds to Cys32 in the active site of hGSTO1-1 and irreversibly inhibits ATP-induced IL-1β posttranslational processing (Laliberte et al. Citation2003); no labeling was observed with a C32A mutant and the labeling of hGSTO1-1 by CRID 2 is blocked by a range of S-substituted GSH derivatives, including S-(4-nitrobenzyl)GSH (152, ), S-(1-adamantyl)GSH, and S-octylGSH. Moreover, a GSH adduct of CRID 2 inhibits ATP-induced IL-1β posttranslational processing. The epoxyketone moiety-containing proteasome inhibitor carfilzomib covalently reacts with Cys32 and Cys192 of hGSTO1 (Federspiel et al. Citation2016); although carfilzomib inhibits GSTO1-1 at micromolar concentrations, the inhibition of GSTO1-1 is expected to be minimal at therapeutic doses of carbfilzomib.
Figure 13. GSTO inhibitors. 156, 1-(1,2,3,5,6,7-hexahydro-s-indacen-4-yl)-3-[2-fluoro-5-oxiranylbenzenesulfonyl]urea (CRID 2); 157, N-[3-[(2-chloroacetyl)(4-nitrophenyl)amino]propyl]-2,2,2-trifluoroacetamide (ML175); 158, 2-chloro-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-2-yl)ethyl)-N-(3-fluorophenyl)acetamide (KT53); 159, 2-chloro-N-(4-chloro-3-(N,N-dimethylsulfamoyl)phenyl)acetamide (C1-27); 160, 9-[2-carboxy-4-[[[2-[[oxo-10-[(phenylsulfonyl)oxy]decyl]amino]ethyl]amino]carbonyl]phenyl]-3,6-bis(dimethylamino)xanthylium; 161, N-[1-oxo-3-[(phenylsulfonyl)oxy]propyl]-l-isoleucylglycyl-4,4,5,5-tetradehydro-l-norvalinamide (NJP2); 162, 1-(4-amino-6-chlorohex-1,5-dione)-11,12-didehydro-5,6-dihydro-dibenz[b,f]azocine; 163, 5-chloromethylfluorescein diacetate; 164, 5-bromomethylfluorescein.
![Figure 13. GSTO inhibitors. 156, 1-(1,2,3,5,6,7-hexahydro-s-indacen-4-yl)-3-[2-fluoro-5-oxiranylbenzenesulfonyl]urea (CRID 2); 157, N-[3-[(2-chloroacetyl)(4-nitrophenyl)amino]propyl]-2,2,2-trifluoroacetamide (ML175); 158, 2-chloro-N-(2-(cyclohexylamino)-2-oxo-1-(pyridin-2-yl)ethyl)-N-(3-fluorophenyl)acetamide (KT53); 159, 2-chloro-N-(4-chloro-3-(N,N-dimethylsulfamoyl)phenyl)acetamide (C1-27); 160, 9-[2-carboxy-4-[[[2-[[oxo-10-[(phenylsulfonyl)oxy]decyl]amino]ethyl]amino]carbonyl]phenyl]-3,6-bis(dimethylamino)xanthylium; 161, N-[1-oxo-3-[(phenylsulfonyl)oxy]propyl]-l-isoleucylglycyl-4,4,5,5-tetradehydro-l-norvalinamide (NJP2); 162, 1-(4-amino-6-chlorohex-1,5-dione)-11,12-didehydro-5,6-dihydro-dibenz[b,f]azocine; 163, 5-chloromethylfluorescein diacetate; 164, 5-bromomethylfluorescein.](/cms/asset/28c6357d-44d2-4a5f-9be9-6fb0124f3a64/itxc_a_1692191_f0013_b.jpg)
Fluorescence-based activity-based protein profiling (Fluopol-ABPP) was used to identify enzyme targets (Bachovchin et al. Citation2009); with this methodology, GSTO1 was found to be a target for the thiol-reactive compounds omeprazole, dioxiram, and an α-chloroacetamide. These studies were extended to focus on GSTO1-1 inhibitors and showed that α-chloroacetamide 157 () inhibits (IC50 = 28 nM) hGSTO1-1 and covalently labels the active-site Cys32 (Tsuboi et al. Citation2010). Additional studies identified α-chloroacetamide 158 () as a potent and selective inactivator of hGSTO1-1 in vitro (IC50 = 21 nM) and of hGSTO1-1 in MDA-MB-435 breast cancer cells (IC50 = 35 nM) (Tsuboi et al. Citation2011); moreover, α-chloroacetamide 158 () enhanced the cytotoxicity of cisplatin in MDA-MB-435 cells.
Screening of α-chloro-N-phenylacetamide derivatives identified α-chloroacetamide 159 () as a potent (IC50 = 31 nM), selective, and irreversible inhibitor of GSTO1-1 (Ramkumar et al. Citation2016). α-Chloroacetamide 159 (), which covalently labels Cys32 in the active site of GSTO1-1, was cytotoxic at low µM concentrations to a panel of cancer cell lines, increased the cells’ sensitivity to cisplatin, and suppressed growth of a human colon cancer xenograft in vivo. Four-hour treatment of HCT116 cells with α-chloroacetamide 159 () causes induction of several stress response-associated genes; 24-h treatment resulted in additional transcriptional changes, many of which also occur in HCT116 GSTO1-1 knockdown cells. These studies were extended to include a panel of acrylamide-based GSTO inhibitors (Dai et al. Citation2019). One compound, N-((3-methylisoxazol-5-yl)methyl)-N-(4-(2-hydroxyphenyl)thiazole-2-yl)acrylamide, is a potent inhibitor of GSTO1-1 (IC50 = 0.22 nM). Its selectivity for GSTO1-1 versus GSTO2-2 and other GSTs was not reported.
In addition to serving as inhibitors, compounds that target Cys32 of GSTO1 have been designed for activity-based protein profiling (ABPP), which allows quantification of protein concentrations. Although inhibition of GSTO1-1 was not explicitly reported in all experiments, adduction of Cys32 would be expected to inhibit its catalytic activity. Rhodamine-tagged phenylsulfonate ester 160 () targets the active-site Cys32 of hGSTO1-1 in estrogen-receptor negative cell lines (Adam et al. Citation2002, Citation2004). A library of peptides bearing electrophilic sulfonate ester or acrylamide groups targeted to Cys residues was synthesized to search for apoptotic-cell-selective inhibitors (Pace et al. Citation2012). Sulfonate ester 161 () selectively inhibits hGSTO1-1 in apoptotic cells compared with control cells. The selective internalization of sulfonate ester 161 () in apoptotic cells was demonstrated by use of click chemistry to attach a rhodamine moiety to the acetylene bond in sulfonate ester 161 () to afford a fluorescent analog; the selectivity for apoptotic cells was attributed to the increased permeability of apoptotic cell membranes.
Li et al. (Citation2019) used the ABPP strategy to search for cellular targets of piperlongumine (103, ). With an alkyne-substituted piperlongumine analog and click-chemistry-based ABPP, GSTO1 was identified as a likely cellular target of piperlongumine (103, ); the C2-C3 olefin of piperlongumine was required for cytotoxicity.
Incubation of GSTO1-1 with dibenzazocine 162 () and MALDI-TOF-MS analysis demonstrated the adduction of GSTO1-1, presumably at Cys32 (Liang et al. Citation2015). Inductively coupled plasma mass spectrometry (ICP-MS) and tagging of the adduct by Cu-free click chemistry allowed quantification of the cellular content of GSTO1-1, which amounted to 2.4 × 10−2 ng/cell in C7721 and MCF-7 tumor cell lines, as well as visualization of GSTO1-1 in cells. The methodology also was used to quantify the increase in GSTO1-1 content after incubation of cells with cisplatin.
A series of chemically reactive fluorescein derivatives, including benzyl halides 163 and 164 (), were screened for their abilities to label proteins in NIH/3T3 cells (Son et al. Citation2010). The cell-permeable benzyl chloride-derived fluorescein diacetate (163, ), which covalently alkylates Cys32, irreversibly and selectively inhibited (IC50 = 51 nM) hGSTO1-1 in HEK293 cells and allowed quantification of catalytically active hGSTO1-1 in intact NIH/3T3 cells and HEK293 cells by fluorescence intensity.
hGSTO1-1 has been proposed for use as a “self-catalytic” tag for study of membrane proteins (Lee et al. Citation2011). A GSTO1-1-epidermal growth factor receptor (EGFR) fusion protein was expressed in EGFR-negative HEK293 cells. Treatment of the cells with bromomethyl fluorescein 164 (), which unlike the diacetylated fluorescein 163 () does not penetrate cells, allowed the visualization of EGFR both on the cell membrane and its subsequent endocytosis upon activation by an EGF ligand. No fluorescence was detected when cells expressing an EGFR fusion protein containing C32A hGSTO1-1 were treated with fluorescein 164 ().
Studies with 4-aminopiperidine-based probes with Cys-reactive moieties identified hGSTO1 in lysates from HEK293T cells overexpressing hGSTO1 (Couvertier and Weerapana Citation2014). The probes also labeled hGSTO1 in intact HEK293T cells expressing hGSTO1.
In contrast to the ABPP approach discussed above to identify hit targets, the “Inverse Drug Discovery” approach uses small molecule probes with latent electrophilic reactivity to identify enzyme and non-enzyme proteins (Mortenson et al. Citation2018). GSTO1 was identified in HEK293T cell lysates as a target for a fluorosulfate-substituted quinoline-based probe; the Tyr229 residue, rather than the Cys32 residue, was modified by the probe. Although inhibition experiments were not reported, the bound fluorosulfate probe is located in the small-molecule binding pocket of GSTO1, which would presumably result in inhibition.
(+)-α-Tocopherol succinate inhibits the hGSTO1-1-catalyzed reduction of methylarsonic acid and dimethylarsinic acid (Sampayo-Reyes and Zakharyan Citation2006). In addition, GSTO1-1 is inhibited by carnosic acid and rifampicin (Board and Menon Citation2016).
2.2.6.6.4. Tissue and species distribution
Northern blot and immunohistochemical analyses show that GSTO1 is widely expressed in mammalian tissues but that expression is highest in liver, heart, and skeletal muscle (Paolicchi et al. Citation1996; Board et al. Citation2000; Rouimi et al. Citation2001; Yin et al. Citation2001; Whitbread et al. Citation2003, Citation2005; Mohana and Achary Citation2017). Northern-blot analysis showed that hGSTO2 is highly expressed in testes, prostate, and pancreas (Whitbread et al. Citation2003; Wang et al. Citation2005); further analysis by RT-PCR revealed detectable expression in most tissues, except skeletal muscle, and high expression in testes and pancreas. The expression of hGSTO2 was examined in seven tumor-derived cell lines but was detectable only in prostatic adenocarcinoma cells (Wang et al. Citation2005); hGSTO2-2 is found in both cytoplasm and nucleus.
In Gsto1-null mice, expression of methylarsonic acid reductase activity was reduced in all tissues studied, whereas in wild-type mice methylarsonic acid reductase activity was the highest in urinary bladder tissue (Chowdhury et al. Citation2006).
2.2.6.6.5. Other biological functions
In addition to its role in xenobiotic metabolism, GSTO has been implicated in several biological processes (Oakley Citation2005; Board and Menon Citation2013, Citation2016).
Chloride intracellular channel proteins (CLIC) are soluble and membrane-bound proteins that function as chloride-selective ion channels (Cromer et al. Citation2002). CLIC proteins show considerable sequence and structural similarity to cytosolic GSTs, particularly to the Omega-class GSTs (Dulhunty et al. Citation2001; Harrop et al. Citation2001; Cromer et al. Citation2007). CLIC proteins lack GST catalytic activity, but do show glutaredoxin-dependent thioltransferase and dehydroascorbic acid reductase activities (Al Khamici et al. Citation2015). GSTO1-1, along with GSTA1-1 and GSTM2-2, inhibits ryanodine receptor calcium ion channels (Dulhunty et al. Citation2011).
Glutathionylation of intracellular protein thiols plays an important role in regulating metabolic pathways. GSTO1-1, but not GSTO2-2, catalyzes deglutathionylation reactions, which may influence protein structure and function (Kim et al. Citation2012; Menon and Board Citation2013).
Bacterial lipopolysaccharide (LPS) induces the TLR4 signaling pathway in macrophages. GSTO1-1 is, however, required for LPS-stimulated signaling in macrophages (Menon et al. Citation2015). Gsto1-null mice exhibit an attenuated inflammatory response to administration of LPS and to the effect of a high-fat diet on glucose tolerance and insulin resistance (Menon et al. Citation2017); moreover, the response to LPS is reduced by inhibition of GSTO1-1. In addition, Gsto1-null mice show increased leakage of bacteria from the colon to the lymphatic system in a model of inflammatory bowel disease. These results indicate that inhibition of GSTO1-1 may be therapeutically useful in inflammatory diseases, including type 2 diabetes.
The demonstration that NEK7, a regulatory kinase in the NLRP3 inflammasome, is activated by GSTO1-1-catalyzed deglutathionylation provided key insight into the role of GSTO1-1 in inflammasome regulation and, ultimately, in the release of inflammatory cytokines from bone marrow-derived macrophages (Hughes et al. Citation2019). When macrophages were primed with lipopolysaccharide and treated with the potent GSTO1-1 inhibitor 159 (), followed by nigericin stimulation, the release of IL-1β was reduced significantly compared with treatment with lipopolysaccharide and nigericin in the absence of the inhibitor. The level of caspase, which converts pro-IL-1β to IL-1β, was also lowered, and macrophage cell death was reduced. Inhibitor 159 () also caused a significant reduction in cytokine release from human peripheral blood mononuclear cells. In an in vivo model of inflammatory bowel disease, the GSTO1-1 inhibitor 159 () caused a lowering of IL-1β release similar to that previously observed in GSTO1-1-deficient mice (Menon et al. Citation2017)
Several studies have investigated the putative role of hGSTO1 and hGSTO2 polymorphisms in the age-of-onset for Parkinson’s disease and increased risk for late-onset Alzheimer’s disease, but inconsistent results have been obtained (Kolsch et al. Citation2004; Whitbread et al. Citation2004; Li et al. Citation2006; Allen et al. Citation2012; Piacentini et al. Citation2012).
Knockdown studies demonstrated a range of pathways regulated by GSTO1 (Ramkumar et al. Citation2016). Gene expression studies in HCT116 cells with siGSTO1 and RNA Bru-Seq analysis (Paulsen et al. Citation2014) showed that a large number of genes are up- or downregulated, including transporter-, stress-, and metabolism-related genes. A significant finding was that GSTO1 inhibition decreases the viability of HCT116 cancer cells.
2.2.6.7. Sigma-class (GSTS, PGDS)4
The Sigma-class GST was discovered through two lines of investigation. One, studies on the enzymology of prostaglandin biotransformation indicated the involvement of GSTs (Christ-Hazelhof et al. Citation1976; Urade et al. Citation1987; Ujihara et al. Citation1988b). Other studies reported that the S-crystallin family of lens proteins found in cephalopods show approximately 30% sequence identity with Alpha-, Mu-, Pi-, and Theta-class GSTs (Ji et al. Citation1995; Meyer and Thomas Citation1995; Tomarev et al. Citation1995; Kanaoka et al. Citation1997). Based on a comparison of the primary structure, gene structure, and crystal structure, the squid proteins were assigned to the Sigma-class GST (Ji et al. Citation1995). Sequence analysis showed that the rat spleen enzyme that catalyzes the isomerization of prostaglandin H2 (PGH2, 165, ) to prostaglandin D2 (PGD2, 166, ) is a Sigma-class GST (Meyer and Thomas Citation1995). A review about Sigma-class GSTs has been published (Flanagan and Smythe Citation2011). (A GSH-independent PGD synthase that catalyzes the isomerization of PGH2 (165, ) to PGD2 (166, ) and is a member of the lipocalin family has also been described (Nagata et al. Citation1991).)
Figure 14. PGDS substrates. 165, prostaglandin H2 (PGH2); 166, prostaglandin D2 (PGD2).
2.2.6.7.1. Structure and properties
As with other cytosolic GSTs, the mammalian GSTS is a homodimeric protein with a molecular mass of approximately 50 kDa; in common with GSTA, GSTM, and GSTP, GSTS has an active-site Tyr residue. One human gene (PGD2), one rat gene (Ptgds2), and one mouse gene (Ptgds2) encoding PGDS have been identified (). The gene encoding hPGD2 has been mapped to chromosome band 4q21-22, and the murine gene encoding Ptgds2 has been mapped to chromosome band 3D–E (Kanaoka et al. Citation2000).
The crystal structures of rat and human PGDS have been solved (Ji et al. Citation1995; Kanaoka et al. Citation1997; Inoue et al. Citation2003). Crystallographic analysis of rat PGDS revealed a prominent cleft, which is the widest and deepest among the GSTs, that facilitates substrate binding and catalysis (Kanaoka et al. Citation1997). Both Ca2+ and Mg2+ increase the activity of hPGDS by ∼150%, apparently by altering the coordinating water structure, an effect not seen with other GSTs (Inoue et al. Citation2003). The effect of Mg2+ on the coordinated water molecules appears to enhance the stabilization of the GSH thiolate by Arg14 and to decrease the Km of GSH; Ca2+, which is a much weaker activator of PGDS, does not affect the binding of GSH.
A single GSTS has been identified in mammals (Kanaoka and Urade Citation2003). Early studies identified a GSH-dependent enzyme from sheep lung that catalyzed the isomerization of prostaglandin H2 (PGH2, 165, ) to prostaglandin D2 (PGD2, 166, ) and the conjugation of known GST substrates, e.g. CDNB (11, ) (Christ-Hazelhof et al. Citation1976); several rat hepatic GSTs also catalyze the isomerization of PGH2 (165, ) to PGD2 (166, ), although the principal product formed by the rat hepatic GSTs was prostaglandin E2 (PGE2).
PGDS activity is increased in human megakaryoblastic CMK cells incubated with phorbol ester (Mahmud et al. Citation1997). The transcription factor MITF is required for the transactivation of PGD2 and the expression of PGDS activity in bone-marrow-derived mast cells (Morii and Oboki Citation2004).
2.2.6.7.2. Substrates
GSTS (PGDS) catalyzes the isomerization of PGH2 (165, ) to PGD2 (166, ) (Kanaoka et al. Citation1997; Jowsey et al. Citation2001). In addition, human, rat, and mouse PGDS catalyze the conjugation of CDNB (11, ), but not 4-nitrobenzyl chloride (125, ), trans-4-phenyl-3-buten-2-one (53, ), 1,2-epoxy-3-(p-nitrophenoxy)propene (116, ), or ethacrynic acid (52, ) (Christ-Hazelhof et al. Citation1976; Urade et al. Citation1987; Flanagan and Smythe Citation2011). Expressed rat and human PGDS catalyze the conjugation of CDNB (11, ), 1-fluoro-2,4-dinitrobenzene, 1-bromo-2,4-dinitrobenzene, 1-iodo-2,4-dinitrobenzene, allyl isothiocyanate, benzyl isothiocyanate, but not the reduction of tert-butyl hydroperoxide (Jowsey et al. Citation2001; Mazari et al. Citation2015).
2.2.6.7.3. Inhibitors
The putative role of PGD2 (166, ) as a proinflammatory mediator of allergic responses and the multiplicity of disease states in which inflammation is an integral component has stimulated discovery research for PGDS inhibitors (for a review, see Thurairatnam Citation2012).
The isomerization of PGH2 (165, ) to PGD2 (166, ) by rat spleen PGDS is inhibited by CDNB (11, ) and by non-substrate ligands, e.g. bilirubin and indocyanine green (Urade et al. Citation1987). Screening of a selection of 1040 drugs or additives as possible hPGDS inhibitors identified 23 drugs for further study (Mazari et al. Citation2015); the most potent inhibitors were erythrosine sodium, suramin, tannic acid, and sanguinarine sulfate (IC50 = 0.2–0.6 μM).
Several PGDS inhibitors have been reported for which interactions with the H-site have been solved by crystallographic analysis. Tetrazolium 167 () inhibits recombinant hPGDS (IC50 = 36.2 μM in the presence of EDTA and 98.1 μM in the presence of Mg2+) (Inoue et al. Citation2004). Tetrazol 168 () is a potent and selective inhibitor of hPGDS (KiPGH2 = 5 μM) and is effective in animal models of allergic disease and suppresses microglial activation and astrogliosis (Matsushita et al. Citation1998; Aritake et al. Citation2006; Mohri et al. Citation2006; Takahashi et al. Citation2010).
Figure 15. PGDS inhibitors. 167, 2-(2′-benzothiazolyl)-5-styryl-3-(4′-phthalhydrazidyl)tetrazolium chloride (BSPT); 168, 4-benzhydryloxy-1-[3-(1H-tetrazol-5-yl)-propyl]-piperidine (HQL-79); 169, 2-phenyl-5-(1H-pyrazol-3-yl)thiazole; 170, 4-methyl-2-phenyl-N-(4-sulfamoylphenyl)-thiazole-5-carboxamide; 171, N-(5-(1H-indol-4-yl)pyridin-3-yl)-3-acetamidopropanamide; 172, 3-(3-cyclopropyl-1H-pyrazol-4-yl)-5,7-dihydropyrrolo[3,4-b]pyridine-6-carboxylic acid cyclopentylamide; 173, N-(1-amino-1-oxo-3-phenylpropan-2-yl)-6-(thiophen-2-yl)nicotinamide; 174, 6-(3-fluorophenyl)-N-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)nicotinamide; 175, 4-(isoquinolin-1-yl)-N-(2-morpholinoethyl)benzamide; 176, 1-amino-4-[[4-[[4-chloro-6-[(2-sulfophenyl)amino]-1,3,5-triazin-2-yl]amino]-3-sulfophenyl]amino]-9,10-dihydro-9,10-dioxo-2-anthracenesulfonic acid (Cibacron Blue 3G-A); 177, nocodazole; 178, 1-amino-4-(4-aminosulphonyl)-phenyl-anthraquinone-2-sulfonic acid (APAS).
![Figure 15. PGDS inhibitors. 167, 2-(2′-benzothiazolyl)-5-styryl-3-(4′-phthalhydrazidyl)tetrazolium chloride (BSPT); 168, 4-benzhydryloxy-1-[3-(1H-tetrazol-5-yl)-propyl]-piperidine (HQL-79); 169, 2-phenyl-5-(1H-pyrazol-3-yl)thiazole; 170, 4-methyl-2-phenyl-N-(4-sulfamoylphenyl)-thiazole-5-carboxamide; 171, N-(5-(1H-indol-4-yl)pyridin-3-yl)-3-acetamidopropanamide; 172, 3-(3-cyclopropyl-1H-pyrazol-4-yl)-5,7-dihydropyrrolo[3,4-b]pyridine-6-carboxylic acid cyclopentylamide; 173, N-(1-amino-1-oxo-3-phenylpropan-2-yl)-6-(thiophen-2-yl)nicotinamide; 174, 6-(3-fluorophenyl)-N-(1-(2,2,2-trifluoroethyl)piperidin-4-yl)nicotinamide; 175, 4-(isoquinolin-1-yl)-N-(2-morpholinoethyl)benzamide; 176, 1-amino-4-[[4-[[4-chloro-6-[(2-sulfophenyl)amino]-1,3,5-triazin-2-yl]amino]-3-sulfophenyl]amino]-9,10-dihydro-9,10-dioxo-2-anthracenesulfonic acid (Cibacron Blue 3G-A); 177, nocodazole; 178, 1-amino-4-(4-aminosulphonyl)-phenyl-anthraquinone-2-sulfonic acid (APAS).](/cms/asset/5d502d5d-9367-48c6-b31d-07c926fd2d68/itxc_a_1692191_f0015_b.jpg)
Fragment-based drug-design strategies have provided useful leads in the search for PGDS inhibitors. A 2D NMR fragment-based discovery approach to the design of PGDS inhibitors was used to identify thiazole 169 () as a potent inhibitor of hPGDS (IC50 = 21 nM) (Hohwy et al. Citation2008). Phenylsulfonamide 170 (), which contains the same phenylthiazole structural component as compound 169 (), was found to be a potent hPGDS inhibitor (IC50 <0.001 µM) by application of a fragment-based strategy involving a high-concentration bioassay and fluorescence-polarization analysis (Hesterkamp et al. Citation2007). Indole 171 (), which inhibits both hPGDS (IC50 = 0.30 μM) and the synthesis of PGD2 (166, ) in MEG-01 cells (EC50 = 0.35 μM), was identified by fragment-based focused screening of 42 000 compounds (Edfeldt et al. Citation2015). Fragment optimization studies led to the discovery of pyrrolopyridine 172 (), which showed favorable pharmacodynamics and pharmacokinetic properties in a rat model (Saxty et al. Citation2014). A 3-methyl-5-fluoroindole-based compound (cf. 171, ) was found to be a potent inhibitor of PGD2 (IC50 = 7 nM) but was a mechanism-based inhibitor of CYP3A4 (Vaz et al. Citation2018); the 4-trifluoromethylindole analog, which does not contain a metabolically oxidizable 3-methyl group, failed to inactivate CYP3A4 and retained the PGD2 inhibitory potency (IC50 = 8 nM).
Screening of an ensemble of potential hPGDS inhibitors, in which the design of the screened structures was based on the hPGDS binding of fragments of known inhibitors, showed that nicotinamide 173 () is a potent inhibitor of hPGDS (IC50 = 1.2 μM) (Christ et al. Citation2010). Information about the active site of PGDS and about important structural elements of previously reported PGDS inhibitors, along with the identification of new substructural components, led to the development of orally effective inhibitors of PGDS (Carron et al. Citation2010): nicotinamide 174 () is a potent and selective inhibitor (IC50 = 0.5–2.3 nM) and shows favorable pharmacokinetic properties in rats. Examination of crystal structures of PGDS complexes with the potent isoquinoline inhibitor 175 () (IC50 = 2.34 nM) and several analogs indicated that a single primary water molecule, which also interacts with Leu199, forms a critical hydrogen bond with the isoquinoline ring nitrogen of inhibitor 175 (Trujillo et al. Citation2012). Other investigators also have noted the significance of PGDS inhibitor interactions with the conserved water that associates with C-terminal Leu199 (Aritake et al. Citation2006; Hohwy et al. Citation2008; Carron et al. Citation2010; Edfeldt et al. Citation2015). Further studies supported the role of water molecules in the active site of hPGDS in substrate and GSH binding (Takaya et al. Citation2018). Substructure analysis of PGDS inhibitors indicated that a phenyl group, a heterocyclic group, e.g. 173, 174, and 175 (), and interaction with a conserved water molecule in the substrate binding site was important for inhibition of PGDS; this information was used to develop a phenyl pyridinylpyrimidine carboxamide with high binding affinity (KD = 0.14 nM) for hPGDS. Analysis of the crystal structure of the hPGDS complex with the phenypyrimidine carboxamide ligand identified six water molecules, including the water that hydrogen bonds with Leu199 that contribute significantly to binding of the ligand in the active site.
A fragment-based, hit-to-lead search for inhibitors of hPGDS identified 3-carboxamide-7-methoxyquinolines as a starting fragment (Deaton et al. Citation2019); aided by crystallographic analysis of inhibitor-PGDS complexes, a 3-carboxamide-3-difluoromethoxyquinoline hPGDS was found to be a potent hPGDS inhibitor (IC50 = 9.9 nM), decreased PGD2 (166, ) production in a murine mast cell degranulation model of inflammation, possessed favorable pharmacokinetic properties, and showed few apparent off-target liabilities.
Screening of a range of GST inhibitors showed that tetrazole 168 () and Cibacron Blue 3G-A (176, ) were the most potent inhibitors of hPGDS (IC50 = 0.2–1.8 μM) (Weber et al. Citation2010). Characterization of the inhibition of hPGDS by nocodazole (177, ) (IC50 ≈65 μM) by docking calculations indicated binding at the prostaglandin H-site. Kinetic and crystallographic analysis of the interaction of Cibacron Blue 3 G-A (176, ) (Ki = 87.5 nM) and APAS (178, ) (Ki = 2.5 μM) with hPGDS indicated that the former compound bound to the H-site whereas the latter compound bound to the G-site (Kado et al. Citation2012).
2.2.6.7.4. Tissue and species distribution
The expression of PGDS varies significantly among species and organs (Ujihara et al. Citation1988a, Citation1988b; Thomson et al. Citation1998; Jowsey et al. Citation2001; Mohri et al. Citation2006; Shimura et al. Citation2010). Immunoblot analysis shows that rat PGDS expression is highest in spleen and bone marrow. Quantitative RT-PCR shows abundant hPGDS mRNA in macrophages and placenta and, in contrast to the rat, little mRNA in spleen and bone marrow. Also, hPGDS mRNA was moderately abundant in lung, but rPGDS was not detected by immunoblot analysis in lung (Jowsey et al. Citation2001). In the rat, PGDS is found in histiocytes and dendritic cells in spleen, thymus, and intestinal Peyer’s patches, indicating a possible role in immune responses (Urade et al. Citation1989).
2.3. Mitochondrial and peroxisomal glutathione transferases (Kappa-class, GSTK, DsbA-L)5
2.3.1. Structure and properties
Early studies indicated the presence of enzymes in liver mitochondrial fractions that catalyzed GSH S-conjugate formation, although rates of conjugation were typically much lower in mitochondria than in cytosol (Booth et al. Citation1961; Suga et al. Citation1967; Kraus and Gross Citation1979; Wahllander et al. Citation1979). Harris et al. (Citation1991) purified a GST (termed GST 13-13) from rat liver mitochondrial matrix whose amino acid sequence was similar to the Theta-class GSTs; the dimeric enzyme catalyzed the conjugation of CDNB (11, ) and ethacrynic acid (52, ) but lacked activity with a panel of known GST substrates. A cDNA clone that encodes GST 13 subunit was isolated from a rat cDNA library (Pemble et al. Citation1996); a search of a human expressed sequence tag (EST) database showed that the rat and human amino acid sequences were similar (77%) but differed from other known GSTs. The novel GST genes were assigned to a new subclass, Kappa. A mouse liver mitochondrial Kappa-class GST has also been characterized (Jowsey et al. Citation2003). Given its putative role in adiponectin polymerization, GSTK1-1 has also been termed disulfide bond-forming oxidoreductase A-like protein (DsbA-L) (Liu et al. Citation2008). A review of the Kappa-class GSTs has been published (Morel and Aninat Citation2011).
The crystal structure of GSTK1-1 is more similar to bacterial disulfide-bond A oxidoreductase (DsbA) and 2-hydroxychromene-2-carboxylic acid isomerase (HCCA) than to members of the canonical GST superfamily (Ladner et al. Citation2004; Robinson et al. Citation2004; Li et al. Citation2005a; Thompson et al. Citation2007). GSTK subunits show a thioredoxin-like domain that is interrupted with an α-helical domain, whereas the canonical GSH transferases have a contiguous thioredoxin-like domain at the N-terminus. A conserved serine (Ser16) is found as an active-site residue in hGSTK1-1 (Wang et al. Citation2011a). Although the G-sites in GSTK1-1 and in the canonical GST superfamily are similar, both GSTK1-1 and the canonical GSTs fail to catalyze DsbA-type reactions, apparently because they lack the CxxC motif found in DsbA (Atkinson and Babbitt Citation2009; Wang et al. Citation2011a). As with the cytosolic GSTs, however, the thioredoxin fold provides residues that serve to recognize GSH (Ladner et al. Citation2004; Li et al. Citation2005a). The H-site of hGSTK1-1 is a hydrophobic pocket that accommodates S-(hexyl)GSH (Wang et al. Citation2011a).
A single gene, GSTK1, loci located on chromosome band 7q34 encodes human GSTK1 (HGNC: 16906; https://www.genenames.org/; accessed 31 October 2019) (Morel et al. Citation2004). The rat (RGD: 735188; https://rgd.mcw.edu/; accessed 31 October 2019) and mouse (MGI: 1923513; http://www.informatics.jax.org/; accessed 31 October 2019; NCBI: 76263) genes Gsk1 encode r- and mGSTK1, respectively (see also ).
The mitochondrial localization of GSTK1-1 in mouse liver and kidney was established by electron microscopy with immunocolloidal gold staining (Thomson et al. Citation2004). In addition to GSTK1-1, Alpha-, Mu-, Pi-, and Zeta-class GSTs have been identified in mitochondria (Soboll et al. Citation1995; Gallagher et al. Citation2006; Goto et al. Citation2009; Li et al. Citation2011; Raza Citation2011; Sun et al. Citation2012).
The presence of GST-catalyzed GSH S-conjugate formation in peroxisomes was reported (Nishino and Ito Citation1990b). The amino acid sequences of rat and hGSTK1 revealed a peroxisomal targeting sequence (Ala–Arg–Leu) in the C-terminus (Pemble et al. Citation1996; Morel et al. Citation2004); Western blot analysis as well as transfection of HepG2 cells with a green fluorescent protein-hGSTK1 fusion construct and fluorescence analysis clearly demonstrated the presence of GSTK1 in peroxisomes. Western blot analysis showed a lack of hGSTK1 expression in cytosol.
In addition to its localization in mitochondria and peroxisomes, DsbA-L is also found in the endoplasmic reticulum (ER) (Liu et al. Citation2015). DsbA-L contains a highly conserved ER targeting sequence in the N-terminal (residues 1–12). The expression of DsbA-L in the ER is largely blocked with a mutant that lacks residues 1–6, but mitochondrial expression is not altered.
2.3.2. Substrates
The physiological function of GSTK1-1 has not been fully established, but human, rat, and mouse GSTK1-1 catalyze the conjugation of several canonical GST substrates (Jowsey et al. Citation2003; Morel et al. Citation2004; Robinson et al. Citation2004; Morel and Aninat Citation2011), including CDNB (11, ), ethacrynic acid (52, ), tert-butyl hydroperoxide, and cumene hydroperoxide (15, ). Significant species differences in substrate selectivities and in rates of biotransformation were observed: human GSTK1-1, but not rat or mouse GSTK1-1, catalyzes the conjugation of 4-nitrobenzyl chloride (125, ), 4-nitrophenyl acetate, and trans,trans-nona-2,4-dienal (Robinson et al. Citation2004). With CDNB (11, ) as the substrate, the relative rates of biotransformation were: mouse > rat > human. Cumene hydroperoxide (15, ) was a substrate for recombinant GSTK1-1 of all three species and the relative rates were: mouse > human > rat; tert-butyl hydroperoxide was a poor substrate for the three GSTKs.
2.3.3. Inhibitors
S-(2,4-Dintrophenyl)GSH (12, ) inhibits hGSTK1-1 (Wang et al. Citation2011a), but studies with other inhibitors have apparently not been reported.
2.3.4. Tissue and species distribution
Western blot analysis showed that mouse GSTK1 expression is the highest in liver and stomach and is also present in kidney, heart, large intestine, testis, and lung (Thomson et al. Citation2004); mGSTK1 is not expressed in small intestine, brain, spleen, or skeletal muscle. It has also been reported that mGSTK1 mRNA is expressed in all tissues examined (Knight et al. Citation2007); expression was higher in female mice than in male mice in heart, kidney, and lung tissue. Human GSTK1 mRNA is expressed in all tissues examined but at differing levels (Morel et al. Citation2004). mGSTK1 is highly expressed in adipose tissue (Liu et al. Citation2008); its expression in 3T3-L1 adipocytes is decreased by TNFα and increased by the PPARγ agonist rosiglitazone.
Gstk1−/− mice lack immunologically detectable GSTK1-1 but liver and kidney mitochondria show moderately reduced enzymatic activity with CDNB (11, ) as the substrate compared with wild-type mice (Blackburn et al. Citation2011); the canonical GSTs found in mitochondria may account for this activity.
2.3.5. Other biological functions
The expression of DsbA-L is negatively correlated with obesity in mice and human subjects (Liu et al. Citation2008). Moreover, DsbA-L overexpression promotes adiponectin multimerization and silencing of DsbA-L expression by RNAi decreases adiponection multimerization and secretion in 3T3-L1 adipocytes. In Gstk−/− mice, serum concentrations of adiponectin as well as adiponectin multimerization are not altered, thereby questioning an absolute requirement of GSTK1-1 for adiponectin multimerization (Theodoratos et al. Citation2012). Later studies showed, however, that colocalization of DsbA-L in both mitochondria and ER is required for promoting adiponectin multimerization and preventing ER oxidative stress (Liu et al. Citation2015).
The DsbA-L -1308 G>T (rs1917760) sequence variant is not associated with type-2 diabetes and obesity in Chinese subjects, whereas T allele carriers showed favorable insulin secretion function (Gao et al. Citation2009). The -1308 G > T genotype is associated with higher body mass indices (BMI) in Japanese male schizophrenic subjects, whereas female subjects with this genotype exhibited a lower BMI; there was no association between the -1038 T genotype and BMI in control subjects (Oniki et al. Citation2016). Later studies showed that the -1308 T allele modulates hGSTK1 transcription by disrupting the RXR/FXR transcription-factor binding site (Shield et al. Citation2010); a -1032G>C sequence variant alters a CpG methylation site and decreases transcription.
Support for the concept that redox communication between peroxisomes and mitochondria is part of a complex signaling process has been reported (Wang et al. Citation2013); mitochondrial GSTK1-1 may play a role in detoxifying lipid peroxides generated in response to peroxisomal oxidative stress. GSTK1 expression is decreased in five mouse models of hypertrophic cardiomyopathy and the decrease may contribute to hypertrophic cardiomyopathy through increased oxidative stress, lipid peroxidation, and mitochondrial dysfunction (Sasagawa et al. Citation2016).
Male Gstk1−/− mice show shortened life span compared with female Gstk1−/− mice; moreover, both male and female Gstk1−/− mice show glomerulopathies similar to those seen in the human nephrotic syndrome (Blackburn et al. Citation2011). The pathophysiological processes that account for the shortened life span and glomerulopathies have not been elucidated.
2.4. Microsomal glutathione transferases (MGST)
2.4.1. Structure and properties
MGSTs are members of the membrane-associated proteins in eicosanoid and glutathione metabolism (MAPEG) superfamily (Jakobsson et al. Citation1999; Bresell et al. Citation2005; Pettersson et al. Citation2005; Martinez Molina et al. Citation2008). The MAPEG superfamily includes MGST1, MGST2, MGST3, leukotriene-C4 synthase (LTC4S, EC4.4.1.20), arachidonate 5-lipoxygenase-activating protein (FLAP, EC1.13.11.34), and prostaglandin-E synthase (PGE synthase, EC5.3.99.3). All MAPEG superfamily members are GSTs and catalyze GST conjugative and peroxidative reactions. Several reviews about the MGSTs are available (Morgenstern and DePierre Citation1985, Citation1988; Andersson et al. Citation1994a; Morgenstern Citation2005; Morgenstern et al. Citation2011). The genes encoding hMGST1, MGST2, and MGST3 are located on chromosome bands 12p13.1-p13.2 (Kelner et al. Citation2000), 1q23 (Jakobsson et al. Citation1997), and 4q28-31 (Jakobsson et al. Citation1996).
Early studies provided evidence for GST activity in hepatic microsomal fractions (Booth et al. Citation1961; Kraus et al. Citation1973; Kraus Citation1975; Nemoto and Gelboin Citation1975; Glatt and Oesch Citation1977; Friedberg et al. Citation1979; Kraus and Gross Citation1979; Lee and McKinney Citation1982). Morgenstern and coworkers (Morgenstern et al. Citation1979, Citation1982) showed that microsomal GST activity was a property of GSTs localized in microsomal membranes rather than contamination by cytoplasmic GSTs. Moreover, with CDNB (11, ) as the substrate, N-ethylmaleimide (NEM) stimulated microsomal GST activity several fold, but had no effect on cytosolic GST activities.
Purification and sequence analysis indicated that rat liver MGST1 is a homotrimer with subunit masses of 17.2 kDa (Morgenstern et al. Citation1982, Citation1985; Boyer et al. Citation1986). Rat MGST1 contains a single Cys residue (Cys49) in each subunit, which was determined to be the site of activation by sulfhydryl reagents, e.g. NEM (Morgenstern and DePierre Citation1983; Morgenstern et al. Citation1985).
Electron crystallography has provided key structural information about MGST1; a review of the application of electron crystallography to the structural determination of MAPEG proteins, including MGST1, is available (Hebert and Jegerschöld Citation2007). Early structural studies of rat liver MGST1 provided details of its trimeric structure and indicated the presence of two α-helices in each subunit (Hebert et al. Citation1995, Citation1997). A 3D electron crystallographic map of rMGST1 confirmed both its trimeric structure as well as the earlier report that the binding sites for GSH and the second substrate are located on the cytoplasmic surface (Andersson et al. Citation1994b; Holm et al. Citation2002; Hebert et al. Citation2005). A proposed topology, based on a projection map at 6-Å resolution, identified four transmembrane α-helices in each monomer; peptides connecting α-helices 1 to 2 and 3 to 4 are located on the cytoplasmic side, and the N- and C-termini are located on the luminal side (Schmidt-Krey et al. Citation2000; Holm et al. Citation2002).
The electron crystallographic structure of the rGSTM1-GSH complex at 3.2-Å resolution revealed the first structural details about the active site and the cytoplasmic domain responsible for activation by sulfhydryl reagents (Holm et al. Citation2006). MGST1 lacks a thioredoxin fold, indicating that its evolutionary origin differs from that of the cytosolic GSTs. Each subunit of the homotrimer contains four left-handed α-helix bundles. Transmembrane helices 2 and 3 are connected by a short loop on the luminal side, whereas helices 3 and 4 are connected by a Pro-rich loop on the cytosolic side; domain E links helices 1 and 2 and contains the stress sensor Cys49. The monomer appears to be stabilized by interactions between transmembrane helices 2 and 3 involving polar residues near the cytoplasmic side, whereas hydrophobic Phe residues located toward the luminal side have a similar function.
Cryoelectron microscopic studies provided additional details about the structure of rat MGST1 (Kuang et al. Citation2017). rMGST1 is present as a dimer of trimers with phospholipids at the interface between the two trimers. The GSH binding site is located at the interface of transmembrane helices 1 and 2. In contrast to the other members of the MPAEG family, GSH is bound in an extended conformation with a hydrogen bond between the thiol group and Arg130, which is located in transmembrane 4 of an adjacent subunit; the R130A mutant is devoid of activity, indicating the critical role of Arg130 in stabilizing the GSH thiolate. Residues that either contribute, or potentially contribute, to GSH binding reside in both transmembrane helices 1 and 2 of the GSH-binding subunit and in transmembrane helices 2 and 4 of a neighboring subunit of the trimer. His76 and Glu81, which are highly conserved in the MAPEG family, are involved in intermolecular contacts between monomers and also contribute to the binding of GSH. The binding site for lipophilic substrates is located near the hydrocarbon region of the bound phospholipids.
A remarkable feature of MGST1 is activation by agents or treatments that modify Cys49. The NEM-treated enzyme was initially isolated and purified in 1982 (Morgenstern et al. Citation1982). Purified native rMGST1 was activated 15-fold by NEM with CDNB (11, ) as the substrate and 10-fold with cumene hydroperoxide (15, ) as the substrate, whereas no activation was seen with methyl iodide, 4-nitrobenzyl chloride (125, ), 1,2-dichloro-4-nitrobenzene, 4-nitropyridine N-oxide, and hexachlorobutadiene as substrates (Morgenstern and DePierre Citation1983).
Studies on the mechanism of activation of rMGST1 showed that the pKa of Cys49 is similar to low molecular-weight thiols, e.g. GSH, that the reactivity of Cys49 is the same in all subunits, and that subunits react independently (Svensson et al. Citation2000). The ability of p-benzoquinones to activate rMGST1 varies with their putative reactivity and steric properties. Reaction of rMGST1 with NEM showed that modification of a single subunit gave no activation and that activation increased as two or three subunits are modified; full activation requires modification of all three subunits. Later studies showed that one GSH thiolate is formed per homotrimer (Morgenstern et al. Citation2001; Svensson et al. Citation2004).
Kinetic analyses of the binding and activation of GSH by rMGST1 showed that GSH thiolate formation, rather than the chemical reaction of the substrate with the thiolate, is the rate-limiting step for chemically reactive substrates such as CDNB (11, ) (Morgenstern et al. Citation2001; Svensson et al. Citation2004). Binding and ionization of GSH involves a slow conformational transition that results in tight binding of the GSH thiolate. Moreover, the reaction of Cys49 of MGST1 with NEM increases the rate of thiolate formation, indicating that activation is associated with a specific chemical event that facilitates the cooperative conformational transition (Busenlehner et al. Citation2004, Citation2007).
Electrospray mass spectrometric data obtained by Ålander et al. (Ålander et al. Citation2009b) showed, as also indicated by electron crystallographic data (Holm et al. Citation2006; Kuang et al. Citation2017), that each rMGST1 trimer binds three GSH molecules, rather than one as reported earlier (Sun and Morgenstern Citation1997; Lengqvist et al. Citation2004). Equilibrium dialysis studies revealed that there is one high-affinity and two low-affinity sites for GSH and that only the high-affinity site is catalytically competent (Ålander et al. Citation2009b); hence, MGST1 shows one-third-of-sites-reactivity. Kinetic studies that evaluated GSH binding, thiolate anion stabilization, and electrophile binding and reaction demonstrated that unactivated MGST1 exists in a dynamic equilibrium between activated and unactivated forms (Spahiu et al. Citation2017). With the chemically unactivated enzyme and reactive electrophilic substrates, the data indicate that a fraction (∼10%) of the enzyme exists in an activated form. Also, the activated enzyme favors catalytic efficiency over stability.
Cys49 serves as a stress sensor in that activation of rMGST1 may favor detoxication of cellular electrophiles (Busenlehner et al. Citation2004). A diverse group of agents or treatments activate MGST1 (for a review, see Shimoji et al. Citation2007): thiol-reactive agents (Morgenstern and DePierre Citation1983; Aniya and Anders Citation1989b), reactive oxygen and nitrogen species (Aniya and Anders Citation1989a, Citation1992; Imaizumi et al. Citation2006), proteolysis (Morgenstern et al. Citation1989; Nakama et al. Citation2010). Activation of MGST1 by hydrogen peroxide is accompanied by dimerization of the transferase (Aniya and Anders Citation1992).
Identification of hMGST2 was accomplished by a TBLASTN search of Gen Bank with the 5-lipoxygenase-activating protein (FLAP) peptide sequence (Jakobsson et al. Citation1996). Similarly, a TBLASTN search with leukotriene C4 synthase (LTC4S) peptides identified rMGST2 (Schröder et al. Citation2003). Both hMGST2 and rMGST2 catalyze the biotransformation of LTA4 (188, ) to LTC4 (189, ) and the biotransformation of 5-HPETE (190, ) to HETE (191, ) (Jakobsson et al. Citation1996; Schröder et al. Citation2003; Ahmad et al. Citation2013). LTC4S, which also catalyzes the conversion of LTA4 (188, ) to LTC4 (189, ), is a protein distinct from MGST2, as MGST2, but not LTC4S, catalyzes the conjugation of CDNB (11, ). Also, in contrast to MGST1, NEM fails to activate hMGST2 (Jakobsson et al. Citation1996). Kinetic studies show that hMGST2 exhibits one-third-of-sites reactivity and that the enzyme lowers the pKa of GSH (Ahmad et al. Citation2013); with reactive substrates, e.g. CDNB (11, ), the rates of GSH thiolate anion formation and the chemical reaction step are similar, but with substrates of lower reactivity, the chemical conjugation step becomes rate limiting. Other studies established the trimeric structure of hMGST2, showed that each subunit possesses a GSH binding site, and confirmed the one-third-of-sites reactivity (Ahmad et al. Citation2015).
Figure 16. MGST substrates. 179, bis-(2,4-dinitrophenylsulfonyl)rhodamine; 180, 2-(2,4-dinitrophenylsulfonyl)rhodamine; 181, rhodamine; 182, (Z)-N-(9-amino-5H-benzo[a]phenoxazin-5-ylidene)-2,4-dinitrophenylsulfonamide (DNs-CV); 183, crystal violet; 184, trichloroethylene; 185, S-(1,2-dichlorovinyl)GSH; 186, chlorotrifluoroethylene; 187, S-(2-chloro-1,1,2-trifluoroethyl)GSH; 188, leukotriene A4 (LTA4); 189, leukotriene C4 (LTC4); 190, 5-hydroperoxyeicosatetraenoic acid (5-HPETE); 191, 5-hydroxyeicosatetraenoic acid (5-HETE).
![Figure 16. MGST substrates. 179, bis-(2,4-dinitrophenylsulfonyl)rhodamine; 180, 2-(2,4-dinitrophenylsulfonyl)rhodamine; 181, rhodamine; 182, (Z)-N-(9-amino-5H-benzo[a]phenoxazin-5-ylidene)-2,4-dinitrophenylsulfonamide (DNs-CV); 183, crystal violet; 184, trichloroethylene; 185, S-(1,2-dichlorovinyl)GSH; 186, chlorotrifluoroethylene; 187, S-(2-chloro-1,1,2-trifluoroethyl)GSH; 188, leukotriene A4 (LTA4); 189, leukotriene C4 (LTC4); 190, 5-hydroperoxyeicosatetraenoic acid (5-HPETE); 191, 5-hydroxyeicosatetraenoic acid (5-HETE).](/cms/asset/237b4941-5329-44d4-9eab-0706ceadc74d/itxc_a_1692191_f0016_b.jpg)
hMGST3 was identified by a TBLASTN search of GenBank with MGST2 peptide sequences (Jakobsson et al. Citation1997). hMGST3 catalyzes the biotransformation of LTA4 (188, ) to LTC4 (189, ) and the reduction of 5-HPETE (190, ) but fails to catalyze the conjugation of CDNB (11, ) in the absence or presence of NEM. Rat MGST3, in contrast to hMGST3, fails to catalyze LTC4 (189, ) synthesis (Schröder et al. Citation2003).
Most inducers of cytosolic GSTs fail to alter the activity of MGST. Treatment of rats with phenobarbital, 3-methylcholanthrene, and trans-stilbene oxide failed to alter microsomal GST activity but increased cytosolic GST activity (Morgenstern et al. Citation1980). Mice fed 2(3)-tert-butyl-4-hydroxyanisole for two weeks showed a significant increase not only in cytosolic GST activity, but also in both unactivated and NEM-activated MGST activity (Morgenstern and Dock Citation1982). Mice fed peroxisome proliferators show an increase in MGST1 activity (Morgenstern and DePierre Citation1988).
MGST2 and MGST3 are also induced by several treatment regimens (Higgins and Hayes Citation2011). Triggering of the endoplasmic reticulum stress-response by brefeldin A and tunicamycin upregulates MGST2 and, thereby, the synthesis of LTC4 (189, ), which results in reactive oxygen species (ROS) production in non-hematopoietic cells (Dvash et al. Citation2015). 5-Fluorouracil (5-FU) activates the MGST2-LTC4 signaling pathway, thus producing ROS; MGST2 null mice are resistant to 5-FU-induced toxicity, and its toxicity in mice is reduced by the MGST inhibitor pranlukast (193, ).
Figure 17. MGST inhibitors. 192, (2S,3R)-3-(2-carboxyethylsulfanyl)-2-hydroxy-3-[2-(8-phenyloctyl)phenyl]propanoic acid (pobilukast, SKF 104353); 193, N-[4-oxo-2-(2H-tetrazol-5-yl)chromen-8-yl]-4-(4-phenylbutoxy)benzamide (pranlukast, ONO-1078); 194, 4-[(4S,5R,6E,8E,10Z,13Z)-1-carboxy-4-hydroxynonadeca-6,8,10,13-tetraen-5-yl]sulfanylbenzoic acid (BAYu9773); 195, 3-[3-tert-butylsulfanyl-1-[(4-chlorophenyl)methyl]-5-propan-2-ylindol-2-yl]-2,2-dimethylpropanoic acid (MK-886).
![Figure 17. MGST inhibitors. 192, (2S,3R)-3-(2-carboxyethylsulfanyl)-2-hydroxy-3-[2-(8-phenyloctyl)phenyl]propanoic acid (pobilukast, SKF 104353); 193, N-[4-oxo-2-(2H-tetrazol-5-yl)chromen-8-yl]-4-(4-phenylbutoxy)benzamide (pranlukast, ONO-1078); 194, 4-[(4S,5R,6E,8E,10Z,13Z)-1-carboxy-4-hydroxynonadeca-6,8,10,13-tetraen-5-yl]sulfanylbenzoic acid (BAYu9773); 195, 3-[3-tert-butylsulfanyl-1-[(4-chlorophenyl)methyl]-5-propan-2-ylindol-2-yl]-2,2-dimethylpropanoic acid (MK-886).](/cms/asset/ef926589-624a-424f-953e-16342518fcb9/itxc_a_1692191_f0017_b.jpg)
No mutations were found in MGST1 in subjects with chronic pancreatitis (Schneider et al. Citation2004). There are four putative promoters of MGST1 (Kelner et al. Citation2000); both the MGST1b and -1d promoters are active, and two variants of the MGST1b promoter lowered the transcription of the luciferase gene 1.4-fold (Iida et al. Citation2001; Guy et al. Citation2004). Thirteen SNPs were identified in an investigation of the association of MGST1 gene locus polymorphisms and colorectal cancer in Han Chinese populations; a risk of colorectal cancer was suggested for individuals having both of two specific risk alleles (Zhang et al. Citation2007).
Treatment of HepG2 cells with 5-aza-2′-deoxycytidine does not alter MGST1 expression, indicating that DNA methylation does not play a role in MGST1 gene expression (Björkhem-Bergman et al. Citation2014).
2.4.2. Substrates
In many substrate-selectivity studies, the identity of the MGST involved was not determined, but given its high expression in the liver, MGST1 was the likely catalyst. It should be noted, however, that in contrast to MGST1, the activities of MGST2 and MGST3 are not altered by the sulfhydryl reagent NEM (Jakobsson et al. Citation1996, Citation1997).
N-Acetyl-L-cysteine (NAC) can replace GSH with CDNB (11, ) as the second substrate (Weinander et al. Citation1994). The use of NAC rather than GSH allows determination of rates of rMGST1-catalzyed reactions in the presence of cytosolic GSTs. The GSH analogs Glutaryl-l-Cys-Gly and α-l-Glu-l-Cys-Gly also serve as substrates for rMGST1 (Mosialou et al. Citation1993).
rMGST1 substrates include many compounds that are substrates for the cytosolic GSTs, including 1-halo-2,4-dinitrobenzenes (Morgenstern and DePierre Citation1988). In contrast, rMGST1 shows low or no activity with several compounds that are known substrates for the cytosolic GSTs, including bromosulfophthalein, 1,2-epoxy-(3-nitrophenoxy)propane (116, ), ethacrynic acid (52, ), 4-nitropyridine N-oxide, trans-4-phenyl-3-buten-2-one (53, ) (Morgenstern et al. Citation1982; Morgenstern and DePierre Citation1983).
Several compounds that release fluorescent S-(2,4-dinitrophenyl)GSH (12, ) have been reported as GST substrates. The fluorogenic substrates 6-chloroacetyl-2-dimethylaminonaphthalene (95, ) and bis-2,4-dinitrophenylsulfonyl-rhodamine (179, ) are substrates for rMGST1 (and for cytosolic GSTs) (Svensson et al. Citation2002; Ålander et al. Citation2009a).
Zhang et al. (Citation2011a) prepared a series of 4-nitro- and 2,4-dinitrophenylsulfonamide derivatives of coumarin, cresyl violet, and rhodamine (181, ) as fluorescent substrates for rMGST1 and for human cytosolic GSTs. The precursor 4-nitro- or 2,4-dinitrophenylsulfonamides (e.g. 182, ) showed very low fluorescent quantum efficiencies but GSTs catalyzed the release of the coumarin, cresyl violet (183, ), and rhodamine (181, ) moieties that showed high fluorescent quantum efficiencies. The highest specific activities for the rMGST1-catalyzed biotransformation of the fluorogenic substrates were seen with the 2,4-dinitrophenylsulfonamide derivatives of cresyl violet (182, ) and rhodamine; the 4-nitrophenylsulfonamide derivatives were poor substrates. Cell lysates of control MCF7 cells (a cell line with low GST and MGST1 activities) showed low activities with the fluorogenic substrates, whereas much higher rates were seen in lysates from rMGST1-overexpressing cells, and the rates were increased by incubation with NEM. Fluorescence microscopic studies of MCF7 cells showed that DNs-cresyl violet (182, ) allowed imaging of expressed rMGST1 in MCF7 cells.
N-(2,4-Dinitrophenylsulfonyl)doxorubicin (32, ) and the 4-nitrophenylsulfonyl analog were prepared as doxorubicin (33, ) prodrugs (Johansson et al. Citation2011); rMGST1-catalyzed the cleavage of the 2,4-dinitro prodrug (32, ) at a higher rate than the less electrophilic mono-nitro analog, N-(4-nitrophenylsulfonyl)doxorubicin [See also GSTA, (2), Substrates, and GSTP, (2), Substrates]. rMGST1 overexpressing MCF7 cells are resistant to doxorubicin-induced cytotoxicity, and this resistance is partially reversed by prodrug 32 (); N-(4-nitrophenylsulfonyl)doxorubicin completely overcame the resistance but was less cytotoxic than prodrug 32 () to MGST1 overexpressing MCF7 cells. These studies were extended by van Gisbergen et al. (van Gisbergen et al. Citation2016) to include an analog of prodrug 32 () in which the 4-nitro group of the N-(2,4-dinitrophenylsulfonyl) moiety is replaced by the less electronegative acetyl group. The 4-acetyl-2-nitrophenylsulfonyl-doxorubicin prodrug was cleaved by rMGST1 at a much slower rate than prodrug 32 () (38.7 versus 463 nmol/mg/min). Similar to the slowly cleaved 4-nitrophenylsulfonyl-doxorubicin prodrug (32, ) (Johansson et al., Citation2011), the 4-acetyl-2-nitro analog completely reversed MGST1-induced resistance to doxorubicin in MCF7 cells. It was proposed that the less readily cleavable sulfonamide prodrugs of doxorubicin capitalize on the overexpression of MGST1 to achieve a more optimal GST-dependent accumulation of doxorubicin in the target cells than prodrugs that undergo a very rapid GST-catalyzed cleavage.
rMGST1 catalyzes the biotransformation of glyceryl trinitrate at rates greater than those observed with the cytosolic GSTs (Ji and Bennett Citation2006); interestingly, however, treatment with agents that activate rMGST1 activity with CDNB as substrate inhibit the biotransformation of glyceryl trinitrate.
Many haloalkenes are substrates for rMGST1 (McLellan et al. Citation1989; Hargus et al. Citation1991; Jolivette and Anders Citation2002); most in vitro investigations of the rMGST1-catalyzed conjugation of haloalkenes, have, however, used hepatic or renal or both microsomal fractions rather than the purified enzyme (for reviews, see Dekant et al. Citation1993; Dekant et al. Citation1994; Anders and Dekant Citation1998). Most chloroalkenes undergo addition–elimination reactions that yield S-(halovinyl)GSH conjugates; trichloroethylene (184, ) and tetrachloroethylene are biotransformed to S-(1,2-dichlorovinyl)GSH (185, ) and S-(trichlorovinyl)GSH, respectively (Dekant et al. Citation1987, Citation1990). Dichloroacetylene undergoes an addition reaction with GSH to give S-(1,2-dichlorovinyl)GSH (185, ) (Kanhai et al. Citation1989). Fluoroalkenes typically undergo addition reactions that give S-(fluoroalkyl)GSH conjugates; for example, 2-chloro-1,1,2-trifluoroethylene (186, ) and tetrafluoroethylene are biotransformed to S-(2-chloro-1,1,2-trifluoroethyl)GSH (187, ) and S-(tetrafluoroethyl)GSH, respectively (Odum and Green Citation1984; Dohn et al. Citation1985). The sevoflurane degradation product 2-(fluoromethoxy)-1,1,3,3,3-pentafluoro-1-propene undergoes both addition and addition–elimination reactions (Altuntas and Kharasch Citation2001).
rMGST1 shows significant peroxidase activity with organic hydroperoxides as substrates: examples include cumene hydroperoxide (15, ), linoleic acid hydroperoxide, linoleic acid ethyl ester hydroperoxide, dilinoleoylphosphatidylcholine hydroperoxide, and cholesteryl linoleate hydroperoxide (Morgenstern and DePierre Citation1983; Mosialou and Morgenstern Citation1989; Mosialou et al. Citation1995).
hMGST2 shows high activity with CDNB (11, ) but much lower specific activities with other substrates, including leukotriene A4 (188, ), EPNP (116, ), several lipid hydroperoxides (e.g. 5-hydroperoxyeicosatetraenoic acid; 190, ), HNE (17, ), 4-chloro-3-nitrobenzaldehyde, 4-chloro-3-nitrobenzamide, and 4-chloro-3-nitrobenzacetophenone (Ahmad et al. Citation2013). HEK293 cells that highly express hMGST2 are resistant to the cytotoxicity of busulfan (21, ), indicating that hMGST2 catalyzes the conjugation of busulfan (21, ) (Harkey et al. Citation2005).
hMGST3 catalyzes the reduction of 5-HPETE (190, ) but CDNB (11, ) is not a substrate (Jakobsson et al. Citation1997).
2.4.3. Inhibitors
Rose bengal, tributyltin acetate, S-hexylglutathione, indomethacin, cibacron blue, hematin, cholic acid, and bromosulphophtalein inhibit MGST1 (Andersson et al. Citation1988; McLellan et al. Citation1989; Mosialou and Morgenstern Citation1990). Ethacrynic acid (52, ) and ethacraplatin (51, ) inhibit MGST1 and reverse cisplatin resistance in rMGST1 overexpressing MCF7 cells (Johansson et al. Citation2011). The GSH S-conjugate of CDNB (12, ) inhibits rMGST1 (Ålander et al. Citation2009b).
Leukotriene C4 (189, ) is a potent (Ki = 5.4 nM), tight-binding inhibitor of rMGST1 (Bannenberg et al. Citation1999); the cysteinyl-leukotriene analogs pobilukast (192, ), pranlukast (193, ), and BAYu9773 (194, ) are also strong inhibitors of rMGST1.
The rat mitochondrial-membrane-bound MGST1 (mtMGST1) is present in both the outer and inner mitochondrial membranes but only the inner mtMGST1 is inhibited by the mitochondrial permeability transition inhibitors, e.g. cyclosporin A and bongkrekic acid (Ulziikhishig et al. Citation2010).
The FLAP inhibitors MK-886 (195, ) and LTC4 (189, ) inhibit the rat MGST2-catalyed biotransformation of LTA4 (188, ), 5-HPETE (190, ), and CDNB (11, ) (Schröder et al. Citation2003). Similarly, MK-886 (195, ) and LTC4 (189, ) inhibit the rat MGST3-catalyed biotransformation of 5-HPETE (190, ) (Schröder et al. Citation2003).
2.4.4. Tissue and species distribution
MGST1 is an abundant enzyme and accounts for 3% of the endoplasmic reticulum protein and 5% of the outer mitochondrial membrane protein (Morgenstern Citation2005). MGST1 activity is widely distributed in rat tissues, but the highest activity is found in liver and testes, followed by intestine and adrenal (DePierre and Morgenstern Citation1983; Morgenstern et al. Citation1984); activation of rMGST1 by NEM varies widely among tissues with about a fivefold activation seen in liver and little activation in extrahepatic tissues. rMGST1 is also found in the inner and outer mitochondrial membranes (Morgenstern et al. Citation1984; Nishino and Ito Citation1990a; Ulziikhishig et al. Citation2010). In the human small intestinal tract, immunohistochemical analysis shows that MGST1 is found only in the crypts (Hayes et al. Citation1989). Immunoblotting studies confirm the widespread distribution of MGST1 in rat tissues (Otieno et al. Citation1997); in liver tissue, staining was more intense in centrilobular regions than in periportal regions. Mouse MGST1 mRNA expression is highest in liver tissue (Kelner et al. Citation2004; Knight et al. Citation2007; Fu et al. Citation2016). In human fetal tissue, the highest level of MGST1 mRNA is found in the adrenal gland (Björkhem-Bergman et al. Citation2014). Neither MGST1 mRNA nor MGST1 protein is detectable in human neuroblastoma cell lines or primary tumor tissue (Kelner et al. Citation2014).
MGST1 is strongly expressed in mouse hematopoietic stem and progenitor cells (HSPC) (Bräutigam et al. Citation2018). Treatment of HSPC cells with MGST1 shRNA significantly reduced MGST1 expression but failed to decrease cell viability.
MGST1 is overexpressed in many cancers that are associated with increased metastatic potentials and has been proposed as a biomarker for clinical staging of tumors; expression of MGST1 is associated with drug resistance in cancer chemotherapy (Morgenstern et al. Citation2011).
Northern blot analysis showed that hMGST2 is expressed in heart, liver, skeletal muscle, pancreas, spleen, prostate, testes, ovary, small intestine, colon, adrenal medulla and cortex, thyroid, bone marrow, mononuclear, and polymorphonuclear peripheral blood cells, and in promyelocytic and myelogenous leukemia cell lines, but not in kidney; hMGST2 is, however, expressed in fetal kidney and liver (Jakobsson et al. Citation1996).
MGST3 mRNA is widely expressed in human tissues, including heart, skeletal muscle, adrenal cortex, brain, placenta, liver, kidney, intestinal tract, bone marrow, and glandular tissues (Jakobsson et al. Citation1997); limited expression was found in lung, thymus, and leukocytes. Rat MGST3 mRNA expression in the brain is broadly distributed and is found only in neurons, although the possibility of expression in other brain cell types could not be excluded; expression was high in the hippocampus and nuclei of cranial nerves, as well as in the spinal cord, and dorsal root ganglia (Fetissov et al. Citation2002).
The administration of LPS failed to alter rMGST3 expression, indicating the lack of a role in the biosynthesis of proinflammatory eicosanoids.
2.4.5. Other biological functions
The MGST1 found in rat liver mitochondrial membranes, i.e. mtMGST1, appears to play a role in apoptosis (Lee et al. Citation2008); modification of mtMGST1 thiol groups by S-glutathionylation or dimer formation leads to cytochrome c release and opening of the mitochondrial permeability transition (MPT) pores. Incubation of mitochondria with peroxynitrite caused activation of MGST1 in the inner mitochondrial membrane and the production of a high molecular-weight protein complex consisting of disulfide-linked mtMGST1, adenine nucleotide translocator, and cyclophillin D; the newly formed protein complex is proposed to function as an MPT pore (Aniya and Imaizumi Citation2011; Imaizumi and Aniya Citation2011). Earlier studies showed that gallic acid-induced oxidative stress activates mtMGST1 and causes mitochondrial swelling and cytochrome c release, which is blocked by GST inhibitors but not by MPT inhibitors, e.g. cyclosporine A (Hossain et al. Citation2009); these findings indicate a role for mtMGST1 in regulating MPT.
MGST1 and mtMGST1 play a role in protecting cells against oxidative-stress-induced cytotoxicity by catalyzing the reduction of hydroperoxides (Maeda et al. Citation2005; Siritantikorn et al. Citation2007; Johansson et al. Citation2010; Aniya and Imaizumi Citation2011; Schaffert Citation2011).
Deletion of Mgst1, in contrast to deletion of cytosolic GSTs, is embryonic lethal in mice (Bräutigam et al. Citation2018). (Mgst2-deficient mice breed normally and are indistinguishable from wild-type mice (Dvash et al. Citation2015).) MGST1 is highly expressed in hematopoietic stem and progenitor cells (HPSC). In HPSC from wild-type mice, transfection with MGST1 shRNA suppresses the expression of MGST1 and the formation of colony-forming units, decreases mitochondrial activity, and increases the expression of genes associated with glycolysis, HIF1 signaling, and NFκB. The authors conclude that impaired hematopoiesis plays a role in the embryonic lethality of Mgst1-null mice.
3. γ-Glutamyltransferase (GGT)6
3.1. Introduction
Mammalian GGTs catalyze the hydrolysis and transfer of the γ-Glu moiety of GSH, GSH S-conjugates, and γ-Glu amides to amino acid or peptide acceptors or to water (for reviews, see Taniguchi and Ikeda Citation1998; Keillor et al. Citation2005; Cooper and Hanigan Citation2010; Castellano and Merlino Citation2012; Castellano and Merlino Citation2013). Although 13 human genes encode GGT sequences, only GGT1 and GGT5 encode catalytically active proteins (Heisterkamp et al. Citation2008); the genes encoding GGT1 and GGT5 map to chromosome 22q11.23 (NCBI Gene ID: 2678 and 2687). Rodents have orthologs of only four of the human GGT genes, including GGT1, GGT5, GGT6, and GGT7 (Heisterkamp et al. Citation2008).
GGTs catalyze three reactions: transpeptidation, autotranspeptidation, and hydrolysis () (Tate and Meister Citation1981). The terms “transpeptidation” and “γ-glutamyl transpeptidase” were first used by Fruton and coworkers (Fruton Citation1950; Fruton et al. Citation1951) and by Hanes et al. (Citation1950, Citation1952). Interestingly, Bergmann and Fraenkel-Conrat (Citation1937) had earlier proposed that proteolytic enzymes could also catalyze transamidation reactions leading to peptide synthesis. Hanes et al. (Citation1952) clearly demonstrated the transfer of the γ-glutamyl moiety of GSH to acceptor amino acids.
Figure 18. γ-Glutamyltransferase-catalyzed transpeptidation, autotranspeptidation, and hydrolysis reactions.
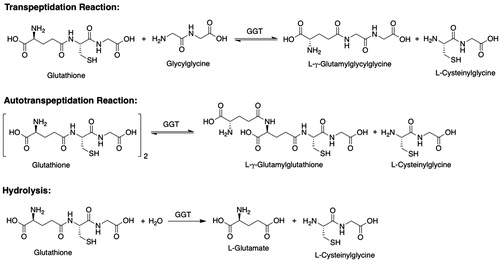
3.2. γ-Glutamyltransferase 1 (GGT1; EC 2.3.2.2) (for GGT5, see Section 3.3 γ-Glutamyltransferase 5 below)
3.2.1. Structure and properties
Mammalian GGT1 is assigned to peptidase Family T3 (MEROPS ID T03.006) (Rawlings et al. Citation2018). GGT2 is an inactive human form assigned to peptidase Family T3 (MEROPS ID T03.015); GGT2 encodes a propeptide that fails to undergo autocleavage into heterodimers (West et al. Citation2013b).
GGT1 is a heterodimeric, glycosylated protein that catalyzes the biotransformation of γ-Glu compounds. GGT1 is a type II transmembrane protein and a member of the N-terminal nucleophile aminohydrolases (Ntn-hydrolase) superfamily (Brannigan et al. Citation1995; Inoue et al. Citation2000; Oinonen and Rouvinen Citation2000; Kinlough et al. Citation2005).
Translation of human GGT1 affords a propeptide (569 amino acids) that undergoes autocatalytic cleavage to give large (amino acids 1–380, 42 kDa) and small (amino acids 381-569, 20 kDa) subunits (Castonguay et al. Citation2007; Heisterkamp et al. Citation2008; West et al. Citation2011; Pica et al. Citation2016). Human GGT1 is anchored to the extracellular face of the plasma membrane by a N-terminal domain (amino acids 5–26 in hGGT1) and a cytoplasmic domain of four polar amino acids (Kinlough et al. Citation2005; West et al. Citation2013a).
The active-site nucleophile responsible for the autocatalytic cleavage of the GGT propeptide and substrate γ-Glu compounds has been identified as a Thr residue (Inoue et al. Citation2000; Kinlough et al. Citation2005). In human GGT1, the active-site nucleophile was identified as Thr381 by use of Hiratake’s phosphonate inhibitor (226, ; see Section 3.2.4, Inhibitors below) and mass spectrometric analysis as well as crystallographic analysis (Han et al. Citation2006; Castonguay et al. Citation2007; West et al. Citation2013a). In the rat, autocatalytic cleavage of the propeptide occurs at Thr380 (Kinlough et al. Citation2005). Rat and mouse GGT1 are the products of a single gene, Ggt1 (Rajagopalan et al. Citation1990; Shi et al. Citation1995; Chikhi et al. Citation1999).
Catalytically competent GGT1 is a membrane-bound heterodimer (Tate and Meister Citation1976). With hGGT1, N-glycosylation is required for maturation of the propeptide. The propeptide may be converted into two subunits either before migration to the cell surface (Nash and Tate Citation1982) or after transport to the brush-border membrane (Kuno et al. Citation1983). Rat GGT propeptide undergoes glycosylation in the Golgi apparatus (Barouki et al. Citation1984). Human GGT1 contains seven glycosylation sites, six in the large subunit and one in the small subunit (Castonguay et al. Citation2007; West et al. Citation2011). The crystal structure of hGGT1 revealed posttranslational modifications, e.g. N-glycosylation sites, disulfide bonds, and active-site configurations, that differ between prokaryotic GGT and hGGT1 (West et al. Citation2013a). Rat GGT1 also contains six potential glycosylation sites in the large subunit and one in the small subunit (Sakamuro et al. Citation1988; Goodspeed et al. Citation1989). Differing degrees of glycosylation may account for the reported range of molecular masses among preparations and species (Goodspeed et al. Citation1989).
Rat renal GGT1 is anchored to the extracellular side of the brush-border membrane by the glycosylated large subunit (Horiuchi et al. Citation1978). The large subunit of rat renal GGT1 contains a membrane-spanning domain with a hydrophobic sequence near the amino terminus, which resides on the cytoplasmic side of the membrane (Tate and Nash Citation1987); the protein lacks a cleavable signal sequence, and translocation and insertion are dependent on a signal recognition particle. The Cys residues in rat GGT1 do not play a role in linking the two subunits (Kinlough et al. Citation2005).
The crystal structure of the soluble ectodomain of deglycosylated, Glu-bound hGGT1 confirms its heterodimeric (small subunit, residues 28–380; large subunit, residues 381–569) structure (West et al. Citation2013a). Access to the active site is located on the small subunit with the active-site Thr381 located deep in the substrate binding site. The “lid loop” that guards the active site in prokaryotic GGT is positioned differently in hGGT1 and allows substrate access. Chloride ions bound near Thr381 may contribute to catalysis. Further studies show that the structures of apo- and Glu-bound hGGT1 are similar (Terzyan et al. Citation2015). These studies show that the product Glu forms hydrogen bonds with the hydroxyl oxygen of Thr381 and with the main-chain Gly474. Although small-subunit residues comprise most of the ligand-binding residues in the active site, a conserved large-subunit residue, Arg107, forms a hydrogen bond with the 5-carboxylate of l-Glu in the hGGT1–Glu complex.
3.2.2. Reactions catalyzed and substrates
GGT1 catalyzes three types of reactions: one, a transpeptidation reaction in which the γ-Glu group of γ-Glu amides is transferred to amino-acid or peptide acceptors; two, an autotranspeptidation reaction in which the γ-Glu group of GSH is transferred to GSH or to GSH S-conjugates; and, three, hydrolysis of γ-Glu amides in which water serves as the acceptor resulting in the formation of Glu ().
The mechanism of γ-Glu transfer or hydrolytic reactions catalyzed by GGT1 is formally similar to the mechanism of the autocleavage of the propeptide that gives rise to the large and small subunits. The active-site Thr (Thr381 in hGGT1) attacks the γ-Glu amide carbonyl of the donor substrate to give a reversible, unstable tetrahedral adduct that converts to an acyl-enzyme intermediate; the acyl-enzyme reacts with water to give L-Glu or with an amino acid or peptide acceptor substrate to give a l-γ-Glu amino acid or peptide (). Crystallographic studies with hGGT1 demonstrate that the Ser-borate complex in the active site of hGGT1 mimics the tetrahedral intermediate formed in the catalytic cycle of hGGT1 (Terzyan et al. Citation2015).
The concept that an acyl-enzyme intermediate is involved in the catalytic cycle of mammalian GGT1 was first proposed by Tate and Meister (1974b, Citation1974a). Support for this concept was provided by the finding that incubation of [glu-14C]GSH and [gly-3H]GSH with rat kidney GGT1 led to greater binding of 14C to GGT1 than of 3H, indicating the formation of an acyl-enzyme intermediate (Elce and Broxmeyer Citation1976). Kinetic studies are supportive of this proposed mechanism (Allison Citation1985). An X-ray crystal structure of the γ-glutamyl-enzyme intermediate formed by E. coli GGT, which contains active-site binding residues and a nucleophilic Thr that are conserved in rat and human GGTs, is available (Okada et al. Citation2006). Kinetic studies with hGGT indicate that the acylation reaction is rate limiting (Castonguay et al. Citation2007).
3.2.2.1. Transpeptidation
Substrate selectivity studies with rat kidney GGT1 show that recognition of the α-amino group and the pKa of the free proton of the γ-peptide bond are critical determinants of donor substrate selectivity (Cook et al. Citation1987). Both l- and d-γ-Glu compounds are substrates (Tate and Meister Citation1985); alkyl groups in the side-chain of the γ-Glu moiety are either poorly tolerated or not tolerated (Griffith and Meister Citation1977), and replacement of the 4-methylene of the γ-Glu moiety with heteroatoms yields inactive donor substrates (Lherbet et al. Citation2003). Moreover, γ-Glu-4-nitrophenyl ester, the ester analog of the widely used assay substrate γ-Glu-4-nitroanilide (196, ), is not a substrate (Castonguay et al. Citation2002).
Figure 19. GGT substrates: 196, γ-Glu-4-nitroanilide; 197, 4-nitroaniline; 198, S-(4-nitrobenzyl)-l-Cys-Gly; 199, S-(4-methylbiphenylyl)GSH; 200, S-(4-methylbiphenylyl)-l-Cys-Gly; 201, clofibryl-S-acyl-GSH (l-γ-Glu-S-[2-(4-chlorophenoxy)-2-methyl-1-oxopropyl]-l-Cys-Gly); 202, clofibryl-S-acyl-l-Cys-Gly (S-[2-(4-chlorophenoxy)-2-methyl-1-oxopropyl]-l-Cys-Gly); 203, 8,9-dihydro-8-(S-glutathionyl)-9-hydroxyaflatoxin B1; 204, 8,9-dihydro-8-(S-l-Cys-Gly)-9-hydroxyaflatoxin B1; 205, l-γ-Glu-S-[(7R,8S,9S,10S)-7,8,9,10-tetrahydro-7,8,9-trihydroxybenzo[a]pyren-10-yl]-l-Cys-Gly ((-)-anti-BPD GSH conjugate); 206, S-[(7R,8S,9S,10S)-7,8,9,10-tetrahydro-7,8,9-trihydroxybenzo[a]pyren-10-yl]-l-Cys-Gly ((−)-anti-BPD l-Cys-Gly conjugate); 207, l-γ-Glu-S-[2-hydroxy-1-(2-oxoethyl)heptyl]- l-Cys-Gly (4-HNE GSH conjugate); 208, S-[2-hydroxy-1-(2-oxoethyl)heptyl]-l-Cys-Gly (4-HNE l-Cys-Gly conjugate); 209, γ-Glu-d,l2-(phenylthiol)Gly-Gly; 210, 2-(phenylthio)Gly-Gly ((2-amino-2-(phenylthio)acetyl)glycine); 211, l-γ-Glu-S-[2-[[4-(dihydroxyarsino)phenyl]amino]-2-oxoethyl]-l-Cys-Gly (4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid; GSAO); 212, S-[2-[[4-(dihydroxyarsino)phenyl]amino]-2-oxoethyl]-l-Cys-Gly (4-(S-cysteinylacetyl)amino)phenylarsonous acid); 213, N-(γ-l-Glu)cysteamine; 214, cysteamine; 215, N5-(6-(benzo[d]thiazol-2-yl)naphthalen-2-yl)Gln; 216, 6-benzo[d]thiazol-2′-yl)-2-aminonaphthalene.
An investigation of the rates of transpeptidation of a series of donor GSH S-conjugates of aromatic compounds showed that the catalytic efficiency varied by a factor of four with bovine kidney GGT (Agblor and Josephy Citation2013). The GSH analog ophthalmic acid (l-γ-Glu-l-α-aminobutyryl-Gly) and several S-substituted GSH analogs also serve as donor substrates for rat kidney GGT (Tate and Meister Citation1974a); GSSG and γ-(dl-α-methylglutamyl)-l-Cys-Gly are poor donor substrates with Met as the acceptor.
Several amino acids and peptides serve as acceptor substrates for rat kidney GGT (for a compilation of donor and acceptor substrates, see Tate and Meister Citation1974a). With l-γ-Glu-4-nitroanilide (196, ) as the γ-Glu donor, l-cystine, l-Gln, and l-Met are the best amino-acid acceptors and Gly–Gly is the best peptide acceptor substrate (Tate and Meister Citation1974a; Thompson and Meister Citation1977). Rat GGT shows high selectivity for l-configuration amino acids or peptides as acceptor substrates but accepts donor substrates with both the l- and l-configuration (Tate and Meister Citation1974a; Thompson and Meister Citation1976, Citation1977).
Meister and coworkers (Orlowski and Meister Citation1970; Meister Citation1973) proposed a role for GGT in amino-acid transport: GGT could serve as a recognition site for amino acids, which could react with GGT-bound Glu; the γ-Glu amino acids would then be translocated into the cell. This proposal was controversial (Bell et al. Citation1987; Sastre et al. Citation1991; Cotgreave and Schuppe-Koistinen Citation1994), and current evidence does not support a role for GGT in amino-acid transport (Inoue Citation2016).
3.2.2.2. Autotranspeptidation
GSH and GSH S-conjugates serve as both donor and acceptor substrates; hence, the term autotranspeptidation. GGT-Catalyzed autotranspeptidation of GSH appears to be a minor reaction, particularly in the presence of alternative acceptor substrates (Elce and Broxmeyer Citation1976; Abbott et al. Citation1986). GGT catalyzes the autotranspeptidation of S-(methyl)GSH to γ-Glu-S-(methyl)GSH, albeit at a low rate (Tate and Meister Citation1974b). γ-Glu-GSH is formed in the isolated rat kidney perfused with GSH (Abbott et al. Citation1986). Although most information about autotranspeptidation reactions has been derived from in vitro studies, autotranspeptidation does occur in vivo: γ-Glu-GSH has been identified in the urine of human subjects with hereditary GGT deficiency and in the urine of mice given a GGT inhibitor (Abbott et al. Citation1986; Oe et al. Citation1998). In addition to 3-(glutathion-S-yl)acetaminophen, a known metabolite of acetaminophen, 3-(γ-Glu-glutathion-S-yl)acetaminophen and 3-(γ-Glu-γ-Glu-glutathion-S-yl)acetaminophen have been identified as minor biliary metabolites of acetaminophen in rats (Mutlib et al. Citation2000). (γ-Glu)n-4-nitroanilide (n = 2, 3, and 4) has been identified in in vitro studies with GGT (Orlowski and Meister Citation1965); these metabolites are presumably formed by an autotranspeptidation reaction. With rat kidney GGT, autotranspeptidation is not observed with d-γ-Glu-4-nitroanilide as the substrate or when the substrate (l-γ-Glu-4-nitroanilide, 196, ) concentration is <10 μM (Thompson and Meister Citation1976).
3.2.2.3. Hydrolysis
The hydrolysis of γ-Glu compounds is typically observed along with transpeptidation and autotranspeptidation. The relative rates of rat renal GGT-catalyzed transpeptidation and hydrolysis are dependent on the concentration of the acceptor (Elce and Broxmeyer Citation1976). With GSH (50 μM) as the donor substrate and Ala (0.5 mM) as the acceptor substrate, rat kidney GGT-catalyzed hydrolysis is greater than transpeptidation from pH 6.0 to 9.0 but decreases above pH 8.5 (McIntyre and Curthoys Citation1979); the rate of transpeptidation increases from pH 6.5 to 9.0. Other studies with l-Glu-4-nitroanilide (196, ) as the donor substrate and a mixture of amino acids (3.11 mM) as acceptor substrates show that at pH 7.5, the rates of rat kidney GGT-catalyzed transpeptidation and hydrolysis are similar (Allison and Meister Citation1981); with GSH (50 μM) as the donor substrate and Ala (0.5 mM) as the acceptor substrate (pH 8.0), approximately 12% of the GSH undergoes transpeptidation. d-γ-Glu-4-nitroanilide undergoes hydrolysis, but not autotranspeptidation; at high substrate concentrations (>10 μM l-γ-Glu-4-nitroanilide, 196, ) autotranspeptidation predominates, but at lower concentrations, only hydrolysis occurs in the absence of acceptors (Thompson and Meister Citation1976). With rat kidney GGT, the relative contributions of transpeptidation, autotranspeptidation, and hydrolysis at saturating donor (l-γ-Glu-4-nitroanilide, 196, ) and acceptor substrate concentrations (Gly–Gly) are 77–80%, 19–22%, and 0.5–1%, respectively; in the absence of an acceptor substrate, the relative contributions of autotranspeptidation and hydrolysis are 96–98% and 2–4%, respectively (Dvoráková et al. Citation1996). The conclusion to be drawn from the above-mentioned studies is that the relative rates of hydrolysis and transpeptidation are highly dependent on reaction conditions but that both reactions occur in vivo.
3.2.3. Substrates
GSH S-conjugate formation is the first step in the mercapturic acid pathway and yields GSH S-conjugates that may serve as substrates for GGT. Although many GSH S-conjugates are biotransformed to mercapturic acids and are, therefore, implicitly biotransformed by GGT, few systematic structure–activity studies of GSH S-conjugates as donor substrates for GGT1 are available. It is well established, however, that a range of structurally diverse GSH S-conjugates are biotransformed by GGT1 (Moss et al. Citation1985; Srivastava et al. Citation1999a; Grillo and Benet Citation2001; Enoiu et al. Citation2003; Agblor and Josephy Citation2013; Grillo et al. Citation2013). A selected representation of that structural diversity includes S-(4-nitrobenzyl)GSH (126, ), and structures 199, 201, 203, 205, and 207 (). Several aza- and oxaglutamyl-4-nitroanilides have been tested as substrates but all are poorer substrates than γ-Glu-4-nitroanilide (196, ) (Lherbet et al. Citation2003).
Studies on GGT-catalyzed hydrolytic reactions are confounded by concurrent transpeptidation reactions. To circumvent this problem and to develop an assay suitable for kinetic analyses of potential GGT inhibitors, the donor substrate γ-Glu-d,l-2-(phenylthiol)Gly–Gly (209, ), which, in the absence of an acceptor substrate, only undergoes a GGT-catalyzed hydrolytic reaction to give 2-(phenylthio)Gly–Gly (210, ) was prepared (Vergauwen et al. Citation2009); the 2-(phenylthio)Gly–Gly (210, ) formed undergoes chemical hydrolysis to give thiophenol, which can be detected by reaction with Ellman’s reagent. The kinetic constants for the hydrolysis of the GSH mimic (209, ) by equine kidney GGT are similar to those of the hydrolysis of GSH.
There is historical and contemporary interest in exploiting the substrate selectivity of GGT to catalyze the bioactivation of prodrugs (Magnan et al. Citation1982; Vamvakas and Anders Citation1994; Keren and Stark Citation1998; Ramsay and Dilda Citation2014). Several studies on GGT-activated prodrugs have focused on chemotherapeutic arsenicals. The trivalent arsenical prodrug 4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid (GSAO, 211, ) was prepared as a potential angiogenesis inhibitor and was shown to inhibit tumor growth in mice (Don et al. Citation2003); the pentavalent arsenic analog was inactive. GSAO (211, ) is biotransformed by GGT to give Cys-Gly S-conjugate 212 (), which is transported into cells by OATP, where it may be converted to the Cys S-conjugate by dipeptidases (Dilda et al. Citation2008). A phase 1 trial (clinicaltrials.gov, NCT01147029) of GSAO (211, ) indicated that the drug was well tolerated (Horsley et al. Citation2013). The arsenical prodrug S-(dimethylarsino)glutathione (darinaparsin) is biotransformed by GGT and dipeptidases to S-(dimethylarsino)-l-Cys, which is transported into cells by a cysteine/cystine transporter (Garnier et al. Citation2014).
Cystinosis is a rare autosomal recessive disorder in which cystine accumulates in lysosomes. Although cysteamine (214, ) is used for the clinical management of cystinosis, its adverse effects limit patient compliance. Hence, the cysteamine (214, ) prodrug N-(γ-l-Glu)cysteamine (213, ) was prepared to attempt to overcome these limitations (Anderson et al. Citation2006); prodrug 213 () and several of its analogs reduce intracellular cystine concentrations in cultured cystinotic cells. The S-acetyl, ethyl ester analog of prodrug 213 () and four structurally related prodrugs are biotransformed to cysteamine (214, ) in human pT37 cells (Frost et al. Citation2016).
There has been considerable interest in developing substrates that allow visualization of GGT activity in cells and in intact mice (for a review, see Luo et al. Citation2018a). The capability of GGT to utilize N5-Substituted l-glutamines, e.g. γ-Glu-4-nitroanilide (196, ) as substrates has been exploited in the design of a variety of GGT-activated probes. Examples of three classes of imaging probes, most of which are γ-Glu amides that are activated by GGT, have been reported: (1) probes that release a fluorescent product or precursor (Iwatate et al. Citation2018; Li et al. Citation2018; Ou-Yang et al. Citation2018; Liu et al. Citation2018a; Luo et al. Citation2018b); (2) probes that release a ratiometric fluorescent product (Liu et al. Citation2018b); and (3) probes that release bioluminescent products (Hai et al. Citation2017). GGT-activated fluorescent probes for imaging intracellular GGT that are suitable for one-photon confocal microscopy have been described but technical issues limit their applicability (Wang et al. Citation2017b, and references cited therein). N5-(6-(Benzo[d]thiazol-2-yl)naphthalen-2-yl)glutamine (215, ), which is biotransformed by GGT to 2-aminonaphthalene 216 (), has been developed as a probe suitable for two-photon microscopy (Wang et al. Citation2017b). γ-N-Substituted Gln 215 () was successfully used to image GGT in HepG2 cells, in normal rat liver slices, and in mouse liver cancer tissue slices.
3.2.4. Inhibitors
Inhibitors have played a central role in elucidating the function of GGT in GSH metabolism, in amino-acid transport, and in drug resistance in tumor cells and are of interest as potential therapeutic agents (Ahluwalia et al. Citation1990; Hanigan Citation2014). The concept that GSH plays a role in tumor resistance to therapy and that GGT may serve to provide l-Cys for GSH synthesis has led to a search for GGT inhibitors with lowered toxicity (Pompella et al. Citation2006; Corti et al. Citation2010; Wickham et al. Citation2013). GGT is, however, variably expressed in tumor tissue: for example, GGT expression is not observed in basal cell carcinomas, Hodgkin’s disease, mesotheliomas, melanomas, non-Hodgkin’s lymphomas, and most soft tissue sarcomas, but epithelium-derived carcinomas retain their GGT-positive phenotype (Hanigan et al. Citation1999).
3.2.4.1. Reversible inhibitors
Early studies showed that the GGT-catalyzed biotransformation of GSH was lower in the presence of l-Ser and borate buffer than in the presence of l-Ser and Tris or Veronal (barbital) buffers (Revel and Ball Citation1959). Other work showed that l-Ser and borate was a competitive inhibitor of GGT (Szewczuk and Connell Citation1965). These studies were extended and showed that both d- and l-Ser in the presence of borate inhibited the transpeptidation of l-γ-Glu-4-nitroanilide (196, ) (Tate and Meister Citation1978); borate and α-methyl-dl-Ser or l-Hse were weak inhibitors of GGT. Binding studies showed that 1 mol of Ser was bound per mol of enzyme; inhibition of GGT by Ser-borate was reversed by dilution or dialysis. Given that borate forms reversible complexes with vicinal hydroxyl groups, the authors proposed that Ser-borate reacts with an active-site hydroxyl group, e.g. Ser, to form an intermediate that resembles the expected tetrahedral transition-state intermediate formed by transpeptidation or hydrolysis of the acyl-enzyme intermediate. It should be noted that a 1:1 mixture of Ser and borate is used in inhibition experiments rather than ester 217 (); crystallographic analysis of the complex formed between hGGT1 and Ser-borate supports the formation of ester 217 () (Terzyan et al. Citation2015).
Figure 20. GGT inhibitors. 217, L-Ser-borate complex (O-(dihydroxyboryl)Ser); 218, 3-amino-3-carboxypropaneboronic acid (γ-boroGlu); 219, 1-γ-l-Glu-2-(2-carboxyphenyl)hydrazide (anthglutin); 220, 2-amino-4-[2-(carboxymethylcarbamoyl)ethylsulfinyl]butyric acid; 221, N-(5-(4-methoxybenzyl)-1,3,4-thiadiazol-2-yl)benzenesulfonamide (OU749); 222, O-(2-diazoacetyl)- l-Ser (azaserine); 223, 6-diazo-5-oxo-l-Nle (DON); 224, (αS,5S)-α-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid (acivicin, AT-125); 225, 2-amino-4-[methyl(4-methylumbelliferyl)phosphono]butanoic acid; 226, 2-amino-4-[mono(4-cyanophenyl)phosphono]butanoic acid (Hiratake′s Phosphonate); 227, 2-amino-4-{[4-(carboxymethyl)phenyl]-(methyl)phosphono}butanoic acid (GGsTop); 228, (2 R,S)-2-amino-4-{(S)-1-[N-(carboxymethyl)carbamoyl]propyl(phenyl)-(Sp*)-phosphono}butanoic acid.
![Figure 20. GGT inhibitors. 217, L-Ser-borate complex (O-(dihydroxyboryl)Ser); 218, 3-amino-3-carboxypropaneboronic acid (γ-boroGlu); 219, 1-γ-l-Glu-2-(2-carboxyphenyl)hydrazide (anthglutin); 220, 2-amino-4-[2-(carboxymethylcarbamoyl)ethylsulfinyl]butyric acid; 221, N-(5-(4-methoxybenzyl)-1,3,4-thiadiazol-2-yl)benzenesulfonamide (OU749); 222, O-(2-diazoacetyl)- l-Ser (azaserine); 223, 6-diazo-5-oxo-l-Nle (DON); 224, (αS,5S)-α-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid (acivicin, AT-125); 225, 2-amino-4-[methyl(4-methylumbelliferyl)phosphono]butanoic acid; 226, 2-amino-4-[mono(4-cyanophenyl)phosphono]butanoic acid (Hiratake′s Phosphonate); 227, 2-amino-4-{[4-(carboxymethyl)phenyl]-(methyl)phosphono}butanoic acid (GGsTop); 228, (2 R,S)-2-amino-4-{(S)-1-[N-(carboxymethyl)carbamoyl]propyl(phenyl)-(Sp*)-phosphono}butanoic acid.](/cms/asset/7d39e963-5e88-42d9-9585-e98a7708c46a/itxc_a_1692191_f0020_b.jpg)
In an attempt to develop more potent inhibitors of GGT than Ser-borate, several arylboronic acids were prepared and tested; two of the monophenyl boronic acids were slightly more potent than Ser-borate (Suenaga et al. Citation1996). The γ-boronic acid analog of L-Glu, 3-amino-3-carboxypropaneboronic acid (γ-boroGlu, 218, ), is a potent, slow-binding inhibitor of pig kidney GGT (Stein et al. Citation2001).
Several γ-Glu hydrazones and hydrazides are reversible inhibitors of GGT. The γ-Glu hydrazones of several α-keto acids inhibit rat kidney GGT, but do not serve as donor or acceptor substrates; the most potent inhibitor of this series is the γ-Glu hydrazone of α-ketoglutarate (Ki = 0.4 mM) (Tate and Meister Citation1974a). Anthglutin (219, ), isolated from the culture medium of Penicillium oxalicum, inhibits GGT (Kinoshita and Minato Citation1978; Minato Citation1979). Both l- and d-anthglutin (219, ) inhibit rat kidney GGT (Griffith and Meister Citation1979). Although both l- and d-anthglutin (219, ) inhibit GGT in vivo in mice, the D isomer is less toxic, perhaps because its hydrolysis to 2-carboxyphenylhydrazine is slower than that of the l isomer or because d-anthglutin interacts with fewer enzymes than l-anthglutin (Griffith and Meister Citation1979; Tate and Meister Citation1981).
Studies on the inhibition of GGT by l-Glu analogs showed that l-Met sulfoxide is a competitive inhibitor of GGT (Lherbet and Keillor Citation2004). Further investigation of l-Met sulfoxide analogs led to the preparation of sulfoxide-bearing GSH analog 220 (), which proved to be a reversible and potent (Ki = 0.053 mM) competitive inhibitor of rat kidney GGT. Further studies showed that the SCSS diasteroisomer of sulfoxide 220 () interacts with GGT (Lherbet et al. Citation2004). The sulfide analog of sulfoxide 220 () is approximately tenfold less potent (Ki = 0.52 mM) than sulfoxide 220 () whereas the sulfone analog is an even weaker inhibitor (Ki = 3.5 mM) of GGT.
OU749 (221, ) inhibits both GGT1-catalyzed transpeptidation and hydrolytic reactions (King et al. Citation2009; Wickham et al. Citation2012). Kinetic analysis showed that OU749 (221, ) exhibits similar potency as an uncompetitive inhibitor of both the transpeptidation and hydrolytic reactions (Wickham et al. Citation2012). Interestingly, OU749 (221, ) is a more potent inhibitor of isolated human kidney GGT than of rat or mouse kidney GGT and is a more potent inhibitor of GGT in human kidney 786-O and HK-2 cells than in rat NRK-52E, pig LLC-PK1, or monkey LLC-MK2 cells (King et al. Citation2009); OU749 (221, ) is 150-fold less cytotoxic than acivicin (224, ) to human kidney 786-O cells. Substitution of the benzenesulfonamide moiety produced divergent effects, increasing or reducing the potency for inhibition of the transpeptidation reaction, whereas substitution with electron withdrawing groups, e.g. nitro or trifluoromethyl, activated the GGT-catalyzed hydrolysis reaction (Wickham et al. Citation2012). The inhibitory selectivity of OU749 (221, ) for hGGT may be associated with Ala472 in the acceptor binding site compared with Ser472 in the rat, mouse, or pig (Hu et al. Citation2012).
3.2.4.2. Irreversible inhibitors
Azaserine (222, ) inhibits a number of glutamine-transforming enzymes, including rat and human kidney GGT, which are inactivated by compound 222 (Tate and Meister Citation1977; Tate and Ross Citation1977). Both transpeptidation and hydrolytic reactions are inhibited by azaserine (222, ); inactivation is blocked by donor, but not acceptor, substrates. The crystal structure of azaserine-inactivated E. coli GGT has been solved and shows that azaserine (222, ) forms an adduct with the active-site Thr (Wada et al. Citation2008). The crystal structure of azaserine-inactivated hGGT has apparently not been reported.
Although not an enzyme-specific inhibitor of GGT (Ahluwalia et al. Citation1990), 6-diazo-5-oxo-l-Nle (DON; 223, ) inactivates rat and human kidney GGT when incubated in the absence of a donor substrate (Inoue et al. Citation1977; Tate and Meister Citation1977; Tate and Ross Citation1977); incubation with the donor substrates γ-Glu-4-nitroanilide (196, ), S-(methyl)GSH and l-Gln blocks DON-induced inactivation of GGT, but the acceptor substrates L-Met and Gly-Gly fail to block inactivation. As with azaserine (222, ), both transpeptidation and hydrolytic reactions are blocked by DON (223, ). The pseudo-first-order rate constant for the inactivation of human GGT1 is approximately 0.13 min−1 (Terzyan et al. Citation2017). When rat kidney GGT was incubated with [6-14C]DON, 1 mole of inhibitor was covalently linked per mole of GGT (Inoue et al. Citation1977). Maleic acid markedly increases the rate of inactivation of rat kidney GGT by DON (Inoue et al. Citation1977). The dipeptide analog of DON (223, ), 6-diazo-5-oxo-l-Nle-Gly, inactivates renal membrane-bound GGT but not purified GGT, indicating that hydrolysis of the dipeptide to give DON (223, ) is required for inactivation (Inoue and Morino Citation1981).
The crystal structure of DON-inactivated and covalently modified hGGT1 has been solved (Terzyan et al. Citation2017). The proposed mechanism of inactivation involves attack of the hydroxyl oxygen of Thr381 on the side-chain carbonyl carbon of DON (223, ) to give a tetrahedral intermediate followed by reaction of the α-amino group of Thr381 on C6 of DON (223, ) to form a protein-bound morpholine ring that contains the α-amino, Cα, Cβ, and Oγ atoms of Thr381 and C5 and C6 of DON (223, ).
Acivicin (AT-125; 224, ), an irreversible inhibitor of GGT, is a fermentation product of Streptomyces sviceus (Martin et al. Citation1973). Several early studies showed that acivicin (224, ) inhibits mammalian GGT (Allen et al. Citation1980; Gardell and Tate Citation1980; Kozak and Tate Citation1980; Reed et al. Citation1980). Studies also showed that acivicin (224, ) exhibited antitumor properties in tumor-derived cell lines and in experimental animals (Allen et al. Citation1980; Thornthwaite and Allen Citation1980; Poster et al. Citation1981; Lui et al. Citation1982; Weber et al. Citation1982). Accordingly, acivicin (224, ) was tested for antitumor activity in human clinical trials (see, for example: Kramer et al. Citation1986; McGuire et al. Citation1986; Earhart et al. Citation1989; Falkson et al. Citation1990); the outcomes were generally unfavorable with acivicin (224, ) showing considerable toxicity and poor tumor response. An ester-based prodrug of acivicin (224, ) that is less cytotoxic than acivicin (224, ) has been reported (Antczak et al. Citation2001).
Acivicin (224, ) stoichiometrically and covalently reacts with purified rat kidney GGT and is a more effective inhibitor of the enzyme than DON (Gardell and Tate Citation1980); l-Ser-borate, a transition-state inhibitor of GGT, protects against acivicin-induced inhibition. Although acivicin (224, ) inhibits several enzymes that utilize L-Gln as a substrate (Ahluwalia et al. Citation1990; Pinto et al. Citation2016), only GGT appears to be covalently modified by [2-3H]acivicin in rat renal brush-border membranes (Kozak and Tate Citation1980). Mutations S451A and S452A caused a drastic reduction in the acivicin-induced hGGT inactivation rates; Ser451 and Ser452 were suggested to be potential active-site nucleophiles (Ikeda et al. Citation1995). Thr381 has, however, been identified unequivocally as the active-site nucleophile in hGGT (Castonguay et al. Citation2007).
Incubation of acivicin-inhibited GGT with hydroxylamine leads to the release of threo-β-hydroxy-l-γ-Glu hydroxamate, which was interpreted as evidence for the displacement of chloride from acivicin (224, ) by a nucleophilic site on GGT (Stole et al. Citation1994; Smith et al. Citation1995). The crystal structures of acivicin-inactivated E. coli and H. pylori GGT have been solved, and both structures show that the adduct with the active site Thr is not an ester, as suggested in an earlier report (Smith et al. Citation1995). Rather, following displacement of chloride by the active-site Thr, the dihydroisoxazole ring of acivicin (224, ) remains bound to the nucleophilic oxygen of the Thr and is retained in the adduct (Wada et al. Citation2008; Williams et al. Citation2009). Other 3-halo-4,5-dihydroisoxazoles also undergo halide displacement by active-site nucleophiles, which results in adducts in which the nucleophile is directly linked to the dihydroisoxazole ring (Watts et al. Citation2006; Pinto et al. Citation2016).
Phosphono derivatives of Glu have been prepared as mechanism-based affinity labeling probes (Inoue et al. Citation2000; Han et al. Citation2006, Citation2007). Indeed, phosphonate diester 225 () is 6000-times more potent than acivicin (224, ) as an inactivator of human GGT1 (Han et al. Citation2007). Phosphonate 226 () was used with expressed hGGT and mass spectrometric analysis to provide unambiguous identification of Thr381 as the active-site nucleophile of human GGT (Castonguay et al. Citation2007).
Phosphonobutanoic acid 227 (GGsTop, ) is more potent and rapid inactivator of hGGT (kon = 57 M−1 s−1) than acivicin (224, ) (kon = 0.40 M−1 s−1) and fails to inhibit glutamine amidotransferases (Han et al. Citation2007; Kamiyama et al. Citation2016). The authors propose that the meta-carboxyl group of the phenylacetic acid moiety of GGsTop (227, ) interacts with Lys562, the recognition site for the C-terminal carboxyl group of Cys-Gly. The mechanism of inactivation of hGGT by GGsTop is similar to that of the other phosphonate inhibitors in that it involves nucleophilic attack of Thr381 on the phosphorous atom with the resulting loss of 3-hydroxyphenylacetic acid; an X-ray crystal structure of the covalently bound enzyme-inhibitor complex is available (Terzyan et al. Citation2015). Of the four possible stereoisomers of GGsTop (227, ), the l-Sp isomer is the most potent (Watanabe et al. Citation2017). Preliminary data show that GGsTop (227, ) lacks acute toxicity in rats given 100 mg/kg i.v., lacks cytotoxicity in human skin fibroblasts and rat hepatic stellate cells (≤1 mM), and is stable in solution (Yamamoto et al. Citation2011; Kamiyama et al. Citation2016). Hence, GGsTop (227, ) appears to lack the toxicity associated with acivicin (224, ), but further studies are needed to assess more fully its toxicity.
In a rat model of renal ischemia-reperfusion injury, GGsTop (227, ) attenuated acute renal injury at single doses of 1 and 10 mg/kg i.v., perhaps by preventing GSH degradation (Yamamoto et al. Citation2011). Similarly, GGsTop (227, ) protects against hepatic ischemic-reperfusion injury in rats, again perhaps by maintaining cellular GSH concentrations (Tamura et al. Citation2016). GGsTop (227, ) also protects GSH degradation in lung lining fluid, indicating a potential use in management of asthma (Joyce-Brady and Hiratake Citation2011; Tuzova et al. Citation2014). The studies with GGsTop (227, ) and analogs were extended to include a series of peptidyl phosphonate ester analogs of GSH (Nakajima et al. Citation2014). Peptidyl phosphonate diester 228 () is a potent (kon = 145 M−1 s−1), irreversible inactivator of hGGT1. Its potency is attributed to the leaving group properties of the phenoxide moiety.
3.2.5. Tissue and species distribution
Mammalian GGT activities differ markedly among tissues. In all species studied, GGT activities are highest in the kidney, typically much higher than in the liver (Goldbarg et al. Citation1960; Hinchman and Ballatori Citation1990). Ultrastructural studies show that GGT is located in rat proximal tubule microvilli, endocytic vacuoles, and basolateral membranes and in the brush border of the jejunal epithelium (Marathe et al. Citation1979; Spater et al. Citation1982). In the rat renal brush-border membrane, the active site of GGT faces the luminal side of the renal tubule (Horiuchi et al. Citation1978). Although hepatic GGT activities are lower than renal activities, GSH secreted into the bile is almost quantitatively degraded to its constituent amino acids in the bile duct (Ballatori et al. Citation1986, Citation1988). The histochemical distribution of GGT in rat and guinea pig hepatocytes has been investigated with an activity stain (Lanca and Israel Citation1991). In the rat, GGT activity is found on both the bile canalicular and sinusoidal sides of hepatocytes, whereas in the guinea pig, GGT activity is strongly localized to the sinusoid side of hepatocytes but also present in the bile canalicular epithelium. Comprehensive immunohistochemical studies show widespread distribution of GGT in normal human tissues, including, for example, the luminal surface of renal proximal tubular cells, but not the glomeruli or distal tubular cells; bronchus; salivary ducts; hepatic bile ducts and canaliculi; gall bladder; pancreas; and testes, epididymis, and seminal vesicles (Hanigan and Frierson Citation1996).
3.2.6. Induction
As with many other drug-metabolizing enzymes, GGT activity in vivo is increased by treatment of animals with carcinogens and other chemicals. There is little hepatic expression of GGT in normal rats. Hepatic activity of GGT is, however, markedly increased after administration of a single dose of 3′-methyl-4-dimethylaminoazobenzene (3-MAB) and is increased exponentially with chronic administration of 3-MAB (Fiala et al. Citation1972, Citation1976). Long-term (≤14 weeks) administration of aflatoxin B1 to rats led to a marked increase in the hepatic expression of GGT well before carcinoma development, indicating that the expression of GGT may serve as a marker for preneoplastic hepatocytes (Kalengayi et al. Citation1975). Although it is often stated that GGT is not expressed in preneoplastic lesions in mice, several reports of carcinogen induction of GGT in preneoplastic mouse hepatocytes have been published (Goldsworthy et al. Citation1986; Hanigan et al. Citation1993). The hepatic GGT induced by carcinogen treatment differs from the renal enzyme in its sugar content (Tsuchida et al. Citation1979).
The mechanism of GGT induction by carcinogens has not been fully elucidated. Incubation of primary cultures of rat, but not mouse, hepatocytes in the absence of carcinogens leads to a marked increase in GGT expression (Gallagher et al. Citation1998). In the rat, the GGT protein precursor is encoded by mRNAIII that is selectively transcribed in the liver from a single copy gene (Brouillet et al. Citation1994). Only mRNAIII is induced indicating that Promoter III is associated with the observed induction by carcinogens (Gallagher et al. Citation1998).
The significance of GGT induction in tumor cells is that it may correlate with drug resistance (Hanigan Citation2014); as indicated above, this has led to a search for safe and effective GGT inhibitors.
3.2.7. GGT as a biomarker and as a risk and prognostic factor in disease states
The measurement of serum GGT activities has long been used as a diagnostic test for liver disease and injury (Orlowski Citation1963; Zein and Discombe Citation1970) and is considered to be a sensitive test for disease- or drug- and chemical-induced liver damage (Sheehan and Haythorn Citation1979).
A relationship between serum GGT activities and a range of human disease states, including cardiovascular disease (Ruttmann et al. Citation2005; Emdin et al. Citation2006; Ndrepepa et al. Citation2016; Wang et al. Citation2017a), atherosclerosis (Paolicchi et al. Citation2006), diabetes (Lee et al. Citation2003; Kim et al. Citation2005; Meisinger et al. Citation2005), Parkinson’s disease (Sian et al. Citation1994; Chinta et al. Citation2006), chronic renal disease (Targher et al. Citation2010), and cancer (Strasak et al. Citation2008; Grimm et al. Citation2013; Preyer et al. Citation2016) has been reported. Oxidative stress has been implicated in a range of human disease states, and it has been proposed that serum GGT activity is a marker for oxidative stress (Lee et al. Citation2004).
The mechanism of the release of GGT from tissues, the association between elevated serum GGT activities and tissue expression of GGT, and the nature of serum GGT have not been fully clarified. Given that colorimetric assays, e.g. γ-Glu-4-nitroanilide (196, ), are commonly used to quantify serum GGT activities, serum GGT appears to retain its catalytic activity. In chemically induced cholestasis in rats, serum GGT activity is of biliary-cell origin rather than from hepatocytes (Bulle et al. Citation1990).
Two forms of GGT are found in human serum (Huseby Citation1982): a major, amphiphilic form that is present in high molecular-mass complexes and a minor, low molecular-mass (80–90 kDa) hydrophilic form that appears to be a proteolytic degradation product of GGT. Fractionation of human serum GGT by gel-filtration chromatography showed four forms (Franzini et al. Citation2008): b-GGT (2000 kDa), m-GGT (940 kDa), s-GGT (140 kDa), and f-GGT (70 kDa); b-, m-, and s-GGT are bound to lipoproteins, and f-GGT is an unbound form. Other studies indicate that b-, m-, and s-GGT are present as complexes with VLDL, LDL, and HDL, respectively (Fornaciari et al. Citation2014); f-GGT is the most abundant form in healthy adults, and b-, m-, and s-GGT are increased in liver diseases. The mechanism of transfer of GGT from plasma membranes to blood has not been fully resolved, although b-GGT is found in exosomes, implicating an exocytotic mechanism. The GGT fractions may prove to be useful in the differential diagnosis of liver disease.
3.3. γ-Glutamyltransferase 5 (GGT5, EC 3.4.19.14, leukotriene-C4 hydrolase, γ-glutamyl leukotrienase)
3.3.1. Structure and properties
Although GGT1 had been considered to be the only enzyme that catalyzes the hydrolysis of the γ-Glu bond of GSH and GSH S-conjugates, a human enzyme with about 40% amino acid similarity to hGGT1 catalyze the hydrolysis of LTC4 (189, ) and GSH, but not γ-Glu-4-nitroanilide (196, ) (Heisterkamp et al. Citation1991). This GGT-related enzyme was termed GGT-rel, but has also been termed GGTLA1 (Morris et al. Citation1993).
Tissue homogenates and partially purified enzyme from Ggt1-null mice catalyze the hydrolysis of leukotriene C4 (189, ) and S-decyl-GSH, but not GSH, GSSG, γ-Glu-4-nitroanilide (196, ), or γ-Glu-4-methoxy-2-naphthylamide, indicating the presence of an enzyme other than mGTT1 that catalyzes the hydrolysis of the γ-Glu bond (Carter et al. Citation1997); this enzyme was termed γ-Glu leukotrienase (GGL). A rat homolog of GGT-rel with 79% identity to human GGT-rel that was weakly expressed in normal trachea has been described but the hydrolysis of γ-Glu substrates was not reported (Potdar et al. Citation1997).
GGT5 has been proposed as the designation for GGTs formerly termed GGT-rel, GGL, and GGTLA1 (Heisterkamp et al. Citation2008). GGT5 (leukotriene C4 hydrolase) is assigned to peptidase Family T3 (MEROPS ID T03.002) (Rawlings et al. Citation2018).
All published reports indicate that like GGT1, GGT5 is a glycosylated, extracellular heterodimeric protein (Heisterkamp et al. Citation1991; Carter et al. Citation1997; Han et al. Citation2002).
3.3.2. Reactions catalyzed and substrates
3.3.2.1. Transpeptidation
With mouse GGT5, addition of Gly-Gly to the reaction mixture failed to increase the hydrolysis of LTC4 (189, ), indicating that mGGT5 may not catalyze transpeptidation reactions (Carter et al. Citation1997). Later studies showed, however, that hGGT5 catalyzes transpeptidation reactions: Gly-Gly decreases the GGT5-catalyzed release of l-Glu from GSH, indicating that, under nonphysiological conditions, hGGT5 can catalyze a transpeptidation reaction (Wickham et al. Citation2011).
3.3.2.2. Autotranspeptidation
GGT5-catalyzed autotranspeptidation reactions have apparently not been reported.
3.3.2.3. Hydrolysis
Given the physiological importance of leukotrienes (Peters-Golden and Henderson Citation2007), the enzymatic hydrolysis of LTC4 (189, ) to LTD4 has been the subject of many investigations. Hence, as indicated above, LTC4 hydrolytic activity assigned to GGT5 has been identified in human, mouse, and rat (Heisterkamp et al. Citation1991; Carter et al. Citation1997; Potdar et al. Citation1997). Furthermore, Ggt5-null mice do not catalyze the hydrolysis of LTC4 (189, ) (Han et al. Citation2002).
Although there is general agreement that GGT5 catalyzes the hydrolysis of LTC4 (189, ) and fails to catalyze the hydrolysis of γ-Glu-4-nitroanilide (196, ), differences in the substrate selectivity of GGT5 have been reported, specifically about its ability to catalyze the hydrolysis of GSH and certain other γ-Glu compounds. It was reported that spleen homogenates from GGT1-null mice as well as partially purified enzyme from GGT1-null mouse spleen failed to hydrolyze GSH or GSSG (Carter et al. Citation1997; Lieberman et al. Citation1999), but expressed hGGT5 has been reported to catalyze the hydrolysis of GSH (Heisterkamp et al. Citation1991).
The substrate selectivity of hGGT5 was investigated with assay conditions that allow selective quantification of expressed hGGT5- or hGGT1-catalyzed hydrolytic reactions (Wickham et al. Citation2011): hGGT5 catalyzes the hydrolysis of GSH, GSSG, LTC4 (189, ), S-(4-nitrobenzyl)GSH (126, ), S-(methyl)GSH, GSH sulfonic acid, but not γ-Glu-Leu. Although both hGGT1 and hGGT5 catalyze the hydrolysis of LTC4 (189, ), Vmax/KmEt is much greater for hGGT1 than for hGGT5 (0.49 versus 0.019 min−1 nM−1) (Wickham et al. Citation2011); as noted below, however, the expression of hGGT5 is high in some tissues so that the contribution of hGGT5 may be physiologically significant in these tissues.
3.3.3. Inhibitors
3.3.3.1. Reversible inhibitors
Ser-borate (217, ) inhibits expressed hGGT5 but is a less potent inhibitor of hGGT5 than of hGGT1 (Wickham et al. Citation2011).
3.3.3.2. Irreversible inhibitors
GGT5 is inhibited by acivicin (224, ) (Heisterkamp et al. Citation1991; Carter et al. Citation1997).
3.3.4. Tissue and species distribution
Northern blot analysis showed that GGT5 is intensely expressed in rat tracheal epithelium, faintly expressed in trachea, and not detectable in brain, heart, kidney, liver, lung spleen, and testis (Potdar et al. Citation1997); after reverse transcriptase polymerase chain reaction amplification, however, a hybridization signal was detected in brain, heart, kidney, liver, lung spleen, testis, and trachea. In the mouse, GGT5 is most highly expressed in the spleen and uterus but is also expressed in thymic lymphocytes, bronchiolar epithelial cells, pulmonary interstitial cells, renal proximal convoluted tubular cells, and crypt cells of the small intestine, and in cerebral, cerebellar, and brain stem neurons but not in glial cells (Carter et al. Citation1998; Han et al. Citation2002).
Immunohistochemical studies show that both GGT5 and GGT1 are highly expressed in human kidney but that only GGT1 is expressed in proximal tubular cells (Hanigan et al. Citation2015). With human liver, GGT5 is strongly expressed in Kupffer cell and macrophages but is weakly expressed in hepatocytes, whereas GGT1 is expressed in bile canalicular surfaces of hepatocytes. Immunohistochemical analysis of normal human tissues showed that GGT5 is detectable in all tissues studied but that the intensity of expression varied among tissues. GGT5 is expressed in primary bronchial epithelial cells (Lukic et al. Citation2016) and in PC-9ER-cells derived from non-small cell lung cancer cells (Serizawa et al. Citation2014).
Although GGT5 is widely distributed in mouse tissues, the highest expression is found in the spleen (Carter et al. Citation1998; Shi et al. Citation2001). GGT5 is also found in the airway tissues of the rat and guinea pig (Funayama et al. Citation1996; Potdar et al. Citation1997). Both GGT1 and GGT5 are widely distributed in human tissues, but the expression in cell types differs (Hanigan et al. Citation2015): GGT5 is expressed in the interstitial cells of the kidney whereas GGT1 is expressed in the apical surface of the proximal tubules.
3.3.5. Induction
The hydrolysis of leukotriene C4 (189, ) is increased in transformed and normal human bronchial epithelial cells incubated with dexamethasone, fluticasone, or beclomethasone (Zaitsu et al. Citation2001, Citation2003). Treatment with glucocorticoids also caused up-regulation of GGT5 mRNA expression, but had no effect on GGT1 activity or GGT1 mRNA expression, suggesting a significant role for GGT5 in the hydrolysis of LTC4 (189, ) in human bronchial epithelial cells.
3.3.6. GGT5 as a biomarker for disease states
In an experimental murine model of peritonitis, GGT5, but not GGT1, is responsible for the generation of LTD4 (189, ) (Shi et al. Citation2001). Induction of airway responsiveness with Aspergillus fumigatus culture filtrate allergen in a murine model of asthma results in an increase in pulmonary GGT5 levels in wild-type and GGT1-null mice and causes a greater increase in airway reactivity in GGT5-null mice than in wild-type mice (Han et al. Citation2002). The level of LTC4 increases substantially in the lungs of GGT5-null mice with allergen-induced asthma, whereas LTD4 is not detectable in either bronchoalveolar lavage fluid or plasma, supporting the conclusion that, in asthmatic inflammation, GGT5 plays an important role in LTD4 generation. In contrast, GGT1 is primarily responsible for the conversion of LTC4 to LTD4 in lung homogenates from mice treated only with saline (Shi et al. Citation2001). GGT5 expression lacks prognostic value in subjects with acute myeloid leukemia (Tian et al. Citation2014).
4. Dipeptidases
4.1. Introduction
Three dipeptidases have been demonstrated to catalyze the hydrolysis of Cys-Gly S-conjugates to Cys S-conjugates and Gly: membrane dipeptidase (DPEP1), aminopeptidase N (APN), and leucyl aminopeptidase (LAP) (Tate Citation1980; Commandeur et al. Citation1995; Cooper and Hanigan Citation2010; Barrett et al. Citation2012; Poon and Josephy Citation2012). DPEP1 and APN are membrane-bound, homodimeric proteins, whereas LAP is a soluble homohexamer. All are metalloenzymes, and zinc is the most commonly found metal; DPEP1 and LAP contain two zinc ions per subunit, whereas APN contains a single zinc ion per subunit; the zinc ions are essential for catalytic activity (Auld Citation2013). The cytosolic nonspecific peptidase (carnosine dipeptidase II, EC3.4.13.18, MEROPS Family M20.005) has apparently not been reported to catalyze the hydrolysis of Cys-Gly S-conjugates, although Cys-Gly is a substrate (Kaur et al. Citation2009).
This discussion of dipeptidases will focus on the roles of DPEP1, APN, and LAP in the mercapturic acid pathway. The biotransformation of few Cys-Gly S-conjugates has been investigated with identified and purified dipeptidases. The cellular and in vivo hydrolysis of Cys-Gly S-conjugates, however, has not been studied extensively enough to allow meaningful conclusions about the relative importance of the three dipeptidases in their metabolism.
Assessment of the role of dipeptidases in the mercapturic acid pathway is complicated by overlapping substrate selectivities and by differing inhibitor susceptibilities: for example, bestatin (267, ) inhibits 12 aminopeptidases (Scornik and Botbol Citation2001). In addition, inhibitor potencies differ among the dipeptidases: bestatin (267, ) is a more potent inhibitor of APN than cilastatin (266, ); cilastatin (266, ) is a more potent inhibitor of DPEP1 than bestatin (267, ) (Hirota et al. Citation1985); amastatin (268, ) is a more potent inhibitor of APN than bestatin (267, ) (Rich et al. Citation1984).
4.2. Membrane-bound dipeptidases
Two membrane-bound dipeptidases catalyze the hydrolysis of Cys-Gly S-conjugates: membrane dipeptidase and aminopeptidase N.
4.2.1. Membrane dipeptidase (DPEP1, EC 3.4.13.19)7
DPEP1 is a zinc-dependent enzyme that catalyzes the hydrolysis of dipeptides, but not tripeptides, with N-terminal amino acids in the l-configuration and C-terminal amino acids in the d- or l-configuration (Campbell et al. Citation1966; Campbell Citation1970). DPEP1 is assigned to Family M19 (MEROPS ID M19.001; https://www.ebi.ac.uk/merops/cgi-bin/pepsum?id=M19.001; accessed 31 October 2019) (Rawlings et al. Citation2018). Early studies demonstrated the presence of an enzyme that catalyzed the hydrolysis of dehydropeptides (Bergmann and Schleich Citation1932a, Citation1932b). The peptidase was later purified from rat kidney and demonstrated to catalyze the hydrolysis of glycyldehydrophenylalanine (229, ), Cys-Gly S-conjugate of N-ethylmaleimide (231, ), l-cystinyl-bis-glycine (233, ), and leukotriene D4 (234, ) (Hirota et al. Citation1985, Citation1987a). For reviews about DPEP1, see Keynan et al. (Citation1996b) and Hooper (Citation2013).
Figure 21. Dipeptidase substrates. 229, glycyldehydrophenylalanine; ((E)-2-[(2-aminoacetyl)amino]-3-phenylprop-2-enoic acid); 230, dehydrophenylalanine; ((Z)-2-amino-3-phenylprop-2-enoic acid); 231, S-(1-ethyl-2,5-dioxopyrrolidin-3-yl)-l-Cys-Gly; 232, S-(1-ethyl-2,5-dioxopyrrolidin-3-yl)-l-Cys; 233, cystinyl-bis-glycine; 234, leukotriene D4 (LTD4); 235, leukotriene E4 (LTE4); 236, S-(2,4-dinitrophenyl)-l-Cys-Gly; 237, S-(2,4-dinitrophenyl)-l-Cys; 238, imipenem (N-formimidoyl thienamycin, MK0787); 239, (αS,2R)-5-carboxy-2,3-dihydro-α-[(1R)-1-hydroxyethyl]-4-[[2-[(iminomethyl)amino]ethyl]thio]-1H-pyrrole-2-acetic acid; 240, S-(benzyl)-l-Cys-Gly; 241, S-(benzyl)-l-Cys; 242, S-(benzyl)-l-Cys-4-nitroanilide; 243, l-leu-4-nitroanilide; 244, 3-cyclohexyl-l-alanyl-O-[[[1-(2-deoxy-2-methylene-β-d-erythro-pentofuranosyl)-1,2-dihydro-2-oxo-4-pyrimidinyl]amino]carbonyl]-d-threonine; 245, 2′-deoxy-2′-methylidenecytidine (DMDC); 246, pomaglumetad methionil; ((1 R,4S,5S,6S)-4-(l-methionylamino)-2-thiabicyclo[3.1.0]hexane-4,6-dicarboxylic acid 2,2-dioxide); 247, LY404039 ((1R,4S,5S,6S)-4-amino-2-thiabicyclo[3.1.0]hexane-4,6-dicarboxylic acid 2,2-dioxide); 248, S-(methyl)-l-Cys-Gly; 249, S-(methyl)-l-Cys; 250, S-(carbamidomethyl)-l-Cys-Gly; 251, S-(carbamidomethyl)-l-Cys; 252, 2-amino-N-(4-((2-oxo-2H-chromen-7-yloxy)methyl)phenyl)propanamide hydrochloride (Ala-PABA-7HC); 253, 7-((4-aminobenzyl)oxy)naphthalen-2(1H)-one; 254, 7-hydroxycoumarin; 255, (2S)-2-amino-N-(6′-aminospiro[isobenzofuran-1(3H),9′-[9H]xanthen]-3′-yl)-4-methylpentanamide (Leu-HMRG); 256, 2-(3,6-diamino-9H-xanthen-9-yl)benzenemethanol (HMRG); 257, l-Ala anilide of amino-hemicyanine (HCAN); 258, amino-hemicyanine (HCA); 259, (R,E)-2-amino-N-(4-(2-(3-(dicyanomethylene)-5,5-dimethylcyclohex-1-en-1-yl)vinyl)phenyl)-4-methylpentanamide (TMN-Leu); 260, (E)-2-(3-(4-aminostyryl)-5,5-dimethylcyclohex-2-en-1-ylidene)malononitrile (TMN-NH2); 261, melflufen (melphalan flufenamide); 4-[bis(2-chloroethyl)amino]-l-phenylalanyl-4-fluoro-l-Phe ethyl ester); 262, 4-fluoroPhe ethyl ester; 263, 5-fu-NGR; 264, N-l-leucylglycine 2-[[5-(dimethylamino-1-naphthalenyl]sulfonyl]hydrazide; 265, glycine 2-[[5-(dimethylamino-1-naphthalenyl]sulfonyl]hydrazide.
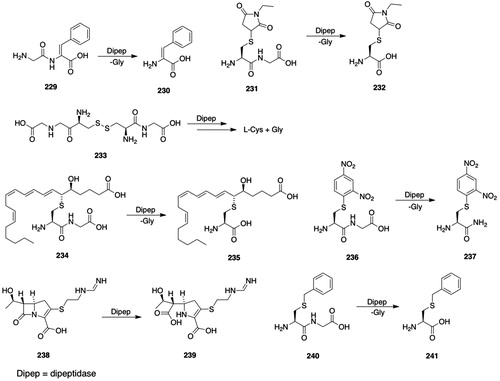
4.2.1.1. Structure and properties
DPEP1 is a zinc-containing homodimer with a subunit Mr of 47 000 to 59 000 that is anchored to the membrane with a glycosyl-phosphatidylinositol moiety (Adachi et al. Citation1989; Littlewood et al. Citation1989; Hooper et al. Citation1990; Adachi et al. Citation1990a; Satoh et al. Citation1993; Brewis et al. Citation1995; Poon and Josephy Citation2012). Murine DPEP3 is a disulfide-linked homodimer with a calculated subunit Mr of 54213 (Yoshitake et al. Citation2011). Differences in subunit Mr among h-, p-, and mDPEP1 are attributed to differing degrees of glycosylation. A “urinary dipeptidase” has been identified as deglycosylated DPEP1 that is formed by the cleavage of glycosylated DPEP1 by a phospholipase C (We et al. Citation1997; Park et al. Citation2001, Citation2002).
Three genes encode human membrane dipeptidases: DPEP1, DPEP2, and DPEP3 (Adachi et al. Citation1990b; Austruy et al. Citation1993). The mouse membrane dipeptidases are encoded by Dpep1, Dpep2, and Dpep3 (Satoh et al. Citation1993; Habib et al. Citation1996, Citation2003); porcine, rabbit, and rat genes encoding the membrane dipeptidase have also been described (Rached et al. Citation1990; Igarashi and Karniski Citation1991; Shimoyama et al. Citation2015).
DPEP1 subunits are linked by interchain disulfide bonds (Keynan et al. Citation1996a; Nitanai et al. Citation2002). Although pDPEP1 contains seven Cys residues, only Cys361 is involved in homodimer formation (Keynan et al. Citation1996a). DPEP1 was initially reported to contain one zinc ion per subunit (Campbell et al. Citation1966; Armstrong et al. Citation1974); the crystal structure, however, clearly shows the presence of two Zn ions along with ligands for each Zn ion (Nitanai et al. Citation2002). DPEP1 lacks the Zn-binding HExxH motif, which is found in many zinc peptidases (Jongeneel et al. Citation1989; Keynan et al. Citation1994).
The crystal structures of oligosaccharide-trimmed, unliganded and cilastin-liganded subunits of hDPEP1 have been solved (Nitanai et al. Citation2002). The subunits consist of α-helices and β-strands organized into (α/β)8 barrel structures that are linked by disulfide bonds between Cys361 to form the homodimer. The active site with its two Zn ions is located in the barrel structure. One Zn ion (Zn1) forms ligands with Asp22 and His20 whereas the other Zn ion (Zn2) forms ligands with His198 and His219; Zn1 and Zn2 are bridged by Glu125. A water molecule (Wat421) forms coordinate bonds with Zn1 and Zn2 and is proposed to undergo deprotonation to form the nucleophile that attacks the carbonyl atom of the scissile bond of the substrate. Previous site-directed mutagenesis studies had demonstrated the importance of Glu125 and His219 for catalytic activity (Adachi et al. Citation1993; Keynan et al. Citation1994, Citation1997).
The selectivity of DPEP1 for dipeptide substrates is demonstrated by the crystal structure of the protein in complex with cilastatin (266, ), which is considered to be a dipeptide analog: the inhibitor completely fills the active site (Nitanai et al. Citation2002); also, superimposition of the structures of glycyldehydrophenylalanine (229, ) and imipenem (238, ) shows that they are accommodated by the active site. An analysis of the cilastatin-DPEP1 complex indicated that the inhibitor binds in a catalytically competent manner, but tight interactions of the alkyl groups at its N- and C-termini with amino acid residues at either end of the active site prevent breakdown of the tetrahedral intermediate (Smyth et al. Citation2003). The substrate selectivity of DPEP1 shows a preference for dipeptides with bulky residues at the N-terminus, which were proposed to interact with a hydrophobic pocket in the active site (Campbell et al. Citation1988); for example, the Vmax/Km for L-Trp-Gly is larger than for Gly-l-Trp.
Figure 22. Dipeptidase inhibitors. 266, cilastatin; 267, bestatin (Ubenimex); 268, amastatin; 269, (+)-(Z)-2-(2,2-dimethylcyclopropane-1-carboxamido)but-2-enoic acid; 270, 2-[(cyanoacetyl)amino]-2-propenoic acid; 271 (2Z)-2-[[(1-amino-2-cyclohexylethyl)hydroxyphosphinyl]methyl]-3-(4-fluorophenyl)-2-propenoic acid; 272, tosedostat (CHR-2797); 2-(2-(hydroxy(hydroxycarbamoyl)methyl)-4-methylpentanoylamino)-2-phenylethanoic acid cyclopentyl ester); 273, α1-amino-3-phenylpropyl(α2-hydroxy-3-phenylpropyl)phosphinic acid; 274, l-Leu thiol; ((S)-2-amino-4-methylpentane-1-thiol); 275, l-leucinal; ((S)-2-amino-4-methylpentanal); 276, Leu-B(OH)2-pinacol; (3-methyl-l-(4,4,5,5-tetramethyl-l,3,2-dioxaborolane-2-yl)-l-butylamine); 277, l-Leu phosphonic acid [(1R)-1-amino-3-methylbutyl]phosphonic acid); 278 (S)-2-[3-(2,6-dimethyl-phenyl)-ureido]-4-methyl-pentanoic acid hydroxyamide; 279, 2-amino-1,4-dihydrophenanthren-3(2H)-one; 280, 7-amino-1-bromo-4-phenyl-5,7,8,9-tetrahydrobenzocyclohepten-6-one; 281, zofenprilat; ((2S,4S)-1-[(2S)-2-methyl-3-sulfanylpropanoyl]-4-(phenylsulfanyl)pyrrolidine-2-carboxylic acid); 282, diethyl 6,8-dibenzyloxy-3,4-dihydroisoquinoline-3,3-dicarboxylate.
![Figure 22. Dipeptidase inhibitors. 266, cilastatin; 267, bestatin (Ubenimex); 268, amastatin; 269, (+)-(Z)-2-(2,2-dimethylcyclopropane-1-carboxamido)but-2-enoic acid; 270, 2-[(cyanoacetyl)amino]-2-propenoic acid; 271 (2Z)-2-[[(1-amino-2-cyclohexylethyl)hydroxyphosphinyl]methyl]-3-(4-fluorophenyl)-2-propenoic acid; 272, tosedostat (CHR-2797); 2-(2-(hydroxy(hydroxycarbamoyl)methyl)-4-methylpentanoylamino)-2-phenylethanoic acid cyclopentyl ester); 273, α1-amino-3-phenylpropyl(α2-hydroxy-3-phenylpropyl)phosphinic acid; 274, l-Leu thiol; ((S)-2-amino-4-methylpentane-1-thiol); 275, l-leucinal; ((S)-2-amino-4-methylpentanal); 276, Leu-B(OH)2-pinacol; (3-methyl-l-(4,4,5,5-tetramethyl-l,3,2-dioxaborolane-2-yl)-l-butylamine); 277, l-Leu phosphonic acid [(1R)-1-amino-3-methylbutyl]phosphonic acid); 278 (S)-2-[3-(2,6-dimethyl-phenyl)-ureido]-4-methyl-pentanoic acid hydroxyamide; 279, 2-amino-1,4-dihydrophenanthren-3(2H)-one; 280, 7-amino-1-bromo-4-phenyl-5,7,8,9-tetrahydrobenzocyclohepten-6-one; 281, zofenprilat; ((2S,4S)-1-[(2S)-2-methyl-3-sulfanylpropanoyl]-4-(phenylsulfanyl)pyrrolidine-2-carboxylic acid); 282, diethyl 6,8-dibenzyloxy-3,4-dihydroisoquinoline-3,3-dicarboxylate.](/cms/asset/7cd5aefa-a253-46de-8292-1e7d800776a5/itxc_a_1692191_f0022_b.jpg)
A computational analysis of the catalytic cycle of DPEP1 revealed substrate binding in which the N-terminal amino acid and carboxylate groups of the dipeptide are coordinated with Zn1 and Zn2, respectively (Liao et al. Citation2010). Deprotonation of the active-site water by Asp288 gives the oxyanion that attacks the substrate carbonyl group to form a tetrahedral intermediate that is stabilized by His152; collapse of the tetrahedral intermediate yields the hydrolysis products. These binding and mechanism results are in agreement with, and expand upon, an earlier proposal (Nitanai et al. Citation2002), and are consistent with the results of docking studies (Smyth et al. Citation2003; Kim et al. Citation2007).
4.2.1.2. Substrates
DPEP1 preferentially catalyzes the hydrolysis of dipeptides with l-amino acids in the N-terminal position and both D- and l-peptides at the C-terminal position, e.g. Gly-l-Ala, Gly-D-Ala (Robinson et al. Citation1953; Semenza Citation1957; Campbell et al. Citation1966; Campbell Citation1970; Kozak and Tate Citation1982; Tate Citation1985; Campbell et al. Citation1988; Watanabe et al. Citation1996; Yamada and Kera Citation1998). A compilation of DPEP1 substrates is available on the MEROPS website (https://www.ebi.ac.uk/merops/cgi-bin/substrates?id=M19.001; accessed 31 October 2019).
Cys-Gly is a substrate for DPEP1 but shows substrate inhibition at elevated substrate concentrations (McIntyre and Curthoys Citation1982). Cys-Gly inhibits the DPEP1-catalyzed hydrolysis of l-Ala-Gly (Kozak and Tate Citation1982). DPEP1 catalyzes the hydrolysis of Cys-Gly S-conjugates, including S-(2,4-dinitrophenyl)- l-Cys-Gly (236, ), the Cys-Gly S-conjugate of NEM (231, ), l-cystinyl-bis-glycine (233, ), and leukotriene D4 (234, ) (Anderson et al. Citation1982; Kozak and Tate Citation1982; Hirota et al. Citation1985; Tate Citation1985; Farrell et al. Citation1987; Hirota et al. Citation1987a; Campbell et al. Citation1988; Adachi et al. Citation1989). The biotransformation of a range of S-aryl-Cys-Gly S-conjugates, e.g. 236 () is catalyzed by DPEP1(Poon and Josephy Citation2012).
The observation that the carbapenem antibiotic imipenem (238, ), which is resistant to bacterial β-lactamase-catalyzed hydrolysis, undergoes renal metabolism in several mammalian species was an unexpected finding: mammals were thought to lack β-lactamase activity. It was demonstrated, however, that DPEP1 catalyzes the hydrolysis of imipenem (238, ) (Kim and Campbell Citation1982; Kropp et al. Citation1982; Kahan et al. Citation1983; Campbell et al. Citation1984). DPEP1 is the only mammalian dipeptidase demonstrated to possess β-lactamase activity: mice with the targeted deletion of the DPEP1 gene lack β-lactamase activity (Habib et al. Citation1998). Formerly, Primaxin® IV, a fixed-dose preparation of imipenem (238, ) and cilastatin (266, ) to prevent DPEP1-catalyzed degradation of imipenem (238, ) was approved for human clinical use; elevated plasma Cys-Gly concentrations are observed in human subjects given Primaxin® IV, which are attributable to the cilastatin-induced inhibition of the DPEP1-catalyzed hydrolysis of Cys-Gly (Badiou et al. Citation2005).
The substrate selectivity of DPEP1 has been exploited to activate prodrugs. DPEP1 is overexpressed in human colorectal and stomach tumor tissue (Kohchi et al. Citation2007). The DPEP1-catalyzed hydrolysis of the dipeptide moiety of prodrug 244 () in these tissues gives an intermediate that spontaneously releases the antitumor agent, 2′-deoxy-2′-methylidenecytidine (245, ). The methionine residue of pomaglumetad methionil (246, ) is cleaved by DPEP1 to give dicarboxylic acid 247 (), which is a selective metabotropic glutamate 2/3 receptor agonist (Moulton et al. Citation2015).
Murine membrane dipeptidase-2 (MEROPS ID M19.002) hydrolyzes leukotriene D4 (234, ), but not cystinyl-bis-glycine (233, ) whereas murine membrane dipeptidase-3 (MEROPS ID M19.004) hydrolyzes cystinyl-bis-glycine (233, ) but not leukotriene D4 (234, ) (Habib et al. Citation2003).
4.2.1.3. Inhibitors
DPEP1 is inhibited by chelating agents, by transition-state analogs, and by substrate analogs. 1,10-Phenanthroline, penicillamine, and dithiothreitol inhibit porcine and human kidney DPEP1, either by chelating zinc or by binding to a zinc coordination site; activity is partially restored by incubation with ZnCl2 (Harper et al. Citation1971; Kozak and Tate Citation1982; McIntyre and Curthoys Citation1982; Campbell et al. Citation1988; Littlewood et al. Citation1989). A compilation of DPEP1 inhibitors is available on the MEROPS website (https://www.ebi.ac.uk/merops/cgi-bin/inhibitors.pl?id=M19.001; accessed 31 October 2019).
Cilastatin (266, ) is a competitive, reversible inhibitor of DPEP1 (Farrell et al. Citation1987) and is a more potent inhibitor of DPEP1 than bestatin (267, ) or amastatin (268, ) (Hirota et al. Citation1985, Citation1986, Citation1987a; Campbell et al. Citation1990a; Watanabe et al. Citation1996).
The crystal structure of DPEP1 in complex with cilastatin (266, ) shows that the active site is completely occupied by the inhibitor (Nitanai et al. Citation2002). This is consistent with the earlier observation that DPEP1 prefers substrates with a bulky, hydrophobic group at the N-terminal position (Campbell et al. Citation1988). The positioning of cilastatin (266, ) in the active site was proposed to prevent the formation of a tetrahedral intermediate (Nitanai et al. Citation2002).
Bestatin (267, ) is a time-dependent, noncompetitive, tight-binding inhibitor of DPEP1 (Campbell et al. Citation1988). Butenoic acid 269 (), which is an analog of cilastatin (266, ), inhibits the DPEP1-catalyzed biotransformation of imipenem (238, ) (Kim and Campbell Citation1982; Graham et al. Citation1987). Propenoic acid 270 () is a mechanism-based inactivator of DPEP1 (Wu and Mobashery Citation1991). Aminophosphinic acid 271 () and related compounds are potent (IC50 ≃ 5 nM) inhibitors of DPEP1 (Gurulingappa et al. Citation2003, Citation2004). An analog of phosphinic acid 271 () is a slow, tight-binding inhibitor of DPEP1 that is significantly more potent (Ki = 0.52 nM) than cilastatin (Ki = 0.11 μM) (266, ) (Parsons et al. Citation1991).
4.2.1.4. Tissue and species distribution
DPEP1 is primarily located in the microsomal fraction of the porcine renal cortex (Harper et al. Citation1971). Porcine DPEP1 is expressed on the lumenal face of renal cortical tubules, but not in the medulla or glomeruli (Littlewood et al. Citation1989). DPEP1 activities are high in rat lung and kidney but much lower in spleen, liver, brain, pancreas, and small intestine (Hirota et al. Citation1986). In rat kidney, DPEP1 is found on both the brush-border and basolateral membranes (Hirota et al. Citation1987b). High levels of human DPEP1 activity are observed in pancreas, renal cortex, and testis, but in contrast to the rat lung, activity is very low in human lung tissue (Kera et al. Citation1999).
Northern-blot analysis shows that the rat DPEP1 is intensely expressed in the small intestine, lung, and kidney, weakly expressed in stomach, pancreas, and testis, and not detectable in brain, heart, liver, or spleen (Adachi et al. Citation1992). DPEP1 is overexpressed in both benign and malignant colon cancer epithelium (Buckhaults et al. Citation2001). DNA-microarray analysis of tumor tissue showed that DPEP1 is highly expressed in human colorectal and stomach tumor tissue but not in adjacent normal tissue (Kohchi et al. Citation2007).
DPEP1 has been identified as the target for the lung-homing peptide GFE-1 (CGFECVRQCPERC) in mice (Rajotte and Ruoslahti Citation1999); given that Cys-Gly is a substrate for DPEP1, the N-terminal Cys-Gly residues of GFE-1 may bind to the active site of DPEP1. These studies were extended to identify DPEP1 binding molecules and their binding mode (Kim et al. Citation2007).
Western-blot analysis showed that murine DPEP3 is expressed in the testicular seminiferous tubules but not in interstitial cells or in heart, liver, lung, kidney, spleen, or ovary (Habib et al. Citation2003; Yoshitake et al. Citation2011).
4.2.2. Aminopeptidase N (APN, EC 3.4.11.2)8,9
The description of a microsomal “cysteinylglycinase” from porcine kidney is apparently the first report of an enzyme that would later be identified as APN (Olson and Binkley Citation1950; Binkley Citation1951; Binkley et al. Citation1957; Semenza Citation1957; Binkley et al. Citation1959). Later studies characterized a microsomal peptidase from porcine kidney that catalyzed the hydrolysis of a range of peptides along with l-Ala and l-Leu-4-nitroanilide (243, ) (Pfleiderer and Celliers Citation1963; Pfleiderer Citation1970). For reviews about APN, see Sjöström et al. (Citation2000), Langner and Ansorge (Citation2002), Lendeckel et al. (Citation2004), Bauvois and Dauzonne (Citation2006), Luan and Xu (Citation2007), Mina-Osorio (Citation2008), and Turner (2013).
4.2.2.1. Structure and properties
APN is a homodimeric, zinc-containing ectoenzyme assigned to MEROPS Family M1 (MEROPS ID M01.001) (Rawlings et al. Citation2018) that catalyzes the release of N-terminal L-amino acids, preferably Ala, from peptides, amides, or arylamides. APN contains one Zn ion per subunit and possesses the HExxH zinc-binding motif (Wacker et al. Citation1971; Lehky et al. Citation1973; Malfroy et al. Citation1989). Mammalian APN is a type-II membrane protein with a short intracellular tail that contains the amino terminus, a transmembrane anchoring sequence, a short extracellular stalk, and a large ectodomain with the carboxy terminus; the active site and the zinc-binding domain are located in the extracellular domain (Sjöström et al. Citation2002; Wong et al. Citation2012; Chen et al. Citation2012a).
ANPEP encodes a peptide of 967 amino acids (Olsen et al. Citation1988; Look et al. Citation1989; Watt and Willard Citation1990). The deduced subunit Mr of APN from several organs and species is approximately 110000, but APN is heavily, but variably, N- and O-glycosylated, which accounts for the range of reported molecular masses (Olsen et al. Citation1988; Malfroy et al. Citation1989; Watt and Yip Citation1989; O'Connell et al. Citation1991; Norén et al. Citation1997; Sjöström et al. Citation2002).
Aminopeptidase N is identical with CD13 and acts, therefore, not only as a hydrolytic enzyme but also plays a number of roles in cancer and virus biology (Olsen et al. Citation1988; Look et al. Citation1989; Riemann et al. Citation1999; Pasqualini et al. Citation2000; Abe and Aoyagi Citation2002; Mina-Osorio Citation2008; Haraguchi et al. Citation2010). APN also serves as a receptor for tumor-homing peptides that bear an Asn-Gly-Arg (NGR) motif (Liu et al. Citation2014a). In addition to its catalytic and tumor-homing peptide receptor functions, APN acts as a cell entry receptor for many coronaviruses (Luan and Xu Citation2007). Mapping of coronavirus binding sites, “virus-binding domains,” revealed their locations on the body and head domains on the surface of the APN protein (Chen et al. Citation2012a). Coronavirus binding may lock APN in the open conformation and, thereby, inhibit catalysis (Santiago et al. Citation2017). Crystal structures of complexes formed by the pig APN ectodomain with the receptor binding proteins of two coronaviruses revealed determinants of coronavirus-APN interactions (Reguera et al. Citation2012). Hence, due to its involvement in a myriad of cell functions, APN/CD13 is an attractive therapeutic target, which has led to a search for modulators and imaging strategies (Bhagwat et al. Citation2002; Langner and Ansorge Citation2002; Su et al. Citation2011; Wickström et al. Citation2011; Hahnenkamp et al. Citation2013; Li et al. Citation2014; Drinkwater et al. Citation2017; He et al. Citation2017a; Amin et al. Citation2018).
Crystal structures of the ectodomains of both human and porcine APN have been solved (Reguera et al. Citation2012; Wong et al. Citation2012; Chen et al. Citation2012a; Joshi et al. Citation2017). hAPN is organized into seven domains in each subunit: (I) cytosolic domain; (II) transmembrane domain; (III) O-glycosylation domain; (IV) N-glycosylation domain; (V and VI) active-site domain with HExxHxE motif; and (VII) dimerization domain (Sjöström et al. Citation2002; Luan and Xu Citation2007; Wong et al. Citation2012; Luan et al. Citation2012b). The ectodomain of pAPN is characterized by four domains (head, side, body, and tail) that form a spacious cavity with a wide opening that is accessible to substrates (Chen et al. Citation2012a); the cavity is sufficiently large to accommodate, for example, substance P (RPKPQQFFGLM). pAPN forms a head-to-head dimer each with a transmembrane anchor and can adopt open or closed conformations, the latter of which is catalytically active. The proposed catalytic mechanism of pAPN involves the binding of zinc by His383, His387, and Glu406 (Chen et al. Citation2012a); an active-site water molecule is activated by the Zn and positioned by Glu350 to effect the transfer of a proton to Glu384. Binding of the peptide substrate is accompanied by activation of the carbonyl oxygen by the Zn atom and Tyr472, and by activation of the scissile bond nitrogen by the carbonyl oxygen of Ala348, all of which facilitate the attack of the zinc-activated water molecule on the carbonyl carbon while a proton is transferred from Glu384 to the leaving scissile bond nitrogen. The catalytic mechanism of APN is similar to that of other metallopeptidases having 2His1Glu (or 1Asp) Zn catalytic sites (Mucha et al. Citation2010; Auld Citation2013).
4.2.2.2. Substrates
APN catalyzes the hydrolysis of N-terminal l-amino acids from a range of peptides (Pfleiderer and Celliers Citation1963; Pfleiderer Citation1970; McDonald and Barrett Citation1986; Turner Citation2013); l-amino acids with neutral, bulky hydrophobic residues are preferred substrates. Although Ala is the N-terminal amino acid most readily cleaved by APN, Phe, Met, Tyr, and Leu are also favored substrates (Huang et al. Citation1997; Mina-Osorio Citation2008). l-Amino acid amides and anilides, e.g. 243 (), are substrates but anilides are hydrolyzed at much higher rates (Pfleiderer and Celliers Citation1963; Wachsmuth et al. Citation1966; Rankin et al. Citation1980; Nakanishi et al. Citation1989; Drag et al. Citation2010; Shibata et al. Citation2016). Glu350 in pAPN may serve as an ionic binding site for recognition of the amino group of substrates and inhibitors, as well as contributing to the stabilization of the transition-state intermediate (Luciani et al. Citation1998). APN catalyzes the hydrolysis of N-terminal, neutral l-amino acids from many polypeptides, including enkephalins, angiotensin III and IV, tuftsin, kinins, chemo- and cytokines, extracellular matrix proteins and more (Ward et al. Citation1990; Abe and Aoyagi Citation2002; Albiston et al. Citation2004; Danziger Citation2008). A compilation of APN substrates is available on the MEROPS website (https://www.ebi.ac.uk/merops/cgi-bin/substrates?id=M01.001; accessed 31 October 2019).
After early studies identified a porcine kidney enzyme that catalyzed the hydrolysis of Cys-Gly (see above), it was proposed that APN might catalyze the hydrolysis of Cys-Gly S-conjugates (Hughey et al. Citation1978). It was subsequently demonstrated that APN catalyzes the hydrolysis of many Cys-Gly S-conjugates, including S-(benzyl)-Cys-Gly (240, ), S-(benzyl)-Cys-4-nitroanilide (242, ), and cystinyl-bis-Gly (233, ), S-(methyl)-L-Cys-Gly (248, ), S-(carbamidomethyl-Cys-Gly (250, ), but shows scant or no activity with leukotriene D4 (234, ) (Hughey et al. Citation1978; Rankin et al. Citation1980; Okajima et al. Citation1981; Anderson et al. Citation1982; Kozak and Tate Citation1982; McIntyre and Curthoys Citation1982).
The role of APN in angiogenesis and tumor biology has led to the development of strategies to image APN in cells and in vivo. Briefly, three types of imaging agents have been described: (1) substrate-based agents that are biotransformed by APN to fluorescent products; (2) inhibitor-based agents, whose inhibitory properties direct them to APN; and (3) targeted agents that exploit the affinity of APN for peptides that contain the NGR motif. Given the large number of imaging agents that has been reported, this section will focus on recently described compounds along with citations to related agents. (Note that some cited authors use the name leucine aminopeptidase (LAP) rather APN.)
l-Ala anilide 252 () represents an early ratiometric fluorescent probe for APN that is biotransformed to the self-immolating intermediate 253 () that yields fluorescent 7-hydroxycoumarin (254, ) (Chen et al. Citation2012b). These studies were extended to develop a naphthalimide-based probe with more favorable spectroscopic properties, which allows ratiometric fluorescent imaging of APN in cells (Chen et al. Citation2013b). The l-Ala anilide of 9-aminocresyl violet was used to quantify APN concentrations in urine and in HepG2 and LO2 cells (He et al. Citation2017b). The l-Leu anilide of hydroxymethyl rhodamine green (Leu-HMRG; 255, ) is hydrolyzed by APN to give rhodamine 256 () and was shown to image APN in HeLa cells (Sakabe et al. Citation2013). The l-Ala and l-Leu anilides of 6-aminoluciferin were used to successfully image APN in xenografted luciferase-expressing tumors in nude mice (Li et al. Citation2014); light production was decreased by administration of the APN inhibitor bestatin (267, ). A boron-dipyrromethylene (BODIPY) core bearing a l-Cys-l-Leu moiety (BODIPY-C-Leu) is biotransformed by APN to the BODIPY-Cys intermediate that undergoes a S to N migration to give the highly fluorescent BODIPY-N-Cys (Zhou et al. Citation2017); BODIPY-Leu successfully imaged APN in HeLa cells and in zebrafish, and light production was decreased by the APN inhibitor bestatin (267, ).
Near-infrared (NIR) probes can offer the advantages of large Stokes shifts, high sensitivity, water solubility, and deep tissue penetrability (Gu et al. Citation2016; Zhang et al. Citation2017; He et al. Citation2017a). The l-Ala anilide of amino-hemicyanine 257 () was used to image APN in HepG2 cells and in tumor-bearing nude mice (He et al. Citation2017a). NIR probe 259 () was used to image APN in HCT116 cells and in HCT116 tumor-bearing nude mice (Zhang et al. Citation2017); the intensity of the image was reduced in APN siRNA-transfected HCT116 cells and in mice given bestatin (267, ).
The selectivity of dipeptidase inhibitors has been exploited for the development of imaging probes. A probe for APN that was based on a bestatin (267, ) scaffold and included a fluorophore, biotin, and an alkyne moiety (for click-chemistry applications) has been described, but imaging results were not reported (Harbut et al. Citation2008). A probe based on bestatin (267, ) linked to 4-amino-1,8-naphthalimide (Bes-Green) was shown to inhibit APN and to image APN in ovarian clear cell carcinoma (ES-2) cells (Chen et al. Citation2015). Cyanine dye Cy 5.5 linked to a hydroxamic acid-based APN inhibitor (Cy 5.5-23) has been reported as an imaging probe (Hahnenkamp et al. Citation2013; Chen et al. Citation2013a); Cy 5.5-23 binds to APN-positive BT-549 breast cancer cells but not to APN-negative BT-20 cells, as shown by fluorescence microscopy.
APN shows high affinity for peptides bearing an NGR motif (Pasqualini et al. Citation2000; Corti et al. Citation2017). The cyclic NGR peptide cyclo-[Cys-Asn-Gly-Arg-Cys]-Gly-Lys linked to cyanine dye Cy 5.5 (NGR-Cy 5.5) was designed to bind to APN (von Wallbrunn et al. Citation2008); NGR-Cy 5.5 was shown by fluorescence microscopy to bind to APN-positive HT-1080 cells, but not to APN-negative MCF-7 cells, and to visualize APN in HT-1080, but not in MCF-7, tumor xenografts in nude mice by 2D planar fluorescence reflectance imaging and by 3D fluorescence-mediated tomography.
APN also catalyzes the activation of prodrugs. The melphalan (26, ) prodrug melflufen (261, ) undergoes APN-catalyzed biotransformation to release melphalan (26, ) (Wickström et al. Citation2010; Strese et al. Citation2013; Ray et al. Citation2016); the cytotoxicity of prodrug 261 () is reduced by the dipeptidase inhibitor bestatin (267, ). Clinical trials to test the efficacy of melflufen (261, ) for the treatment of multiple myeloma are currently in progress (clinicaltrials.gov) (Wickström et al. Citation2017). Hybrid prodrugs of 5-fluorouracil (5-FU) and gemcitabine linked to bestatin (267, ) are expected to be cleaved by plasma hydrolases to release the active antitumor agents and the APN inhibitor bestatin (267, ) (Jiang et al. Citation2016a, Citation2016b); the compounds inhibited APN and exhibited antitumor and antiangiogenic effects in several cellular and in vivo models. APN-activated glycinamide-based prodrugs of a panel of vascular disrupting agents that inhibit tubulin polymerization have been described (Devkota et al. Citation2016); several of these compounds were cytotoxic in cancer cell lines.
The 5-fluorouracil (5-FU) prodrug 263 (5-FU-NGR, ) is based on the affinity of NGR peptides for APN (Luan et al. Citation2012a; Cui et al. Citation2014b). The labile acyloxy-methylene linker is designed to release 5-FU (Menger and Rourk Citation1997); moreover, the NGR peptide was modified to include an Arg-(ω-NO2)-COOCH3 moiety to inhibit nitric oxide synthase. 5-FU-NGR (263, ) inhibited growth of Colo205 cells, delayed expansion of Colo205 xenografts in nude mice, induced apoptosis in Colo205 cells and xenografts, and inhibited APN expression. The affinity of APN for NGR peptides has been exploited to target peptide-conjugates, e.g. TNFα (Corti and Ponzoni Citation2004; Gregorc et al. Citation2010; Corti et al. Citation2017). Indeed, the NGR-hTNF conjugate is being investigated in clinical trials for the treatment of hepatocellular carcinoma (Santoro et al. Citation2010).
4.2.2.3. Inhibitors
The putative role of APN in tumor progression metastasis has stimulated the search for inhibitors, particularly small-molecule inhibitors. Consequently, a large number of APN inhibitors have been reported. These inhibitors should also find use in exploring the role of APN in the mercapturic acid pathway. This section will focus on more recently reported compounds. Reviews of APN inhibitors are available (Shimazawa et al. Citation1999; Abe and Aoyagi Citation2002; Fournié-Zaluski and Roques Citation2002; Xu and Li Citation2005; Bauvois and Dauzonne Citation2006; Luan and Xu Citation2007; Zhang and Xu Citation2008; Mucha et al. Citation2010; Su et al. Citation2011; Wickström et al. Citation2011; Zhang et al. Citation2011b; Luan et al. Citation2012b; Hitzerd et al. Citation2014; Drinkwater et al. Citation2017; Amin et al. Citation2018). A compilation of APN inhibitors is available on the MEROPS website (https://www.ebi.ac.uk/merops/cgi-bin/pepsum?id=M01.001; accessed 31 October 2019).
The structural requirements for APN inhibitors have been summarized (Drag et al. Citation2010; Halder et al. Citation2013; Amin et al. Citation2018): briefly, APN inhibitors are typically characterized by hydrophobic groups that are accommodated by the S1, S1′, and S2′ sites and by a zinc-binding group (ZBG), including hydroxamic acids, thiols, boronic acid, phosphonic acids, and β-dicarbonyl compounds. These requirements are met by a range of scaffolds that have been exploited to identify potent and selective APN inhibitors.
APN is inhibited by a range of natural-product inhibitors, including bestatin (267, ), amastatin (268, ), actinonin, leuhistin, and probestin (Bauvois and Dauzonne Citation2006). Co-crystallization of zinc-bound hAPN with the inhibitors bestatin (267, ) and amastatin (268, ), both of which contain a nonhydrolyzable α-hydroxy-β-amino acid moiety, showed that the α-hydroxyl group of amastatin (268, ) coordinates with the zinc ion whereas bestatin (267, ) resides deep in the active-site pocket, where its α-hydroxyl, amino, and carbonyl groups form only water-mediated hydrogen bonds with hAPN amino-acid residues (Wong et al. Citation2012). LYP, the dimethylaminoethyl ester of bestatin (267, ), is a more potent inhibitor of APN than bestatin (267, ), inhibits the growth of ovarian carcinoma ES-2 and SKOV-3 cells, inhibits angiogenesis in ES-2 xenografts in mice, and delays growth of the ES-2 xenografts (Luan et al. Citation2009; Gao et al. Citation2011a, Citation2011b). hAPN is inhibited by the bile acids chenodeoxycholic acid and cholic acid, particularly the unconjugated acids, and by metal chelating agents (Nakanishi et al. Citation1989; Vanderheyden et al. Citation2006).
The conjugated β-dicarbonyl compound, curcumin (69, ), is a noncompetitive, irreversible inhibitor of purified APN and also inhibits APN activity in human umbilical vein endothelial cells and in HT1080 fibrosarcoma cells; curcumin (69, ) inhibits both angiogenesis and tumor cell invasion by APN-positive cells (Shim et al. Citation2003). Other β-dicarbonyl compounds, which were designed to act as zinc-binding agents and to interact with key active-site residues, also are effective APN inhibitors (Ma et al. Citation2013).
Hydroxamic acids, because of their zinc-ligand-forming ability, have been studied as inhibitors of APN (Chan et al. Citation1982; Flipo et al. Citation2007; Mucha et al. Citation2010). The hydroxamic acid tosedostat (CHR-2797; 272, ) is an orally bioavailable APN inhibitor, as well as an inhibitor of several other aminopeptidases. Tosedostat is biotransformed to a carboxylic acid metabolite that is also an effective aminopeptidase inhibitor. Tosedostat has been investigated in clinical trials for its antitumor activity (clinicaltrials.gov) (DiNardo and Cortes Citation2014). Hydroxamic acid moieties have been incorporated into a wide variety of chemical scaffolds that have yielded effective APN inhibitors (Inagaki et al. Citation2010; Li et al. Citation2012; Su et al. Citation2012a; Hahnenkamp et al. Citation2013; Jin et al. Citation2013; Su et al. Citation2013; Zhang et al. Citation2013; Cui et al. Citation2014a; Hou et al. Citation2016; Ma et al. Citation2016; Cao et al. Citation2018).
Phosphorous-containing inhibitors, particularly α-aminoalkylphosphonic acid and -phosphinic acids have been studied as APN inhibitors (Mucha et al. Citation2010; Amin et al. Citation2018). Phosphinic acids (phosphinates) are of interest because they function as transition-state analog inhibitors (Amin et al. Citation2018). For example, phosphinic acid 273 () is a potent inhibitor of hAPN (IC50 = 60 nM) (Grzywa et al. Citation2010a). Other phosphinic and phosphonic acid inhibitors of APN have been reported, some of which exhibit IC50 or Ki values in the low nM range (Chen et al. Citation1999; Drag et al. Citation2010; Grzywa et al. Citation2010b; Mucha et al. Citation2011; Mucha Citation2012; Niu et al. Citation2015).
Some early studies of APN inhibitors focused on agents that are Leu-derived amino acid analogs, including l-Leu thiol 274 (), l-leucinal (275, ), and Leu-B(OH)2-pinacol (276, ) (Andersson et al. Citation1982; Shenvi Citation1986). There has been considerable interest in l-leucine-ureido derivatives as APN inhibitors. These compounds build on the structure of bestatin (267, ), which also contains a l-Leu component whose sidechain is accommodated by the S1′ site of APN, along with the addition of a hydroxamic acid moiety as a ZBG (Wong et al. Citation2012). l-Leucine-ureido analog 278 (), for example, is a potent in vitro inhibitor of pAPN (IC50 = 20 nM), inhibits hAPN in ES-2 human ovarian cells, blocks invasion by ES-2 cells, and blocks pulmonary metastasis in mice bearing H22 tumor cells (Ma et al. Citation2016). A number of other l-Leu-ureido-based APN inhibitors, many of which contain a hydroxamic acid zinc binding group, have been reported (Su et al. Citation2012a, Citation2012b, Citation2013).
3-Amino-2-tetralone derivatives have been identified as APN inhibitors. Tetralone 279 () is a potent, slow-binding inhibitor of APN (Ki = 20 nM) but is a much weaker inhibitor of LAP (Ki = 3 μM) (Schalk et al. Citation1994). The structurally related aminobenzosuberones are also selective inhibitors of APN (Albrecht et al. Citation2011; Maiereanu et al. Citation2011; Albrecht et al. Citation2012; Revelant et al. Citation2015). Aminobenzosuberone 280 () is a selective and potent inhibitor of pAPN and hAPN (Ki = 0.06 and 0.035 nM, respectively) (Peng et al. Citation2017). The amino group is required for inhibition, and binding interactions of the hydrated ketone group mimic stabilization interactions of the transition state for peptide bond hydrolysis. In hAPN, a C-Br⋅⋅⋅π interaction with F896 is thought to contribute to the high binding affinity. Studies on the cancer chemotherapeutic potential of aminobenzosuberone 280 () show that it inhibits capillary tube formation, lowers cell migration, and retards growth of xenograft tumors (Schmitt et al. Citation2013); the maximum total tolerated dose (MTTD) in mice was estimated to be 40 mg/kg when given every third day for a total of eight doses.
An additional number of APN inhibitors have been reported, but most are less potent than the compounds described above. These include flavone derivatives, cinnamoyl pyrrolidines, cyclic imides, β-amino-α-hydroxyphenylbutanoic acids, indoline-2,3-diones, and 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acids (for details about these compounds, see Mucha et al. Citation2010; Amin et al. Citation2018). Dihydroisoquinoline 282 () inhibits APN and shows antiproliferative effects in several cell lines (Ziemska et al. Citation2016).
4.2.2.4. Tissue and species distribution
APN is widely distributed across mammalian species and is also found in avian species (Sjöström et al. Citation2002). The membrane-bound APN is present in nearly all tissues, including kidney, intestine, heart, lung, eye, and thymus, skin and respiratory tract (Hughey et al. Citation1978; Sjöström et al. Citation1978; Hansen et al. Citation1987; Nakanishi et al. Citation1989; Dixon et al. Citation1994; Danielsen et al. Citation1995; Jardinaud et al. Citation2004). APN is widely expressed in brain tissue, which has led to the proposal that it may play a role in hypertension (Danziger Citation2008). APN is expressed in the brush-border of fetal porcine enterocytes (Danielsen et al. Citation1995). Immunohistochemical studies showed the presence of APN in bile canaliculi (Röcken et al. Citation2004, Citation2005). APN is found in rat and guinea pig kidney brush-border membranes (Rankin et al. Citation1980; Terashima et al. Citation1992). In rabbit kidney, in situ hybridization studies demonstrated the presence of APN mRNA in proximal convoluted tubules, but not in glomeruli (Yang et al. Citation1993). Immunoelectron microscopic analysis demonstrated a high level of APN expression on the lumenal surfaces of normal human proximal tubular cells and renal cancer cells (Stange et al. Citation2000).
4.2.2.5. Other biological functions
Given the range of functions associated with APN in addition to catalysis of the hydrolysis of Cys-Gly and Cys-Gly S-conjugates, APN (CD13) has been branded as a “…moonlighting enzyme…” (Mina-Osorio Citation2008). Upon ligand binding, CD13 functions as an enzyme, receptor, and signaling molecule, and these functions depend on peptide cleavage, endocytosis, and signaling. APN also serves as a receptor for a range of viruses (Yeager et al. Citation1992; Delmas et al. Citation1994). The enzymatic activity of APN is not required for interaction of the transmissible gastroenteritis virus with the virus receptor (Delmas et al. Citation1994).
APN was considered to be a possible binding target of the cholesterol absorption inhibitor ezetimibe (Kramer et al. Citation2005). Subsequent studies showed conclusively that Niemann-Pick C1-Like 1 (NPC1L1) protein is the molecular target of ezetimibe (Garcia-Calvo et al. Citation2005; Betters and Yu Citation2010; Jia et al. Citation2011).
4.3. Cytosolic dipeptidases
One cytosolic dipeptidase has been demonstrated to catalyze the hydrolysis of both Cys-Gly and Cys-Gly S-conjugates: leucyl aminopeptidase.
4.3.1. Leucyl aminopeptidase (LAP, EC 3.4.11.1)10
The first reference to an enzyme that would later be identified as LAP was the description of an intestinal peptidase that catalyzed the hydrolysis of Leu-Gly and other leucyl dipeptides (Linderstrøm-Lang and Sato Citation1929; Holter Citation1979).
Although APN catalyzes the hydrolysis of Cys-Gly (see above), other studies show that LAP is the major liver enzyme that catalyzes the hydrolysis of Cys-Gly and Cys-Gly S-conjugates, but its activity in other organs was not reported (Jösch et al. Citation1998, Citation2003). LAP is a metallopeptidase assigned to the Family M17 (MEROPS ID M17.001) (Rawlings et al. Citation2018). Reviews about LAP are available (Taylor Citation1993a, Citation1993b; Kim and Lipscomb Citation1994; Sträter and Lipscomb Citation2013).
4.3.1.1. Structure and properties
Enzymes that catalyze the hydrolysis of Leu dipeptides and Leu amides and anilides have been purified from several sources, including porcine intestine and kidney (Smith and Bergmann Citation1944; Spackman et al. Citation1955), rat liver (Tamura et al. Citation1973), and bovine lens (Glässer and Hanson Citation1964; Hanson et al. Citation1965; Kretschmer and Hanson Citation1965; Melbye and Carpenter Citation1971; Hanson and Frohne Citation1976; Cuypers et al. Citation1982). Human, bovine, and porcine lens LAP (hl-, bl-, and plLAP) and bovine and porcine kidney LAP (bk- and pkLAP) show similar immunoreactivity (Taylor et al. Citation1984a, Citation1984b; Oettgen and Taylor Citation1985). LAP protein amounts to approximately 0.05% of human, bovine, and porcine lens protein (Taylor et al. Citation1984a). LAP3 and Lap3 encode the human and murine LAP proteins, respectively (Cunningham et al. Citation2015).
LAP is a hexameric protein, whose subunit molecular mass is 52–54 kDa (Melbye and Carpenter Citation1971; Carpenter and Harrington Citation1972; Kohno et al. Citation1986; Burley et al. Citation1992); protein analysis, cross-linking studies, and X-ray crystallographic studies indicate that LAP is arranged as a dimer of two trimers. There is one active site per subunit, as shown by bestatin (267, ) binding (Taylor et al. Citation1993). Each of the six blLAP subunits binds two divalent metal ions: one of the metal binding sites (site 1, Zn488 in blLAP) is readily exchangeable and can bind Zn2+, Mn2+, Mg2+, or Co2+, whereas the tight-binding site (site 2, Zn489 in blLAP) binds only Zn2+ or Co2+ (Carpenter and Vahl Citation1973; Kim and Lipscomb Citation1993a; Lowther and Matthews Citation2002; Cappiello et al. Citation2006). Metal-ion occupation of both sites is required for activity, and both metal-binding sites are occupied by Zn2+ in native blLAP (Carpenter and Vahl Citation1973; Allen et al. Citation1983; Kim and Lipscomb Citation1993b). In native blLAP, Zn488 forms coordinate bonds with the side-chain carboxylate oxygens of Asp255, Asp332, and Glu334 and the carbonyl oxygen of Asp332; Zn489 forms coordinate bonds with the side-chain carboxylates of Asp255, Asp273, and Glu334 and to the side-chain amine of Lys250 (Burley et al. Citation1992). The distance between the zinc ions is 2.88 Å (Burley et al. Citation1990).
The substrate selectivity and turnover and susceptibility to inhibition of LAP is influenced by the species of origin, i.e. bovine lens or porcine kidney, and by the metals that occupy the metal-binding sites (Lowther and Matthews Citation2002). Purified blLAP contains two Zn2+ ions per subunit, whereas purified pkLAP contains only one Zn2+ ion per subunit (Himmelhoch Citation1969; Carpenter and Vahl Citation1973; Van Wart and Lin Citation1981). Although there are two metal binding sites in pkLAP, the metal affinity of the second site is apparently sufficiently low to prevent purification of the protein with two bound metals (Lin et al. Citation1988). A particularly interesting finding is that the hydrolysis of Cys-Gly is catalyzed by Mn2+Zn2+ blLAP but not by Zn2+Zn2+ blLAP (Cappiello et al. Citation2006). The kcat for the hydrolysis of l-Leu-Gly-NHNH-Dns (264, ) is approximately threefold higher with Zn2+Mn2+ pkLAP than with Zn2+Zn2+ pkLAP (Lin et al. Citation1988). Metal-ion substitution affects both Km and kcat (Allen et al. Citation1983). As indicated above, rates of biotransformation vary with the metal substitution but under in vivo conditions where [S] ≪ Km the effects of metal substitution are likely to be small (Lin et al. Citation1988; Lowther and Matthews Citation2002).
Crystal structures of native pk- and blLAP (Jurnak et al. Citation1977; Taylor et al. Citation1984b; Burley et al. Citation1992) and of blLAP in complex with bestatin (267, ) (Burley et al. Citation1990, Citation1991, Citation1992; Kim et al. Citation1993), amastatin (268, ) (Kim and Lipscomb Citation1993b), l-leucinal (275, ) (Sträter and Lipscomb Citation1995b; Sträter et al. Citation1999), l-leu phosphonic acid (277, ) (Sträter and Lipscomb Citation1995a; Sträter et al. Citation1999; Grembecka et al. Citation2003), zofenoprilat (281, ) (Cappiello et al. Citation2006), and microginin FR1 (Kraft et al. Citation2006) have been solved. X-Ray crystallographic studies confirm the hexameric structure of pkLAP (Taylor et al. Citation1984b). The N-terminal domain (160 amino acid residues) of blLAP is associated with trimer-trimer interactions but is not involved in catalysis (Burley et al. Citation1992); the C-terminal domain (327 amino acid residues) contains the two zinc ions and is responsible for catalysis. The N- and C-terminal domains are linked by an α-helix. The active sites are located on the interior of the hexamer and line a disk-shaped cavity (Burley et al. Citation1990); access to the cavity is provided by solvent channels. There are two solvent channels near the zinc ions, the larger of which affords substrate access to the active site (Erhardt et al. Citation2005; Jaime et al. Citation2007 (pt iii)).
The proposed catalytic mechanism of LAP involves the interaction of both Zn2+ ions, which function in a co-catalytic manner (Kim and Lipscomb Citation1994; Sträter and Lipscomb Citation1995b; Lipscomb and Sträter Citation1996; Lowther and Matthews Citation2002; Schürer et al. Citation2002; Weston Citation2005; Zhu et al. Citation2012). The two Zn2+ ions are located in close proximity (<3 Å) and are associated with a bridging water or a hydroxyl ligand that functions as a nucleophile (Paul et al. Citation2016). The Zn1 atom serves to polarize the substrate carbonyl group (Sträter and Lipscomb Citation1995b; Sträter et al. Citation1999). Zn2 functions to position the substrate in the active site through coordinate bonding with the terminal amino group of the substrate. Attack of the nucleophile on the carbonyl group of the substrate leads to the formation of a gem-diolate transition state, resulting in product formation (Jaime et al. Citation2007 (pt iii)). The nature of the metallohybrids affect only the formation of the gem-diolate but not the subsequent steps in the catalytic cycle (Zhu et al. Citation2012). An active site bicarbonate ion functions as a proton donor and protonates the newly released amino group of the product.
4.3.1.2. Substrates
The hydrolysis of amino terminal prolyl di- and tripeptides and polypeptides, e.g. melanostatin (Pro-Leu-Gly-NH2), by enzymes partially purified from bovine and porcine kidney led to the proposal that a distinct prolyl aminopeptidase (EC 3.4.11.5) catalyzes these reactions (Nordwig and Mayer Citation1973; Khilji and Bailey Citation1978). Later studies showed, however, that the putative prolyl aminopeptidase is identical with LAP (Turzynski and Mentlein Citation1990; Matsushima et al. Citation1991).
LAP shows high selectivity for peptides with N-terminal, hydrophobic l-amino acid substituents, e.g. l-Met >l-Leu > l-Phe > l-Val > l-Ala > Gly (Hanson and Frohne Citation1976; Kohno et al. Citation1986; Cappiello et al. Citation2004). Peptides with d-amino acids in the P1′ site, e.g. l-Leu-d-Leu, are cleaved but at very low rates (Smith and Spackman Citation1955; Yamada and Kera Citation1998). Although early studies demonstrated the role of APN and DPEP1 in particulate fractions of kidney in the metabolism of Cys-Gly and Cys-Gly S-conjugates, the finding that rat liver cytosolic LAP also catalyzes the hydrolysis of Cys-Gly and Cys-Gly S-conjugates showed that it plays a role in the γ-Glu cycle (Jösch et al. Citation2003). blLAP catalyzes the hydrolysis of Cys-Gly, which is attributed to the Zn2+Mn2+ metallohybrid (Cappiello et al. Citation2004, Citation2006). The results of a molecular modeling analysis attribute the failure of (Zn/Zn)blLAP to catalyze the hydrolysis of Cys-Gly to the interaction of the thiol group of Cys-Gly with Zn1, which displaces the water molecule required for catalysis; in contrast, the Mn2+ ion in (Mn/Zn)blLAP prefers to interact with oxygen- or nitrogen-containing ligands rather than sulfur-containing ligands, which allows catalysis to proceed.
In addition to catalyzing the hydrolysis of Cys-Gly and Cys-Gly S-conjugates, LAP shows broad substrate selectivity for dipeptides and some tripeptides (for compilations of LAP substrates, see Smith and Bergmann Citation1944; Smith and Slonim Citation1948; Smith and Polglase Citation1949; Smith and Spackman Citation1955; Kohno et al. Citation1986). Leucinamide is a specific substrate for porcine liver and ELD mouse ascites carcinoma cells LAP (Patterson et al. Citation1963; Ledeme et al. Citation1981). The chromogenic substrates l-Leu-4-nitroanilide (243, ) and l-Leu-β-naphthylamide are poor substrates for human or porcine liver LAP compared with l-leucinamide (Ledeme et al. Citation1981; Kohno et al. Citation1986).
Jösch et al. (Citation1998) demonstrated the hydrolysis of the Cys-Gly S-conjugate of monochlorobimane (3, ) by rat and guinea pig liver homogenates. Studies with rat and guinea pig liver subcellular fractions showed that the highest specific activity for the hydrolysis of bimane-Cys-Gly (3, ) is found in the cytosolic fraction, although lower activity is present in the microsomal fraction (Jösch et al. Citation2003); immunoprecipitation, substrate selectivity, and inhibition studies identified LAP as the enzyme that catalyzes the hydrolysis of the Cys-Gly S-conjugate of monochlorobimane (3, ). The specific activity for the hydrolysis of bimane-Cys-Gly (3, ) in sinusoidal plasma membranes was low, whereas the specific activity of canalicular plasma membranes amounted to 52–67% of the cytosolic fraction.
pkLAP catalyzes the hydrolysis of Leu-Gly-NHNH-Dns (264, ) (Lin and Van Wart Citation1988); the fluorescence changes are attributed to direct excitation of the dansyl group rather than energy transfer between pkLAP and the substrate. The (kcat/Km) × 10−6 (min−1 M−1) for the biotransformation of hydrazide 264 () varies little with metal substitution, i.e. Zn, Mg, Mn, Ni, Cu (Lin et al. Citation1988).
4.3.1.3. Inhibitors
Bestatin (267, ) is a potent, nonhydrolyzable inhibitor of LAP (Suda et al. Citation1976; Umezawa et al. Citation1976; Rich et al. Citation1984; Wilkes and Prescott Citation1985; Kohno et al. Citation1986). Kinetic analysis shows that bestatin (267, ) is a slow, very tight binding inhibitor of pkLAP (Ki (koff/kon = 0.58 nM) (Wilkes and Prescott Citation1985). The crystal structure of blLAP in complex with bestatin (267, ) shows that the α-amino group of the inhibitor interacts with Zn2 and the 2-hydroxyl group binds to both Zn1 and Zn2, and that the Phe and Leu sidechains occupy the S1 and S1′ hydrophobic pockets near the active site (Burley et al. Citation1991, Citation1992; Kim et al. Citation1993; Kim and Lipscomb Citation1994). The photoaffinity label, p-azidobestatin is also a slow, tight binding inhibitor of blLAP (Taylor et al. Citation1992).
Amastatin (268, ) is a strong inhibitor of LAP (Rich et al. Citation1984; Wilkes and Prescott Citation1985); kinetic analysis shows that it is a slow, tight-binding inhibitor of pkLAP (Ki (koff/kon = 3.0 × 10−2 μM) (Wilkes and Prescott Citation1985). The slow binding of bestatin (267, ) and amastatin (268, ) to blLAP may be attributed interactions between the α-hydroxyl-β-amino acid residue and the P1–P3′ residues of the inhibitor (Rich et al. Citation1984). The crystal structure of amastatin (268, ) in complex with blLAP is similar to that of bestatin (267, ) and is supportive of a catalytic mechanism in which the transition state involves the formation of a tetrahedral gem-diolate (Kim and Lipscomb Citation1993b).
l-Leucinal (275, ) is a potent transition-state inhibitor of pkLAP (Ki = 6 × 10−2 μM) (Andersson et al. Citation1982). The crystal structure of blLAP complexed with l-leucinal (275, ) shows that the transition-state inhibitor forms a gem-diolate intermediate with one hydroxyl group bridging the active site metal ions and the other hydroxyl group binds to Zn1 (Sträter and Lipscomb Citation1995b).
L-Leu phosphonic acid (277, ) is a strong inhibitor of pkLAP (Ki = 0.23 μM) and a weak inhibitor of APN (Ki = 53 μM) whereas the phosphinic acid analog is much weaker (Ki = 87 μM) (Giannousis and Bartlett Citation1987; Lejczak et al. Citation1989). The crystal structure of blLAP in complex with phosphonic acid 277 () shows that the amino group interacts with Zn2 (Zn489), that one phosphoryl oxygen forms a bridge with both Zn ions, and that one phosphoryl oxygen binds to Zn1 (Zn488) (Sträter and Lipscomb Citation1995a). The availability of the crystal structure of blLAP in complex with phosphonic acid 277 () (PDB 1LCP) was exploited in a Ligand Design (LUDI) approach to design new blLAP inhibitors (Grembecka et al. Citation2000). An extension of these studies identified phosphinate dipeptide transition-state analogs as selective inhibitors of blLAP compared with APN (Grembecka et al. Citation2003). A range of theoretical studies aimed at predicting characteristics of LAP inhibitors have been reported (Grembecka et al. Citation1999, Citation2001, Citation2002).
3D-QSAR, docking, and toxicology prediction studies on a large panel of isoquinoline and phosphonic and phosphinic acid derivatives as inhibitors of bl- and pkLAP have been conducted, but no biological studies have apparently been reported (Ziemska et al. Citation2017).
Thio and thiono analogs of LAP substrates and inhibitors have been investigated. l-Leu thiol (274, ) and l-lysine thiol are weak inhibitors of pkLAP (Ki = >500 μM) (Ocain and Rich Citation1987). α-Thiolbestatin is a less potent inhibitor of LAP than bestatin (267, ) (Ocain and Rich Citation1988). The thiono analog of anilide 243 () is a weak inhibitor of pkLAP (Ki = 0.19 mM) but is resistant to pkLAP-catalyzed hydrolysis (Beattie et al. Citation1987). The effect of metallohybrid composition on the inhibitory action of the ACE inhibitor zofenprilat (281, ) on blLAP has been investigated (Cappiello et al. Citation2006). Zofenprilat (281, ) is a poor inhibitor (Ki = 1.3 mM) of the (Mn/Zn)blLAP-catalyzed hydrolysis of Cys-Gly but is a potent inhibitor (Ki = 0.8 μM) of the (Zn/Zn)blLAP-catalyzed hydrolysis of Leu-Gly; the inhibition of (Zn/Zn)blLAP by zofenprilat (281, ) is attributed to the interaction of the thiol group with both Zn2+ ions. Indeed, the crystal structure of blLAP in complex with zofenprilat (281, ) shows that its thiol group displaces the water molecule required for catalysis.
The pentapeptide microginin FR1 inhibits blLAP (IC50 = 1.3 μM) (Kraft et al. Citation2006). The crystal structure of blLAP in complex with microginin FR1 shows that the amino group of the 3-amino-2-hydroxydecanoic acid sidechain coordinates with one of the active-site Zn ions and that the hydroxyl group interacts with Zn2 and with the active-site water molecule.
4.3.1.4. Tissue and species distribution
LAP is present in all mammalian species studied. The LAP found in bovine and porcine lens and kidney and in human lens all share antigenic determinants, but are not identical (Taylor et al. Citation1984a); within organs of a single species, however, the LAP proteins appear to be identical.
Human LAP is present in kidney, liver, duodenum, ileum, colon, rectal tissue, submaxillary gland, and lung (Nachlas et al. Citation1960; Tamura et al. Citation1975). LAP activities in hepatocellular carcinoma and lung squamous cell carcinoma tissue are lower than that in normal liver and lung tissue, whereas activity in stomach adenocarcinoma tissue is higher than in normal stomach tissue (Tamura et al. Citation1975). LAP is also found in human peripheral lymphocytes and polymorphonuclear leukocytes (Smith et al. Citation1983; Kohno and Kanno Citation1985). LAP is present in porcine muscle but the activity is low compared with kidney (Joseph and Sanders Citation1966). Rabbit LAP is present in renal proximal tubules, but not in glomeruli (Yang et al. Citation1993).
4.4. Cellular and in vivo hydrolysis of Cys-Gly and Cys-Gly S-conjugates
Although xenobiotic-derived Cys-Gly S-conjugates are obligate intermediates in the mercapturic acid pathway, few reports are available that identify the dipeptidases that catalyze their in vivo hydrolysis to Cys S-conjugates. Given that at least three peptidases catalyze the hydrolysis of Cys-Gly and Cys-Gly S-conjugates, there is often some uncertainty about the roles of these peptidases in Cys-Gly S-conjugate metabolism.
Jones et al. (Jones et al. Citation1978; Moldéus et al. Citation1978; Jones et al. Citation1979) investigated the fate of GSH conjugates in isolated rat liver and kidney cells: incubation of acetaminophen with liver cells led to the formation of a GSH S-conjugate; when the liver cells were removed by centrifugation and kidney cells were added, there was a rapid formation of the Cys S-conjugate of acetaminophen, which was gradually converted to the mercapturic acid. Addition of Phe-Gly, as a competitive dipeptidase inhibitor, to the incubation mixture inhibited the conversion of the GSH S-conjugate to the cysteine conjugate and led to the accumulation of the Cys-Gly adduct, indicating a role for one or more dipeptidases in the biotransformation of GSH S-conjugates.
Studies of MBDm1/MBDm1 mice with the targeted deletion of the membrane dipeptidase show that multiple dipeptidases catalyze the hydrolysis of leukotriene D4 (162, ) and cystinyl-bis-glycine (161, ): rates of hydrolysis of leukotriene D4 (162, ) are 86% in small intestine, approximately 35% in lung, heart, and pancreas, and 16% in kidney of the rates observed in wild-type mice (Habib et al. Citation1998). Similarly, the rate of hydrolysis of cystinyl-bis-glycine (161, ) in MBDm1/MBDm1 mice is about 40% of that observed in wild-type mice.
Perfusion of rat liver with bimane-Cys-Gly (3, ) led to the formation of bimane-mercapturate (5, ) (30% of infused amount) and a trace of bimane-Cys (4, ), indicating the dipeptidase-dependent biotransformation of the Cys-Gly S-conjugate (Jösch et al. Citation1998).
5. Cysteine S-conjugate N-acetyltransferase (NAT8)11
5.1. Introduction
NAT8 (EC 2.3.1.80) catalyzes the reaction of a Cys S-conjugate (4, ) with acetyl-CoA to give a mercapturic acid (5, ). The N-acetylation of Cys S-conjugates is a common reaction for the terminal processing of GSH S-conjugates (Chasseaud Citation1976). Moreover, biotransformation of nephrotoxic Cys S-conjugates to mercapturic acids is an important detoxication reaction, because the N-acetylated Cys S-conjugates do not undergo Cys S-conjugate β-lyase-dependent bioactivation (Anders and Dekant Citation1998).
Early studies provided indirect evidence for an N-acetylation step in mercapturic acid formation: dogs given bromobenzene (6, ) excreted S-(bromophenyl)mercapturic acid (10, ) (Baumann and Preusse Citation1879; Jaffé Citation1879). The conversion of benzyl chloride and S-(benzyl)- l-Cys (241, ) to S-(benzyl)mercapturic acid (283, ) by dogs, rabbits, and rats was also reported (Stekol Citation1938). When S-(4-bromophenyl)-l-Cys or S-(benzyl)-l-Cys (241, ) was given to human subjects, the corresponding mercapturic acids were excreted in the urine (Stekol Citation1946). The administration of several Cys S-conjugates to rats, rabbits, or guinea pigs also led to the excretion of the corresponding mercapturic acids (West and Mathura Citation1954; Bray et al. Citation1959b); moreover, incubation of the S-conjugates with liver slices also resulted in mercapturic acid formation. Elce (Citation1970) was apparently the first to demonstrate the role of acetyl-CoA in mercapturic acid formation.
Figure 23. NAT8 and aminoacylase substrates. 283, N-acetyl-S-(benzyl)-l-Cys; 284, S-(1,2-dichlorovinyl)-l-Cys (DCVC); 285, N-acetyl-S-(1,2-dichlorovinyl)-l-Cys; 286, S-(2-chloro-1,1,2-trifluoroethyl)-l-Cys (CTFC); 287, N-acetyl-S-(2-chloro-1,1,2-trifluoroethyl)-l-Cys; 288, S-[2-(fluoromethoxy)-1,1,3,3,3-pentafluoropropyl]-l-Cys; 289, N-acetyl-S-[2-(fluoromethoxy)-1,1,3,3,3-pentafluoropropyl]-l-Cys; 290, S-[2-(fluoromethoxy)-1,3,3,3-tetrafluoro-1-propenyl]-l-Cys; 291, N-acetyl-S-[2-(fluoromethoxy)-1,3,3,3-tetrafluoro-1-propenyl]-l-Cys; 292, 2-bromo-6-(l-cystein-S-yl)hydroquinone; 293, N-acetyl-2-bromo-6-(l-cystein-S-yl)hydroquinone, 294, S-[2-hydroxy-1-(2-oxoethyl)heptyl]-l-Cys (4-HNE l-Cys conjugate); 295, N-acetyl-S-[2-hydroxy-1-(2-oxoethyl)heptyl]-l-Cys (4-HNE l-Cys mercapturic acid); 296; N-[3-(2-furyl)acryloyl]-l-Met; 297, 3-(2-furyl)acrylic acid; 298, 2-acetamidoacrylic acid; 299, 2-aminoacrylic acid.
![Figure 23. NAT8 and aminoacylase substrates. 283, N-acetyl-S-(benzyl)-l-Cys; 284, S-(1,2-dichlorovinyl)-l-Cys (DCVC); 285, N-acetyl-S-(1,2-dichlorovinyl)-l-Cys; 286, S-(2-chloro-1,1,2-trifluoroethyl)-l-Cys (CTFC); 287, N-acetyl-S-(2-chloro-1,1,2-trifluoroethyl)-l-Cys; 288, S-[2-(fluoromethoxy)-1,1,3,3,3-pentafluoropropyl]-l-Cys; 289, N-acetyl-S-[2-(fluoromethoxy)-1,1,3,3,3-pentafluoropropyl]-l-Cys; 290, S-[2-(fluoromethoxy)-1,3,3,3-tetrafluoro-1-propenyl]-l-Cys; 291, N-acetyl-S-[2-(fluoromethoxy)-1,3,3,3-tetrafluoro-1-propenyl]-l-Cys; 292, 2-bromo-6-(l-cystein-S-yl)hydroquinone; 293, N-acetyl-2-bromo-6-(l-cystein-S-yl)hydroquinone, 294, S-[2-hydroxy-1-(2-oxoethyl)heptyl]-l-Cys (4-HNE l-Cys conjugate); 295, N-acetyl-S-[2-hydroxy-1-(2-oxoethyl)heptyl]-l-Cys (4-HNE l-Cys mercapturic acid); 296; N-[3-(2-furyl)acryloyl]-l-Met; 297, 3-(2-furyl)acrylic acid; 298, 2-acetamidoacrylic acid; 299, 2-aminoacrylic acid.](/cms/asset/426acd04-b723-499d-a203-395161038d58/itxc_a_1692191_f0023_b.jpg)
5.2. Structure and properties
NAT8 was purified approximately three-fold from rat kidney microsomes, but the lability of the enzyme prevented further purification (Duffel and Jakoby Citation1982, Citation1985). An approximately 6-fold purification of the enzyme with a detergent-solubilization procedure has been reported, but again considerable activity was lost during purification (Aigner et al. Citation1996). Protein analysis indicated that NAT8 appeared to be a homodimer with a subunit molecular mass of 34 kDa, although the results of a later study indicated that NAT8 was likely a minor component of the enzyme preparation analyzed by Aigner et al. (Veiga-da-Cunha et al. Citation2010). An improved, detergent-based purification procedure that yielded an approximately 60-fold purification of pig kidney NAT8 has been reported, but the preparation contained considerable acetyl-CoA hydrolase activity (Kraus et al. Citation2000).
A search for the N-acetyltransferase that catalyzes the N-acetylation of Lys residues in β-site amyloid precursor protein cleaving enzyme 1 (BACE1) led to the identification of NAT8B (ATase1) and a similar protein designated as NAT8 (ATase2) (Ko and Puglielli Citation2009). NAT8B and NAT8 catalyze the N-acetylation of Lys residues in CD133 (Mak et al. Citation2014). Veiga-da-Cunha et al. (Citation2010) cloned NAT8, and its expression in HEK293T cells yielded a 25 kDa protein (227 amino acids) that catalyzed the N-acetylation of S-(benzyl)-l-Cys (241, ) and LTE4 (235, ). hNAT8 has three domains (UniProtKB: Q9UHE5): cytoplasmic domain (residues 1–42); helical transmembrane domain (residues 43–63); and C-terminal topological domain (residues 64–227) that contains the active site. Expression of tagged NAT8 in CHO cells and confocal microscopic analysis with immunolabeling confirmed the ER location of NAT8 (Veiga-da-Cunha et al. Citation2010).
As many as eight NAT8-related sequences are found in some genomes and some may catalyze the N-acetylation of Cys S-conjugates (Veiga-da-Cunha et al. Citation2010). Two SNPs, E104K and F143S, were identified in NAT8 and their expression and assay shows activities that differ from the wild-type by no more than two-fold. The gene encoding hNAT8 is located on chromosome bands 2p12-p13.1 (Ozaki et al. Citation1998).
The N-acetylation of the Cys S-conjugates of the sevoflurane degradation products 287 and 289 () shows considerable interindividual variability: with renal tissue from 20 human subjects there were 6- and 11-fold differences in cytosolic (see below) and microsomal N-acetylation, respectively (Altuntas and Kharasch Citation2002).
NAT8L (NAT8-like), which shares 30% sequence identity with NAT8, catalyzes the N-acetylation of L-Asp, but Cys S-conjugates are not substrates (Wiame et al. Citation2010).
5.3. Substrates
A large number of diverse chemicals are metabolized to mercapturic acids in vivo (Chasseaud Citation1976). NAT8 catalyzes the N-acetylation of many l-Cys S-conjugates, but d-Cys S-conjugates are poor substrates (Green and Elce Citation1975; Duffel and Jakoby Citation1982). Representative examples of Cys S-conjugates that are substrates for NAT8 are shown in along with the corresponding product mercapturic acids (Commandeur and Vermeulen Citation1990; Commandeur et al. Citation1991; Lau et al. Citation1995; Birner et al. Citation1997; Kraus et al. Citation2000; Altuntas and Kharasch Citation2002). Although free l-Lys is not a substrate for NAT8, protein-bound l-Lys, e.g. BACE1, is acetylated (Ko and Puglielli Citation2009; Veiga-da-Cunha et al. Citation2010).
A good correlation between Vmax/Km for N-acetylation and the substituent lipophilicity of alkyl Cys S-conjugates, as modeled by Hansch π constants, was observed (Duffel and Jakoby Citation1982). The relevance of substrate lipophilicity in the NAT8-catalyzed acetylation reaction is supported by the linear correlation between logP and Vmax/Km for S-conjugate 286 () and two structurally related haloalkyl Cys S-conjugates (Kraus et al. Citation2000).
LTE4 (235, ) is biotransformed to N-acetyl-LTE4 in rats and in isolated rat hepatocytes (Denzlinger et al. Citation1985; Örning et al. Citation1986; Stene and Murphy Citation1988). In human subjects given LTE4 (235, ), N-acetyl-LTE4 is formed as a minor metabolite, and its formation was not detected in human hepatocytes (Huber et al. Citation1990; Sala et al. Citation1990). Similarly, LTE4 (235, ) undergoes N-acetylation by expressed hNAT8 but at a rate about 100-fold lower than S-(benzyl)- l-Cys (241, ) (Veiga-da-Cunha et al. Citation2010). Human NAT8 expressed in HEK293T cells catalyzes the N-acetylation of three aromatic Cys S-conjugates, S-(benzyl)- l-Cys (241, ), S-(4-nitrobenzyl)- l-Cys, and S-(1-menaphthyl)- l-Cys, which exhibit similar Km and Vmax values, although the aromatic groups differ in size and electronic properties (Deol and Josephy Citation2017).
HNE (17, ) is biotransformed to a GSH S-conjugate (207, ), which is hydrolyzed to Cys S-conjugate 294 () that undergoes N-acetylation to give mercapturic acid 295 () (Alary et al. Citation2003; Mol et al. Citation2017).
The synthetic homocysteine S-conjugate of propachlor (2-(homocysteine-S-yl)-N-isopropyl-N-phenylacetamide), is metabolized in rats to the mercapturic acid analog 2-(N-acetylhomocysteine-S-yl)-N-isopropyl-N-phenylacetamide (Davison et al. Citation1992). The metabolism in rats of a GSH S-conjugate to a product that is N-acetylated on the Glu moiety has been reported (Yin et al. Citation2004); the reaction required microsomal enzymes and acetyl-CoA, but it was not determined that NAT8 catalyzed the reaction.
5.4. Inhibitors
Sulfhydryl reactive agents, e.g. p-chloromercuribenzoate, and N-ethylmaleimide, inhibit NAT8 (Green and Elce Citation1975; Aigner et al. Citation1996). N-Acetyl-S-(benzyl)-l-Cys (283, ) is a product inhibitor of NAT8 (Ki = 310 μM), but the GSH S-conjugate of sulfobromophthalein is not an inhibitor (Okajima et al. Citation1984). Several anions, including probenecid, iodipamide, sulfobromophthalein, and p-aminohippurate, are weak inhibitors of NAT8 (Inoue et al. Citation1984; Okajima et al. Citation1984; Aigner et al. Citation1996).
A high-throughput screen of 14 400 compounds led to the identification of two compounds, phenoxazine 300 () and naphthoquinone 301 (), that inhibit NAT8 (Ding et al. Citation2012). Both compounds exhibited noncompetitive inhibition kinetics with respect to acetyl-CoA, and both inhibited the acetylation of BACE1 and decreased its level of expression in H4 (human neuroglioma) cells. Inhibitors 300 and 301 () apparently have not been tested with Cys S-conjugates as NAT8 substrates.
Figure 24. NAT8 inhibitors. 300, (6-chloro-5H-benzo[a]phenoxazin-5-one, 301, 2-chloro-3-(2-ethoxyanilino)-1,4-dihydronaphthalene-1,4-dione.
![Figure 24. NAT8 inhibitors. 300, (6-chloro-5H-benzo[a]phenoxazin-5-one, 301, 2-chloro-3-(2-ethoxyanilino)-1,4-dihydronaphthalene-1,4-dione.](/cms/asset/b59ed7cf-1fe1-45af-b2b7-d37ada972cb4/itxc_a_1692191_f0024_b.jpg)
5.5. Tissue and species distribution
Mercapturic acid formation has been demonstrated in several species, including man, dogs, rats, mice, goats, and rabbits, but little mercapturic acid formation is observed in guinea pigs and calves (Stekol Citation1938, Citation1946; Bray et al. Citation1959b; Brüsewitz et al. Citation1977; Bakke et al. Citation1988, Citation1990; Skiles et al. Citation1991).
The rate of N-acetylation of 2-bromo-6-(l-cystein-S-yl)hydroquinone (292, ) shows significant species and strain differences: B6C3F1 mice ≈ F344 rats ≈ Sprague-Dawley rats > hamsters > BALB/c mice ≫ guinea pigs (Lau et al. Citation1995). The N-acetylation of S-(1,2,3,4,4-pentachloro-1,3-butadienyl)- l-Cys is somewhat greater in rat kidney microsomes than in human kidney microsomes (Green et al. Citation2003).
NAT8 activity is located in liver and kidney microsomes but activity is greater in the kidney than in the liver (Elce Citation1970; Green and Elce Citation1975; Hughey et al. Citation1978; Ko and Puglielli Citation2009; Veiga-da-Cunha et al. Citation2010), although the N-acetylation of Cys S-conjugates 288 and 290 () in human kidney cytosol has been reported (Altuntas and Kharasch Citation2002).
An enzyme located in rat liver and kidney microsomal fractions catalyzes the N-acetylation of several Cys S-conjugates, including S-(benzyl)- l-Cys (241, ), S-(butyl)-l-Cys, and S-(diphenylmethyl)- l-Cys (Green and Elce Citation1975). The microsomal location of NAT8 indicates that the cytosolic arylamine N-acetyltransferases NAT1 and NAT2 (EC 2.3.1.5) do not catalyze the N-acetylation of Cys S-conjugates (Weber and King Citation1981). The human genes encoding NAT8 are expressed predominantly in kidney and liver (Ozaki et al. Citation1998). NAT8 and NAT8B are expressed in mouse primary neurons and cerebral cortex and in human brain tissue (Ding et al. Citation2012).
The ER localization of NAT8 has been confirmed (Veiga-da-Cunha et al., Citation2010). Treatment of rat liver and kidney microsomal fractions with trypsin or chymotrypsin significantly decreases NAT8 activity, indicating that protease-sensitive domains of NAT8 are located on the cytoplasmic surface of the endoplasmic reticulum (ER) (Okajima et al. Citation1984). Furthermore, nonpermeant S-acetyldextran derivatives of Cys inhibit NAT8 activity, indicating that the active site of NAT8 faces the cytoplasmic surface of the ER. NAT8 catalyzed N-acetylation of lysine residues located in the N-terminal and globular regions of BACE1 has been localized to the lumen of the ER (Costantini et al. Citation2007); the luminal location requires a transporter for the polar acetyl-CoA, which has been identified as acetyl-coenzyme A transporter 1 (AT-1) (Hirabayashi et al. Citation2004; Jonas et al. Citation2010).
NAT8 activity is found, along with GGT and dipeptidase activities in the outer stripe region of the medulla of the rat kidney, which contains proximal tubular cells (Hughey et al. Citation1978). Immunohistochemical studies show that NAT8 is highly expressed in the proximal tubular cells of the inner and outer cortices of human kidney (Chambers et al. Citation2010, Supplementary Figures 4 and 5).
NAT8 activity in rat kidney is concentrated in the rat renal proximal tubule (Hughey et al. Citation1978; Heuner et al. Citation1991). Microinfusion studies with rat renal tubules and S-(benzyl)-l-Cys (241, ) showed that N-acetyl-S-(benzyl)-l-Cys (283, ) formation was greater in late proximal tubules than in early proximal tubules and that no mercapturic acid formation was detected in early distal tubules (Heuner et al. Citation1991). Infusion of Cys S-conjugate 241 () into the rat renal artery resulted in 90% conversion to mercapturic acid 283 ().
5.6. Other biological functions
Apart from its importance in the mercapturic acid pathway, NAT8 may also play a role in regulation of blood pressure and renal function (Juhanson et al. Citation2008). Genome-wide association studies implicate NAT8 with susceptibility to chronic kidney disease (Chambers et al. Citation2010; Köttgen et al. Citation2010; Tin et al. Citation2013). A genome-wide association study revealed that a genetically determined metabotype is linked to a variation at the NAT8 locus and that this variation is associated with N-acetyl-Orn (Suhre et al. Citation2011); elevated blood N-acetyl-Orn concentrations are associated with decreased GFR chronic kidney disease. A missense mutation in NAT8 is a known susceptibility locus for chronic kidney disease in African Americans (Yu et al. Citation2014). The missense mutation is significantly associated with N-acetyl-Orn and N-acetyl-1-methylhistidine levels, which exhibit a predictive relationship with chronic kidney disease. It has been proposed that the mercapturic acid pathway may play a role in the development of diabetic kidney disease due to the tubular cell toxicity of cysteine S-conjugates; NAT8-catalyzed N-acetylation of the conjugates may provide a protective effect (Gonçalves-Dias et al. Citation2019a).
Hepatic NAT8 activities are decreased after hepatic ischemia/reperfusion injury in rats and can be restored by giving mesenchymal stem cells intravenously (Fu et al. Citation2014); furthermore, NAT8 overexpression in human hepatic L02 cells blocks hydrogen peroxide-induced apoptosis.
NAT8B- and NAT8-catalyzed transient N-acetylation of Lys residues has been proposed to regulate levels of BACE1 and, thereby, the levels of neurotoxic amyloid β-peptide (Aβ) in the brain. Both NAT8B and NAT8 levels are upregulated in the brains of Alzheimer’s Disease patients and may represent potential drug targets for the prevention of Aβ accumulation (Ding et al. Citation2012). Evidence also supports a role for NAT8B/NAT8-catalyzed transient N-acetylation of Lys residues in the regulation of expression and trafficking of CD133 protein to the surface of cells (Mak et al. Citation2014).
6. Aminoacylases
6.1. Introduction
Aminoacylases catalyze the hydrolysis of N-acyl-L-α-amino acids, including mercapturic acids (), to yield cysteine S-conjugates, alkanoic acids, and amino acids (for a review, see Anders and Dekant Citation1994). Most early reports dealt with the hydrolysis of protein N-acetyl amino acids and with the use of aminoacylases to catalyze the enantiospecific hydrolysis of N-acetyl l-amino acids (Birnbaum et al. Citation1952; Fones and Lee Citation1953; Gade and Brown Citation1981). Birnbaum et al. (Citation1952) were apparently the first to use the term “acylase” to describe the enzymatic hydrolysis of N-acyl-l-α-amino acids. Aminoacylases may serve to salvage N-acyl amino acids formed by protein degradation (Endo Citation1980; Giardina et al. Citation1997; Pittelkow et al. Citation1998; Perrier et al. Citation2005; Lindner et al. Citation2008b). The spectrum of N-acetyl amino acids found in the urine of human subjects with aminoacylase 1 deficiency (https://www.omim.org/entry/609924; accessed 31 October 2019) affords insight into the role of the aminoacylases in the processing of N-acetyl amino acids derived from N-acetylated peptides (Engelke et al. Citation2008; Alessandri et al. Citation2014, Citation2018).
Early in vivo studies provided inconsistent evidence for the deacetylation of mercapturic acids (Parke and Williams Citation1951; Marsden and Young Citation1958), although the hydrolysis of S-(benzyl)mercapturic acid (283, ), S-(2-chloro-4-nitrophenyl)mercapturic acid, and S-(butyl)mercapturic acid to the corresponding cysteine S-conjugates was demonstrated in rat, rabbit, and guinea pig liver tissue extracts (Bray and James Citation1960). In vivo studies show, however, that mercapturic acids undergo hydrolysis (and reacetylation): when [acetyl-2H3]-labeled mercapturic acids were given to rats, a variable fraction of the dose was excreted as the unlabeled mercapturic acid (Commandeur et al. Citation1991; Uttamsingh et al. Citation1998a).
Three mammalian enzymes catalyze the hydrolysis of N-acyl α-l-amino acids: aminoacylase-1, aspartoacylase, and aminoacylase-3; all are zinc hydrolases (Hernick and Fierke Citation2005). Aminoacylase-1 and -3 show a preference for N-acyl aliphatic-l-amino acids and N-acyl aromatic-l-amino acids, respectively, but mercapturic acids may be substrates for either enzyme. Aspartoacylase (EC 3.5.1.15) is highly selective for N-acetyl-l-Asp and has not been reported to catalyze the hydrolysis of xenobiotic-derived mercapturic acids.
6.2. Aminoacylase-1 (Acy1, EC 3.5.1.14)12
Acy1 preferentially catalyzes the hydrolysis of aliphatic N-acyl α-l-amino acids, many mercapturic acids, and the l-Cys prodrug N-acetyl-l-Cys.
6.2.1. Structure and properties
An enzyme purified to homogeneity from pig kidney was termed “acylase 1” (Bruns and Schulze Citation1962). N-Acetyl-l-Met was the preferred substrate but a range of N-acyl-l-aliphatic amino acids were also substrates; N-Acyl-l-aromatic amino acids, e.g. N-acetyl-l-Trp, were poor substrates.
Porcine kidney and human and bovine liver Acy1 is a homodimeric, zinc metallopeptidase (Kördel and Schneider Citation1976a, Citation1977b; Gade and Brown Citation1981; Mitta et al. Citation1992; Cook et al. Citation1993; Palm and Röhm Citation1995; D'Ambrosio et al. Citation2003). A trimeric aggregate of rat kidney Acy1 has been reported (Giardina et al. Citation2000). Aminoacylase-1 is assigned to Family M20 (MEROPS ID M20.973) (Rawlings et al. Citation2018), whose subclasses represent degradative dipeptidases and exopeptidases as well as amidohydrolases (Biagini and Puigserver Citation2001).
The subunit Mr of Acy1 of human, pork, and rat kidney acylases is approximately 45 kDa (Mitta et al. Citation1992; Cook et al. Citation1993; Giardina et al. Citation2000; D'Ambrosio et al. Citation2003). Each porcine Acy1 monomer contains two domains: a catalytic domain (residues 1–188 and 311–399 or 311–406) and a dimerization domain (residues 189–310) (Palm and Röhm Citation1995; D'Ambrosio et al. Citation2003; Liu et al. Citation2006). The nucleotide sequences of human, porcine, mouse, and rat Acy1 have been reported (Mitta et al. Citation1992, Citation1993; Giardina et al. Citation2000; Durand et al. Citation2003; Sass et al. Citation2006); the deduced amino acid sequences of h-, p-, and rAcy1 show 80–90% identity. ACY1 and Acy1 encode the human and murine proteins, respectively (Cook et al. Citation1993; Visel et al. Citation2004). A baculovirus expression system for h- and pAcy1 has been described (Pittelkow et al. Citation1998).
The gene encoding human Acy1 is assigned to chromosome 3p21.1 (Miller et al. Citation1990; Cook et al. Citation1993). All human tissue culture cells and all mouse tissues and cells from a single strain show one electrophoretic form of Acy1 (Naylor et al. Citation1979). Human subjects with Acy1 deficiency express inactive Acy1 proteins and exhibit clinical symptoms associated with impaired development (van Coster et al. Citation2005; Sass et al. Citation2006, Citation2007; Ferri et al. Citation2014) (see Section 6.2.5, Other biological functions).
Early studies demonstrated the presence of several Cys residues in Acy1 and that chemical modification of the thiol residues inactivated the enzyme (Kördel and Schneider Citation1976a; Frey et al. Citation1977; Szajáni et al. Citation1980; Heese and Röhm Citation1989). Later analysis of pAcy1 by several techniques shows the presence of five Cys residues, which most studies show are present as thiols rather than disulfides (Mitta et al. Citation1992; Miyagi et al. Citation1992; Palm and Röhm Citation1995; Wang et al. Citation1995; D'Ambrosio et al. Citation2003). Rat and human Acy1 contain five and four Cys residues, respectively (Durand et al. Citation2003; Perrier et al. Citation2004). Although certain of the four Cys residues in hAcy1 are conserved in p- and rAcy1, there are differences in the location of the remaining Cys residues within the amino acid sequences. The Cys residues may play a structural or catalytic role (Szajáni et al. Citation1980; Löffler et al. Citation1986; Henseling and Röhm Citation1988; Heese and Röhm Citation1989). Chemical modification of Cys residues by chloromethyl ketone analogs of l-Ala and l-Leu inactivate pAcy1 (Frey et al. Citation1977). Cys116 in h- and pAcy1 is thought to play a role in catalysis, but Cys116 is missing in rat renal Acy1, which is much less sensitive to inhibition by mercuric chloride than h- or pAcy1.
Dialysis of Acy1 against 1,10-phenanthroline inactivates the enzyme, but activity is restored by addition of Zn2+ or Co2+ (Kördel and Schneider Citation1977b; Kumpe et al. Citation1981; Wu and Tsou Citation1993). Replacement of the Zn2+ ion with Mn2+, Co2+, Ni2+, or Cd2+ in Acy1 results in significant alterations in the kinetic properties of porcine kidney Acy1 with a panel of four N-acetyl-l-amino acids (Gilles et al. Citation1984). With N-chloroacetyl-l-Leu as the substrate, the Km and Vmax for the native enzyme and the Co2+-reconstituted (after inactivation with 1,10-phenanthroline) Acy1 are similar (Wu and Tsou Citation1993). The sensitivity of native and Mn2+-substituted Acy1 to a panel of inhibitors is similar (Heese et al. Citation1990).
Zinc may play a structural (Heese et al. Citation1990; Tang et al. Citation1995) or a catalytic (Szajáni et al. Citation1980; Lindner et al. Citation2003) role in Acy1-catalyzed reactions, although the latter is favored (Hernick and Fierke Citation2005; Auld Citation2013). Metal analysis indicates that Acy1 contains one Zn2+ ion per subunit (Kördel and Schneider Citation1977b; Löffler et al. Citation1986; Palm and Röhm Citation1995; Pittelkow et al. Citation1998), whereas the crystal structure of the zinc-binding domain of the T347G mutant of hAcy1 clearly shows the presence of two Zn2+ ions per subunit (Lindner et al. Citation2003). The crystal structure of the Zn-binding domain also shows a dinuclear zinc-binding site with a His-Asp-Glu-Glu-His motif characteristic of clan MH members (Lindner et al. Citation2003). Accordingly, photooxidation or chemical modification of the His residues in pAcy1 results in loss of activity (Kördel and Schneider Citation1977a). Chemical modification of Trp residues with 5-diazo-1H-tetrazole and N-bromosuccinimide, which do not react with His residues, also inactivates pAcy1 (Kördel and Schneider Citation1976b).
The reaction mechanism for Acy1 has not been fully elucidated but may resemble that found for other Zn-containing hydrolases (Hernick and Fierke Citation2005; Auld Citation2013). The rate-determining step for the Acy1-catalyed hydrolysis of a series of N-acyl-amino acids is consistent with a nucleophilic attack, perhaps assisted by a zinc ion, on the acyl carbonyl carbon of the N-acyl-amino group (Ötvös et al. Citation1971; Szajáni et al. Citation1980; Denton Citation2000). On the basis of mutational studies and homology modeling, Liu et al. (Citation2006) suggest a mechanism for pAcy1 in which the active-site Glu146 acts as a general base to abstract a proton from the zinc-coordinated water; the resulting hydroxide ion may then attack the substrate acyl carbonyl to form a tetrahedral intermediate that eliminates the acyl residue; hence, the Zn2+ provides the water-derived nucleophile and stabilizes the active site. The proposed mechanism does not include a role in catalysis for a second active-site Zn2+ ion.
6.2.2. Substrates
Acy1 preferentially catalyzes the hydrolysis of N-acyl α-l-amino acids with neutral aliphatic N-acyl substituents, e.g. N-acetyl-l-Met (Birnbaum et al. Citation1952; Bruns and Schulze Citation1962; Lorentz et al. Citation1975; Gade and Brown Citation1981; Giardina et al. Citation2000; Durand et al. Citation2003; Lindner et al. Citation2008a); N-acetyl-l-Asp, N-acetyl-l-Phe, N-acetyl-l-Tyr, N-acetyl-l-His, and Nα-Acetyl-l-Lys are scarcely hydrolyzed. Dipeptides with uncharged N-terminal amino acids, e.g. Gly-l-Ala, are substrates (Rao et al. Citation1952; Kördel and Schneider Citation1975), but α-N-acetyl-peptides, e.g. N-acetyl-Ala-Ala-Ala, are not substrates (Gade and Brown Citation1978). For compilations of rates of hydrolysis of N-acyl-l-amino acids, see (Birnbaum et al. Citation1952; Fones and Lee Citation1953; Fu and Birnbaum Citation1953; Ötvös et al. Citation1971; Lorentz et al. Citation1975; Chenault et al. Citation1989; Durand et al. Citation2003).
The synthetic substrate N-[3-(2-furyl)acryloyl]- l-Met (296, ) is used for the assay of pAcy1 activity but is not biotransformed by r- or mAcy1(Heese et al. Citation1988; Giardina et al. Citation1997, Citation2000; Durand et al. Citation2003). Acetamidoacrylic acid (298, ) has been used as an assay substrate for pAcy1 (Cho et al. Citation1987).
Rat and porcine kidney Acy1 catalyze the hydrolysis of the l-Cys prodrug N-acetyl-l-Cys (Uttamsingh et al. Citation1998b; Yamauchi et al. Citation2002). The rate of hydrolysis of N-acetyl-l-Cys is highest in kidney tissue in rabbit, rat, monkey, and dog, but low activity is also present in liver, heart, small intestine, lung, brain, and pancreas in rat, monkey, and dog, but not in the rabbit (Yamauchi et al. Citation2002). The rate of hydrolysis of S-alkyl-N-acetyl-l-Cys compounds varies considerably: for example, the Vmax/Km (mL/mg/min) ranged from 56.9 for S-(n-propyl)-N-acetyl-l-Cys to 5 for S-(n-butyl)-N-acetyl-l-Cys, whereas the Vmax/Km for N-acetyl-l-Cys is 10.2 (Uttamsingh et al. Citation1998b). Moreover, the rate of hydrolysis of several S-alkyl-N-acetyl-l-Cys compounds is related to their calculated molecular volumes and log P values.
Xenobiotic-derived mercapturic acids are substrates for Acy1. N-Acetyl-S-(1,1,2,2-tetrafluoroethyl)-l-Cys, N-acetyl-S-(2-chloro-1,1,2-trifluoroethyl)- l-Cys (287, ), and N-acetyl-S-(2-bromo-1,1,2-trifluoroethyl)- l-Cys are hydrolyzed by purified rat kidney Acy1 (Uttamsingh et al. Citation1998b).
Rat kidney and liver homogenates catalyze the hydrolysis of N-acetyl-S-(1,2,2-trichlorovinyl)-l-Cys, N-acetyl-S-(1,2-dichlorovinyl)-l-Cys (285, ), N-acetyl-S-(1,2,3,4,4-pentachlorobutadienyl)-l-Cys, N-acetyl-S-(1,1,2,2-tetrafluoroethyl)-l-Cys, N-acetyl-S-(2,2-dichloro-1,1-difluoroethyl)-l-Cys, and N-acetyl-S-(2,2-dibromo-1,1-difluoroethyl)-l-Cys, but these results do not indicate whether Acy1 or Acy3 (see below) catalyzes the reaction (Vamvakas et al. Citation1987; Commandeur et al. Citation1988; Pratt and Lock Citation1988; Commandeur et al. Citation1989);
Studies with mercapturic acids that are formed as metabolites of the sevoflurane degradation product 1,1,3,3,3-pentafluoro-2-(fluoromethoxy)-1-propene (Compound A) showed that human kidney cytosol catalyzes the hydrolysis of N-acetyl-S-[1,3,3,3-tetrafluoro-2-(fluoromethoxy)-1-propenyl]- l-Cys (291, ), but not N-acetyl-S-[1,1,3,3,3-pentafluoro-2-(fluoromethoxy)propyl]- l-Cys (289, ) (Uttamsingh et al. Citation1998a); other studies showed, however, that human kidney cytosol catalyzed the hydrolysis of both mercapturic acids 289 and 291 () and that rates of N-deacetylation were greater than rates of N-acetylation (Altuntas and Kharasch Citation2002).
Human and porcine Acy1 also catalyze the hydrolysis of a mercapturic acid formed from 3,4-epoxyprecocene II epoxide as well as several other synthetic mercapturic acids. (Stocker et al. Citation2012).
6.2.3. Inhibitors
Acy1 is inhibited by zinc chelating agents, e.g. 1,10-phenanthroline and EDTA, by thiol reagents, e.g. 4-chloromercuribenzoic acid, 2-chloromercuri-4-nitrophenol, mercuric chloride, and 2-mercaptoethanol, by diethyl pyrocarbonate (a histidine reagent), and by 2-ethoxy-1-(ethoxycarbonyl)-1,2-dihydroquinoline (a carboxyl reagent), but not by benzamidine (a trypsin inhibitor), phenylmethanesulfonyl fluoride, iodoacetate, or iodoacetamide (Kördel and Schneider Citation1977b; Szajáni et al. Citation1980; Löffler and Schneider Citation1987b; Heese et al. Citation1988; Wang et al. Citation1995; Giardina et al. Citation1997; Durand et al. Citation2003). Porcine Acy1 is much more sensitive to inhibition by mercuric chloride than rat Acy1 (Durand et al. Citation2003). Poorly hydrolyzed Acy1 substrates, e.g. N-isovaleroyl-l-Nva, are competitive inhibitors of rapidly hydrolyzed substrates, e.g. N-chloroacetyl-l-Nva (Ötvös et al. Citation1971). l-α-Hydroxy- and l-α-fluorofatty acids are noncompetitive, reversible inhibitors of pAcy1 with inhibition constants in the mM range (Tamura et al. Citation2000).
n-Butylmalonic acid (302, ) is a slow-binding inhibitor of porcine renal Acy1, whereas benzylmalonic acid (303, ) is a weak, competitive inhibitor (Röhm Citation1989). Malonyl glucoside 304 () is a strong competitive inhibitor of pAcy1 (Ki = 11 μM) (Stocker et al. Citation2005). N-Hydroxyamino acids inhibit Acy1 (Löffler and Schneider Citation1987a; Heese et al. Citation1990); N-hydroxyaminobutanoic acid (305, ) is a potent inhibitor (Ki = 0.07 nM) of pAcy1.
Figure 25. Aminoacylase inhibitors. 302, n-butylmalonic acid; 303, benzylmalonic acid; 304, maakiain 3-O-(6′-O-malonyl-β-d-glucoside (malonyl glucoside); 305, N-hydroxy-α-aminobutanoic acid; 306, 2-(4-ethoxyphenyl)-2-methylsuccinic acid; 307, ebselen; 308, N-(2,3,5-trichloro-4-oxocyclohexa-2,5-dien-1-ylidene)benzenesulfonamide; 309, 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF).
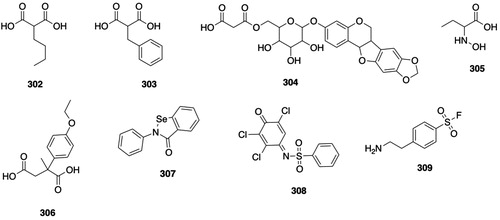
6.2.4. Tissue and species distribution
Acy1 is a cytosolic enzyme (Greenhough and Turner Citation1991; Cook et al. Citation1993; Lindner et al. Citation2000). Although Acy1 has been reported to be associated with cellular membranes, apparently due to the presence of the hydrophobic PWW(L,A) motif (Heese et al. Citation1988; Palm and Röhm Citation1995); Acy1 is, however, not anchored to membranes (Greenhough and Turner Citation1991).
Investigation of the distribution of Acy1 in mammalian tissues by several techniques shows that expression is highest in the kidney (Albert and Szewczuk Citation1972; Naylor et al. Citation1979; Qavi and Kit Citation1980; Csaikl et al. Citation1986; Miller et al. Citation1989; Miller and Kao Citation1989; Lindner et al. Citation2000; Yamauchi et al. Citation2002; Cigna et al. Citation2007; Zhong et al. Citation2009). Acy1 is also present in liver (bile duct epithelium, hepatocytes), lung (bronchiolar epithelium), brain (gray, but not white, matter), skeletal muscle, stomach, small intestine, adrenal glands, pancreas, and testes, but not in serum or erythrocytes (Naylor et al. Citation1979; Miller and Kao Citation1989; Uttamsingh et al. Citation2000). In contrast to most species studied, little Acy1 activity or protein was found in canine liver tissue (Yamauchi et al. Citation2002). Acy1 is present in the distal tubules of the porcine kidney and in the glomeruli and proximal and distal convoluted tubules of the rat (Löffler et al. Citation1982; Uttamsingh et al. Citation2000).
In man and cynomolgus monkey, Acy1 is localized in the straight and convoluted renal proximal tissues (Yamauchi et al. Citation2002). Transcriptomic analysis (RNA-Seq) shows high expression of hAcy1 in kidney, liver, small intestine, and duodenum but detectable transcripts are found in most tissues (Fagerberg et al. Citation2014). Acy1 is detectable in human-mouse somatic cell hybrids but not in mitochondria from these cells (Qavi and Kit Citation1980).
Acy1 is an abundant protein: the immunoelectrophoretically determined concentration of Acy1 protein amounts to 1.5 mg/g fresh tissue in the porcine kidney compared with 0.4 mg/g fresh tissue in the liver (Löffler et al. Citation1982); moreover, in the distal proximal tubule the concentration of Acy1 is estimated to amount to 15 mg/g fresh tissue.
Acy1 expression is decreased in ferric nitrilotriacetate-induced renal cell carcinoma tissue in rats (Zhong et al. Citation2009) but is upregulated in SK-N-SH neuroblastoma cells exposed to neural differentiation agents (Long et al. Citation2011).
6.2.5. Other biological functions
Aminoacylase-1 deficiency is an autosomal-recessive inborn error of metabolism (entry 609924; https://www.omim.org/; accessed 31 October 2019) characterized by the increased excretion of N-acetyl-L-amino acids and by several developmental disabilities, including muscular hypotonia, seizures, delayed psychomotor development, and autistic behavior (van Coster et al. Citation2005; Sass et al. Citation2007; Engelke et al. Citation2008; Tylki-Szymanska et al. Citation2010; Sommer et al. Citation2011; Alessandri et al. Citation2014; Ferri et al. Citation2014). Mutational analysis shows loss-of-function mutations associated with abnormal splicing (Sass et al. Citation2006; Sommer et al. Citation2011; Ferri et al. Citation2014). (For a summary of the clinical findings and mutations in aminoacylase 1 deficiency subjects, see Sass et al. Citation2007; Alessandri et al. Citation2018).
hAcyl may play a role in cancer-cell growth regulation. Immunoassay shows that hAcy1 expression varies widely in several small cell lung cancer (SCLC)- and non-small cell lung cancer (NSCLC) cell lines (Miller et al. Citation1989; Scaloni et al. Citation1992); similarly, hAcy1 activity is positive, reduced, or undetectable in SCLC and NSCLC tissue. Inactivation of ACY1 may afford a selective growth advantage in small cell lung cancer (Cook et al. Citation1998). A decreased expression of Acy1 is found in lung and renal-cell carcinoma tissues and the decrease may be associated with deletions in the DNF15S2 locus on chromosome 3 (Miller et al. Citation1989; Naylor et al. Citation1989; Jones et al. Citation1991; Balabanov et al. Citation2001; Hwa et al. Citation2005). Acy1 may exert a tumor suppressor function in an iron-induced rat model of renal-cell carcinoma (Zhong et al. Citation2009); transfection of Acy1 into rat renal-cell carcinoma cell lines retards proliferation and colony formation. hAcy1 is overexpressed in colorectal cancer tissue, and its expression in colorectal cancer tissue and its presence in serum is positively correlated with tumor stage, indicating that it may play a role in tumor progression (Shi et al. Citation2013; Yu et al. Citation2017). Acy1 mRNA and protein expression are decreased in human hepatocellular carcinoma tissue (Wei et al. Citation2014); knockdown of ACY1 in human SMC7721 cells promotes cell viability and invasiveness whereas overexpression of Acy1 in human BEL7402 cells inhibits growth. Although Acy1 is scarcely detectable serum of normal subjects, elevated serum Acy1 concentrations are associated with delayed graft function in subjects undergoing renal transplantation (Welberry Smith et al. Citation2013).
Acy1 may regulate the activity of sphingosine kinase 1 (SPHK1), which promotes cell proliferation and suppresses apoptosis (Maceyka et al. Citation2004); Acy1 increases the anti-apoptotic effect of SPHK1 in NIH 3T3 cells. Transfection of renal-cell carcinoma-derived cell lines with Acy1 decreases cell proliferation, an effect that may be associated with stimulation of SPHK1 (Zhong et al. Citation2009).
6.3. Aminoacylase-3 (Acy3, EC 3.5.1.114)13
Acy3 preferentially catalyzes the hydrolysis of aromatic N-acetyl α-l-amino acids and some mercapturic acids. Acy3 has not been explicitly assigned to a MEROPS Family or Clan.
6.3.1. Structure and properties
An aminoacylase that preferentially catalyzes the hydrolysis of N-acetyl-l-Tyr and N-acetyl-l-Phe was purified from rat liver and kidney (Endo Citation1978); the enzyme was designated as Acylase III and was demonstrated to be distinct from Acy1.
An enzyme purified from rat liver and kidney that catalyzed the hydrolysis of N-acetyl aryl amino acids and S-aryl- and S-aralkyl-mercapturic acids was stated to resemble Acy3 (Suzuki and Tateishi Citation1981); the rat liver enzyme was likely a homotetramer. Subsequent studies identified an Acy3-like enzyme from rat kidney that catalyzed the hydrolysis of haloalkene-derived mercapturic acids (Uttamsingh and Anders Citation1999).
Murine Acy3 is a homotetramer of approximately 140 kDa with a subunit molecular mass of approximately 35 kDa and catalyzes the hydrolysis of N-acetyl-S-benzyl-l-Cys (283, ), N-acetyl-l-His, N-acetyl-l-Tyr, and N-acetyl-l-Phe (Pushkin et al. Citation2004). Murine Acy3 shows 43% and 5% amino acid sequence identity with murine aspartoacylase and Acy1, respectively. Additional studies showed that expressed murine Acy3 is present as both a homodimer and homotetramer (Newman et al. Citation2007). Acy3 monomers of approximately 75 kDa have been detected in human neuroblastoma-derived cell lines and in rat cortical neurons (Long et al. Citation2011; Tsirulnikov et al. Citation2012a). The specific activities of the dimeric and tetrameric forms of Acy3 were similar with N-acetyl-S-benzyl-l-Cys (283, ) as the substrate (Ryazantsev et al. Citation2007); electron microscopic analysis and modeling studies at 16 Å resolution indicated that tetrameric Acy3 may be attributed to a back-to-back stacking of dimeric forms of Acy3.
The gene encoding hAcy3 is located on chromosome 11q13.2 (entry 614413; www.omim.org; accessed 31 October 2019). A genome-wide association study found three SNPs on chromosome 11 that mapped to the coding and regulatory regions for ACY3 (Hiltunen et al. Citation2015).
The mAcy3 active-site residue Glu177 is catalytically essential (Tsirulnikov et al. Citation2009): the E177A mutant is catalytically inactive. The crystal structure of the E177A mutant of mAcy3 (the structure of wild-type mAcy3 was not resolvable) shows the presence of two domains: a hydrolytic domain (residues 1–120) that contains the catalytic site and a shielding domain (residues 211-318) that limits access to the active site (Hsieh et al. Citation2010). Acy3 possesses catalytic residues similar to aspartoacylase, and the single zinc ion is coordinated with His21, Glu24, and His116 (Tsirulnikov et al. Citation2009; Hsieh et al. Citation2010). In E177A-Acy3 in complex with N-acetyl-l-Tyr, the substrate carbonyl (acetyl) coordinates with the zinc ion and the N-acetyl-α-amino carboxylic acid moiety of the substrate forms hydrogen bonds and van der Waals interactions with active-site residues. Similar interactions are seen with N-acetyl-S-(1,2-dichlorovinyl)-l-Cys (285, ) in complex with E177A-Acy3. The substrate selectivity of Acy3 is dependent on van der Waals interactions with the substrate side-chain (Hsieh et al. Citation2010).
Metal analysis indicates that wild-type murine Acy3 expressed in E. coli contains 0.35 zinc ions per subunit (Tsirulnikov et al. Citation2009). Dialysis against o-phenanthroline inactivates Acy3, whereas incubation with Co2+ or Ni2+ activates Acy3. Consistent with the X-ray crystal structure of E177A-Acy3, which shows that the zinc ion is coordinated with His21, Glu24, and His116, mutation of these residues to Ala abolishes Acy3 activity, which is not restored by addition of cobalt (Tsirulnikov et al. Citation2009; Hsieh et al. Citation2010). It is likely that enzyme-bound zinc participates in mercapturic acid hydrolysis, perhaps by the promoted-water pathway described for carboxypeptidase A (Hsieh et al. Citation2010; Wu et al. Citation2010).
6.3.2. Substrates
Acy3 catalyzes the hydrolysis of many N-acyl amino acids, S-aryl and S-aralkyl mercapturic acids, and haloalkene-derived mercapturic acids. With purified rat kidney and liver Acy3, the substrates with the highest specific activities include: N-acetyl-l-Tyr, N-chloroacetyl-l-Trp, N-acetyl-S-(benzyl)- l-Cys, N-acetyl-S-(4-bromophenyl)-l-Cys, and N-acetyl-S-(4-nitrobenzyl)- l-Cys (Endo Citation1978; Suzuki and Tateishi Citation1981). With expressed murine Acy3, the substrates with the highest Vmax/Km ratio include: N-acetyl-S-(2,2-dichlorovinyl)- l-Cys, N-acetyl-S-(4-chlorobenzyl)- l-Cys, N-acetyl-S-(4-bromobenzyl)-l-Cys, N-acetyl-S-(pentachlorobutadienyl)-l-Cys, and N-acetyl-S-(1,2,2-trichlorovinyl)-l-Cys (Newman et al. Citation2007); the Acy1 substrate N-acetyl-l-Cys is not a substrate for Acy3.
Purified or expressed Acy3 catalyzes the hydrolysis of a several haloalkene-derived mercapturic acids (Uttamsingh and Anders Citation1999; Newman et al. Citation2007); mercapturic acids 285, 287, 289, and 291 () are representative examples. Cytosolic enzymes catalyze the hydrolysis of bromophenol-derived mercapturic acid 293 (), but the enzyme involved was not specifically identified as Acy1 or Acy3 (Lau et al. Citation1995); given the structure of mercapturic acid 293 (), it is likely that Acy3 is involved.
Acy3, but not Acy1, catalyzes the hydrolysis of the mercapturic acids of 4-hydroxynonenal (295, ) and acrolein (N-acetyl-S-(3-oxopropyl)-l-Cys) (Tsirulnikov et al. Citation2012a). N-Acetyl-S-farnesyl-l-Cys and N-acetyl-S-geranylgeranyl-l-Cys are substrates for hAcy3, but not hAcy1 (Tsirulnikov et al. Citation2018); inhibition of Acy3 in human hepatocellular carcinoma cell lines increases the levels of N-acetyl-S-farnesyl-l-Cys.
hAcy3 has been identified as a core binding protein of hepatitis C virus (Chen et al. Citation2009). Incubation of murine Acy3 with N-acetylated and non-acetylated “short” HCVCP peptides that correspond to the N-terminal sequence (residues 2–11) of the HCVCP and the N-terminal sequence (residues 2–11) of mAcy3 led to the formation of chain-length-shortened peptides, indicating that mAcy3 may exhibit endopeptidase activity (Tsirulnikov et al. Citation2012b); mAcy3 failed, however, to catalyze the N-deacetylation of N-acetylated-HCVCP and a panel of N-acetylated peptides.
6.3.3. Inhibitors
No selective inhibitors for Acy3 have apparently been reported. 4-Chloromercuribenzoic acid and Cu2+ inhibit, but dithiothreitol activates, rat and murine kidney Acy3 (Endo Citation1978; Newman et al. Citation2007; Tsirulnikov et al. Citation2009). A small-molecule screen identified inhibitors of mouse Acy3 with potencies in the nanomolar range (Tsirulnikov et al. Citation2012a): 2-(4-ethoxyphenyl)-2-methylsuccinate (306, ) (I50 = 0.63 nM), ebselen (307, ) (I50 = 1.0 nM), and N-(2,3,5-trichloro-4-oxocyclohexa-2,5-dien-1-ylidene)benzenesulfonamide (308 ) (I50 = 3.20 nM) inhibited deacetylation of N-acetyl-l-Tyr; in addition, ibuprofen (Ki = 0.04 mM) is a comparatively weak inhibitor of Acy3. Molecular docking experiments indicate that these inhibitors interact with the active site of Acy3. Acy3 inhibitors 306 and 307 () decrease the cytotoxicity of the mercapturic acid of HNE (295, ) in primary rat cortex neurons, indicating a role for Acy3 in the cytotoxicity of HNE (Tsirulnikov et al. Citation2012a).
The endopeptidase activity of mAcy3 is inhibited by the serine protease inhibitor 4-(2-aminoethyl)benzenesulfonyl fluoride 309 () (Tsirulnikov et al. Citation2012b).
6.3.4. Tissue and species distribution
Acy3 is a cytosolic enzyme (Suzuki and Tateishi Citation1981; Lau et al. Citation1995; Uttamsingh and Anders Citation1999; Chen et al. Citation2009). Acy3 activity is highest in the kidney but is also present in liver, spleen, brain, and thymus (Endo Citation1980). Northern blot analysis shows that murine Acy3 is abundantly expressed in kidney and to a lesser extent in liver (Pushkin et al. Citation2004); low levels of transcripts are also present in brain, heart, lung, stomach, small intestine, and testis. Immunohistochemical studies show that Acy3 is most highly expressed in the apical membrane of the S1 segment of the renal proximal tubule, although it is detectable in the cytosol of the S2 and S3 segments of mouse kidney (Pushkin et al. Citation2004). Immunolocalization studies show that Acy3 is found primarily in neurons but is also present in mouse brain blood vessels (Tsirulnikov et al. Citation2012a). Acy3 is expressed in the nuclei of several neuroblastoma-derived cell lines, in the human adrenal gland, and in neuroblastoma tumor tissue (Long et al. Citation2011); Acy3 may play a role in neuroblastoma promotion, and high expression of Acy3 in human neuroblastoma tissue is associated with poor prognosis. Transcriptomic analysis (RNA-Seq) shows high expression of hAcy3 in kidney, small intestine, and duodenum but detectable transcripts are found in most tissues (Fagerberg et al. Citation2014).
6.3.5. Other biological functions
Acy3 is expressed in human hepatocytes but is highly expressed in hepatocellular carcinoma cell lines and in human hepatocellular carcinoma tissue (Tsirulnikov et al. Citation2018). Inhibition of Acy3 and suppression of Acy3 expression with siRNA decrease the viability of HuH7 and HepG2 cells. The authors propose that inhibition or silencing of Acy3 prevents Ras membrane association, which is required for cell growth.
SNPs in ACY3 are associated with the ambulatory systolic blood pressure response to bisoprolol, although ACY3 is not known to be associated with blood pressure regulation (Hiltunen et al. Citation2015). An extension of these studies showed that systolic and diastolic blood pressure responses to bisoprolol were significantly correlated with plasma concentrations of Phe and N-acetyl-l-Phe (Rimpelä et al. Citation2017); ACY3 variation rs2514036 may serve as a biomarker for response to β-blockers. A genome-wide association study and metabolomic profiling identified a variant in ACY3 that is associated with serum N-acetylphenylalanine levels in African American men (Yu et al. Citation2014); further analysis showed that the ACY3 SNP is associated with systolic and diastolic blood pressure responses to bisoprolol (Rimpelä et al. Citation2017).
7. Mercapturic acids: transport and elimination; mercapturic acids as biomarkers; bioactivation by the mercapturic acid pathway
7.1. Transport and elimination
The enzymatic synthesis of mercapturic acids and their renal elimination play central roles in the detoxication of drugs and chemicals. The enzymatic synthesis and processing of mercapturic acids has been discussed in preceding sections of this review. Brief comment about the renal transport and elimination of mercapturic acids and other S-conjugates is merited.
The terms Phase I and Phase II drug metabolism were coined by R. Tecwyn Williams (Williams Citation1959). Subsequently, the terms Phase 0, Phase III, and Phase IV have been proposed to describe the transport processes that surround the metabolic Phases I and II (for a review, see Döring and Petzinger Citation2014).
Mercapturic acid and S-conjugate transport involves at least four families of transporters: organic anion transporters (OAT), organic anion transporting polypeptide transporters (OATP), multidrug resistance ABC transporters (MDR), and amino acid transporters (for reviews, see Commandeur et al. Citation1995; Roth et al. Citation2012; Makrides et al. Citation2014; Yin and Wang Citation2016).
Members of the organic anion transport (OAT) family play a major role in the transport of organic anions in the kidney and other organs (Sweet Citation2005; Rizwan and Burckhardt Citation2007; Burckhardt and Burckhardt Citation2011; Wang and Sweet Citation2013; McLean et al. Citation2016; Lozano et al. Citation2018; Nigam Citation2018). Both organic anion transporter-1 (OAT1; SLC22A6) and -3 (OAT3; SLC22A8), which are located on the basolateral membrane of epithelial cells, may transport mercapturic acids. There are, however, few explicit examples of mercapturic acid transport by OAT1 or OAT3, although in some studies the inhibition of mercapturic acid transport by probenecid indicated the involvement of a member of the OAT transport family (Lock and Ishmael Citation1985; Zhang and Stevens Citation1989). Rat OAT1-expressing X. laevis oocytes facilitate the transport of the mercapturic acid N-acetyl-S-(2,4-dinitrophenyl)- l-Cys (Pombrio et al. Citation2001); several mercapturic acids, e.g. N-acetyl-S-(2,4-dinitrophenyl)-l-Cys and 283 (), are competitive inhibitors of 4-aminohippuric acid transport, indicating that they are likely substrates for OAT1. The transport of mercapturic acid metabolites of alkyl polycyclic aromatic hydrocarbons in HEK293 cells expressing hOAT1 and hOAT3 has been reported (Bakhiya et al. Citation2007); 1-methylpyrenyl mercapturic acid was transported by both hOAT1 and -3, whereas 1,8-dimethylpyrenyl mercapturic acid was not a substrate for hOAT1 and was a poor substrate for hOAT3. The N-acetyl-l-Cys conjugate of cisplatin (NAC-1) is transported in HEK293 cells overexpressing OAT1 and -3 and is inhibited by probenecid (Hu et al. Citation2017); moreover, the nephrotoxicity of NAC-1 is decreased in OAT1−/− and OAT3−/− mice. The Tyr kinase inhibitor nilotinib and several other Tyr kinase inhibitors inhibit 4-aminohippuric acid and estrone-3-sulfate uptake in HEK293 cells overexpressing hOAT1 and hOAT3, respectively. The mercapturic acid pathway plays a significant role in the disposition of inorganic and alkyl mercury compounds and involves several transport systems, including OAT1 and OAT3 (for reviews, see Bridges and Zalups Citation2010; Bridges and Zalups Citation2017; Zalups and Bridges Citation2018). In addition to mercapturic acids, the Cys S-conjugates S-(1,2-dichlorovinyl)-l-cysteine (284, ) and S-(2-chloro-1,1,2-trifluoroethyl)- l-cysteine (286, ) are transported by OAT1 (Dantzler et al. Citation1998; Groves and Morales Citation1999; Groves et al. Citation2003).
Members of the organic anion transporting polypeptide transporter family (OATP; SLCO) are involved in mediating the transport of many drugs (for reviews, see Kalliokoski and Niemi Citation2009; Hagenbuch and Stieger Citation2013; Stieger and Hagenbuch Citation2014; Kovacsics et al. Citation2017). Given the preference of OATPs for substrates greater than 350 Da, most mercapturic acids are unlikely substrates (Roth et al. Citation2012). Taurocholate uptake is trans-stimulated by S-(2,4-dinitrophenyl)GSH (12, ), S-sulfobromophthalein-glutathione (BSP-SG), and S-methyl-GSH, but not by N-acetyl-l-Cys in Xenopus laevis oocytes expressing rat Oatp2 (rOATP1A4) (Li et al. Citation2000).
Several members of the ATP-binding cassette [ABC] family (multidrug resistant proteins, MRP; P-glycoproteins) facilitate the transport of Cys, Cys-Gly, and GSH S-conjugates (for reviews, see Deeley et al. Citation2006; Borst et al. Citation2007; Ballatori et al. Citation2009; Keppler Citation2011; Nies Citation2013). For example, leukotriene C4 (189, ), S-(2,4-dinitrophenyl)GSH (12. ), and GSH S-conjugates of aflatoxin (20, ), melphalan (27, ), chlorambucil (29, ), doxorubicin, and daunorubicin serve as MRP substrates, whereas leukotriene D4 (234, ), leukotriene E4 (235, ), and the Cys-Gly S-conjugates of dietary isothiocyanates are apparently the only examples of Cys and Cys-Gly S-conjugates serving as MRP substrates (Keppler et al. Citation1998; Priebe et al. Citation1998; Wijnholds et al. Citation2000; Paumi et al. Citation2001; Jedlitschky and Keppler Citation2002; Callaway et al. Citation2004). The chelating agents 2,3-dimercaptopropane-1-sulfonic acid (DMPS) and 2,3-dimercaptosuccinic acid (DMSA) are employed to promote the elimination of mercury; MRP2 transports the DMPS- and DMSA-S-conjugates of mercury (Bridges et al. Citation2008a, Citation2008b). MRP2 plays a role in the efflux of inorganic and methylmercury S-conjugates from renal proximal tubular cells (Bridges et al. Citation2011). Several Tyr kinase inhibitors inhibit ABC transporters and have been investigated as targets for anticancer chemotherapy, but inhibition of S-conjugate transport has apparently not been reported (Beretta et al. Citation2017; Robey et al. Citation2018).
Renal amino acid transporters facilitate the renal transport of protein amino acids and Cys S-conjugates (Bröer Citation2008; Silbernagl and Gekle Citation2008; Makrides et al. Citation2014). The Na+-dependent uptake of S-(1,2-dichlorovinyl)- l-Cys (DCVC) (284, ) has been demonstrated in rat luminal brush border membrane preparations and in rat renal proximal tubules (Schaeffer and Stevens Citation1987a; Zhang and Stevens Citation1989); the basolateral uptake of DCVC (284, ) in LLC-PK1 cells is attributable to a Na+-independent l-like amino acid transport system (Schaeffer and Stevens Citation1987b). In isolated rat renal proximal tubular cells, both the Na+-dependent OAT system and the Na+-independent amino acid transport system L are involved in the uptake of DCVC (284, ), whereas the uptake of the homocysteine (Hcy) analog S-(1,2-dichlorovinyl)- l-Hcy is attributable to transport system L and also to the Na+-dependent A and ASC amino acid transport systems (Lash and Anders Citation1989).
The Na+-dependent System B0,+ and the Na+-independent System b0,+ amino acid transporters catalyze the renal transport of several mercury S-conjugates (Bridges and Zalups Citation2017). Some methylmercury S-conjugates are substrates for the l-type large neutral amino acid transporters, LAT1 and LAT2, which are expressed in the kidney (Simmons-Willis et al. Citation2002; Park et al. Citation2005).
There are a few reports of the urinary excretion of Cys S-conjugates. The luminal uptake of Cys S-conjugates may explain the paucity of reports of the renal excretion of Cys S-conjugates. Calves given GSH S-conjugates excrete Cys S-conjugates, but not mercapturic acids, because of an apparent lack of NAT8 activity (Bakke et al. Citation1990; Davison Citation1991).
RLIP76, a Ral-effector binding protein, catalyzes the ATP-dependent transport of S-(2,4-dinitrophenyl)GSH (12, ), HNE-GSH (18, ), and several anticancer drugs (Awasthi et al. Citation2000; Singhal et al. Citation2009a). RLIP76 protects cancer cells from electrophile-induced apoptosis, and its inhibition or depletion may find therapeutic use in cancer chemotherapy (Singhal et al. Citation2009b, Citation2015, Citation2017; Awasthi et al. Citation2018; Nagaprashantha et al. Citation2018).
7.2. Mercapturic acids as biomarkers
Mercapturic acids are formed as metabolites of a host of chemicals and their formation and excretion may serve as biomarkers for exposure (De Rooij et al. Citation1998; Perbellini et al. Citation2002; Haufroid and Lison Citation2005; Kotapati et al. Citation2015). Reviews of LC-MS methods for quantifying the excretion of mercapturic acids demonstrates the potential utility of mercapturic acid formation and excretion as biomarkers (Mathias and B'Hymer Citation2014, Citation2016). LC-MS combined with metabonomic analysis has also been used to detect mercapturic acids as biomarkers (Wagner et al. Citation2006). A review of the methodologies for mercapturomic profiling has been published (Gonçalves-Dias et al. Citation2019b). Exposure may, however, be underestimated if several metabolites are excreted and the mercapturic acid is the only metabolite measured (Barr et al. Citation2007). Moreover, the utility of mercapturic acids as biomarkers depends on their reliable urinary excretion in surveyed populations. Considerable information is available about transporter polymorphisms and studies of associations between transporter polymorphisms and drug disposition have been published, but the effect of transporter polymorphisms on mercapturic acid excretion has apparently not been reported (Fujita et al. Citation2005; Erdman et al. Citation2006; Zhou et al. Citation2015; McLean et al. Citation2016; Yee et al. Citation2018). The usefulness of mercapturic acids as excreted biomarkers depends not only on the activities of renal transporters but also on the activities of Phase I and II enzymes, which could increase or decrease the metabolism of a compound to an electrophilic metabolite that undergoes GSH S-conjugate formation and processing by the enzymes of the mercapturic acid pathway.
7.3. Bioactivation by the mercapturic acid pathway
Toxicity associated with the mercapturic acid pathway often depends on the initial formation of GSH S-conjugates and further enzymatic processing by enzymes of the mercapturic acid pathway or by other xenobiotic metabolizing enzymes or both. Some S-conjugates may serve as transport or delivery agents and release toxic compounds or metabolites at the target organ (Baillie and Kassahun Citation1994). Cys S-conjugates, formed as intermediates in the mercapturic acid pathway or by the aminoacylase-catalyzed hydrolysis of mercapturic acids, may undergo bioactivation by Cys S-conjugate β-lyase to give nephrotoxic metabolites. An extended discussion about the bioactivation of mercapturic acids ispathway is beyond the scope of this review, but several reviews are available (Lock and Berndt Citation1988; Bakke Citation1990; Anders Citation1991; Monks and Lau Citation1992; Anders and Dekant Citation1994; Guengerich Citation2003; Anders Citation2008; Cooper and Hanigan Citation2018).
8. Conclusions
The enzymes of the mercapturic acid pathway are well characterized. DNA and protein sequences, including SNPs, along with protein crystal structures are available, as are reports on substrate selectivity, inhibitor sensitivity, and tissue, organ, and species distribution. The roles of the MA pathway enzymes in detoxication are well established.
Future investigations of the MA pathway enzymes are likely to focus on their regulatory functions in biochemical and physiological processes. The regulatory functions may involve catalysis, as in the case of the regulation of inflammasome activation by GSTO1-1 (Hughes et al. Citation2019), or they may involve non-catalytic processes, such as protein-protein interactions. The elucidation of the mechanisms and of the potential clinical relevance of inhibiting or activating such regulatory processes is likely to involve the design of both active site and non-active site directed inhibitors, which may also be required to be both isozyme-selective and either tissue or cell-selective.
Examples of GSTs that exhibit regulatory functions distinct from their metabolism functions include GSTM2-2, which inhibits calcium release through the cardiac RyR2 ryanodine receptor by an apparently non-catalytic mechanism (Samarasinghe et al. Citation2015) and GSTP1-1, which exhibits a variety of regulatory functions, including inhibition of apoptosis by forming a non-catalytic complex with non-phosphorylated JNK1α2 (De Luca et al. Citation2012).
Aminopeptidase N (APN, CD13) is a highly multifunctional enzyme that has been termed a “moonlighting” protein. Its regulatory, signaling, receptor, and hydrolytic functions make it an intriguing potential therapeutic target for more than a dozen human cancers, as well as lymphedema, autoimmune disorders, rheumatoid arthritis, and other pathologies associated with APN overexpression (Amin et al. Citation2018).
Cysteine S-conjugate N-acetyltransferase (NAT8) has been implicated in the regulation of blood pressure and kidney function, and several studies support a role for aminoacylase-1 (Acy1) as a regulator of tumor growth.
Although MA pathway research is a mature field of investigation, the above-mentioned representative examples indicate that much remains to be learned about the biological significance of MA pathway enzymes.
Declaration of interest
The authors report no conflict of interest. The authors participated in the development of the paper as individual professionals and have sole responsibility for the writing and content of the paper. Neither of the authors has been involved in the last 5 years with regulatory or legal proceedings related to the contents of the paper and neither has received funding in the last 5 years in support of the preparation of the paper.
Supplemental material
Supplemental material for this article is available online here.
Acknowledgments
The authors acknowledge with thanks the helpful comments of the independent reviewers who were selected by the Editor and who were anonymous to the authors. The authors thank Dr. Baojian Wu, Jinan University, for provision of images shown in .
Notes
Notes
1 The accepted name is glutathione transferase (https://www.qmul.ac.uk/sbcs/iubmb/enzyme/EC2/5/1/18.html; accessed 31 October 2019), but the term glutathione S-transferase is, however, frequently used. The abbreviation GST for glutathione transferases is retained [Mannervik B, Board PG, Hayes JD, Listowsky I, Pearson WR. 2005. Nomenclature for mammalian soluble glutathione transferases. Methods Enzymol. 401: 1–8.]
2 The GSTs were designated as the Molecule of the Month for August, 2017, by the Protein Data Bank (http://pdb101.rcsb.org/motm/212; accessed 31 October 2019).
3 This section describes properties of the cytosolic GST that are similar among the several classes; properties that are distinctive of the several cytosolic GST classes are discussed in Sections 2.2.6.1 to 2.2.6.6.
4 Although identified as a Sigma-class GST, the accepted name for the enzyme (EC 5.3.99.2) that catalyzes the isomerization of PGH2 to PGD2 is prostaglandin-D synthase (PGDS) (https://www.qmul.ac.uk/sbcs/iubmb/enzyme/EC5/3/99/2.html; accessed 31 October 2019).
5 The terms GSTK and DsbA-L are used synonymously in the scientific literature. Their use in this review reflects their use in the cited publications.
6 γ-Glutamyltransferase is the IUBMB accepted name (https://www.qmul.ac.uk/sbcs/iubmb/enzyme/EC2/3/2/2.html; accessed 31 October 2019); γ-glutamyl transpeptidase is also widely used.
7 Membrane dipeptidase is the accepted name, but renal dipeptidase and dehydropeptidase I are also frequently used; for other names, see: http://www.sbcs.qmul.ac.uk/iubmb/enzyme/EC3/4/13/19.html; accessed 31 October 2019; https://www.ebi.ac.uk/merops/cgi-bin/pepsum?id=M19.001; accessed 31 October 2019.
8 Although membrane alanyl aminopeptidase is the accepted name, aminopeptidase N is widely used in the literature and is used in this review; aminopeptidase M is also used. For other names, see: http://www.sbcs.qmul.ac.uk/iubmb/enzyme/EC3/4/11/2.html; accessed 31 October 2019; https://www.ebi.ac.uk/merops/cgi-bin/pepsum?id=M01.001; accessed 31 October 2019.
9 A commercial preparation of APN (https://www.sigmaaldrich.com/catalog/substance/leucineaminopeptidasemicrosomalfromporcinekidney12345905463111?lang=en®ion=US; accessed 31 October 2019) is listed as leucine aminopeptidase but is also identified as aminopeptidase N and M.
10 Leucyl aminopeptidase is the accepted name; for other names, see: http://www.sbcs.qmul.ac.uk/iubmb/enzyme/EC3/4/11/1.html, accessed 31 October 2019.
11 Although cysteine S-conjugate N-acetyltransferase is the recommended name, NAT8 is widely used in the literature and is used in this review; a range of names is found in the literature, including ATase 2, GLA, Camello-like proteins 1 and 4, and TSC501 (https://www.genenames.org/cgi-bin/gene_symbol_report?hgnc_id=HGNC:18069; accessed 31 October 2019).
12 The accepted name is N-acyl-aliphatic-l-amino acid amidohydrolase (http://www.sbcs.qmul.ac.uk/iubmb/enzyme/EC3/5/1/14.html; accessed 31 October 2019). Aminoacylase-1 and acylase-1 are, however, widely used in the literature and will be used in this review.
13 The accepted name is N-acyl-aromatic-l-amino acid amidohydrolase (http://www.sbcs.qmul.ac.uk/iubmb/enzyme/EC3/5/1/114.html; accessed 31 October 2019) but the name aminoacylase 3 is used in the literature and in this review.
References
- Abbott WA, Griffith OW, Meister A. 1986. γ-Glutamyl-glutathione. Natural occurrence and enzymology. J Biol Chem. 261:13657–13661.
- Abdellatif Y, Liu D, Gallant EM, Gage PW, Board PG, Dulhunty AF. 2007. The Mu class glutathione transferase is abundant in striated muscle and is an isoform-specific regulator of ryanodine receptor calcium channels. Cell Calcium. 41:429–440.
- Abe F, Aoyagi T. 2002. Physiological roles of ectoenzymes indicated by the use of aminopeptidase inhibitors. In: Langner J, Ansorge J, editors. Ectopeptidases CD13/aminopeptidase N and CD26/dipeptidylpeptidase IV in medicine and biology. New York: Springer; p. 95–122.
- Abel EL, Bammler TK, Eaton DL. 2004. Biotransformation of methyl parathion by glutathione S-transferases. Toxicol Sci. 79:224–232.
- Adachi H, Ishida N, Tsujimoto M. 1992. Primary structure of rat renal dipeptidase and expression of its mRNA in rat tissues and COS-1 cells. Biochim Biophys Acta. 1132:311–314.
- Adachi H, Katayama T, Inuzuka C, Oikawa S, Tsujimoto M, Nakazato H. 1990a. Identification of membrane anchoring site of human renal dipeptidase and construction and expression of a cDNA for its secretory form. J Biol Chem. 265:15341–15345.
- Adachi H, Katayama T, Nakazato H, Tsujimoto M. 1993. Importance of Glu-125 in the catalytic activity of human renal dipeptidase. Biochim Biophys Acta. 1163:42–48.
- Adachi H, Kubota I, Okamura N, Iwata H, Tsujimoto M, Nakazato H, Nishihara T, Noguchi T. 1989. Purification and characterization of human microsomal dipeptidase. J Biochem. 105:957–961.
- Adachi H, Tawaragi Y, Inuzuka C, Kubota I, Tsujimoto M, Nishihara T, Nakazato H. 1990b. Primary structure of human microsomal dipeptidase deduced from molecular cloning. J Biol Chem. 265:3992–3995.
- Adam GC, Burbaum J, Kozarich JW, Patricelli MP, Cravatt BF. 2004. Mapping enzyme active sites in complex proteomes. J Am Chem Soc. 126:1363–1368.
- Adam GC, Sorensen EJ, Cravatt BF. 2002. Proteomic profiling of mechanistically distinct enzyme classes using a common chemotype. Nat Biotechnol. 20:805–809.
- Adams DJ, Dai M, Pellegrino G, Wagner BK, Stern AM, Shamji AF, Schreiber SL. 2012. Synthesis, cellular evaluation, and mechanism of action of piperlongumine analogs. Proc Natl Acad Sci USA. 109:15115–15120.
- Adang AEP, Brussee J, Meyer DJ, Coles B, Ketterer B, van der Gen A, Mulder GJ. 1988a. Substrate specificity of rat liver glutathione S-transferase isoenzymes for a series of glutathione analogues, modified at the γ-glutamyl moiety. Biochem J. 255:721–724.
- Adang AEP, Brussee J, van der Gen A, Mulder GJ. 1990. The glutathione-binding site in glutathione S-transferases. Investigation of the cysteinyl, glycyl and γ-glutamyl domains. Biochem J. 269:47–54.
- Adang AEP, Brussee J, van der Gen A, Mulder GJ. 1991. Inhibition of rat liver glutathione S-transferase isoenzymes by peptides stabilized against degradation by γ-glutamyl transpeptidase. J Biol Chem. 266:830–836.
- Adang AEP, Duindam AJ, Brussee J, Mulder GJ, van der Gen A. 1988b. Synthesis and nucleophilic reactivity of a series of glutathione analogues, modified at the γ-glutamyl moiety. Biochem J. 255:715–720.
- Adang AEP, Meyer DJ, Brussee J, van der Gen A, Ketterer B, Mulder GJ. 1989. Interaction of rat glutathione S-transferases 7-7 and 8-8 with γ-glutamyl- or glycyl-modified glutathione analogues. Biochem J. 264:759–764.
- Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, et al. 1999. Regulation of JNK signaling by GSTp. EMBO J. 18:1321–1334.
- Agblor AA, Josephy PD. 2013. Donor substrate specificity of bovine kidney γ-glutamyltransferase. Chem Biol Interact. 203:480–485.
- Agúndez JA, Ladero JM. 2008. Glutathione S-transferase GSTT1 and GSTM1 allozymes: beyond null alleles. Pharmacogenomics. 9:359–363.
- Ahluwalia GS, Grem JL, Hao Z, Cooney DA. 1990. Metabolism and action of amino acid analog anti-cancer agents. Pharmacol Ther. 46:243–271.
- Ahmad S, Niegowski D, Wetterholm A, Haeggström JZ, Morgenstern R, Rinaldo-Matthis A. 2013. Catalytic characterization of human microsomal glutathione S-transferase 2: identification of rate-limiting steps. Biochemistry. 52:1755–1764.
- Ahmad S, Thulasingam M, Palombo I, Daley DO, Johnson KA, Morgenstern R, Haeggström JZ, Rinaldo-Matthis A. 2015. Trimeric microsomal glutathione transferase 2 displays one third of the sites reactivity. Biochim Biophys Acta. 1854:1365–1371.
- Ahmed AE, Anders MW. 1978. Metabolism of dihalomethanes to formaldehyde and inorganic halide – II. Studies on the mechanism of the reaction. Biochem Pharmacol. 27:2021–2025.
- Aigner A, Jäger M, Pasternack R, Weber P, Wienke D, Wolf S. 1996. Purification and characterization of cysteine-S-conjugate N-acetyltransferase from pig kidney. Biochem J. 317:213–218.
- Al Khamici H, Brown LJ, Hossain KR, Hudson AL, Sinclair-Burton A, Ng JPM, Daniel EL, Hare JE, Cornell BA, Curmi PMG, et al. 2015. Members of the chloride intracellular ion channel protein family demonstrate glutaredoxin-like enzymatic activity. PLoS One. 10:e115699.
- Ålander J, Johansson K, Heuser VD, Farebo H, Järvliden J, Abe H, Shibata A, Ito M, Ito Y, Morgenstern R. 2009a. Characterization of a new fluorogenic substrate for microsomal glutathione transferase 1. Anal Biochem. 390:52–56.
- Ålander J, Lengqvist J, Holm PJ, Svensson R, Gerbaux P, Heuvel RH, Hebert H, Griffiths WJ, Armstrong RN, Morgenstern R. 2009b. Microsomal glutathione transferase 1 exhibits one-third-of-the-sites-reactivity towards glutathione. Arch Biochem Biophys. 487:42–48.
- Alary J, Fernandez Y, Debrauwer L, Perdu E, Guéraud F. 2003. Identification of intermediate pathways of 4-hydroxynonenal metabolism in the rat. Chem Res Toxicol. 16:320–327.
- Albert Z, Szewczuk A. 1972. Histochemical and biochemical studies on Co++-activated acylase in rodent tissues. Acta Histochem. 42:68–76.
- Albiston AL, Ye SY, Chai SY. 2004. Membrane bound members of the M1 family: more than aminopeptidases. Protein Pept Lett. 11:491–500.
- Albrecht S, Al-Lakkis-Wehbe M, Orsini A, Defoin A, Pale P, Salomon E, Tarnus C, Weibel JM. 2011. Amino-benzosuberone: a novel warhead for selective inhibition of human aminopeptidase-N/CD13. Bioorg Med Chem. 19:1434–1449.
- Albrecht S, Salomon E, Defoin A, Tarnus C. 2012. Rapid and efficient synthesis of a novel series of substituted aminobenzosuberone derivatives as potent, selective, non-peptidic neutral aminopeptidase inhibitors. Bioorg Med Chem. 20:4942–4953.
- Alessandri MG, Casarano M, Pezzini I, Doccini S, Nesti C, Cioni G, Battini R. 2014. Isolated mild intellectual disability expands the aminoacylase 1 phenotype spectrum. JIMD Rep. 16:81–87.
- Alessandri MG, Milone R, Casalini C, Nesti C, Cioni G, Battini R. 2018. Four years follow up of ACY1 deficient patient and pedigree study. Brain Dev. 40:570–575.
- Ålin P, Danielson UH, Mannervik B. 1985. 4-Hydroxyalk-2-enals are substrates for glutathione transferase. FEBS Lett. 179:267–270.
- Ali-Osman F, Akande O, Antoun G, Mao JX, Buolamwini J. 1997a. Molecular cloning, characterization, and expression in Escherichia coli of full-length cDNAs of three human glutathione S-transferase Pi gene variants. Evidence for differential catalytic activity of the encoded proteins. J Biol Chem. 272:10004–10012.
- Ali-Osman F, Brunner JM, Kutluk TM, Hess K. 1997b. Prognostic significance of glutathione S-transferase Pi expression and subcellular localization in human gliomas. Clin Cancer Res. 3:2253–2261.
- Al-Kassab S, Boyland E, Williams K. 1963. An enzyme from rat liver catalysing conjugations with glutathione. 2. Replacement of nitro groups. Biochem J. 87:4–9.
- Allen TC, Granville LA, Cagle PT, Haque A, Zander DS, Barrios R. 2007. Expression of glutathione S-transferase Pi and glutathione synthase correlates with survival in early stage non-small cell carcinomas of the lung. Hum Pathol. 38:220–227.
- Allen L, Meck R, Yunis A. 1980. The inhibition of γ-glutamyl transpeptidase from human pancreatic carcinoma cells by (αS,5S)-α-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid (AT-125; NSC-163501). Res Commun Chem Pathol Pharmacol. 27:175–182.
- Allen MP, Yamada AH, Carpenter FH. 1983. Kinetic parameters of metal-substituted leucine aminopeptidase from bovine lens. Biochemistry. 22:3778–3783.
- Allen M, Zou F, Chai HS, Younkin CS, Miles R, Nair AA, Crook JE, Pankratz VS, Carrasquillo MM, Rowley CN, et al. 2012. Glutathione S-transferase Omega genes in Alzheimer and Parkinson disease risk, age-at-diagnosis and brain gene expression: an association study with mechanistic implications. Mol Neurodegener. 7:13.
- Allison RD. 1985. gamma-Glutamyl transpeptidase: kinetics and mechanism. Meth Enzymol. 113:419–437.
- Allison RD, Meister A. 1981. Evidence that transpeptidation is a significant function of γ-glutamyl transpeptidase. J Biol Chem. 256:2988–2992.
- Allocati N, Masulli M, Di Ilio C, Federici L. 2018. Glutathione transferases: substrates, inihibitors and pro-drugs in cancer and neurodegenerative diseases. Oncogenesis. 7:8.
- Alparslan MM, Daniş Ö. 2015. In vitro inhibition of human placental glutathione S-transferase by 3-arylcoumarin derivatives. Arch Pharm Chem Life Sci. 348:635–642.
- Altuntas TG, Kharasch ED. 2001. Glutathione S-conjugation of the sevoflurane degradation product, fluoromethyl-2,2-difluoro-1-(trifluoromethyl)vinyl ether (Compound A) in human liver, kidney, and blood in vitro. Toxicol Appl Pharmacol. 177:85–93.
- Altuntas TG, Kharasch ED. 2002. Biotransformation of L-cysteine S-conjugates and N-acetyl-L-cysteine S-conjugates of the sevoflurane degradation product fluoromethyl-2,2-difluoro-1-(trifluoromethyl)vinyl ether (compound A) in human kidney in vitro: interindividual variability in N-acetylation, N-deacetylation, and β-lyase-catalyzed metabolism. Drug Metab Dispos. 30:148–154.
- Alves CS, Kuhnert DC, Sayed Y, Dirr HW. 2006. The intersubunit lock-and-key motif in human glutathione transferase A1-1: role of the key residues Met51 and Phe52 in function and dimer stability. Biochem J. 393:523–528.
- Amin SA, Adhikari N, Jha T. 2018. Design of aminopeptidase N inhibitors as anti-cancer agents. J Med Chem. 61:6468–6490.
- Anders MW. 1991. Glutathione-dependent bioactivation of xenobiotics: implications for mutagenicity and carcinogenicity. In: Ernster L, Esumi H, Fujii Y, editors. Xenobiotics and cancer implications for chemical carcinogenesis and cancer chemotherapy. Tokyo: Japan Scientific Societies Press; p. 89–99.
- Anders MW. 2008. Chemical toxicology of reactive intermediates formed by the glutathione-dependent bioactivation of halogen-containing compounds. Chem Res Toxicol. 21:145–159.
- Andorfer JH, Tchaikovskaya T, Listowsky I. 2004. Selective expression of glutathione S-transferase genes in the murine gastrointestinal tract in response to dietary organosulfur compounds. Carcinogenesis. 25:359–367.
- Anders MW, Anderson WB, Tzeng HF, Board PG. 2001. Glutathione transferase Zeta: novel xenobiotic substrates and enzyme inactivation. Chem Biol Interact. 133:211–216.
- Anders MW, Dekant W. 1994. Aminoacylases. Adv Pharmacol. 27:433–448.
- Anders MW, Dekant W. 1998. Glutathione-dependent bioactivation of haloalkenes. Annu Rev Pharmacol Toxicol. 38:501–537.
- Anderson ME, Allison RD, Meister A. 1982. Interconversion of leukotrienes catalyzed by purified γ-glutamyl transpeptidase: concomitant formation of leukotriene D4 and γ-glutamyl amino acids. Proc Natl Acad Sci USA. 79:1088–1091.
- Anderson WB, Board PG, Gargano B, Anders MW. 1999. Inactivation of glutathione transferase Zeta by dichloroacetic acid and other fluorine-lacking α-haloalkanoic acids. Chem Res Toxicol. 12:1144–1149.
- Anderson RJ, Cairns D, Cardwell WA, Case M, Groundwater PW, Hall AG, Hogarth L, Jones AL, Meth-Cohn O, Suryadevara P. 2006. Design, synthesis and initial in vitro evaluation of novel prodrugs for the treatment of cystinosis. Lett Drug Des Discov. 3:336–345.
- Anderson WB, Liebler DC, Board PG, Anders MW. 2002. Mass spectral characterization of dichloroacetic acid-modified human glutathione transferase Zeta. Chem Res Toxicol. 15:1387–1397.
- Andersson L, Isley TC, Wolfenden R. 1982. α-Aminoaldehydes: transition state analogue inhibitors of leucine aminopeptidase. Biochemistry. 21:4177–4180.
- Andersson C, Mosialou E, Weinander R, Morgenstern R. 1994a. Enzymology of microsomal glutathione S-transferase. Adv Pharmacol. 27:19–35.
- Andersson C, Söderström M, Mannervik B. 1988. Activation and inhibition of microsomal glutathione transferase from mouse liver. Biochem J. 249:819–823.
- Andersson C, Weinander R, Lundqvist G, DePierre JW, Morgenstern R. 1994b. Functional and structural membrane topology of rat liver microsomal glutathione transferase. Biochim Biophys Acta. 1204:298–304.
- Ang WH, Khalaila I, Allardyce CS, Juillerat-Jeanneret L, Dyson PJ. 2005. Rational design of platinum(IV) compounds to overcome glutathione-S-transferase mediated drug resistance. J Am Chem Soc. 127:1382–1383.
- Aniya Y, Anders MW. 1989a. Activation of rat liver microsomal glutathione S-transferase by reduced oxygen species. J Biol Chem. 264:1998–2002.
- Aniya Y, Anders MW. 1989b. Regulation of rat liver microsomal glutathione S-transferase activity by thiol/disulfide exchange. Arch Biochem Biophys. 270:330–334.
- Aniya Y, Anders MW. 1992. Activation of rat liver microsomal glutathione S-transferase by hydrogen peroxide: role for protein-dimer formation. Arch Biochem Biophys. 296:611–616.
- Aniya Y, Imaizumi N. 2011. Mitochondrial glutathione transferases involving a new function for membrane permeability transition pore regulation. Drug Metab Rev. 43:292–299.
- Antczak C, Bauvois B, Monneret C, Florent JC. 2001. A new acivicin prodrug designed for tumor-targeted delivery. Bioorg Med Chem. 9:2843–2848.
- Appiah-Opong R, Commandeur JN, Istyastono E, Bogaards JJ, Vermeulen NP. 2009. Inhibition of human glutathione S-transferases by curcumin and analogues. Xenobiotica. 39:302–311.
- Arakawa S, Fujimoto K, Kato A, Endo S, Fukahori A, Shinagawa A, Fischer T, Mueller J, Takasaki W. 2012. Evaluation of hepatic glutathione transferase Mu 1 and Theta 1 activities in humans and mice using genotype information. Drug Metab Dispos. 40:497–503.
- Aritake K, Kado Y, Inoue T, Miyano M, Urade Y. 2006. Structural and functional characterization of HQL-79, an orally selective inhibitor of human hematopoietic prostaglandin D synthase. J Biol Chem. 281:15277–15286.
- Armstrong RN. 1997. Structure, catalytic mechanism, and evolution of the glutathione transferases. Chem Res Toxicol. 10:2–18.
- Armstrong RN. 2000. Mechanistic diversity in a metalloenzyme superfamily. Biochemistry. 39:13625–13632.
- Armstrong RN. 2010. Glutathione transferases. In: McQueen CA, editor. Comprehensive toxicology. 2nd ed. Vol. 4. Oxford: Elsevier; p. 295–321.
- Armstrong RN, Morgenstern R, Board PG. 2018. Glutathione transferases. In: McQueen CA, editor. Comprehensive toxicology. 3rd ed. Vol. 10. Oxford: Elsevier; p. 326–362.
- Armstrong DJ, Mukhopadhyay SK, Campbell BJ. 1974. Physicochemical characterization of renal dipeptidase. Biochemistry. 13:1745–1750.
- Arttamangkul S, Bhalgat MK, Haugland RP, Diwu Z, Liu J, Klaubert DH, Haugland RP. 1999. 5-(Pentafluorobenzoylamino)fluorescein: a selective substrate for the determination of glutathione concentration and glutathione S-transferase activity. Anal Biochem. 269:410–417.
- Atkinson HJ, Babbitt PC. 2009. Glutathione transferases are structural and functional outliers in the thioredoxin fold. Biochemistry. 48:11108–11116.
- Auld DS. 2013. Catalytic mechanisms for metallopeptidases. In: Rawlings ND, Salvesen G, editors. Handbook of proteolytic enzymes. 3rd ed. Vol. 1. San Diego: Elsevier; p. 370–396.
- Austruy E, Jeanpierre C, Antignac C, Whitmore SA, Van Cong N, Bernheim A, Callen DF, Junien C. 1993. Physical and genetic mapping of the dipeptidase gene DPEP1 to 16q24.3. Genomics. 15:684–687.
- Awasthi S, Cheng J, Singhal SS, Saini MK, Pandya U, Pikula S, Bandorowicz-Pikula J, Singh SV, Zimniak P, Awasthi YC. 2000. Novel function of human RLIP76: ATP-dependent transport of glutathione conjugates and doxorubicin. Biochemistry. 39:9327–9334.
- Awasthi YC, Sharma R, Singhal SS. 1994. Human glutathione S-transferases. Int J Biochem. 26:295–308.
- Awasthi S, Singhal SS, Singhal J, Nagaprashantha L, Li H, Yuan YC, Liu Z, Berz D, Igid H, Green WC, et al. 2018. Anticancer activity of 2′-hydroxyflavanone towards lung cancer. Oncotarget. 9:36202–36219.
- Axarli I, Labrou NE, Petrou C, Rassias N, Cordopatis P, Clonis YD. 2009. Sulphonamide-based bombesin prodrug analogues for glutathione transferase, useful in targeted cancer chemotherapy. Eur J Med Chem. 44:2009–2016.
- Baars AJ, Mukhtar H, Zoetemelk CE, Jansen M, Breimer DD. 1981. Glutathione S-transferase activity in rat and human tissues and organs. Comp Biochem Physiol C Comp Pharmacol. 70:285–288.
- Bachovchin DA, Brown SJ, Rosen H, Cravatt BF. 2009. Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes. Nat Biotechnol. 27:387–394.
- Badiou S, Bellet H, Lehmann S, Cristol JP, Jaber S. 2005. Elevated plasma cysteinylglycine levels caused by cilastatin-associated antibiotic treatment. Clin Chem Lab Med. 43:332–334.
- Baillie TA, Kassahun K. 1994. Reversibility in glutathione-conjugate formation. Adv Pharmacol. 27:163–181.
- Bakhiya N, Batke M, Laake J, Monien BH, Frank H, Seidel A, Engst W, Glatt H. 2007. Directing role of organic anion transporters in the excretion of mercapturic acids of alkylated polycyclic aromatic hydrocarbons. Drug Metab Dispos. 35:1824–1831.
- Bakke JE. 1990. Biochemical and physiological dispositions of glutathione conjugates. Drug Metab Rev. 22:637–643.
- Bakke JE, Davison KL, Larsen GL. 1990. Evidence for the absence of cysteine S-conjugate N-acetyltransferase activity in the metabolism of propachlor, naphthalene, and dichlobanil in calves. Xenobiotica. 20:801–807.
- Bakke JE, Larsen GL, Struble C, Feil VJ, Brandt I, Brittebo EB. 1988. Metabolism of 2,6-dichlorobenzonitrile, 2,6-dichlorothiobenzamide in rodents and goats. Xenobiotica. 18:1063–1075.
- Balabanov S, Zimmermann U, Protzel C, Scharf C, Klebingat KJ, Walther R. 2001. Tumour-related enzyme alterations in the clear cell type of human renal cell carcinoma identified by two-dimensional gel electrophoresis. Eur J Biochem. 268:5977–5980.
- Balchin D, Fanucchi S, Achilonu I, Adamson RJ, Burke J, Fernandes M, Gildenhuys S, Dirr HW. 2010. Stability of the domain interface contributes towards the catalytic function at the H-site of class Alpha glutathione transferase A1-1. Biochim Biophys Acta. 1804:2228–2233.
- Ballatori N, Jacob R, Barrett C, Boyer JL. 1988. Biliary catabolism of glutathione and differential reabsorption of its amino acid constituents. Am J Physiol Gastrointest Liver Physiol. 254:G1–G7.
- Ballatori N, Jacob R, Boyer JL. 1986. Intrabiliary glutathione hydrolysis. A source of glutamate in bile. J Biol Chem. 261:7860–7865.
- Ballatori N, Krance SM, Marchan R, Hammond CL. 2009. Plasma membrane glutathione transporters and their roles in cell physiology and pathophysiology. Mol Aspects Med. 30:13–28.
- Balogh LM, Atkins WM. 2011. Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metab Rev. 43:165–178.
- Bannenberg G, Dahlén SE, Luijerink M, Lundqvist G, Morgenstern R. 1999. Leukotriene C4 is a tight-binding inhibitor of microsomal glutathione transferase-1 – effects of leukotriene pathway modifiers. J Biol Chem. 274:1994–1999.
- Barnes MM, James SP, Wood PB. 1959. The formation of mercapturic acids. 1. Formation of mercapturic acid and the levels of glutathione in tissues. Biochem J. 71:680–690.
- Barouki R, Finidori J, Chobert MN, Aggerbeck M, Laperche Y, Hanoune J. 1984. Biosynthesis and processing of γ-glutamyl transpeptidase in hepatoma tissue culture cells. J Biol Chem. 259:7970–7974.
- Barr DB, Panuwet P, Nguyen JV, Udunka S, Needham LL. 2007. Assessing exposure to atrazine and its metabolites using biomonitoring. Environ Health Perspect. 115:1474–1478.
- Barrett AJ, Rawlings ND, Woessner J, editors. 2012. Handbook of proteolytic enzymes. San Diego (CA): Academic Press.
- Bass NM, Kirsch RE, Tuff SA, Marks I, Saunders SJ. 1977. Ligandin heterogeneity: evidence that the two non-identical subunits are the monomers of two distinct proteins. Biochim Biophys Acta. 492:163–175.
- Batist G, Tulpule A, Sinha BK, Katki AG, Myers CE, Cowan KH. 1986. Overexpression of a novel anionic glutathione transferase in multidrug-resistant human breast cancer cells. J Biol Chem. 261:15544–15549.
- Bauer M, Herbarth O, Aust G, Hengstler JG, Dotzauer A, Graebsch C, Schmuecking E. 2006. Expression patterns and novel splicing variants of glutathione-S-transferase isoenzymes of human lung and hepatocyte cell lines. Cell Tissue Res. 324:423–432.
- Baumann E, Preusse C. 1879. Ueber Bromphenylmercaptursäure. Ber Dtsch Chem Ges. 12:806–810.
- Bauvois B, Dauzonne D. 2006. Aminopeptidase-N/CD13 (EC 3.4.11.2) inhibitors: chemistry, biological evaluations, and therapeutic prospects. Med Res Rev. 26:88–130.
- Beattie RE, Elmore DT, Williams CH, Guthrie DJ. 1987. The behaviour of leucine aminopeptidase towards thionopeptides. Biochem J. 245:285–288.
- Bell JG, Buddington RK, Walton MJ, Cowey CB. 1987. Studies on the putative role of gamma-glutamyl transpeptidase in intestinal transport of amino acids in Atlantic salmon. J Comp Physiol B Biochem Syst Environ Physiol. 157:161–169.
- Benson AM, Talalay P, Keen JH, Jakoby WB. 1977. Relationship between the soluble glutathione-dependent Δ5-3-ketosteroid isomerase and the glutathione S-transferases of the liver. Proc Natl Acad Sci USA. 74:158–162.
- Beretta GL, Cassinelli G, Pennati M, Zuco V, Gatti L. 2017. Overcoming ABC transporter-mediated multidrug resistance: the dual role of tyrosine kinase inhibitors as multitargeting agents. Eur J Med Chem. 142:271–289.
- Bergmann M, Fraenkel-Conrat H. 1937. The rôle of specificity in the enzymatic synthesis of proteins: syntheses with intracellular enzymes. J Biol Chem. 119:707–720.
- Bergmann M, Schleich H. 1932a. Über die enzymatische Spaltung dehydrierter Peptide. Auffindung einer Dehydrodipeptidase. Hoppe Seylers Z Physiol Chem. 205:65–82.
- Bergmann M, Schleich H. 1932b. Weiteres über Dehydro-dipeptidase. Über die enzymatische Angreifbarkeit von Verbindungen aus Brenztraubensäure und Aminosäure. Hoppe Seylers Z Physiol Chem. 207:235–240.
- Berhane K, Mannervik B. 1990. Inactivation of the genotoxic aldehyde acrolein by human glutathione transferases of classes Alpha, Mu, and Pi. Mol Pharmacol. 37:251–254.
- Betters JL, Yu LQ. 2010. NPC1L1 and cholesterol transport. FEBS Lett. 584:2740–2747.
- Bhagwat SV, Mullick J, Avadhani NG, Raza H. 1998. Differential response of cytosolic, microsomal, and mitochondrial glutathione S-transferases to xenobiotic inducers. Int J Oncol. 13:281–288.
- Bhagwat SV, Okamoto Y, Shapiro LH. 2002. CD13/APN as a target for inhibiting tumor angiogenesis. In: Langner J, Ansorge J, editors. Ectopeptidases CD13/aminopeptidase N and CD26/dipeptidylpeptidase IV in medicine and biology. New York: Springer.
- Biagini A, Puigserver A. 2001. Sequence analysis of the aminoacylase-1 family. A new proposed signature for metalloexopeptidases. Comp Biochem Physiol B Biochem Mol Biol. 128:469–481.
- Binkley F. 1951. Metabolism of glutathione. Nature. 167:888–889.
- Binkley F, Alexander V, Bell FE, Lea C. 1957. Peptidases and alkaline phosphatases of swine kidney. J Biol Chem. 228:559–567.
- Binkley F, Davenport J, Eastall F. 1959. Localization of enzymes responsible for the hydrolysis of glutathione. Biochem Biophys Res Commun. 1:206–208.
- Birnbaum SM, Levintow L, Kingsley RB, Greenstein JP. 1952. Specificity of amino acid acylases. J Biol Chem. 194:455–470.
- Birner G, Bernauer U, Werner M, Dekant W. 1997. Biotransformation, excretion and nephrotoxicity of haloalkene-derived cysteine S-conjugates. Arch Toxicol. 72:1–8.
- Björkhem-Bergman L, Johansson M, Morgenstern R, Rane A, Ekström L. 2014. Prenatal expression of thioredoxin reductase 1 (TRXR1) and microsomal glutathione transferase 1 (MGST1) in humans. FEBS Open Bio. 4:886–891.
- Björnestedt R, Stenberg G, Widersten M, Board PG, Sinning I, Jones TA, Mannervik B. 1995. Functional significance of arginine 15 in the active site of human class Alpha glutathione transferase A1-1. J Mol Biol. 247:765–773.
- Blackburn AC, Coggan M, Shield AJ, Cappello J, Theodoratos A, Murray TP, Rooke M, Larter CZ, Koina ME, Dahlstrom JE, et al. 2011. Glutathione transferase Kappa deficiency causes glomerular nephropathy without overt oxidative stress. Lab Invest. 91:1572–1583.
- Blackburn AC, Coggan M, Tzeng HF, Lantum H, Polekhina G, Parker MW, Anders MW, Board PG. 2001. GSTZ1d: a new allele of glutathione transferase Zeta and maleylacetoacetate isomerase. Pharmacogenetics. 11:671–678.
- Blackburn AC, Matthaei KI, Lim C, Taylor MC, Cappello JY, Hayes JD, Anders MW, Board PG. 2006. Deficiency of glutathione transferase Zeta causes oxidative stress and activation of antioxidant response pathways. Mol Pharmacol. 69:650–657.
- Blackburn AC, Tzeng HF, Anders MW, Board PG. 2000. Discovery of a functional polymorphism in human glutathione transferase Zeta by expressed sequence tag database analysis. Pharmacogenetics. 10:49–57.
- Blackburn AC, Woollatt E, Sutherland GR, Board PG. 1998. Characterization and chromosome location of the gene GSTZ1 encoding the human Zeta class glutathione transferase and maleylacetoacetate isomerase. Cytogenet Genome Res. 83:109–114.
- Board PG. 2011. The Omega-class glutathione transferases: structure, function, and genetics. Drug Metab Rev. 43:226–235.
- Board PG, Anders MW. 2005. Human glutathione transferase Zeta. Meth Enzymol. 401:61–77.
- Board PG, Anders MW. 2007. Glutathione transferase Omega 1 catalyzes the reduction of S-(phenacyl)glutathiones to acetophenones. Chem Res Toxicol. 20:149–154.
- Board PG, Anders MW. 2011. Glutathione transferase Zeta: discovery, polymorphic variants, catalysis, inactivation, and properties of Gstz1-/- mice. Drug Metab Rev. 43:215–225.
- Board PG, Baker RT, Chelvanayagam G, Jermiin LS. 1997. Zeta, a novel class of glutathione transferases in a range of species from plants to humans. Biochem J. 328:929–935.
- Board PG, Chelvanayagam G, Jermiin LS, Tetlow N, Tzeng HF, Anders MW, Blackburn AC. 2001. Identification of novel glutathione transferases and polymorphic variants by expressed sequence tag database analysis. Drug Metab Dispos. 29:544–547.
- Board PG, Coggan M, Cappello J, Zhou H, Oakley AJ, Anders MW. 2008. S-(4-Nitrophenacyl)glutathione is a specific substrate for glutathione transferase Omega 1-1. Anal Biochem. 374:25–30.
- Board PG, Coggan M, Chelvanayagam G, Easteal S, Jermiin LS, Schulte GK, Danley DE, Hoth LR, Griffor CM, Kamath AV, et al. 2000. Identification, characterization and crystal structure of the Omega class glutathione transferases. J Biol Chem. 275:24798–24806.
- Board PG, Coggan M, Wilce MC, Parker MW. 1995. Evidence for an essential serine residue in the active site of the Theta class glutathione transferases. Biochem J. 311:247–250.
- Board P, Harris M, Flanagan J, Langton L, Coggan M. 1998. Genetic heterogeneity of the structure and function of GSTT2 and GSTP1. Chem Biol Interact. 111–112:83–89.
- Board PG, Menon D. 2013. Glutathione transferases, regulators of cellular metabolism and physiology. Biochim Biophys Acta. 1830:3267–3288.
- Board PG, Menon D. 2016. Structure, function and disease relevance of Omega-class glutathione transferases. Arch Toxicol. 90:1049–1067.
- Board PG, Suzuki T, Shaw DC. 1988. Human muscle glutathione S-transferase (GST-4) shows close homology to human liver GST-1. Biochim Biophys Acta. 953:214–217.
- Board PG, Taylor MC, Coggan M, Parker MW, Lantum HB, Anders MW. 2003. Clarification of the role of key active site residues of glutathione transferase Zeta/maleylacetoacetate isomerase by a new spectrophotometric technique. Biochem J. 374:731–737.
- Bocedi A, Fabrini R, Farrotti A, Stella L, Ketterman AJ, Pedersen JZ, Allocati N, Lau PC, Grosse S, Eltis LD, et al. 2013. The impact of nitric oxide toxicity on the evolution of the glutathione transferase superfamily. A proposal for an evolutionary driving force. J Biol Chem. 288:24936–24947.
- Bocedi A, Fabrini R, Lo Bello M, Caccuri AM, Federici G, Mannervik B, Cornish-Bowden A, Ricci G. 2016. Evolution of negative cooperativity in glutathione transferase enabled preservation of enzyme function. J Biol Chem. 291:26739–26749.
- Bogaards JJ, van Ommen B, van Bladeren PJ. 1992. Purification and characterization of eight glutathione S-transferase isoenzymes of hamster. Comparison of subunit composition of enzymes from liver, kidney, testis, pancreas and trachea. Biochem J. 286:383–388.
- Bolton MG, Hilton J, Robertson KD, Streeper RT, Colvin OM, Noe DA. 1993. Kinetic analysis of the reaction of melphalan with water, phosphate, and glutathione. Drug Metab Dispos. 21:986–996.
- Boone CD, Zhong G, Smeltz M, James MO, McKenna R. 2014. Preliminary X-ray crystallographic analysis of glutathione transferase Zeta 1 (GSTZ1a-1a). Acta Crystallogr F Struct Biol Commun. 70:187–189.
- Booth J, Boyland E, Sims P. 1961. An enzyme from rat liver catalyzing conjugations with glutathione. Biochem J. 79:516–524.
- Borst P, de Wolf C, van de Wetering K. 2007. Multidrug resistance-associated proteins 3, 4, and 5. Pflugers Arch. 453:661–673.
- Boušová I, Skálová L. 2012. Inhibition and induction of glutathione S-transferases by flavonoids: possible pharmacological and toxicological consequences. Drug Metab Rev. 44:267–286.
- Boyer TD, Vessey DA, Kempner E. 1986. Radiation inactivation of microsomal glutathione S-transferase. J Biol Chem. 261:16963–16968.
- Boyland E, Chasseaud LF. 1969. The role of glutathione and glutathione S-transferases in mercapturic acid biosynthesis. Adv Enzymol Relat Areas Mol Biol. 32:173–219.
- Boyland E, Sims P. 1958. Metabolism of polycyclic compounds. 12. An acid-labile precursor of 1-naphthylmercapturic acid and naphthol: an N-acetyl-S-(1:2-dihydrohydroxy-naphthyl)-L-cysteine. Biochem J. 68:440–447.
- Boyland E, Sims P, Soloman JP. 1957. An acid-labile precursor of 1-naphthylmercapturic acid and 1-naphthol. Biochem J. 66:41–42.
- Boyland E, Williams K. 1965. An enzyme catalysing the conjugation of epoxides with glutathione. Biochem J. 94:190–197.
- Brannigan JA, Dodson G, Duggleby HJ, Moody PC, Smith JL, Tomchick DR, Murzin AG. 1995. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature. 378:416–419.
- Bräutigam M, Teusch N, Schenk T, Sheikh M, Aricioglu RZ, Borowski SH, Neudörfl JM, Baumann U, Griesbeck AG, Pietsch M. 2015. Selective inhibitors of glutathione transferase P1 with trioxane structure as anticancer agents. ChemMedChem. 10:629–639.
- Bräutigam L, Zhang J, Dreij K, Spahiu L, Holmgren A, Abe H, Tew KD, Townsend DM, Kelner MJ, Morgenstern R, et al. 2018. MGST1, a GSH transferase/peroxidase essential for development and hematopoietic stem cell differentiation. Redox Biol. 17:171–179.
- Bray HG, Franklin TJ, James SP. 1959a. The formation of mercapturic acids. 2. The possible role of glutathionase. Biochem J. 71:690–696.
- Bray HG, Franklin TJ, James SP. 1959b. The formation of mercapturic acids. 3. N-Acetylation of S-substituted cysteines in the rabbit, rat and guinea pig. Biochem J. 73:465–473.
- Bray HG, James SP. 1960. The formation of mercapturic acids. 4. Deacetylation of mercapturic acids by the rabbit, rat and guinea pig. Biochem J. 74:394–397.
- Bresell A, Weinander R, Lundqvist G, Raza H, Shimoji M, Sun TH, Balk L, Wiklund R, Eriksson J, Jansson C, et al. 2005. Bioinformatic and enzymatic characterization of the MAPEG superfamily. FEBS J. 272:1688–1703.
- Brewis IA, Ferguson MA, Mehlert A, Turner AJ, Hooper NM. 1995. Structures of the glycosyl-phosphatidylinositol anchors of porcine and human renal membrane dipeptidase. Comprehensive structural studies on the porcine anchor and interspecies comparison of the glycan core structures. J Biol Chem. 270:22946–22956.
- Bridges CC, Joshee L, Zalups RK. 2008a. MRP2 and the DMPS- and DMSA-mediated elimination of mercury in TR– and control rats exposed to thiol S-conjugates of inorganic mercury. Toxicol Sci. 105:211–220.
- Bridges CC, Joshee L, Zalups RK. 2008b. Multidrug resistance proteins and the renal elimination of inorganic mercury mediated by 2,3-dimercaptopropane-1-sulfonic acid and meso-2,3-dimercaptosuccinic acid. J Pharmacol Exp Ther. 324:383–390.
- Bridges CC, Joshee L, Zalups RK. 2011. MRP2 and the handling of mercuric ions in rats exposed acutely to inorganic and organic species of mercury. Toxicol Appl Pharmacol. 251:50–58.
- Bridges CC, Zalups RK. 2010. Transport of inorganic mercury and methylmercury in target tissues and organs. J Toxicol Environ Health B Crit Rev. 13:385–410.
- Bridges CC, Zalups RK. 2017. Mechanisms involved in the transport of mercuric ions in target tissues. Arch Toxicol. 91:63–81.
- Brock J, Board PG, Oakley AJ. 2013. Structural insights into Omega-class glutathione transferases: a snapshot of enzyme reduction and identification of a non-catalytic ligandin site. PLoS One. 8:e60324.
- Bröer S. 2008. Amino acid transport across mammalian intestinal and renal epithelia. Physiol Rev. 88:249–286.
- Brouillet A, Darbouy M, Okamoto T, Chobert MN, Lahuna O, Garlatti M, Goodspeed D, Laperche Y. 1994. Functional characterization of the rat γ-glutamyl transpeptidase promoter that is expressed and regulated in the liver and hepatoma cells. J Biol Chem. 269:14878–14884.
- Brown SD, Babbitt PC. 2012. Inference of functional properties from large-scale analysis of enzyme superfamilies. J Biol Chem. 287:35–42.
- Brundin A, Ratnayake JH, Sunram JM, Anders MW. 1982. Glutathione-dependent reductive dehalogenation of 2,2′,4′-trichloroacetophenone to 2′,4′-dichloroacetophenone. Biochem Pharmacol. 31:3885–3890.
- Bruns CM, Hubatsch I, Ridderstrom M, Mannervik B, Tainer JA. 1999. Human glutathione transferase A4-4 crystal structures and mutagenesis reveal the basis of high catalytic efficiency with toxic lipid peroxidation products. J Mol Biol. 288:427–439.
- Bruns FH, Schulze C. 1962. Acylase I. Reindarstellung, physikalisch-chemische Eigenschaften and Identifizieren mit “Hippurikase”. Biochem Z. 336:162–181.
- Brüsewitz G, Cameron BD, Chasseaud LF, Görler K, Hawkins DR, Koch H, Mennicke WH. 1977. The metabolism of benzyl isothiocyanate and its cysteine conjugate. Biochem J. 162:99–107.
- Buckhaults P, Rago C, St Croix B, Romans KE, Saha S, Zhang L, Vogelstein B, Kinzler KW. 2001. Secreted and cell surface genes expressed in benign and malignant colorectal tumors. Cancer Res. 61:6996–7001.
- Bulle F, Mavier P, Zafrani ES, Preaux AM, Lescs MC, Siegrist S, Dhumeaux D, Guellaen G. 1990. Mechanism of gamma-glutamyl transpeptidase release in serum during intrahepatic and extrahepatic cholestasis in the rat: a histochemical, biochemical and molecular approach. Hepatology. 11:545–550.
- Burckhardt G, Burckhardt BC. 2011. In vitro and in vivo evidence of the importance of organic anion transporters (OATs) in drug therapy. In: Fromm M, Kim R, editors. Hamdbook of experimental pharmacology. Vol. 201. Berlin: Springer; p. 29–104.
- Burg D, Mulder GJ. 2002. Glutathione conjugates and their synthetic derivatives as inhibitors of glutathione-dependent enzymes involved in cancer and drug resistance. Drug Metab Rev. 34:821–863.
- Burley SK, David PR, Lipscomb WN. 1991. Leucine aminopeptidase: bestatin inhibition and a model for enzyme-catalyzed peptide hydrolysis. Proc Natl Acad Sci USA. 88:6916–6920.
- Burley SK, David PR, Sweet RM, Taylor A, Lipscomb WN. 1992. Structure determination and refinement of bovine lens leucine aminopeptidase and its complex with bestatin. J Mol Biol. 224:113–140.
- Burley SK, David PR, Taylor A, Lipscomb WN. 1990. Molecular structure of leucine aminopeptidase at 2.7-Å resolution. Proc Natl Acad Sci USA. 87:6878–6882.
- Busenlehner LS, Ålander J, Jegerscohld C, Holm PJ, Bhakat P, Hebert H, Morgenstern R, Armstrong RN. 2007. Location of substrate binding sites within the integral membrane protein microsomal glutathione transferase-1. Biochemistry. 46:2812–2822.
- Busenlehner LS, Codreanu SG, Holm PJ, Bhakat P, Hebert H, Morgenstern R, Armstrong RN. 2004. Stress sensor triggers conformational response of the integral membrane protein microsomal glutathione transferase 1. Biochemistry. 43:11145–11152.
- Byington KH, Hansbrough E. 1979. Inhibition of the enzymatic activity of ligandin by organogermanium, organolead or organotin compounds and the biliary excretion of sulfobromophthalein by the rat. J Pharmacol Exp Ther. 208:248–253.
- Cacciatore I, Caccuri AM, Cocco A, De Maria F, Di Stefano A, Luisi G, Pinnen F, Ricci G, Sozio P, Turella P. 2005. Potent isozyme-selective inhibition of human glutathione S-transferase A1-1 by a novel glutathione S-conjugate. Amino Acids. 29:255–261.
- Caccuri AM, Antonini G, Board PG, Parker MW, Nicotra M, Lo Bello M, Federici G, Ricci G. 1999. Proton release on binding of glutathione to Alpha, Mu and Delta class glutathione transferases. Biochem J. 344:419–425.
- Caccuri AM, Antonini G, Nicotra M, Battistoni A, Bello ML, Board PG, Parker MW, Ricci G. 1997. Catalytic mechanism and role of hydroxyl residues in the active site of Theta class glutathione S-transferases. Investigation of Ser-9 and Tyr-113 in a glutathione S-transferase from the Australian sheep blowfly, Lucilia cuprina. J Biol Chem. 272:29681–29686.
- Caccuri AM, Ascenzi P, Antonini G, Parker MW, Oakley AJ, Chiessi E, Nuccetelli M, Battistoni A, Bellizia A, Ricci G. 1996. Structural flexibility modulates the activity of human glutathione transferase P1-1. Influence of a poor co-substrate on dynamics and kinetics of human glutathione transferase. J Biol Chem. 271:16193–16198.
- Callaway EC, Zhang Y, Chew W, Chow HH. 2004. Cellular accumulation of dietary anticarcinogenic isothiocyanates is followed by transporter-mediated export as dithiocarbamates. Cancer Lett. 204:23–31.
- Calvaresi M, Stenta M, Garavelli M, Altoé P, Bottoni A. 2012. Computational evidence for the catalytic mechanism of human glutathione S-transferase A3-3: a QM/MM investigation. ACS Catal. 2:280–286.
- Campbell BJ. 1970. Renal dipeptidase. Methods Enzymol. 19:722–729.
- Campbell BJ, Baker SF, Shukla SD, Forrester LJ, Zahler WL. 1990a. Bioconversion of leukotriene D4 by lung dipeptidase. Biochim Biophys Acta. 1042:107–112.
- Campbell JA, Corrigall AV, Guy A, Kirsch RE. 1991. Immunohistologic localization of Alpha, Mu, and Pi class glutathione S-transferases in human tissues. Cancer. 67:1608–1613.
- Campbell BJ, Forrester LJ, Zahler WL, Burks M. 1984. β-Lactamase activity of purified and partially characterized human renal dipeptidase. J Biol Chem. 259:14586–14590.
- Campbell BJ, Lin YC, Davis RV, Ballew E. 1966. The purification and properties of a particulate renal dipeptidase. Biochim Biophys Acta. 118:371–386.
- Campbell BJ, Shih YD, Forrester LJ, Zahler WL. 1988. Specificity and inhibition studies of human renal dipeptidase. Biochim Biophys Acta. 956:110–118.
- Campbell E, Takahashi Y, Abramovitz M, Peretz M, Listowsky I. 1990b. A distinct human testis and brain Mu-class glutathione S-transferase. Molecular cloning and characterization of a form present even in individuals lacking hepatic type Mu isoenzymes. J Biol Chem. 265:9188–9193.
- Cao J, Zang J, Ma C, Li X, Hou J, Li J, Huang Y, Xu W, Wang B, Zhang Y. 2018. Design, synthesis, and biological evaluation of pyrazoline-based hydroxamic acid derivatives as aminopeptidase N (APN) inhibitors. ChemMedChem. 13:431–436.
- Cappiello M, Alterio V, Amodeo P, Del Corso A, Scaloni A, Pedone C, Moschini R, De Donatis GM, De Simone G, Mura U. 2006. Metal ion substitution in the catalytic site greatly affects the binding of sulfhydryl-containing compounds to leucyl aminopeptidase. Biochemistry. 45:3226–3234.
- Cappiello M, Lazzarotti A, Buono F, Scaloni A, D'Ambrosio C, Amodeo P, Méndez BL, Pelosi P, Del Corso A, Mura U. 2004. New role for leucyl aminopeptidase in glutathione turnover. Biochem J. 378:35–44.
- Carpenter FH, Harrington KT. 1972. Intermolecular cross-linking of monomeric proteins and cross-linking of oligomeric proteins as a probe of quaternary structure. Application to leucine aminopeptidase (bovine lens). J Biol Chem. 247:5580–5586.
- Carpenter FH, Vahl JM. 1973. Leucine aminopeptidase (Bovine Lens). Mechanism of activation by Mg2+ and Mn2+ of the zinc metalloenzyme, amino acid composition, and sulfhydryl content. J Biol Chem. 248:294–304.
- Carron CP, Trujillo JI, Olson KL, Huang W, Hamper BC, Dice T, Neal BE, Pelc MJ, Day JE, Rohrer DC. 2010. Discovery of an oral potent selective inhibitor of hematopoietic prostaglandin D synthase (HPGDS). ACS Med Chem Lett. 1:59–63.
- Carter BZ, Shi ZZ, Barrios R, Lieberman MW. 1998. Gamma-glutamyl leukotrienase, a gamma-glutamyl transpeptidase gene family member, is expressed primarily in spleen. J Biol Chem. 273:28277–28285.
- Carter BZ, Wiseman AL, Orkiszewski R, Ballard KD, Ou CN, Lieberman MW. 1997. Metabolism of leukotriene C4 in γ-glutamyl transpeptidase-deficient mice. J Biol Chem. 272:12305–12310.
- Castellano I, Merlino A. 2012. γ-Glutamyltranspeptidases: sequence, structure, biochemical properties, and biotechnological applications. Cell Mol Life Sci. 69:3381–3394.
- Castellano I, Merlino A. 2013. Gamma-glutamyl transpeptidases: structure and function. New York: Springer.
- Castonguay R, Halim D, Morin M, Furtos A, Lherbet C, Bonneil E, Thibault P, Keillor JW. 2007. Kinetic characterization and identification of the acylation and glycosylation sites of recombinant human γ-glutamyltranspeptidase. Biochemistry. 46:12253–12262.
- Castonguay R, Lherbet C, Keillor JW. 2002. Mapping of the active site of rat kidney γ-glutamyl transpeptidase using activated esters and their amide derivatives. Bioorg Med Chem. 10:4185–4191.
- Cesareo E, Parker LJ, Pedersen JZ, Nuccetelli M, Mazzetti AP, Pastore A, Federici G, Caccuri AM, Ricci G, Adams JJ. 2005. Nitrosylation of human glutathione transferase P1-1 with dinitrosyl diglutathionyl iron complex in vitro and in vivo. J Biol Chem. 280:42172–42180.
- Chambers JC, Zhang W, Lord GM, van der Harst P, Lawlor DA, Sehmi JS, Gale DP, Wass MN, Ahmadi KR, Bakker SJ. 2010. Genetic loci influencing kidney function and chronic kidney disease. Nat Genet. 42:373–375.
- Chan WWC, Dennis P, Demmer W, Brand K. 1982. Inhibition of leucine aminopeptidase by amino acid hydroxamates. J Biol Chem. 257:7955–7957.
- Chasseaud LF. 1974. Glutathione S-transferases. In: Flohé L, Benöhr HC, Sies H, Waller, HD, Wendel, A editors. Glutathione. Stuttgart: Thieme; p. 90–109.
- Chasseaud LF. 1973. The nature and distribution of enzymes catalyzing the conjugation of glutathione with foreign compounds. Drug Metab Rev. 2:185–220.
- Chasseaud LF. 1976. Conjugation with glutathione and mercapturic acid excretion. In: Arias IM, Jakoby W, editors. Glutathione: metabolism and function. New York: Raven Press; p. 77–114.
- Chen H, Roques BP, Fournié-Zaluski MC. 1999. Design of the first highly potent and selective aminopeptidase N (EC 3.4.11.2) inhibitor. Bioorg Med Chem Lett. 9:1511–1516.
- Chen H, Wang H, Qin XJ, Chen C, Feng LL, Chen LZ, Du LP, Li MY. 2015. A bestatin-based fluorescent probe for aminopeptidase N cell imaging. Chin Chem Lett. 26:513–516.
- Chen J, Armstrong RN. 1995. Stereoselective catalysis of a retro-Michael reaction by class Mu glutathione transferases. Consequences for the internal distribution of products in the active site. Chem Res Toxicol. 8:580–585.
- Chen J, Schenker S, Henderson GI. 2002. 4-Hydroxynonenal detoxification by mitochondrial glutathione S-transferase is compromised by short-term ethanol consumption in rats. Alcoholism Clin Exp Res. 26:1252–1258.
- Chen L, Du L, Li M. 2013a. The first inhibitor-based fluorescent imaging probe for aminopeptidase N. Drug Discoveries Ther. 7:124–125.
- Chen L, Lin YL, Peng G, Li F. 2012a. Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc Natl Acad Sci USA. 109:17966–17971.
- Chen L, Sun W, Li J, Liu Z, Ma Z, Zhang W, Du L, Xu W, Fang H, Li M. 2013b. The first ratiometric fluorescent probes for aminopeptidase N cell imaging. Org Biomol Chem. 11:378–382.
- Chen L, Sun W, Li W, Li J, Du L, Xu W, Fang H, Li M. 2012b. The first ratiometric fluorescent probe for aminopeptidase N. Anal Methods. 4:2661–2663.
- Chen WJ, Graminski GF, Armstrong RN. 1988. Dissection of the catalytic mechanism of isozyme 4-4 of glutathione S-transferase with alternative substrates. Biochemistry. 27:647–654.
- Chen YR, Chen TY, Zhang SL, Lin SM, Zhao YR, Ye F, Zhang X, Shi L, Dang SS, Liu M. 2009. Identification of a novel protein binding to hepatitis C virus core protein. J Gastroenterol Hepatol. 24:1300–1304.
- Chenault HK, Dahmer J, Whitesides GM. 1989. Kinetic resolution of unnatural and rarely occurring amino acids: enantioselective hydrolysis of N-acyl amino acids catalyzed by acylase I. J Am Chem Soc. 111:6354–6364.
- Chikhi N, Holic N, Guellaen G, Laperche Y. 1999. Gamma-glutamyl transpeptidase gene organization and expression: a comparative analysis in rat, mouse, pig and human species. Comp Biochem Physiol B Biochem Mol Biol. 122:367–380.
- Chinta SJ, Kumar JM, Zhang H, Forman HJ, Andersen JK. 2006. Up-regulation of γ-glutamyl transpeptidase activity following glutathione depletion has a compensatory rather than an inhibitory effect on mitochondrial complex I activity: implications for Parkinson’s disease. Free Radical Biol Med. 40:1557–1563.
- Cho HY, Tanizawa K, Tanaka H, Soda K. 1987. A spectrophotometric rate assay of aminoacylase. Anal Biochem. 165:142–146.
- Chowdhury UK, Zakharyan RA, Hernandez A, Avram MD, Kopplin MJ, Aposhian HV. 2006. Glutathione-S-transferase-omega [MMA(V) reductase] knockout mice: enzyme and arsenic species concentrations in tissues after arsenate administration. Toxicol Appl Pharmacol. 216:446–457.
- Christ AN, Labzin L, Bourne GT, Fukunishi H, Weber JE, Sweet MJ, Smythe ML, Flanagan JU. 2010. Development and characterization of new inhibitors of the human and mouse hematopoietic prostaglandin D2 synthases. J Med Chem. 53:5536–5548.
- Christ-Hazelhof E, Nugteren DH, Van Dorp DA. 1976. Conversions of prostaglandin endoperoxides by glutathione-S-transferases and serum albumins. Biochim Biophys Acta. 450:450–461.
- Ciaccio PJ, Tew KD, LaCreta FP. 1990. The spontaneous and glutathione S-transferase-mediated reaction of chlorambucil with glutathione. Cancer Commun. 2:279–285.
- Cigna N, Nicoletti C, Durand A, Chaix JC, Giardina T, Perrier J. 2007. Acylase 1 expression in rat intestinal crypt-villus axis. Cell Biol Int. 31:966–973.
- Coggan M, Flanagan JU, Parker MW, Vichai V, Pearson WR, Board PG. 2002. Identification and characterization of GSTT3, a third murine Theta class glutathione transferase. Biochem J. 366:323–332.
- Coles B, Ketterer B, Hinson JA. 1990. The role of glutathione and glutathione transferases in chemical carcinogenesis. Crit Rev Biochem Mol Biol. 25:47–70.
- Coles BF, Kadlubar FF. 2005. Human Alpha class glutathione S-transferases: genetic polymorphism, expression, and susceptibility to disease. Meth Enzymol. 401:9–42.
- Combes B, Stakelum GS. 1961. A liver enzyme that conjugates sulfobromophthalein sodium with glutathione. J Clin Invest. 40:981–988.
- Commandeur JN, Brakenhoff JP, De Kanter FJ, Vermeulen NP. 1988. Nephrotoxicity of mercapturic acids of three structurally related 2,2-difluoroethylenes in the rat. Indications for different bioactivation mechanisms. Biochem Pharmacol. 37:4495–4504.
- Commandeur JN, De Kanter FJ, Vermeulen NP. 1989. Bioactivation of the cysteine-S-conjugate and mercapturic acid of tetrafluoroethylene to acylating reactive intermediates in the rat: dependence of activation and deactivation activities on acetyl coenzyme A availability. Mol Pharmacol. 36:654–663.
- Commandeur JN, Stijntjes GJ, Wijngaard J, Vermeulen NP. 1991. Metabolism of L-cysteine S-conjugates and N-(trideuteroacetyl)-L-cysteine S-conjugates of four fluoroethylenes in the rat. Role of balance of deacetylation and acetylation in relation to the nephrotoxicity of mercapturic acids. Biochem Pharmacol. 42:31–38.
- Commandeur JN, Vermeulen NP. 1990. Identification of N-acetyl(2,2-dichlorovinyl)- and N-acetyl(1,2-dichlorovinyl)-L-cysteine as two regioisomeric mercapturic acids of trichloroethylene in the rat. Chem Res Toxicol. 3:212–218.
- Commandeur JNM, Stijntjes GJ, Vermeulen N. 1995. Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics. Pharmacol Rev. 47:271–330.
- Comstock KE, Widersten M, Hao XY, Henner WD, Mannervik B. 1994. A comparison of the enzymatic and physicochemical properties of human glutathione transferase M4-4 and three other human Mu class enzymes. Arch Biochem Biophys. 311:487–495.
- Conklin DJ, Haberzettl P, Lesgards JF, Prough RA, Srivastava S, Bhatnagar A. 2009. Increased sensitivity of glutathione S-transferase P-null mice to cyclophosphamide-induced urinary bladder toxicity. J Pharmacol Exp Ther. 331:456–469.
- Cook ND, Upperton KP, Challis BC, Peters TJ. 1987. The donor specificity and kinetics of the hydrolysis reaction of γ-glutamyltransferase. Biochim Biophys Acta. 914:240–245.
- Cook RM, Burke BJ, Buchhagen DL, Minna JD, Miller YE. 1993. Human aminoacylase-1. Cloning, sequence, and expression analysis of a chromosome 3p21 gene inactivated in small cell lung cancer. J Biol Chem. 268:17010–17017.
- Cook RM, Franklin WA, Moore MD, Johnson BE, Miller YE. 1998. Mutational inactivation of aminoacylase-I in a small cell lung cancer cell line. Genes Chromosom Cancer. 21:320–325.
- Cooper AJL, Hanigan MH. 2010. Enzymes involved in processing glutathione conjugates. In: McQueen CA, editor. Comprehensive toxicology. 2nd ed. Vol. 4. Oxford: Elsevier; p. 323–366.
- Cooper AJL, Hanigan MH. 2018. Metabolism of glutathione S-conjugates: multiple pathways. In: McQueen CA, editor. Comprehensive toxicology. 3rd ed. Vol. 10. Oxford: Elsevier; p. 363–406.
- Corti A, Fiocchi M, Curnis F. 2017. Targeting CD13 with Asn-Gly-Arg (NGR) peptide-drug conjugates. In: Mina-Osorio P, editor. Next-generation therapies and technologies for immune-mediated inflammatory diseases. Springer; p. 101–122.
- Corti A, Franzini M, Paolicchi A, Pompella A. 2010. Gamma-glutamyltransferase of cancer cells at the crossroads of tumor progression, drug resistance and drug targeting. Anticancer Res. 30:1169–1181.
- Corti A, Ponzoni M. 2004. Tumor vascular targeting with tumor necrosis factor alpha and chemotherapeutic drugs. Ann NY Acad Sci. 1028:104–112.
- Costantini C, Ko MH, Jonas MC, Puglielli L. 2007. A reversible form of lysine acetylation in the ER and Golgi lumen controls the molecular stabilization of BACE1. Biochem J. 407:383–395.
- Cotgreave IA, Schuppe-Koistinen I. 1994. A role for γ-glutamyl transpeptidase in the transport of cystine into human endothelial cells: relationship to intracellular glutathione. Biochim Biophys Acta. 1222:375–382.
- Couvertier SM, Weerapana E. 2014. Cysteine-reactive chemical probes based on a modular 4-aminopiperidine scaffold. MedChemCommun. 5:357–362.
- Crawford LA, Weerapana E. 2016. A tyrosine-reactive irreversible inhibitor for glutathione S-transferase Pi (GSTP1). Mol Biosyst. 12:1768–1771.
- Cromer BA, Gorman MA, Hansen G, Adams JJ, Coggan M, Littler DR, Brown LJ, Mazzanti M, Breit SN, Curmi PMG, et al. 2007. Structure of the Janus protein human CLIC2. J Mol Biol. 374:719–731.
- Cromer BA, Morton CJ, Board PG, Parker MW. 2002. From glutathione transferase to pore in a CLIC. Eur Biophys J. 31:356–364.
- Csaikl F, Reimer G, Csaikl U. 1986. Aminoacylase-1 isoenzymes: a comparative study. Heredity. 56:157–160.
- Cui DX, Wang XJ, Zhang XR, Zhao TK, Guo XX, Liu SM, Wang B, Ma WQ, Gao ZQ. 2014a. Preliminary detection of the antitumour activity of indoline-2,3-dione derivative DH-12a targeting aminopeptidase N. Mol Med Rep. 10:2681–2688.
- Cui SX, Zhang HL, Xu WF, Qu XJ. 2014b. 13F-1, a novel 5-fluorouracil prodrug containing an Asn-Gly-Arg (NO2) COOCH3 tripeptide, inhibits human colonic carcinoma growth by targeting aminopeptidase N (APN/CD13). Eur J Pharmacol. 734:50–59.
- Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fitzgerald S, et al. 2015. Ensembl 2015. Nucleic Acids Res. 43:D662–D669.
- Curry SH, Chu PI, Baumgartner TG, Stacpoole PW. 1985. Plasma concentrations and metabolic effects of intravenous sodium dichloroacetate. Clin Pharmacol Ther. 37:89–93.
- Curry SH, Lorenz A, Chu PI, Limacher M, Stacpoole PW. 1991. Disposition and pharmacodynamics of dichloroacetate (DCA) and oxalate following oral DCA doses. Biopharm Drug Dispos. 12:375–390.
- Cuypers HT, van Loon-Klaassen LA, Egberts WT, de Jong WW, Bloemendal H. 1982. The primary structure of leucine aminopeptidase from bovine eye lens. J Biol Chem. 257:7077–7085.
- Czerwinski M, Gibbs JP, Slattery JT. 1996. Busulfan conjugation by glutathione S-transferases α, μ, and π. Drug Metab Dispos. 24:1015–1019.
- Dai WY, Samanta S, Xue D, Petrunak EM, Stuckey JA, Han YY, Sun DX, Wu Y, Neamati N. 2019. Structure-based design of N-(5-phenylthiazol-2-yl)acrylamides as novel and potent glutathione S-transferase Omega 1 inhibitors. J Med Chem. 62:3068–3087.
- D'Ambrosio C, Talamo F, Vitale RM, Amodeo P, Tell G, Ferrara L, Scaloni A. 2003. Probing the dimeric structure of porcine aminoacylase 1 by mass spectrometric and modeling procedures. Biochemistry. 42:4430–4443.
- Danielsen EM, Hansen GH, Niels-Christiansen LL. 1995. Localization and biosynthesis of aminopeptidase N in pig fetal small-intestine. Gastroenterology. 109:1039–1050.
- Danielson UH, Mannervik B. 1985. Kinetic independence of the subunits of cytosolic glutathione transferase from the rat. Biochem J. 231:263–267.
- Dantzler WH, Evans KK, Groves CE, Welborn JR, North J, Stevens JL, Wright SH. 1998. Relation of cysteine conjugate nephrotoxicity to transport by the basolateral organic anion transport system in isolated S2 segments of rabbit proximal renal tubules. J Pharmacol Exp Ther. 286:52–60.
- Danziger RS. 2008. Aminopeptidase N in arterial hypertension. Heart Fail Rev. 13:293–298.
- Davies SJ, D'Sousa R, Philips H, Mattey D, Hiley C, Hayes JD, Aber GM, Strange RC. 1993. Localisation of α, μ and π class glutathione S-transferases in kidney: comparison with CuZn superoxide dismutase. Biochim Biophys Acta. 1157:204–208.
- Davison KL. 1991. Propachlor-S-glutathione metabolism by kidneys and ureters of calves. J Anim Sci. 69:1116–1121.
- Davison KL, Bakke JE, Larsen GL. 1992. Comparative metabolism of the cysteine and homocysteine conjugates of propachlor by in situ perfused kidneys of rats. Xenobiotica. 22:479–485.
- De Luca A, Federici L, De Canio M, Stella L, Caccuri AM. 2012. New insights into the mechanism of JNK1 inhibition by glutathione transferase P1-1. Biochemistry. 51:7304–7312.
- De Luca A, Mei G, Rosato N, Nicolai E, Federici L, Palumbo C, Pastore A, Serra M, Caccuri AM. 2014. The fine-tuning of TRAF2-GSTP1-1 interaction: effect of ligand binding and in situ detection of the complex. Cell Death Dis. 5:e1015.
- De Luca A, Rotili D, Carpanese D, Lenoci A, Calderan L, Scimeca M, Mai A, Bonanno E, Rosato A, Geroni C, et al. 2015. A novel orally active water-soluble inhibitor of human glutathione transferase exerts a potent and selective antitumor activity against human melanoma xenografts. Oncotarget. 6:4126–4143.
- de Oliveira DM, de Farias MT, Teles ALB, dos Santos MC, de Cerqueira MD, Lima RMF, El-Bachaa RS. 2014. 8-Methoxypsoralen is a competitive inhibitor of glutathione S-transferase P1-1. Front Cell Neurosci. 8:308.
- De Rooij BM, Commandeur JNM, Vermeulen N. 1998. Mercapturic acids as biomarkers of exposure to electrophilic chemicals: applications to environmental and industrial chemicals. Biomarkers. 3:239–303.
- Deaton DN, Do Y, Holt J, Jeune MR, Kramer HF, Larkin AL, Orband-Miller LA, Peckham GE, Poole C, Price DJ, et al. 2019. The discovery of quinoline-3-carboxamides as hematopoietic prostaglandin D synthase (H-PGDS) inhibitors. Bioorg Med Chem. 27:1456–1478.
- Deeley RG, Westlake C, Cole S. 2006. Transmembrane transport of endo- and xenobiotics by mammalian ATP-binding cassette multidrug resistance proteins. Physiol Rev. 86:849–899.
- Dekant W, Anders MW, Monks TJ, et al. 1993. Bioactivation of halogenated xenobiotics by S-conjugate formation. In: Anders MW, Dekant W, Henschler D, Oberleithner H, Silbernagl S, editors. Renal disposition and nephrotoxicity of xenobiotics. San Diego (CA): Academic Press; p. 187–215.
- Dekant W, Koob M, Henschler D. 1990. Metabolism of trichloroethene – in vivo and in vitro evidence for activation by glutathione conjugation. Chem Biol Interact. 73:89–101.
- Dekant W, Martens G, Vamvakas S, Metzler M, Henschler D. 1987. Bioactivation of tetrachloroethylene. Role of glutathione S-transferase-catalyzed conjugation versus cytochrome P-450-dependent phospholipid alkylation. Drug Metab Dispos. 15:702–709.
- Dekant W, Vamvakas S, Anders MW. 1994. Formation and fate of nephrotoxic and cytotoxic glutathione S-conjugates: cysteine conjugate beta-lyase pathway. Adv Pharmacol. 27:115–162.
- Delmas B, Gelfi J, Kut E, Sjöström H, Noren O, Laude H. 1994. Determinants essential for the transmissible gastroenteritis virus-receptor interaction reside within a domain of aminopeptidase-N that is distinct from the enzymatic site. J Virol. 68:5216–5224.
- Denton P. 2000. A structure-activity relationship for the hydrolysis of acetylamino acids by porcine aminoacylase. Bioorg Chem. 28:205–213.
- Denzlinger C, Rapp S, Hagmann W, Keppler D. 1985. Leukotrienes as mediators in tissue trauma. Science. 230:330–332.
- Deol R, Josephy PD. 2017. Acetylation of aromatic cysteine conjugates by recombinant human N-acetyltransferase 8. Xenobiotica. 47:202–207.
- DePierre JW, Morgenstern R. 1983. Comparison of the distribution of microsomal and cytosolic glutathione S-transferase activities in different organs of the rat. Biochem Pharmacol. 32:721–723.
- Deponte M. 2013. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim Biophys Acta. 1830:3217–3266.
- Desmots F, Rissel M, Loyer P, Turlin B, Guillouzo A. 2001. Immunohistological analysis of glutathione transferase A4 distribution in several human tissues using a specific polyclonal antibody. J Histochem Cytochem. 49:1573–1580.
- Devkota L, Lin CM, Strecker TE, Wang YF, Tidmore JK, Chen Z, Guddneppanavar R, Jelinek CJ, Lopez R, Liu L, et al. 2016. Design, synthesis, and biological evaluation of water-soluble amino acid prodrug conjugates derived from combretastatin, dihydronaphthalene, and benzosuberene-based parent vascular disrupting agents. Bioorg Med Chem. 24:938–956.
- Dhanani S, Awasthi YC. 2007. Glutathione S-transferase isozyme composition of human tissues. In: Awasthi YC, editor. Toxicology of glutathione transferases. Boca Raton (FL): Taylor & Francis; p. 321–338.
- Di Cianni F, Campa D, Tallaro F, Rizzato C, De Rango F, Barale R, Passarino G, Canzian F, Gemignani F, Montesanto A, et al. 2013. MAP3K7 and GSTZ1 are associated with human longevity: a two-stage case-control study using a multilocus genotyping. Age. 35:1357–1366.
- Di Paolo V, Fulci C, Rotili D, Sciarretta F, Lucidi A, Morozzo Della Rocca B, De Luca A, Rosato A, Quintieri L, Caccuri AM. 2019. Synthesis and characterisation of a new benzamide-containing nitrobenzoxadiazole as a GSTP1-1 inhibitor endowed with high stability to metabolic hydrolysis. J Enzyme Inhib Med Chem. 34:1131–1139.
- Dilda PJ, Ramsay EE, Corti A, Pompella A, Hogg PJ. 2008. Metabolism of the tumor angiogenesis inhibitor 4-(N-(S-glutathionylacetyl)amino)phenylarsonous acid. J Biol Chem. 283:35428–35434.
- DiNardo CD, Cortes JE. 2014. Tosedostat for the treatment of relapsed and refractory acute myeloid leukemia. Curr Opin Investig Drugs. 23:265–272.
- Ding Y, Ko MH, Pehar M, Kotch F, Peters NR, Luo Y, Salamat SM, Puglielli L. 2012. Biochemical inhibition of the acetyltransferases ATase1 and ATase2 reduces β-secretase (BACE1) levels and Aβ generation. J Biol Chem. 287:8424–8433.
- Dirr H, Reinemer P, Huber R. 1994a. Refined crystal structure of porcine class Pi glutathione S-trsansferase (pGST P1-1) at 2.1 Å resolution. J Mol Biol. 243:72–92.
- Dirr H, Reinemer P, Huber R. 1994b. X-ray crystal structures of cytosolic glutathione S-transferases. Implications for protein architecture, substrate recognition and catalytic function. Eur J Biochem. 220:645–661.
- Dixon J, Kaklamanis L, Turley H, Hickson ID, Leek RD, Harris AL, Gatter KC. 1994. Expression of aminopeptidase-n (CD 13) in normal tissues and malignant neoplasms of epithelial and lymphoid origin. J Clin Pathol. 47:43–47.
- Dohn DR, Quebbemann AJ, Borch RF, Anders MW. 1985. Enzymatic reaction of chlorotrifluoroethene with glutathione: 19F NMR evidence for stereochemical control of the reaction. Biochemistry. 24:5137–5143.
- Don AS, Kisker O, Dilda P, Donoghue N, Zhao X, Decollogne S, Creighton B, Flynn E, Folkman J, Hogg PJ. 2003. A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell. 3:497–509.
- Döring B, Petzinger E. 2014. Phase 0 and phase III transport in various organs: combined concept of phases in xenobiotic transport and metabolism. Drug Metab Rev. 46:261–282.
- Dourado DF, Fernandes PA, Mannervik B, Ramos MJ. 2010a. Glutathione transferase A1-1: catalytic importance of arginine 15. J Phys Chem B. 114:1690–1697.
- Dourado DF, Fernandes PA, Ramos MJ. 2008a. Mammalian cytosolic glutathione transferases. Curr Protein Pept Sci. 9:325–337.
- Dourado DF, Fernandes PA, Ramos MJ. 2009. Glutathione transferase A1-1: catalytic role of water. Theor Chem Acc. 124:71–83.
- Dourado D, Fernandes PA, Mannervik B, Ramos MJ. 2008b. Glutathione transferase: new model for glutathione activation. Chem Eur J. 14:9591–9598.
- Dourado D, Fernandes PA, Mannervik B, Ramos MJ. 2014. Isomerization of Δ5-androstene-3,17-dione into Δ4-androstene-3,17-dione catalyzed by human glutathione transferase A3-3: a computational study identifies a dual role for glutathione. J Phys Chem A. 118:5790–5800.
- Dourado D, Fernandes PA, Ramos MJ. 2010b. Glutathione transferase classes Alpha, Pi, and Mu: GSH activation mechanism. J Phys Chem B. 114:12972–12980.
- Drag M, Bogyo M, Ellman JA, Salvesen GS. 2010. Aminopeptidase fingerprints, an integrated approach for identification of good substrates and optimal inhibitors. J Biol Chem. 285:3310–3318.
- Drinkwater N, Lee J, Yang W, Malcolm TR, McGowan S. 2017. M1 aminopeptidases as drug targets: broad applications or therapeutic niche? FEBS J. 284:1473–1488.
- Duffel MW, Jakoby WB. 1982. Cysteine S-conjugate N-acetyltransferase from rat kidney microsomes. Mol Pharmacol. 21:444–448.
- Duffel MW, Jakoby WB. 1985. Cysteine S-conjugate N-acetyltransferase. Meth Enzymol. 113:516–520.
- Dulhunty A, Gage P, Curtis S, Chelvanayagam G, Board P. 2001. The glutathione transferase structural family includes a nuclear chloride channel and a ryanodine receptor calcium release channel modulator. J Biol Chem. 276:3319–3323.
- Dulhunty AF, Hewawasam R, Liu D, Casarotto MG, Board PG. 2011. Regulation of the cardiac muscle ryanodine receptor by glutathione transferases. Drug Metab Rev. 43:236–252.
- Dulik DM, Fenselau C, Hilton J. 1986. Characterization of melphalan-glutathione adducts whose formation is catalyzed by glutathione transferases. Biochem Pharmacol. 35:3405–3409.
- Durand A, Giardina T, Villard C, Roussel A, Puigserver A, Perrier J. 2003. Rat kidney acylase I: further characterisation and mutation studies on the involvement of Glu 147 in the catalytic process. Biochimie. 85:953–962.
- Dvash E, Har-Tal M, Barak S, Meir O, Rubinstein M. 2015. Leukotriene C-4 is the major trigger of stress-induced oxidative DNA damage. Nat Commun. 6:10112.
- Dvoráková L, Krůsek J, Stastný F, Lisý V. 1996. Analysis of kinetic properties of γ-glutamyl transpeptidase from rat kidney. Gen Physiol Biophys. 15:403–413.
- Earhart RH, Khandekar JD, Faraggi D, Schinella RA, Davis TE. 1989. Phase II trial of continuous drug infusions in advanced ovarian carcinoma: acivicin versus vinblastine. Invest New Drugs. 7:255–260.
- Eaton DL, Bammler TK. 1999. Concise review of the glutathione S-transferases and their significance to toxicology. Toxicol Sci. 49:156–164.
- Edfeldt F, Evenäs J, Lepistö M, Ward A, Petersen J, Wissler L, Rohman M, Sivars U, Svensson K, Perry M, et al. 2015. Identification of indole inhibitors of human hematopoietic prostaglandin D2 synthase (hH-PGDS). Bioorg Med Chem Lett. 25:2496–2500.
- Edwards SW, Knox WE. 1956. Homogentisate metabolism: the isomerization of maleylacetoacetate by an enzyme which requires glutathione. J Biol Chem. 220:79–91.
- Eklund BI, Moberg M, Bergquist J, Mannervik B. 2006. Divergent activities of human glutathione transferases in the bioactivation of azathioprine. Mol Pharmacol. 70:747–754.
- Elce JS. 1970. Metabolism of a glutathione conjugate of 2-hydroxyoestradiol by rat liver and kidney preparations in vitro. Biochem J. 116:913–917.
- Elce JS, Broxmeyer B. 1976. γ-Glutamyltransferase of rat kidney. Simultaneous assay of the hydrolysis and transfer reactions with (glutamate-14C)glutathione. Biochem J. 153:223–232.
- Emdin M, Passino C, Pompella A, Paolicchi A. 2006. Gamma-glutamyltransferase as a cardiovascular risk factor. Eur Heart J. 27:2145–2146.
- Endo Y. 1978. N-Acyl-L-aromatic amino acid deacylase in animal tissues. Biochim Biophys Acta. 523:207–214.
- Endo Y. 1980. In vivo deacetylation of N-acetyl amino acids by kidney acylases in mice and rats. A possible role of acylase system in mammalian kidneys. Biochim Biophys Acta. 628:13–18.
- Engelke UF, Sass JO, Van Coster RN, Gerlo E, Olbrich H, Krywawych S, Calvin J, Hart C, Omran H, Wevers RA. 2008. NMR spectroscopy of aminoacylase 1 deficiency, a novel inborn error of metabolism. NMR Biomed. 21:138–147.
- Enoiu M, Herber R, Leroy P, Wellman M. 2003. The role of gamma-glutamyltranspeptidase in the metabolism and cytotoxicity of 4-hydroxynonenal-glutathione conjugate: evidence and hypothesis. Biofactors. 17:175–185.
- Erdman AR, Mangravite LM, Urban TJ, Lagpacan LL, Castro RA, de la Cruz M, Chan W, Huang CC, Johns SJ, Kawamoto M, et al. 2006. The human organic anion transporter 3 (OAT3; SLC22A8): genetic variation and functional genomics. Am J Physiol Renal Fluid Electrolyte Physiol. 290:F905–F912.
- Erhardt S, Jaime E, Weston J. 2005. A water sluice is generated in the active site of bovine lens leucine aminopeptidase. J Am Chem Soc. 127:3654–3655.
- Escobar-García DM, Del Razo LM, Sanchez-Peña LC, Mandeville PB, Lopez-Campos C, Escudero-Lourdes C. 2012. Association of glutathione S-transferase Omega 1-1 polymorphisms (A140D and E208K) with the expression of interleukin-8 (IL-8), transforming growth factor beta (TGF-β), and apoptotic protease-activating factor 1 (Apaf-1) in humans chronically exposed to arsenic in drinking water. Arch Toxicol. 86:857–868.
- Fabrini R, De Luca A, Stella L, Mei G, Orioni B, Ciccone S, Federici G, Lo Bello M, Ricci G. 2009. Monomer-dimer equilibrium in glutathione transferases: a critical re-examination. Biochemistry. 48:10473–10482.
- Fagerberg L, Hallström BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, et al. 2014. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics. 13:397–406.
- Falkson G, Cnaan A, Simson IW, Dayal Y, Falkson H, Smith TJ, Haller DG. 1990. A randomized phase II study of acivicin and 4′-deoxydoxorubicin in patients with hepatocellular carcinoma in an Eastern Cooperative Oncology Group study. Am J Clin Oncol. 13:510–515.
- Fang YY, Kashkarov U, Anders MW, Board PG. 2006. Polymorphisms in the human glutathione transferase Zeta promoter. Pharmacogenet Genomics. 16:307–313.
- Farrell CA, Allegretto NJ, Hitchcock M. 1987. Cilastatin-sensitive dehydropeptidase I enzymes from three sources all catalyze carbapenem hydrolysis and conversion of leukotriene D4 to leukotriene E4. Arch Biochem Biophys. 256:253–259.
- Federici L, Lo Sterzo C, Pezzola S, Di Matteo A, Scaloni F, Federici G, Caccuri AM. 2009. Structural basis for the binding of the anticancer compound 6-(7-nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol to human glutathione S-transferases. Cancer Res. 69:8025–8034.
- Federspiel JD, Codreanu SG, Goyal S, Albertolle ME, Lowe E, Teague J, Wong H, Guengerich FP, Liebler DC. 2016. Specificity of protein covalent modification by the electrophilic proteasome inhibitor carfilzomib in human cells. Mol Cell Proteomics. 15:3233–3242.
- Fernández-Cañón JM, Peñalva MA. 1998. Characterization of a fungal maleylacetoacetate isomerase gene and identification of its human homologue. J Biol Chem. 273:329–337.
- Ferri L, Funghini S, Fioravanti A, Biondi EG, la Marca G, Guerrini R, Donati MA, Morrone A. 2014. Aminoacylase I deficiency due to ACY1 mRNA exon skipping. Clin Genet. 86:367–372.
- Fetissov SO, Schröder O, Jakobsson PJ, Samuelsson B, Haeggström JZ, Hökfelt T. 2002. Expression of microsomal glutathione S-transferase type 3 mRNA in the rat nervous system. Neuroscience. 115:891–897.
- Fiala S, Fiala AE, Dixon B. 1972. γ-Glutamyl transpeptidase in transplantable, chemically induced rat hepatomas and “spontaneous” mouse hepatomas. J Natl Cancer Inst. 48:1393–1401.
- Fiala S, Mohindru A, Kettering WG, Fiala AE, Morris HP. 1976. Glutathione and gamma glutamyl transpeptidase in rat liver during chemical carcinogenesis. J Natl Cancer Inst. 57:591–598.
- Findlay VJ, Townsend DM, Saavedra JE, Buzard GS, Citro ML, Keefer LK, Ji X, Tew KD. 2004. Tumor cell responses to a novel glutathione S-transferase-activated nitric oxide-releasing prodrug. Mol Pharmacol. 65:1070–1079.
- Fjellstedt TA, Allen RH, Duncan BK, Jakoby WB. 1973. Enzymatic conjugation of epoxides with glutathione. J Biol Chem. 248:3702–3707.
- Flanagan JU, Rossjohn J, Parker MW, Board PG, Chelvanayagam G. 1998. A homology model for the human Theta-class glutathione transferase T1-1. Proteins. 33:444–454.
- Flanagan JU, Rossjohn J, Parker MW, Board PG, Chelvanayagam G. 1999. Mutagenic analysis of conserved arginine residues in and around the novel sulfate binding pocket of the human Theta class glutathione transferase T2-2. Protein Sci. 8:2205–2212.
- Flanagan JU, Smythe ML. 2011. Sigma-class glutathione transferases. Drug Metab Rev. 43:194–214.
- Flatgaard JE, Bauer KE, Kauvar LM. 1993. Isozyme specificity of novel glutathione-S-transferase inhibitors. Cancer Chemother Pharmacol. 33:63–70.
- Flipo M, Beghyn T, Charton J, Leroux VA, Deprez BP, Deprez-Poulain RF. 2007. A library of novel hydroxamic acids targeting the metallo-protease family: design, parallel synthesis and screening. Bioorg Med Chem. 15:63–76.
- Fones WS, Lee M. 1953. Hydrolysis of N-acyl derivatives of alanine and phenylalanine by acylase I and carboxypeptidase. J Biol Chem. 201:847–856.
- Forkert PG, D'Costa D, El-Mestrah M. 1999. Expression and inducibility of Alpha, Pi, and Mu glutathione S-transferase protein and mRNA in murine lung. Am J Respir Cell Mol Biol. 20:143–152.
- Fornaciari I, Fierabracci V, Corti A, Aziz Elawadi H, Lorenzini E, Emdin M, Paolicchi A, Franzini M. 2014. Gamma-glutamyltransferase fractions in human plasma and bile: characteristic and biogenesis. PLoS One. 9:e88532.
- Fournié-Zaluski MC, Roques BP. 2002. New selective aminopeptidase N inhibitors as potential therapeutics. In: Langner J, Ansorge J, editors. Ectopeptidases CD13/aminopeptidase N and CD26/dipeptidylpeptidase IV in medicine and biology. New York: Springer.
- Fowler J, Seltzer S. 1970. The synthesis of model compounds for maleylacetoacetic acid. Maleylacetone. J Org Chem. 35:3529–3532.
- Franzini M, Bramanti E, Ottaviano V, Ghiri E, Scatena F, Barsacchi R, Pompella A, Donato L, Emdin M, Paolicchi A. 2008. A high performance gel filtration chromatography method for γ-glutamyltransferase fraction analysis. Anal Biochem. 374:1–6.
- Frey J, Kördel W, Schneider F. 1977. The reaction of aminoacylase with chloromethylketone analogs of amino acids. Z Naturforsch C Biosci. 32:769–776.
- Friedberg T, Bentley P, Stasiecki P, Glatt HR, Raphael D, Oesch F. 1979. The identification, solubilization, and characterization of microsome-associated glutathione S-transferases. J Biol Chem. 254:12028–12033.
- Frost L, Suryadevara P, Cannell SJ, Groundwater PW, Hambleton PA, Anderson RJ. 2016. Synthesis of diacylated γ-glutamyl-cysteamine prodrugs, and in vitro evaluation of their cytotoxicity and intracellular delivery of cysteamine. Eur J Med Chem. 109:206–215.
- Frova C. 2006. Glutathione transferases in the genomics era: new insights and perspectives. Biomol Eng. 23:149–169.
- Fruton JS. 1950. The role of proteolytic enzymes in the biosynthesis of peptide bonds. Yale J Biol Med. 22:263–271.
- Fruton JS, Johnston RB, Fried M. 1951. Elongation of peptide chains in enzyme-catalyzed transamidation reactions. J Biol Chem. 190:39–53.
- Fu J, Zhang H, Zhuang Y, Liu H, Shi Q, Li D, Ju X. 2014. The role of N-acetyltransferase 8 in mesenchymal stem cell-based therapy for liver ischemia/reperfusion injury in rats. PLoS One. 9:e103355.
- Fu JJ, Liu L, Huang ZJ, Lai YS, Ji H, Peng SX, Tian JD, Zhang YH. 2013. Hybrid molecule from O2-(2,4-dinitrophenyl)diazeniumdiolate and oleanolic acid: a glutathione S-transferase π-activated nitric oxide prodrug with selective anti-human hepatocellular carcinoma activity and improved stability. J Med Chem. 56:4641–4655.
- Fu SC, Birnbaum SM. 1953. The hydrolytic action of acylase I on N-acylamino acids. J Am Chem Soc. 75:918–920.
- Fu ZD, Selwyn FP, Cui JY, Klaassen CD. 2016. RNA sequencing quantification of xenobiotic-processing genes in various sections of the intestine in comparison to the liver of male mice. Drug Metab Dispos. 44:842–856.
- Fujikawa Y, Morisaki F, Ogura A, Morohashi K, Enya S, Niwa R, Goto S, Kojima H, Okabe T, Nagano T, et al. 2015. A practical fluorogenic substrate for high-throughput screening of glutathione S-transferase inhibitors. Chem Commun. 51:11459–11462.
- Fujikawa Y, Nampo T, Mori M, Kikkawa M, Inoue H. 2018. Fluorescein diacetate (FDA) and its analogue as substrates for Pi-class glutathione S-transferase (GSTP1) and their biological application. Talanta. 179:845–852.
- Fujikawa Y, Urano Y, Komatsu T, Hanaoka K, Kojima H, Terai T, Inoue H, Nagano T. 2008. Design and synthesis of highly sensitive fluorogenic substrates for glutathione S-transferase and application for activity imaging in living cells. J Am Chem Soc. 130:14533–14543.
- Fujimoto K, Arakawa S, Watanabe T, Yasumo H, Ando Y, Takasaki W, Manabe S, Yamoto T, Oda S. 2007. Generation and functional characterization of mice with a disrupted glutathione S-transferase, Theta 1 gene. Drug Metab Dispos. 35:2196–2202.
- Fujita T, Brown C, Carlson EJ, Taylor T, de la Cruz M, Johns SJ, Stryke D, Kawamoto M, Fujita K, Castro R, et al. 2005. Functional analysis of polymorphisms in the organic anion transporter, SLC22A6 (OAT1). Pharmacogenet Genomics. 15:201–209.
- Fulci C, Rotili D, De Luca A, Stella L, della Rocca BM, Forgione M, Di Paolo V, Mai A, Falconi M, Quintieri L, et al. 2017. A new nitrobenzoxadiazole-based GSTP1-1 inhibitor with a previously unheard of mechanism of action and high stability. J Enzyme Inhib Med Chem. 32:240–247.
- Funayama T, Sekizawa K, Yamaya M, Yamauchi K, Ohno I, Ohrui T, Terajima M, Okinaga S, Sasaki H. 1996. Role of leukotriene-degrading enzymes in pulmonary response to antigen infusion in sensitized guinea pigs in vivo. Am J Respir Cell Mol Biol. 15:260–267.
- Gade W, Brown JL. 1978. Purification and partial characterization of α-N-acylpeptide hydrolase from bovine liver. J Biol Chem. 253:5012–5018.
- Gade W, Brown JL. 1981. Purification, characterization and possible function of α-N-acylamino acid hydrolase from bovine liver. Biochim Biophys Acta. 662:86–93.
- Gale L, Tew KD. 2001. Glutathione S-transferases as emerging therapeutic targets. Expert Opin Ther Targets. 5:477–489.
- Gallagher BC, Rudolph DB, Hinton BT, Hanigan MH. 1998. Differential induction of γ-glutamyl transpeptidase in primary cultures of rat and mouse hepatocytes parallels induction during hepatocarcinogenesis. Carcinogenesis. 19:1251–1255.
- Gallagher EP, Gardner JL, Barber DS. 2006. Several glutathione S-transferase isozymes that protect against oxidative injury are expressed in human liver mitochondria. Biochem Pharmacol. 71:1619–1628.
- Gao F, Fang Q, Zhang R, Lu J, Lu H, Wang C, Ma X, Xu J, Jia W, Xiang K. 2009. Polymorphism of DsbA-L gene associates with insulin secretion and body fat distribution in Chinese population. Endocr J. 56:487–494.
- Gao JJ, Gao ZH, Zhao CR, Yuan Y, Cui SX, Zhang XF, Cheng YN, Xu WF, Tang W, Qu XJ. 2011a. LYP, a novel bestatin derivative, inhibits cell growth and suppresses APN/CD13 activity in human ovarian carcinoma cells more potently than bestatin. Investig New Drugs. 29:574–582.
- Gao JJ, Xue X, Gao ZH, Cui SX, Cheng YN, Xu WF, Tang W, Qu XJ. 2011b. LYP, a bestatin dimethylaminoethyl ester, inhibited cancer angiogenesis both in vitro and in vivo. Microvasc Res. 82:122–130.
- Garcia-Calvo M, Lisnock JM, Bull HG, Hawes BE, Burnett DA, Braun MP, Crona JH, Davis HR, Dean DC, Detmers PA, et al. 2005. The target of ezetimibe is Niemann-Pick Cl-Like 1 (NPC1L1). Proc Natl Acad Sci USA. 102:8132–8137.
- Gardell SJ, Tate SS. 1980. Affinity labeling of γ-glutamyl transpeptidase by glutamine antagonists. Effects of the γ-glutamyl transferase and proteinase activities. FEBS Lett. 122:171–174.
- Gardner JL, Gallagher EP. 2001. Development of a peptide antibody specific to human glutathione S-transferase alpha 4-4 (hGSTA4-4) reveals preferential localization in human liver mitochondria. Arch Biochem Biophys. 390:19–27.
- Garnier N, Redstone GGJ, Dahabieh MS, Nichol JN, del Rincon SV, Gu YX, Bohle DS, Sun Y, Conklin DS, Mann KK, et al. 2014. The novel arsenical darinaparsin is transported by cystine importing systems. Mol Pharmacol. 85:576–585.
- Georgakis ND, Karagiannopoulos DA, Thireou TN, Eliopoulos EE, Labrou NE, Tsoungas PG, Koutsilieris MN, Clonis YD. 2017. Concluding the trilogy: the interaction of 2,2′-dihydroxy-benzophenones and their carbonyl N-analogues with human glutathione transferase M1-1 face to face with the P1-1 and A1-1 isoenzymes involved in MDR. Chem Biol Drug Des. 90:900.
- Geroni C, Marchini S, Cozzi P, Galliera E, Ragg E, Colombo T, Battaglia R, Howard M, D'Incalci M, Broggini M. 2002. Brostallicin, a novel anticancer agent whose activity is enhanced upon binding to glutathione. Cancer Res. 62:2332–2336.
- Giannousis PP, Bartlett PA. 1987. Phosphorus amino acid analogues as inhibitors of leucine aminopeptidase. J Med Chem. 30:1603–1609.
- Giardina T, Biagini A, Dalle Ore F, Ferre E, Reynier M, Puigserver A. 1997. The hog intestinal mucosa acylase I: subcellular localization, isolation, kinetic studies and biological function. Biochimie. 79:265–273.
- Giardina T, Perrier J, Puigserver A. 2000. The rat kidney acylase I, characterization and molecular cloning. Differences with other acylases I. Eur J Biochem. 267:6249–6255.
- Gildenhuys S, Dobreva M, Kinsley N, Sayed Y, Burke J, Pelly S, Gordon GP, Sayed M, Sewell T, Dirr HW. 2010a. Arginine 15 stabilizes an SNAr reaction transition state and the binding of anionic ligands at the active site of human glutathione transferase A1-1. Biophys Chem. 146:118–125.
- Gildenhuys S, Wallace LA, Burke JP, Balchin D, Sayed Y, Dirr HW. 2010b. Class Pi glutathione transferase unfolds via a dimeric and not monomeric intermediate: functional implications for an unstable monomer. Biochemistry. 49:5074–5081.
- Gilles I, Löffler HG, Schneider F. 1984. Ein neues Isolierungsverfahren fur Schweinenierenacylase. Kinetik des Co2+, Mn2+, Ni2+ und Cd2+-enzyms. Z Naturforsch C Biosci. 39:1017–1020.
- Gillham B. 1971. The reaction of aralkyl sulphate esters with glutathione catalysed by rat liver preparations. Biochem J. 121:667–672.
- Gillham B. 1973. The mechanism of the reaction between glutathione and 1-menaphthyl sulphate catalysed by a glutathione S-transferase from rat liver. Biochem J. 135:797–804.
- Glässer D, Hanson H. 1964. Ein weiteres Verfahren zur Gewinnung kristallisierter Leucinaminopeptidase aus Rinderaugenlinsen. Naturwissenschaften. 51:110–111.
- Glatt H, Oesch F. 1977. Inactivation of electrophilic metabolites by glutathione S-transferases and limitation of the system due to subcellular localization. Arch Toxicol. 39:87–96.
- Goldbarg JA, Friedman OM, Pineda EP, Smith EE, Chatterji R, Stein EH, Rutenburg AM. 1960. The colorimetric determination of γ-glutamyl transpeptidase with a synthetic substrate. Arch Biochem Biophys. 91:61–70.
- Goldsworthy TL, Hanigan MH, Pitot HC, Shinozuka H. 1986. Models of hepatocarcinogenesis in the rat–contrasts and comparisons. Crit Rev Toxicol. 17:61–89.
- Gonçalves-Dias C, Morello J, Correia MJ, Coelho NR, Antunes AMM, Macedo MP, Monteiro EC, Soto K, Pereira SA. 2019a. Mercapturate pathway in the tubulocentric perspective of diabetic kidney disease. Nephron. 143:17.
- Gonçalves-Dias C, Morello J, Semedo V, Correia MJ, Coelho NR, Monteiro EC, Antunes AMM, Pereira SA. 2019b. The mercapturomic profile of health and non-communicable diseases. High Throughput. 8.
- Goodspeed DC, Dunn TJ, Miller CD, Pitot HC. 1989. Human γ-glutamyl transpeptidase cDNA: comparison of hepatoma and kidney mRNA in the human and rat. Gene. 76:1–9.
- Goto S, Kawakatsu M, Izumi S, Urata Y, Kageyama K, Ihara Y, Koji T, Kondo T. 2009. Glutathione S-transferase Pi localizes in mitochondria and protects against oxidative stress. Free Radical Biol Med. 46:1392–1403.
- Graham DW, Ashton WT, Barash L, Brown JE, Brown RD, Canning LF, Chen A, Springer JP, Rogers EF. 1987. Inhibition of the mammalian β-lactamase renal dipeptidase (dehydropeptidase-I) by Z-2-(acylamino)-3-substituted-propenoic acids. J Med Chem. 30:1074–1090.
- Grahn E, Novotny M, Jakobsson E, Gustafsson A, Grehn L, Olin B, Madsen D, Wahlberg M, Mannervik B, Kleywegt GJ. 2006. New crystal structures of human glutathione transferase A1-1 shed light on glutathione binding and the conformation of the C-terminal helix. Acta Crystallogr D Biol Crystallogr. 62:197–207.
- Green RM, Elce JS. 1975. Acetylation of S-substituted cysteines by a rat liver and kidney microsomal N-acetyltransferase. Biochem J. 147:283–289.
- Green T, Lee R, Farrar D, Hill J. 2003. Assessing the health risks following environmental exposure to hexachlorobutadiene. Toxicol Lett. 138:63–73.
- Greenhough KJ, Turner AJ. 1991. Aminoacylase I is not a glycolipid-anchored ectoenzyme in pig kidney. Biochim Biophys Acta. 1076:364–368.
- Gregorc V, Citterio G, Vitali G, Spreafico A, Scifo P, Borri A, Donadoni G, Rossoni G, Corti A, Caligaris-Cappio F, et al. 2010. Defining the optimal biological dose of NGR-hTNF, a selective vascular targeting agent, in advanced solid tumours. Eur J Cancer. 46:198–206.
- Grembecka J, Kędzierski P, Sokalski WA. 1999. Non-empirical analysis of the nature of the inhibitor-active-site interactions in leucine aminopeptidase. Chem Phys Lett. 313:385–392.
- Grembecka J, Kȩdzierski P, Sokalski WA, Leszczyński J. 2001. Electrostatic models of inhibitory activity. Int J Quantum Chem. 83:180–192.
- Grembecka J, Mucha A, Cierpicki T, Kafarski P. 2002. Structure-based design and synthesis of dipeptide analogs as new inhibitors of leucine aminopeptidase. Phosphorus Sulfur Silicon Relat Elem. 177:1739–1743.
- Grembecka J, Mucha A, Cierpicki T, Kafarski P. 2003. The most potent organophosphorus inhibitors of leucine aminopeptidase. Structure-based design, chemistry, and activity. J Med Chem. 46:2641–2655.
- Grembecka J, Sokalski WA, Kafarski P. 2000. Computer-aided design and activity prediction of leucine aminopeptidase inhibitors. J Comput Aided Mol Des. 14:531–544.
- Griffith OW, Meister A. 1977. Selective inhibition of γ-glutamyl-cycle enzymes by substrate analogs. Proc Natl Acad Sci USA. 74:3330–3334.
- Griffith OW, Meister A. 1979. Translocation of intracellular glutathione to membrane-bound γ-glutamyl transpeptidase as a discrete step in the γ-glutamyl cycle: glutathionuria after inhibition of transpeptidase. Proc Natl Acad Sci USA. 76:268–272.
- Grillo MP, Benet LZ. 2001. Interaction of γ-glutamyltranspeptidase with clofibryl-S-acyl-glutathione in vitro and in vivo in rat. Chem Res Toxicol. 14:1033–1040.
- Grillo MP, Lohr MT, Khera S. 2013. Interaction of γ-glutamyltranspeptidase with ibuprofen-S-acyl-glutathione in vitro and in vivo in human. Drug Metab Dispos. 41:111–121.
- Grimm C, Hofstetter G, Aust S, Mutz-Dehbalaie I, Bruch M, Heinze G, Rahhal-Schupp J, Reinthaller A, Concin N, Polterauer S. 2013. Association of gamma-glutamyltransferase with severity of disease at diagnosis and prognosis of ovarian cancer. Br J Cancer. 109:610–614.
- Groom H, Lee M, Patil P, Josephy PD. 2014. Inhibition of human glutathione transferases by dinitronaphthalene derivatives. Arch Biochem Biophys. 555–556:71–76.
- Groves CE, Morales MN. 1999. Chlorotrifluoroethylcysteine interaction with rabbit proximal tubule cell basolateral membrane organic anion transport and apical membrane amino acid transport. J Pharmacol Exp Ther. 291:555–561.
- Groves CE, Muñoz L, Bahn A, Burckhardt G, Wright SH. 2003. Interaction of cysteine conjugates with human and rabbit organic anion transporter 1. J Pharmacol Exp Ther. 304:560–566.
- Grzywa R, Oleksyszyn J, Salvesen GS, Drąg M. 2010a. Identification of very potent inhibitor of human aminopeptidase N (CD13). Bioorg Med Chem Lett. 20:2497–2499.
- Grzywa R, Sokol AM, Sieńczyk M, Radziszewicz M, Kościołek B, Carty MP, Oleksyszyn J. 2010b. New aromatic monoesters of α-aminoaralkylphosphonic acids as inhibitors of aminopeptidase N/CD13. Bioorg Med Chem. 18:2930–2936.
- Gu K, Liu Y, Guo Z, Lian C, Yan CC, Shi P, Tian H, Zhu WH. 2016. In situ ratiometric quantitative tracing of intracellular leucine aminopeptidase activity via an activatable near-infrared fluorescent probe. ACS Appl Mater Interfaces. 8:26622–26629.
- Gu Y, Guo J, Pal A, Pan SS, Zimniak P, Singh SV, Ji X. 2004. Crystal structure of human glutathione S-transferase A3-3 and mechanistic implications for its high steroid isomerase activity. Biochemistry. 43:15673–15679.
- Guengerich FP. 2003. Activation of dihaloalkanes by thiol-dependent mechanisms. J Biochem Mol Biol. 36:20–27.
- Guo X, Dixit V, Liu HP, Shroads AL, Henderson GN, James MO, Stacpoole PW. 2006. Inhibition and recovery of rat hepatic glutathione S-transferase Zeta and alteration of tyrosine metabolism following dichloroacetate exposure and withdrawal. Drug Metab Dispos. 34:36–42.
- Gurulingappa H, Buckhalts P, Kinzler KW, Vogelstein B, Khan SR. 2004. Synthesis and evaluation of aminophosphinic acid derivatives as inhibitors of renal dipeptidase. Bioorg Med Chem Lett. 14:3531–3533.
- Gurulingappa H, Buckhaults P, Kumar SK, Kinzler KW, Vogelstein B, Khan SR. 2003. Design, synthesis and evaluation of new RDP inhibitors. Tetrahedron Lett. 44:1871–1873.
- Guthenberg C, Akerfeldt K, Mannervik B. 1979. Purification of glutathione-S-transferase from human placenta. Acta Chem Scand. 33:595–596.
- Guthenberg C, Mannervik B. 1979. Purification of glutathione S-transferases from rat lung by affinity chromatography. Evidence for an enzyme form absent in rat liver. Biochem Biophys Res Commun. 86:1304–1310.
- Guy CA, Hoogendoorn B, Smith SK, Coleman S, O'Donovan MC, Buckland PR. 2004. Promoter polymorphisms in glutathione-S-transferase genes affect transcription. Pharmacogenetics. 14:45–51.
- Habib GM, Barrios R, Shi ZZ, Lieberman MW. 1996. Four distinct membrane-bound dipeptidase RNAs are differentially expressed and show discordant regulation with γ-glutamyl transpeptidase. J Biol Chem. 271:16273–16280.
- Habib GM, Shi ZZ, Cuevas AA, Guo Q, Matzuk MM, Lieberman MW. 1998. Leukotriene D4 and cystinyl-bis-glycine metabolism in membrane-bound dipeptidase-deficient mice. Proc Natl Acad Sci USA. 95:4859–4863.
- Habib GM, Shi ZZ, Cuevas AA, Lieberman MW. 2003. Identification of two additional members of the membrane-bound dipeptidase family. FASEB J. 17:1313–1315.
- Habig WH, Pabst MJ, Fleischner G, Gatmaitan Z, Arias IM, Jakoby WB. 1974a. The identity of glutathione S-transferase B with ligandin, a major binding protein of liver. Proc Natl Acad Sci USA. 71:3879–3882.
- Habig WH, Pabst MJ, Jakoby WB. 1974b. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J Biol Chem. 249:7130–7139.
- Habig WH, Pabst MJ, Jakoby WB. 1976. Glutathione S-transferase AA from rat liver. Arch Biochem Biophys. 175:710–716.
- Hagenbuch B, Stieger B. 2013. The SLCO (former SLC21) superfamily of transporters. Mol Aspects Med. 34:396–412.
- Hahnenkamp A, Schäfers M, Bremer C, Höltke C. 2013. Design and synthesis of small-molecule fluorescent photoprobes targeted to aminopeptdase N (APN/CD13) for optical imaging of angiogenesis. Bioconjugate Chem. 24:1027–1038.
- Hai Z, Wu J, Wang L, Xu J, Zhang H, Liang G. 2017. Bioluminescence sensing of γ-glutamyltranspeptidase activity in vitro and in vivo. Anal Chem. 89:7017–7021.
- Halder AK, Saha A, Jha T. 2013. Exploration of structural and physicochemical requirements and search of virtual hits for aminopeptidase N inhibitors. Mol Divers. 17:123–137.
- Hall A, Robson CN, Hickson ID, Harris AL, Proctor SJ, Cattan AR. 1989. Possible role of inhibition of glutathione S-transferase in the partial reversal of chlorambucil resistance by indomethacin in a Chinese hamster ovary cell line. Cancer Res. 49:6265–6268.
- Hall AG, Matheson E, Hickson ID, Foster SA, Hogarth L. 1994a. Purification of an Alpha class glutathione S-transferase from melphalan-resistant Chinese hamster ovary cells and demonstration of its ability to catalyze melphalan-glutathione adduct formation. Cancer Res. 54:3369–3372.
- Hall AG, McGuckin AG, Pearson AD, Cattan AR, Malcolm AJ, Reid MM. 1994b. Glutathione S-transferase in bone marrow metastases of disseminated neuroblastoma. J Clin Pathol. 47:468–469.
- Han B, Luo G, Shi ZZ, Barrios R, Atwood D, Liu W, Habib GM, Sifers RN, Corry DB, Lieberman MW. 2002. Gamma-glutamyl leukotrienase, a novel endothelial membrane protein, is specifically responsible for leukotriene D(4) formation in vivo. Am J Pathol. 161:481–490.
- Han L, Hiratake J, Kamiyama A, Sakata K. 2007. Design, synthesis, and evaluation of gamma-phosphono diester analogues of glutamate as highly potent inhibitors and active site probes of gamma-glutamyl transpeptidase. Biochemistry. 46:1432–1447.
- Han L, Hiratake J, Tachi N, Suzuki H, Kumagai H, Sakata K. 2006. γ-(Monophenyl)phosphono glutamate analogues as mechanism-based inhibitors of γ-glutamyl transpeptidase. Bioorg Med Chem. 14:6043–6054.
- Hanes CS, Hird FJ, Isherwood FA. 1950. Synthesis of peptides in enzymic reactions involving glutathione. Nature. 166:288–292.
- Hanes CS, Hird FJ, Isherwood FA. 1952. Enzymic transpeptidation reactions involving γ-glutamyl peptides and α-amino-acyl peptides. Biochem J. 51:25–35.
- Hanigan MH. 2014. Gamma-glutamyl transpeptidase: redox regulation and drug resistance. Adv Cancer Res. 122:103–141.
- Hanigan MH, Frierson HF. 1996. Immunohistochemical detection of γ-glutamyl transpeptidase in normal human tissue. J Histochem Cytochem. 44:1101–1108.
- Hanigan MH, Frierson HF Jr, Swanson PE, De Young BR. 1999. Altered expression of γ-glutamyl transpeptidase in human tumors. Hum Pathol. 30:300–305.
- Hanigan MH, Gillies EM, Wickham S, Wakeham N, Wirsig-Wiechmann CR. 2015. Immunolabeling of γ-glutamyl transferase 5 in normal human tissues reveals that expression and localization differ from γ-glutamyl transferase 1. Histochem Cell Biol. 143:505–515.
- Hanigan MH, Winkler ML, Drinkwater NR. 1993. Induction of three histochemically distinct populations of hepatic foci in C57BL/6J mice. Carcinogenesis. 14:1035–1040.
- Hansen GH, Sjöström H, Norén O, Dabelsteen E. 1987. Immunomicroscopic localization of aminopeptidase N in the pig enterocyte. Implications for the route of intracellular transport. Eur J Cell Biol. 43:253–259.
- Hanson H, Frohne M. 1976. Crystalline leucine aminopeptidase from lens (α-aminoacyl-peptide hydrolase; EC 3.4.11.1). Methods Enzymol. 45:504–521.
- Hanson H, Gläßer D, Kirschke H. 1965. Leucinaminopeptidase aus Rinderaugenlinsen. Kristallisation, Eigenschaften und optimale Wirkungsbedingungen des Enzyms. Biol Chem. 340:107–125.
- Hansson LO, Bolton-Grob R, Massoud T, Mannervik B. 1999a. Evolution of differential substrate specificities in Mu class glutathione transferases probed by DNA shuffling. J Mol Biol. 287:265–276.
- Hansson LO, Bolton-Grob R, Widersten M, Mannervik B. 1999b. Structural determinants in domain II of human glutathione transferase M2-2 govern the characteristic activities with aminochrome, 2-cyano-1,3-dimethyl-1-nitrosoguanidine, and 1,2-dichloro-4-nitrobenzene. Prot Sci. 8:2742–2750.
- Haraguchi N, Ishii H, Mimori K, Tanaka F, Ohkuma M, Kim HM, Akita H, Takiuchi D, Hatano H, Nagano H, et al. 2010. CD13 is a therapeutic target in human liver cancer stem cells. J Clin Invest. 120:3326–3339.
- Harbut MB, Velmourougane G, Reiss G, Chandramohanadas R, Greenbaum DC. 2008. Development of bestatin-based activity-based probes for metallo-aminopeptidases. Bioorg Med Chem Lett. 18:5932–5936.
- Hargus SJ, Fitzsimmons ME, Aniya Y, Anders MW. 1991. Stereochemistry of the microsomal glutathione S-transferase-catalyzed addition of glutathione to chlorotrifluoroethene. Biochemistry. 30:717–721.
- Harkey MA, Czerwinski M, Slattery J, Kiem HP. 2005. Overexpression of glutathione-S-transferase, MGSTII, confers resistance to busulfan and melphalan. Cancer Invest. 23:19–25.
- Harper C, Rene A, Campbell BJ. 1971. Renal dipeptidase: localization and inhibition. Biochim Biophys Acta. 242:446–458.
- Harris JM, Meyer DJ, Coles B, Ketterer B. 1991. A novel glutathione transferase (13-13) isolated from the matrix of rat liver mitochondria having structural similarity to class Theta enzymes. Biochem J. 278:137–141.
- Harris MJ, Coggan M, Langton L, Wilson SR, Board PG. 1998. Polymorphism of the Pi class glutathione S-transferase in normal populations and cancer patients. Pharmacogenetics. 8:27–31.
- Harrop SJ, DeMaere MZ, Fairlie WD, Reztsova T, Valenzuela SM, Mazzanti M, Tonini R, Qiu MR, Jankova L, Warton K, et al. 2001. Crystal structure of a soluble form of the intracellular chloride ion channel CLIC1 (NCC27) at 1.4-Å resolution. J Biol Chem. 276:44993–45000.
- Harshbarger W, Gondi S, Ficarro SB, Hunter J, Udayakumar D, Gurbani D, Singer WD, Liu Y, Li L, Marto JA, et al. 2017. Structural and biochemical analyses reveal the mechanism of glutathione S-transferase Pi 1 inhibition by the anti-cancer compound piperlongumine. J Biol Chem. 292:112–120.
- Hashmi M, Vamvakas S, Anders MW. 1992. Bioactivation mechanism of S-(3-oxopropyl)-N-acetyl-L-cysteine, the mercapturic acid of acrolein. Chem Res Toxicol. 5:360–365.
- Haufroid V, Lison D. 2005. Mercapturic acids revisited as biomarkers of exposure to reactive chemicals in occupational toxicology: a minireview. Int Arch Occup Environ Health. 78:343–354.
- Hayes JD, Flanagan JU, Jowsey IR. 2005. Glutathione transferases. Annu Rev Pharmacol Toxicol. 45:51–88.
- Hayes JD, Judah DJ, Neal GE, Nguyen T. 1992. Molecular cloning and heterologous expression of a cDNA encoding a mouse glutathione S-transferase Yc subunit possessing high catalytic activity for aflatoxin B1-8,9-epoxide. Biochem J. 285:173–180.
- Hayes JD, Mantle TJ. 1986. Use of immuno-blot techniques to discriminate between the glutathione S-transferase Yf, Yk, Ya, Yn/Yb and Yc subunits and to study their distribution in extrahepatic tissues. Evidence for three immunochemically distinct groups of transferase in the rat. Biochem J. 233:779–788.
- Hayes JD, Pulford DJ. 1995. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 30:445–600.
- Hayes JD, Pulford DJ, Ellis EM, McLeod R, James RF, Seidegård J, Mosialou E, Jernström B, Neal GE. 1998. Regulation of rat glutathione S-transferase A5 by cancer chemopreventive agents: mechanisms of inducible resistance to aflatoxin B1. Chem Biol Interact. 111–112:51–67.
- Hayes PC, Harrison DJ, Bouchier IA, McLellan LI, Hayes JD. 1989. Cytosolic and microsomal glutathione S-transferase isoenzymes in normal human liver and intestinal epithelium. Gut. 30:854–859.
- Hayeshi R, Mutingwende I, Mavengere W, Masiyanise V, Mukanganyama S. 2007. The inhibition of human glutathione S-transferases activity by plant polyphenolic compounds ellagic acid and curcumin. Food Chem Toxicol. 45:286–295.
- He P, Moran GR. 2011. Structural and mechanistic comparisons of the metal-binding members of the vicinal oxygen chelate (VOC) superfamily. J Inorg Biochem. 105:1259–1272.
- He X, Hu Y, Shi W, Li X, Ma H. 2017a. Design, synthesis and application of a near-infrared fluorescent probe for in vivo imaging of aminopeptidase N. Chem Commun. 53:9438–9441.
- He X, Xu Y, Shi W, Ma H. 2017b. Ultrasensitive detection of aminopeptidase N activity in urine and cells with a ratiometric fluorescence probe. Anal Chem. 89:3217–3221.
- Hebert H, Jegerschöld C. 2007. The structure of membrane associated proteins in eicosanoid and glutathione metabolism as determined by electron crystallography. Curr Opin Struct Biol. 17:396–404.
- Hebert H, Jegerschöld C, Bhakat P, Holm PJ. 2005. Two-dimensional crystallization and electron crystallography of MAPEG proteins. Meth Enzymol. 401:161–168.
- Hebert H, Schmidt-Krey I, Morgenstern R. 1995. The projection structure of microsomal glutathione transferase. EMBO J. 14:3864–3869.
- Hebert H, Schmidt-Krey I, Morgenstern R, Murata K, Hirai T, Mitsuoka K, Fujiyoshi Y. 1997. The 3.0 Å projection structure of microsomal glutathione transferase as determined by electron crystallography of p21212 two-dimensional crystals. J Mol Biol. 271:751–758.
- Heese D, Berger S, Röhm KH. 1990. Nuclear magnetic relaxation studies of the role of the metal ion in Mn2+-substituted aminoacylase I. Eur J Biochem. 188:175–180.
- Heese D, Löffler HG, Röhm KH. 1988. Further characterization of porcine kidney aminoacylase I reveals close similarity to ‘renal dipeptidase’. Biol Chem Hoppe Seyler. 369:559–566.
- Heese D, Röhm KH. 1989. Reactivities of sulfhydryl groups in native and metal-free aminoacylase I. Biol Chem Hoppe Seyler. 370:607–612.
- Hegazy UM, Mannervik B, Stenberg G. 2004. Functional role of the lock and key motif at the subunit interface of glutathione transferase P1-1. J Biol Chem. 279:9586–9596.
- Heisterkamp N, Groffen J, Warburton D, Sneddon TP. 2008. The human gamma-glutamyltransferase gene family. Hum Genet. 123:321–332.
- Heisterkamp N, Rajpert-De Meyts E, Uribe L, Forman HJ, Groffen J. 1991. Identification of a human gamma-glutamyl cleaving enzyme related to, but distinct from, gamma-glutamyl transpeptidase. Proc Natl Acad Sci USA. 88:6303–6307.
- Henseling J, Röhm KH. 1988. Aminoacylase I from hog kidney: anion effects and the pH dependence of kinetic parameters. Biochim Biophys Acta. 959:370–3777.
- Hernick M, Fierke CA. 2005. Zinc hydrolases: the mechanisms of zinc-dependent deacetylases. Arch Biochem Biophys. 433:71–84.
- Hesterkamp T, Barker J, Davenport A, Whittaker M. 2007. Fragment based drug discovery using fluorescence correlation: spectroscopy techniques: challenges and solutions. CTMC. 7:1582–1591.
- Heuner A, Dekant W, Schwegler JS, Silbernagl S. 1991. Localization and capacity of the last step of mercapturic acid biosynthesis and the reabsorption and acetylation of cysteine S-conjugates in the rat kidney. Pflugers Arch. 417:523–527.
- Higgins LG, Hayes JD. 2011. Mechanisms of induction of cytosolic and microsomal glutathione transferase (GST) genes by xenobiotics and pro-inflammatory agents. Drug Metab Rev. 43:92–137.
- Hiltunen TP, Donner KM, Sarin AP, Saarela J, Ripatti S, Chapman AB, Gums JG, Gong Y, Cooper-DeHoff RM, Frau F, et al. 2015. Pharmacogenomics of hypertension: a genome‐wide, placebo‐controlled cross‐over study, using four classes of antihypertensive drugs. J Am Heart Assoc. 4:e001521.
- Himmelhoch SR. 1969. Leucine aminopeptidase: a zinc metalloenzyme. Arch Biochem Biophys. 134:597–602.
- Hinchman CA, Ballatori N. 1990. Glutathione-degrading capacities of liver and kidney in different species. Biochem Pharmacol. 40:1131–1135.
- Hirabayashi Y, Kanamori A, Nomura KH, Nomura K. 2004. The acetyl-CoA transporter family SLC33. Pflügers Arch. 447:760–762.
- Hiratsuka A, Okada T, Nishiyama T, Fujikawa M, Ogura K, Okuda H, Watabe T, Watabe T. 1994. Novel Theta class glutathione S-transferases Yrs-Yrs' and Yrs'-Yrs' in rat liver cytosol: their potent activity toward 5-sulfoxymethylchrysene, a reactive metabolite of the carcinogen 5-hydroxymethylchrysene. Biochem Biophys Res Commun. 202:278–284.
- Hiratsuka A, Sebata N, Kawashima K, Okuda H, Ogura K, Watabe T, Satoh K, Hatayama I, Tsuchida S, Ishikawa T. 1990. A new class of rat glutathione S-transferase Yrs-Yrs inactivating reactive sulfate esters as metabolites of carcinogenic arylmethanols. J Biol Chem. 265:11973–11981.
- Hirota T, Nishikawa Y, Komai T, Igarashi T, Kitagawa H. 1987a. Role of dehydropeptidase-I in the metabolism of glutathione and its conjugates in the rat kidney. Res Commun Chem Pathol Pharmacol. 56:235–242.
- Hirota T, Nishikawa Y, Takahagi H, Igarashi T, Kitagawa H. 1985. Simultaneous purification and properties of dehydropeptidase-I and aminopeptidase-M from rat kidney. Res Commun Chem Pathol Pharmacol. 49:435–445.
- Hirota T, Nishikawa Y, Tanaka M, Fukuda K, Igarashi T, Kitagawa H. 1987b. Localization of dehydropeptidase-I, an enzyme processing glutathione, in the rat kidney. J Biochem. 102:547–550.
- Hirota T, Nishikawa Y, Tanaka M, Igarashi T, Kitagawa H. 1986. Characterization of dehydropeptidase I in the rat lung. Eur J Biochem. 160:521–525.
- Hitzerd SM, Verbrugge SE, Ossenkoppele G, Jansen G, Peters GJ. 2014. Positioning of aminopeptidase inhibitors in next generation cancer therapy. Amino Acids. 46:793–808.
- Hoensch H, Morgenstern I, Petereit G, Siepmann M, Peters WH, Roelofs HM, Kirch W. 2002. Influence of clinical factors, diet, and drugs on the human upper gastrointestinal glutathione system. Gut. 50:235–240.
- Hohwy M, Spadola L, Lundquist B, Hawtin P, Dahmén J, Groth-Clausen I, Nilsson E, Persdotter S, von Wachenfeldt K, Folmer RH, et al. 2008. Novel prostaglandin D synthase inhibitors generated by fragment-based drug design. J Med Chem. 51:2178–2186.
- Holley SL, Fryer AA, Carroll W, Hoban PR, Strange RC. 2007. GST polymorphism: where to now? Clinical applications and functional analysis. In: Awasthi YC, editor. Toxicology of glutathione transferases. Boca Raton (FL): Taylor & Francis; p. 129–154.
- Holm PJ, Bhakat P, Jegerschold C, Gyobu N, Mitsuoka K, Fujiyoshi Y, Morgenstern R, Hebert H. 2006. Structural basis for detoxification and oxidative stress protection in membranes. J Mol Biol. 360:934–945.
- Holm PJ, Morgenstern R, Hebert H. 2002. The 3-D structure of microsomal glutathione transferase 1 at 6 Å resolution as determined by electron crystallography of p22121 crystals. Biochim Biophys Acta. 1594:276–285.
- Holter H. 1979. Linderstrøm-Lang and leucylaminopeptidase. Trends Biochem Sci. 4:239–240.
- Hooper NM, Keen JN, Turner AJ. 1990. Characterization of the glycosyl-phosphatidylinositol-anchored human renal dipeptidase reveals that it is more extensively glycosylated than the pig enzyme. Biochem J. 265:429–433.
- Hooper NM. 2013. Membrane dipeptidase. In: Rawlings ND, Salvesen G, editors. Handbook of proteolytic enzymes. 3rd ed. Vol. 2. San Diego: Elsevier Science; p. 1670–1673.
- Horiuchi S, Inoue M, Morino Y. 1978. γ-Glutamyl transpeptidase: sidedness of its active site on renal brush-border membrane. Eur J Biochem. 87:429–437.
- Horsley L, Cummings J, Middleton M, Ward T, Backen A, Clamp A, Dawson M, Farmer H, Fisher N, Halbert G, et al. 2013. A phase 1 trial of intravenous 4-(N-(S-glutathionylacetyl)amino) phenylarsenoxide (GSAO) in patients with advanced solid tumours. Cancer Chemother Pharmacol. 72:1343–1352.
- Hossain QS, Ulziikhishig E, Lee KK, Yamamoto H, Aniya Y. 2009. Contribution of liver mitochondrial membrane-bound glutathione transferase to mitochondrial permeability transition pores. Toxicol Appl Pharmacol. 235:77–85.
- Hou J, Jin K, Li J, Jiang Y, Li X, Wang X, Huang Y, Zhang Y, Xu W. 2016. LJNK, an indoline-2,3-dione-based aminopeptidase N inhibitor with promising antitumor potency. Anticancer Drugs. 27:496–507.
- Hsieh JM, Tsirulnikov K, Sawaya MR, Magilnick N, Abuladze N, Kurtz I, Abramson J, Pushkin A. 2010. Structures of aminoacylase 3 in complex with acetylated substrates. Proc Natl Acad Sci USA. 107:17962–17967.
- Hu S, Leblanc AF, Gibson AA, Hong KW, Kim JY, Janke LJ, Li L, Vasilyeva A, Finkelstein DB, Sprowl JA, et al. 2017. Identification of OAT1/OAT3 as contributors to cisplatin toxicity. Clin Transl Sci. 10:412–420.
- Hu X, Legler PM, Khavrutskii I, Scorpio A, Compton JR, Robertson KL, Friedlander AM, Wallqvist A. 2012. Probing the donor and acceptor substrate specificity of the γ-glutamyl transpeptidase. Biochemistry. 51:1199–1212.
- Hu X, Srivastava SK, Xia H, Awasthi YC, Singh SV. 1996. An Alpha class mouse glutathione S-transferase with exceptional catalytic efficiency in the conjugation of glutathione with 7β,8α-dihydroxy-9α,10α-oxy-7,8,9,10-tetrahydrobenzo(a)pyrene. J Biol Chem. 271:32684–32688.
- Huang J, Tan PH, Thiyagarajan J, Bay BH. 2003. Prognostic significance of glutathione S-transferase-Pi in invasive breast cancer. Mod Pathol. 16:558–565.
- Huang K, Takahara S, Kinouchi T, Takeyama M, Ishida T, Ueyama H, Nishi K, Ohkubo I. 1997. Alanyl aminopeptidase from human seminal plasma: purification, characterization, and immunohistochemical localization in the male genital tract. J Biochem. 122:779–787.
- Hubatsch I, Ridderström M, Mannervik B. 1998. Human glutathione transferase A4-4: an Alpha class enzyme with high catalytic efficiency in the conjugation of 4-hydroxynonenal and other genotoxic products of lipid peroxidation. Biochem J. 330:175–179.
- Huber M, Müller J, Leier I, Jedlitschky G, Ball HA, Moore KP, Taylor GW, Williams R, Keppler D. 1990. Metabolism of cysteinyl leukotrienes in monkey and man. Eur J Biochem. 194:309–315.
- Hughes MM, Hooftman A, Angiari S, Tummala P, Zaslona Z, Runtsch MC, McGettrick AF, Sutton CE, Diskin C, Rooke M, et al. 2019. Glutathione transferase Omega-1 regulates NLRP3 inflammasome activation through NEK7 deglutathionylation. Cell Rep. 29:151–161.e155.
- Hughey RP, Rankin BB, Elce JS, Curthoys NP. 1978. Specificity of a particulate rat renal peptidase and its localization along with other enzymes of mercapturic acid synthesis. Arch Biochem Biophys. 186:211–217.
- Hurst R, Bao Y, Jemth P, Mannervik B, Williamson G. 1998. Phospholipid hydroperoxide glutathione peroxidase activity of human glutathione transferases. Biochem J. 332:97–100.
- Huseby NE. 1982. Hydrophilic form of γ-glutamyltransferase: proteolytic formation in liver homogenates and its estimation in serum. Clin Chim Acta. 124:113–121.
- Hussey AJ, Hayes JD. 1992. Characterization of a human class-Theta glutathione S-transferase with activity towards 1-menaphthyl sulphate. Biochem J. 286:929–935.
- Hutson DH, Holmes DS, Crawford MJ. 1976. The involvement of glutathione in the reductive dechlorination of a phenacyl halide. Chemosphere. 5:79–84.
- Hwa JS, Park HJ, Jung JH, Kam SC, Park HC, Kim CW, Kang KR, Hyun JS, Chung KH. 2005. Identification of proteins differentially expressed in the conventional renal cell carcinoma by proteomic analysis. J Korean Med Sci. 20:450–455.
- Igarashi P, Karniski LP. 1991. Cloning of cDNAs encoding a rabbit renal brush border membrane protein immunologically related to band 3. Sequence similarity with microsomal dipeptidase. Biochem J. 280:71–78.
- Iida A, Saito S, Sekine A, Harigae S, Osawa S, Mishima C, Kondo K, Kitamura Y, Nakamura Y. 2001. Catalog of 46 single-nucleotide polymorphisms (SNPs) in the microsomal glutathione S-transferase 1 (MGST1) gene. J Hum Genet. 46:590–594.
- Ikeda Y, Fujii J, Anderson ME, Taniguchi N, Meister A. 1995. Involvement of Ser-451 and Ser-452 in the catalysis of human γ-glutamyl transpeptidase. J Biol Chem. 270:22223–22228.
- Imaizumi N, Aniya Y. 2011. The role of a membrane-bound glutathione transferase in the peroxynitrite-induced mitochondrial permeability transition pore: formation of a disulfide-linked protein complex. Arch Biochem Biophys. 516:160–167.
- Imaizumi N, Miyagi S, Aniya Y. 2006. Reactive nitrogen species derived activation of rat liver microsomal glutathione S-transferase. Life Sci. 78:2998–3006.
- Inagaki Y, Tang W, Zhang L, Du G, Xu W, Kokudo N. 2010. Novel aminopeptidase N (APN/CD13) inhibitor 24F can suppress invasion of hepatocellular carcinoma cells as well as angiogenesis. Biosci Trends. 4:56–60.
- Inoue M. 2016. Glutathionists in the battlefield of γ-glutamyl cycle. Arch Biochem Biophys. 595:61–63.
- Inoue M, Hiratake J, Suzuki H, Kumagai H, Sakata K. 2000. Identification of catalytic nucleophile of Escherichia coli γ-glutamyltranspeptidase by γ-monofluorophosphono derivative of glutamic acid: N-terminal thr-391 in small subunit is the nucleophile. Biochemistry. 39:7764–7771.
- Inoue M, Horiuchi S, Morino Y. 1977. Affinity labeling of rat-kidney γ-glutamyl transpeptidase. Eur J Biochem. 73:335–342.
- Inoue M, Morino Y. 1981. Inactivation of renal gamma-glutamyl transferase by 6-diazo-5-oxo-L-norleucylglycine, an inactive precursor of affinity-labeling reagent. Proc Natl Acad Sci USA. 78:46–49.
- Inoue M, Okajima K, Morino Y. 1984. Hepato-renal cooperation in biotransformation, membrane transport, and elimination of cysteine S-conjugates of xenobiotics. J Biochem. 95:247–254.
- Inoue T, Irikura D, Okazaki N, Kinugasa S, Matsumura H, Uodome N, Yamamoto M, Kumasaka T, Miyano M, Kai Y, et al. 2003. Mechanism of metal activation of human hematopoietic prostaglandin D synthase. Nat Struct Mol Biol. 10:291–296.
- Inoue T, Okano Y, Kado Y, Aritake K, Irikura D, Uodome N, Okazaki N, Kinugasa S, Shishitani H, Matsumura H, et al. 2004. First determination of the inhibitor complex structure of human hematopoietic prostaglandin D synthase. J Biochem. 135:279–283.
- Ishikawa T, Casini AF, Nishikimi M. 1998. Molecular cloning and functional expression of rat liver glutathione-dependent dehydroascorbate reductase. J Biol Chem. 273:28708–28712.
- Ito M, Shibata A, Zhang J, Hiroshima M, Sako Y, Nakano Y, Kojima-Aikawa K, Mannervik B, Shuto S, Ito Y, et al. 2012. Universal caging group for the in-cell detection of glutathione transferase applied to 19F NMR and bioluminogenic probes. ChemBioChem. 13:1428–1432.
- Ito M, Shuto S, Ito Y, Abe H. 2014. Development of molecular probe targeting on glutathione transferase. J Synth Org Chem Jpn. 72:822–831.
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et al. 1997. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 236:313–322.
- Iwatate RJ, Kamiya M, Umezawa K, Kashima H, Nakadate M, Kojima R, Urano Y. 2018. Silicon rhodamine-based near-infrared fluorescent probe for γ-glutamyltransferase. Bioconjugate Chem. 29:241–244.
- Jaffé M. 1879. Ueber die nach Einführung von Brombenzol und Chlorbenzol im Organismus entstehenden schwefelhaltigen Säuren. Ber Dtsch Chem Ges. 12:1092–1098.
- Jaime E, Kluge S, Weston J. 2007. On the origin of the broad-band selectivity of bovine-lens-leucine-aminopeptidase. ARKIVOC. 2007:77–95.
- Jakobson I, Askelöf P, Warholm M, Mannervik B. 1977. A steady-state-kinetic random mechanism for glutathione S-transferase A from rat liver. A model involving kinetically significant enzyme-product complexes in the forward reaction. Eur J Biochem. 77:253–262.
- Jakobsson PJ, Mancini JA, Ford-Hutchinson AW. 1996. Identification and characterization of a novel human microsomal glutathione S-transferase with leukotriene C4 synthase activity and significant sequence identity to 5-lipoxygenase-activating protein and leukotriene C4 synthase. J Biol Chem. 271:22203–22210.
- Jakobsson PJ, Mancini JA, Riendeau D, Ford-Hutchinson AW. 1997. Identification and characterization of a novel microsomal enzyme with glutathione-dependent transferase and peroxidase activities. J Biol Chem. 272:22934–22939.
- Jakobsson PJ, Morgenstern R, Mancini J, Ford-Hutchinson A, Persson B. 1999. Common structural features of MAPEG – a widespread superfamily of membrane associated proteins with highly divergent functions in eicosanoid and glutathione metabolism. Protein Sci. 8:689–692.
- Jakoby WB. 1978. The glutathione S-transferases: a group of multifunctional detoxification proteins. Adv Enzymol Relat Areas Mol Biol. 46:383–414.
- Jakoby WB, Ketterer B, Mannervik B. 1984. Glutathione transferases: nomenclature. Biochem Pharmacol. 33:2539–2540.
- Jardim BV, Moschetta MG, Gelaleti GB, Leonel C, Regiani VR, de Santi Neto D, Bordin-Junior NA, Perea SA, Zuccari DA. 2012. Glutathione transferase Pi (GSTpi) expression in breast cancer: an immunohistochemical and molecular study. Acta Histochem. 114:510–517.
- Jardinaud F, Banisadr G, Noble F, Mélik-Parsadaniantz S, Chen H, Dugave C, Laplace H, Rostène W, Fournié-Zaluski MC, Roques BP, et al. 2004. Ontogenic and adult whole body distribution of aminopeptidase N in rat investigated by in vitro autoradiography. Biochimie. 86:105–113.
- Jedlitschky G, Keppler D. 2002. Transport of leukotriene C4 and structurally related conjugates. Vitam Horm. 64:153–184.
- Jeffery AM, Jerina DM. 1975. Novel rearrangements during dehydration of nucleophile adducts of arene oxides. A reappraisal of premercapturic acid structures. J Am Chem Soc. 97:4427–4428.
- Jeffery CJ. 1999. Moonlighting proteins. Trends Biochem Sci. 24:8–11.
- Jeffery CJ. 2018. Protein moonlighting: what is it, and why is it important? Philos Trans R Soc B. 373:20160523.
- Jemth P, Mannervik B. 1997. Kinetic characterization of recombinant human glutathione transferase T1-1, a polymorphic detoxication enzyme. Arch Biochem Biophys. 348:247–254.
- Jemth P, Mannervik B. 2000. Active site serine promotes stabilization of the reactive glutathione thiolate in rat glutathione transferase T2-2. Evidence against proposed sulfatase activity of the corresponding human enzyme. J Biol Chem. 275:8618–8624.
- Jernström B, Funk M, Frank H, Mannervik B, Seidel A. 1996. Glutathione S-transferase A1-1-catalysed conjugation of bay and fjord region diol epoxides or polycyclic aromatic hydrocarbons with glutathione. Carcinogenesis. 17:1491–1498.
- Jernström B, Seidel A, Funk M, Oesch F, Mannervik B. 1992. Glutathione conjugation of trans-3,4-dihydroxy 1,2-epoxy 1,2,3,4-tetrahydrobenzo[c]phenanthrene isomers by human glutathione transferases. Carcinogenesis. 13:1549–1555.
- Ji X, Johnson WW, Sesay MA, Dickert L, Prasad SM, Ammon HL, Armstrong RN, Gilliland GL. 1994. Structure and function of the xenobiotic substrate binding site of a glutathione S-transferase as revealed by X-ray crystallographic analysis of product complexes with the diastereomers of 9-(S-glutathionyl)-10-hydroxy-9,10-dihydrophenanthrene. Biochemistry. 33:1043–1052.
- Ji X, von Rosenvinge EC, Johnson WW, Tomarev SI, Piatigorsky J, Armstrong RN, Gilliland GL. 1995. Three-dimensional structure, catalytic properties, and evolution of a Sigma class glutathione transferase from squid, a progenitor of the lens S-crystallins of cephalopods. Biochemistry. 34:5317–5328.
- Ji X, Zhang P, Armstrong RN, Gilliland GL. 1992. The three-dimensional structure of a glutathione S-transferase from the Mu gene class. Structural analysis of the binary complex of isoenzyme 3-3 and glutathione at 2.2-Å resolution. Biochemistry. 31:10169–10184.
- Ji Y, Bennett BM. 2006. Biotransformation of glyceryl trinitrate by rat hepatic microsomal glutathione S-transferase 1. J Pharmacol Exp Ther. 318:1050–1056.
- Jia L, Betters JL, Yu L. 2011. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu Rev Physiol. 73:239–259.
- Jiang YQ, Hou JN, Li XY, Huang YX, Wang XJ, Wu JD, Zhang J, Xu WF, Zhang YJ. 2016a. Discovery of a novel chimeric ubenimex-gemcitabine with potent oral antitumor activity. Bioorg Med Chem. 24:5787–5795.
- Jiang YQ, Li XY, Hou JN, Huang YX, Jia YP, Zou MM, Zhang J, Wang XJ, Xu WF, Zhang YJ. 2016b. Discovery of BC-01, a novel mutual prodrug (hybrid drug) of ubenimex and fluorouracil as anticancer agent. Eur J Med Chem. 121:649–657.
- Jin K, Zhang X, Ma C, Xu Y, Yuan Y, Xu W. 2013. Novel indoline-2,3-dione derivatives as inhibitors of aminopeptidase N (APN). Bioorg Med Chem. 21:2663–2670.
- Johansson AS, Mannervik B. 2001. Human glutathione transferase A3-3, a highly efficient catalyst of double-bond isomerization in the biosynthetic pathway of steroid hormones. J Biol Chem. 276:33061–33065.
- Johansson AS, Mannervik B. 2002. Active-site residues governing high steroid isomerase activity in human glutathione transferase A3-3. J Biol Chem. 277:16648–16654.
- Johansson K, Ito M, Schophuizen CM, Thengumtharayil SM, Heuser VD, Zhang J, Shimoji M, Vahter M, Ang WH, Dyson PJ, et al. 2011. Characterization of new potential anticancer drugs designed to overcome glutathione transferase mediated resistance. Mol Pharm. 8:1698–1708.
- Johansson K, Järvliden J, Gogvadze V, Morgenstern R. 2010. Multiple roles of microsomal glutathione transferase 1 in cellular protection: a mechanistic study. Free Radic Biol Med. 49:1638–1645.
- Johnson MK. 1966. Studies on glutathione S-alkyltransferase of the rat. Biochem J. 98:44–56.
- Jolivette LJ, Anders MW. 2002. Structure-activity relationship for the biotransformation of haloalkenes by rat liver microsomal glutathione transferase 1. Chem Res Toxicol. 15:1036–1041.
- Jonas MC, Pehar M, Puglielli L. 2010. AT-1 is the ER membrane acetyl-CoA transporter and is essential for cell viability. J Cell Sci. 123:3378–3388.
- Jones DP, Moldfus P, Stead AH, Ormstad K, Jörnvall H, Orrenius S. 1979. Metabolism of glutathione and a glutathione conjugate by isolated kidney cells. J Biol Chem. 254:2787–2792.
- Jones DP, Stead AH, Moldéus P, Orrenius S. 1978. Glutathione and glutathione conjugate metabolism in isolated liver and kidney cells. In: Sies H, Wendel A, editors. Functions of glutathione in liver and kidney. New York: Springer-Verlag; p. 194–200.
- Jones WM, Scaloni A, Bossa F, Popowicz AM, Schneewind O, Manning JM. 1991. Genetic relationship between acylpeptide hydrolase and acylase, two hydrolytic enzymes with similar binding but different catalytic specificities. Proc Natl Acad Sci USA. 88:2194–2198.
- Jongeneel CV, Bouvier J, Bairoch A. 1989. A unique signature identifies a family of zinc-dependent metallopeptidases. FEBS Lett. 242:211–214.
- Jösch C, Klotz LO, Sies H. 2003. Identification of cytosolic leucyl aminopeptidase (EC 3.4.11.1) as the major cysteinylglycine-hydrolysing activity in rat liver. Biol Chem. 384:213–218.
- Jösch C, Sies H, Akerboom T. 1998. Hepatic mercapturic acid formation: involvement of cytosolic cysteinylglycine S-conjugate dipeptidase activity. Biochem Pharmacol. 56:763–771.
- Joseph RL, Sanders WJ. 1966. Leucine aminopeptidase in extracts of swine muscle. Biochem J. 100:827–832.
- Josephy DP, Mannervik B. 2006. Glutathione transferases, molecular toxicology. 2nd ed. New York: Oxford; p. 333–364.
- Josephy PD. 2010. Genetic variations in human glutathione transferase enzymes: significance for pharmacology and toxicology. Hum Genomics Proteomics. 2:876940.
- Josephy PD, Kent M, Mannervik B. 2009. Single-nucleotide polymorphic variants of human glutathione transferase T1-1 differ in stability and functional properties. Arch Biochem Biophys. 490:24–29.
- Josephy PD, Pan D, Ianni MD, Mannervik B. 2011. Functional studies of single-nucleotide polymorphic variants of human glutathione transferase T1-1 involving residues in the dimer interface. Arch Biochem Biophys. 513:87–93.
- Joshi S, Chen L, Winter MB, Lin YL, Yang Y, Shapovalova M, Smith PM, Liu C, Li F, LeBeau AM. 2017. The rational design of therapeutic peptides for aminopeptidase N using a substrate-based approach. Sci Rep. 7:1424.
- Jowsey IR, Thomson AM, Flanagan JU, Murdock PR, Moore GB, Meyer DJ, Murphy GJ, Smith SA, Hayes JD. 2001. Mammalian class Sigma glutathione S-transferases: catalytic properties and tissue-specific expression of human and rat GSH-dependent prostaglandin D2 synthases. Biochem J. 359:507–516.
- Jowsey IR, Thomson RE, Orton TC, Elcombe CR, Hayes JD. 2003. Biochemical and genetic characterization of a murine class Kappa glutathione S-transferase. Biochem J. 373:559–569.
- Joyce-Brady M, Hiratake J. 2011. Inhibiting glutathione metabolism in lung lining fluid as a strategy to augment antioxidant defense. Curr Enzyme Inhib. 7:71–78.
- Juhanson P, Kepp K, Org E, Veldre G, Kelgo P, Rosenberg M, Viigimaa M, Laan M. 2008. N-Acetyltransferase 8, a positional candidate for blood pressure and renal regulation: resequencing, association and in silico study. BMC Med Genet. 9:25.
- Jurnak F, Rich A, van Loon-Klaassen A, Bloemendal H, Taylor A, Carpenter FH. 1977. Preliminary X-ray study of leucine aminopeptidase (bpvine lens), and oligomeric metalloenzyme. J Mol Biol. 112:149–153.
- Juronen E, Tasa G, Uusküla M, Pooga M, Mikelsaar AV. 1996. Purification, characterization and tissue distribution of human class Theta glutathione S-transferase T1-1. Biochem Mol Biol Int. 39:21–29.
- Kado Y, Aritake K, Uodome N, Okano Y, Okazaki N, Matsumura H, Urade Y, Inoue T. 2012. Human hematopoietic prostaglandin D synthase inhibitor complex structures. J Biochem. 151:447–455.
- Kahan FM, Kropp H, Sundelof JG, Birnbaum J. 1983. Thienamycin: development of imipenen-cilastatin. J Antimicrob Chemother. 12:1–35.
- Kalengayi MM, Ronchi G, Desmet VJ. 1975. Histochemistry of gamma-glutamyl transpeptidase in rat liver during aflatoxin B1-induced carcinogenesis. J Natl Cancer Inst. 55:579–588.
- Kalliokoski A, Niemi M. 2009. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 158:693–705.
- Kamiyama A, Nakajima M, Han L, Wada K, Mizutani M, Tabuchi Y, Kojima-Yuasa A, Matsui-Yuasa I, Suzuki H, Fukuyama K, et al. 2016. Phosphonate-based irreversible inhibitors of human γ-glutamyl transpeptidase (GGT). GGsTop is a non-toxic and highly selective inhibitor with critical electrostatic interaction with an active-site residue Lys562 for enhanced inhibitory activity. Bioorg Med Chem. 24:5340–5352.
- Kanaoka Y, Ago H, Inagaki E, Nanayama T, Miyano M, Kikuno R, Fujii Y, Eguchi N, Toh H, Urade Y, et al. 1997. Cloning and crystal structure of hematopoietic prostaglandin D synthase. Cell. 90:1085–1095.
- Kanaoka Y, Fujimori K, Kikuno R, Sakaguchi Y, Urade Y, Hayaishi O. 2000. Structure and chromosomal localization of human and mouse genes for hematopoietic prostaglandin D synthase. Conservation of the ancestral genomic structure of Sigma-class glutathione S-transferase. Eur J Biochem. 267:3315–3322.
- Kanaoka Y, Urade Y. 2003. Hematopoietic prostaglandin D synthase. Prostaglandins Leukot Essent Fatty Acids. 69:163–167.
- Kanhai W, Dekant W, Henschler D. 1989. Metabolism of the nephrotoxin dichloroacetylene by glutathione conjugation. Chem Res Toxicol. 2:51–56.
- Karpusas M, Axarli I, Chiniadis L, Papakyriakou A, Bethanis K, Scopelitou K, Clonis YD, Labrou NE. 2013. The interaction of the chemotherapeutic drug chlorambucil with human glutathione transferase A1-1: kinetic and structural analysis. PLoS One. 8:e56337.
- Karshikoff A, Reinemer P, Huber R, Ladenstein R. 1993. Electrostatic evidence for the activation of the glutathione thiol by Tyr7 in Pi-class glutathione transferases. Eur J Biochem. 215:663–670.
- Kaur H, Kumar C, Junot C, Toledano MB, Bachhawat AK. 2009. Dug1p is a Cys-Gly peptidase of the γ-glutamyl cycle of Saccharomyces cerevisiae and represents a novel family of Cys-Gly peptidases. J Biol Chem. 284:14493–14502.
- Keen JH, Jakoby WB. 1978. Glutathione transferases. Catalysis of nucleophilic reactions of glutathione. J Biol Chem. 253:5654–5657.
- Keillor JW, Castonguay R, Lherbet C. 2005. Gamma-glutamyl transpeptidase substrate specificity and catalytic mechanism. Meth Enzymol. 401:449–467.
- Kelner MJ, Bagnell RD, Montoya MA, Estes LA, Forsberg L, Morgenstern R. 2000. Structural organization of the microsomal glutathione S-transferase gene (MGST1) on chromosome 12p13.1–13.2: identificatiion of the correct promoter region and demonstration of transcriptional regulation in response to oxidative stress. J Biol Chem. 275:13000–13006.
- Kelner MJ, Bagnell RD, Morgenstern R. 2004. Structural organization of the murine microsomal glutathione S-transferase gene (MGST1) from the 129/SvJ strain: identification of the promoter region and a comprehensive examination of tissue expression. Biochim Biophys Acta. 1678:163–169.
- Kelner MJ, Diccianni MB, Yu AL, Rutherford MR, Estes LA, Morgenstern R. 2014. Absence of MGST1 mRNA and protein expression in human neuroblastoma cell lines and primary tissue. Free Radic Biol Med. 69:167–171.
- Keppler D. 2011. Multidrug resistance proteins (MRPs, ABCCs): importance for pathophysiology and drug therapy. In: Fromm MF, Kim RB, editors. Drug transporters, handbook of experimental pharmacology. Vol. 201. New York: Springer; p. 299–323.
- Keppler D, Leier I, Jedlitschky G, König J. 1998. ATP-dependent transport of glutathione S-conjugates by the multidrug resistance protein MRP1 and its apical isoform MRP2. Chem Biol Interact. 111–112:153–161.
- Kera Y, Liu Z, Matsumoto T, Sorimachi Y, Nagasaki H, Yamada RH. 1999. Rat and human membrane dipeptidase: tissue distribution and developmental changes. Comp Biochem Physiol B Biochem Mol Biol. 123:53–58.
- Keren R, Stark AA. 1998. γ-Glutamyl transpeptidase-dependent mutagenicity and cytotoxicity of γ-glutamyl derivatives: a model for biochemical targeting of chemotherapeutic agents. Environ Mol Mutagen. 32:377–386.
- Ketley JN, Habig WH, Jakoby WB. 1975. Binding of nonsubstrate ligands to the glutathione S-transferases. J Biol Chem. 250:8670–8673.
- Keynan S, Habgood NT, Hooper NM, Turner AJ. 1996a. Site-directed mutagenesis of conserved cysteine residues in porcine membrane dipeptidase. Cys 361 alone is involved in disulfide-linked dimerization. Biochemistry. 35:12511–12517.
- Keynan S, Hooper NM, Turner AJ. 1994. Directed mutagenesis of pig renal membrane dipeptidase. His219 is critical but the DHXXH motif is not essential for zinc binding or catalytic activity. FEBS Lett. 349:50–54.
- Keynan S, Hooper NM, Turner AJ. 1996b. Molecular and functional aspects of membrane dipeptidase. In: Hooper NM, editor. Zinc metalloproteases in health and disease. London: Taylor & Francis; p. 285–309.
- Keynan S, Hooper NM, Turner AJ. 1997. Identification by site-directed mutagenesis of three essential histidine residues in membrane dipeptidase, a novel mammalian zinc peptidase. Biochem J. 326:47–51.
- Khilji MA, Bailey GS. 1978. The purification of a bovine kidney enzyme which cleaves melanocyte-stimulating hormone-release inhibiting factor. Biochim Biophys Acta. 527:282–288.
- Kim DJ, Noh JH, Cho NH, Lee BW, Choi YH, Jung JH, Min YK, Lee MS, Lee MK, Kim KW. 2005. Serum γ-glutamyltransferase within its normal concentration range is related to the presence of diabetes and cardiovascular risk factors. Diabet Med. 22:1134–1140.
- Kim H, Burley SK, Lipscomb WN. 1993a. Re-refinement of the X-ray crystal structure of bovine lens leucine aminopeptidase complexed with bestatin. J Mol Biol. 230:722–724.
- Kim H, Lipscomb WN. 1993b. Differentiation and identification of the two catalytic metal binding sites in bovine lens leucine aminopeptidase by x-ray crystallography. Proc Natl Acad Sci USA. 90:5006–5010.
- Kim H, Lipscomb WN. 1993. X-ray crystallographic determination of the structure of bovine lens leucine aminopeptidase complexed with amastatin: formulation of a catalytic mechanism featuring a gem-diolate transition state. Biochemistry. 32:8465–8478.
- Kim H, Lipscomb WN. 1994. Structure and mechanism of bovine lens leucine aminopeptidase. Adv Enzymol Relat Areas Mol Biol. 68:153–213.
- Kim HS, Campbell BJ. 1982. β-Lactamase activity of renal dipeptidase against N-formimidoyl-thienamycin. Biochem Biophys Res Commun. 108:1638–1642.
- Kim K, Kim SH, Kim J, Kim H, Yim J. 2012. Glutathione S-transferase Omega 1 activity is sufficient to suppress neurodegeneration in a Drosophila model of Parkinson disease. J Biol Chem. 287:6628–6641.
- Kim M, Kim J, Jung E, Choi K, Shin JM, Kang SK, Kim MK, Choi YJ, Choi SH. 2007. Binding mode analysis between membrane dipeptidase and its substrates. Mol Simul. 33:495–503.
- Kim Y, Maciag AE, Cao Z, Deschamps JR, Saavedra JE, Keefer LK, Holland RJ. 2015. PABA/NO lead optimization: improved targeting of cytotoxicity to glutathione S-transferase P1-overexpressing cancer cells. Bioorg Med Chem. 23:4980–4988.
- King JB, West MB, Cook PF, Hanigan MH. 2009. A novel, species-specific class of uncompetitive inhibitors of gamma-glutamyl transpeptidase. J Biol Chem. 284:9059–9065.
- Kinlough CL, Poland PA, Bruns JB, Hughey RP. 2005. Gamma-glutamyltranspeptidase: disulfide bridges, propeptide cleavage, and activation in the endoplasmic reticulum. Meth Enzymol. 401:426–449.
- Kinoshita T, Minato S. 1978. Structural studies on anthglutin, an inhibitor of γ-glutamyl transpeptidase, from Penicillium oxalicum. Bull Chem Soc Jpn. 51:3282–3285.
- Kitada M, McLenithan JC, Anders MW. 1985. Purification and characterization of S-phenacylglutathione reductase from rat liver. J Biol Chem. 260:11749–11754.
- Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li CQ, Trudel LJ, Yasui H, Vallet S, et al. 2007. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood. 110:709–718.
- Knight RH, Young L. 1957. The urinary excretion of premercapturic acids. Biochem J. 66:55p–56p.
- Knight RH, Young L. 1958. Biochemical studies of toxic agents. 11. The occurrence of premercapturic acids. Biochem J. 70:111–119.
- Knight TR, Choudhuri S, Klaassen CD. 2007. Constitutive mRNA expression of various glutathione S-transferase isoforms in different tissues of mice. Toxicol Sci. 100:513–524.
- Knight TR, Choudhuri S, Klaassen CD. 2008. Induction of hepatic glutathione S-transferases in male mice by prototypes of various classes of microsomal enzyme inducers. Toxicol Sci. 106:329–338.
- Ko MH, Puglielli L. 2009. Two endoplasmic reticulum (ER)/ER Golgi intermediate compartment-based lysine acetyltransferases post-translationally regulate BACE1 levels. J Biol Chem. 284:2482–2492.
- Kodera Y, Isobe K, Yamauchi M, Kondo K, Akiyama S, Ito K, Nakashima I, Takagi H. 1994. Expression of glutathione-S-transferases Alpha and Pi in gastric cancer: a correlation with cisplatin resistance. Cancer Chemother Pharmacol. 34:203–208.
- Kodym R, Calkins P, Story M. 1999. The cloning and characterization of a new stress response protein. A mammalian member of a family of Theta class glutathione S-transferase-like proteins. J Biol Chem. 274:5131–5137.
- Koeplinger KA, Zhao Z, Peterson T, Leone JW, Schwende FS, Heinrikson RL, Tomasselli AG. 1999. Activated sulfonamides are cleaved by glutathione-S-transferases. Drug Metab Dispos. 27:986–991.
- Kogias E, Osterberg N, Baumer B, Psarras N, Koentges C, Papazoglou A, Saavedra JE, Keefer LK, Weyerbrock A. 2012. Growth-inhibitory and chemosensitizing effects of the glutathione-S-transferase-π-activated nitric oxide donor PABA/NO in malignant gliomas. Int J Cancer. 130:1184–1194.
- Kohchi Y, Hattori K, Oikawa N, Mizuguchi E, Isshiki Y, Aso K, Yoshinari K, Shirai H, Miwa M, Inagaki Y. 2007. Design and synthesis of novel prodrugs of 2′-deoxy-2′-methylidenecytidine activated by membrane dipeptidase overexpressed in tumor tissues. Bioorg Med Chem Lett. 17:2241–2245.
- Kohno H, Kanda S, Kanno T. 1986. Immunoaffinity purification and characterization of leucine aminopeptidase from human liver. J Biol Chem. 261:10744–10748.
- Kohno H, Kanno T. 1985. Intracellular localization and properties of aminopeptidases in human peripheral lymphocytes. Int J Biochem. 17:1143–1148.
- Kolsch H, Linnebank M, Lutjohann D, Jessen F, Wullner U, Harbrecht U, Thelen KM, Kreis M, Hentschel F, Schulz A, et al. 2004. Polymorphisms in glutathione S-transferase Omega-1 and AD, vascular dementia, and stroke. Neurology. 63:2255–2260.
- Kördel W, Schneider F. 1975. The pH-dependence of the peptidase activity of aminoacylase. Hoppe Seyler’s Z Physiol Chem. 356:915–920.
- Kördel W, Schneider F. 1976a. Chemical investigations on pig kidney aminoacylase. Biochim Biophys Acta. 445:446–457.
- Kördel W, Schneider F. 1976b. Chemical modification of two tryptophan residues abolishes the catalytic activity of aminoacylase. Hoppe Seylers. Z Physiol Chem. 357:1109–1115.
- Kördel W, Schneider F. 1977a. Identification of essential histidine residues of aminoacylase by photooxidation and by reaction with diethylpyrocarbonate. Z Naturforsch C Biosci. 32:337–341.
- Kördel W, Schneider F. 1977b. Renal aminoacylase, a zinc enzyme. Z Naturforsch C Biosci. 32:342–344.
- Kotapati S, Esades A, Matter B, Le C, Tretyakova N. 2015. High throughput HPLC-ESI(-)-MS/MS methodology for mercapturic acid metabolites of 1,3-butadiene: biomarkers of exposure and bioactivation. Chem Biol Interact. 241:23–31.
- Köttgen A, Pattaro C, Boger CA, Fuchsberger C, Olden M, Glazer NL, Parsa A, Gao X, Yang Q, Smith AV, et al. 2010. New loci associated with kidney function and chronic kidney disease. Nat Genet. 42:376–384.
- Koutsoumpli GE, Dimaki VD, Thireou TN, Eliopoulos EE, Labrou NE, Varvounis GI, Clonis YD. 2012. Synthesis and study of 2-(pyrrolesulfonylmethyl)-N-arylimines: a new class of inhibitors for human glutathione transferase A1-1. J Med Chem. 55:6802–6813.
- Kovacsics D, Patik I, Özvegy-Laczka C. 2017. The role of organic anion transporting polypeptides in drug absorption, distribution, excretion and drug-drug interactions. Expert Opin Drug Metab Toxicol. 13:409–424.
- Kozak EM, Tate SS. 1980. Interaction of the antitumor drug, L-αS, 5S)-α-amino-3-chloro-4,5-dihydro-5-isoxazoleacetic acid (AT-125) with renal brush border membranes. Specific labeling of γ-glutamyl transpeptidase. FEBS Lett. 122:175–178.
- Kozak EM, Tate SS. 1982. Glutathione-degrading enzymes of microvillus membranes. J Biol Chem. 257:6322–6327.
- Kraft M, Schleberger C, Weckesser J, Schulz GE. 2006. Binding structure of the leucine aminopeptidase inhibitor microginin FR1. FEBS Lett. 580:6943–6947.
- Kramer BS, Birch R, Greco A, Prestridge K, Johnson R. 1986. Phase II evaluation of acivicin in lung cancer: a Southeastern Cancer Study Group Trial. Cancer Treat Rep. 70:1031–1032.
- Kramer W, Girbig F, Corsiero D, Pfenninger A, Frick W, Jähne G, Rhein M, Wendler W, Lottspeich F, Hochleitner EO, et al. 2005. Aminopeptidase N (CD13) is a molecular target of the cholesterol absorption inhibitor ezetimibe in the enterocyte brush border membrane. J Biol Chem. 280:1306–1320.
- Kraus P. 1975. Biodegradation of α-hexachlorocyclohexane. VI. The dechlorination of α-hexachlorocyclohexane by microsomes and cytosol of rat liver. Naunyn Schmiedebergs Arch Pharmacol. 291:79–87.
- Kraus P, Gross B. 1979. Particle-bound glutathione-S-transferases. Enzyme. 24:205–208.
- Kraus P, Noack G, Portig J. 1973. Biodegradation of α-hexachlorocyclohexane. II. Glutathione-mediated conversion to hydrophilic substance by particulate fractions of rat liver and by homogenates of various rat organs. Naunyn Schmiedebergs Arch Pharmacol. 279:199–202.
- Kraus T, Uttamsingh V, Anders MW, Wolf S. 2000. Porcine kidney microsomal cysteine S-conjugate N-acetyltransferase-catalyzed N-acetylation of haloalkene-derived cysteine S-conjugates. Drug Metab Dispos. 28:440–445.
- Kretschmer K, Hanson H. 1965. Kristallisierte leucinaminopeptidase aus rinderaugenlinsen. Physikalische konstanten, I. Hoppe Seylers Z Physiol Chem. 340:126–137.
- Kropp H, Sundelof JG, Hajdu R, Kahan FM. 1982. Metabolism of thienamycin and related carbapenem antibiotics by the renal dipeptidase, dehydropeptidase. Antimicrob Agents Chemother. 22:62–70.
- Kuang Q, Purhonen P, Ålander J, Svensson R, Hoogland V, Winerdal J, Spahiu L, Ottosson-Wadlund A, Jegerschöld C, Morgenstern R, et al. 2017. Dead-end complex, lipid interactions and catalytic mechanism of microsomal glutathione transferase 1, an electron crystallography and mutagenesis investigation. Sci Rep. 7:7897.
- Kumpe E, Löffler HG, Schneider F. 1981. Studies on the Zn2+/Co2+ exchange with acylamino acid amidohydrolase from pig kidney. Z Naturforsch C Biosci. 36:951–955.
- Kuno T, Matsuda Y, Katunuma N. 1983. The conversion of the precursor form of γ-glutamyltranspeptidase to its subunit form takes place in brush border membranes. Biochem Biophys Res Commun. 114:889–895.
- Kunze T. 1996. Phosphono analogues of glutathione as new inhibitors of glutathione S-transferases. Arch Pharm Pharm Med Chem. 329:503–509.
- Kunze T, Heps S. 2000. Phosphono analogs of glutathione: inhibition of glutathione transferases, metabolic stability, and uptake by cancer cells. Biochem Pharmacol. 59:973–981.
- Kurtovic S, Modén O, Shokeer A, Mannervik B. 2008. Structural determinants of glutathione transferases with azathioprine activity identified by DNA shuffling of Alpha class members. J Mol Biol. 375:1365–1379.
- Laborde E. 2010. Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death. Cell Death Differ. 17:1373–1380.
- Lack L. 1961. Enzymic cis-trans isomerization of maleylpyruvic acid . J Biol Chem. 236:2835–2840.
- Ladner JE, Parsons JF, Rife CL, Gilliland GL, Armstrong RN. 2004. Parallel evolutionary pathways for glutathione transferases: structure and mechanism of the mitochondrial class Kappa enzyme rGSTK1-1. Biochemistry. 43:352–361.
- Lage H. 2008. An overview of cancer multidrug resistance: a still unsolved problem. Cell Mol Life Sci. 65:3145–3167.
- Laliberte RE, Perregaux DG, Hoth LR, Rosner PJ, Jordan CK, Peese KM, Eggler JF, Dombroski MA, Geoghegan KF, Gabel CA. 2003. Glutathione S-transferase Omega 1-1 is a target of cytokine release inhibitory drugs and may be responsible for their effect on interleukin-1β posttranslational processing. J Biol Chem. 278:16567–16578.
- Lanca AJ, Israel Y. 1991. Histochemical demonstration of sinusoidal γ-glutamyltransferase activity by substrate protection fixation: comparative studies in rat and guinea pig liver. Hepatology. 14:857–863.
- Landi S. 2000. Mammalian class Theta GST and differential susceptibility to carcinogens: a review. Mutat Res. 463:247–283.
- Langner J, Ansorge J, editors. 2002. Ectopeptidases. CD13/aminopeptidase N and CD26I/dipeptidylpeptidase IV in medicine and biology. New York: Springer.
- Lannutti F, Marrone A, Re N. 2012. Binding of GSH conjugates to π-GST: a cross-docking approach. J Mol Graph Model. 32:9–18.
- Lantum HB, Baggs RB, Krenitsky DM, Board PG, Anders MW. 2002a. Immunohistochemical localization and activity of glutathione transferase Zeta (GSTZ1-1) in rat tissues. Drug Metab Dispos. 30:616–625.
- Lantum HB, Board PG, Anders MW. 2002b. Kinetics of the biotransformation of maleylacetone and chlorofluoroacetic acid by polymorphic variants of human glutathione transferase Zeta (hGSTZ1-1). Chem Res Toxicol. 15:957–963.
- Larsson AK, Emrén LO, Bardsley WG, Mannervik B. 2004. Directed enzyme evolution guided by multidimensional analysis of substrate-activity space. Protein Eng Des Sel. 17:49–55.
- Lash LH, Anders MW. 1989. Uptake of nephrotoxic S-conjugates by isolated rat renal proximal tubular cells. J Pharmacol Exp Ther. 248:531–537.
- Lau SS, Kleiner HE, Monks TJ. 1995. Metabolism as a determinant of species susceptibility to 2,3,5-(triglutathion-S-yl)hydroquinone-mediated nephrotoxicity. The role of N-acetylation and N-deacetylation. Drug Metab Dispos. 23:1136–1142.
- Le Trong I, Stenkamp RE, Ibarra C, Atkins WM, Adman ET. 2002. 1.3-Å resolution structure of human glutathione S-transferase with S-hexyl glutathione bound reveals possible extended ligandin binding site. Proteins. 48:618–627.
- Ledeme N, Hennon G, Vincent-Fiquet O, Plaquet R. 1981. Purification and enzymatic properties of an L-leucine aminopeptidase from swine liver. Biochim Biophys Acta. 660:262–270.
- Lee CY, McKinney JD. 1982. Identity of microsomal glutathione S-transferases. Mol Cell Biochem. 48:91–96.
- Lee DH, Blomhoff R, Jacobs DR Jr. 2004. Is serum gamma glutamyltransferase a marker of oxidative stress?. Free Radic Res. 38:535–539.
- Lee DH, Ha MH, Kim JH, Christiani DC, Gross MD, Steffes M, Blomhoff R, Jacobs DR Jr. 2003. Gamma-glutamyltransferase and diabetes–a 4 year follow-up study. Diabetologia. 46:359–364.
- Lee HE, Seltzer S. 1989. cis-β-Acetylacrylate is a substrate for maleylacetoacetate cis-trans isomerase. Mechanistic implications. Biochem Int. 18:91–97.
- Lee JJ, Son J, Ha HH, Chang YT. 2011. Fluorescent labeling of membrane proteins on the surface of living cells by a self-catalytic glutathione S-transferase Omega 1 tag. Mol Biosyst. 7:1270–1276.
- Lee KK, Shimoji M, Hossain QS, Sunakawa H, Aniya Y. 2008. Novel function of glutathione transferase in rat liver mitochondrial membrane: role for cytochrome c release from mitochondria. Toxicol Appl Pharmacol. 232:109–118.
- Lehky P, Lisowski J, Wolf DP, Wacker H, Stein EA. 1973. Pig kidney particulate aminopeptidase. A zinc metalloenzyme. Biochim Biophys Acta. 321:274–281.
- Lejczak B, Kafarski P, Zygmunt J. 1989. Inhibition of aminopeptidases by aminophosphonates. Biochemistry. 28:3549–3555.
- Lendeckel U, Bukowska A, Laettig JH, Brandt W. 2004. Alanyl-aminopeptidases in human T cells. Structures and functions. In: Hooper NM, Lendeckel U, editors. Aminopeptidases in biology and disease. Vol. 2. New York: Springer; p. 201–227.
- Lengqvist J, Svensson R, Evergren E, Morgenstern R, Griffiths WJ. 2004. Observation of an intact noncovalent homotrimer of detergent-solubilized rat microsomal glutathione transferase-1 by electrospray mass spectrometry. J Biol Chem. 279:13311–13316.
- Levi AJ, Gatmaitan Z, Arias IM. 1969. Two hepatic cytoplasmic protein fractions, Y and Z, and their possible role in the hepatic uptake of bilirubin, sulfobromophthalein, and other anions. J Clin Invest. 48:2156–2167.
- Lherbet C, Gravel C, Keillor JW. 2004. Synthesis of S-alkyl L-homocysteine analogues of glutathione and their kinetic studies with γ-glutamyl transpeptidase. Bioorg Med Chem Lett. 14:3451–3455.
- Lherbet C, Keillor JW. 2004. Probing the stereochemistry of the active site of γ-glutamyl transpeptidase using sulfur derivatives of L-glutamic acid. Org Biomol Chem. 2:238–245.
- Lherbet C, Morin M, Castonguay R, Keillor JW. 2003. Synthesis of aza and oxaglutamyl-p-nitroanilide derivatives and their kinetic studies with γ-glutamyltranspeptidase. Bioorg Med Chem Lett. 13:997–1000.
- Li J, Chen L, Wu W, Zhang W, Ma Z, Cheng Y, Du L, Li M. 2014. Discovery of bioluminogenic probes for aminopeptidase N imaging. Anal Chem. 86:2747–2751.
- Li J, Xia Z, Ding J. 2005a. Thioredoxin-like domain of human Kappa class glutathione transferase reveals sequence homology and structure similarity to the Theta class enzyme. Protein Sci. 14:2361–2369.
- Li L, Meier PJ, Ballatori N. 2000. Oatp2 mediates bidirectional organic solute transport: a role for intracellular glutathione. Mol Pharmacol. 58:335–340.
- Li L, Shi W, Wu X, Li X, Ma H. 2018. In vivo tumor imaging by a γ-glutamyl transpeptidase-activatable near-infrared fluorescent probe. Anal Bioanal Chem. 410:26.
- Li L, Zhao Y, Cao R, Li L, Cai GH, Li JJ, Qi XB, Chen S, Zhang ZY. 2019. Activity- based protein profiling reveals GSTO1 as the covalent target of piperlongumine and a promising target for combination therapy for cancer. Chem Commun. 55:4407–4410.
- Li QB, Fang H, Wang XJ, Hu GY, Wang Q, Xu WF. 2012. Novel potent 2,5-pyrrolidinedione peptidomimetics as aminopeptidase N inhibitors. Design, synthesis and activity evaluation. Bioorg Med Chem Lett. 22:850–853.
- Li W, James MO, McKenzie SC, Calcutt NA, Liu C, Stacpoole PW. 2011. Mitochondrion as a novel site of dichloroacetate biotransformation by glutathione transferase Zeta 1. J Pharmacol Exp Ther. 336:87–94.
- Li WS, Lam WS, Liu KC, Wang CH, Chang HC, Jen YC, Hsu YT, Shivatare SS, Jao SC. 2010. Overcoming the drug resistance in breast cancer cells by rational design of efficient glutathione S-transferase inhibitors. Org Lett. 12:20–23.
- Li Y, Cao Z, Zhu H, Trush MA. 2005b. Differential roles of 3H-1,2-dithiole-3-thione-induced glutathione, glutathione S-transferase and aldose reductase in protecting against 4-hydroxy-2-nonenal toxicity in cultured cardiomyocytes. Arch Biochem Biophys. 439:80–90.
- Li YJ, Scott WK, Zhang L, Lin PI, Oliveira SA, Skelly T, Doraiswamy MP, Welsh-Bohmer KA, Martin ER, Haines JL, et al. 2006. Revealing the role of glutathione S-transferase omega in age-at-onset of Alzheimer and Parkinson diseases. Neurobiol Aging. 27:1087–1093.
- Liang Y, Jiang X, Tang N, Yang L, Chen H, Wang Q. 2015. Quantification and visualization of glutathione S-transferase omega 1 in cells using inductively coupled plasma mass spectrometry (ICP-MS) and fluorescence microscopy. Anal Bioanal Chem. 407:2373–2381.
- Liao RZ, Himo F, Yu JG, Liu RZ. 2010. Dipeptide hydrolysis by the dinuclear zinc enzyme human renal dipeptidase: mechanistic insights from DFT calculations. J Inorg Biochem. 104:37–46.
- Lieberman MW, Shields JE, Will Y, Reed DJ, Carter BZ. 1999. Gamma-glutamyl leukotrienase cleavage of leukotriene C4 . Adv Exp Med Biol. 469:301–306.
- Lien S, Gustafsson A, Andersson AK, Mannervik B. 2001. Human glutathione transferase A1-1 demonstrates both half-of-the-sites and all-of-the-sites reactivity. J Biol Chem. 276:35599–35605.
- Lien S, Larsson AK, Mannervik B. 2002. The polymorphic human glutathione transferase T1-1, the most efficient glutathione transferase in the denitrosation and inactivation of the anticancer drug 1,3-bis(2-chloroethyl)-1-nitrosourea. Biochem Pharmacol. 63:191–197.
- Lim CEL, Matthaei KI, Blackburn AC, Davis RP, Dahlstrom JE, Koina ME, Anders MW, Board PG. 2004. Mice deficient in glutathione transferase Zeta/maleylacetoacetate isomerase exhibit a range of pathological changes and elevated expression of Alpha, Mu, and Pi class glutathione transferases. Am J Pathol. 165:679–693.
- Lin WY, Lin SH, Van Wart HE. 1988. Steady-state kinetics of hydrolysis of dansyl-peptide substrates by leucine aminopeptidase. Biochemistry. 27:5062–5068.
- Lin WY, Van Wart HE. 1988. Hydrolysis of dansyl-peptide substrates by leucine aminopeptidase: origin of dansyl fluorescence changes during hydrolysis. Biochemistry. 27:5054–5061.
- Lindblad B, Lindstedt S, Steen G. 1977. On the enzymic defects in hereditary tyrosinemia. Proc Natl Acad Sci USA. 74:4641–4645.
- Linderstrøm-Lang K, Sato M. 1929. Die spaltung von glycylglycin, alanylglycin und leucylglycin durch darm- und malzpeptidasen. Hoppe Seylers Z Physiol Chem. 184:83–92.
- Lindner H, Höpfner S, Täfler-Naumann M, Miko M, Konrad L, Röhm KH. 2000. The distribution of aminoacylase I among mammalian species and localization of the enzyme in porcine kidney. Biochimie. 82:129–137.
- Lindner HA, Alary A, Wilke M, Sulea T. 2008a. Probing the acyl-binding pocket of aminoacylase-1. Biochemistry. 47:4266–4275.
- Lindner HA, Lunin VV, Alary A, Hecker R, Cygler M, Ménard R. 2003. Essential roles of zinc ligation and enzyme dimerization for catalysis in the aminoacylase-1/M20 family. J Biol Chem. 278:44496–44504.
- Lindner HA, Tafler-Naumann M, Röhm KH. 2008b. N-Acetylamino acid utilization by kidney aminoacylase-1. Biochimie. 90:773–780.
- Lipscomb JC, Mahle DA, Brashear WT, Barton HA. 1995. Dichloroacetic acid: metabolism in cytosol. Drug Metab Dispos. 23:1202–1205.
- Lipscomb WN, Sträter N. 1996. Recent advances in zinc enzymology. Chem Rev. 96:2375–2434.
- Listowsky I. 2005. A subclass of Mu glutathione S-transferases selectively expressed in testis and brain . Meth Enzymol. 401:278–287.
- Listowsky I, Rowe JD, Patskovsky YV, Tchaikovskaya T, Shintani N, Novikova E, Nieves E. 1998. Human testicular glutathione S-transferases: insights into tissue-specific expression of the diverse subunit classes. Chem Biol Interact. 111–112:103–112.
- Littlewood GM, Hooper NM, Turner AJ. 1989. Ectoenzymes of the kidney microvillar membrane. Affinity purification, characterization and localization of the phospholipase C-solubilized form of renal dipeptidase. Biochem J. 257:361–367.
- Litwack G, Ketterer B, Arias IM. 1971. Ligandin: a hepatic protein which binds steroids, bilirubin, carcinogens and a number of exogenous organic anions. Nature. 234:466–467.
- Liu C, Yang Y, Chen L, Lin YL, Li F. 2014a. A unified mechanism for aminopeptidase N-based tumor cell motility and tumor-homing therapy. J Biol Chem. 289:34520–34529.
- Liu D, Hewawasam R, Pace SM, Gallant EM, Casarotto MG, Dulhunty AF, Board PG. 2009. Dissection of the inhibition of cardiac ryanodine receptors by human glutathione transferase GSTM2-2. Biochem Pharmacol. 77:1181–1193.
- Liu F, Wang Z, Wang W, Luo JG, Kong L. 2018a. Red-emitting fluorescent probe for detection of γ-glutamyltranspeptidase and its application of real-time imaging under oxidative stress in cells and in vivo. Anal Chem. 90:7467–7473.
- Liu M, Chen H, Wei L, Hu D, Dong K, Jia W, Dong LQ, Liu F. 2015. Endoplasmic reticulum (ER) localization is critical for DsbA-L protein to suppress ER stress and adiponectin down-regulation in adipocytes. J Biol Chem. 290:10143–10148.
- Liu M, Zhou L, Xu A, Lam KS, Wetzel MD, Xiang R, Zhang J, Xin X, Dong LQ, Liu F. 2008. A disulfide-bond A oxidoreductase-like protein (DsbA-L) regulates adiponectin multimerization. Proc Natl Acad Sci USA. 105:18302–18307.
- Liu S, Zhang P, Ji X, Johnson WW, Gilliland GL, Armstrong RN. 1992. Contribution of tyrosine 6 to the catalytic mechanism of isoenzyme 3-3 of glutathione S-transferase. J Biol Chem. 267:4296–4299.
- Liu X, An BH, Kim MJ, Park JH, Kang YS, Chang M. 2014b. Human glutathione S-transferase P1-1 functions as an estrogen receptor alpha signaling modulator. Biochem Biophys Res Commun. 452:840–844.
- Liu Y, Tan J, Zhang Y, Zhuang J, Ge M, Shi B, Li J, Xu G, Xu S, Fan C, et al. 2018b. Visualizing glioma margins by real-time tracking of γ-glutamyltranspeptidase activity. Biomaterials. 173:1–10.
- Liu Z, Zhen Z, Zuo Z, Wu Y, Liu A, Yi Q, Li W. 2006. Probing the catalytic center of porcine aminoacylase 1 by site-directed mutagenesis, homology modeling and substrate docking. J Biochem. 139:421–430.
- Lo HW, Ali-Osman F. 2007. Genetic polymorphism and function of glutathione S-transferases in tumor drug resistance. Curr Opin Pharmacol. 7:367–374.
- Lock EA, Berndt WO. 1988. Studies on the mechanism of nephrotoxicity and nephrocarcinogenicity of halogenated alkenes. Crit Rev Toxicol. 19:23–42.
- Lock EA, Ishmael J. 1985. Effect of the organic acid transport inhibitor probenecid on renal cortical uptake and proximal tubular toxicity of hexachloro-1,3-butadiene and its conjugates. Toxicol Appl Pharmacol. 81:32–42.
- Löffler HG, Röhm KH, Schneider F. 1986. Studies on the metal ion dependence, kinetics and SH (S-S) groups of acylamino acid amidohydrolase. In: Bertini I, Luchinat C, Maret W, editors. Zinc enzymes. Vol. 1. Boston (MA): Birkhäuser; p. 281–288.
- Löffler HG, Schneider F. 1987a. Aminoacylase from hog kidney: essential carboxyl groups, inhibition by α-N-OH-acids, lack of esterase activity. Biol Chem Hoppe Seyler. 368:1072–1073.
- Löffler HG, Schneider F. 1987b. Inhibition of aminoacylase from hog kidney by 2-ethoxy-1-(ethoxycarbonyl)-1,2-dihydroquinoline. Biol Chem Hoppe Seyler. 368:481–485.
- Löffler HG, Schneider F, Aumuller G, Unsicker K. 1982. Immunzytochemische untersuchungen zur lokalisation der aminoacylase I (EC 3.5.1.14) in der schweineniere. Acta Histochem. 25:57–60.
- Long PM, Stradecki HM, Minturn JE, Wesley UV, Jaworski DM. 2011. Differential aminoacylase expression in neuroblastoma. Int J Cancer. 129:1322–1330.
- Look AT, Ashmun RA, Shapiro LH, Peiper SC. 1989. Human myeloid plasma membrane glycoprotein CD13 (gp150) is identical to aminopeptidase N. J Clin Invest. 83:1299–1307.
- Lorentz K, Voss J, Flatter B. 1975. A new method for the assay of aminoacylase: elaboration of a fixed-incubation method for routine measurements. Clin Chim Acta. 63:263–269.
- Lowther WT, Matthews BW. 2002. Metalloaminopeptidases: common functional themes in disparate structural surroundings. Chem Rev. 102:4581–4608.
- Lozano E, Briz O, Macias RIR, Serrano MA, Marin JJG, Herraez E. 2018. Genetic heterogeneity of SLC22 family of transporters in drug disposition. J Pers Med. 8:14.
- Lu WD, Atkins WM. 2004. A novel antioxidant role for ligandin behavior of glutathione S-transferases: attenuation of the photodynamic effects of hypericin. Biochemistry. 43:12761–12769.
- Luan Y, Jing F, Zhang J, Zou M, Wang X, Jia Y, Liu N, Mou J, Xu W. 2012a. Design, synthesis, and activity evaluation of a new 5-fluorouracil prodrug containing an Asn-Gly-Arg(NO2)COOCH3 tripeptide. Protein Pept Lett. 19:1122–1131.
- Luan Y, Ma C, Wang Y, Fang H, Xu W. 2012b. The characteristics, functions and inhibitors of three aminopeptidases belonging to the M1 family. Curr Protein Pept Sci. 13:490–500.
- Luan Y, Wang X, Zhu H, Qu X, Fang H, Xu W. 2009. The synthesis and activity evaluation of a bestatin derivative (LYP) as an aminopeptidase N inhibitor. Lett Drug Des Discov. 6:420–423.
- Luan Y, Xu W. 2007. The structure and main functions of aminopeptidase N. Curr Med Chem. 14:639–647.
- Lucena MI, Andrade RJ, Martínez C, Ulzurrun E, García-Martín E, Borraz Y, Fernández MC, Romero-Gomez M, Castiella A, Planas R, et al. 2008. Glutathione S-transferase M1 and T1 null genotypes increase susceptibility to idiosyncratic drug-induced liver injury. Hepatology. 48:588–596.
- Luciani N, Marie-Claire C, Ruffet E, Beaumont A, Roques BP, Fournié-Zaluski MC. 1998. Characterization of Glu(350) as a critical residue involved in the N-terminal amine binding site of aminopeptidase N (EC 3.4.11.2): insights into its mechanism of action. Biochemistry. 37:686–692.
- Lui MS, Kizaki H, Weber G. 1982. Biochemical pharmacology of acivicin in rat hepatoma cells. Biochem Pharmacol. 31:3469–3473.
- Luisi G, Mollica A, Carradori S, Lenoci A, De Luca A, Caccuri AM. 2016. Nitrobenzoxadiazole-based GSTP1-1 inhibitors containing the full peptidyl moiety of (pseudo)glutathione. J Enzyme Inhib Med Chem. 31:924–930.
- Lukic A, Ji J, Idborg H, Samuelsson B, Palmberg L, Gabrielsson S, Rådmark O. 2016. Pulmonary epithelial cancer cells and their exosomes metabolize myeloid cell-derived leukotriene C4 to leukotriene D4. J Lipid Res. 57:1659–1669.
- Luo Z, An R, Ye D. 2018a. Recent advances in the development of optical imaging probes for γ-glutamyltranspeptidase. ChemBioChem. 20(4):474–487.
- Luo Z, Huang Z, Li K, Sun Y, Lin J, Ye D, Chen HY. 2018b. Targeted delivery of a γ-glutamyl transpeptidase activatable near-infrared-fluorescent probe for selective cancer imaging. Anal Chem. 90:2875–2883.
- Lyttle MH, Hocker MD, Hui HC, Caldwell CG, Aaron DT, Engqvist-Goldstein A, Flatgaard JE, Bauer KE. 1994a. Isozyme-specific glutathione-S-transferase inhibitors: design and synthesis. J Med Chem. 37:189–194.
- Lyttle MH, Satyam A, Hocker MD, Bauer KE, Caldwell CG, Hui HC, Morgan AS, Mergia A, Kauvar LM. 1994b. Glutathione-S-transferase activates novel alkylating agents. J Med Chem. 37:1501–1507.
- Ma C, Cao J, Liang X, Huang Y, Wu P, Li Y, Xu W, Zhang Y. 2016. Novel leucine ureido derivatives as aminopeptidase N inhibitors. Design, synthesis and activity evaluation. Eur J Med Chem. 108:21–27.
- Ma C, Li X, Liang X, Jin K, Cao J, Xu W. 2013. Novel β-dicarbonyl derivatives as inhibitors of aminopeptidase N (APN). Bioorg Med Chem Lett. 23:4948–4952.
- Ma Q, He XQ. 2012. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol Rev. 64:1055–1081.
- Maceyka M, Nava VE, Milstien S, Spiegel S. 2004. Aminoacylase 1 is a sphingosine kinase 1-interacting protein. FEBS Lett. 568:30–34.
- Maciag AE, Holland RJ, Kim Y, Kumari V, Luthers CE, Sehareen WS, Biswas D, Morris NL, Ji X, Anderson LM, et al. 2014. Nitric oxide (NO) releasing poly ADP-ribose polymerase 1 (PARP-1) inhibitors targeted to glutathione S-transferase P1-overexpressing cancer cells. J Med Chem. 57:2292–2302.
- Maeda A, Crabb JW, Palczewski K. 2005. Microsomal glutathione S-transferase 1 in the retinal pigment epithelium: protection against oxidative stress and a potential role in aging. Biochemistry. 44:480–489.
- Maellaro E, Del Bello B, Sugherini L, Santucci A, Comporti M, Casini AF. 1994. Purification and characterization of glutathione-dependent dehydroascorbate reductase from rat liver. Biochem J. 301:471–476.
- Magnan SD, Shirota FN, Nagasawa HT. 1982. Drug latentiation by γ-glutamyl transpeptidase. J Med Chem. 25:1018–1021.
- Mahajan S, Atkins WM. 2005. The chemistry and biology of inhibitors and pro-drugs targeted to glutathione S-transferases. Cell Mol Life Sci. 62:1221–1233.
- Mahmud I, Ueda N, Yamaguchi H, Yamashita R, Yamamoto S, Kanaoka Y, Urade Y, Hayaishi O. 1997. Prostaglandin D synthase in human megakaryoblastic cells. J Biol Chem. 272:28263–28266.
- Maiereanu C, Schmitt C, Schifano-Faux N, Le Nouen D, Defoin A, Tarnus C. 2011. A novel amino-benzosuberone derivative is a picomolar inhibitor of mammalian aminopeptidase N/CD13. Bioorg Med Chem. 19:5716–5733.
- Mainwaring GW, Foster JR, Green T. 1998. Nuclear and cellular immunolocalization of Theta-class glutathione S-transferase GSTT1-1 in the liver and lung of the mouse. Biochem J. 329:431–432.
- Mainwaring GW, Williams SM, Foster JR, Tugwood J, Green T. 1996. The distribution of Theta-class glutathione S-transferases in the liver and lung of mouse, rat and human. Biochem J. 318:297–303.
- Mak AB, Pehar M, Nixon AM, Williams RA, Uetrecht AC, Puglielli L, Moffat J. 2014. Post-translational regulation of CD133 by ATase1/ATase2-mediated lysine acetylation. J Mol Biol. 426:2175–2182.
- Makrides V, Camargo SMR, Verrey F. 2014. Transport of amino acids in the kidney. Compr Physiol. 4:367–403.
- Malfroy B, Kado-Fong H, Gros C, Giros B, Schwartz JC, Hellmiss R. 1989. Molecular cloning and amino acid sequence of rat kidney aminopeptidase M: a member of a super family of zinc-metallohydrolases. Biochem Biophys Res Commun. 161:236–241.
- Mannervik B. 1985. The isoenzymes of glutathione transferase. Adv Enzymol Relat Areas Mol Biol. 57:357–417.
- Mannervik B, Ålin P, Guthenberg C, Jensson H, Tahir MK, Warholm M, Jörnvall H. 1985. Identification of three classes of cytosolic glutathione transferase common to several mammalian species: correlation between structural data and enzymatic properties. Proc Natl Acad Sci USA. 82:7202–7206.
- Mannervik B, Awasthi YC, Board PG, Hayes JD, Di Ilio C, Ketterer B, Listowsky I, Morgenstern R, Muramatsu M, Pearson WR. 1992. Nomenclature for human glutathione transferases. Biochem J. 282:305–306.
- Mannervik B, Board PG, Hayes JD, Listowsky I, Pearson WR. 2005. Nomenclature for mammalian soluble glutathione transferases. Meth Enzymol. 401:1–8.
- Mannervik B, Castro VM, Danielson UH, Tahir MK, Hansson J, Ringborg U. 1987. Expression of class Pi glutathione transferase in human malignant melanoma cells. Carcinogenesis. 8:1929–1932.
- Mannervik B, Danielson UH. 1988. Glutathione transferases–structure and catalytic activity. Crit Rev Biochem. 23:283–337.
- Mannervik B, Guthenberg C, Jakobson I, Warholm M. 1978. Glutathione conjugation: reaction mechanism of glutathione S-transferase A. In: Aitio A, editor. Conjugation reactions in drug biotransformation. Amsterdam: Elsevier; p. 101–110.
- Mannervik B, Jensson H. 1982. Binary combinations of four protein subunits with different catalytic specificities explain the relationship between six basic glutathione S-transferases in rat liver cytosol. J Biol Chem. 257:9909–9912.
- Mannervik B, Widersten M. 1995. Human glutathione transferases: classification, tissue distribution, structure, and functional properties. In: Pacifici GM, Fracchia GN, editors. Advances in drug metabolism in man. Luxemburg: Commission of the European Communities; p. 407–459.
- Marathe GV, Nash B, Haschemeyer RH, Tate SS. 1979. Ultrastructural localization of gamma-glutamyl transpeptidase in rat kidney and jejunum. FEBS Lett. 107:436–440.
- Marnell LL, Garcia-Vargas GG, Chowdhury UK, Zakharyan RA, Walsh B, Avram MD, Kopplin MJ, Cebrian ME, Silbergeld EK, Aposhian HV. 2003. Polymorphisms in the human monomethylarsonic acid (MMA V) reductase/hGSTO1 gene and changes in urinary arsenic profiles. Chem Res Toxicol. 16:1507–1513.
- Marsden CM, Young L. 1958. Biochemical studies of toxic agents. 10. Observations on the metabolism of 35S-labelled mercapturic acids. Biochem J. 69:257–265.
- Martin DG, Duchamp DJ, Chidester CG. 1973. The isolation, structure, and absolute configuration of U-42,126 a novel antitumor antibiotic. Tetrahedron Lett. 14:2549–2552.
- Martin JL. 1995. Thioredoxin–a fold for all reasons. Structure. 3:245–250.
- Martinez Molina D, Eshaghi S, Nordlund P. 2008. Catalysis within the lipid bilayer-structure and mechanism of the MAPEG family of integral membrane proteins. Curr Opin Struct Biol. 18:442–449.
- Mathew N, Kalyanasundaram H, Balaraman K. 2006. Glutathione S-transferase (GST) inhibitors. Expert Opin Ther Patents. 16:431–444.
- Mathias PI, B’Hymer C. 2014. A survey of liquid chromatographic-mass spectrometric analysis of mercapturic acid biomarkers in occupational and environmental exposure monitoring. J Chromatogr B Analyt Technol Biomed Life Sci. 964:136–145.
- Mathias PI, B’hymer C. 2016. Mercapturic acids: recent advances in their determination by liquid chromatography/mass spectrometry and their use in toxicant metabolism studies and in occupational and environmental exposure studies. Biomarkers. 21:293–315.
- Matsushima M, Takahashi T, Ichinose M, Miki K, Kurokawa K, Takahashi K. 1991. Structural and immunological evidence for the identity of prolyl aminopeptidase with leucyl aminopeptidase. Biochem Biophys Res Commun. 178:1459–1464.
- Matsushita N, Hizue M, Aritake K, Hayashi K, Takada A, Mitsui K, Hayashi M, Hirotsu I, Kimura Y, Tani T, et al. 1998. Pharmacological studies on the novel antiallergic drug HQL-79: I. Antiallergic and antiasthmatic effects in various experimental models. Jpn J Pharmacol. 78:1–10.
- Mazari AMA, Hegazy UM, Mannervik B. 2015. Identification of new inhibitors for human hematopoietic prostaglandin D2 synthase among FDA-approved drugs and other compounds. Chem Biol Interact. 229:91–99.
- McDonald JK, Barrett AJ. 1986. Microsomal alanyl aminotransferase. Mammalian proteases: a glossary and bibliography. London: Academic Press; p. 59–71.
- McGuire WP, Blessing JA, DiSaia PJ, Buchsbaum HJ. 1986. Phase II trial of acivicin in patients with advanced epithelial ovarian carcinoma. A gynecologic oncology group study. Invest New Drugs. 4:49–52.
- McIntyre T, Curthoys NP. 1982. Renal catabolism of glutathione. Characterization of a particulate rat renal dipeptidase that catalyzes the hydrolysis of cysteinylglycine. J Biol Chem. 257:11915–11921.
- McIntyre TM, Curthoys NP. 1979. Comparison of the hydrolytic and transfer activities of rat renal γ-glutamyltranspeptidase. J Biol Chem. 254:6499–6504.
- McKinstry WJ, Oakley AJ, Rossjohn J, Verger D, Tan KL, Board PG, Parker MW. 1998. Preliminary X-ray crystallographic studies of a newly defined human Theta-class glutathione transferase. Acta Crystallogr D Biol Crystallogr. D54:148–150.
- McLean C, Wilson A, Kim RB. 2016. Impact of transporter polymorphisms on drug development: is it clinically significant? J Clin Pharmacol. 56:S40–S58.
- McLellan LI, Hayes JD. 1989. Differential induction of class Alpha glutathione S-transferases in mouse liver by the anticarcinogenic antioxidant butylated hydroxyanisole. Purification and characterization of glutathione S-transferase Ya1Ya1. Biochem J. 263:393–402.
- McLellan LI, Wolf CR, Hayes JD. 1989. Human microsomal glutathione S-transferase. Its involvement in the conjugation of hexachlorobuta-1,3-diene with glutathione. Biochem J. 258:87–93.
- Meisinger C, Lowel H, Heier M, Schneider A, Thorand B, Group KS. 2005. Serum γ-glutamyltransferase and risk of type 2 diabetes mellitus in men and women from the general population. J Intern Med. 258:527–535.
- Meister A. 1973. On the enzymology of amino acid transport. Science. 180:33–39.
- Meister A. 1988. On the discovery of glutathione. Trends Biochem Sci. 13:185–188.
- Melbye SW, Carpenter FH. 1971. Leucine aminopeptidase (bovine lens). Stability and size of subunits. J Biol Chem. 246:2459–2463.
- Menger FM, Rourk MJ. 1997. Synthesis and reactivity of 5-fluorouracil/cytarabine mutual prodrugs. J Org Chem. 62:9083–9088.
- Menon D, Board PG. 2013. A role for glutathione transferase Omega 1 (GSTO1-1) in the glutathionylation cycle. J Biol Chem. 288:25769–25779.
- Menon D, Coll R, O’Neill LAJ, Board PG. 2014. Glutathione transferase Omega 1 is required for the LPS stimulated induction of NADPH oxidase 1 and the production of reactive oxygen species in macrophages. Free Radic Biol Med. 73:318–327.
- Menon D, Coll R, O'Neill LA, Board PG. 2015. Glutathione transferase Omega 1 modulates metabolism in LPS/TLR4 activated macrophages. J Cell Sci. 128:1982–1990.
- Menon D, Innes A, Oakley AJ, Dahlstrom JE, Jensen LM, Brüstle A, Tummala P, Rooke M, Casarotto MG, Baell JB, et al. 2017. GSTO1-1 plays a pro-inflammatory role in models of inflammation, colitis and obesity. Sci Rep. 7:17832.
- Meyer DJ. 1993. Significance of an unusually low Km for glutathione in glutathione transferases of the Alpha, Mu and Pi classes. Xenobiotica. 23:823–834.
- Meyer DJ, Beale D, Tan KH, Coles B, Ketterer B. 1985. Glutathione transferases in primary rat hepatomas: the isolation of a form with GSH peroxidase activity. FEBS Lett. 184:139–143.
- Meyer DJ, Christodoulides LG, Hong Tan K, Ketterer B. 1984. Isolation, properties and tissue distribution of rat glutathione transferase E. FEBS Lett. 173:327–330.
- Meyer DJ, Coles B, Pemble SE, Gilmore KS, Fraser GM, Ketterer B. 1991. Theta, a new class of glutathione transferases purified from rat and man. Biochem J. 274:409–414.
- Meyer DJ, Gilmore KS, Harris JM, Hartley JA, Ketterer B. 1992. Chlorambucil-monoglutathionyl conjugate is sequestered by human Alpha class glutathione S-transferases. Br J Cancer. 66:433–438.
- Meyer DJ, Thomas M. 1995. Characterization of rat spleen prostaglandin H D-isomerase as a Sigma-class GSH transferase. Biochem J. 311:739–742.
- Miene C, Klenow S, Veeriah S, Richling E, Glei M. 2009. Impact of apple polyphenols on GSTT2 gene expression, subsequent protection of DNA and modulation of proliferation using LT97 human colon adenoma cells. Mol Nutr Food Res. 53:1254–1262.
- Miller YE, Drabkin H, Jones C, Fisher JH. 1990. Human aminoacylase-1: cloning, regional assignment to distal chromosome 3p21.1, and identification of a cross-hybridizing sequence on chromosome 18. Genomics. 8:149–154.
- Miller YE, Kao B. 1989. Monoclonal antibody based immunoassay for human aminoacylase-1. J Immunoassay. 10:129–152.
- Miller YE, Minna JD, Gazdar AF. 1989. Lack of expression of aminoacylase-1 in small cell lung cancer. Evidence for inactivation of genes encoded by chromosome 3p. J Clin Invest. 83:2120–2124.
- Mina-Osorio P. 2008. The moonlighting enzyme CD13: old and new functions to target. Trends Mol Med. 14:361–371.
- Minato S. 1979. Isolation of anthglutin, an inhibitor of gamma-glutamyl transpeptidase from Penicillum oxalicum . Arch Biochem Biophys. 192:235–240.
- Mitchell AE, Zheng J, Hammock BD, Lo Bello M, Jones AD. 1998. Structural and functional consequences of haloenol lactone inactivation of murine and human glutathione S-transferase. Biochemistry. 37:6752–6759.
- Mitta M, Kato I, Tsunasawa S. 1993. The nucleotide sequence of human aminoacylase-1. Biochim Biophys Acta. 1174:201–203.
- Mitta M, Ohnogi H, Yamamoto A, Kato I, Sakiyama F, Tsunasawa S. 1992. The primary structure of porcine aminoacylase 1 deduced from cDNA sequence. J Biochem. 112:737–742.
- Miyagi M, Ohnogi H, Mitta M, Umeda Y, Kato I, Sakiyama F, Tsunasawa S. 1992. Determination of the covalent structure of aminoacylase from porcine kidney by ion-spray ionization mass spectrometry. J Protein Chem. 11:378–379.
- Modén O, Mannervik B. 2014. Glutathione transferases in the bioactivation of azathioprine. Adv Cancer Res. 122:199–244.
- Mohana K, Achary A. 2017. Human cytosolic glutathione-S-transferases: quantitative analysis of expression, comparative analysis of structures and inhibition strategies of isozymes involved in drug resistance. Drug Metab Rev. 49:318–337.
- Mohri I, Taniike M, Taniguchi H, Kanekiyo T, Aritake K, Inui T, Fukumoto N, Eguchi N, Kushi A, Sasai H. 2006. Prostaglandin D2-mediated microglia/astrocyte interaction enhances astrogliosis and demyelination in twitcher. J Neurosci. 26:4383–4393.
- Mol M, Regazzoni L, Altomare A, Degani G, Carini M, Vistoli G, Aldini G. 2017. Enzymatic and non-enzymatic detoxification of 4-hydroxynonenal: methodological aspects and biological consequences. Free Radic Biol Med. 111:328–344.
- Moldéus P, Jones DP, Ormstad K, Orrenius S. 1978. Formation and metabolism of a glutathione-S-conjugate in isolated rat liver and kidney cells. Biochem Biophys Res Commun. 83:195–200.
- Monks TJ, Lau SS. 1992. Toxicology of quinone-thioethers. Crit Rev Toxicol. 22:243–270.
- Morales GA, Laborde E. 2007. Small-molecule inhibitors of glutathione S-transferase P1-1 as anticancer therapeutic agents. In: John EM, editor. Annual Reports in Medicinal Chemistry. Vol. 42. London: Academic Press; p. 321–335.
- Morel F, Aninat C. 2011. The glutathione transferase Kappa family. Drug Metab Rev. 43:281–291.
- Morel F, Rauch C, Coles B, Le Ferrec E, Guillouzo A. 2002. The human glutathione transferase Alpha locus: genomic organization of the gene cluster and functional characterization of the genetic polymorphism in the hGSTA1 promoter. Pharmacogenetics. 12:277–286.
- Morel F, Rauch C, Petit E, Piton A, Theret N, Coles B, Guillouzo A. 2004. Gene and protein characterization of the human glutathione S-transferase Kappa and evidence for a peroxisomal localization. J Biol Chem. 279:16246–16253.
- Morel F, Schulz WA, Sies H. 1994. Gene structure and regulation of expression of human glutathione S-transferases Alpha. Biol Chem Hoppe Seyler. 375:641–649.
- Morgan AS, Ciaccio PJ, Tew KD, Kauvar LM. 1996. Isozyme-specific glutathione S-transferase inhibitors potentiate drug sensitivity in cultured human tumor cell lines. Cancer Chemother Pharmacol. 37:363–370.
- Morgan AS, Sanderson PE, Borch RF, Tew KD, Niitsu Y, Takayama T, Von Hoff DD, Izbicka E, Mangold G, Paul C, et al. 1998. Tumor efficacy and bone marrow-sparing properties of TER286, a cytotoxin activated by glutathione S-transferase. Cancer Res. 58:2568–2575.
- Morgenstern R. 2005. Microsomal glutathione transferase 1. Meth Enzymol. 401:136–146.
- Morgenstern R, DePierre JW. 1983. Microsomal glutathione transferase. Purification in unactivated form and further characterization of the activation process, substrate specificity and amino acid composition. Eur J Biochem. 134:591–597.
- Morgenstern R, DePierre JW. 1985. Microsomal glutathione transferase. Rev Biochem Toxicol. 7:67–104.
- Morgenstern R, DePierre JW. 1988. Membrane-bound glutathione transferases. In: Sies H, Ketterer B, editors. Glutathione conjugation mechanisms and biological significance. London: Academic Press; p. 157–174.
- Morgenstern R, DePierre JW, Ernster L. 1979. Activation of microsomal glutathione S-transferase activity by sulfhydryl reagents. Biochem Biophys Res Commun. 87:657–663.
- Morgenstern R, DePierre JW, Jörnvall H. 1985. Microsomal glutathione transferase. Primary structure. J Biol Chem. 260:13976–13983.
- Morgenstern R, Dock L. 1982. A comparison of induction of microsomal glutathione S-transferase activity in the liver of the mouse and rat by dietary 2(3)-tert-butyl-4-hydroxyanisole (BHA). Acta Chem Scand. 36:255–256.
- Morgenstern R, Guthenberg C, DePierre JW. 1982. Microsomal glutathione S-transferase. Purification, initial characterization and demonstration that it is not identical to the cytosolic glutathione S-transferases A, B and C. Eur J Biochem. 128:243–248.
- Morgenstern R, Lundquist G, Jörnvall H, DePierre JW. 1989. Activation of rat liver microsomal glutathione transferase by limited proteolysis. Biochem J. 260:577–582.
- Morgenstern R, Lundqvist G, Andersson G, Balk L, DePierre JW. 1984. The distribution of microsomal glutathione transferase among different organelles, different organs, and different organisms. Biochem Pharmacol. 33:3609–3614.
- Morgenstern R, Meijer J, Depierre JW, Ernster L. 1980. Characterization of rat-liver microsomal glutathione S-transferase activity. Eur J Biochem. 104:167–174.
- Morgenstern R, Svensson R, Bernat BA, Armstrong RN. 2001. Kinetic analysis of the slow ionization of glutathione by microsomal glutathione transferase MGST1. Biochemistry. 40:3378–3384.
- Morgenstern R, Zhang J, Johansson K. 2011. Microsomal glutathione transferase 1: mechanism and functional roles. Drug Metab Rev. 43:300–306.
- Morii E, Oboki K. 2004. MITF is necessary for generation of prostaglandin D-2 in mouse mast cells. J Biol Chem. 279:48923–48929.
- Morris C, Courtay C, van Kessel AG, ten Hoeve J, Heisterkamp N, Groffen J. 1993. Localization of a gamma-glutamyl-transferase-related gene family on chromosome 22. Hum Genet. 91:31–36.
- Morrow CS, Smitherman PK, Diah SK, Schneider E, Townsend AJ. 1998. Coordinated action of glutathione S-transferases (GSTs) and multidrug resistance protein 1 (MRP1) in antineoplastic drug detoxification. Mechanism of GST A1-1- and MRP1-associated resistance to chlorambucil in MCF7 breast carcinoma cells. J Biol Chem. 273:20114–20120.
- Mortenson DE, Brighty GJ, Plate L, Bare G, Chen W, Li S, Wang H, Cravatt BF, Forli S, Powers ET, et al. 2018. Inverse Drug Discovery” strategy to identify proteins that are targeted by latent electrophiles as exemplified by aryl fluorosulfates. J Am Chem Soc. 140:200–210.
- Mosialou E, Ekstrom G, Adang AE, Morgenstern R. 1993. Evidence that rat liver microsomal glutathione transferase is responsible for glutathione-dependent protection against lipid peroxidation. Biochem Pharmacol. 45:1645–1651.
- Mosialou E, Morgenstern R. 1989. Activity of rat liver microsomal glutathione transferase toward products of lipid peroxidation and studies of the effect of inhibitors on glutathione-dependent protection against lipid peroxidation. Arch Biochem Biophys. 275:289–294.
- Mosialou E, Morgenstern R. 1990. Inhibition studies on rat liver microsomal glutathione transferase. Chem Biol Interact. 74:275–280.
- Mosialou E, Piemonte F, Andersson C, Vos RM, van Bladeren PJ, Morgenstern R. 1995. Microsomal glutathione transferase: lipid-derived substrates and lipid dependence. Arch Biochem Biophys. 320:210–216.
- Moss EJ, Neal GE, Judah DJ. 1985. The mercapturic acid pathway metabolites of a glutathione conjugate of aflatoxin B1. Chem Biol Interact. 55:139–155.
- Motohashi H, O'Connor T, Katsuoka F, Engel JD, Yamamoto M. 2002. Integration and diversity of the regulatory network composed of Maf and CNC families of transcription factors. Gene. 294:1–12.
- Moulton RD, Ruterbories KJ, Bedwell DW, Mohutsky MA. 2015. In vitro characterization of the bioconversion of pomaglumetad methionil, a novel metabotropic glutamate 2/3 receptor agonist peptide prodrug. Drug Metab Dispos. 43:756–761.
- Moyer AM, Salavaggione OE, Hebbring SJ, Moon I, Hildebrandt MA, Eckloff BW, Schaid DJ, Wieben ED, Weinshilboum RM. 2007. Glutathione S-transferase T1 and M1: gene sequence variation and functional genomics. Clin Cancer Res. 13:7207–7216.
- Mucha A. 2012. Synthesis and modifications of phosphinic dipeptide analogues. Molecules. 17:13530–13568.
- Mucha A, Drag M, Dalton JP, Kafarski P. 2010. Metallo-aminopeptidase inhibitors. Biochimie. 92:1509–1529.
- Mucha A, Kafarski P, Berlicki Ł. 2011. Remarkable potential of the α-aminophosphonate/phosphinate structural motif in medicinal chemistry. J Med Chem. 54:5955–5980.
- Mukanganyama S, Bezabih M, Robert M, Ngadjui BT, Kapche GF, Ngandeu F, Abegaz B. 2011. The evaluation of novel natural products as inhibitors of human glutathione transferase P1-1. J Enzyme Inhib Med Chem. 26:460–467.
- Mukherjee B, Salavaggione OE, Pelleymounter LL, Moon I, Eckloff BW, Schaid DJ, Wieben ED, Weinshilboum RM. 2006. Glutathione S-transferase omega 1 and omega 2 pharmacogenomics. Drug Metab Dispos. 34:1237–1246.
- Mulder GJ, Ouwerkerk-Mahadevan S. 1997. Modulation of glutathione conjugation in vivo: how to decrease glutathione conjugation in vivo or in intact cellular systems in vitro. Chem Biol Interact. 105:17–34.
- Musdal Y, Hegazy UM, Aksoy Y, Mannervik B. 2013. FDA-approved drugs and other compounds tested as inhibitors of human glutathione transferase P1-1. Chem Biol Interact. 205:53–62.
- Mutlib AE, Shockcor J, Espina RJ, Graciani N, Du A, Gan LS. 2000. Disposition of glutathione conjugates in rats by a novel glutamic acid pathway: characterization of unique peptide conjugates by liquid chromatography/mass spectrometry and liquid chromatography/NMR. J Pharmacol Exp Ther. 294:735–745.
- Nachlas MM, Monis B, Rosenbatt D, Seligman AM. 1960. Improvement in the histochemical localization of leucine aminopeptidase with a new substrate, L-leucyl-4-methoxy-2-naphthylamide. J Biophys Biochem Cytol. 7:261–264.
- Nafissi S, Saadat I, Farrashbandi H, Saadat M. 2011. Association between three genetic polymorphisms of glutathione S-transferase Z1 (GSTZ1) and susceptibility to schizophrenia. Psychiatry Res. 187:314–315.
- Nagaprashantha LD, Singhal J, Li H, Warden C, Liu X, Horne D, Awasthi S, Salgia R, Singhal SS. 2018. 2′-Hydroxyflavanone effectively targets RLIP76-mediated drug transport and regulates critical signaling networks in breast cancer. Oncotarget. 9:18053–18068.
- Nagata A, Suzuki Y, Igarashi M, Eguchi N, Toh H, Urade Y, Hayaishi O. 1991. Human brain prostaglandin D synthase has been evolutionarily differentiated from lipophilic-ligand carrier proteins. Proc Natl Acad Sci USA. 88:4020–4024.
- Nakajima M, Watanabe B, Han L, Shimizu B, Wada K, Fukuyama K, Suzuki H, Hiratake J. 2014. Glutathione-analogous peptidyl phosphorus esters as mechanism-based inhibitors of γ-glutamyl transpeptidase for probing cysteinyl-glycine binding site. Bioorg Med Chem. 22:1176–1194.
- Nakama S, Oshiro N, Aniya Y. 2010. Activation of rat liver microsomal glutathione transferase by hepsin. Biol Pharm Bull. 33:561–567.
- Nakanishi M, Moriyama A, Narita Y, Sasaki M. 1989. Aminopeptidase M from human liver. I. Solubilization, purification, and some properties of the enzyme. J Biochem. 106:818–825.
- Nash B, Tate SS. 1982. Biosynthesis of rat renal γ-glutamyl transpeptidase. Evidence for a common precursor of the two subunits. J Biol Chem. 257:585–588.
- Naylor SL, Marshall A, Hensel C, Martinez PF, Holley B, Sakaguchi AY. 1989. The DNF15S2 locus at 3p21 is transcribed in normal lung and small cell lung cancer. Genomics. 4:355–361.
- Naylor SL, Shows TB, Klebe RJ. 1979. Bioautographic visualization of aminoacylase-1: assignment of the structural gene ACY-1 to chromosome 3 in man. Somat Cell Mol Genet. 5:11–21.
- Ndrepepa G, Braun S, Schunkert H, Laugwitz KL, Kastrati A. 2016. Gamma-glutamyl transferase and prognosis in patients with coronary artery disease. Clin Chim Acta. 452:155–160.
- Nebert DW, Vasiliou V. 2004. Analysis of the glutathione S-transferase (GST) gene family. Hum Genomics. 1:460–464.
- Németi B, Gregus Z. 2009. Mechanism of thiol-supported arsenate reduction mediated by phosphorolytic-arsenolytic enzymes: I. The role of arsenolysis. Toxicol Sci. 110:270–281.
- Németi B, Gregus Z. 2013. Reduction of dimethylarsinic acid to the highly toxic dimethylarsinous acid by rats and rat liver cytosol. Chem Res Toxicol. 26:432–443.
- Németi B, Poor M, Gregus Z. 2015a. Reduction of the pentavalent arsenical dimethylarsinic acid and the GSTO1 substrate S-(4-nitrophenacyl)glutathione by rat liver cytosol: analyzing the role of GSTO1 in arsenic reduction. Chem Res Toxicol. 28:2199–2209.
- Németi B, Poór M, Gregus Z. 2015b. A high-performance liquid chromatography-based assay of glutathione transferase Omega 1 supported by glutathione or non-physiological reductants. Anal Biochem. 469:12–18.
- Nemoto N, Gelboin H. 1975. Assay and properties of glutathione-S-benzo(a)pyrene-4,5-oxide transferase. Arch Biochem Biophys. 170:739–742.
- Newman D, Abuladze N, Scholz K, Dekant W, Tsuprun V, Ryazantsev S, Bondar G, Sassani P, Kurtz I, Pushkin A. 2007. Specificity of aminoacylase III-mediated deacetylation of mercapturic acids. Drug Metab Dispos. 35:43–50.
- Nies AT. 2013. MRP2 (ABCC2) and MRP3 (ABCC3). In: Ishikawa T, Kim RB, König J, editors. Pharmacogenomics of human drug transporters: clinical impacts. 1st ed. Hoboken (NJ): Wiley; p. 345–364.
- Nigam SK. 2018. The SLC22 transporter family: a paradigm for the impact of drug transporters on metabolic pathways, signaling, and disease. Annu Rev Pharmacol Toxicol. 58:663–687.
- Ning B, Wang C, Morel F, Nowell S, Ratnasinghe DL, Carter W, Kadlubar FF, Coles B. 2004. Human glutathione S-transferase A2 polymorphisms: variant expression, distribution in prostate cancer cases/controls and a novel form. Pharmacogenetics. 14:35–44.
- Nishino H, Ito A. 1990a. Purification and properties of glutathione S-transferase from outer mitochondrial membrane of rat liver. Biochem Int. 20:1059–1066.
- Nishino H, Ito A. 1990b. Subcellular distribution of N-ethylmaleimide-stimulatable glutathione S-transferase activity in rat liver. Evidence of localization of glutathione S-transferase in peroxisomal membrane. Biochem Int. 21:331–340.
- Nitanai Y, Satow Y, Adachi H, Tsujimoto M. 2002. Crystal structure of human renal dipeptidase involved in β-lactam hydrolysis. J Mol Biol. 321:177–184.
- Niu MM, Wang FZ, Li F, Dong YR, Gu YQ. 2015. Establishment of a screening protocol for identification of aminopeptidase N inhibitors. J Taiwan Inst Chem Eng. 49:19–26.
- Nordwig A, Mayer H. 1973. The cleavage of prolyl peptides by kidney peptidases. Detection of a new peptidase capable of removing N-terminal proline. Hoppe Seylers Z Physiol Chem. 354:380–383.
- Norén K, Hansen GH, Clausen H, Noren O, Sjöström H, Vogel LK. 1997. Defectively N-glycosylated and non-O-glycosylated aminopeptidase N (CD13) is normally expressed at the cell surface and has full enzymatic activity. Exp Cell Res. 231:112–118.
- Nuccetelli M, Mazzetti AP, Rossjohn J, Parker MW, Board P, Caccuri AM, Federici G, Ricci G, Lo Bello M. 1998. Shifting substrate specificity of human glutathione transferase (from class Pi to class Alpha) by a single point mutation. Biochem Biophys Res Commun. 252:184–189.
- Oakley A. 2011. Glutathione transferases: a structural perspective. Drug Metab Rev. 43:138–151.
- Oakley AJ. 2005. Glutathione transferases: new functions. Curr Opin Struct Biol. 15:716–723.
- Oakley AJ, Bello ML, Battistoni A, Ricci G, Rossjohn J, Villar HO, Parker MW. 1997. The structures of human glutathione transferase P1-1 in complex with glutathione and various inhibitors at high resolution. J Mol Biol. 274:84–100.
- Oakley AJ, Lo Bello M, Nuccetelli M, Mazzetti AP, Parker MW. 1999. The ligandin (non-substrate) binding site of human Pi class glutathione transferase is located in the electrophile binding site (H-site). J Mol Biol. 291:913–926.
- O'Brien ML, Tew KD. 1996. Glutathione and related enzymes in multidrug resistance. Eur J Cancer. 32A:967–978.
- Ocain TD, Rich DH. 1987. L-lysinethiol: a subnanomolar inhibitor of aminopeptidase B. Biochem Biophys Res Commun. 145:1038–1042.
- Ocain TD, Rich DH. 1988. Synthesis of sulfur-containing analogues of bestatin. Inhibition of aminopeptidases by α-thiolbestatin analogues. J Med Chem. 31:2193–2199.
- O'Connell PJ, Gerkis V, d'Apice AJ. 1991. Variable O-glycosylation of CD13 (aminopeptidase N). J Biol Chem. 266:4593–4597.
- Odum J, Green T. 1984. The metabolism and nephrotoxicity of tetrafluoroethylene in the rat. Toxicol Appl Pharmacol. 76:306–318.
- Oe T, Ohyagi T, Naganuma A. 1998. Determination of γ-glutamylglutathione and other low-molecular-mass biological thiol compounds by isocratic high-performance liquid chromatography with fluorimetric detection. J Chromatogr B Biomed Sci Appl. 708:285–289.
- Oettgen HC, Taylor A. 1985. Purification, preliminary characterization, and immunological comparison of hog lens leucine aminopeptidase (EC 3.4.11.1) with hog kidney and beef lens aminopeptidases. Anal Biochem. 146:238–245.
- Ogura K, Nishiyama T, Okada T, Kajita J, Narihata H, Watabe T, Hiratsuka A, Watabe T. 1991. Molecular cloning and amino acid sequencing of rat liver class Theta glutathione S-transferase Yrs-Yrs inactivating reactive sulfate esters of carcinogenic arylmethanols. Biochem Biophys Res Commun. 181:1294–1300.
- Oinonen C, Rouvinen J. 2000. Structural comparison of Ntn-hydrolases. Protein Sci. 9:2329–2337.
- Okada T, Suzuki H, Wada K, Kumagai H, Fukuyama K. 2006. Crystal structures of gamma-glutamyltranspeptidase from Escherichia coli, a key enzyme in glutathione metabolism, and its reaction intermediate . Proc Natl Acad Sci USA. 103:6471–6476.
- Okajima K, Inoue M, Morino Y. 1981. Topology and some properties of the renal brush border membrane-bound peptidase(s) participating in the metabolism of S-carbamidomethyl glutathione. Biochim Biophys Acta Gen Subj. 675:379–385.
- Okajima K, Inoue M, Morino Y, Itoh K. 1984. Topological aspects of microsomal N-acetyltransferase, an enzyme responsible for the acetylation of cysteine S-conjugates of xenobiotics. Eur J Biochem. 142:281–286.
- Olsen J, Cowell GM, Kønigshøfer E, Danielsen EM, Møller J, Laustsen L, Hansen OC, Welinder KG, Engberg J, Hunziker W, et al. 1988. Complete amino acid sequence of human intestinal aminopeptidase N as deduced from cloned cDNA. FEBS Lett. 238:307–314.
- Olson CK, Binkley F. 1950. Metabolism of glutathione. III. Enzymatic hydrolysis of cysteinylglycine. J Biol Chem. 186:731–735.
- Ong LK, Clark AG. 1986. Inhibition of rat liver glutathione S-transferases by glutathione conjugates and corresponding L-cysteines and mercapturic acids. Biochem Pharmacol. 35:651–654.
- Oniki K, Kamihashi R, Tomita T, Ishioka M, Yoshimori Y, Osaki N, Tsuchimine S, Sugawara N, Kajiwara A, Morita K, et al. 2016. Glutathione S-transferase K1 genotype and overweight status in schizophrenia patients: a pilot study. Psychiatry Res. 239:190–195.
- Orlowski M. 1963. The role of gamma-glutamyl transpeptidase in the internal diseases clinic. Arch Immunol Ther Exp (Warsz). 11:1–61.
- Orlowski M, Meister A. 1965. Isolation of γ-glutamyl transpeptidase from hog kidney. J Biol Chem. 240:338–347.
- Orlowski M, Meister A. 1970. The γ-glutamyl cycle: a possible transport system for amino acids. Proc Natl Acad Sci USA. 67:1248–1255.
- Örning L, Norin E, Gustafsson B, Hammarström S. 1986. In vivo metabolism of leukotriene C4 in germ-free and conventional rats. Fecal excretion of N-acetylleukotriene E4. J Biol Chem. 261:766–771.
- Othman H, Saadat I, Farvardin-Jahromi M, Saadat M. 2012. Susceptibility to exudative age-related macular degeneration and three genetic polymorphisms of glutathione S-transferase Z1 (GSTZ1). Eur J Ophthalmol. 22:431–435.
- Otieno MA, Baggs RB, Hayes JD, Anders MW. 1997. Immunolocalization of microsomal glutathione S-transferase in rat tissues. Drug Metab Dispos. 25:12–20.
- Ötvös L, Moravcsik E, Mády G. 1971. Investigation on the mechanism of acylase-I-catalyzed acylamino acid hydrolysis. Biochem Biophys Res Commun. 44:1056–1064.
- Ouwerkerk-Mahadevan S, Mulder GJ. 1998. Inhibition of glutathione conjugation in the rat in vivo by analogues of glutathione conjugates. Chem Biol Interact. 111–112:163–176.
- Ouwerkerk-Mahadevan S, van Boom JH, Dreef-Tromp MC, Ploemen JH, Meyer DJ, Mulder GJ. 1995. Glutathione analogues as novel inhibitors of rat and human glutathione S-transferase isoenzymes, as well as of glutathione conjugation in isolated rat hepatocytes and in the rat in vivo. Biochem J. 308:283–290.
- Ouwerkerk-Mahadevan S, van Boom JH, Mulder GJ. 1996. Isoenzyme-selective inhibition of glutathione conjugation in vivo: selective inhibition of the conjugation of S-2-bromoisovalerylurea in the rat. J Pharmacol Exp Ther. 276:923–928.
- Ou-Yang J, Li YF, Wu P, Jiang WL, Liu HW, Li CY. 2018. Detecting and imaging of γ-glutamytranspeptidase activity in serum, live cells, and pathological tissues with a high signal-stability probe by releasing a precipitating fluorochrome. ACS Sens. 3:1354–1361.
- Ozaki K, Fujiwara T, Nakamura Y, Takahashi E. 1998. Isolation and mapping of a novel human kidney- and liver-specific gene homologous to the bacterial acetyltransferases. J Hum Genet. 43:255–258.
- Pabst MJ, Habig WH, Jakoby WB. 1973. Mercapturic acid formation: the several glutathione transferases of rat liver. Biochem Biophys Res Commun. 52:1123–1128.
- Pabst MJ, Habig WH, Jakoby WB. 1974. Glutathione S-transferase A. A novel kinetic mechanism in which the major reaction pathway depends on substrate concentration. J Biol Chem. 249:7140–7147.
- Pace NJ, Pimental DR, Weerapana E. 2012. An inhibitor of glutathione S-transferase omega 1 that selectively targets apoptotic cells. Angew Chem Int Ed Engl. 51:8365–8368.
- Paiva L, Marcos R, Creus A, Coggan M, Oakley AJ, Board PG. 2008. Polymorphism of glutathione transferase omega 1 in a population exposed to a high environmental arsenic burden. Pharmacogenet Genomics. 18:1–10.
- Palm GJ, Röhm KH. 1995. Aminoacylase I from porcine kidney: identification and characterization of two major protein domains. J Protein Chem. 14:233–240.
- Pandya U, Srivastava SK, Singhal SS, Pal A, Awasthi S, Zimniak P, Awasthi YC, Singh SV. 2000. Activity of allelic variants of Pi class human glutathione S-transferase toward chlorambucil. Biochem Biophys Res Commun. 278:258–262.
- Paolicchi A, Emdin M, Passino C, Lorenzini E, Titta F, Marchi S, Malvaldi G, Pompella A. 2006. Beta-lipoprotein- and LDL-associated serum gamma-glutamyltransferase in patients with coronary atherosclerosis. Atherosclerosis. 186:80–85.
- Paolicchi A, Pezzini A, Saviozzi M, Piaggi S, Andreuccetti M, Chieli E, Malvaldi G, Casini AF. 1996. Localization of a GSH-dependent dehydroascorbate reductase in rat tissues and subcellular fractions. Arch Biochem Biophys. 333:489–495.
- Park JB, Levine M. 1996. Purification, cloning and expression of dehydroascorbic acid-reducing activity from human neutrophils: identification as glutaredoxin. Biochem J. 315:931–938.
- Park SW, Choi K, Kim IC, Lee HH, Hooper NM, Park HS. 2001. Endogenous glycosylphosphatidylinositol-specific phospholipase C releases renal dipeptidase from kidney proximal tubules in vitro. Biochem J. 353:339–344.
- Park SW, Choi K, Lee HB, Park SK, Turner AJ, Hooper NM, Park HS. 2002. Glycosyl-phosphatidylinositol (GPI)-anchored renal dipeptidase is released by a phospholipase C in vivo. Kidney Blood Press Res. 25:7–12.
- Park SY, Kim JK, Kim IJ, Choi BK, Jung KY, Lee S, Park KJ, Chairoungdua A, Kanai Y, Endou H, et al. 2005. Reabsorption of neutral amino acids mediated by amino acid transporter LAT2 and TAT1 in the basolateral membrane of proximal tubule. Arch Pharm Res. 28:421–432.
- Parke DV, Williams RT. 1951. Metabolism of benzene. Excretion of phenylsulfuric acid after administration of phenylmercapturic acid and phenylcysteine. Biochem J. 48:27–28.
- Parker LJ, Bocedi A, Ascher DB, Aitken JB, Harris HH, Lo Bello M, Ricci G, Morton CJ, Parker MW. 2017. Glutathione transferase P1-1 as an arsenic drug-sequestering enzyme. Protein Sci. 26:317–326.
- Parker LJ, Ciccone S, Italiano LC, Primavera A, Oakley AJ, Morton CJ, Hancock NC, Bello ML, Parker MW. 2008. The anti-cancer drug chlorambucil as a substrate for the human polymorphic enzyme glutathione transferase P1-1: kinetic properties and crystallographic characterisation of allelic variants. J Mol Biol. 380:131–144.
- Parker LJ, Italiano LC, Morton CJ, Hancock NC, Ascher DB, Aitken JB, Harris HH, Campomanes P, Rothlisberger U, De Luca A, et al. 2011. Studies of glutathione transferase P1-1 bound to a platinum(IV)-based anticancer compound reveal the molecular basis of its activation. Chem Eur J. 17:7806–7816.
- Parraga A, Garcia-Saez I, Walsh SB, Mantle TJ, Coll M. 1998. The three-dimensional structure of a class-Pi glutathione S-transferase complexed with glutathione: the active-site hydration provides insights into the reaction mechanism. Biochem J. 333:811–816.
- Parsons WH, Hajdu R, Schoen WR, Combs PL, Sundelof J, Patchett AA. 1991. A new class of potent, slowly reversible dehydropeptidase inhibitors. Biochem Int. 23:1107–1115.
- Pasqualini R, Koivunen E, Kain R, Lahdenranta J, Sakamoto M, Stryhn A, Ashmun RA, Shapiro LH, Arap W, Ruoslahti E. 2000. Aminopeptidase N Is a receptor for tumor-homing peptides and a target for inhibiting angiogenesis. Cancer Res. 60:722–727.
- Patskovsky Y, Patskovska L, Almo SC, Listowsky I. 2006. Transition state model and mechanism of nucleophilic aromatic substitution reactions catalyzed by human glutathione S-transferase M1a-1a. Biochemistry. 45:3852–3862.
- Patterson EK, Hsiao SH, Keppel A. 1963. Studies on dipeptidases and aminopeptidases. I. Distinction between leucine aminopeptidase and enzymes that hydrolyze L-leucyl-beta-naphthylamide . J Biol Chem. 238:3611–3620.
- Paul TJ, Barman A, Ozbil M, Bora RP, Zhang T, Sharma G, Hoffmann Z, Prabhakar R. 2016. Mechanisms of peptide hydrolysis by aspartyl and metalloproteases. Phys Chem Chem Phys. 18:24790–24801.
- Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Magnuson B, Wilson TE, Ljungman M. 2014. Use of Bru-Seq and BruChase-Seq for genome-wide assessment of the synthesis and stability of RNA. Methods. 67:45–54.
- Paumi CM, Ledford BG, Smitherman PK, Townsend AJ, Morrow CS. 2001. Role of multidrug resistance protein 1 (MRP1) and glutathione S-transferase A1-1 in alkylating agent resistance. Kinetics of glutathione conjugate formation and efflux govern differential cellular sensitivity to chlorambucil versus melphalan toxicity. J Biol Chem. 276:7952–7956.
- Păunescu E, Soudani M, Martin P, Scopelliti R, Lo Bello M, Dyson PJ. 2017. Organometallic glutathione S-transferase inhibitors. Organometallics. 36:3313–3321.
- Pearson WR. 2005. Phylogenies of glutathione transferase families. Meth Enzymol. 401:186–204.
- Pemble S, Schroeder KR, Spencer SR, Meyer DJ, Hallier E, Bolt HM, Ketterer B, Taylor JB. 1994. Human glutathione S-transferase Theta (GSTT1): cDNA cloning and the characterization of a genetic polymorphism. Biochem J. 300:271–276.
- Pemble SE, Taylor JB. 1992. An evolutionary perspective on glutathione transferases inferred from class-Theta glutathione transferase cDNA sequences. Biochem J. 287:957–963.
- Pemble SE, Taylor JB, Ketterer B. 1986. Tissue distribution of rat glutathione transferase subunit 7, a hepatoma marker. Biochem J. 240:885–889.
- Pemble SE, Wardle AF, Taylor JB. 1996. Glutathione S-transferase class Kappa: characterization by the cloning of rat mitochondrial GST and identification of a human homologue. Biochem J. 319:749–754.
- Peng G, McEwen AG, Olieric V, Schmitt C, Albrecht S, Cavarelli J, Tarnus C. 2017. Insight into the remarkable affinity and selectivity of the aminobenzosuberone scaffold for the M1 aminopeptidases family based on structure analysis. Proteins. 85:1413–1421.
- Perbellini L, Veronese N, Princivalle A. 2002. Mercapturic acids in the biological monitoring of occupational exposure to chemicals. J Chromatogr B: Anal Technol Biomed Life Sci. 781:269–290.
- Perperopoulou FD, Tsoungas PG, Thireou TN, Rinotas VE, Douni EK, Eliopoulos EE, Labrou NE, Clonis YD. 2014. 2,2′-Dihydroxybenzophenones and their carbonyl N-analogues as inhibitor scaffolds for MDR-involved human glutathione transferase isoenzyme A1-1. Bioorg Med Chem. 22:3957–3970.
- Perrier J, Durand A, Giardina T, Puigserver A. 2004. The rat kidney acylase 1. Evidence for a new cDNA form and comparisons with the porcine intestinal enzyme. Comp Biochem Physiol B Biochem Mol Biol. 138:277–283.
- Perrier J, Durand A, Giardina T, Puigserver A. 2005. Catabolism of intracellular N-terminal acetylated proteins: involvement of acylpeptide hydrolase and acylase. Biochimie. 87:673–685.
- Peter H, Deutschmann S, Reichel C, Hallier E. 1989. Metabolism of methyl chloride by human erythrocytes. Arch Toxicol. 63:351–355.
- Petermann A, Miene C, Schulz-Raffelt G, Palige K, Hölzer J, Glei M, Böhmer FD. 2009. GSTT2, a phase II gene induced by apple polyphenols, protects colon epithelial cells against genotoxic damage. Mol Nutr Food Res. 53:1245–1253.
- Peters WH, Nagengast FM, Wobbes T. 1989. Glutathione S-transferases in normal and cancerous human colon tissue. Carcinogenesis. 10:2371–2374.
- Peters-Golden M, Henderson WR. 2007. Leukotrienes. N Engl J Med. 357:1841–1854.
- Pettersson PL, Johansson AS, Mannervik B. 2002. Transmutation of human glutathione transferase A2-2 with peroxidase activity into an efficient steroid isomerase. J Biol Chem. 277:30019–30022.
- Pettersson PL, Thorén S, Jakobsson PJ. 2005. Human microsomal prostaglandin E synthase 1: a member of the MAPEG protein superfamily. Meth Enzymol. 401:147–161.
- Pezzola S, Antonini G, Geroni C, Beria I, Colombo M, Broggini M, Mongelli N, Leboffe L, MacArthur R, Mozzi AF. 2010. Role of glutathione transferases in the mechanism of brostallicin activation. Biochemistry. 49:226–235.
- Pfleiderer G. 1970. Particle-bound aminopeptidase from pig kidney. Methods Enzymol. 19:514–521.
- Pfleiderer G, Celliers PG. 1963. Isolierung einer aminopeptidase aus nierenpartikeln. Biochem Z. 339:186–189.
- Piacentini S, Polimanti R, De Angelis F, Iorio A, Fuciarelli M. 2013. Phenotype versus genotype methods for copy number variant analysis of glutathione S-transferases M1. Ann Hum Genet. 77:409–415.
- Piacentini S, Polimanti R, Squitti R, Mariani S, Migliore S, Vernieri F, Rossini PM, Manfellotto D, Fuciarelli M. 2012. GSTO1*E155del polymorphism associated with increased risk for late-onset Alzheimer’s disease: association hypothesis for an uncommon genetic variant. Neurosci Lett. 506:203–207.
- Pica A, Chi MC, Chen YY, d'Ischia M, Lin LL, Merlino A. 2016. The maturation mechanism of γ-glutamyl transpeptidases: insights from the crystal structure of a precursor mimic of the enzyme from Bacillus licheniformis and from site-directed mutagenesis studies. Biochim Biophys Acta. 1864:195–203.
- Pinto A, Tamborini L, Cullia G, Conti P, De Micheli C. 2016. Inspired by nature: the 3-halo-4,5-dihydroisoxazole moiety as a novel molecular warhead for the design of covalent inhibitors. ChemMedChem. 11:10–14.
- Pitot HC, Dragan YP, Teeguarden J, Hsia S, Campbell H. 1996. Quantitation of multistage carcinogenesis in rat liver. Toxicol Pathol. 24:119–128.
- Pittelkow S, Lindner H, Röhm KH. 1998. Human and porcine aminoacylase I overproduced in a baculovirus expression vector system: evidence for structural and functional identity with enzymes isolated from kidney. Protein Expression Purif. 12:269–276.
- Polekhina G, Board PG, Blackburn AC, Parker MW. 2001. Crystal structure of maleylacetoacetate isomerase/glutathione transferase Zeta reveals the molecular basis for its remarkable catalytic promiscuity. Biochemistry. 40:1567–1576.
- Pombrio JM, Giangreco A, Li L, Wempe MF, Anders MW, Sweet DH, Pritchard JB, Ballatori N. 2001. Mercapturic acids (N-acetylcysteine S-conjugates) as endogenous substrates for the renal organic anion transporter-1. Mol Pharmacol. 60:1091–1099.
- Pompella A, De Tata V, Paolicchi A, Zunino F. 2006. Expression of γ-glutamyltransferase in cancer cells and its significance in drug resistance. Biochem Pharmacol. 71:231–238.
- Poon JC, Josephy PD. 2012. Hydrolysis of S-aryl-cysteinylglycine conjugates catalyzed by porcine kidney cortex membrane dipeptidase. Xenobiotica. 42:1178–1186.
- Poster DS, Bruno S, Penta J, Neil GL, McGovren JP. 1981. Acivicin. An antitumor antibiotic. Cancer Clin Trials. 4:327–330.
- Potdar PD, Andrews KL, Nettesheim P, Ostrowski LE. 1997. Expression and regulation of γ-glutamyl transpeptidase-related enzyme in tracheal cells. Am J Physiol Lung Cell Mol Physiol. 273:L1082–L1089.
- Pouliou FM, Thireou TN, Eliopoulos EE, Tsoungas PG, Labrou NE, Clonis YD. 2015. Isoenzyme- and allozyme-specific inhibitors: 2,2′-dihydroxybenzophenones and their carbonyl N-analogues that discriminate between human glutathione transferase A1-1 and P1-1 allozymes. Chem Biol Drug Des. 86:1055–1063.
- Prabhu KS, Reddy PV, Gumpricht E, Hildenbrandt GR, Scholz RW, Sordillo LM, Reddy CC. 2001. Microsomal glutathione S-transferase A1-1 with glutathione peroxidase activity from sheep liver: molecular cloning, expression and characterization. Biochem J. 360:345–354.
- Prabhu KS, Reddy PV, Jones EC, Liken AD, Reddy CC. 2004. Characterization of a class Alpha glutathione-S-transferase with glutathione peroxidase activity in human liver microsomes. Arch Biochem Biophys. 424:72–80.
- Pratt IS, Lock EA. 1988. Deacetylation and further metabolism of the mercapturic acid of hexachloro-1,3-butadiene by rat kidney cytosol in vitro. Arch Toxicol. 62:341–345.
- Preyer O, Johansen D, Holly J, Stocks T, Pompella A, Nagel G, Concin H, Ulmer H, Concin N. 2016. γ-Glutamyltransferase and breast cancer risk beyond alcohol consumption and other life style factors – a pooled cohort analysis. PLoS One. 11:e0149122.
- Priebe W, Krawczyk M, Kuo MT, Yamane Y, Savaraj N, Ishikawa T. 1998. Doxorubicin- and daunorubicin-glutathione conjugates, but not unconjugated drugs, competitively inhibit leukotriene C4 transport mediated by MRP/GS-X pump. Biochem Biophys Res Commun. 247:859–863.
- Pushkin A, Carpenito G, Abuladze N, Newman D, Tsuprun V, Ryazantsev S, Motemoturu S, Sassani P, Solovieva N, Dukkipati R, et al. 2004. Structural characterization, tissue distribution, and functional expression of murine aminoacylase III. Am J Physiol Cell Physiol. 286:C848–C856.
- Qavi H, Kit S. 1980. Electrophoretic patterns of aminoacylase-1 (ACY-1) isozymes in vertebrate cells and histochemical procedure for detecting ACY-1 activity. Biochem Genet. 18:669–679.
- Qiang L, Farmer SR. 2006. C/EBPα-dependent induction of glutathione S-transferase ζ/maleylacetoacetate isomerase (GSTζ/MAAI) expression during the differentiation of mouse fibroblasts into adipocytes. Biochem Biophys Res Commun. 340:845–851.
- Quesada-Soriano I, Primavera A, Casas-Solvas JM, Téllez-Sanz R, Barón C, Vargas-Berenguel A, Lo Bello M, Garcia-Fuentes L. 2012. Identifying and characterizing binding sites on the irreversible inhibition of human glutathione S-transferase P1-1 by S-thiocarbamoylation. ChemBioChem. 13:1594–1604.
- Rached E, Hooper NM, James P, Semenza G, Turner AJ, Mantei N. 1990. cDNA cloning and expression in Xenopus laevis oocytes of pig renal dipeptidase, a glycosyl-phosphatidylinositol-anchored ectoenzyme. Biochem J. 271:755–760.
- Raffalli-Mathieu F, Mannervik B. 2005. Human glutathione transferase A3-3 active as steroid double-bond isomerase. Methods Enzymol. 401:265–278.
- Rajagopalan S, Park JH, Patel PD, Lebovitz RM, Lieberman MW. 1990. Cloning and analysis of the rat γ-glutamyltransferase gene. J Biol Chem. 265:11721–11725.
- Rajotte D, Ruoslahti E. 1999. Membrane dipeptidase is the receptor for a lung-targeting peptide identified by in vivo phage display. J Biol Chem. 274:11593–11598.
- Ramkumar K, Samanta S, Kyani A, Yang S, Tamura S, Ziemke E, Stuckey JA, Li S, Chinnaswamy K, Otake H, et al. 2016. Mechanistic evaluation and transcriptional signature of a glutathione S-transferase Omega 1 inhibitor. Nat Commun. 7:13084.
- Ramsay EE, Dilda PJ. 2014. Glutathione S-conjugates as prodrugs to target drug-resistant tumors. Front Pharmacol. 5:181.
- Rankin BB, McIntyre TM, Curthoys NP. 1980. Brush border membrane hydrolysis of S-benzyl-cysteine-p-nitroanilide, and activity of aminopeptidase M. Biochem Biophys Res Commun. 96:991–996.
- Rao KR, Birnbaum SM, Kingsley RB, Greenstein JP. 1952. Enzymatic susceptibility of corresponding chloroacetyl- and glycyl-L-amino acids. J Biol Chem. 198:507–524.
- Rautio J, Vernerova M, Aufderhaar I, Huttunen KM. 2014. Glutathione-S-transferase selective release of metformin from its sulfonamide prodrug. Bioorg Med Chem Lett. 24:5034–5036.
- Rawlings ND, Barrett AJ, Thomas PD, Huang X, Bateman A, Finn RD. 2018. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 46:D624–D632.
- Ray A, Ravillah D, Das DS, Song Y, Nordström E, Gullbo J, Richardson PG, Chauhan D, Anderson KC. 2016. A novel alkylating agent melflufen induces irreversible DNA damage and cytotoxicity in multiple myeloma cells. Br J Haematol. 174:397–409.
- Raza H. 2011. Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease. FEBS J. 278:4243–4251.
- Raza H, Robin MA, Fang JK, Avadhani NG. 2002. Multiple isoforms of mitochondrial glutathione S-transferases and their differential induction under oxidative stress. Biochem J. 366:45–55.
- Reed DJ, Ellis WW, Meck RA. 1980. The inhibition of γ-glutamyl transpeptidase and glutathione metabolism of isolated rat kidney cells by L-(αS,5S)-α-amino-3-chloro-4, 5-dihydro-5-isoxazoleacetic acid (AT-125; NSC-163501). Biochem Biophys Res Commun. 94:1273–1277.
- Reguera J, Santiago C, Mudgal G, Ordoño D, Enjuanes L, Casasnovas JM. 2012. Structural bases of coronavirus attachment to host aminopeptidase N and its inhibition by neutralizing antibodies. PLoS Pathogens. 8:e1002859.
- Reinemer P, Dirr HW, Ladenstein R, Huber R, Lo Bello M, Federici G, Parker MW. 1992. Three-dimensional structure of class π glutathione S-transferase from human placenta in complex with S-hexylglutathione at 2.8 Å resolution. J Mol Biol. 227:214–226.
- Revel JP, Ball EG. 1959. The reaction of glutathione with amino acids and related compounds as catalyzed by γ-glutamyl transpeptidase. J Biol Chem. 234:577–582.
- Revelant G, Al-Lakkis-Wehbe M, Schmitt M, Alavi S, Schmitt C, Roux L, Al-Masri M, Schifano-Faux N, Maiereanu C, Tarnus C, et al. 2015. Exploring S1 plasticity and probing S1′ subsite of mammalian aminopeptidase N/CD13 with highly potent and selective aminobenzosuberone inhibitors. Bioorg Med Chem. 23:3192–3207.
- Ricci G, Caccuri AM, Lo Bello M, Pastore A, Piemonte F, Federici G. 1994. Colorimetric and fluorometric assays of glutathione transferase based on 7-chloro-4-nitrobenzo-2-oxa-1,3-diazole. Anal Biochem. 218:463–465.
- Ricci G, De Maria F, Antonini G, Turella P, Bullo A, Stella L, Filomeni G, Federici G, Caccuri AM. 2005. 7-Nitro-2,1,3-benzoxadiazole derivatives, a new class of suicide inhibitors for glutathione S-transferases. Mechanism of action of potential anticancer drugs. J Biol Chem. 280:26397–26405.
- Ricci G, Turella P, De Maria F, Antonini G, Nardocci L, Board PG, Parker MW, Carbonelli MG, Federici G, Caccuri AM. 2004. Binding and kinetic mechanisms of the Zeta class glutathione transferase. J Biol Chem. 279:33336–33342.
- Rich DH, Moon BJ, Harbeson S. 1984. Inhibition of aminopeptidases by amastatin and bestatin derivatives. Effect of inhibitor structure on slow-binding processes. J Med Chem. 27:417–422.
- Riemann D, Kehlen A, Langner J. 1999. CD13–not just a marker in leukemia typing. Immunol Today. 20:83–88.
- Rimpelä JM, Kontula KK, Fyhrquist F, Donner KM, Tuiskula AM, Sarin AP, Mohney RP, Stirdivant SM, Hiltunen TP. 2017. Replicated evidence for aminoacylase 3 and nephrin gene variations to predict antihypertensive drug responses. Pharmacogenomics. 18:445–458.
- Rizwan AN, Burckhardt G. 2007. Organic anion transporters of the SLC22 family: biopharmaceutical, physiological, and pathological roles. Pharm Res. 24:450–470.
- Robertson GJ, Stoychev SH, Sayed Y, Achilonu I, Dirr HW. 2017. The effects of mutating Tyr9 and Arg15 on the structure, stability, conformational dynamics and mechanism of GSTA3-3. Biophys Chem. 224:40–48.
- Robertson IG, Jernström B. 1986. The enzymatic conjugation of glutathione with bay-region diol-epoxides of benzo[a]pyrene, benz[a]anthracene and chrysene. Carcinogenesis. 7:1633–1636.
- Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM. 2018. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat Rev Cancer. 18:452–464.
- Robin MA, Prabu SK, Raza H, Anandatheerthavarada HK, Avadhani NG. 2003. Phosphorylation enhances mitochondrial targeting of GSTA4-4 through increased affinity for binding to cytoplasmic Hsp70. J Biol Chem. 278:18960–18970.
- Robinson A, Huttley GA, Booth HS, Board PG. 2004. Modelling and bioinformatics studies of the human Kappa-class glutathione transferase predict a novel third glutathione transferase family with similarity to prokaryotic 2-hydroxychromene-2-carboxylate isomerases. Biochem J. 379:541–552.
- Robinson DS, Birnbaum SM, Greenstein JP. 1953. Purification and properties of an aminopeptidase from kidney cellular particulates. J Biol Chem. 202:1–26.
- Röcken C, Carl-McGrath S, Gräntzdörffer I, Mantke R, Roessner A, Lendeckel U. 2004. Ectopeptidases are differentially expressed in hepatocellular carcinomas. Int J Oncol. 24:487–495.
- Röcken C, Licht J, Roessner A, Carl-McGrath S. 2005. Canalicular immunostaining of aminopeptidase N (CD13) as a diagnostic marker for hepatocellular carcinoma. J Clin Pathol. 58:1069–1075.
- Röhm KH. 1989. Butylmalonate is a transition state analogue for aminocylase I. FEBS Lett. 250:191–194.
- Rolland D, Raharijaona M, Barbarat A, Houlgatte R, Thieblemont C. 2010. Inhibition of GST-Pi nuclear transfer increases mantle cell lymphoma sensitivity to cisplatin, cytarabine, gemcitabine, bortezomib and doxorubicin. Anticancer Res. 30:3951–3957.
- Rose PW, Prlic A, Altunkaya A, Bi C, Bradley AR, Christie CH, Costanzo LD, Duarte JM, Dutta S, Feng Z, et al. 2017. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 45:D271–D281.
- Rossjohn J, McKinstry WJ, Oakley AJ, Verger D, Flanagan J, Chelvanayagam G, Tan KL, Board PG, Parker MW. 1998. Human Theta class glutathione transferase: the crystal structure reveals a sulfate-binding pocket within a buried active site. Structure. 6:309–322.
- Roth MB, Obaidat A, Hagenbuch B. 2012. OATPs, OATs and OCTs: the organic anion and cation transporters of the SLCO and SLC22A gene superfamilies. Br J Pharmacol. 165:1260–1287.
- Rotili D, De Luca A, Tarantino D, Pezzola S, Forgione M, Morozzo Della Rocca B, Falconi M, Mai A, Caccuri AM. 2015. Synthesis and structure-activity relationship of new cytotoxic agents targeting human glutathione-S-transferases. Eur J Med Chem. 89:156–171.
- Rouimi P, Anglade P, Benzekri A, Costet P, Debrauwer L, Pineau T, Tulliez J. 2001. Purification and characterization of a glutathione S-transferase Omega in pig: evidence for two distinct organ-specific transcripts. Biochem J. 358:257–262.
- Rouimi P, Debrauwer L, Tulliez J. 1995. Electrospray ionization-mass spectrometry as a tool for characterization of glutathione S-transferase isozymes. Anal Biochem. 229:304–312.
- Rowe JD, Nieves E, Listowsky I. 1997. Subunit diversity and tissue distribution of human glutathione S-transferases: interpretations based on electrospray ionization-MS and peptide sequence-specific antisera. Biochem J. 325:481–486.
- Rowe JD, Patskovsky YV, Patskovska LN, Novikova E, Listowsky I. 1998. Rationale for reclassification of a distinctive subdivision of mammalian class Mu glutathione S-transferases that are primarily expressed in testis. J Biol Chem. 273:9593–9601.
- Rozell B, Hansson HA, Guthenberg C, Tahir MK, Mannervik B. 1993. Glutathione transferases of classes alpha, mu and pi show selective expression in different regions of rat kidney. Xenobiotica. 23:835–849.
- Rudolf JL, Bauerly KA, Tchaparian E, Rucker RB, Mitchell AE. 2008. The influence of diet composition on phase I and II biotransformation enzyme induction. Arch Toxicol. 82:893–901.
- Rushmore TH, King RG, Paulson KE, Pickett CB. 1990. Regulation of glutathione S-transferase Ya subunit gene expression: identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic compounds. Proc Natl Acad Sci USA. 87:3826–3830.
- Rushmore TH, Pickett CB. 1990. Transcriptional regulation of the rat glutathione S-transferase Ya subunit gene. Characterization of a xenobiotic-responsive element controlling inducible expression by phenolic antioxidants. J Biol Chem. 265:14648–14653.
- Ruttmann E, Brant LJ, Concin H, Diem G, Rapp K, Ulmer H; Vorarlberg Health Monitoring Promotion Program Study Group. 2005. γ-Glutamyltransferase as a risk factor for cardiovascular disease mortality: an epidemiological investigation in a cohort of 163,944 Austrian adults. Circulation. 112:2130–2137.
- Ruzza P, Calderan A. 2013. Glutathione transferase (GST)-activated prodrugs. Pharmaceutics. 5:220–231.
- Ryazantsev S, Abuladze N, Newman D, Bondar G, Kurtz I, Pushkin A. 2007. Structural characterization of dimeric murine aminoacylase III. FEBS Lett. 581:1898–1902.
- Saadat I, Khalili M, Nafissi S, Omidvari S, Saadat M. 2012. Susceptibility to breast cancer and three polymorphisms of GSTZ1. DNA Cell Biol. 31:337–341.
- Sakabe M, Asanuma D, Kamiya M, Iwatate RJ, Hanaoka K, Terai T, Nagano T, Urano Y. 2013. Rational design of highly sensitive fluorescence probes for protease and glycosidase based on precisely controlled spirocyclization. J Am Chem Soc. 135:409–414.
- Sakamuro D, Yamazoe M, Matsuda Y, Kangawa K, Taniguchi N, Matsuo H, Yoshikawa H, Ogasawara N. 1988. The primary structure of human gamma-glutamyl transpeptidase. Gene. 73:1–9.
- Sala A, Voelkel N, Maclouf J, Murphy RC. 1990. Leukotriene E4 elimination and metabolism in normal human subjects. J Biol Chem. 265:21771–21778.
- Salinas AE, Wong MG. 1999. Glutathione S-transferases–a review. Curr Med Chem. 6:279–309.
- Samarasinghe K, Liu D, Tummala P, Cappello J, Pace SM, Arnolda L, Casarotto MG, Dulhunty AF, Board PG. 2015. Glutathione transferase M2 variants inhibit ryanodine receptor function in adult mouse cardiomyocytes. Biochem Pharmacol. 97:269–280.
- Sampayo-Reyes A, Zakharyan RA. 2006. Inhibition of human glutathione S-transferase Omega by tocopherol succinate. Biomed Pharmacother. 60:238–244.
- Santiago C, Mudgal G, Reguera J, Recacha R, Albrecht S, Enjuanes L, Casasnovas JM. 2017. Allosteric inhibition of aminopeptidase N functions related to tumor growth and virus infection. Sci Rep. 7:46045.
- Santoro A, Pressiani T, Citterio G, Rossoni G, Donadoni G, Pozzi F, Rimassa L, Personeni N, Bozzarelli S, Rossoni G, et al. 2010. Activity and safety of NGR-hTNF, a selective vascular-targeting agent, in previously treated patients with advanced hepatocellular carcinoma. Br J Cancer. 103:837–844.
- Sasagawa S, Nishimura Y, Okabe S, Murakami S, Ashikawa Y, Yuge M, Kawaguchi K, Kawase R, Okamoto R, Ito M, et al. 2016. Downregulation of GSTK1 Is a common mechanism underlying hypertrophic cardiomyopathy. Front Pharmacol. 7:162.
- Sass JO, Mohr V, Olbrich H, Engelke U, Horvath J, Fliegauf M, Loges NT, Schweitzer-Krantz S, Moebus R, Weiler P, et al. 2006. Mutations in ACY1, the gene encoding aminoacylase 1, cause a novel inborn error of metabolism. Am J Hum Genet. 78:401–409.
- Sass JO, Olbrich H, Mohr V, Hart C, Woldseth B, Krywawych S, Bjurulf B, Lakhani PK, Buchdahl RM, Omran H. 2007. Neurological findings in aminoacylase 1 deficiency. Neurology. 68:2151–2153.
- Sastre J, Sweiry JH, Doolabh K, Vina J, Mann GE. 1991. Significance of gamma-glutamyltranspeptidase in exocrine pancreatic amino acid transport. Biochim Biophys Acta, Biomembr. 1065:213–216.
- Satoh S, Keida Y, Konta Y, Maeda M, Matsumoto Y, Niwa M, Kohsaka M. 1993. Purification and molecular cloning of mouse renal dipeptidase. Biochim Biophys Acta. 1163:234–242.
- Satyam A, Hocker MD, Kane-Maguire KA, Morgan AS, Villar HO, Lyttle MH. 1996. Design, synthesis, and evaluation of latent alkylating agents activated by glutathione S-transferase. J Med Chem. 39:1736–1747.
- Sau A, Filomeni G, Pezzola S, D'Aguanno S, Tregno FP, Urbani A, Serra M, Pasello M, Picci P, Federici G, et al. 2012. Targeting GSTP1-1 induces JNK activation and leads to apoptosis in cisplatin-sensitive and -resistant human osteosarcoma cell lines. Mol BioSyst. 8:994–1006.
- Sau A, Tregno FP, Valentino F, Federici G, Caccuri AM. 2010. Glutathione transferases and development of new principles to overcome drug resistance. Arch Biochem Biophys. 500:116–122.
- Saxty G, Norton D, Affleck K, Clapham D, Cleasby A, Coyle J, Day P, Frederickson M, Hancock A, Hobbs H, et al. 2014. Identification of orally bioavailable small-molecule inhibitors of hematopoietic prostaglandin D2 synthase using X-ray fragment based drug discovery. MedChemComm. 5:134–141.
- Sayed Y, Wallace LA, Dirr HW. 2000. The hydrophobic lock-and-key intersubunit motif of glutathione transferase A1-1: implications for catalysis, ligandin function and stability. FEBS Lett. 465:169–172.
- Scaloni A, Jones W, Pospischil M, Sassa S, Schneewind O, Popowicz AM, Bossa F, Graziano SL, Manning JM. 1992. Deficiency of acylpeptide hydrolase in small-cell lung carcinoma cell lines. J Lab Clin Med. 120:546–552.
- Schaeffer VH, Stevens JL. 1987a. Mechanism of transport for toxic cysteine conjugates in rat kidney cortex membrane vesicles. Mol Pharmacol. 32:293–298.
- Schaeffer VH, Stevens JL. 1987b. The transport of S-cysteine conjugates in LLC-PK1 cells and its role in toxicity. Mol Pharmacol. 31:506–512.
- Schaffert CS. 2011. Role of MGST1 in reactive intermediate-induced injury. World J Gastroenterol. 17:2552–2557.
- Schalk C, Dorchymont H, Jauch MF, Tarnus C. 1994. 3-Amino-2-tetralone derivatives: novel potent and selective inhibitors of aminopeptidase-M (EC 3.4.11.2). Arch Biochem Biophys. 311:42–46.
- Schmidt-Krey I, Mitsuoka K, Hirai T, Murata K, Cheng Y, Fujiyoshi Y, Morgenstern R, Hebert H. 2000. The three-dimensional map of microsomal glutathione transferase 1 at 6 Å resolution. EMBO J. 19:6311–6316.
- Schmitt C, Voegelin M, Marin A, Schmitt M, Schegg F, Hénon P, Guenot D, Tarnus C. 2013. Selective aminopeptidase-N (CD13) inhibitors with relevance to cancer chemotherapy. Bioorg MedChem. 21:2135–2144.
- Schmuck E, Cappello J, Coggan M, Brew J, Cavanaugh JA, Blackburn AC, Baker RT, Eyre HJ, Sutherland GR, Board PG. 2008. Deletion of Glu155 causes a deficiency of glutathione transferase omega 1-1 but does not alter sensitivity to arsenic trioxide and other cytotoxic drugs. Int J Biochem Cell Biol. 40:2553–2559.
- Schmuck EM, Board PG, Whitbread AK, Tetlow N, Cavanaugh JA, Blackburn AC, Masoumi A. 2005. Characterization of the monomethylarsonate reductase and dehydroascorbate reductase activities of Omega class glutathione transferase variants: implications for arsenic metabolism and the age-at-onset of Alzheimer’s and Parkinson’s diseases. Pharmacogenet Genomics. 15:493–501.
- Schneider A, Tögel S, Barmada MM, Whitcomb DC. 2004. Genetic analysis of the glutathione s-transferase genes MGST1, GSTM3, GSTT1, and GSTM1 in patients with hereditary pancreatitis. J Gastroenterol. 39:783–787.
- Schröder KR, Hallier E, Meyer DJ, Wiebel FA, Muller AM, Bolt HM. 1996. Purification and characterization of a new glutathione S-transferase, class Theta, from human erythrocytes. Arch Toxicol. 70:559–566.
- Schröder KR, Hallier E, Peter H, Bolt HM. 1992. Dissociation of a new glutathione S-transferase activity in human erythrocytes. Biochem Pharmacol. 43:1671–1674.
- Schröder O, Sjöström M, Qiu H, Stein J, Jakobsson PJ, Haeggström JZ. 2003. Molecular and catalytic properties of three rat leukotriene C4 synthase homologs. Biochem Biophys Res Commun. 312:271–276.
- Schürer G, Horn AHC, Gedeck P, Clark T. 2002. The reaction mechanism of bovine lens leucine aminopeptidase. J Phys Chem B. 106:8815–8830.
- Scornik OA, Botbol V. 2001. Bestatin as an experimental tool in mammals. Curr Drug Metab. 2:67–85.
- Seltzer S. 1989a. Maleylacetoacetate cis-trans isomerase. Coenzymes Cofactors. 3:733–751.
- Seltzer S. 1989b. Maleylacetoacetate cis-trans isomerase. In: Dolphin D, Avramovic O, Poulson R, editors. Glutathione: chemical, biochemical and medical aspects, Part A. New York: Wiley; p. 733–751.
- Seltzer S, Lin M. 1979. Maleylacetone cis-trans-isomerase. Mechanism of the interaction of coenzyme glutathione and substrate maleylacetone in the presence and absence of enzyme. J Am Chem Soc. 101:3091–3097.
- Semenza G. 1957. Chromatographic purification of cysteinyl-glycinase. Biochim Biophys Acta. 24:401–413.
- Serizawa M, Kusuhara M, Zangiacomi V, Urakami K, Watanabe M, Takahashi T, Yamaguchi K, Yamamoto N, Koh Y. 2014. Identification of metabolic signatures associated with erlotinib resistance of non-small cell lung cancer cells. Anticancer Res. 34:2779–2787.
- Shami PJ, Saavedra JE, Wang LY, Bonifant CL, Diwan BA, Singh SV, Gu Y, Fox SD, Buzard GS, Citro ML, et al. 2003. JS-K, a glutathione/glutathione S-transferase-activated nitric oxide donor of the diazeniumdiolate class with potent antineoplastic activity. Mol Cancer Ther. 2:409–417.
- Sharma R, Ansari GAS, Awasthi YC. 2007. Physiological substrates of glutathione S-transferases. In: Aswathi YC, editor. Toxicology of glutathione transferases. Boca Raton (FL): Taylor & Francis.
- Sheehan D, Meade G, Foley VM, Dowd CA. 2001. Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem J. 360:1–16.
- Sheehan M, Haythorn P. 1979. Predictive values of various liver function tests with respect to the diagnosis of liver disease. Clin Biochem. 12:262–263.
- Shenvi AB. 1986. α-Aminoboronic acid derivatives: effective inhibitors of aminopeptidases. Biochemistry. 25:1286–1291.
- Sherratt PJ, Hayes JD. 2002. Glutathione S-transferases. In: Ioannides C, editor. Enzyme systems that metabolise drugs and other xenobiotics. New York: Wiley; p. 319–352.
- Sherratt PJ, Manson MM, Thomson AM, Hissink EA, Neal GE, van Bladeren PJ, Green T, Hayes JD. 1998. Increased bioactivation of dihaloalkanes in rat liver due to induction of class Theta glutathione S-transferase T1-1. Biochem J. 335:619–630.
- Sherratt PJ, Pulford DJ, Harrison DJ, Green T, Hayes JD. 1997. Evidence that human class Theta glutathione S-transferase T1-1 can catalyse the activation of dichloromethane, a liver and lung carcinogen in the mouse. Comparison of the tissue distribution of GST T1-1 with that of classes Alpha, Mu and Pi GST in human. Biochem J. 326:837–846.
- Sherratt PJ, Williams S, Foster J, Kernohan N, Green T, Hayes JD. 2002. Direct comparison of the nature of mouse and human GST T1-1 and the implications on dichloromethane carcinogenicity. Toxicol Appl Pharmacol. 179:89–97.
- Shi H, Hayes MT, Kirana C, Miller RJ, Keating JP, Stubbs RS. 2013. Overexpression of aminoacylase 1 is associated with colorectal cancer progression. Hum Pathol. 44:1089–1097.
- Shi ZZ, Habib GM, Lebovitz RM, Lieberman MW. 1995. Cloning of cDNA and genomic structure of the mouse γ-glutamyl transpeptidase-encoding gene. Gene. 167:233–237.
- Shi ZZ, Han B, Habib GM, Matzuk MM, Lieberman MW. 2001. Disruption of γ-glutamyl leukotrienase results in disruption of leukotriene D4 synthesis in vivo and attenuation of the acute inflammatory response. Mol Cell Biol. 21:5389–5395.
- Shibata A, Nakano Y, Ito M, Araki M, Zhang J, Yoshida Y, Shuto S, Mannervik B, Mogenstern R, Ito Y, et al. 2013. Fluorogenic probes using 4-substituted-2-nitrobenzenesulfonyl derivatives as caging groups for the analysis of human glutathione transferase catalyzed reactions. Analyst. 138:7326–7330.
- Shibata M, Koike M, Kusumi S, Sato N, Uchiyama Y. 2016. A specific tripeptidyl substrate for tripeptidyl peptidase activity is effectively hydrolyzed by alanyl aminopeptidase/aminopeptidase N/CD13 in the rat kidney. Arch Histol Cytol. 76:1–8.
- Shield AJ, Murray TP, Cappello JY, Coggan M, Board PG. 2010. Polymorphisms in the human glutathione transferase Kappa (GSTK1) promoter alter gene expression. Genomics. 95:299–305.
- Shim JS, Kim JH, Cho HY, Yum YN, Kim SH, Park HJ, Shim BS, Choi SH, Kwon HJ. 2003. Irreversible inhibition of CD13/aminopeptidase N by the antiangiogenic agent curcumin. Chem Biol. 10:695–704.
- Shimazawa R, Takayama H, Fujimoto Y, Komoda M, Dodo K, Yamasaki R, Shirai R, Koiso Y, Miyata K, Kato F, et al. 1999. Novel small molecule nonpeptide aminopeptidase N inhibitors with a cyclic imide skeleton. J Enzyme Inhib. 14:259–275.
- Shimoji M, Aniya A, Morgenstern R. 2007. Activation of microsomal glutathione transferase 1 in toxicology. In: Aswathi YC, editor. Toxicology of glutathione transferases. Boca Raton (FL): Taylor & Francis; p. 293–319.
- Shimoyama M, De Pons J, Hayman GT, Laulederkind SJ, Liu W, Nigam R, Petri V, Smith JR, Tutaj M, Wang SJ, et al. 2015. The Rat Genome Database 2015: genomic, phenotypic and environmental variations and disease. Nucleic Acids Res. 43:D743–D750.
- Shimura C, Satoh T, Igawa K, Aritake K, Urade Y, Nakamura M, Yokozeki H. 2010. Dendritic cells express hematopoietic prostaglandin D synthase and function as a source of prostaglandin D2 in the skin. Am J Pathol. 176:227–237.
- Shishido Y, Tomoike F, Kimura Y, Kuwata K, Yano T, Fukui K, Fujikawa H, Sekido Y, Murakami-Tonami Y, Kameda T, et al. 2017. A covalent G-site inhibitor for glutathione S-transferase Pi (GSTP1-1). Chem Commun (Camb). 53:11138–11141.
- Shokeer A, Larsson AK, Mannervik B. 2005. Residue 234 in glutathione transferase T1-1 plays a pivotal role in the catalytic activity and the selectivity against alternative substrates. Biochem J. 388:387–392.
- Shokeer A, Mannervik B. 2010a. Minor modifications of the C-terminal helix reschedule the favored chemical reactions catalyzed by Theta class glutathione transferase T1-1. J Biol Chem. 285:5639–5645.
- Shokeer A, Mannervik B. 2010b. Residue 234 is a master switch of the alternative-substrate activity profile of human and rodent Theta class glutathione transferase T1-1. Biochim Biophys Acta. 1800:466–473.
- Shroads AL, Coats BS, McDonough CW, Langaee T, Stacpoole PW. 2015. Haplotype variations in glutathione transferase Zeta 1 influence the kinetics and dynamics of chronic dichloroacetate in children. J Clin Pharmacol. 55:50–55.
- Shroads AL, Langaee T, Coats BS, Kurtz TL, Bullock JR, Weithorn D, Gong Y, Wagner DA, Ostrov DA, Johnson JA, et al. 2012. Human polymorphisms in the glutathione transferase Zeta 1/maleylacetoacetate isomerase gene influence the toxicokinetics of dichloroacetate. J Clin Pharmacol. 52:837–849.
- Shyam K, Penketh PG, Shapiro M, Belcourt MF, Loomis RH, Rockwell S, Sartorelli AC. 1999. Hypoxia-selective nitrobenzyloxycarbonyl derivatives of 1,2-bis(methylsulfonyl)-1-(2-chloroethyl)hydrazines. J Med Chem. 42:941–946.
- Sian J, Dexter DT, Lees AJ, Daniel S, Jenner P, Marsden CD. 1994. Glutathione-related enzymes in brain in Parkinson’s disease. Ann Neurol. 36:356–361.
- Silbernagl S, Gekle M. 2008. Chapter 72, Amino acids, oligopeptides, and hyperaminoacidurias. In: Alpern RJ, Hebert SC, editors. Seldin and Giebisch’s The Kidney. 4th ed. San Diego (CA): Academic Press; p. 2021–2044.
- Simmons-Willis TA, Koh AS, Clarkson TW, Ballatori N. 2002. Transport of a neurotoxicant by molecular mimicry: the methylmercury-L-cysteine complex is a substrate for human L-type large neutral amino acid transporter (LAT) 1 and LAT2. Biochem J. 367:239–246.
- Simons PC, Vander Jagt DL. 1977. Purification of glutathione S-transferases from human liver by glutathione-affinity chromatography. Anal Biochem. 82:334–341.
- Singh S. 2015. Cytoprotective and regulatory functions of glutathione S-transferases in cancer cell proliferation and cell death. Cancer Chemother Pharmacol. 75:1–15.
- Singh SP, Zimniak L, Zimniak P. 2010. The human hGSTA5 gene encodes an enzymatically active protein. Biochim Biophys Acta, Gen Subj. 1800:16–22.
- Singh SV, Varma V, Zimniak P, Srivastava SK, Marynowski SW, Desai D, Amin S, Ji XH. 2004. Structural basis for catalytic differences between Alpha class human glutathione transferases hGSTA1-1 and hGSTA2-2 for glutathione conjugation of environmental carcinogen benzo[a]pyrene-7,8-diol-9,10-epoxide. Biochemistry. 43:9708–9715.
- Singhal SS, Jain D, Singhal P, Awasthi S, Singhal J, Horne D. 2017. Targeting the mercapturic acid pathway and vicenin-2 for prevention of prostate cancer. Biochim Biophys Acta Rev Cancer. 1868:167–175.
- Singhal SS, Sehrawat A, Mehta A, Sahu M, Awasthi S. 2009a. Functional reconstitution of RLIP76 catalyzing ATP-dependent transport of glutathione-conjugates. Int J Oncol. 34:191–199.
- Singhal SS, Singhal J, Figarola J, Horne D, Awasthi S. 2015. RLIP76 targeted therapy for kidney cancer. Pharm Res. 32:3123–3136.
- Singhal SS, Yadav S, Roth C, Singhal J. 2009b. RLIP76: a novel glutathione-conjugate and multi-drug transporter. Biochem Pharmacol. 77:761–769.
- Sinning I, Kleywegt GJ, Cowan SW, Reinemer P, Dirr HW, Huber R, Gilliland GL, Armstrong RN, Ji X, Board PG, et al. 1993. Structure determination and refinement of human Alpha class glutathione transferase A1-1, and a comparison with the Mu and Pi class enzymes. J Mol Biol. 232:192–212.
- Siritantikorn A, Johansson K, Åhlen K, Rinaldi R, Suthiphongchai T, Wilairat P, Morgenstern R. 2007. Protection of cells from oxidative stress by microsomal glutathione transferase 1. Biochem Biophys Res Commun. 355:592–596.
- Sjöström H, Norén O, Jeppesen L, Staun M, Svensson B, Christiansen L. 1978. Purification of different amphiphilic forms of a microvillus aminopeptidase from pig small intestine using immunoadsorbent chromatography. Eur J Biochem. 88:503–511.
- Sjöström H, Norén O, Olsen J. 2000. Structure and function of aminopeptidase N. Adv Exp Med Biol. 477:25–34.
- Sjöström H, Norén O, Olsen J. 2002. Chapter 2, Structure and function of aminopeptidase N. In: Langner J, Ansorge S, editors. Cellular peptidases in immune functions and diseases. Vol. 477. Boston (MA): Springer; p. 25–34.
- Skiles GL, Smith DJ, Appleton ML, Carlson JR, Yost GS. 1991. Isolation of a mercapturate adduct produced subsequent to glutathione conjugation of bioactivated 3-methylindole. Toxicol Appl Pharmacol. 108:531–537.
- Smith EL, Bergmann M. 1944. The peptidases of intestinal mucosa. J Biol Chem. 153:627–651.
- Smith EL, Polglase WJ. 1949. The specificity of leucine aminopeptidase; optical and side chain specificity. J Biol Chem. 180:1209–1223.
- Smith EL, Slonim NB. 1948. The specificity of leucine aminopeptidase. J Biol Chem. 176:835–841.
- Smith EL, Spackman DH. 1955. Leucine aminopeptidase. V. Activation, specificity, and mechanism of action. J Biol Chem. 212:271–299.
- Smith GP, MacGregor RR, Peters TJ. 1983. Subcellular localisation of leucine aminopeptidase in human polymorphonuclear leukocytes. Biochim Biophys Acta Biomembr. 728:222–227.
- Smith TK, Ikeda Y, Fujii J, Taniguchi N, Meister A. 1995. Different sites of acivicin binding and inactivation of γ-glutamyl transpeptidases. Proc Natl Acad Sci USA. 92:2360–2364.
- Smitherman PK, Townsend AJ, Kute TE, Morrow CS. 2004. Role of multidrug resistance protein 2 (MRP2, ABCC2) in alkylating agent detoxification: MRP2 potentiates glutathione S-transferase A1-1-mediated resistance to chlorambucil cytotoxicity. J Pharmacol Exp Ther. 308:260–267.
- Smyth TP, Wall JG, Nitanai Y. 2003. A substrate variant as a high-affinity, reversible inhibitor: insight from the X-ray structure of cilastatin bound to membrane dipeptidase. Bioorg Med Chem. 11:991–998.
- Soboll S, Gründel S, Harris J, Kolb-Bachofen V, Ketterer B, Sies H. 1995. The content of glutathione and glutathione S-transferases and the glutathione peroxidase activity in rat liver nuclei determined by a non-aqueous technique of cell fractionation. Biochem J. 311:889–894.
- Soh Y, Goto S, Kitajima M, Moriyama S, Kotera K, Nakayama T, Nakajima H, Kondo T, Ishimaru T. 2005. Nuclear localisation of glutathione S-transferase Pi is an evaluation factor for drug resistance in gynaecological cancers. Clin Oncol. 17:264–270.
- Soiefer AI, Kostyniak PJ. 1983. The enzymatic defluorination of fluoroacetate in mouse liver cytosol: the separation of defluorination activity from several glutathione S-transferases of mouse liver. Arch Biochem Biophys. 225:928–935.
- Soiefer AI, Kostyniak PJ. 1984. Purification of a fluoroacetate-specific defluorinase from mouse liver cytosol. J Biol Chem. 259:10787–10792.
- Sommer A, Christensen E, Schwenger S, Seul R, Haas D, Olbrich H, Omran H, Sass JO. 2011. The molecular basis of aminoacylase 1 deficiency. Biochim Biophys Acta. 1812:685–690.
- Son J, Lee J-J, Lee J-S, Schüller A, Chang Y-T. 2010. Isozyme-specific fluorescent inhibitor of glutathione S-transferase Omega 1. ACS Chem Biol. 5:449–453.
- Spackman DH, Smith EL, Brown DM. 1955. Leucine aminopeptidase. IV. Isolation and properties of the enzyme from swine kidney. J Biol Chem. 212:255–269.
- Spahiu L, Ålander J, Ottosson-Wadlund A, Svensson R, Lehmer C, Armstrong RN, Morgenstern R. 2017. Global kinetic mechanism of microsomal glutathione transferase 1 and insights into dynamic enzyme activation. Biochemistry. 56:3089–3098.
- Spater HW, Poruchynsky MS, Quintana N, Inoue M, Novikoff AB. 1982. Immunocytochemical localization of γ-glutamyltransferase in rat kidney with protein A-horseradish peroxidase. Proc Natl Acad Sci USA. 79:3547–3550.
- Sreerama L, Hedge MW, Sladek NE. 1995. Identification of a class 3 aldehyde dehydrogenase in human saliva and increased levels of this enzyme, glutathione S-transferases, and DT-diaphorase in the saliva of subjects who continually ingest large quantities of coffee or broccoli. Clin Cancer Res. 1:1153–1163.
- Srivastava SK, Hu X, Xia H, Awasthi S, Amin S, Singh SV. 1999a. Metabolic fate of glutathione conjugate of benzo[a]pyrene-(7R,8S)-diol (9S,10R)-epoxide in human liver. Arch Biochem Biophys. 371:340–344.
- Srivastava SK, Singhal SS, Hu X, Awasthi YC, Zimniak P, Singh SV. 1999b. Differential catalytic efficiency of allelic variants of human glutathione S-transferase Pi in catalyzing the glutathione conjugation of thiotepa. Arch Biochem Biophys. 366:89–94.
- Stacpoole PW, Kurtz TL, Han Z, Langaee T. 2008. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev. 60:1478–1487.
- Stange T, Kettmann U, Holzhausen HJ. 2000. Immunoelectron microscopic demonstration of the membrane proteases aminopeptidase N/CD13 and dipeptidyl peptidase IV/CD26 in normal and neoplastic renal parenchymal tissues and cells. Eur J Histochem. 44:157–164.
- Stein RL, DeCicco C, Nelson D, Thomas B. 2001. Slow-binding inhibition of γ-glutamyl transpeptidase by γ-boroGlu. Biochemistry. 40:5804–5811.
- Stekol JA. 1938. Studies on the mercapturic acid synthesis in animals. IX. The conversion of benzyl chloride and S-benzylcysteine into benzylmercapturic acid in the organism of the dog, rabbit, and rat. J Biol Chem. 124:129–133.
- Stekol JA. 1946. Studies on the mercapturic acid synthesis in animals. XIV. On the synthesis of mercapturic acids in man. J Biol Chem. 164:651–656.
- Stene DO, Murphy RC. 1988. Metabolism of leukotriene E4 in isolated rat hepatocytes. Identification of β-oxidation products of sulfidopeptide leukotrienes. J Biol Chem. 263:2773–2778.
- Stevens JM, Hornby JA, Armstrong RN, Dirr HW. 1998. Class Sigma glutathione transferase unfolds via a dimeric and a monomeric intermediate: impact of subunit interface on conformational stability in the superfamily. Biochemistry. 37:15534–15541.
- Stieger B, Hagenbuch B. 2014. Organic anion-transporting polypeptides. In: Bevensee MO, editor. Current topics in membranes exchangers. Vol. 73. San Diego (CA): Academic Press; p. 205–232.
- Stocker P, Brunel JM, de Rezende L, Tavares -do Amaral A, Morelli X, Roche P, Vidal N, Giardina T, Perrier J. 2012. Aminoacylase 1-catalysed deacetylation of bioactives epoxides mycotoxin-derived mercapturates; 3,4-epoxyprecocenes as models of cytotoxic epoxides. Biochimie. 94:1668–1675.
- Stocker P, Yousfi M, Salmi C, Perrier J, Brunel JM, Moulin A. 2005. Maackiain 3-O-(6′-O-malonyl-beta-D-glucopyranoside) from Oudneya africana, a powerful inhibitor of porcine kidney acylase I. Biochimie. 87:507–512.
- Stoddard EG, Killinger B, Nair RN, Sadler N, Volk RF, Purvine SO, Shukla AK, Smith JN, Wright AT. 2017. Activity-based probes for isoenzyme- and site-specific functional characterization of glutathione S-transferases. J Am Chem Soc. 139:16032–16035.
- Stoddard EG, Killinger BJ, Nag SA, Martin J, Corley R, Smith JN, Wright AT. 2019. Benzo[a]pyrene induction of glutathione S-transferases: an activity-based protein profiling investigation. Chem Res Toxicol. 32:1259–1267.
- Stole E, Smith TK, Manning JM, Meister A. 1994. Interaction of γ-glutamyl transpeptidase with acivicin. J Biol Chem. 269:21435–21439.
- Strange RC, Spiteri MA, Ramachandran S, Fryer AA. 2001. Glutathione-S-transferase family of enzymes. Mutat Res. 482:21–26.
- Strasak AM, Rapp K, Brant LJ, Hilbe W, Gregory M, Oberaigner W, Ruttmann E, Concin H, Diem G, Pfeiffer KP. 2008. Association of γ-glutamyltransferase and risk of cancer incidence in men: a prospective study. Cancer Res. 68:3970–3977.
- Sträter N, Lipscomb WN. 1995a. Transition state analogue L-leucinephosphonic acid bound to bovine lens leucine aminopeptidase: X-ray structure at 1.65 Å resolution in a new crystal form. Biochemistry. 34:9200–9210.
- Sträter N, Lipscomb WN. 1995b. Two-metal ion mechanism of bovine lens leucine aminopeptidase: active site solvent structure and binding mode of L-leucinal, a gem-diolate transition state analogue, by X-ray crystallography. Biochemistry. 34:14792–14800.
- Sträter N, Lipscomb WN. 2013. Leucyl aminopeptidase (animal). In: Rawlings ND, Salvesen G, editors. Handbook of proteolytic enzymes. 3rd ed. Vol. 2. San Diego (CA): Elsevier Science; p. 1465–1470.
- Sträter N, Sun L, Kantrowitz ER, Lipscomb WN. 1999. A bicarbonate ion as a general base in the mechanism of peptide hydrolysis by dizinc leucine aminopeptidase. Proc Natl Acad Sci USA. 96:11151–11155.
- Strese S, Wickström M, Fuchs PF, Fryknäs M, Gerwins P, Dale T, Larsson R, Gullbo J. 2013. The novel alkylating prodrug melflufen (J1) inhibits angiogenesis in vitro and in vivo. Biochem Pharmacol. 86:888–895.
- Su L, Cao J, Jia Y, Zhang X, Fang H, Xu W. 2012a. Development of synthetic aminopeptidase N/CD13 inhibitors to overcome cancer metastasis and angiogenesis. ACS Med Chem Lett. 3:959–964.
- Su L, Fang H, Xu WF. 2011. Aminopeptidase N (EC 3.4.11.2) inhibitors (2006-2010): a patent review. Expert Opin Ther Pat. 21:1241–1265.
- Su L, Jia Y, Wang X, Zhang L, Fang H, Xu W. 2013. Discovery of a synthetic aminopeptidase N inhibitor LB-4b as a potential anticancer agent. Bioorg Med Chem Lett. 23:2512–2517.
- Su L, Jia Y, Zhang L, Xu Y, Fang H, Xu W. 2012b. Design, synthesis and biological evaluation of novel amino acid ureido derivatives as aminopeptidase N/CD13 inhibitors. Bioorg Med Chem. 20:3807–3815.
- Suda H, Aoyagi T, Takeuchi T, Umezawa H. 1976. Inhibition of aminopeptidase B and leucine aminopeptidase by bestatin and its stereoisomer. Arch Biochem Biophys. 177:196–200.
- Suenaga H, Nakashima K, Mikami M, Yamamoto H, James TD, Sandanayake K, Shinkai S. 1996. Screening of arylboronic acids to search for a strong inhibitor for γ-glutamyl transpeptidase (γ-GTP). Recl Trav Chim Pays Bas. 115:44–48.
- Suga T, Ohata I, Kumaoka H, Akagi M. 1967. Studies on mercapturic acids. Investigation of glutathione-conjugating enzymes by the thin-layer chromatography method. Chem Pharm Bull. 15:1059–1064.
- Suhre K, Shin SY, Petersen AK, Mohney RP, Meredith D, Wägele B, Altmaier E, Deloukas P, Erdmann J, Grundberg E. 2011. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 477:54–60.
- Sun HD, Ru YW, Zhang DJ, Yin SY, Yin L, Xie YY, Guan YF, Liu SQ. 2012. Proteomic analysis of glutathione S-transferase isoforms in mouse liver mitochondria. World J Gastroenterol. 18:3435–3442.
- Sun TH, Morgenstern R. 1997. Binding of glutathione and an inhibitor to microsomal glutathione transferase. Biochem J. 326:193–196.
- Sundberg AG, Nilsson R, Appelkvist EL, Dallner G. 1993. Immunohistochemical localization of α and π class glutathione transferases in normal human tissues. Pharmacol Toxicol. 72:321–331.
- Sundberg K, Widersten M, Seidel A, Mannervik B, Jernström B. 1997. Glutathione conjugation of bay- and fjord-region diol epoxides of polycyclic aromatic hydrocarbons by glutathione transferases M1-1 and P1-1. Chem Res Toxicol. 10:1221–1227.
- Suzuki S, Tateishi M. 1981. Purification and characterization of a rat liver enzyme catalyzing N-deacetylation of mercapturic acid conjugates. Drug Metab Dispos. 9:573–577.
- Svensson R, Ålander J, Armstrong RN, Morgenstern R. 2004. Kinetic characterization of thiolate anion formation and chemical catalysis of activated microsomal glutathione transferase 1. Biochemistry. 43:8869–8877.
- Svensson R, Grenö C, Johansson AS, Mannervik B, Morgenstern R. 2002. Synthesis and characterization of 6-chloroacetyl-2-dimethylaminonaphthalene as a fluorogenic substrate and a mechanistic probe for glutathione transferases. Anal Biochem. 311:171–178.
- Svensson R, Rinaldi R, Swedmark S, Morgenstern R. 2000. Reactivity of cysteine-49 and its influence on the activation of microsomal glutathione transferase 1: evidence for subunit interaction. Biochemistry. 39:15144–15149.
- Sweet DH. 2005. Organic anion transporter (Slc22a) family members as mediators of toxicity. Toxicol Appl Pharmacol. 204:198–215.
- Szajáni B, Kiss A, Boross L. 1980. Investigation of the active center and catalytic mechanism of porcine kidney aminoacylase: a model of the active center. Acta Biochim Biophys Acad Sci Hung. 15:29–37.
- Szewczuk A, Connell GE. 1965. The reaction of iodoacetamide with the active center of γ-glutamyl transpeptidase. Biochim Biophys Acta. 105:352–367.
- Takahashi S, Tsurumura T, Aritake K, Furubayashi N, Sato M, Yamanaka M, Hirota E, Sano S, Kobayashi T, Tanaka T. 2010. High-quality crystals of human haematopoietic prostaglandin D synthase with novel inhibitors. Acta Crystallogr F Struct Biol Cryst Commun. 66:846–850.
- Takahashi Y, Campbell EA, Hirata Y, Takayama T, Listowsky I. 1993. A basis for differentiating among the multiple human Mu-glutathione S-transferases and molecular cloning of brain GSTM5. J Biol Chem. 268:8893–8898.
- Takaya D, Inaka K, Omura A, Takenuki K, Kawanishi M, Yabuki Y, Nakagawa Y, Tsuganezawa K, Ogawa N, Watanabe C, et al. 2018. Characterization of crystal water molecules in a high-affinity inhibitor and hematopoietic prostaglandin D synthase complex by interaction energy studies. Bioorg Med Chem. 26:4726–4734.
- Tamura K, Hayashi N, George J, Toshikuni N, Arisawa T, Hiratake J, Tsuchishima M, Tsutsumi M. 2016. GGsTop, a novel and specific γ-glutamyl transpeptidase inhibitor, protects hepatic ischemia-reperfusion injury in rats. Am J Physiol Gastrointest Liver Physiol. 311:G305–G312.
- Tamura T, Oki Y, Yoshida A, Kuriyama T, Kawakami H, Inoue H, Inagaki K, Tanaka H. 2000. Noncompetitive, reversible inhibition of aminoacylase-1 by a series of L-alpha-hydroxyl and L-alpha-fluoro fatty acids: ligand specificity of aspergillus oryzae and porcine kidney enzymes. Arch Biochem Biophys. 379:261–266.
- Tamura Y, Niinobe M, Arima T, Okuda H, Fujii S. 1973. Studies on aminopeptidases in rat liver and plasma. Biochim Biophys Acta. 327:437–445.
- Tamura Y, Niinobe M, Arima T, Okuda H, Fujii S. 1975. Aminopeptidases and arylamidases in normal and cancer tissues in humans. Cancer Res. 35:1030–1034.
- Tan KL, Board PG. 1996. Purification and characterization of a recombinant human Theta-class glutathione transferase (GSTT2-2). Biochem J. 315:727–732.
- Tan KL, Chelvanayagam G, Parker MW, Board PG. 1996. Mutagenesis of the active site of the human Theta-class glutathione transferase GSTT2-2: catalysis with different substrates involves different residues. Biochem J. 319:315–321.
- Tan KL, Webb GC, Baker RT, Board PG. 1995. Molecular cloning of a cDNA and chromosomal localization of a human Theta-class glutathione S-transferase gene (GSTT2) to chromosome 22. Genomics. 25:381–387.
- Tanaka-Kagawa T, Jinno H, Hasegawa T, Makino Y, Seko Y, Hanioka N, Ando M. 2003. Functional characterization of two variant human GSTO 1-1s (Ala140Asp and Thr217Asn). Biochem Biophys Res Commun. 301:516–520.
- Tang ZY, Yu JY, Zhou Q, He B, Wang ZF, Zhou HM. 1995. Secondary structure of holo- and apo-aminoacylase from prediction, circular dichroism, and FT-Raman spectroscopy. J Biochem. 118:706–709.
- Taniguchi N, Ikeda Y. 1998. Gamma-glutamyl transpeptidase: catalytic mechanism and gene expression. Adv Enzymol Relat Areas Mol Biol. 72:239–278.
- Targher G, Kendrick J, Smits G, Chonchol M. 2010. Relationship between serum gamma-glutamyltransferase and chronic kidney disease in the United States adult population. Findings from the National Health and Nutrition Examination Survey 2001–2006. Nutr Metab Cardiovasc Dis. 20:583–590.
- Tars K, Larsson AK, Shokeer A, Olin B, Mannervik B, Kleywegt GJ. 2006. Structural basis of the suppressed catalytic activity of wild-type human glutathione transferase T1-1 compared to its W234R mutant. J Mol Biol. 355:96–105.
- Tars K, Olin B, Mannervik B. 2010. Structural basis for featuring of steroid isomerase activity in Alpha class glutathione transferases. J Mol Biol. 397:332–340.
- Tate SS. 1980. Enzymes of mercapturic acid formation. In: Jakoby WB, editor. Enzymatic basis of detoxication. Vol. II. New York: Academic Press; p. 95–120.
- Tate SS. 1985. Microvillus membrane peptidases that catalyze hydrolysis of cysteinylglycine and its derivatives. Meth Enzymol. 113:471–484.
- Tate SS, Meister A. 1974a. Interaction of γ-glutamyl transpeptidase with amino acids, dipeptides, and derivatives and analogs of glutathione. J Biol Chem. 249:7593–7602.
- Tate SS, Meister A. 1974b. Stimulation of the hydrolytic activity and decrease of the transpeptidase activity of γ-glutamyl transpeptidase by maleate; identity of a rat kidney maleate-stimulated glutaminase and γ-glutamyl transpeptidase. Proc Natl Acad Sci USA. 71:3329–3333.
- Tate SS, Meister A. 1976. Subunit structure and isozymic forms of γ-glutamyl transpeptidase. Proc Natl Acad Sci USA. 73:2599–2603.
- Tate SS, Meister A. 1977. Affinity labeling of γ-glutamyl transpeptidase and location of the γ-glutamyl binding site on the light subunit. Proc Natl Acad Sci USA. 74:931–935.
- Tate SS, Meister A. 1978. Serine-borate complex as a transition-state inhibitor of γ-glutamyl transpeptidase. Proc Natl Acad Sci USA. 75:4806–4809.
- Tate SS, Meister A. 1981. Gamma-glutamyl transpeptidase: catalytic, structural and functional aspects. Mol Cell Biochem. 39:357–368.
- Tate SS, Meister A. 1985. Gamma-glutamyl transpeptidase from kidney. Meth Enzymol. 113:400–419.
- Tate SS, Nash B. 1987. Membrane translocation and insertion of NH2-terminally anchored γ-glutamyl transpeptidase require a signal recognition particle. FEBS Lett. 211:133–136.
- Tate SS, Ross ME. 1977. Human kidney γ-glutamyl transpeptidase. Catalytic properties, subunit structure, and localization of the γ-glutamyl binding site on the light subunit. J Biol Chem. 252:6042–6045.
- Taylor A. 1993a. Aminopeptidases: structure and function. FASEB J. 7:290–298.
- Taylor A. 1993b. Aminopeptidases: towards a mechanism of action. Trends Biochem Sci. 18:167–171.
- Taylor A, Peltier CZ, Jahngen EG Jr, Laxman E, Szewczuk Z, Torre FJ. 1992. Use of azidobestatin as a photoaffinity label to identify the active site peptide of leucine aminopeptidase. Biochemistry. 31:4141–4150.
- Taylor A, Peltier CZ, Torre FJ, Hakamian N. 1993. Inhibition of bovine lens leucine aminopeptidase by bestatin: number of binding sites and slow binding of this inhibitor. Biochemistry. 32:784–790.
- Taylor A, Surgenor T, Thomson DK, Graham RJ, Oettgen H. 1984a. Comparison of leucine aminopeptidase from human lens, beef lens and kidney, and hog lens and kidney. Exp Eye Res. 38:217–229.
- Taylor A, Volz KW, Lipscomb WN, Takemoto LJ. 1984b. Leucine aminopeptidase from bovine lens and hog kidney. Comparison using immunological techniques, electron microscopy, and X-ray diffraction. J Biol Chem. 259:14757–14761.
- Taylor MC, Board PG, Blackburn AC, Mellick GD, Le Couteur DG. 2001. Zeta class glutathione transferase polymorphisms and Parkinson’s disease. J Neurol Neurosurg Psychiatry. 70:407.
- Tecle B, Casida JE. 1989. Enzymatic defluorination and metabolism of fluoroacetate, fluoroacetamide, fluoroethanol, and (-)-erythro-fluorocitrate in rats and mice examined by 19F and 13C NMR. Chem Res Toxicol. 2:429–435.
- Terashima H, Wong H, Kobayashi R, Bunnett NW. 1992. Immunochemical localization of aminopeptidase M in the alimentary tract of the guinea-pig and rat. Gastroenterology. 102:1867–1876.
- Terzyan SS, Burgett AW, Heroux A, Smith CA, Mooers BH, Hanigan MH. 2015. Human γ-glutamyl transpeptidase 1: structures of the free enzyme, inhibitor-bound tetrahedral transition states, and glutamate-bound enzyme reveal novel movement within the active site during catalysis. J Biol Chem. 290:17576–17586.
- Terzyan SS, Cook PF, Heroux A, Hanigan MH. 2017. Structure of 6-diazo-5-oxo-norleucine-bound human gamma-glutamyl transpeptidase 1, a novel mechanism of inactivation. Protein Sci. 26:1196–1205.
- Tew KD. 1994. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 54:4313–4320.
- Tew KD. 2016. Glutathione-associated enzymes in anticancer drug resistance. Cancer Res. 76:7–9.
- Tew KD, Dutta S, Schultz M. 1997. Inhibitors of glutathione S-transferases as therapeutic agents. Adv Drug Deliv Rev. 26:91–104.
- Tew KD, Manevich Y, Grek C, Xiong Y, Uys J, Townsend DM. 2011. The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer. Free Radic Biol Med. 51:299–313.
- Tew KD, Monks A, Barone L, Rosser D, Akerman G, Montali JA, Wheatley JB, Schmidt DE Jr. 1996. Glutathione-associated enzymes in the human cell lines of the National Cancer Institute Drug Screening Program. Mol Pharmacol. 50:149–159.
- Tew KD, Townsend DM. 2012. Glutathione-S-transferases as determinants of cell survival and death. Antioxid Redox Signal. 17:1728–1737.
- Thakker DR, Yagi H, Levin W, Wood AW, Conney AH, Jerina DM. 1985. Polycyclic aromatic hydrocarbons: metabolic activation to ultimage carcinogens. In: Anders MW, editor. Bioactivation of foreign compounds. Orlando (FL): Academic Press; p. 177–242.
- Theodoratos A, Blackburn AC, Coggan M, Cappello J, Larter CZ, Matthaei KI, Board PG. 2012. The impact of glutathione transferase Kappa deficiency on adiponectin multimerisation in vivo. Int J Obes. 36:1366–1369.
- Theodoratos A, Tu WJ, Cappello J, Blackburn AC, Matthaei K, Board PG. 2009. Phenylalanine-induced leucopenia in genetic and dichloroacetic acid generated deficiency of glutathione transferase Zeta. Biochem Pharmacol. 77:1358–1363.
- Thier R, Wiebel FA, Hinkel A, Burger A, Brüning T, Morgenroth K, Senge T, Wilhelm M, Schulz TG. 1998. Species differences in the glutathione transferase GSTT1-1 activity towards the model substrates methyl chloride and dichloromethane in liver and kidney. Arch Toxicol. 72:622–629.
- Thompson GA, Meister A. 1976. Hydrolysis and transfer reactions catalyzed by γ-glutamyl transpeptidase; evidence for separate substrate sites and for high affinity of L-cystine. Biochem Biophys Res Commun. 71:32–36.
- Thompson GA, Meister A. 1977. Interrelationships between the binding sites for amino acids, dipeptides, and γ-glutamyl donors in γ-glutamyl transpeptidase. J Biol Chem. 252:6792–6798.
- Thompson LC, Ladner JE, Codreanu SG, Harp J, Gilliland GL, Armstrong RN. 2007. 2-Hydroxychromene-2-carboxylic acid isomerase: a Kappa class glutathione transferase from Pseudomonas putida. Biochemistry. 46:6710–6722.
- Thompson LC, Walters J, Burke J, Parsons JF, Armstrong RN, Dirr HW. 2006. Double mutation at the subunit interface of glutathione transferase rGSTM1-1 results in a stable, folded monomer. Biochemistry. 45:2267–2273.
- Thomson AM, Meyer DJ, Hayes JD. 1998. Sequence, catalytic properties and expression of chicken glutathione-dependent prostaglandin D2 synthase, a novel class Sigma glutathione S-transferase. Biochem J. 333:317–325.
- Thomson RE, Bigley AL, Foster JR, Jowsey IR, Elcombe CR, Orton TC, Hayes JD. 2004. Tissue-specific expression and subcellular distribution of murine glutathione S-transferase class Kappa. J Histochem Cytochem. 52:653–662.
- Thorn CF, Ji Y, Weinshilboum RM, Altman RB, Klein TE. 2012. PharmGKB summary: very important pharmacogene information for GSTT1. Pharmacogenet Genomics. 22:646–651.
- Thornthwaite JT, Allen LM. 1980. The effect of the glutamine analog, AT-125, on the cell cycle of MCF-7 and BT-20 human breast carcinoma cells using DNA flow cytometry. Res Commun Chem Pathol Pharmacol. 29:393–396.
- Thurairatnam S. 2012. Hematopoietic prostaglandin D synthase inhibitors. Prog Med Chem. 51:97–133.
- Tian Y, Huang Z, Wang Z, Yin C, Zhou L, Zhang L, Huang K, Zhou H, Jiang X, Li J, et al. 2014. Identification of novel molecular markers for prognosis estimation of acute myeloid leukemia: over-expression of PDCD7, FIS1 and Ang2 may indicate poor prognosis in pretreatment patients with acute myeloid leukemia. PLoS One. 9:e84150.
- Tin A, Colantuoni E, Boerwinkle E, Kottgen A, Franceschini N, Astor BC, Coresh J, Kao WH. 2013. Using multiple measures for quantitative trait association analyses: application to estimated glomerular filtration rate. J Hum Genet. 58:461–466.
- Tipping E, Ketterer B, Christodoulides L, Elliott BM, Aldridge WN, Bridges JW. 1979. The interactions of triethyltin with rat glutathione-S-transferases A, B and C. Enzyme-inhibition and equilibrium-dialysis studies. Chem Biol Interact. 24:317–327.
- Tomarev SI, Chung S, Piatigorsky J. 1995. Glutathione S-transferase and S-crystallins of cephalopods: evolution from active enzyme to lens-refractive proteins. J Mol Evol. 41:1048–1056.
- Tomarev SI, Zinovieva RD. 1988. Squid major lens polypeptides are homologous to glutathione S-transferases subunits. Nature. 336:86–88.
- Tong Z, Board PG, Anders MW. 1998a. Glutathione transferase Zeta catalyses the oxygenation of the carcinogen dichloroacetic acid to glyoxylic acid. Biochem J. 331:371–374.
- Tong Z, Board PG, Anders MW. 1998b. Glutathione transferase Zeta-catalyzed biotransformation of dichloroacetic acid and other α-haloacids. Chem Res Toxicol. 11:1332–1338.
- Townsend DM, Manevich Y, He L, Hutchens S, Pazoles CJ, Tew KD. 2009. Novel role for glutathione S-transferase Pi. Regulator of protein S-glutathionylation following oxidative and nitrosative stress. J Biol Chem. 284:436–445.
- Townsend DM, Shen H, Staros AL, Gaté L, Tew KD. 2002. Efficacy of a glutathione S-transferase π-activated prodrug in platinum-resistant ovarian cancer cells. Mol Cancer Ther. 1:1089–1095.
- Townsend DM, Tew KD. 2003. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 22:7369–7375.
- Trujillo JI, Kiefer JR, Huang W, Day JE, Moon J, Jerome GM, Bono CP, Kornmeier CM, Williams ML, Kuhn C, et al. 2012. Investigation of the binding pocket of human hematopoietic prostaglandin (PG) D2 synthase (hH-PGDS): a tale of two waters. Bioorg Med Chem Lett. 22:3795–3799.
- Tsirulnikov K, Abuladze N, Bragin A, Faull K, Cascio D, Damoiseaux R, Schibler MJ, Pushkin A. 2012a. Inhibition of aminoacylase 3 protects rat brain cortex neuronal cells from the toxicity of 4-hydroxy-2-nonenal mercapturate and 4-hydroxy-2-nonenal. Toxicol Appl Pharmacol. 263:303–314.
- Tsirulnikov K, Abuladze N, Newman D, Ryazantsev S, Wolak T, Magilnick N, Koag MC, Kurtz I, Pushkin A. 2009. Mouse aminoacylase 3: a metalloenzyme activated by cobalt and nickel. Biochim Biophys Acta. 1794:1049–1057.
- Tsirulnikov K, Abuladze N, Vahi R, Hasnain H, Phillips M, Ryan CM, Atanasov I, Faull KF, Kurtz I, Pushkin A. 2012b. Aminoacylase 3 binds to and cleaves the N-terminus of the hepatitis C virus core protein. FEBS Lett. 586:3799–3804.
- Tsirulnikov K, Duarte S, Ray A, Datta N, Zarrinpar A, Hwang L, Faull K, Pushkin A, Kurtz I. 2018. Aminoacylase 3 is a new potential marker and therapeutic target in hepatocellular carcinoma. J Cancer. 9:1–12.
- Tsuboi K, Bachovchin DA, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, Ferguson J, Cravatt BF, Hodder P, Rosen H. 2010. Optimization and characterization of an inhibitor for glutathione S-transferase Omega 1 (GSTO1). Probe Reports from the NIH Molecular Libraries Program. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22834034.
- Tsuboi K, Bachovchin DA, Speers AE, Spicer TP, Fernandez-Vega V, Hodder P, Rosen H, Cravatt BF. 2011. Potent and selective inhibitors of glutathione S-transferase Omega 1 that impair cancer drug resistance. J Am Chem Soc. 133:16605–16616.
- Tsuchida S, Hoshino K, Sato T, Ito N, Sato K. 1979. Purification of γ-glutamyltransferases from rat hepatomas and hyperplastic hepatic nodules, and comparison with the enzyme from rat kidney. Cancer Res. 39:4200–4205.
- Tsuchida S, Sekine Y, Shineha R, Nishihira T, Sato K. 1989. Elevation of the placental glutathione I-transferase form (GST-Pi) in tumor tissues and the levels in sera of patients with cancer. Cancer Res. 49:5225–5229.
- Tu CP, Reddy CC. 1985. On the multiplicity of rat liver glutathione S-transferases. J Biol Chem. 260:9961–9964.
- Tu LQ, Chen YY, Wright PF, Rix CJ, Bolton-Grob R, Ahokas JT. 2005. Characterization of the fluoroacetate detoxication enzymes of rat liver cytosol. Xenobiotica. 35:989–1002.
- Tu LQ, Wright PF, Rix CJ, Ahokas JT. 2006. Is fluoroacetate-specific defluorinase a glutathione S-transferase? Comp Biochem Physiol C Toxicol Pharmacol. 143:59–66.
- Turner AJ. 2013. Aminopeptidase N. In: Rawlings ND, Salvesen GS, editors. Handbook of proteolytic enzymes. 3rd ed. Vol. 1. San Diego (CA): Elsevier Science; p. 397–403.
- Turzynski A, Mentlein R. 1990. Prolyl aminopeptidase from rat brain and kidney. Action on peptides and identification as leucyl aminopeptidase. Eur J Biochem. 190:509–515.
- Tuzova M, Jean JC, Hughey RP, Brown LA, Cruikshank WW, Hiratake J, Joyce-Brady M. 2014. Inhibiting lung lining fluid glutathione metabolism with GGsTop as a novel treatment for asthma. Front Pharmacol. 5:179.
- Tylki-Szymanska A, Gradowska W, Sommer A, Heer A, Walter M, Reinhard C, Omran H, Sass JO, Jurecka A. 2010. Aminoacylase 1 deficiency associated with autistic behavior. J Inherit Metab Dis. 33:S211–S214.
- Tzeng HF, Blackburn AC, Board PG, Anders MW. 2000. Polymorphism- and species-dependent inactivation of glutathione transferase Zeta by dichloroacetate. Chem Res Toxicol. 13:231–236.
- Ujihara M, Horiguchi Y, Ikai K, Urade Y. 1988a. Characterization and distribution of prostaglandin D synthetase in rat skin. J Invest Dermatol. 90:448–451.
- Ujihara M, Tsuchida S, Satoh K, Sato K, Urade Y. 1988b. Biochemical and immunological demonstration of prostaglandin D2, E2, and F2α formation from prostaglandin H2 by various rat glutathione S-transferase isozymes. Arch Biochem Biophys. 264:428–437.
- Ulziikhishig E, Lee KK, Hossain QS, Higa Y, Imaizumi N, Aniya Y. 2010. Inhibition of mitochondrial membrane bound-glutathione transferase by mitochondrial permeability transition inhibitors including cyclosporin A. Life Sci. 86:726–732.
- Umezawa H, Aoyagi T, Suda H, Hamada M, Takeuchi T. 1976. Bestatin, an inhibitor of aminopeptidase B, produced by actinomycetes. J Antibiot. 29:97–99.
- Uno Y, Murayama N, Kato M, Tanaka S, Ohkoshi T, Yamazaki H. 2018. Genetic variants of glutathione S-Transferase GSTT1 and GSTT2 in cynomolgus macaques: identification of GSTT substrates and functionally relevant alleles. Chem Res Toxicol. 32:333–340.
- Uno Y, Murayama N, Kunori M, Yamazaki H. 2013. Characterization of microsomal glutathione S-transferases MGST1, MGST2, and MGST3 in cynomolgus macaque. Drug Metab Dispos. 41:1621–1625.
- Urade Y, Fujimoto N, Ujihara M, Hayaishi O. 1987. Biochemical and immunological characterization of rat spleen prostaglandin D synthetase. J Biol Chem. 262:3820–3825.
- Urade Y, Ujihara M, Horiguchi Y, Ikai K, Hayaishi O. 1989. The major source of endogenous prostaglandin D2 production is likely antigen-presenting cells. Localization of glutathione-requiring prostaglandin D synthetase in histiocytes, dendritic, and Kupffer cells in various rat tissues. J Immunol. 143:2982–2989.
- Usta M, Wortelboer HM, Vervoort J, Boersma MG, Rietjens IM, van Bladeren PJ, Cnubben NH. 2007. Human glutathione S-transferase-mediated glutathione conjugation of curcumin and efflux of these conjugates in Caco-2 cells. Chem Res Toxicol. 20:1895–1902.
- Uttamsingh V, Anders MW. 1999. Acylase-catalyzed deacetylation of haloalkene-derived mercapturates. Chem Res Toxicol. 12:937–942.
- Uttamsingh V, Baggs RB, Krenitsky DM, Anders MW. 2000. Immunohistochemical localization of the acylases that catalyze the deacetylation of N-acetyl-L-cysteine and haloalkene-derived mercapturates. Drug Metab Dispos. 28:625–632.
- Uttamsingh V, Iyer RA, Baggs RB, Anders MW. 1998a. Fate and toxicity of 2-(fluoromethoxy)-1,1,3,3,3-pentafluoro-1-propene (compound A)-derived mercapturates in male, Fischer 344 rats. Anesthesiology. 89:1174–1183.
- Uttamsingh V, Keller DA, Anders MW. 1998b. Acylase I-catalyzed deacetylation of N-acetyl-L-cysteine and S-alkyl-N-acetyl-L-cysteines. Chem Res Toxicol. 11:800–809.
- Vamvakas S, Anders MW. 1994. Bioconversion of prodrugs by conjugate-processing enzymes. Adv Pharmacol. 27:481–501.
- Vamvakas S, Dekant W, Berthold K, Schmidt S, Wild D, Henshler D. 1987. Enzymatic transformation of mercapturic acids derived from halogenated alkenes to reactive and mutagenic intermediates. Biochem Pharmacol. 36:2741–2748.
- van Bladeren PJ, van Ommen B. 1991. The inhibition of glutathione S-transferases: mechanisms, toxic consequences and therapeutic benefits. Pharmacol Ther. 51:35–46.
- Van Coster RN, Gerlo EA, Giardina TG, Engelke UF, Smet JE, De Praeter CM, Meersschaut VA, De Meirleir LJ, Seneca SH, Devreese B, et al. 2005. Aminoacylase I deficiency: a novel inborn error of metabolism. Biochem Biophys Res Commun. 338:1322–1326.
- van Gisbergen MW, Cebula M, Zhang J, Ottosson-Wadlund A, Dubois L, Lambin P, Tew KD, Townsend DM, Haenen GR, Drittij-Reijnders MJ, et al. 2016. A chemical reactivity window determines prodrug efficiency towards glutathione transferase overexpressing cancer cells. Mol Pharm. 13:2010–2025.
- van Iersel M, Ploemen JP, Lo Bello M, Federici G, van Bladeren PJ. 1997. Interactions of alpha, beta-unsaturated aldehydes and ketones with human glutathione S-transferase P1-1. Chem Biol Interact. 108:67–78.
- van Lieshout EMM, Tiemessen DM, Roelofs HMJ, Peters W. 1998. Nonsteroidal anti-inflammatory drugs enhance glutathione S-transferase Theta levels in rat colon. Biochim Biophys Acta, Gen Subj. 1381:305–311.
- van Ommen B, den Besten C, Rutten AL, Ploemen JH, Vos RM, Muller F, van Bladeren PJ. 1988. Active site-directed irreversible inhibition of glutathione S-transferases by the glutathione conjugate of tetrachloro-1,4-benzoquinone. J Biol Chem. 263:12939–12942.
- van Ommen B, Ploemen JH, Bogaards JJ, Monks TJ, Gau SS, van Bladeren PJ. 1991. Irreversible inhibition of rat glutathione S-transferase 1-1 by quinones and their glutathione conjugates. Structure-activity relationship and mechanism. Biochem J. 276:661–666.
- Van Ommen B, Ploemen JHTM, Ruven HJ, Vos RME, Bogaards JJP, Berkel WJH, Bladeren PJ. 1989. Studies on the active site of rat glutathione S-transferase isoenzyme 4-4. Chemical modification by tetrachloro-1,4-benzoquinone and its glutathione conjugate. Eur J Biochem. 181:423–429.
- Van Wart HE, Lin SH. 1981. Metal binding stoichiometry and mechanism of metal ion modulation of the activity of porcine kidney leucine aminopeptidase. Biochemistry. 20:5682–5689.
- Vanderheyden PM, Demaegdt H, Swales J, Lenaerts PJ, De Backer JP, Vogel LK, Vauquelin G. 2006. Synergistic inhibition of the enzymatic activity of aminopeptidase N by divalent metal ion chelators. Fundam Clin Pharmacol. 20:613–619.
- Vasieva O. 2011. The many faces of glutathione transferase Pi. Curr Mol Med. 11:129–139.
- Vaz RJ, Li Y, Munson M, Elliot M, Thurairatnam S. 2018. Amelioration of mechanism-based inactivation of CYP3A4 by a H-PGDS inhibitor. Bioorg Med Chem Lett. 28:3046–3049.
- Veiga-da-Cunha M, Tyteca D, Stroobant V, Courtoy PJ, Opperdoes FR, Van Schaftingen E. 2010. Molecular identification of NAT8 as the enzyme that acetylates cysteine S-conjugates to mercapturic acids. J Biol Chem. 285:18888–18898.
- Vergauwen B, Dudycz LW, Dansercoer A, Devreese B. 2009. A direct spectrophotometric γ-glutamyltransferase inhibitor screening assay targeting the hydrolysis-only mode. Biochem Biophys Res Commun. 380:591–596.
- Visel A, Thaller C, Eichele G. 2004. GenePaint.org: an atlas of gene expression patterns in the mouse embryo. Nucleic Acids Res. 32:D552–D556.
- von Wallbrunn A, Waldeck J, Höltke C, Zühlsdorf M, Mesters R, Heindel W, Schäfers M, Bremer C, 2008. In vivo optical imaging of CD13/APN-expression in tumor xenografts. J Biomed Opt. 13:011007.
- Vorachek WR, Pearson WR, Rule GS. 1991. Cloning, expression, and characterization of a class-Mu glutathione transferase from human muscle, the product of the GST4 locus. Proc Natl Acad Sci USA. 88:4443–4447.
- Vos RME, Van Ommen B, Hoekstein MSJ, De Goede JHM, Van Bladeren PJ. 1989. Irreversible inhibition of rat hepatic glutathione S-transferase isoenzymes by a series of structurally related quinones. Chem Biol Interact. 71:381–392.
- Wachsmuth ED, Fritze I, Pfleiderer G. 1966. An aminopeptidase occurring in pig kidney. II. A study on the mechanism of the hydrolysis. Biochemistry. 5:175–182.
- Wacker H, Lehky P, Fischer EH, Stein EA. 1971. Physical and chemical characterization of pig kidney particulate aminopeptidase. Helv Chim Acta. 54:473–485.
- Wada K, Hiratake J, Irie M, Okada T, Yamada C, Kumagai H, Suzuki H, Fukuyama K. 2008. Crystal structures of Escherichia coli γ-glutamyltranspeptidase in complex with azaserine and acivicin: novel mechanistic implication for inhibition by glutamine antagonists. J Mol Biol. 380:361–372.
- Wagner S, Scholz K, Donegan M, Burton L, Wingate J, Völkel W. 2006. Metabonomics and biomarker discovery: LC-MS metabolic profiling and constant neutral loss scanning combined with multivariate data analysis for mercapturic acid analysis. Anal Chem. 78:1296–1305.
- Wahllander A, Soboll S, Sies H, Linke I, Muller M. 1979. Hepatic mitochondrial and cytosolic glutathione content and the subcellular distribution of GSH-S-transferases. FEBS Lett. 97:138–140.
- Wang B, Peng Y, Zhang T, Ding J. 2011a. Crystal structures and kinetic studies of human Kappa class glutathione transferase provide insights into the catalytic mechanism. Biochem J. 439:215–225.
- Wang B, Van Veldhoven PP, Brees C, Rubio N, Nordgren M, Apanasets O, Kunze M, Baes M, Agostinis P, Fransen M. 2013. Mitochondria are targets for peroxisome-derived oxidative stress in cultured mammalian cells. Free Radic Biol Med. 65:882–894.
- Wang CH, Wu HT, Cheng HM, Yen TJ, Lu IH, Chang HC, Jao SC, Shing TK, Li WS. 2011b. Inhibition of glutathione S-transferase M1 by new gabosine analogues is essential for overcoming cisplatin resistance in lung cancer cells. J Med Chem. 54:8574–8581.
- Wang J, Zhang D, Huang R, Li X, Huang W. 2017a. Gamma-glutamyltransferase and risk of cardiovascular mortality: a dose-response meta-analysis of prospective cohort studies. PLoS One. 12:e0172631.
- Wang L, Sweet DH. 2013. Renal organic anion transporters (SLC22 family): expression, regulation, roles in toxicity, and impact on injury and disease. AAPS J. 15:53–69.
- Wang L, Xu J, Ji C, Gu S, Lv Y, Li S, Xu Y, Xie Y, Mao Y. 2005. Cloning, expression and characterization of human glutathione S-transferase Omega 2. Int J Mol Med. 16:19–27.
- Wang P, Zhang J, Liu HW, Hu XX, Feng LL, Yin X, Zhang XB. 2017b. An efficient two-photon fluorescent probe for measuring γ-glutamyltranspeptidase activity during the oxidative stress process in tumor cells and tissues. Analyst. 142:1813–1820.
- Wang SL, Rice SA, Serra MT, Gross B. 1986. Purification and identification of rat hepatic cytosolic enzymes responsible for defluorination of methoxyflurane and fluoroacetate. Drug Metab Dispos. 14:392–398.
- Wang ZX, Wang HR, Zhou HM. 1995. Kinetics of inactivation of aminoacylase by 2-chloromercuri-4-nitrophenol: a new type of complexing inhibitor. Biochemistry. 34:6863–6868.
- Ward PE, Benter IF, Dick L, Wilk S. 1990. Metabolism of vasoactive peptides by plasma and purified renal aminopeptidase M. Biochem Pharmacol. 40:1725–1732.
- Warholm M, Jensson H, Tahir MK, Mannervik B. 1986. Purification and characterization of three distinct glutathione transferases from mouse liver. Biochemistry. 25:4119–4125.
- Watanabe B, Tabuchi Y, Wada K, Hiratake J. 2017. Synthesis and evaluation of the inhibitory activity of the four stereoisomers of the potent and selective human γ-glutamyl transpeptidase inhibitor GGsTop. Bioorg Med Chem Lett. 27:4920–4924.
- Watanabe T, Hirano S. 2013. Metabolism of arsenic and its toxicological relevance. Arch Toxicol. 87:969–979.
- Watanabe T, Kera Y, Matsumoto T, Yamada Rh. 1996. Purification and kinetic properties of a D-amino-acid peptide hydrolyzing enzyme from pig kidney cortex and its tentative identification with renal membrane dipeptidase. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1298:109–118.
- Watt VM, Willard HF. 1990. The human aminopeptidase N gene: isolation, chromosome localization, and DNA polymorphism analysis. Hum Genet. 85:651–654.
- Watt VM, Yip CC. 1989. Amino acid sequence deduced from a rat kidney cDNA suggests it encodes the Zn-peptidase aminopeptidase N. J Biol Chem. 264:5480–5487.
- Watts RE, Siegel M, Khosla C. 2006. Structure-activity relationship analysis of the selective inhibition of transglutaminase 2 by dihydroisoxazoles. J Med Chem. 49:7493–7501.
- We JS, Kang BY, Lee JC, Lee HB, Park HS. 1997. Identification of urinary dipeptidase as the released form of renal dipeptidase. Kidney Blood Press Res. 20:411–415.
- Webb G, Vaska V, Coggan M, Board P. 1996. Chromosomal localization of the gene for the human Theta class glutathione transferase (GSTT1). Genomics. 33:121–123.
- Weber G, Prajda N, Lui MS, Denton JE, Aoki T, Sebolt J, Zhen YS, Burt ME, Faderan MA, Reardon MA. 1982. Multi-enzyme-targeted chemotherapy by acivicin and actinomycin. Adv Enzyme Regul. 20:75–96.
- Weber JE, Oakley AJ, Christ AN, Clark AG, Hayes JD, Hall R, Hume DA, Board PG, Smythe ML, Flanagan JU. 2010. Identification and characterisation of new inhibitors for the human hematopoietic prostaglandin D2 synthase. Eur J Med Chem. 45:447–454.
- Weber WW, King CM. 1981. N-Acetyltransferase and arylhydroxamic acid acyltransferase. Meth Enzymol. 77:272–280.
- Wei X, Li J, Xie H, Ling Q, Wang J, Lu D, Zhou L, Xu X, Zheng S. 2014. Proteomics-based identification of the tumor suppressor role of aminoacylase 1 in hepatocellular carcinoma. Cancer Lett. 351:117–125.
- Weinander R, Anderson C, Morgenstern R. 1994. Identification of N-acetylcysteine as a new substrate for rat liver microsomal glutathione transferase. A study of thiol ligands. J Biol Chem. 269:71–76.
- Welberry Smith MP, Zougman A, Cairns DA, Wilson M, Wind T, Wood SL, Thompson D, Messenger MP, Mooney A, Selby PJ, et al. 2013. Serum aminoacylase-1 is a novel biomarker with potential prognostic utility for long-term outcome in patients with delayed graft function following renal transplantation. Kidney Int. 84:1214–1225.
- Wells WW, Xu DP, Yang YF, Rocque PA. 1990. Mammalian thioltransferase (glutaredoxin) and protein disulfide isomerase have dehydroascorbate reductase activity. J Biol Chem. 265:15361–15364.
- Wempe MF, Anderson WB, Tzeng HF, Board PG, Anders MW. 1999. Glutathione transferase Zeta-catalyzed biotransformation of deuterated dihaloacetic acids. Biochem Biophys Res Commun. 261:779–783.
- West HD, Mathura GR. 1954. Synthesis of some aryl-substituted L-cysteines and their fate in the animal body. J Biol Chem. 208:315–318.
- West MB, Chen Y, Wickham S, Heroux A, Cahill K, Hanigan MH, Mooers BH. 2013a. Novel insights into eukaryotic γ-glutamyl transpeptidase 1 from the crystal structure of the glutamate-bound human enzyme. J Biol Chem. 288:31902–31913.
- West MB, Wickham S, Parks EE, Sherry DM, Hanigan MH. 2013b. Human GGT2 does not autocleave into a functional enzyme: a cautionary tale for interpretation of microarray data on redox signaling. Antioxid Redox Signal. 19:1877–1888.
- West MB, Wickham S, Quinalty LM, Pavlovicz RE, Li C, Hanigan MH. 2011. Autocatalytic cleavage of human γ-glutamyl transpeptidase is highly dependent on N-glycosylation at asparagine 95. J Biol Chem. 286:28876–28888.
- Weston J. 2005. Mode of action of bi- and trinuclear zinc hydrolases and their synthetic analogues. Chem Rev. 105:2151–2174.
- Whitbread AK, Masoumi A, Tetlow N, Schmuck E, Coggan M, Board PG. 2005. Characterization of the Omega class of glutathione transferases. Meth Enzymol. 401:78–99.
- Whitbread AK, Mellick GD, Silburn PA, Le Couteur DG, Board PG. 2004. Glutathione transferase Omega class polymorphisms in Parkinson disease. Neurology. 62:1910–1911.
- Whitbread AK, Tetlow N, Eyre HJ, Sutherland GR, Board PG. 2003. Characterization of the human Omega class glutathione transferase genes and associated polymorphisms. Pharmacogenetics. 13:131–144.
- Whittington A, Vichai V, Webb G, Baker R, Pearson W, Board P. 1999. Gene structure, expression and chromosomal localization of murine Theta class glutathione transferase mGSTT1-1. Biochem J. 337:141–151.
- Wiame E, Tyteca D, Pierrot N, Collard F, Amyere M, Noel G, Desmedt J, Nassogne MC, Vikkula M, Octave JN, et al. 2010. Molecular identification of aspartate N-acetyltransferase and its mutation in hypoacetylaspartia. Biochem J. 425:127–136.
- Wickham S, Regan N, West MB, Kumar VP, Thai J, Li PK, Cook PF, Hanigan MH. 2012. Divergent effects of compounds on the hydrolysis and transpeptidation reactions of γ-glutamyl transpeptidase. J Enzyme Inhib Med Chem. 27:476–489.
- Wickham S, Regan N, West MB, Thai J, Cook PF, Terzyan SS, Li PK, Hanigan MH. 2013. Inhibition of human γ-glutamyl transpeptidase: development of more potent, physiologically relevant, uncompetitive inhibitors. Biochem J. 450:547–557.
- Wickham S, West MB, Cook PF, Hanigan MH. 2011. Gamma-glutamyl compounds: substrate specificity of gamma-glutamyl transpeptidase enzymes. Anal Biochem. 414:208–214.
- Wickström M, Larsson R, Nygren P, Gullbo J. 2011. Aminopeptidase N (CD13) as a target for cancer chemotherapy. Cancer Sci. 102:501–508.
- Wickström M, Nygren P, Larsson R, Harmenberg J, Lindberg J, Sjöberg P, Jerling M, Lehmann F, Richardson P, Anderson K, et al. 2017. Melflufen – a peptidase-potentiated alkylating agent in clinical trials. Oncotarget. 8:66641–66655.
- Wickström M, Viktorsson K, Lundholm L, Aesoy R, Nygren H, Sooman L, Fryknäs M, Vogel LK, Lewensohn R, Larsson R, et al. 2010. The alkylating prodrug J1 can be activated by aminopeptidase N, leading to a possible target directed release of melphalan. Biochem Pharmacol. 79:1281–1290.
- Widersten M, Pearson WR, Engström Å, Mannervik B. 1991. Heterologous expression of the allelic variant Mu-class glutathione transferases μ and ψ. Biochem J. 276:519–524.
- Wijnholds J, Mol C, van Deemter L, de Haas M, Scheffer GL, Baas F, Beijnen JH, Scheper RJ, Hatse S, De Clercq E, et al. 2000. Multidrug-resistance protein 5 is a multispecific organic anion transporter able to transport nucleotide analogs. Proc Natl Acad Sci USA. 97:7476–7481.
- Wilce MC, Board PG, Feil SC, Parker MW. 1995. Crystal structure of a Theta-class glutathione transferase. EMBO J. 14:2133–2143.
- Wilce MC, Parker MW. 1994. Structure and function of glutathione S-transferases. Biochim Biophys Acta, Protein Struct Mol Enzymol. 1205:1–18.
- Wilkes SH, Prescott JM. 1985. The slow, tight binding of bestatin and amastatin to aminopeptidases. J Biol Chem. 260:13154–13162.
- Williams K, Cullati S, Sand A, Biterova EI, Barycki JJ. 2009. Crystal structure of acivicin-inhibited γ-glutamyltranspeptidase reveals critical roles for its C-terminus in autoprocessing and catalysis. Biochemistry. 48:2459–2467.
- Williams RT. 1959. Detoxication mechanisms. 2nd ed. London: Chapman & Hall.
- Winayanuwattikun P, Ketterman AJ. 2005. An electron-sharing network involved in the catalytic mechanism is functionally conserved in different glutathione transferase classes. J Biol Chem. 280:31776–31782.
- Wong AH, Zhou D, Rini JM. 2012. The X-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing. J Biol Chem. 287:36804–36813.
- Wood JL. 1970. Biochemistry of mercapturic acid formation. In: Fishman WH, editor. Metabolic conjugation and metabolic hydrolysis. Vol. 2. New York: Academic Press; p. 261–300.
- Wu B, Dong D. 2012. Human cytosolic glutathione transferases: structure, function, and drug discovery. Trends Pharmacol Sci. 33:656–668.
- Wu HB, Tsou CL. 1993. A comparison of Zn(II) and Co(II) in the kinetics of inactivation of aminoacylase by 1,10-phenanthroline and reconstitution of the apoenzyme. Biochem J. 296:435–441.
- Wu JH, Batist G. 2013. Glutathione and glutathione analogues; therapeutic potentials. Biochim Biophys Acta. 1830:3350–3353.
- Wu S, Zhang C, Xu D, Guo H. 2010. Catalysis of carboxypeptidase A: promoted-water versus nucleophilic pathways. J Phys Chem B. 114:9259–9267.
- Wu YQ, Mobashery S. 1991. Targeting renal dipeptidase (dehydropeptidase I) for inactivation by mechanism-based inactivators. J Med Chem. 34:1914–1916.
- Xia H, Gu Y, Pan SS, Ji X, Singh SV. 1999. Amino acid substitutions at positions 207 and 221 contribute to catalytic differences between murine glutathione S-transferase Al-1 and A2-2 toward (+)-anti-7,8-dihydroxy-9,10-epoxy-7,8,9, 10-tetrahydrobenzo[a]pyrene. Biochemistry. 38:9824–9830.
- Xia H, Pan SS, Hu X, Srivastava SK, Pal A, Singh SV. 1998. Cloning, expression, and biochemical characterization of a functionally novel alpha class glutathione S-transferase with exceptional activity in the glutathione conjugation of (+)-anti-7,8-dihydroxy-9,10-oxy-7,8,9,10-tetrahydrobenzo(a)pyrene. Arch Biochem Biophys. 353:337–348.
- Xiang ZD, Snouwaert JN, Kovarova M, Nguyen M, Repenning PW, Latour AM, Cyphert JM, Koller BH. 2014. Mice lacking three loci encoding 14 glutathione transferase genes: a novel tool for assigning function to the GSTP, GSTM, and GSTT families. Drug Metab Dispos. 42:1074–1083.
- Xiao G, Liu S, Ji X, Johnson WW, Chen J, Parsons JF, Stevens WJ, Gilliland GL, Armstrong RN. 1996. First-sphere and second-sphere electrostatic effects in the active site of a class Mu glutathione transferase. Biochemistry. 35:4753–4765.
- Xiao Q, Deng DH, Li HY, Ye FH, Huang LL, Zhang B, Ye BB, Mo ZN, Yang XB, Liu ZF. 2014. GSTT1 and GSTM1 polymorphisms predict treatment outcome for acute myeloid leukemia: a systematic review and meta-analysis. Ann Hematol. 93:1381–1390.
- Xie Y, Dahlin JL, Oakley AJ, Casarotto MG, Board PG, Baell JB. 2018. Reviewing hit discovery literature for difficult targets: glutathione transferase Omega-1 as an example. J Med Chem. 61:7448–7470.
- Xu W, Li Q. 2005. Progress in the development of aminopeptidase N (APN/CD13) inhibitors. Curr Med Chem Anticancer Agents. 5:281–301.
- Yälçin S, Jensson H, Mannervik B. 1983. A set of inhibitors for discrimination between the basic isozymes of glutathione transferase in rat liver. Biochem Biophys Res Commun. 114:829–834.
- Yamada R, Kera Y. 1998. D-Amino acid hydrolysing enzymes. In: Jollès P, editor. D-amino acids in sequences of secreted peptides of multicellular organisms. Vol. 85. Basel: Birkhäuser; p. 145–155.
- Yamamoto S, Watanabe B, Hiratake J, Tanaka R, Ohkita M, Matsumura Y. 2011. Preventive effect of GGsTop, a novel and selective γ-glutamyl transpeptidase inhibitor, on ischemia/reperfusion-induced renal injury in rats. J Pharmacol Exp Ther. 339:945–951.
- Yamauchi A, Ueda N, Hanafusa S, Yamashita E, Kihara M, Naito S. 2002. Tissue distribution of and species differences in deacetylation of N-acetyl-L-cysteine and immunohistochemical localization of acylase I in the primate kidney. J Pharm Pharmacol. 54:205–212.
- Yang X, Liu G, Li H, Zhang Y, Song D, Li C, Wang R, Liu B, Liang W, Jing Y, et al. 2010. Novel oxadiazole analogues derived from ethacrynic acid: design, synthesis, and structure-activity relationships in inhibiting the activity of glutathione S-transferase P1-1 and cancer cell proliferation. J Med Chem. 53:1015–1022.
- Yang XF, Milhiet PE, Gaudoux F, Crine P, Boileau G. 1993. Complete sequence of rabbit kidney aminopeptidase N and mRNA localization in rabbit kidney by in situ hybridization. Biochem Cell Biol. 71:278–287.
- Yeager CL, Ashmun RA, Williams RK, Cardellichio CB, Shapiro LH, Look AT, Holmes KV. 1992. Human aminopeptidase N is a receptor for human coronavirus 229E. Nature. 357:420–422.
- Yee SW, Brackman DJ, Ennis EA, Sugiyama Y, Kamdem LK, Blanchard R, Galetin A, Zhang L, Giacomini KM. 2018. Influence of transporter polymorphisms on drug disposition and response: a perspective from the international transporter consortium. Clin Pharmacol Ther. 104:803–817.
- Yin J, Wang J. 2016. Renal drug transporters and their significance in drug-drug interactions. Acta Pharm Sin B. 6:363–373.
- Yin W, Doss GA, Stearns RA, Kumar S. 2004. N-Acetylation of the glutamate residues of intact glutathione conjugates in rats: a novel pathway for the metabolic processing of thiol adducts of xenobiotics. Drug Metab Dispos. 32:43–48.
- Yin ZL, Dahlstrom JE, Le Couteur DG, Board PG. 2001. Immunohistochemistry of Omega class glutathione S-transferase in human tissues. J Histochem Cytochem. 49:983–987.
- Yoshitake H, Yanagida M, Maruyama M, Takamori K, Hasegawa A, Araki Y. 2011. Molecular characterization and expression of dipeptidase 3, a testis-specific membrane-bound dipeptidase: complex formation with TEX101, a germ-cell-specific antigen in the mouse testis. J Reprod Immunol. 90:202–213.
- Yu B, Liu XZ, Cao XZ, Zhang MY, Chang H. 2017. Study of the expression and function of ACY1 in patients with colorectal cancer. Oncol Lett. 13:2459–2464.
- Yu B, Zheng Y, Alexander D, Morrison AC, Coresh J, Boerwinkle E. 2014. Genetic determinants influencing human serum metabolome among African Americans. PLoS Genet. 10:e1004212.
- Yu L, Kalla K, Guthrie E, Vidrine A, Klimecki WT. 2003. Genetic variation in genes associated with arsenic metabolism: glutathione S-transferase Omega 1-1 and purine nucleoside phosphorylase polymorphisms in European and indigenous Americans. Environ Health Perspect. 111:1421–1427.
- Zaitsu M, Hamasaki Y, Aoki Y, Miyazaki S. 2001. A novel pharmacologic action of glucocorticosteroids on leukotriene C4 catabolism. J Allergy Clin Immunol. 108:122–124.
- Zaitsu M, Hamasaki Y, Tsuji K, Matsuo M, Fujita I, Aoki Y, Ishii E, Kohashi O. 2003. Dexamethasone accelerates catabolism of leukotriene C4 in bronchial epithelial cells. Eur Respir J. 22:35–42.
- Zakharyan RA, Aposhian HV. 1999. Enzymatic reduction of arsenic compounds in mammalian systems: the rate-limiting enzyme of rabbit liver arsenic biotransformation is MMAV reductase. Chem Res Toxicol. 12:1278–1283.
- Zakharyan RA, Sampayo-Reyes A, Healy SM, Tsaprailis G, Board PG, Liebler DC, Aposhian HV. 2001. Human monomethylarsonic acid (MMAV) reductase is a member of the glutathione-S-transferase superfamily. Chem Res Toxicol. 14:1051–1057.
- Zalups RK, Bridges CC. 2018. Mechanisms involved in the renal handling and toxicity of mercury. In: McQueen CA, editor. Comprehensive toxicology. 3rd ed. Vol. 10. Oxford: Elsevier; p. 410–435.
- Zein M, Discombe G. 1970. Serum gamma-glutamyl transpeptidase as a diagnostic aid. Lancet. 296:748–750.
- Zhang GH, Stevens JL. 1989. Transport and activation of S-(1,2-dichlorovinyl)-L-cysteine and N-acetyl-S-(1,2-dichlorovinyl)-L-cysteine in rat kidney proximal tubules. Toxicol Appl Pharmacol. 100:51–61.
- Zhang H, Liao LH, Liu SM, Lau KW, Lai AC, Zhang JH, Wang Q, Chen XQ, Wei W, Liu H, et al. 2007. Microsomal glutathione S-transferase gene polymorphisms and colorectal cancer risk in a Han Chinese population. Int J Colorectal Dis. 22:1185–1194.
- Zhang J, Grek C, Ye ZW, Manevich Y, Tew KD, Townsend DM. 2014a. Pleiotropic functions of glutathione S-transferase P. Adv Cancer Res. 122:143–175.
- Zhang J, Shibata A, Ito M, Shuto S, Ito Y, Mannervik B, Abe H, Morgenstern R. 2011a. Synthesis and characterization of a series of highly fluorogenic substrates for glutathione transferases, a general strategy. J Am Chem Soc. 133:14109–14119.
- Zhang R, Kang KA, Piao MJ, Kim KC, Zheng J, Yao CW, Cha JW, Maeng YH, Chang WY, Moon PG, et al. 2014b. Epigenetic alterations are involved in the overexpression of glutathione S-transferase Pi-1 in human colorectal cancers. Int J Oncol. 45:1275–1283.
- Zhang W, Liu F, Zhang C, Luo JG, Luo J, Yu W, Kong L. 2017. Near-infrared fluorescent probe with remarkable large Stokes shift and favorable water solubility for real-time tracking leucine aminopeptidase in living cells and in vivo. Anal Chem. 89:12319–12326.
- Zhang W, Moden O, Mannervik B. 2010. Differences among allelic variants of human glutathione transferase A2-2 in the activation of azathioprine. Chem Biol Interact. 186:110–117.
- Zhang X, Fang H, Zhang J, Yuan Y, Xu W. 2011b. Recent advance in aminopeptidase N (APN/CD13) inhibitor research. Curr Med Chem. 18:5011–5021.
- Zhang X, Xu W. 2008. Aminopeptidase N (APN/CD13) as a target for anti-cancer agent design. Curr Med Chem. 15:2850–2865.
- Zhang X, Zhang L, Zhang J, Feng J, Yuan Y, Fang H, Xu W. 2013. Design, synthesis and preliminary activity evaluation of novel 3-amino-2-hydroxyl-3-phenylpropanoic acid derivatives as aminopeptidase N/CD13 inhibitors. J Enzyme Inhib Med Chem. 28:545–551.
- Zhao GM, Wang X. 2006. Advance in antitumor agents targeting glutathione-S-transferase. Curr Med Chem. 13:1461–1471.
- Zhao T, Singhal SS, Piper JT, Cheng J, Pandya U, Clark-Wronski J, Awasthi S, Awasthi YC. 1999a. The role of human glutathione S-transferases hGSTA1-1 and hGSTA2-2 in protection against oxidative stress. Arch Biochem Biophys. 367:216–224.
- Zhao Z, Koeplinger KA, Peterson T, Conradi RA, Burton PS, Suarato A, Heinrikson RL, Tomasselli AG. 1999b. Mechanism, structure-activity studies, and potential applications of glutathione S-transferase-catalyzed cleavage of sulfonamides. Drug Metab Dispos. 27:992–998.
- Zheng J, Liu G, Tozkoparan B, Wen D. 2005. Mechanistic studies of inactivation of glutathione S-transferase Pi isozyme by a haloenol lactone derivative. Med Chem. 1:191–198.
- Zheng J, Mitchell AE, Jones AD, Hammock BD. 1996. Haloenol lactone is a new isozyme-selective and active site-directed inactivator of glutathione S-transferase. J Biol Chem. 271:20421–20425.
- Zhong Y, Onuki J, Yamasaki T, Ogawa O, Akatsuka S, Toyokuni S. 2009. Genome-wide analysis identifies a tumor suppressor role for aminoacylase 1 in iron-induced rat renal cell carcinoma. Carcinogenesis. 30:158–164.
- Zhou H, Brock J, Liu D, Board PG, Oakley AJ. 2012. Structural insights into the dehydroascorbate reductase activity of human Omega-class glutathione transferases. J Mol Biol. 420:190–203.
- Zhou W, Shultz JW, Murphy N, Hawkins EM, Bernad L, Good T, Moothart L, Frackman S, Klaubert DH, Bulleit RF, et al. 2006. Electrophilic aromatic substituted luciferins as bioluminescent probes for glutathione S-transferase assays. Chem Commun (Camb). 44:4620–4622.
- Zhou Y, Yuan J, Li Z, Wang Z, Cheng D, Du Y, Li W, Kan Q, Zhang W. 2015. Genetic polymorphisms and function of the organic anion-transporting polypeptide 1A2 and its clinical relevance in drug disposition. Pharmacology. 95:201–208.
- Zhou Z, Wang F, Yang G, Lu C, Nie J, Chen Z, Ren J, Sun Q, Zhao C, Zhu WH. 2017. A ratiometric fluorescent probe for monitoring leucine aminopeptidase in living cells and zebrafish model. Anal Chem. 89:11576–11582.
- Zhu X, Barman A, Ozbil M, Zhang T, Li S, Prabhakar R. 2012. Mechanism of peptide hydrolysis by co-catalytic metal centers containing leucine aminopeptidase enzyme: a DFT approach. J Biol Inorg Chem. 17:209–222.
- Ziemska J, Guśpiel A, Jarosz J, Nasulewicz-Goldeman A, Wietrzyk J, Kawęcki R, Pypowski K, Jarończyk M, Solecka J. 2016. Molecular docking studies, biological and toxicity evaluation of dihydroisoquinoline derivatives as potential anticancer agents. Bioorg Med Chem. 24:5302–5314.
- Ziemska J, Solecka J, Jarończyk M. 2017. QSAR, docking studies and toxicology prediction of isoquinoline derivatives as leucine aminopeptidase inhibitors. Chem Pap. 71:2557–2568.
- Zimniak P, Singh SP. 2007. Families of glutathione transferases. In: Awasthi YC, editor. Toxicology of the glutathione transferases. Boca Raton (FL): Taylor & Francis; p. 11–26.
- Zoi OG, Thireou TN, Rinotas VE, Tsoungas PG, Eliopoulos EE, Douni EK, Labrou NE, Clonis YD. 2013. Designer xanthone: an inhibitor scaffold for MDR-involved human glutathione transferase isoenzyme A1-1. J Biomol Screen. 18:1092–1102.
- Zompra A, Georgakis N, Pappa E, Thireou T, Eliopoulos E, Labrou N, Cordopatis P, Clonis Y. 2016. Glutathione analogues as substrates or inhibitors that discriminate between allozymes of the MDR-involved human glutathione transferase P1-1. Biopolymers. 106:330–344.
- Zusterzeel PL, Peters WH, De Bruyn MA, Knapen MF, Merkus HM, Steegers EA. 1999. Glutathione S-transferase isoenzymes in decidua and placenta of preeclamptic pregnancies. Obstet Gynecol. 94:1033–1038.