
Abstract
Organ-on-chip (OoC) systems are microfabricated cell culture devices designed to model functional units of human organs by harboring an in vitro generated organ surrogate. In the present study, we reviewed issues and opportunities related to the application of OoC in the safety and efficacy assessment of chemicals and pharmaceuticals, as well as the steps needed to achieve this goal. The relative complexity of OoC over simple in vitro assays provides advantages and disadvantages in the context of compound testing. The broader biological domain of OoC potentially enhances their predictive value, whereas their complexity present issues with throughput, standardization and transferability. Using OoCs for regulatory purposes requires detailed and standardized protocols, providing reproducible results in an interlaboratory setting. The extent to which interlaboratory standardization of OoC is feasible and necessary for regulatory application is a matter of debate. The focus of applying OoCs in safety assessment is currently directed to characterization (the biology represented in the test) and qualification (the performance of the test). To this aim, OoCs are evaluated on a limited scale, especially in the pharmaceutical industry, with restricted sets of reference substances. Given the low throughput of OoC, it is questionable whether formal validation, in which many reference substances are extensively tested in different laboratories, is feasible for OoCs. Rather, initiatives such as open technology platforms, and collaboration between OoC developers and risk assessors may prove an expedient strategy to build confidence in OoCs for application in safety and efficacy assessment.
General introduction to organ-on-chip systems
Assessing the potential toxic effects of chemicals, as well as the potential toxic and therapeutic effects of pharmaceuticals, on human health remains challenging, because of the complexity of biological processes involved and the poor experimental accessibility to informative in vivo systems. This leads researchers to make use of model systems, which range from single cell lines to animals including rats, mice, dogs or monkeys as well as more simple model organisms, such as the fruit fly Drosophila melanogaster (Meneely et al. Citation2019) and the nematode Caenorhabditis elegans (Mirzoyan et al. Citation2019). The most common animal models, while extremely valuable for studying (patho)physiology in an in vivo setting and for supporting pre-clinical development of therapeutics, are rather costly, difficult to handle, and in case of preclinical testing of pharmaceuticals, frequently of poor translational value due to interspecies differences. Conversely, models based on two-dimensional cell culture of primary or immortalized cell lines are easily accessible for experiments, but generally lack key features of in vivo settings, including spatial organization, the dimensionality of the extracellular matrix (ECM), dynamic signaling environments, and systemic interactions (Jackson and Lu Citation2016; Duval et al. Citation2017). Microphysiological systems, an umbrella term that bundles diverse laboratory methodologies to reproduce aspects of human physiology and disease in small scale, provide opportunities to combine the advantages of both worlds and are seen as an extremely promising approach to improve biomedical research, including toxicology and pharmaceutical development, and thereby advance consumer/patient benefit and safety as well as animal welfare (Marx et al. Citation2020). The most prominent representatives of such methods are organoids and organ-on-chip (OoC) systems, two initially independent, but now increasingly interlinked approaches (Park et al. Citation2019). Organoids are stem cell-derived and self-organizing structures that replicate key structural and functional characteristics of functional units of human organs (Kretzschmar and Clevers Citation2016; Rossi et al. Citation2018). While fascinating in their ability to emulate fundamental aspects of human organs, organoids clearly have room for improvement regarding the (rather random) tissue configuration and control of biological and physicochemical environment. These are, conversely, the strengths of OoC systems, microfabricated cell culture devices designed to model functional units of human organs in vitro by harboring an in vitro generated organ surrogate, which may be an organoid (Park et al. Citation2019; Parrish et al. Citation2019). The European Organ-on-Chip Society endorses the following definition: “An Organ-on-Chip (OoC) is a fit for purpose fabricated microfluidic-based device, containing living engineered organ substructures in a controlled micro- or nano-environment, that recapitulate one or more aspects of the dynamics, functionality and (patho)physiological response of an organ in vivo, in real-time monitoring mode” (https://euroocs.eu/organ-on-chip). Microfluidics culture conditions means on the one hand that only tiny amounts of culture medium (and consequently of test substances – for pharmaceuticals an economic advantage not to be underestimated) are necessary for operating the system. On the other hand, it implies that several physical aspects of the culture itself are different compared to standard cell culture. These include a pronounced and tightly controlled laminar flow of the culture medium (in contrast to absence of flow or uncontrolled, turbulent flow in standard cell culture) mimicking the continuous supply of oxygen and nutrients via blood and the dominant role played by surface tension and capillary forces (Sackmann et al. Citation2014).
The rather misleading use of the term “chip” refers to the fact that microfabrication methods derived from the computer microchip industry have been – and still are – used to produce the small-scale culture devices. In its basic form, OoC systems include at least two microfluidic channels separated by membranes with different degrees of permeability, therefore permitting the culture and the (curated) communication between different cells types. Frequent additional features include special, ECM-like surface coating, controlled flow and further innovative features, such as setting controlled and more physiological oxygen conditions, and applying pneumatic compression or stretching to better reproduce specific cues particularly relevant for the organ in question. Importantly, miniaturized sensors allow real-time monitoring (and, if necessary, adjustment) of parameters as pH, flow regime, mechanical stimulation and metabolic activity (e.g. measurement of glucose, lactate and a wide range of additional organic substrates and products). Moreover, OoC systems can be broadly classified into two different types: single-organ systems, emulating the main functions of single tissues or organs, and multi-organ systems, linking various tissues/organs to mimic tissue interactions present in vivo, including systemic effects.
In most cases, the 3 D organ-like structures are constructed using immortalized cell lines, primary cells, or stem cells, including embryonic stem cells and induced pluripotent stem cells (iPSC). Notably, generation of new cell types via differentiation of stem cells allows incorporating time-dependent developmental aspects in cell culture. In most cases, it will be clearly advantageous to avoid using non-human cells, as using cells of human origin will prevent detecting misleading toxicological or pharmacological effects of substances due to species-specific differences. Immortalized cells are rather easy to handle, but it is essential to regularly check their identity and characteristics, as they are usually cultured for long time periods and may (often gradually) loose functional relevance for the original organ and therefore the experimental question (Gillet et al. Citation2013; Ben-David, Siranosian et al. Citation2018; van der Hee et al. Citation2020). Primary cells, on the other hand, while having a rather higher biological relevance due to their immediate relationship to a specific organ (Czekanska et al. Citation2014), are more demanding in their handling (need to plan resections, limited culture capacity) and their availability may be limited. In addition, for each batch of primary cells detailed characterization, for instance regarding proliferation, differentiation, and metabolic ability is required. This is due to the interindividual variation shown by organ and cell donors, and potentially having chronic or acute diseases that will affect cell behavior ex vivo. However, this potential disadvantage can be turned into an advantage as it may allow generating disease models and assessing the effects of drugs administered to patients. The most promising cell type, however, are iPSCs. Specifically, human iPSCs (hiPSCs) can be obtained by introducing specific transcription factors into somatic cells derived from adult tissue, inducing a program that allows the cells to regain stem cell characteristics, thereby endowing them with the ability to differentiate into various cell types such as cardiomyocytes, adipocytes, skeletal muscle progenitor cells, neural cells, pancreatic β-cells, and hematopoietic progenitor cells similar to embryonic stem cells (Chambers et al. Citation2009; Choi et al. Citation2009; Huang et al. Citation2009; Zhang et al. Citation2009; Tanaka et al. Citation2013; Pagliuca et al. Citation2014). If isolated from patients, they provide new perspectives in the area of personalized medicine (Junying et al. Citation2009) as they can be used to engineer personalized healthy or diseased tissue constructs (Takahashi et al. Citation2007; Lancaster and Knoblich Citation2014). This personalized pharmaceutical screening recapitulates the patient’s physiology much closer than animal models. Safety testing of pharmaceutical candidates, reducing preclinical research time, and the possibility of developing drugs specific for an individual patient’s genome and disease are important reasons for the use of hiPSCs. Nonetheless, in 2 D or 3 D scaffolds controlled hiPSC differentiation is challenging; it is therefore essential to increase the efficiency and reproducibility of hiPSC differentiation. A possible solution may be the generation of cell stocks that represent a specific patient population. Due to their enormous renewability and their potency to differentiate into major cell types, various types of tissues or organoids can be created, facilitating the development of multiple organ OoCs to capture complex drug interactions. Moreover, hiPSCs do not suffer from the ethical issues that limit the use human embryonic stem cells. Earlier concerns that hiPSCs may have epigenetic memory have now been largely invalidated (Roost et al. Citation2017).
A key advancement in generating organ-like structures is the use of structural (as a scaffold) and biologically active substances, such as coating of surfaces with components derived from the extracellular matrix (Nikolova and Chavali Citation2019). A further improvement of scaffolds can be reached by bioprinting methods, which for instance may enable the creation of channels for better perfusion of the 3 D structure as well as improved standardization of experiments by allowing the generation of structures with the same form and size over time (Kolesky et al. Citation2018).
Different lung-on-chip models provide a particularly illustrative example of OoC systems. As it would not be feasible to reconstruct a whole lung due to the variety and complexity of its cellular composition, different groups have focused on specific aspects of the organ. This includes a human airway musculature model as a pre-requisite for bronchoconstriction and bronchodilation (Nesmith et al. Citation2014) and an epithelial airway model to simulate the architecture and pressure issues in relation to mucus distribution (Tavana et al. Citation2011). Another system reproduces the alveolar environment, the key site for pulmonary gas exchange and pathogen invasion (Huh et al. Citation2010). Ideally, such systems could model diverse lung diseases including asthma, chronic obstructive pulmonary disease or cystic fibrosis. It also seems natural to use these systems to assess the local toxic effects of inhaled substances, as exemplified by a micro-engineered breathing lung-on-chip (Benam et al. Citation2016; Li et al. Citation2019).
The use of animals for safety testing of chemicals and pharmaceuticals is common practice, and is even required by many legislative frameworks. It is, however, ethically questionable. OoCs promise to contribute to a future without animal testing or at least to the replacement, reduction and refinement of animal experiments for regulatory toxicity testing, in line with directive 2010/63/EU. From an animal welfare perspective, one may thus consider the use of OoCs for toxicity testing more ethical than the use of animals. OoCs can, however, be associated with a different set of ethical issues. The use of human (stem) cells may raise issues of ownership and use, and the need for informed consent by the donor of the cells. This may affect the availability of cells.
In order to maintain transparency with regard to protected elements in Organization for Economic Co-operation and Development (OECD) test guidelines and to ensure global availability of the method itself, the OECD developed guidance for good practices for licensing according to the F/RAND principles (OECD Citation2019). Fair, reasonable, and nondiscriminatory (F/RAND) terms, denote a voluntary licensing commitment from the owner of an intellectual property right (IPR) in case the IPR is essential to a technical standard. Transparency with regard to IPR issues is already required when a method is submitted to the test guideline programme. In addition, to avoid monopoly situations when IPR issues are related to a single reference method, performance standards have to be developed to allow and promote the development of similar, so-called me-too methods. It is also important that, despite of IP elements, the description of a method enables adequate scientific validation and independent review since regulatory acceptance always depends on a comprehensive assessment of relevance and reliability of each method. Validation of OoC systems is discussed in Section “Validation of organs-on-chips.”
Toward coordinated development and implementation of organ-on-chips
In order to mimic tissue, organ or organ system structure and function for the development of OoC, multiple disciplines need to be combined, including human biology, engineering and biotechnology. To fully capitalize on the value of OoC for toxicology and pharmacology and to define the next steps for actual application of OoC in safety testing of chemicals and efficacy and safety testing of pharmaceuticals, multiple stakeholders need to be involved. Implementation of OoC requires effective partnerships between academia, biotech companies, regulatory agencies, chemical and pharmaceutical companies, patient groups, and other governmental agencies (Beilmann et al. Citation2019; Tagle Citation2019; Heringa et al. Citation2020; Tetsuka et al. Citation2020).
To stimulate interdisciplinary research toward OoC development, worldwide, several platforms and programs have been established. An example of a national platform is The Institute for Human Organ and Disease Model Technologies (hDMT, the Netherlands). hDMT integrates state-of-the-art human stem cell technologies with top level engineering, physics, chemistry, biology, clinical and pharmaceutical expertise from academia and industry to develop and valorize human organ and disease models-on-a-chip (https://www.hdmt.technology/). In 2017, the two-year EU-funded organ-on-chip development (ORCHID) project started, to establish a European infrastructure to enable coordinated development, production and implementation of OoCs. Aims of the project were to build an ecosystem that will move OoCs from the laboratory into real-life medical care for citizens of Europe and beyond and to create a roadmap for OoCs to stimulate acceptance of the technology by end-users and regulators (Mastrangeli et al. Citation2019). Another key initiative is EUROoC, an interdisciplinary training network for advancing OoC technology in Europe funded by the European Union’s Horizon 2020 research and innovation programme. Its main objective is to create a network of application-oriented researchers fully trained in development and application of the OoC technology and thereby support innovative research projects, which together promote the development of advanced OoC systems (https://www.eurooc.eu/). As a result of ORCHID and EUROoC, the European Organ-on-Chip society (EUROoCS) was developed. EUROoCS is a sustainable forum for information exchange among experts, development of end-user guidelines and the formation of training networks. EUROoCS facilitates the implementation of the ORCHID roadmap in an ongoing dialogue between developers, end users and regulators (https://euroocs.eu).
Similar initiatives have been developed in the US. To streamline the therapeutic development pipeline of pharmaceuticals, the US National Institutes of Health (NIH) Center for Advancing Translational Sciences (NCATS), in collaboration with the Defense Advanced Research Projects Agency (DARPA) and the US Food and Drug Administration (FDA), leads the Tissue Chip for Drug Screening program. A major focus of the program is the development of OoC to more accurately predict drug safety and efficacy in humans. Public-private partnerships between NCATS and other stakeholders, such as regulators from the FDA and the pharmaceutical industry were formed to move OoC toward acceptance and implementation in regulatory decision making (Fitzpatrick and Sprando Citation2019; Tagle Citation2019).
An important aspect of such partnerships is the involvement of regulatory scientists from the beginning and during all stages of OoC development (Fitzpatrick and Sprando Citation2019). With the aim to improve toxicological safety and risk assessment and preclinical safety predictions in pharmacology, regulatory scientists should keep pace with advances in basic and applied science and technology. At the same time, regulatory scientists should critically appraise new technologies for their fitness-for-purpose and use, in line with the regulatory needs for information relevant to human health (Fitzpatrick and Sprando Citation2019; Bos et al. Citation2020). Using OoC to generate information for demonstrating safety, discussions are needed on how to gain confidence in the performance of the technology. This may result in modification of OoC technology to conform to the regulatory needs. A positive side-effect is that this will stimulate the discussion on modernizing toxicology, creating space for innovation.
In this review, we will discuss the application of OoC in assessment of chemical toxicology and pharmacological safety and the requirements and challenges to attain this goal.
Current and developing approaches in regulatory pharmacology and toxicology
The classical road toward assessing safety of industrial chemicals, and safety and efficacy of pharmaceuticals has been and still is through animal experiments. Since the 1980s, these dedicated animal studies have been thoroughly globally standardized under the OECD for chemicals, and under the International Conference on Harmonization (ICH) for pharmaceuticals. Regional laws regulate their use and application for different sectors, such as plant protection products, biocides, industrial chemicals, cosmetic ingredients, food additives and pharmaceuticals, under the supervision of organizations such as the Environmental Protection Agency (EPA) and the Food and Drug Administration (FDA) in the USA, and the European Food Safety Authority (EFSA), the European Chemicals Agency (ECHA) and the European Medicines Agency (EMA), respectively, in Europe. The widespread use and expanding application of animal studies over the years has prompted ethical concerns as to the appropriateness of using animals for human efficacy and safety assessment, in line with Directive 2010/63/EU. To address these concerns, also regulatory activities have been taken up, e.g. by the EMA Working Group on the Application of the 3Rs in Regulatory Testing of Medicinal Products (https://www.ema.europa.eu/en/committees/working-parties-other-groups/chmp/expert-group-3rs). In addition, differences between human and animal physiology and toxicology have shown that animal studies are not always good predictors for human health. This might lead to an under-/overestimation of hazard (for chemicals) and risk as well as efficacy (for pharmaceuticals); thus potentially compromising consumer protection and benefit. In addition, it might also lead to the identification of potential hazards that are irrelevant for humans. It is well-known that both false-negative and false-positive results limit considerably the predictivity of animal experimentation and compromise the development of beneficial drugs for human use (Van Meer et al. Citation2013; Van Meer et al. Citation2015; Van Norman Citation2019; Ferreira et al. Citation2020). An avenue to circumvent part of these issues is the use of micro physiological systems (MPS). Due to the early stage of MPS technologies, specific guidance on safety assessment by MPS models is not established at the level of regulatory bodies. However, initial steps are being taken by pharmaceutical industry. Qualification of OoC systems is discussed in Section “Validation of organs-on-chips.”
The extrapolation of findings in animal studies to the human situation has by default been done conservatively. In the domain of industrial chemicals, animal studies provide the final information level for hazard and risk assessment. These may include acute, sub-chronic and chronic exposure testing, with specific additional testing protocols for e.g. carcinogenicity using lifetime exposures and for reproductive and developmental toxicity using mating, pregnancy and generation studies. These hazard assessment investigations might in some cases directly lead to a ban of the substance but in most cases an overall No Observed Adverse Effect Level (NOAEL) is derived, representing the highest exposure level at which no adverse health effect is observed. This NOAEL is then used to calculate a safe exposure level for humans, in which by default a hundred-fold uncertainty factor is applied, representing a factor of 10 for intraspecies differences and another factor of 10 for interspecies variation. However, dependent on the quality and extent of information available, other factors may be used case by case. Kinetic considerations may be considered in human safety assessment. Absorption, Distribution, Metabolism and Excretion (ADME) of a given substance may differ significantly among species, and may therefore affect the relevance of animal findings and require specific uncertainty factors for interspecies extrapolation. Increasingly, computational kinetic modeling is used to assess internal exposure, to underpin human safety assessments (Paini et al. Citation2017, Citation2019).
In the case of pharmaceuticals, animal studies are usually followed by careful clinical testing in healthy human volunteers, followed by testing in patients. Safety testing is a tiered, step-wise approach. Safety data are established in repeated-dose toxicity studies, which are used to inform starting doses in first-in-human trials. Exposure levels (at NOAELs) in animal studies are compared to exposure in human at the expected maximum recommended human dose (MRHD) to establish a first indication of human relevance. In general, it is preferred to use a pharmacologically relevant model, i.e. a model that expresses the drug target and modulates downstream effects similarly to humans, is pharmacokinetically compatible (has a favorable bioavailability, comparable metabolic profile), and is tolerated by the test species. Product-specific considerations should be taken into account, e.g. monoclonal antibodies do not metabolize, and the target sequence generally restricts the model of choice to non-human primates (ICH Citation2013). Subsequent sub-acute and chronic toxicity testing is initiated to support increasingly longer clinical trials (i.e. phase 2 and 3 trials). For acute, sub-chronic and chronic treatment in animals, limit dose selection up to 1000 mg/kg/day is generally considered sufficient (ICH Citation2013). It should be noted that single-dose toxicity studies intended to investigate acute toxicity (and lethality) are not considered relevant (ICH Citation2013). However, a dose providing a 50-fold margin of exposure compared to estimated exposure at the MRHD is also considered acceptable as the maximum dose for acute and repeated-dose toxicity studies in any species (ICH Citation2013). For other aspects of pharmaceutical safety for which no human data can be obtained, including genotoxicity, carcinogenicity and reproductive toxicity, non-clinical studies are often the only means of establishing information on safety for risk assessment before marketing. These largely rely on establishing safety margins. For example, in the ICH S5(R3) guideline, for embryo-foetal developmental toxicity a maximum exposure-based dose limit is established at 25 fold exposure at the MRHD for testing of small molecules (ICH Citation2020). The addition of this limit for setting a high dose compared to earlier established limits (e.g. testing up to 1000 mg/kg/day, maximum feasible dose, or the presence of maternal toxicity) was based on the findings that known human teratogens were detected in rodent and non-rodent embryo-foetal developmental toxicity studies at exposure margins below 25 fold exposure at MRHD (Andrews et al. Citation2019). In addition, compound class-specific guidance provides avenues to reduce the non-clinical package. For instance ICH S6(R1) facilitates the use of a single responder species for chronic toxicity testing of biologicals, when sufficiently justified (ICH Citation2011). It is expected that similar studies comparing animal exposure data to human exposure data in combination with epidemiological data for other areas in toxicology may provide a basis to set more data-driven and less conservative dose-limiting endpoints.
Whereas classical hazard and risk assessment of chemicals and safety evaluation of pharmaceuticals as described above has been largely based on adverse health effects and the exposures at which they are observed in animal studies, in recent decades mechanistic characteristics have gained importance in human safety assessment. Knowledge of modes and mechanisms of action of chemicals allows a more detailed consideration of interspecies differences and the relevance of animal findings for the human situation. Studies in isolated organs, organoids and cell cultures have proven instrumental in revealing mechanistic properties of chemicals, enabling more refined human safety assessment and support read-across or grouping approaches to facilitate the assessment of the enormous number of substances on the market (Ball et al. Citation2016; ECHA Citation2016; Patlewicz et al. Citation2019). These approaches are based on the concept of Quantitative structure-activity relationships (QSARs) correlating chemical structure with biological activity using statistical approaches and commonly used in drug discovery as well as toxicology (Perkins et al. Citation2003). Read-across and grouping is applied to predict toxicity information for one substance by using available data for another, structural sufficiently similar substance since testing on a substance-by-substance approach require huge resources. For grouping, read-across is applied based on a mechanistic understanding of the physicochemical and toxicological properties, of a group of many structurally related chemicals for which toxicity information is limited. Such an approach is currently discussed e.g. for perfluorinated compounds (Cousins et al. Citation2020). Read-across can, and probably should, be supported by in vitro data demonstrating that source and target chemicals indeed share toxicological profiles, since small structural variations can have a huge impact in toxicity (Moné et al. Citation2020). These developments gained further momentum through growing societal resistance to animal studies for ethical reasons. Alternatives to animal testing have by now acquired a steady status as useful tools in the process of chemical and pharmaceutical hazard assessment. In case of pharmaceuticals, their current application in safety assessment is primarily in prescreening for unacceptable characteristics prior to decisions on whether a substance is promising to the extent that it can enter into animal safety testing (EMA Citation2015). In addition, in vitro studies have been increasingly used in mechanistic studies for the specific functional characteristics of substances and for pharmacodynamic investigations for pharmaceuticals.
To date, the most extensive application of in vitro assays in toxicology is represented by the ToxCast program of the US EPA. In this effort, several hundreds of high throughput assays have been combined to provide an overview of mechanistic properties of the substances tested. The assays in this battery have been primarily collected from available high throughput assays, and updated with additional and novel assays deemed essential in view of missing mechanistic aspects. This program has been instrumental in advancing the use of in vitro assays in toxicological hazard assessment and has been used for substance prioritization (Auerbach et al. Citation2016; Karmaus et al. Citation2016). The interpretation of the wealth of information on individual parameters emerging from this testing program is intrinsically complex and challenging. In particular, the integration into the broader physiological landscape and the (quantitative) extrapolation of assay results to adverse health effects and even risk assessment is not straightforward. It requires models that combine effects on the different parameters resulting in an integrated assessment based on physiology. The Adverse Outcome Pathway (AOP) approach provides a descriptive model for such an integration.
Originally defined in the area of environmental safety assessment, the AOP approach offers a modular framework to visually organize detailed information on mechanisms of toxicity (). It proposes that a substance triggers a Molecular Initiating Event (MIE), which in turn triggers a cascade of subsequent Key Events (KE) that stretch from the molecular to the cellular, tissue, organ and multi-organ integration level, ultimately leading to an Adverse Outcome (AO) at the level of the intact organism (Pittman et al. Citation2018). The different stages in the process are connected by so-called Key Event Relationships (KERs), each describing the kinetics of the relation between two adjacent components of the AOP. Whilst initially AOPs were simply described as linear and one-directional, the nature of physiology has required the definition to be broadened to include equilibrium KER, feedback loops, and supra-linear network relationships that allow connecting multiple MIEs with multiple AOs. Thus, for various toxicities, in principle AOP networks can be defined describing all relevant MIEs and AOs with their KERs in the network of KEs connecting them (Knapen et al. Citation2018; Villeneuve et al. Citation2018). Ad ultimo, these AOP networks could theoretically be combined into a single all-encompassing AOP network describing the complete set of toxicologically relevant physiological pathways. In addition to necessarily expanding the AOP concept to the network version, another aspect essential for its application in chemical risk assessment is to describe the KER in quantitative terms, resulting in a quantitative AOP (qAOP) network (Spinu et al. Citation2020). The ability to quantitatively assess the consequences at the level of the AOs of being triggered by a certain KE depends on the magnitude and duration of its modulation, and the way in which the modulation is processed through the downstream KEs and KERs in the AOP network. Not every KE trigger results in an AO, given that physiology supports our survival by providing homeostatic control (Conolly et al. Citation2017). Thus, KERs need to be described in terms of mathematic equations similar to what is common practice in kinetic models of the fate of chemicals in the body. Finally, one can envisage that in computational risk assessment based on batteries of in vitro assays, quantitative mechanistic toxicodynamics represented in the qAOP network are combined with kinetic modeling of (internal) exposure to achieve integrated hazard and risk assessment.
Figure 1. Example of an AOP. The MIE (in green) is linked to the AO (in red) by a series of KE (in yellow). This AOP describes the linkage between hepatic injury caused by protein alkylation and the formation of liver fibrosis. Taken from: https://aopwiki.org/aops/38.
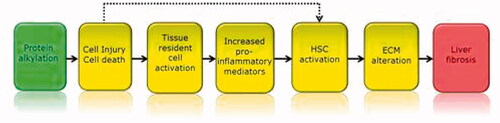
The AOP concept can be used in the development of an Integrated Approach to Testing and Assessment (IATA). IATAs are pragmatic, science-based approaches for chemical hazard characterization that rely on an integrated analysis of existing information coupled with the generation of new information using testing strategies. They follow an iterative approach to answer a defined question in a specific regulatory context, taking into account the acceptable level of uncertainty associated with the decision context. There is a range of IATAs; from more flexible, non-formalised judgment based approaches (e.g. grouping and read-across) to more structured, prescriptive, rule-based approaches (e.g. Integrated Testing Strategy). IATAs can include a combination of methods and can be informed by integrating results from one or many methodological approaches (http://www.oecd.org/chemicalsafety/risk-assessment/iata-integrated-approaches-to-testing-and-assessment.htm). In these IATAs (OECD Citation2016a), OoC systems can be instrumental in detecting the effects of substances on MIEs and KEs, and to establish potential links between AOPs as well as quantitative KER. As such, it is not likely that OoC systems will model a complete AO, but will rather become part of multi-method approaches such as an IATA.
With the increasing knowledge of physiological differences among species, awareness has grown that human safety assessment should ideally be based on testing against human physiology. Of course, with the exception of pharmaceuticals, it is considered unethical to test man-made chemicals in humans. However, computational approaches are arising in which human physiology is modeled and applied in e.g. clinical diagnostics and treatment, allowing personalized approaches that are increasingly gaining application in actual clinical practice. The “Virtual Physiological Human” (https://www.vph-institute.org/) may also be used in human chemical and pharmaceutical safety assessment (Piersma et al. Citation2019). It can be supported by cell culture assays based on human derived cells, each of which covering the essential elements of human physiology that need assessment of chemical effects. The virtual human model can be expanded in its details with any knowledge of human physiology as well as knowledge of adaptive and adverse effects of substances thereon. As to its sufficiency for reliable human safety assessment, the virtual human should in principle contain the comprehensive qAOP network, quantitatively covering all mechanisms of toxicity. This qAOP network is basically a description of that particular section of human physiology that is prone to chemical modulation, potentially leading to adverse health effects if homeostasis is challenged beyond its ability to regain a physiologic equilibrium.
The challenge for in vitro testing underlying virtual human safety assessment is to design a testing strategy that sufficiently covers the qAOP network. This may be achieved by selecting those KEs and KERs that are rate-limiting for the response of the intact individual and by representing those in in vitro assays combined in a testing strategy. The results of all in vitro assays can then be entered into the computational human model to be integrated to the level of the virtual human, resulting in a safety assessment at the level of the intact individual. This approach is still conceptual, and many steps are needed to translate this work in actual practice. Especially, critics tend to refer to limitations in current knowledge on mechanisms of human physiology and toxicity, of profound differences in physiology and vulnerability within the human population, related e.g. to age and gender, and of related challenges as to the impact of dose, timing and duration of exposure on the nature, magnitude and severity of adverse health effects. These complexities indeed show that significant challenges are ahead. However, current progress in mechanistic knowledge of human physiology and toxicology, combined with rapid developments in computational approaches such as big data analysis, machine learning and artificial intelligence may indicate that such an outlook is not merely utopic.
Multi‐OoC models have been developed mimicking interaction between different organs in order to advance the development of adequate in vitro models for both drug and toxicity testing. In drug screening, OoCs can confirm both the absence of toxic effects (e.g. by assessing cardiovascular, liver, or skin toxicity) and the effectiveness in treating the intended target pathways. For example, metabolism-dependent drug toxicity and anti-cancer efficacy have been demonstrated by combining liver and tumor models.
How can organ-on-chips improve safety prediction and reduce animal use?
The question comes up whether and how OoC can be useful as tools in the innovating arena of toxicological hazard and risk assessment. OoC models are increasingly proving useful in providing opportunities to study complex biological systems in a controlled in vitro environment. Clearly, as compared to simple single-cell type in vitro assays, OoC provide added complexity. OoC will usually contain a larger biological domain, covering a larger segment of the qAOP network, which may facilitate studying directly the connection between KEs that are rather remotely related in the network. Downsides of OoC as to their application in regulatory toxicology are their relatively low throughput, the complexity of their kinetics (volume, time, interactions between cell types), and the difficulty of interlaboratory standardization, feeding into issues on validation. In addition, similar to simple 2 D in vitro assays, OoC do not give ultimate answers about adverse health effects at the level of the intact individual. OoC results still need interpretation in the wider context of physiology and toxicology when used for hazard and risk assessment.
Two major scenarios that allow the transition to more predictive and 3 R-compliant drug development and regulatory safety assessment can be identified, an evolutionary and a revolutionary one (Scialli et al. Citation2018; Burgdorf et al. Citation2019). The evolutionary approach represents the current practice of replacing one animal test at a time by a set of non-animal methods that together predict the endpoint for which the animal test was designed (Heringa et al. Citation2020). The evolutionary approach is carried out in a stepwise fashion as standardized and validated non-animal methods become available and the relevance or predictivity of an alternative approach is still defined by comparison to (reference) animal data. Several non-animal methods for assessing toxicity endpoints as irritation and corrosion, sensitization and genotoxicity have already been validated and accepted by the corresponding authorities such as the OECD (for chemicals and pharmaceuticals), the ICH (for pharmaceuticals), and in EU guidelines (e.g. the EMA). However, there is a lack of accepted non-animal methods for more complex endpoints as acute toxicity, repeated-dose toxicity, carcinogenicity, reproductive toxicity, and neurotoxicity. The revolutionary approach, on the other hand, starts with the identification of crucial molecular or biochemical mechanisms that are relevant for human disease. Tests are developed to map critical rate-limiting steps in (networks of) physiological pathways that lead to toxicity through chemical perturbation (Piersma et al. Citation2019). This approach is obviously tightly linked to the development of AOPs and AOP networks (Knapen et al. Citation2018; Villeneuve et al. Citation2018). Consequently, the coverage of central KEs that are studied by a battery of complementary assays, should primarily determine the relevance of testing strategies (Hartung et al. Citation2017; Piersma, van Benthem, et al. Citation2018). This approach defines relevance as physiological relevance and is a measure of the extent a method or testing strategy is covering central events of a molecular network mediating toxicity. The predictive capacity of a test system relates to its ability to determine whether a chemical can perturb this network to an extent that leads to irreversible adverse effects.
Opportunities and limitations of OoCs in the context of the transition to a more predictive, non-animal regulatory safety assessment have been discussed (Heringa et al. Citation2020). OoC can play a role in both the evolutionary and the revolutionary transition of regulatory safety assessment. The main advantage of OoC technologies lies in the possibility to mimic in vivo processes more closely and, thus, increase biological relevance for both specific pathways and broader endpoints (Huh et al. Citation2010).
Both the evolutionary and the revolutionary approach aim to transform toxicology and pharmacology from being predominantly based on animal testing to relying on human physiology. One of the major benefits of OoCs is their great potential to recapitulate human physiology. This is especially relevant with the use of intact tissues instead of individual cells, enabling interaction between cells through direct contact, and between cells and extracellular components such as the extracellular matrix. These interactions should provide a pharmacologic or toxic response that better resembles the in vivo situation compared to a suspension culture with individual cells having no direct contact. Also, the dynamic supply of nutrients, drainage of waste products, and the possibility to apply shear stress should add to a better agreement with the in vivo situation. Moreover, the integration of sensors in the OoC enables continuous monitoring of the response. Using a range of sensors provides a more comprehensive picture of the pharmacologic or toxic response, for instance by simultaneously measuring effects on cell metabolism (Kieninger et al. Citation2018). Not only can temperature, pH, and oxygen be continuously monitored, sensor technology also enables online electrochemical measurement of e.g. the liver biomarkers albumin and glutathione S-transferase, and the cardiac biomarker creatine kinase (Zhang, Aleman et al. Citation2017). On the other hand, dilution of signaling molecules, small volumes and concentrations might challenge subsequent analytics (dynamic range, background problem).
A principle advantage of OoC over animal models is the possibility to introduce human specific targets and thus identify compounds that exert their toxicological or pharmacological effect only in humans.
Even in case an OoC has some degree of predictivity, this does not necessarily lead to a reduction in the number of experimental animals, but may rather add another layer of knowledge on safety or reduce the attrition rate in drug discovery. Therefore, in case the use an OoC does not lead to reduction in animal use, the OoC still has merit in a better-founded safety profile or a reduced attrition rate.
Another practical advantage of OoC is the possibility to integrate cells from different tissues into a single OoC, e.g. combining hepatocytes and cardiomyocytes to identify cardiotoxic pharmaceuticals that require liver metabolism to exert this effect (Oleaga, Riu et al. Citation2018). It is technically challenging, however, to integrate multiple organs into a single coupled system and to apply biological scaling. To replicate human physiology or toxicology and drug response with interconnected human OoC, it is critical that each OoC has the correct relative size. Otherwise, organ compartments may lead to underproduction or overproduction of metabolites, which can affect other organs in the device. An approach to scale the organ size is to construct it according to the fluid residence time in each organ since the blood residence time within an organ is correlated with the organ size, the perfusion rate, and the tissue composition. Not only the organ-chip volume but also the media volume need to be physiologically scaled both compared with their in vivo counterpart as well as relative to the other organs on the chip. Otherwise non physiological retention times of circulating media could falsify the test results. Also the volume-to-cell density scaling is important in order to avoid dilution of metabolites.
A further major benefit of OoC is the possibility to integrate (i) exposure/treatment, (ii) kinetics, and (iii) effect. In a suspension cell culture, exposure is directly linked to effect without the possibility of kinetics being evaluated. For quantitative prediction of dose-dependent adverse outcome in vivo it is essential that in vitro results are expressed in a quantitative way, allowing extrapolation of concentration-response in vitro to dose-response in vivo. This requires more detailed assessment in vitro than the common positive-negative determination of compound effects. It also requires kinetic modeling to extrapolate in vitro to in vivo exposures. An intermediate step between suspension cell culture and OoC is the use of barrier models, such as the blood-brain barrier, but also intestinal, respiratory, and vascular barriers. The barrier passage of the chemical or pharmaceutical can be evaluated, next to the effects of the compound on the cells making up the barrier, including the toxic effect on the barrier function itself. In such models, exposure, kinetics and effect may be integrated. In an OoC, advantages such as continuous monitoring and nutrient supply, apply (Arık et al. Citation2018). In case the actual adverse effect occurs in another tissue than the barrier, information on the kinetics beyond the barrier may be required. In OoC, the tissue comprising the barrier may be combined with the tissue in which the adverse effect takes place. The development of (quantitative) in vitro-in vivo extrapolation (QIVIVE) and physiologically based toxicokinetic (PBTK) or pharmacokinetic (PBPK) models is obviously as important as challenging and subject of various large-scale research programmes (Fragki et al. Citation2017; Bell et al. Citation2018; Punt et al. Citation2020).
The applicability of a certain OoC for chemical or pharmaceutical safety evaluation may be evaluated by two different approaches. The first approach may be regarded as tissue-based. In this approach the tissue that is mimicked in the OoC, is assumed to recapitulate as much as possible the characteristics of the tissue in vivo. This implies that the response to a drug or chemical is regarded as typical for that tissue, e.g. liver toxicants in liver parenchymal tissue. The qualification of the OoC is based on a large series of chemicals or drugs (with some of them requiring metabolism to exert its effect). Here, effects are monitored at a tissue level. Agreement with in vivo rodent data or human data is evaluated, firstly by commonly described and applied validation (EURL ECVAM) or qualification (EMA/FDA) approaches, and secondly within an IATA.
The second approach may be regarded as mechanism-based. This approach is based on AOPs. The OoC is investigated to assess whether the MIE is operative, and in addition to that which KEs. Clearly, downstream KE and the AO are less likely to be operative in the OoC because, especially for the AO, a more complex system (organ or intact organism) is often required. This exercise provides an in-depth understanding of the extent to which the AOP is captured in the OoC. Since AOPs are not single entities but rather part of a network, KEs in “neighbouring” AOPs should be considered as well.
Clearly, both approaches have their merit and so a combined approach should be taken. Knowledge on the KEs operative in the OoC will inform on the most downstream KE operative, this being the first candidate to test for toxic or pharmacological effects. Assaying reference compounds is required to map the OoC and the KEs operative. These reference compounds serve different goals, and a comprehensive and well-chosen set to evaluate the OoC at hand is imperative. Several groups of reference compounds can be identified that may of course partly overlap. The first group is required to delineate the chemical and biological applicability domains (chemical classes, mechanism/mode of action, MIE/KEs). The second group is required to evaluate the transferability (robustness, reproducibility) of the OoC model. The third group comprises compounds of known clinical effect, preferably in humans but also in experimental animals. A fourth group may consist of compounds that require special attention, such as compounds requiring metabolism for their effect, and “false-positives” defined here as exerting an adverse effect at a relatively low dose, that is not seen in humans. Together, these reference compounds should map the toxicological domain of the OoC.
When a developed OoC is sufficiently characterized as delineated above, and has proven to meet its requirements (e.g. one or more KE in a certain AOP can be reliably measured), thereby preferably filling a regulatory gap, the OoC needs to be standardized. Standardization of an OoC involves describing the preparation and use of an OC in such detail in an SOP that the results of the readouts are robust, reproducible, and transferable. For complex systems such as an OoC, a rigorous and strict standardization of the methods will be necessary to achieve sufficient reproducibility. Standardization also comprises computer control and measurement of flow, pH, lactate, etc. Clearly, a system where computer control is applied is less prone to variability than manual control. Helpful guidance documents to facilitate standardization are the OECD Guidance Documents (GD) 211 (OECD Citation2014) and Good In Vitro Method Practices (OECD Citation2018) on how to describe an in vitro assay and on what aspects and possible artifacts to consider when developing an in vitro method, respectively. While it is core business for academics to regularly develop new and improved OoCs, this is unlikely to aid in their regulatory use. There should be a balance between academic progress and precipitation in standard OoCs. Preferably, the generation of an OoC, both regarding the engineering part (chips, sensors, computer control and measurement) and the biology (cells, tissues, hiPSC) should be transparent and available to the interested community following FRAND (Fair, Reasonable and Nondiscriminatory) principles. There should also be a balance between the level of advancement of the OoC and its robustness, to have the OoC find its way to the regulatory arena.
Beyond within-laboratory standardization and reproducibility, the issue of transferability between laboratories is increasingly challenging with in vitro systems becoming more complex. Standardization between laboratories is complicated by differences in infrastructure, equipment, culture medium (and serum) batches, and sources of biological material. In the ECVAM validation study of rat whole embryo culture assay, it was decided that local differences in culture media, rat strain, and incubators were allowed, as long as the standardized morphological scoring system employed was used similarly and would show results of testing control compounds within certain limits of acceptability (Piersma et al. Citation2004). Likewise, it is important to qualify the essentials of standardization of OoC, and their transferability may have to be judged based on the performance of positive and negative control compounds, and novel test compounds be scored relative to these controls in each laboratory rather than requiring absolute similarity in readouts between laboratories.
Standardization and quality assurance as a prerequisite for reproducibility in the context of validation is of central importance from an industrial as well as from a regulatory point of view. Standardization of OoCs is deemed challenging since its technology is complex and interdisciplinary and involves not only defined cells or tissue culture technologies used but also encompasses the standardization of equipment, such as pumps, valves, and computer interfaces. Nonetheless, international efforts are recommended to generate reliable, reproducible and robust methods (Marx et al. Citation2016; Mastrangeli et al. Citation2019). With regard to OoCs, the focus is currently on the qualification or characterization rather than on their validation. Here, reproducibility and transferability can be assessed with a limited set of reference chemicals. However, full validation relies on a more comprehensive reference set which, again, can hardly be defined (Mastrangeli et al. Citation2019). Regarding the current validation process, a practical barrier would be that different components from the complex OoC systems are marketed by different laboratories which together would have to organize the validation process (Heringa et al. Citation2020). It is agreed, however, that regulatory acceptance will require confidence that only strict quality control can create (Smirnova et al. Citation2018). Thus, it seems expedite to involve regulatory agencies in the early stages of OoC development.
To reduce uncertainties in predictions for human health risk assessment which are based on in vitro methods, the need for guidance on Good Cell Culture Practice (GCCP) as a tool for standardization is generally recognized, not only in academic research but also in regulatory implementation, and led to several guidance documents also comprising OoC technologies (e.g. (Pamies and Hartung Citation2017; Pamies et al. Citation2018). In 2018, the OECD published a Guidance Document on Good in Vitro Method Practices (GIVIMP (OECD Citation2018)). It describes the scientific, technical and quality practices needed at all stages between in vitro method development and implementation for regulatory use. The GIVIMP document has been written for various users, including also method developers. GIVIMP tackles the following key aspects related to in vitro work: (1) roles and responsibilities, (2) quality considerations, (3) facilities, (4) apparatus, material and reagents, (5) test systems, (6) test and reference/control items, (7) standard operating procedures (SOPs), (8) performance of the method, (9) reporting of results, and (10) storage and retention of records and materials. The document also identified quality requirements for equipment, material and reagents (Mastrangeli et al. Citation2019).
The complexity of OoC technologies also increases the number of parameters to control, thereby introducing new challenges for reproducibility, quality control and interpretation of results. Establishment and maintenance of a sufficient understanding of any in vitro system and of the relevant factors it could affect, is a prerequisite for adequate quality assurance (Alépée et al. Citation2014; Pamies and Hartung Citation2017). Ongoing standardization in the field of microfluidics and cell culture, and existing standards in the electronics industry can provide the basis for developing guidance in the OoC field. Standardization can be a step toward open technology platforms, as criteria defined in a guidance can result in common design environments and building blocks (Mastrangeli et al. Citation2019).
Major challenges with regard to harmonization and standardization in the OoC field are (1) the lack of detailed understanding of some human organs and tissues, (2) complexity of protocols, and (3) reproducibility of the systems (Mastrangeli et al. Citation2019). Collecting samples on the chip may interfere with its operation, resulting in changes in the concentration of various metabolites (Wu et al. Citation2020). Other drawbacks of chip-based systems can be adsorption effects due to high surface to volume ratio (Campana and Wlodkowic Citation2018) or precipitation of test chemicals. In addition, the higher operational complexity requires adequate personnel training (Campana and Wlodkowic Citation2018), a particular challenge with respect to transferability.
In sum, the development of OoC currently focusses on the establishment of standardized protocols, quality assurance of the various components and the demonstration of reproducibility in the lab of the test method developer. Already transferability, step 3 of the six steps in the modular approach of the current validation scheme (Hartung et al. Citation2004; OECD Citation2005), is hardly addressed. Therefore, it seems unlikely that standardized test guidelines based on these methods will be developed in the near future. However, OECD Guidance Document No. 211 provides a harmonized template for assay annotations of non-guideline in vitro methods (OECD Citation2014) and criteria for testing strategies (i.e. Defined Approaches) are described in OECD GD 255 (OECD Citation2016b). Following the criteria depicted in these documents, can facilitate acceptance of the produced data (by the regulator) in particular in a weight-of-evidence approach (Bal-Price et al. Citation2018). Transferability is not part of qualification of OoC for testing of pharmaceuticals. Although currently OECD GD 211 and 255 are not used for testing of pharmaceuticals, they could in fact be used, as they describe more general harmonization of terms and testing strategies.
Due to their ability to mimic key features of in vivo settings, advanced OoC systems, especially when combined in a set as part of a testing strategy have tremendous potential to assess complex toxicity endpoints that cannot be addressed by the two-dimensional cell culture models used for assessing toxicity endpoints today. However, their improved value over current methods needs to be demonstrated on a case-by-case basis, without compromising the demonstration of safety for humans. Thus, endpoints that cannot be studied in 2 D systems, such as glomerulus or biliary system toxicity, are more likely accepted to be modeled by OoC (Fabre et al. Citation2020). OoC systems are also discussed in compliance with drug screening and pharmacological studies (Mittal et al. Citation2019; Shrestha et al. Citation2020). Furthermore, OoCs may provide relevant and early evidence of toxicity or pharmacodynamics, which can, in the interim, subsequently be confirmed in vivo with either less animals or fewer studies, in line with directive 2010/63/EU on the protection of animals used for scientific purposes.
Validation and qualification of organs-on-chips
Validation is a process to demonstrate the reproducibility and relevance of a test method. Validation is the interface between the development and optimization of a test method and its regulatory acceptance and permits a knowledge-based evaluation of the suitability of a method for a specific regulatory purpose. In 2005, the OECD published internationally agreed principles and criteria for carrying out validation studies. Based on Hartung et al. (Citation2004), this guidance describes a modular approach to validation defining six central modules: (1) test method definition, (2) within-laboratory reproducibility, (3) transferability, (4) between-laboratory reproducibility, (5) predictive capacity and (6) applicability domain as well as an optional definition of performance standards to facilitate the development of similar methods (OECD Citation2005). The first four modules define reproducibility whereas the following modules target relevance. Testing for reproducibility ensures that the method delivers comparable results regardless of location, time and operator as a basic principle in science. Relevance describes the relationship of a test to the (biological) effect of interest and whether a test is meaningful and useful for a particular (regulatory) purpose. Following the modular approach, it is based on the definition of the limitations of the method (applicability domain) with respect to certain chemical classes (viz. the first group of chemicals in Section “How can organ-on-chips improve safety prediction and reduce animal use?”) as well as the predictive capacity of a method based on qualitative as well as, if possible, quantitative in vitro-in vivo comparisons with animal or human data (viz. the third group of chemicals in Section “How can organ-on-chips improve safety prediction and reduce animal use?”). It should be noted that in pharmacology, transferability and between-laboratory reproducibility (viz. the second group of chemicals in Section “How can organ-on-chips improve safety prediction and reduce animal use?”) are not required for qualification of a test method (see also below).
Both, reproducibility and relevance are assessed using an appropriate set of reference chemicals. The selection of reference chemicals is of particular importance since this set should allow to determine the dependence of the method’s relevance on chemical properties and, thus, to define its domain of applicability with regard to chemical category and mechanism of action. However, the number of reference chemicals is obviously always limited, and, therefore, it might become a matter of debate if the method is indeed suitable for a specific chemical undergoing risk assessment and if it is comprised in the biological domain. In other words, a chemical could score as a false-negative in an in vitro test if the mechanism of toxic action of that chemical is not within the biological domain of the assay. This points to the need for combining assays with different biological domains, to make sure that all relevant mechanisms are being included in the testing strategy. In addition, animal data as a gold standard has itself intrinsic variability (Hoffmann Citation2015; Luechtefeld et al. Citation2016; Luechtefeld et al. Citation2018) and might be limited by species differences (Martignoni et al. Citation2006; Tralau et al. Citation2015) adding another level of complexity on the assessment of in vitro methods, in particular if they are based on human cells or tissues. Where applicable, to improve qualification of human cell or tissue based models human based gold standard data may be preferred in the future, although probably human intraspecies differences may provide similar limitations in variability as animal to human species differences.
In addition, a single alternative test method cannot replace an in vivo study, and different in silico, in chemico and in vitro tools have to be combined in testing strategies due to the inherent biological complexity of adverse effects that might also involve tissue interactions. In this context, the question arises of how to define (biological) relevance for single methods or testing strategies, i.e. which steps have to be addressed in a test system in order to be able to cover relevant mechanisms of toxicity (Piersma, Burgdorf, et al. Citation2018; Piersma, van Benthem, et al. Citation2018). Again, the mechanistic concept of AOPs comes into play (Vinken et al. Citation2017) to address the need of combining assays with complementary biological domains to improve predictivity compared to single assays.
What remains valid of the original validation paradigm, however, is the need to assess reproducibility and a thorough description of technical limitations (Piersma, van Benthem, et al. Citation2018). This, again, relies on a set of appropriate reference chemicals eliciting well-characterized in vivo effects, which remains a challenge since at least for most industrial chemicals the mechanism of action remains elusive.
Concepts of model validity and credibility were explored (Patterson et al. Citation2021), resulting in a set of credibility factors to compare different kinds of predictive toxicology approaches.
The FDA defines qualification as a conclusion that within the stated context of use, the model can be relied upon to have a specific interpretation and application in drug development and regulatory review (FDA Citation2020). The ICH S5 (R3) guideline on reproductive toxicology lists requirements for qualification of a test method. Qualification within this guideline includes (1) a description and justification of the predictive model, (2) an evaluation of the biological plausibility of the model, (3) an assessment of the accuracy and ability for the method to detect embryo-foetal toxicity related endpoints, (4) a discussion determining whether an effect is negative or positive in the assay, (5) a definition and justification of the threshold for molecular and metabolic markers, (6) details of the algorithm employed for determining positive and negative outcomes to the current golden standard, (7) a list of compounds in each of the training sets and test sets, (8) data sources for compounds in the qualification data set, (9) data demonstrating the test method’s performance covering a range of biological and chemical domains justified for the intended use of the method (context of use), (10) data demonstrating the sensitivity, specificity, positive and negative predictive values, and reproducibility, (11) description of the performance of each method, next to the integrated assessment used for the predictive model, and (12) historical data of the assay including data on positive controls.
When a developed OoC is sufficiently characterized as delineated above, and has proven to meet its requirements (e.g. one or more KE in a certain AOP can be reliably measured), thereby preferably filling a regulatory gap, the OoC needs to be standardized. Standardization of an OoC involves describing the preparation and use of an OC in such detail in an SOP that the results of the readouts are robust, reproducible, and transferable. For complex systems such as an OoC, a rigorous and strict standardization of the methods will be necessary to achieve sufficient reproducibility. Standardization also comprises computer control and measurement of flow, pH, lactate, etc. Clearly, a system where computer control is applied is less prone to variability than manual control. Helpful guidance documents to facilitate standardization are the OECD Guidance Documents (GD) 211 (OECD Citation2014) and Good In Vitro Method Practices (OECD Citation2018) on how to describe an in vitro assay and on what aspects and possible artifacts to consider when developing an in vitro method, respectively. While it is core business for academics to regularly develop new and improved OoCs, this is unlikely to aid in their regulatory use. There should be a balance between academic progress and precipitation in standard OoCs. Preferably, the generation of an OoC, both regarding the engineering part (chips, sensors, computer control and measurement) and the biology (cells, tissues, hiPSC) should be transparent and available to the interested community following FRAND (Fair, Reasonable and Nondiscriminatory) principles. There should also be a balance between the level of advancement of the OoC and its robustness, to have the OoC find its way to the regulatory arena.
Beyond within-laboratory standardization and reproducibility, the issue of transferability between laboratories is increasingly challenging with in vitro systems becoming more complex. Standardization between laboratories is complicated by differences in infrastructure, equipment, culture medium (and serum) batches, and sources of biological material. In the ECVAM validation study of rat whole embryo culture assay, it was decided that local differences in culture media, rat strain, and incubators were allowed, as long as the standardized morphological scoring system employed was used similarly and would show results of testing control compounds within certain limits of acceptability (Piersma et al. Citation2004). Likewise, it is important to qualify the essentials of standardization of OoC, and their transferability may have to be judged based on the performance of positive and negative control compounds, and novel test compounds be scored relative to these controls in each laboratory rather than requiring absolute similarity in readouts between laboratories.
In the pharmaceutical field there is no need for standardized test guidelines, since many non-animal methods are developed in-house, and their variability cannot be captured in such a guideline. Furthermore, for pharmacological characterization there is purposefully no or very limited guidance as this would lock any innovation. It can be imagined that testing paradigms are discussed in product-specific guidelines or testing paradigm-related guidance (e.g. ICH S5R3). In addition, it is expected that innovation will go too fast for standardization of test guidelines. So, when a guideline is fully developed and published, it would not be relevant anymore. Due to staggered product development in the pharmaceutical industry, OoC may be used to defer testing in vivo to a later stage in development and therefore through attrition lead to less animal testing. Applicability of mature OoCs within the regulatory context are most viable in the drug discovery/early development phase where multiple candidates can be screened for relevant pharmacodynamic or toxicity endpoints. These OoCs can continue on to supplement or replace pharmacological mode of action and proof of concept studies. OoCs can also help to identify relevant pharmacodynamic and/or toxicity markers to monitor in animal toxicology studies, which could result using fewer animals because more targeted information is available via OoCs. Also, in certain cases OoCs could be used to waive certain toxicity studies (e.g. in case of positive evidence of toxicity, or confirmation of within class toxicity profiles). Similarly, OoCs may provide relevant monitoring markers in clinical studies.
In the pharmaceutical field standardized test guidelines for novel alternative assays are not optimal, since many different non-animal methods are developed in-house by pharmaceutical companies, and variability between these in-house assays cannot be captured in a generalized guideline. Furthermore, for pharmacological characterization there is purposefully no or very limited guidance as this would lock innovation. It can be imagined that testing paradigms are discussed in product-specific guidelines or testing paradigm-related guidance (e.g. ICH S5R3). In addition, it is expected that innovation will go too fast for standardization of test guidelines for specific alternative assays. In such a case, when a guideline is fully developed over the course of multiple years, upon publication the scientific field would have progressed at such a rate the guideline would not be relevant anymore. Due to staggered product development in the pharmaceutical industry, OoC may be used to defer testing in vivo to a later stage in development and therefore through attrition lead to less animal testing. Applicability of mature OoCs within the regulatory context are most viable in the drug discovery/early development phase where multiple candidates can be screened for relevant pharmacodynamic or toxicity endpoints. These OoCs can continue on to supplement or replace pharmacological mode of action and proof of concept studies. OoCs can also help to identify relevant pharmacodynamic and/or toxicity markers to monitor in animal toxicology studies, which could result in using fewer animals as more targeted information is available through OoCs. Furthermore, in certain cases, OoCs could be used to waive certain toxicity studies (e.g. in case of positive evidence of toxicity, or confirmation of within class toxicity profiles). Similarly, OoCs may provide relevant monitoring bio-markers in clinical studies.
Guidance on benchmarking safety assessment in liver MPS (Baudy et al. Citation2020), for development and qualification of lung MPS for safety assessment (Ainslie et al. Citation2019), and for development and qualification of skin MPS (Hardwick et al. Citation2020) have been reported, all from a pharmaceutical industry perspective.
Outlook
In a recent survey, many method developers and end users agreed that thorough and consistent validation is not reached yet, and that further work is needed to reach broad acceptance by both pharmaceutical companies and regulatory agencies (Allwardt et al. Citation2020). An important factor is the availability of high-quality SOPs, in order for the OoC to be robust and transferable. A harmonized questionnaire as provided by ToxTemp (Krebs, Waldmann et al. Citation2019) can aid in a comprehensive test method and SOP description to define the readiness of the OoC technology for regulatory purposes (Krebs, Waldmann et al. Citation2019). The technology underlying the OoC, including engineering and computer software, should be open source. A data repository comprising effects of a wide range of compounds on the applicable MIE and KEs is very useful to underline the OoC specifications. The biology, engineering, and software should be standardized, in order to build up such a data repository. The time required from the accepted OoC to an ICH or OECD guideline may take several years.
Generally, OoC models are not only being developed for regulatory purposes. However, it is still essential that in case of a possible regulatory application, with the developer deciding to fill that regulatory need and market the model for regulatory purposes, developers interact with regulators already from the start of the OoC development. The developer may wish to have exclusive IP rights for some time to get a return on investment and after that make the IP matters public. In that period of IP coverage, regulators may assist in characterizing the OoC according to their needs, meanwhile setting up a data repository. The developer, in turn, gets expert information on the way the OoC should be interrogated for important OoC characteristics. Regulators of course remain confidential about the OoC in the period of IP coverage.
As is often the case with new technologies, critical aspects regarding the suitability of OoC systems for regulatory purposes will only be revealed when put into practice. For this purpose, the assessment of the regulatory relevance of the individual OoC systems developed within the training network EUROoC may represent a promising beginning. The EUROoC project has training as important goal. The Validation Board of the project aims to train OoC developers (PhD students) in validation requirements as a basis for their future career. The Validation Board members are experts from industry as well as regulatory bodies. Specifically, by answering a standardized questionnaire, EUROoC OoC model developers will summarize the relevant information about their systems, including typical toxicological or pharmacologic mechanisms to be assessed, relevant toxicological endpoints and the specific assays used to measure them, and characteristics and availability of positive and negative chemicals/pharmaceuticals, among others. In the next step, based on the information they provided, system developers will receive advice from a board composed by representatives of regulatory agencies (including some of the authors of this article) on first steps toward (pre-) validation and regulatory relevance. This may prove a useful trial balloon for assessing key aspects, potential pitfalls, as well as unforeseen issues regarding the regulatory applicability of OoC systems. We look forward to sharing the obtained insights with the scientific community, such as EUROoCS, in the near future.
Acknowledgements
The manuscript was approved by the German Federal Institute for Risk Assessment, and by the Dutch Medicines Evaluation Board; in both cases by anonymous review. The reviewers are kindly acknowledged. Dr. Jacqueline van Engelen and Dr. Jelle Vriend, both from the National Institute of Public Health and the Environment, The Netherlands, are acknowledged for critical review of the manuscript. The manuscript was sent to all EUROoC project partners, and no objections were received. Both individuals who reviewed the manuscript on behalf of the Journal are acknowledged for their useful comments.
Declaration of interest
The authors’ affiliations are shown above. All experts that provided input or review are included as coauthors or listed in the acknowledgments. The authors have sole responsibility for the writing and content of the paper. The content was not subject to the funders’ control. There are no conflicts of interest to disclose for any of the authors. None of the authors have participated in the last 5 years in any legal, regulatory or advocacy proceedings related to the contents of the paper.
The work has been initiated by EUROoC, an interdisciplinary training network for advancing Organ-on-a-chip technology in Europe (https://www.eurooc.eu/). This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 812954.
ASK, AHP and RJV were supported by the Dutch Ministry of Agriculture, Nature and Food Quality, project 10B.5.1-5.
References
- Ainslie GR, Davis M, Ewart L, Lieberman LA, Rowlands DJ, Thorley AJ, Yoder G, Ryan AM. 2019. Microphysiological lung models to evaluate the safety of new pharmaceutical modalities: a biopharmaceutical perspective. Lab Chip. 19(19):3152–3161.
- Alépée N, Bahinski A, Daneshian M, De Wever B, Fritsche E, Goldberg A, Hansmann J, Hartung T, Haycock J, Hogberg HT, et al. 2014. State-of-the-art of 3D cultures (organs-on-a-chip) in safety testing and pathophysiology. ALTEX. 31(4):441–477.
- Allwardt V, Ainscough AJ, Viswanathan P, Sherrod SD, McLean JA, Haddrick M, Pensabene V. 2020. Translational roadmap for the organs-on-a-chip industry toward broad adoption. Bioengineering. 7(3):112–127.
- Andrews PA, Blanset D, Costa PL, Green M, Green ML, Jacobs A, Kadaba R, Lebron JA, Mattson B, McNerney ME, et al. 2019. Analysis of exposure margins in developmental toxicity studies for detection of human teratogens. Regul Toxicol Pharm. 105:62–68.
- Arık YB, van der Helm MW, Odijk M, Segerink LI, Passier R, van den Berg A, van der Meer AD. 2018. Barriers-on-chips: measurement of barrier function of tissues in organs-on-chips. Biomicrofluidics. 12(4):042218.
- Auerbach S, Filer D, Reif D, Walker V, Holloway AC, Schlezinger J, Srinivasan S, Svoboda D, Judson R, Bucher JR, et al. 2016. Prioritizing environmental chemicals for obesity and diabetes outcomes research: a screening approach using ToxCast™ high-throughput data. Environ Health Perspect. 124(8):1141–1154.
- Ball N, Cronin MTD, Shen J, Blackburn K, Booth ED, Bouhifd M, Donley E, Egnash L, Hastings C, Juberg DR, et al. 2016. Toward good read-across practice (GRAP) guidance. ALTEX. 33(2):149–166.
- Bal-Price A, Hogberg HT, Crofton KM, Daneshian M, FitzGerald RE, Fritsche E, Heinonen T, Hougaard Bennekou S, Klima S, Piersma AH, et al. 2018. Recommendation on test readiness criteria for new approach methods in toxicology: exemplified for developmental neurotoxicity. ALTEX. 35(3):306–352.
- Baudy AR, Otieno MA, Hewitt P, Gan J, Roth A, Keller D, Sura R, Van Vleet TR, Proctor WR. 2020. Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry. Lab Chip. 20(2):215–225.
- Bell SM, Chang X, Wambaugh JF, Allen DG, Bartels M, Brouwer KLR, Casey WM, Choksi N, Ferguson SS, Fraczkiewicz G, et al. 2018. In vitro to in vivo extrapolation for high throughput prioritization and decision making. Toxicology in Vitro. 47:213–227.
- Beilmann M, Boonen H, Czich A, Dear G, Hewitt P, Mow T, Newham P, Oinonen T, Pognan F, Roth A, et al. 2019. Optimizing drug discovery by investigative toxicology: current and future trends. ALTEX. 36(2):289–313.
- Benam KH, Novak R, Nawroth J, Hirano-Kobayashi M, Ferrante TC, Choe Y, Prantil-Baun R, Weaver JC, Bahinski A, Parker KK, et al. 2016. Matched-comparative modeling of normal and diseased human airway responses using a microengineered breathing lung chip. Cell Syst. 3(5):456–466.e454.
- Ben-David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, Strathdee CA, Dempster J, Lyons NJ, Burns R, et al. 2018. Genetic and transcriptional evolution alters cancer cell line drug response. Nature. 560(7718):325–330.
- Bos PMJ, Geraets L, De Wit-Bos L, Heringa M, Van Engelen J. 2020. Towards an animal-free human health assessment: starting from the current regulatory needs. ALTEX. 37(3):395–408.
- Burgdorf T, Piersma AH, Landsiedel R, Clewell R, Kleinstreuer N, Oelgeschläger M, Desprez B, Kienhuis A, Bos P, de Vries R, et al. 2019. Workshop on the validation and regulatory acceptance of innovative 3R approaches in regulatory toxicology – evolution versus revolution. Toxicol in Vitro. 59:1–11.
- Campana O, Wlodkowic D. 2018. Ecotoxicology goes on a chip: embracing miniaturized bioanalysis in aquatic risk assessment. Environ Sci Technol. 52(3):932–946.
- Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. 2009. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 27(3):275–280.
- Choi KD, Yu J, Smuga-Otto K, Salvagiotto G, Rehrauer W, Vodyanik M, Thomson J, Slukvin I. 2009. Hematopoietic and endothelial differentiation of human induced pluripotent stem cells. Stem Cells. 27(3):559–567.
- Conolly RB, Ankley GT, Cheng W, Mayo ML, Miller DH, Perkins EJ, Villeneuve DL, Watanabe KH. 2017. Quantitative adverse outcome pathways and their application to predictive toxicology. Environ Sci Technol. 51(8):4661–4672.
- Cousins IT, Dewitt JC, Glüge J, Goldenman G, Herzke D, Lohmann R, Miller M, Ng CA, Scheringer M, Vierke L, et al. 2020. Strategies for grouping per- and polyfluoroalkyl substances (PFAS) to protect human and environmental health . Environ Sci Process Impacts. 22(7):1444–1460.
- Czekanska EM, Stoddart MJ, Ralphs JR, Richards RG, Hayes JS. 2014. A phenotypic comparison of osteoblast cell lines versus human primary osteoblasts for biomaterials testing. J Biomed Mater Res A. 102(8):2636–2643.
- Duval K, Grover H, Han LH, Mou Y, Pegoraro AF, Fredberg J, Chen Z. 2017. Modeling physiological events in 2D vs. 3D cell culture. Physiology. 32(4):266–277.
- ECHA. 2016. Practical guide: how to use alternatives to animal testing. Available from https://echa.europa.eu/documents/10162/13655/practical_guide_how_to_use_alternatives_en.pdf
- EMA. 2015. Reflection paper providing an overview of the current regulatory testing requirements for medicinal products for human use and opportunities for implementation of the 3Rs. Available from https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-providing-overview-current-regulatory-testing-requirements-medicinal-products-human_en.pdf.
- Fabre K, Berridge B, Proctor WR, Ralston S, Will Y, Baran SW, Yoder G, Van Vleet TR. 2020. Introduction to a manuscript series on the characterization and use of microphysiological systems (MPS) in pharmaceutical safety and ADME applications. Lab Chip. 20(6):1049–1057.
- FDA. 2020. Qualification process for drug development tools. Guidance for industry and FDA staff. Available from https://www.fda.gov/regulatory-information/search-fda-guidance-documents/qualification-process-drug-development-tools-guidance-industry-and-fda-staff.
- Ferreira GS, Veening-Griffioen DH, Boon WPC, Moors EHM, van Meer PJK. 2020. Levelling the translational gap for animal to human efficacy data. Animals. 10(7):1199–1113.
- Fitzpatrick S, Sprando R. 2019. Advancing regulatory science through innovation: in vitro microphysiological systems. Cell Mol Gastroenterol Hepatol. 7(1):239–240.
- Fragki S, Piersma AH, Rorije E, Zeilmaker MJ. 2017. In vitro to in vivo extrapolation of effective dosimetry in developmental toxicity testing: Application of a generic PBK modelling approach. Toxicology and Applied Pharmacology. 332:109–120.
- Gillet JP, Varma S, Gottesman MM. 2013. The clinical relevance of cancer cell lines. J Natl Cancer Inst. 105(7):452–458.
- Hardwick RN, Betts CJ, Whritenour J, Sura R, Thamsen M, Kaufman EH, Fabre K. 2020. Drug-induced skin toxicity: gaps in preclinical testing cascade as opportunities for complex in vitro models and assays. Lab Chip. 20(2):199–214.
- Hartung T, FitzGerald RE, Jennings P, Mirams GR, Peitsch MC, Rostami-Hodjegan A, Shah I, Wilks MF, Sturla SJ. 2017. Systems toxicology: real world applications and opportunities. Chem Res Toxicol. 30(4):870–882.
- Hartung T, Bremer S, Casati S, Coecke S, Corvi R, Fortaner S, Gribaldo L, Halder M, Hoffmann S, Roi AJ, et al. 2004. A modular approach to the ECVAM principles on test validity. Altern Lab Anim. 32(5):467–472.
- Heringa MB, Park MVDZ, Kienhuis AS, Vandebriel RJ. 2020. The value of organs-on-chip for regulatory safety assessment. ALTEX. 37(2):208–222.
- Hoffmann S. 2015. LLNA variability: an essential ingredient for a comprehensive assessment of non-animal skin sensitization test methods and strategies. ALTEX. 32(4):379–383.
- Huang H, Song TJ, Li X, Hu L, He Q, Liu M, Lane MD, Tang QQ. 2009. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proc Natl Acad Sci U S A. 106(31):12670–12675.
- Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Yuan Hsin H, Ingber DE. 2010. Reconstituting organ-level lung functions on a chip. Science. 328(5986):1662–1668.
- ICH. 2011. ICH S6 (R1) guideline on preclinical safety evaluation of biotechnology-derived pharmaceuticals. Amsterdam: EMA.
- ICH. 2013. ICH M3 (R2) guideline on non-clinical safety studies for the conduct of human clinical trials for pharmaceuticals. Amsterdam: EMA.
- ICH. 2020. ICH S5 (R3) guideline on reproductive toxicology: detection of toxicity to reproduction for human pharmaceuticals – step 5. Amsterdam: EMA.
- Jackson EL, Lu H. 2016. Three-dimensional models for studying development and disease: moving on from organisms to organs-on-a-chip and organoids. Integr Biol. 8(6):672–683.
- Junying Y, Kejin H, Kim SO, Shulan T, Stewart R, Slukvin II, Thomson JA. 2009. Human induced pluripotent stem cells free of vector and transgene sequences. Science. 324(5928):797–801.
- Karmaus AL, Filer DL, Martin MT, Houck KA. 2016. Evaluation of food-relevant chemicals in the ToxCast high-throughput screening program. Food Chem Toxicol. 92:188–196.
- Kieninger J, Weltin A, Flamm H, Urban GA. 2018. Microsensor systems for cell metabolism - from 2D culture to organ-on-chip. Lab Chip. 18(9):1274–1291.
- Knapen D, Angrish MM, Fortin MC, Katsiadaki I, Leonard M, Margiotta-Casaluci L, Munn S, O'Brien JM, Pollesch N, Smith LC, et al. 2018. Adverse outcome pathway networks I: development and applications. Environ Toxicol Chem. 37(6):1723–1733.
- Kolesky DB, Homan KA, Skylar-Scott M, Lewis JA. 2018. In vitro human tissues via multi-material 3-D bioprinting. Altern Lab Anim. 46(4):209–215.
- Krebs A, Waldmann T, Wilks MF, van Vugt-Lussenburg BMA, van der Burg B, Terron A, Steger-Hartmann T, Ruegg J, Rovida C, Pedersen E, et al. 2019. Template for the description of cell-based toxicological test methods to allow evaluation and regulatory use of the data. ATLEX. 36(4):682–699.
- Kretzschmar K, Clevers H. 2016. Organoids: modeling development and the stem cell niche in a dish. Dev Cell. 38(6):590–600.
- Lancaster MA, Knoblich JA. 2014. Organogenesisin a dish: modeling development and disease using organoid technologies. Science. 345(6194):1247125.
- Li K, Yang X, Xue C, Zhao L, Zhang Y, Gao X. 2019. Biomimetic human lung-on-a-chip for modeling disease investigation. Biomicrofluidics. 13(3):031501.
- Luechtefeld T, Maertens A, Russo DP, Rovida C, Zhu H, Hartung T. 2016. Analysis of draize eye irritation testing and its prediction by mining publicly available 2008-2014 REACH data. ALTEX. 33(2):123–134.
- Luechtefeld T, Marsh D, Rowlands C, Hartung T. 2018. Machine learning of toxicological big data enables read-across structure activity relationships (RASAR) outperforming animal test reproducibility. Toxicol Sci. 165(1):198–212.
- Martignoni M, Groothuis GMM, de Kanter R. 2006. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2(6):875–894.
- Marx U, Akabane T, Andersson TB, Baker E, Beilmann M, Beken S, Brendler-Schwaab S, Cirit M, David R, Dehne EM, et al. 2020. Biology-inspired microphysiological systems to advance patient benefit and animal welfare in drug development. ALTEX. 37(3):364–394.
- Marx U, Andersson TB, Bahinski A, Beilmann M, Beken S, Cassee FR, Cirit M, Daneshian M, Fitzpatrick S, Frey O, et al. 2016. Biology-inspired microphysiological system approaches to solve the prediction dilemma of substance testing. ALTEX. 33(3):272–321.
- Mastrangeli M, Millet S, Mummery C, Loskill P, Braeken D, Eberle W, Cipriano M, Fernandez L, Graef M, Gidrol X, et al. 2019. Building blocks for a European Organ-on-Chip roadmap. ALTEX. 36(3):481–492.
- Meneely PM, Dahlberg CL, Rose JK. 2019. Working with worms: Caenorhabditis elegans as a model organism. Curr Protoc Essent Lab Tech. 19(1):e35.
- Mirzoyan Z, Sollazzo M, Allocca M, Valenza AM, Grifoni D, Bellosta P. 2019. Drosophila melanogaster: a model organism to study cancer. Front Genet. 10:10.
- Mittal R, Woo FW, Castro CS, Cohen MA, Karanxha J, Mittal J, Chhibber T, Jhaveri VM. 2019. Organ-on-chip models: implications in drug discovery and clinical applications. J Cell Physiol. 234(6):8352–8380.
- Moné MJ, Pallocca G, Escher SE, Exner T, Herzler M, Bennekou SH, Kamp H, Kroese ED, Leist M, Steger-Hartmann T, et al. 2020. Setting the stage for next-generation risk assessment with non-animal approaches: the EU-ToxRisk project experience. Arch Toxicol. 94(10):3581–3592.
- Nesmith AP, Agarwal A, McCain ML, Parker KK. 2014. Human airway musculature on a chip: an in vitro model of allergic asthmatic bronchoconstriction and bronchodilation. Lab Chip. 14(20):3925–3936.
- Nikolova MP, Chavali MS. 2019. Recent advances in biomaterials for 3D scaffolds: a review. Bioact Mater. 4:271–292.
- OECD. 2005. Guidance document on the validation and international acceptance of new or updated test methods for hazard assessment. OECD Series on Testing and Assessment. No. 34. Paris: OECD.
- OECD. 2014. Guidance document for describing non-guideline in vitro test methods. OECD Series on Testing and Assessment. No. 211. Paris: OECD.
- OECD. 2016a. Guidance document for the use of adverse outcome pathways in developing integrated approaches to testing and assessment (IATA). OECD Series on Testing and Assessment. No. 260. Paris: OECD.
- OECD. 2016b. Guidance document on the reporting of defined approaches to be used within integrated approaches to testing and assessment. OECD Series on Testing and Assessment. No. 255. Paris: OECD.
- OECD. 2018. Guidance document on good in vitro method practices (GIVIMP). OECD Series on Testing and Assessment. No. 286. Paris: OECD.
- OECD. 2019. Guiding principles on good practices for the availability/distribution of protected elements in OECD test guidelines. OECD Series on Testing and Assessment. No. 298. Paris: OECD.
- Oleaga C, Riu A, Rothemund S, Lavado A, McAleer CW, Long CJ, Persaud K, Narasimhan NS, Tran M, Roles J, et al. 2018. Investigation of the effect of hepatic metabolism on off-target cardiotoxicity in a multi-organ human-on-a-chip system. Biomaterials. 182:176–190.
- Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. 2014. Generation of functional human pancreatic β cells in vitro. Cell. 159(2):428–439.
- Paini A, Leonard JA, Joossens E, Bessems JGM, Desalegn A, Dorne JL, Gosling JP, Heringa MB, Klaric M, Kliment T, et al. 2019. Next generation physiologically based kinetic (NG-PBK) models in support of regulatory decision making. Comput Toxicol. 9:61–72.
- Paini A, Leonard JA, Kliment T, Tan YM, Worth A. 2017. Investigating the state of physiologically based kinetic modelling practices and challenges associated with gaining regulatory acceptance of model applications. Regul Toxicol Pharmacol. 90:104–115.
- Pamies D, Hartung T. 2017. 21st century cell culture for 21st century toxicology. Chem Res Toxicol. 30(1):43–52.
- Pamies D, Bal-Price A, Chesné C, Coecke S, Dinnyes A, Eskes C, Grillari R, Gstraunthaler G, Hartung T, Jennings P, et al. 2018. Advanced Good Cell Culture Practice for human primary, stem cell-derived and organoid models as well as microphysiological systems. ALTEX. 35(3):353–378.
- Park SE, Georgescu A, Huh D. 2019. Organoids-on-a-chip. Science. 364(6444):960–965.
- Parrish J, Lim K, Zhang B, Radisic M, Woodfield TBF. 2019. New frontiers for biofabrication and bioreactor design in microphysiological system development. Trends Biotechnol. 37(12):1327–1343.
- Patlewicz G, Lizarraga LE, Rua D, Allen DG, Daniel AB, Fitzpatrick SC, Garcia-Reyero N, Gordon J, Hakkinen P, Howard AS, et al. 2019. Exploring current read-across applications and needs among selected U.S. Federal Agencies. Regul Toxicol Pharmacol. 106:197–209.
- Patterson EA, Whelan MP, Worth AP. 2021. The role of validation in establishing the scientific credibility of predictive toxicology approaches intended for regulatory application. Comput Toxicol. 17:100144.
- Perkins R, Fang H, Tong W, Welsh WJ. 2003. Quantitative structure-activity relationship methods: Perspectives on drug discovery and toxicology. Environ Toxicol Chem. 22(8):1666–1679.
- Piersma AH, van Benthem J, Ezendam J, Kienhuis AS. 2018. Validation redefined. Toxicol in Vitro. 46:163–165.
- Piersma AH, van Benthem J, Ezendam J, Staal YC, Kienhuis AS. 2019. The virtual human in chemical safety assessment. Curr Opin Toxicol. 15:26–32.
- Piersma AH, Burgdorf T, Louekari K, Desprez B, Taalman R, Landsiedel R, Barroso J, Rogiers V, Eskes C, Oelgeschläger M, et al. 2018. Workshop on acceleration of the validation and regulatory acceptance of alternative methods and implementation of testing strategies. Toxicol in Vitro. 50:62–74.
- Piersma AH, Genschow E, Verhoef A, Spanjersberg MQI, Brown NA, Brady M, Burns A, Clemann N, Seiler A, Spielmann H. 2004. Validation of the postimplantation rat whole-embryo culture test in the international ECVAM validation study on three in vitro embryotoxicity tests. ATLA Alternatives to Laboratory Animals. 32(3):275–307.
- Pittman ME, Edwards SW, Ives C, Mortensen HM. 2018. AOP-DB: a database resource for the exploration of Adverse Outcome Pathways through integrated association networks. Toxicol Appl Pharmacol. 343:71–83.
- Punt A, Bouwmeester H, Blaauboer BJ, Coecke S, Hakkert B, Hendriks DFG, Jennings P, Kramer NI, Neuhoff S, Masereeuw R, et al. 2020. New approach methodologies (NAMs) for human-relevant biokinetics predictions. Meeting the paradigm shift in toxicology towards an animal-free chemical risk assessment. ALTEX. 37(4):607–622.
- Roost MS, Slieker RC, Bialecka M, Van Iperen L, Gomes Fernandes MM, He N, Suchiman HED, Szuhai K, Carlotti F, De Koning EJP, et al. 2017. DNA methylation and transcriptional trajectories during human development and reprogramming of isogenic pluripotent stem cells. Nat Commun. 8(1):908.
- Rossi G, Manfrin A, Lutolf MP. 2018. Progress and potential in organoid research. Nat Rev Genet. 19(11):671–687.
- Sackmann EK, Fulton AL, Beebe DJ. 2014. The present and future role of microfluidics in biomedical research. Nature. 507(7491):181–189.
- Scialli AR, Daston G, Chen C, Coder PS, Euling SY, Foreman J, Hoberman AM, Hui J, Knudsen T, Makris SL, et al. 2018. Rethinking developmental toxicity testing: evolution or revolution? Birth Defects Res. 110(10):840–850.
- Shrestha J, Razavi Bazaz S, Aboulkheyr Es H, Yaghobian Azari D, Thierry B, Warkiani ME, Ghadiri M. 2020. Lung-on-a-chip: the future of respiratory disease models and pharmacological studies. Crit Rev Biotechnol. 40(2):213–230.
- Smirnova L, Kleinstreuer N, Corvi R, Levchenko A, Fitzpatrick SC, Hartung T. 2018. 3S – systematic, systemic, and systems biology and toxicology. ALTEX. 35(2):139–162.
- Spinu N, Cronin MTD, Enoch SJ, Madden JC, Worth AP. 2020. Quantitative adverse outcome pathway (qAOP) models for toxicity prediction. Arch Toxicol. 94(5):1497–1510.
- Tagle DA. 2019. The NIH microphysiological systems program: developing in vitro tools for safety and efficacy in drug development. Curr Opin Pharmacol. 48:146–154.
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. 2007. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 131(5):861–872.
- Tanaka A, Woltjen K, Miyake K, Hotta A, Ikeya M, Yamamoto T, Nishino T, Shoji E, Sehara-Fujisawa A, Manabe Y, et al. 2013. Efficient and reproducible myogenic differentiation from human iPS cells: prospects for modeling miyoshi myopathy in vitro. PLoS One. 8(12):61540.
- Tavana H, Zamankhan P, Christensen PJ, Grotberg JB, Takayama S. 2011. Epithelium damage and protection during reopening of occluded airways in a physiologic microfluidic pulmonary airway model. Biomed Microdevices. 13(4):731–742.
- Tetsuka K, Ohbuchi M, Kawabe T, Goto T, Kiyonaga F, Takama K, Yamazaki S, Fujimori A. 2020. Reconstituted human organ models as a translational tool for human organ response: Definition, expectations, cases, and strategies for implementation in drug discovery and development. Biol Pharm Bull. 43(3):375–383.
- Tralau T, Oelgeschläger M, Gürtler R, Heinemeyer G, Herzler M, Höfer T, Itter H, Kuhl T, Lange N, Lorenz N, et al. 2015. Regulatory toxicology in the twenty-first century: challenges, perspectives and possible solutions. Arch Toxicol. 89(6):823–850.
- van der Hee B, Madsen O, Vervoort J, Smidt H, Wells JM. 2020. Congruence of transcription programs in adult stem cell-derived jejunum organoids and original tissue during long-term culture. Front Cell Dev Biol. 8:375.
- Van Meer PJK, Kooijman M, Van Der Laan JW, Moors EHM, Schellekens H. 2013. The value of non-human primates in the development of monoclonal antibodies. Nat Biotechnol. 31(10):882–883.
- Van Meer PJK, Graham ML, Schuurman HJ. 2015. The safety, efficacy and regulatory triangle in drug development: impact for animal models and the use of animals. Eur J Pharmacol. 759:3–13.
- Van Norman GA. 2019. Limitations of animal studies for predicting toxicity in clinical trials: is it time to rethink our current approach? JACC Basic Transl Sci. 4(7):845–854.
- Villeneuve DL, Angrish MM, Fortin MC, Katsiadaki I, Leonard M, Margiotta-Casaluci L, Munn S, O'Brien JM, Pollesch NL, Smith LC, et al. 2018. Adverse outcome pathway networks II: network analytics. Environ Toxicol Chem. 37(6):1734–1748.
- Vinken M, Knapen D, Vergauwen L, Hengstler JG, Angrish M, Whelan M. 2017. Adverse outcome pathways: a concise introduction for toxicologists. Arch Toxicol. 91(11):3697–3707.
- Wu Q, Liu J, Wang X, Feng L, Wu J, Zhu X, Wen W, Gong X. 2020. Organ-on-a-chip: recent breakthroughs and future prospects. Biomed Eng Online. 19(1):9.
- Zhang J, Wilson GF, Soerens AG, Koonce CH, Yu J, Palecek SP, Thomson JA, Kamp TJ. 2009. Functional cardiomyocytes derived from human induced pluripotent stem cells. Circ Res. 104(4):e30–e41.
- Zhang YS, Aleman J, Shin SR, Kilic T, Kim D, Shaegh SAM, Massa S, Riahi R, Chae S, Hu N, et al. 2017. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc Natl Acad Sci U S A. 114(12):E2293–E2302.