
Abstract
Chemical regulatory authorities around the world require systemic toxicity data from acute exposures via the oral, dermal, and inhalation routes for human health risk assessment. To identify opportunities for regulatory uses of non-animal replacements for these tests, we reviewed acute systemic toxicity testing requirements for jurisdictions that participate in the International Cooperation on Alternative Test Methods (ICATM): Brazil, Canada, China, the European Union, Japan, South Korea, Taiwan, and the USA. The chemical sectors included in our review of each jurisdiction were cosmetics, consumer products, industrial chemicals, pharmaceuticals, medical devices, and pesticides. We found acute systemic toxicity data were most often required for hazard assessment, classification, and labeling, and to a lesser extent quantitative risk assessment. Where animal methods were required, animal reduction methods were typically recommended. For many jurisdictions and chemical sectors, non-animal alternatives are not accepted, but several jurisdictions provide guidance to support the use of test waivers to reduce animal use for specific applications. An understanding of international regulatory requirements for acute systemic toxicity testing will inform ICATM’s strategy for the development, acceptance, and implementation of non-animal alternatives to assess the health hazards and risks associated with acute toxicity.
Introduction
Regulatory authorities are tasked with managing the safe use of chemicals and products, which often requires the submission of acute toxicity data to estimate human hazard or health risk associated with acute exposures. Typically, standardized acute systemicFootnote1 toxicity tests use rodents as the testing species and lethality as an endpoint to determine the potential systemic toxicity of chemicals via oral, dermal, or inhalation routes (OECD Citation2002a, Citation2002b, Citation2008, Citation2009a, Citation2009b, Citation2017, Citation2018). The dose or concentration expected to produce lethality in 50% of the animals tested, characterized as the LD50 (oral/dermal) or LC50 (inhalation), respectively, is used as a measure of relative potency and to map hazard classifications. Data collected from these tests potentially have multiple regulatory uses, depending on the chemical sector and regulatory authority. These uses include hazard classification and labeling, setting exposure levels, dose setting for repeated dose toxicity, risk assessment, and identifying mechanism of toxic action.
For many substances, the first in vivo toxicity tests performed may be single-dose lethality tests. These tests can use 13–61 animals per substance if all three routes are tested according to Organisation for Economic Co-operation and Development (OECD) test guidelines for acute systemic toxicity (). The current OECD test guidelines for acute toxicity are animal reduction methods compared with analogous guidelines from the 1980s. In addition, test guidelines for the fixed dose procedures for oral (420) and dermal toxicity (402) and the fixed concentration procedure for inhalation (433) offer animal use refinements. These guidelines use evident toxicity as the toxic endpoint, which is more humane than lethality. Although OECD guidance recommends that death, severe pain, and distress be avoided as test endpoints (OECD Citation2000), multiple regulatory guidelines and hazard classification systems use the lethality endpoint as a benchmark. The majority of the OECD acute systemic toxicity test guidelines, including those with the evident toxicity endpoint, result in an LD50 or LC50 range associated with the acute toxicity hazard categories of the Globally Harmonized System of Classification and Labelling of Chemicals (GHS) (UN Citation2021a) (). In addition, those that result in a point estimate of the LD50 or LC50 can also be readily applied to other hazard classification systems.
Table 1. OECD acute toxicity test guidelines.
We have undertaken a review to catalog the global acute systemic toxicity data needs, requirements, and uses for human health hazard evaluations to: (1) assess where available non-animal approaches can be readily incorporated to meet scientific and regulatory demands, (2) gain a clear understanding where development of alternative approaches are most urgently needed, (3) identify areas for international harmonization, and (4) shape international strategies that increase the likelihood of implementing new approach methodologies for acute systemic toxicity testing for hazard and risk assessments.
To fulfill the scope of this review, data were collected by countries and regions collaborating under the International Cooperation on Alternative Test Methods (ICATM). Accordingly, regulatory requirements discussed in this review apply to regulatory jurisdictions within the influence of the following organizations that participate in ICATM: Brazilian Center for the Validation of Alternative Methods, Health Canada, Guangdong Provincial Center for Disease Control and Prevention, European Union (EU) Reference Laboratory for Alternatives to Animal Testing, Japanese Center for the Validation of Alternative Methods, Korean Center for the Validation of Alternative Methods, U.S. Interagency Coordinating Committee on the Validation of Alternative Methods (ICCVAM), and Taiwan National Health Research Institutes. Previous publications focused specifically on the status of acute toxicity testing requirements and data uses by U.S. regulatory agencies (Strickland et al. Citation2018) or on current regulatory requirements for the human health assessment of chemicals and cosmetic products in the EU (Pistollato et al. Citation2021). ICATM was created to foster dialog among national validation organizations to facilitate international cooperation in the critical areas of validation studies, independent peer review, and development of harmonized recommendations for alternative toxicity test methods. Characterizing regulatory needs for specific toxicity endpoints is an important step toward developing and implementing alternatives that fulfill those needs (ICCVAM Citation2018).
Acute systemic toxicity testing requirements by sector
For this review, acute systemic toxicity is defined as adverse effects that are observed within a short time, up to 14 days, for example, after a single dose via oral, dermal, or inhalation exposures. The following sections contain regulatory information for acute systemic toxicity testing by chemical sector, including current testing needs for oral, dermal, and inhalation exposures; data uses; recommended test guidelines; and currently accepted non-animal methods. The chemical sectors covered include cosmetics, consumer products, industrial chemicals, medical devices, pesticides, and pharmaceuticals. We provide a comprehensive characterization of the regulatory requirements for each chemical sector wherever information was available; gaps in coverage indicate that we found no published documentation or applicable regulatory statutes. Relevant regulatory agencies and the laws and regulations that govern each sector are provided in .
Table 2. Acute systemic toxicity regulatory agencies and relevant statutes and regulations.
To aid in public health and safety decisions, jurisdictions may use acute systemic toxicity data to classify substances according to the acute systemic toxicity hazards for each exposure route. Unless otherwise indicated, chemical manufacturers and importers, who may be referred to as chemical sponsors, are responsible for providing the appropriate documentation or testing to evaluate the safety of final products and ingredients manufactured, imported, exported, or sold. Regulatory authorities may also accept suggested hazard classifications for the substance from the manufacturer or importer. Alternatively, the regulator may have sole responsibility for determining a hazard classification.
Documentation provided by manufacturers or importers is used to classify substances based on the classification system of the applicable regulatory jurisdiction or the GHS (UN Citation2021a). Because GHS has yet to be adopted globally, in many countries and all chemical sectors (UN Citation2021b), present a comparison of GHS with other classification systems used by the regulatory jurisdictions covered in this review for oral, dermal, and inhalation hazard categories, respectively. GHS is applied to consumer products in Brazil, China, South Korea, and Taiwan; to industrial chemicals in Brazil and Taiwan; and to pesticides in Brazil, China, Japan, South Korea, and Taiwan. In the EU, the GHS is implemented through the Classification, Labelling and Packaging (CLP) Regulation No. 1272/2008, which is applied to all substances and mixtures supplied in the EU except to chemicals that are in the finished state intended for the final user (medicines, medical devices, cosmetics, veterinary medicines, food and feeding stuff such as food additives, food flavoring, and feeding stuffs used in animal nutrition). However, the EU CLP does not use the least toxic, and optional, category 5. In most countries and regions covered in this review, the medical device and pharmaceutical sectors typically use some form of risk assessment and do not use the GHS or other toxicity classification systems. The exception is the pharmaceutical sector in Japan, which uses Japanese regulatory oral and dermal hazard categories specifically for toxic pharmaceuticals.
Figure 1. Acute oral hazard classifications. CA: Canada; EU: European Union; GHS: Globally Harmonized System of Classification and Labeling of Chemicals; JP: Japan; SK: South Korea; US: United States.

Figure 2. Acute dermal hazard classifications. CA: Canada; EU: European Union; GHS: Globally Harmonized System of Classification and Labeling of Chemicals; JP: Japan; SK: South Korea; US: United States.

Figure 3. Acute inhalation hazard classifications. CA: Canada; EU: European Union; GHS: Globally Harmonized System of Classification and Labeling of Chemicals; JP: Japan; SK: South Korea; US: United States.

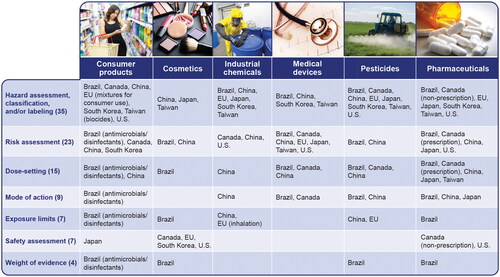
shows other applications of acute systemic toxicity data in addition to hazard classification. The three most frequent applications of acute systemic toxicity data include hazard assessment, classification, and/or labeling (35 cases); risk assessment (23 cases); and dose setting for longer-term studies (15 cases). For countries and sectors that use acute systemic toxicity data for only one purpose, hazard assessment/classification/labeling was the most frequent application (15). Some of the less frequent uses include characterizing mode of action, setting safe exposure limits, safety assessment, and weight-of-evidence (WoE) assessment (overall assessment of acute toxicity characteristics or overall toxicity profile).
Figure 4. Uses for acute systemic toxicity data by chemical sector and country or region. EU: European Union; U.S.: United States.
Consumer products
For this review, consumer products refer to those chemical products packaged for use in the home or school. These might include paints, glue, cleaning products, solvents, paint strippers, treated textiles, laundry detergent, air fresheners, anti-corrosives, colorants, and printer toner. Brazil, China, South Korea, and Taiwan also regulate biocides such as disinfectants or antimicrobials under consumer safety legislation. In fact, the only consumer products regulated by South Korea and Taiwan are those that contain biocides. In China, food, cosmetics, medicine, and tobacco are classified as special consumer products. For the purposes of this review, Chinese information requirements for consumer products will be limited to general consumer products, which include items such as daily chemical hygiene products (i.e. soaps, shampoos, etc.), textiles, detergents, and food-contact materials. Brazil also regulates specific substances in school supplies such as paint, glue, watercolors, and powder material. In the EU, consumer products are articles such as textiles treated with chemicals, or mixtures such as cleaning products and paints.
The most frequent uses of acute systemic toxicity data for consumer products include hazard assessment, hazard classification, and hazard labeling (). Some authorities also use such data for risk assessment, dose setting for longer-term studies, setting safe exposure limits, safety assessment, and defining mechanism of toxic action.
Brazil
In Brazil, the consumer products that require acute systemic toxicity testing are antimicrobials, disinfectants, toys, and school supplies. Brazil requires that acute systemic toxicity by oral, dermal, and inhalation routes be assessed using test methods that are internationally recognized, such as OECD test guidelines (). The information needed from acute systemic toxicity tests includes LD50, clinical observations (mortality, nature, severity, and duration of effects), body weight, gross necropsy (necropsy and histopathological findings), and GHS classification (). Brazil does not currently accept any non-animal methods for assessment of acute systemic toxicity of antimicrobials and disinfectants. However, such methods will be considered for their applicability under current regulations when published by an international authority such as OECD.
Acute oral systemic toxicity information is required for toys and school supplies to determine hazard classification, i.e. whether they are nontoxic (having an oral LD50 equal to or greater than 2000 mg/kg). Testing for school supplies should be those described in the Brazilian Association of National Standards No. 15236, but OECD test guidelines are also acceptable. School items containing paint, glue, watercolors, and powder material in quantities of greater than 3 g/unit of use should be tested while substances less than 3 g/unit of use are exempt. However, chemical substances used in school supplies must be GHS category 5 or unclassified (LD50 greater than or equal to 2000 mg/kg) for acute oral toxicity. For school supplies, non-animal methods may only be used for specific situations. For example, information from an in vitro assay can be used to eliminate further testing if such information supports a prediction that the product’s LD50 is greater than 2000 mg/kg and there are historical data about the safety of the product.
Canada
Canadian manufacturers and importers use acute toxicity information from all possible routes of exposure to classify the hazards present in their products, which trigger requirements to display labeling with hazard symbols and warning statements, instructions for safe use and first-aid treatment, and packaging requirements when applicable (see classifications in ) (Minister of Justice Canada Citation2019). The label must also disclose all hazardous ingredients present at a concentration of 1% (by weight) or more (Minister of Justice Canada Citation2019) that contribute to the classification results. Consumer chemicals in the “very toxic” category are prohibited and any “toxic” product must be packaged in a child-resistant container. Health Canada does not require submission of premarket acute toxicity test results for consumer chemical products, but they may require this information to verify compliance (Health Canada Citation2007).
When classifying the hazards of a consumer product, Health Canada suggests that human data take precedence over animal data. Animal study data should be generated using methods described in OECD test guidelines (), but methods from other national or international standards may be acceptable. Available data from the scientific literature may also be acceptable in lieu of testing. If no data from animal tests are available, non-animal data from in silico or in vitro methods may be considered. When data for a product are lacking, data from individual product ingredients or similar formulations may be used to assess acute toxicity hazard information. In cases where a consumer product contains an ingredient of concern, Health Canada may conduct a quantitative risk assessment using data from the scientific literature.
China
For general consumer products, regulatory authorities in China require acute oral and inhalation, but not dermal, toxicity information for GHS hazard classification and labeling, risk assessment, and dose setting for longer-term studies (General Administration of Quality Supervision Inspection and Quarantine of the People’s Republic of China Citation2008). Acute oral study results should provide the LD50 for finished products. Guidance for performing acute oral testing recommends animal reduction and refinement methods such as the up-and-down method, the acute toxic class method, and the fixed dose procedure (Standardization Administration of China Citation2008a, Citation2008b, Citation2012b). Guidance for performing inhalation testing recommends animal tests with multiple dose groups (Standardization Administration of China Citation2008c) and two animal reduction and refinement alternatives: the acute toxic class method and the fixed concentration procedure (Standardization Administration of China Citation2011, Citation2012a). No non-animal approaches are accepted for acute oral or inhalation toxicity of consumer products in China.
European Union
In the EU, consumer products are articles (products with a shape, surface, or design that determines function to a greater degree than chemical composition), which include textiles treated with chemicals, newspapers, plastic packaging, etc., or mixtures such as cleaning products and paints. The main pieces of chemical legislation are the CLP regulation for classification and labeling and the Registration, Evaluation, Authorization and Restriction of Chemicals (REACH) Regulation No. 1907/2006 (EC Citation2006) for setting data requirements for registration of chemicals and the rules for risk assessment and chemical management. Both legislations apply to industrial and consumer uses of chemicals. The relevant information with regard to information requirements for acute systemic toxicity is described in the section on the requirements for industrial chemicals in the EU.
Japan
No acute systemic toxicity information is required in Japan. Manufacturers, importers, and sellers of household products are responsible for the conduct of any toxicity tests deemed necessary to ascertain the safety of their products. Methods used should be adequate to demonstrate the safety or hazard of the tested substance; there is neither a requirement that animal testing be used nor a limitation on animal testing. There are 21 hazardous substances in household products regulated for the ordinary use of general consumers. The substances include methanol, trichloroethylene in aerosol products, hydrochloric acid and sodium hydroxide in cleaners, formaldehyde, tributyltin compounds, triphenyltin compounds, and azo compounds in textile products. Manufacturers, importers, and sellers must conform to specific limitations on the amounts of these substances in household products.
South Korea
In South Korea, acute oral, dermal, and inhalation toxicity data are required for the purpose of hazard classification and labeling for consumer products that are biocides or formulations containing biocides. The primary information needed from these tests is LD50 and LC50 values. The GHS system is used for classification. All the current acute toxicity test guidelines from OECD are acceptable (). In addition, the non-animal approaches considered in a WoE approach to support a classification decision include read-across, in silico predictions, and in vitro and in chemico data.
Taiwan
In Taiwan, products containing biocides are the only consumer products that require acute systemic toxicity data. The primary acute systemic toxicity information needed for products containing biocides are LD50 and LC50 values or ranges, mortality and moribundity, clinical observations, gross necropsy findings, and body weight. Information for dermal acute systemic toxicity tests should include the lethality range and skin irritation (Draize score). This information is used for hazard classification according to the GHS. Data are accepted from in vivo acute oral, dermal, and inhalation toxicity test methods described in current guidelines from OECD (). New testing is not required if data are available from the scientific literature or from available databases. Data from non-animal methods are not currently accepted.
United States
In the USA, manufacturers and sellers of consumer products use acute toxicity information to determine hazard classification and labeling (). In addition to animal tests for acute oral, dermal, and inhalation toxicity described in the regulations (CPSC Citation2019), the U.S. Consumer Product Safety Commission (CPSC) may also consider data generated using OECD test guidelines and other scientifically supported methods. CPSC has no requirements for animal testing and strongly encourages manufacturers to base labeling on existing data and alternatives to animal testing whenever possible (CPSC Citation2015). CPSC lists acceptable alternatives to animal tests on its website; however, no non-animal methods for acute systemic toxicity are currently listed (CPSC Citation2012). CPSC will consider, on a case-by-case basis, data from methods not previously approved. To assist in this effort, the agency has recently published draft information needs for the evaluation of alternative method nominations (CPSC Citation2021).
Cosmetics
Cosmetics are typically defined as substances applied either externally to the human body or to the teeth and the mucous membranes of the oral cavity for the purpose of cleaning, perfuming, protecting, changing appearance, keeping in good condition, or correcting odors. Thus, they typically include makeup, skin lotions, soaps, perfumes, deodorants and antiperspirants, hair care products, nail care products, and oral care products. For cosmetic products, acute systemic toxicity data are most frequently used for safety assessments ().
Brazil
Acute oral toxicity testing is required in Brazil for premarket approval of cosmetics, perfumes, and children’s toiletries. Any cosmetics that straighten or curl hair must have acute toxicity information for oral, inhalation, and dermal routes on the active ingredient, but for finished products a theoretical calculation for acute oral toxicity is allowed. Acute systemic toxicity information is used for setting exposure levels, determining doses for subsequent tests, risk assessment, and WoE assessments. Regulators use in vivo information such as dose response, clinical observation, body weight, gross necropsy, histopathology, and LD50. Information can be generated using OECD methods or any alternative method validated for regulatory purposes. Test waivers may be acceptable with technical justification. Alternative methods recognized by the National Council for Control of Animal Experimentation (CONCEA) are generally accepted (CONCEA Citation2014); however, no non-animal alternatives for acute systemic toxicity are recognized. Outside of CONCEA and OECD methods, other non-animal alternatives may be accepted on a case-by-case basis, provided that the information generated is sufficient to support a decision on product safety.
Canada
There are no specific toxicity testing requirements for cosmetic products under the Cosmetics Regulations in Canada. Manufacturers and importers must notify Health Canada within 10 days after first selling a cosmetic in Canada and provide information about the form and function of the cosmetic, and the identity and concentrations of its ingredients. Health Canada may request evidence to establish the safety of a cosmetic if safety concerns are identified. In most circumstances, safety can be established through non-animal testing approaches such as in vitro testing, computer-based models, and the use of existing information on similar chemicals. However, new substances used in cosmetics may have acute toxicity data requirements under the New Substances Notification Regulations of the Canadian Environmental Protection Act (see the section on industrial chemicals for Canada).
China
In China, cosmetics are classified as special or general cosmetics. Special cosmetics, which include products such as hair dye, chemical hair waving solutions, whitening, sunscreen, products to reduce hair loss, or new products with efficacy claims are considered higher risk and are subject to registration, which expires after five years, with the National Medical Products Administration. Conversely, general cosmetics are subject to notification, which has no expiration. Safety assessments, which include acute oral and dermal toxicity information, must be performed on individual cosmetic ingredients for both types of products (National Medical Products Administration Citation2021). Acute inhalation toxicity information is also needed if inhalation is an important route of exposure.
Acute systemic toxicity data for cosmetics are used to inform hazard classification and labeling and perform human health risk assessment. Regulators generally desire the dermal LD50 to make regulatory decisions for cosmetics safety; however, animal tests are not required for general-use cosmetics or any cosmetics shipped from overseas.
European Union
Manufacturers of cosmetics must submit a product safety report to the European Commission prior to placing a product on the market. The Scientific Committee on Consumer Safety (SCCS) performs a safety evaluation on specific cosmetic substances identified in annexes to the cosmetics regulation as being of potential concern for human health (namely colorants, preservatives, hair dyes, and chemical UV filters). Animal tests for the safety evaluation of cosmetic ingredients are prohibited. Animal data are only accepted if (1) tests were performed before the testing and marketing bans were applied on 11 March 2013 or (2) the data were obtained in order to be in compliance with legislation regulating substances other than cosmetics (SCCS Citation2021).
Data on acute systemic toxicity are not mandatory for the safety assessment of cosmetic ingredients for consumer use. Acute toxicity plays a limited role in cosmetics assessments because appropriately used ingredients pose little risk of acute toxicity, and sufficient information about toxicity is often available from repeat-dose studies if conducted before 2013 (EC Citation2013c; Pistollato et al. Citation2021).
If acute toxicity information is needed, a WoE approach using chemical grouping or read-across, QSAR, in vitro studies, or repeated dose toxicity studies may be sufficient (Pistollato et al. Citation2021). The approaches outlined in OECD Guidance Document 237 on bridging or waiving should be considered (OECD Citation2016). Bridging, also referred to as read-across, is the use of existing data from one substance to characterize the hazard of another substance of similar composition for which there are little or no data. To date, there are no validated non-animal methods for acute dermal or inhalation toxicity. The only validated non-animal method for acute systemic toxicity is the 3T3 neutral red uptake test, which is an in vitro approach that has been determined to be sufficient, when used with additional data, to identify substances that are non-classified (i.e. having an LD50 greater than 2000 mg/kg) (JRC Citation2013; Prieto et al. Citation2013). The EU will consider scientifically justified non-animal methods that have not yet been officially validated for assessing acute systemic toxicity on a case-by-case basis.
Japan
In Japan, “general cosmetics” are products such as moisturizing lotion and makeup that are applied to the body for cleansing, for enhancing attractiveness, or for maintaining a healthy condition, while “quasi-drugs” are medicated cosmetics that include products applied to correct skin or hair conditions, such as acne or hair loss. Acute toxicity information is not required to register new ingredients of general cosmetics in Japan. The Standards for Cosmetics (Ministry of Health and Welfare Citation2000) generates a positive and negative list of cosmetic ingredients, where ingredients on the positive list are permitted within a maximum limit and ingredients on the negative list are not allowed. Acute oral toxicity information is needed for new ingredients on the positive list and for quasi-drugs to inform hazard classification.
For a new cosmetic ingredient to be considered for inclusion on the positive list, an approximate oral LD50 is required but dermal and inhalation toxicity information are not needed (MHLW Citation2017). For new quasi-drugs, both ingredients and finished products must be tested for acute oral toxicity, unless the active ingredient has an oral LD50 of at least 2000 mg/kg, in which case no additional tests are required for products that contain the active ingredient. Data are accepted from any acute oral test described in an OECD test guideline.
Use of the neutral red uptake cytotoxicity assay (OECD Citation2010) to predict acute oral LD50 of at least 2000 mg/kg is accepted in a WoE approach as an alternative method for acute oral toxicity testing for the ingredients of general cosmetics and additives, but not active ingredients, of quasi-drugs.
South Korea
As of 2016, animal testing is not permitted by South Korea for ingredients used solely in cosmetics. The need for acute systemic toxicity test data for evaluating the safety of functional cosmetics is now waived. According to the Regulation on the Examination of Functional Cosmetics, non-animal methods must be used for the safety evaluation of new functional cosmetics ingredients (Ministry of Food and Drug Safety Citation2021).
Taiwan
In Taiwan, animal testing for cosmetic safety evaluation has been banned since 2019. Data for cosmetic safety from animal tests are not accepted unless an explanation and related supporting documents for the necessity to conduct animal tests are provided. For data from an animal test to be accepted, the 2019 Regulations Governing the Application of Animal Testing for the Safety Assessment of Cosmetics or Cosmetic Ingredients requires the submission of documentation indicating that the cosmetic ingredient may be acutely harmful and that there are no non-animal methods available to evaluate its acute systemic toxicity (Ministry of Health and Welfare Citation2019). If animal testing is deemed necessary, methods described in OECD Test Guideline 423 for acute oral toxicity, Test Guideline 402 for acute dermal toxicity, and Test Guideline 436 for acute inhalation toxicity can be used. Clinical studies in humans are also recommended. No non-animal approaches are accepted for acute systemic toxicity.
United States
No acute systemic toxicity testing or data are required for cosmetics in the USA. Companies and individuals who manufacture or market cosmetics are legally responsible for ensuring the safety of their products. The U.S. Food and Drug Administration (FDA) Center for Food Safety and Applied Nutrition advises that the safety of a product can be adequately substantiated through (1) reliance on available toxicological test data for individual ingredients and for product formulations that are similar in composition to the product of interest and (2) performance of additional toxicological and other tests that are appropriate in light of existing data and information (FDA Citation1975). In cases where the safety of a cosmetic product has not been determined, the product must be labeled as such (FDA Citation2016).
Industrial chemicals
Industrial chemicals are typically those that are used in manufacturing and commerce and not regulated by statutes for other substances such as medicines, pesticides, food or feed additives. The most frequent uses of acute systemic toxicity data for industrial chemicals are hazard assessment, classification, and labeling ().
Brazil
Currently, Brazil does not have legislation or specific regulations for managing industrial chemicals. However, acute toxicity data are needed for all industrial chemicals used in the workplace to support GHS classification and labeling and to enable documentation of hazards in safety data sheets (Brazil Ministry of Labor and Employment. Secretary of Labor Inspection Citation2011). Because the chemical industry in Brazil rarely produces new substances, acute toxicity data needed for classification and labeling are usually available from other sources. Thus, the industrial chemical sector in Brazil has no impact on animal testing for acute systemic toxicity.
Canada
In Canada, acute systemic toxicity data for industrial chemicals are used for risk assessment within the regulatory framework to control substances of concern. Industrial substances fall into two main groups: existing substances and new substances. Existing substances are on the Canadian Domestic Substances List and are manufactured or imported into Canada on a commercial scale. Acute toxicity information is not needed for existing substances. New substances are those not on the Domestic Substances List. New substances that are in commerce internationally are on the Non-domestic Substances List. New substances on the Non-domestic Substances List are subject to the notification requirements set out in the Regulations; however, they are subject to fewer information requirements in comparison to new substances that are not on the Non-domestic Substances List.
Mammalian acute toxicity data are required prior to import or manufacture of new chemicals and polymers beyond specified quantities. A mammalian oral, dermal, or inhalation acute toxicity test, selected on the basis of the most significant route of potential human exposure, is required for new chemicals produced or imported in quantities greater than 1000 kg/year and for those on the Non-domestic Substances List produced or imported in quantities greater than 10 000 kg/year (Government of Canada Citation2005). For new substances produced or imported in quantities greater than 10 000 kg/year, mammalian acute toxicity data for a second route are required unless the boiling point of the substance is less than 0 °C and acute inhalation data are available. Mammalian acute oral toxicity data are required for new polymers and biopolymers produced or imported in quantities greater than 10 000 kg/year.
All mammalian toxicity testing must be compliant with Good Laboratory Practices (GLP) and conducted according to, or consistent with, OECD test guidelines. In lieu of mammalian acute toxicity test data, waiver requests for specific tests and read-across data may be submitted for new substances. Other alternatives, such as predictive models, technical tools, and professional judgment, can be used to assess potential risks in a WoE approach. Any alternative to OECD GLP testing is subject to review and approval by the New Substances program.
China
Acute toxicity information on industrial chemicals for oral and inhalation routes is required in China for setting exposure levels, hazard classification and labeling, risk assessment, dose setting in longer-term studies, and determining mechanism of toxic action.
The information needed from acute oral and inhalation tests includes LD50 and gross pathology. Both active ingredients and finished products containing active ingredients must be tested. Guidance for performing acute oral testing recommends animal reduction and refinement methods such as the acute toxic class method, the fixed dose procedure, and the up-and-down method (Standardization Administration of China Citation2008a, Citation2008b, Citation2012b). Guidance for performing inhalation testing recommends a standard inhalation test with multiple dose groups tested simultaneously (Standardization Administration of China Citation2008c) and two animal reduction and refinement methods, the fixed concentration procedure and the acute toxic class method (Standardization Administration of China Citation2011, Citation2012a). No non-animal approaches are accepted for assessing acute oral and inhalation toxicity of industrial chemicals.
European Union
In the EU, REACH is connected to CLP. Acute systemic toxicity data for industrial chemicals generated according to REACH information requirements (which are tonnage triggered) are used to classify substances or mixtures into four hazard categories for acute systemic toxicity aligned to the GHS (UN Citation2021a, Citation2021b) for oral, dermal, and inhalation exposure (). New testing is not required under CLP Regulation and, whenever possible, classifications should be based on the available relevant information. Results of quantitative structure–activity relationships (QSARs), read-across, and in vitro testing may be used in a WoE approach to establish hazard classification (ECHA Citation2017b). If new data are generated, studies must be conducted in accordance with the EU test methods (EC Citation2008) or other validated international test methods such as those described in OECD test guidelines ().
Acute inhalation data may be used to establish maximum limits for human exposure if the substance is classified as an acute toxicity hazard and there is a potential for high peak exposures, which may occur for example, when transferring volatile substances from one vessel to another (ECHA Citation2012, Citation2017a).
Dermal testing is not necessary for substances manufactured or imported at quantities of greater than 10 tons per year that are not classified by the oral route (i.e. oral LD50 is greater than or equal to 2000 mg/kg) for which no systemic effects have been observed in in vivo studies involving dermal exposure (e.g. skin irritation or sensitization) or are predicted by non-testing approaches such as read-across and QSAR. If there are available test data from a 28-day oral study including doses up to 1000 mg/kg/day and no effects are seen, it can be concluded that the substance is not acutely toxic and no further testing is needed (ECHA Citation2017b).
Testing in animals is to be a last resort (EC Citation2006; ECHA Citation2017b). An in vivo acute oral toxicity study can potentially be avoided using a WoE approach that includes the results of a 28-day oral study (Gissi et al. Citation2017, Citation2018; Buesen et al. Citation2018) combined with non-animal testing methods such as read-across. The WoE approach is intended for substances manufactured or imported at quantities of greater than 10 tons per year for which an oral repeated dose 28-day toxicity study or a repeated dose study combined with the reproduction/developmental toxicity screening test is required (ECHA Citation2017b). If the WoE evaluation leads to the assumption of low or no expected acute oral toxicity (i.e. that LD50 would be greater than 2000 mg/kg), animal testing can be avoided. Data from the in vitro neutral red uptake cytotoxicity assay may be used in a WoE approach for predicting acute oral toxicity or to determine starting doses for in vivo oral studies, but this assay cannot be used as a stand-alone test. Also, the OECD guidance on bridging or waiving should be considered (OECD Citation2016).
Any new acute systemic toxicity studies must be conducted in accordance with the EU test methods per Commission Regulation (EC) No. 440/2008 regarding test methods accepted under the REACH regulation (EC Citation2008). Alternatively, they may use other internationally accepted test methods such as those described in OECD test guidelines (). The fixed dose/concentration procedures that use signs of non-lethal toxicity are preferred due to the animal welfare advantages (ECHA Citation2017b).
Japan
Japan uses acute toxicity information for hazard classification to assist in regulating the manufacture and use of new and existing chemicals and the import of chemicals ().
Domestically manufactured and imported chemicals must be tested for acute oral, dermal, and inhalation toxicity. Pure substances, as well as dilutions containing active ingredients, must be tested, but testing is not required for mixtures or final formulations. Testing may be performed according to any of the current OECD acute toxicity test guidelines (). No non-animal toxicity tests are accepted for acute systemic toxicity, however, dermal tests may be waived for substances that are positive for in vitro skin corrosivity, unless there is compelling evidence to suggest that the substance would be dermally absorbed and produce toxicity. In such cases, the test substance may potentially be assessed as either a poisonous or deleterious substance based on toxic potency per the oral or inhalation routes.
South Korea
For industrial chemicals, South Korea requires data for at least one acute systemic toxicity route to determine whether a chemical should be designated as a toxic substance. A chemical is designated as toxic if the oral LD50 is less than 300 mg/kg, the dermal LD50 is less than 1000 mg/kg, or the inhalation LC50 is less than 1 mg/L (after a 4 h exposure to mist or dust). Oral toxicity data are required for chemicals produced in quantities over one ton/year and dermal or inhalation toxicity data are required for substances produced at greater than 10 tons/year (Ha et al. Citation2016).
The primary information needed from acute systemic toxicity tests is LD50 and LC50 values. Methods described in current OECD acute toxicity test guidelines are acceptable for generation of acute data (). The non-animal methods accepted, when used in a WoE approach, include read-across, in silico predictions, and in vitro and in chemico tests.
Taiwan
Acute systemic toxicity data are required in Taiwan for hazard assessment and for the hazard classification and labeling of industrial chemicals. The testing required depends upon the amount of chemical produced or imported. Chemicals produced or imported in quantities greater than 10 tons/year must be tested by two routes of incidental exposure to humans; those produced or imported in quantities between 1 and 10 tons/year must be tested by one exposure route; and those produced or imported in quantities less than 1 ton/year need only physicochemical information. The primary information needed from acute systemic toxicity tests are LD50 or LC50 values or ranges, signs of toxicity (mortality and/or moribund status), clinical observations, gross necropsy findings, body weight, and GHS hazard classification. Information for dermal acute systemic toxicity tests should include the lethality range and degree of skin irritation as characterized by Draize score. Data generated using the current acute toxicity test guidelines from OECD are acceptable. No non-animal approaches are accepted. When acute systemic toxicity data are available, from the scientific literature or databases, for example, no new acute toxicity study shall be performed for registration.
United States
Acute toxicity information for industrial chemicals is used for suggesting language for safety data sheets, for conducting occupational risk assessments, and for determining appropriate levels of personal protective equipment (Scarano Citation2015). While there are no specific requirements for the submission of acute systemic toxicity data, chemical sponsors must submit all available toxicity data to the U.S. Environmental Protection Agency (EPA). Rodent data for acute systemic toxicity endpoints are usually submitted with premanufacture notices, but any data provided to predict acute toxicity are reviewed, including data from non-animal methods if accompanied by adequate scientific justification (Scarano Citation2015). Data generated using methods described in EPA and OECD test guidelines are accepted. Non-guideline animal tests and non-animal tests may also be used. Examples of accepted non-animal methods are read-across for predicting acute toxicity of a target substance and the use of physicochemical data for determining the likelihood of absorption by all routes. The information generated by any animal or non-animal method must be sufficient for EPA to judge the safety of a chemical with respect to acute effects, including lethality. EPA may use predictive models, technical tools, and professional judgment to assess potential risks.
Medical devices
For the purposes of this review, a medical device is a product such as an instrument, machine, or implant that is intended for use in the diagnosis, prevention, or treatment of diseases or other medical conditions. In vitro diagnostics are not considered to be medical devices. The regulatory authorities for all countries or regions surveyed recognize the 10993 series of medical device safety standards from the International Organization for Standardization (ISO) or provide similar guidance. However, approaches to the application of ISO guidances may differ among jurisdictions.
According to the ISO 10993 standards, toxicity evaluations fall within the broader category of biocompatibility evaluations, which consider the ability of a material to perform with an appropriate host response in a specific application. ISO 10993-1:2018, Biological Evaluation of Medical Devices – Part 1: Evaluation and Testing within a Risk Management Process, provides the framework for the biological evaluation of medical devices, emphasizing a tiered approach based on risk of systemic toxicity to limit the need for in vivo testing (ISO Citation2018). Thus, the most frequent use of acute systemic toxicity data for medical devices is to assess the risk of potential systemic toxins that may leach from medical devices ().
ISO guidance recommends acute toxicity testing for implanted devices and for those that have components outside the body but have direct or indirect contact with internal body fluids or tissues (referred to as “externally communicating” devices). However, characterization of the acute toxicity of the materials and the processes for manufacturing the finished device using existing data is recommended before any animal testing is performed (ISO Citation2018). The purpose of this information is to determine the potential toxicity of substances that could be leached or extracted from a device. If new acute systemic toxicity data are needed, tests are conducted in accordance with ISO 10993-11: 2017, Biological Evaluation of Medical Devices – Part 11: Tests for Systemic Toxicity (ISO Citation2017). This standard recommends documenting observed effects such as body weight change, changes in food and water consumption, clinical observations and adverse clinical signs, and mortality. Furthermore, hematology and clinical chemistry data, urinalysis, gross pathology, and organ weights may also be considered in specific cases, such as for device materials with expected or observed toxicity in previous studies or for new device materials with no previous clinical experience. Few alternatives to animal testing are recommended, but ISO 10993-11 does recommend using in vitro cytotoxicity data to determine starting doses for acute oral toxicity tests to reduce animal use for oral systemic toxicity evaluations (ISO Citation2017) and for determining whether substances are non-toxic by the oral route.
Brazil
Brazilian authorities require animal tests for acute and other systemic toxicity data to inform the overall risk assessment that may impact the labeling of medical devices. Acute toxicity studies of finished products or their extracts may be used to establish a dosage regimen for subacute, subchronic, and other studies, and may provide information on the mode of toxic action by the intended clinical exposure route. Thus, oral, dermal, or inhalation tests may be required, depending on the medical device’s intended use and its route of exposure.
Test waivers are the only acceptable non-animal approach for acute systemic toxicity. Toxicity studies may be waived with technical justification that (1) documents the impracticality of carrying out the animal study or (2) verifies that toxicological outcomes from alternative methods are sufficient to make a regulatory decision on product safety. The acceptance of non-animal methods for medical device testing requires that they be validated specifically for medical devices (which includes the testing of extracts) or recognized internationally for regulatory purposes, such as ISO standards or OECD methods.
Canada
Medical devices are generally tested using the final, sterilized device or, if not feasible, representative components of a device may be considered. As part of the biocompatibility evaluation, Health Canada accepts the outcomes from acute and longer term toxicity studies to provide information on safety and effectiveness of the device and its mode of toxic action. To obtain this information, animals, usually rodents, receive single or multiple exposures to medical devices or their extracts for a 24-h period by the intended clinical exposure route (as per the ISO 10993 standard with reference to OECD test guidelines). Blood or tissue samples may be collected for assessment and, following an observation period, the animals may be euthanized for the evaluation of clinical pathology, gross pathology, and histopathology.
Biocompatibility testing may be waived if the device under consideration is similar in design and materials to an existing licensed device.
China
Acute systemic toxicity is one of the biocompatibility endpoints evaluated during preclinical safety testing of medical devices in China (Shan and Liu Citation2020). Acute oral and dermal systemic toxicity information is required for medical devices to classify and label hazards, document lethality potential, conduct human health risk assessments, and to set doses for longer-term studies. Only extracts from finished products must be tested.
The National Medical Products Administration’s guidance, Biological Evaluation of Medical Devices, provides recommendations for required tests (Shan and Liu Citation2020). This guidance is provided in multiple parts, similar to the multipart ISO 10993 guideline, Biological Evaluation of Medical Devices, which recommends submitting acute systemic toxicity information for all externally communicating and implanted devices (ISO Citation2009). Rodents are typically tested by administering single or multiple medical device extracts within one 24-h period by the oral and dermal routes. Animals are observed for clinical signs for at least three days, and then euthanized for the evaluation of gross pathology. LD50 values for oral and dermal exposure routes are calculated. Clinical pathology and histopathology may also be examined when deemed appropriate.
No non-animal methods are accepted for acute oral and dermal systemic toxicity information; however, an in vitro cytotoxicity test is recommended for selecting starting doses for acute oral toxicity to reduce the number of animals used.
European Union
All marketed devices in the EU must meet general safety and performance requirements (Keene Citation2020). The requirements do not explicitly mention biocompatibility or acute systemic toxicity; however, general safety and performance requirements can typically be met by following the ISO guidelines for the biological evaluation of medical devices.
Acute systemic toxicity testing is conducted to determine whether substances that can be leached or extracted from a device could cause toxicity. Depending on the expected clinical exposures, there are multiple routes that could be evaluated. For oral, intravenous, dermal, or inhalation studies, rodents typically receive single or multiple administrations of medical device extracts within one 24-h period. Rabbits may be used for dermal or implantation studies. Following an observation period of at least three days for clinical signs, symptoms, and body weight, the animals are euthanized; gross pathology and potentially clinical pathology and histopathology may be evaluated.
ISO 10993-11 recommends using in vitro cytotoxicity data to determine starting doses for acute oral toxicity tests to reduce animal use for oral systemic toxicity evaluations, but no alternatives to replace animal use are recommended (ISO Citation2017).
Japan
In Japan, acute systemic toxicity testing is used to confirm that a medical device does not release chemicals (Kojima and Sakaguchi Citation2020). The testing is performed following ISO guidelines. Typically, a single dose of device extract is administered to mice intravenously (for a saline extract) or intraperitoneally (for an oil extract) to evaluate whether any systemic effects occur within 72 h (Kojima and Sakaguchi Citation2020). Clinical effects and body weights are noted and compared to control animals to determine whether the extracts produce an effect. Evaluation of gross pathology, clinical pathology, organ weights, and histopathology is performed if clinically indicated or if longer exposure testing is not anticipated. No non-animal approaches are accepted.
South Korea
Medical device applications in South Korea must include acute systemic toxicity information to assess chemical hazard. Acute systemic toxicity information is required for medical devices for the routes relevant to clinical exposure and test protocols must use both polar and nonpolar eluates (Ministry of Food and Drug Safety Citation2014). Endpoints evaluated during testing should include body weight change, gross pathology, and clinical effects, as well as a dose–response assessment. Methods described in ISO test guidelines are recommended to evaluate toxicity for these exposure routes. Inhalation toxicity information is not needed. Individual chemical components of devices and extract mixtures must be tested. No non-animal methods are accepted for testing; however, chemical sponsors are encouraged to apply the approaches for reducing animal use described in the ISO test guidelines for acute systemic toxicity.
Taiwan
Acute systemic toxicity information is one of the biocompatibility tests required for medical device registration in Taiwan. The requirement guidance typically follows the ISO 10993 guideline (ISO Citation2018).
Acute toxicity studies are conducted using two types of extractions prepared from devices or materials (usually those prepared using both polar and nonpolar solvents) and injected into an animal by the intravenous (polar extract) and intraperitoneal (nonpolar extract) routes. The information required includes body weights, clinical observations, mortality, and gross necropsy findings. The biological reactivity of treated animals must be less than those of animals treated with the vehicle control. No non-animal approaches are accepted.
United States
In 2020, the FDA Center for Devices and Radiological Health (CDRH) has recently updated its guidance for using ISO 10993-1 (FDA Citation2020). CDRH recommends submitting acute systemic toxicity information for surface devices that contact breached or compromised skin (regardless of duration of contact) or have prolonged or permanent contact with mucosal membranes, in addition to the recommendations in the ISO guidance, which recommend testing externally communicating and implanted devices.
Acute systemic toxicity testing conducted to support a submission to CDRH based on ISO 10993 usually involves dosing mice with single or multiple injections of medical device extracts (often intraperitoneally or intravenously) within one 24-h period. Following an observation period of at least three days, the animals are euthanized and clinical pathology, gross pathology, and histopathology may be evaluated (ISO Citation2006).
Chemistry information considered in conjunction with literature data can be used to waive testing for acute systemic toxicity endpoints if the dose, route, and frequency of exposure from the literature report(s) are consistent with the proposed device use. For example, no-observed-adverse-effect levels and lowest-observed-adverse-effect levels from published systemic toxicity studies can often be used to waive acute systemic toxicity testing if exposure data for device extractables or leachables demonstrate that exposure will be below these effect levels. Similarly, a separate biocompatibility assessment of acute toxicity may not be recommended if these acute endpoints can be included in another in vivo study design. CDRH’s guidance for the use of ISO 10993-1 states that an alternative approach can be used if it “satisfies the requirements of the applicable statutes and regulations” (FDA Citation2020). Sponsors who want to use an alternative approach are encouraged to contact CDRH prior to conducting any testing. New testing may be avoided if a sponsor can explain why the materials and manufacturing are not expected to adversely impact biocompatibility.
Pesticides
Pesticides are typically defined as agents for pest management, prevention, or mitigation. These include, but are not limited to, substances such as fungicides, insecticides, herbicides, rodenticides, disinfectants, attractants, plant defoliants, and plant growth regulators. The most frequent uses of acute systemic toxicity data for pesticides are hazard classification and labeling ().
Brazil
In Brazil, pesticide sponsors submit acute systemic toxicity information to the Brazil National Health Surveillance Agency for hazard classification and labeling (MAPA, ANVISA, IBAMA Citation2013). Acute systemic toxicity data are also used for risk assessment, transport regulations, dose setting for longer-term studies, information on mechanism of toxic action, and WoE assessments for acute toxicity or toxicity in general.
Technical ingredients and formulated products must undergo toxicological assessment via the acute oral, dermal, and inhalation routes (ANVISA Citation2019). For acute inhalation toxicity, formulations may be exempt from testing when there are: (1) reliable existing inhalation data for all ingredients, (2) reliable data from validated methods that predict lesser or equal toxicity to that of a comparable technical product, and (3) documentation that at least 99% of a solid formulation is compose of particles of a least 50 µm in diameter. Regarding pesticide impurities, acute oral toxicity testing is only required when there is evidence that an impurity could result in a more hazardous technical product. QSARs or in silico tools may be used to determine toxicity outcomes of new impurities, and their use is encouraged for situations where there is no existing information about the impurity.
The information needed from acute systemic toxicity tests includes clinical observations, body weight changes, gross pathology, histopathology, and LD50. Acute inhalation studies must also provide an assessment of inhaled fraction from nominal and effective air concentrations and particle size distribution (e.g. mass median aerodynamic diameter and geometric standard deviation). OECD test methods are preferred. For all acute toxicity endpoints, tests may be required for the registration of both new technical ingredients and formulated products. Additional tests may also be required for acute oral and inhalation toxicity in cases of post-registration formulation changes.
Toxicological studies may be waived by presenting a substantiated technical justification about the impossibility of carrying out the study or providing documentation to verify that toxicological outcomes from alternative methods are sufficient to make a regulatory decision on product safety.
Canada
Health Canada typically requires acute oral and inhalation data for active ingredients and finished pesticide products (Health Canada Citation2018b). Acute toxicity studies may also be required for manufacturing concentrates, as well as for metabolites or degradation products found in plants, livestock, or drinking water, for example.
LD50 or LC50 values are used for hazard classification and labeling of pesticides and may be used to determine personal protective equipment and classification as restricted use, commercial use, or domestic use () (Health Canada Citation2013). Acute systemic toxicity data, including clinical signs (behavioral and physiological), dose response, body weight changes, and gross pathology, can be used to inform considerations regarding the adequacy of dose selection in other toxicity studies. Acute toxicity data can also be used to inform the interpretation of other toxicology data. For all exposure routes, the recommended methods are those adopted by OECD () and EPA (EPA Citation1998a, Citation1998b, Citation2002).
Acute oral studies are not required for technical-grade active ingredients and end-use products if the test substance is a gas or highly volatile liquid (Health Canada Citation2018b). As of 2017, acute dermal studies are typically not required, but may be requested in rare circumstances, including the use of new technology or presence of unique characteristics, that would warrant a more comprehensive assessment of acute dermal hazard for labeling purposes (Health Canada Citation2017). Inhalation information is required if a respirable material (e.g. gas, vapor, aerosol, or particulate) is produced under conditions of use.
Acute toxicity studies may not be required when the criteria provided in the Pest Management Regulatory Agency’s guidance for waiving or bridging of mammalian acute toxicity tests for pesticides are met () (Health Canada Citation2013). For example, acute oral toxicity tests may be waived for exposure-based considerations for an end-use product that is non-friable and too large to be ingested. Waivers of inhalation tests may be considered if the test substance is corrosive to skin or has a pH less than two or greater than 11.5. Waiver criteria in the equivalent OECD guidance are also recognized (OECD Citation2016). Subsequent guidance allowed the routine requirement for acute dermal systemic toxicity tests to be removed for both active ingredients and end-use products in the majority of circumstances (Health Canada Citation2017).
Table 3. Canadian PMRA criteria for waiving acute systemic toxicity studies.
Non-animal methods such as read-across, WoE assessments, calculation methods (e.g. additivity approach for mixtures), and new approach methodologies may be submitted to fulfill acute toxicity data requirements along with a scientifically valid rationale for consideration. Although alternatives such as in vitro assays, in silico (QSAR) assessments, and chemical grouping are not considered full replacements for in vivo acute toxicity studies, Health Canada will consider these submissions in a WoE assessment for any pesticide product. Alternative approaches can be discussed during pre-submission meetings with Health Canada.
China
To make regulatory decisions on pesticides, regulatory authorities in China generally desire oral LD50 and gross pathology information. Data from acute systemic toxicity studies can be used to inform GHS hazard classification and labeling, exposure levels, and transport regulations, as well as for activities such as dose setting for longer-term studies, human and ecological risk assessment, and determining mechanism of toxic action.
Acute dermal, oral, and inhalation assessments are required for new active ingredients and new formulated pesticide products. Guidance for acute toxicity testing is found in the GB 15760-2017 series Toxicological Test Methods for Pesticides Registration (Ministry of Agriculture Citation2017), which includes three oral toxicity tests (Horn’s method, the up-and-down-procedure, and Miller and Tainter’s method); one dermal toxicity test; and one inhalation toxicity test. OECD test guidelines and practices are not automatically accepted. Data generated using methods described in OECD test guidelines may be accepted if the tests are conducted in a GLP-compliant laboratory in China or if the generating organization outside of China has a signed Mutual Acceptance of Data agreement with China. No non-animal alternatives are accepted for acute systemic toxicity testing of pesticides.
European Union
In the EU, pesticidal substances are categorized either as plant protection products, which protect crops and other desirable or useful plants from damage or disease, or as biocidal products, which include disinfectants, preservatives, and non-crop pest control agents such as antifouling products and rodenticides. Although the marketing and use of plant protection products and biocidal products are regulated by different laws, each requires an assessment of acute systemic toxicity for both active ingredients and final marketed products. Acute toxicity information is used for hazard assessment and classification according to the CLP classification system, which is based on GHS, and calculation of acceptable exposure levels. However, acute toxicity information is not typically used for risk assessment, partially because of the limitations in study design that prevent the identification of a no-observed-adverse-effect level and provide little information on nature, severity, and reversibility of effects (ECHA Citation2017c).
Plant protection products
Acute systemic toxicity information is mandatory for both active ingredients and finished plant protection products (EC Citation2013a, Citation2013b). In additional to hazard classification and labeling, data may also be used to derive acute reference doses for active ingredients (EC Citation2013a). Acute dermal toxicity information is needed for active ingredients unless waiving is scientifically justified (e.g. when oral LD50 is greater than 2000 mg/kg). Inhalation information is needed if (1) the active ingredient has a vapor pressure greater than 0.01 Pa at 20 °C or (2) is a powder containing a significant proportion of particles with diameter less than 50 μm and comprises at least 1% by weight of the finished product or is included in products that are powders or are applied by spraying.
For final products, acute oral toxicity testing is needed unless an alternative approach, such as bridging or additivity calculations can be justified (EC Citation2013b). Dermal acute toxicity information is needed unless the product is orally nontoxic (oral LD50 is greater than 2000 mg/kg) per CLP guidance (ECHA Citation2017a). Acute inhalation toxicity information for the product or the smoke it generates is needed if the product is a gas or liquified gas, is a smoke-generating product or fumigant, is used with fogging or misting equipment, releases vapor, is supplied in an aerosol dispenser, is a powder containing a significant proportion of particles with diameter less than 50 μm (>1% by weight), is applied by aircraft where inhalation exposure is anticipated, contains an active ingredient with vapor pressure greater than 0.01 Pa, is used in enclosed spaces, or is applied by spraying.
Tests of active ingredients using vertebrate animals shall be undertaken only where no other validated methods are available (EC Citation2013a). Alternative methods to be considered include in vitro and in silico methods. Reduction and refinement methods for in vivo testing, such as those in OECD test guidelines (), are encouraged if animal testing of active ingredients or products must be performed.
Biocidal products
Assessment of acute toxicity by the oral and one other route is required for all active biocide ingredients and finished biocide products that are not gases. All available human, animal, and alternative data are evaluated to determine whether additional testing is needed (ECHA Citation2017c). Acute oral tests are not needed if the active ingredient is corrosive to skin, or if the substance is a gas or highly volatile. The preferred method for acute oral testing is the fixed dose procedure; however, data generated using any method described in an OECD test guideline for acute oral toxicity are acceptable (ECHA Citation2017c, Citation2022). Testing by the inhalation route is appropriate if inhalation exposure to humans is likely and the active ingredient (1) has a vapor pressure greater than 0.01 Pa at 20 °C, (2) is a powder with a significant proportion of particles with median mass aerodynamic diameter less than 5 μm, or (3) is included in products that are powders or are applied in a manner than generates exposure particles or droplets that are inhalable (i.e. having a median mass aerodynamic diameter less than 50 μm). The acute toxic class method for acute inhalation toxicity (OECD Citation2009b) is preferred but either protocol described in OECD Test Guideline 403 for acute inhalation toxicity is also acceptable (OECD Citation2009a). Testing by the dermal route is necessary only if inhalation is unlikely, skin contact is likely, and high dermal absorption and bioavailability are indicated either by the substance’s physicochemical and toxicological properties or results of a dermal penetration study.
For biocidal products, a tiered approach is used to classify mixtures for acute toxicity (ECHA Citation2017a). If available, acute toxicity data for the mixture are used. Otherwise, bridging principles or calculations can be used to predict the acute toxicity of the mixture if data are available for the components. In vivo testing is not necessary for mixtures when there are enough available data on each component to allow classification using additivity calculations and when synergistic effects between any of the components are unlikely (EC Citation2012). The biocides regulation (No. 528/2012) encourages the generation of information by alternative methods that do not use animals; however, no non-animal approaches, other than waivers and bridging, have been adopted.
Japan
In Japan, acute systemic toxicity testing is required for the oral, dermal, and inhalation routes for both pesticide active ingredients and final formulations. The information from these tests is used for hazard assessment, classification, and labeling per the GHS ().
Test data may be generated using any current OECD acute toxicity test guideline (). No non-animal toxicity tests are accepted as replacements for animal acute systemic toxicity tests; however, the requirement for information on dermal toxicity may be waived for active ingredients and final formulations if the test substance is corrosive or if its oral LD50 is greater than 2000 mg/kg.
South Korea
Testing of pesticides in South Korea is required to classify toxicity and evaluate safety of pesticide residues. Thus, information derived from acute oral, dermal, and inhalation studies is desired by Korean regulators to establish LD50 and provide GHS hazard classification and labeling of pesticide products.
OECD test guidelines, which provide for reduction of animal use, are acceptable for acute systemic toxicity. Acute dermal toxicity testing is only performed when absolutely necessary after analyzing all information related to the potential for dermal toxicity of the test substance. Dermal toxicity testing requirements may be waived when serious irritation, such as dermal corrosion, or death is expected. No non-animal toxicity tests are accepted as replacements for animal acute systemic toxicity tests.
Taiwan
Taiwan regulates pesticide formulations and active ingredients. Registrants must provide documentation of acute toxicity studies to inform hazard assessment, GHS hazard classification, and labeling. Acute systemic toxicity tests can be waived for pesticide formulations according to Taiwanese regulations. Oral acute toxicity tests can be waived when the pesticide is a gas or is highly volatile. Dermal acute toxicity tests can be waived when the oral LD50 is greater than 2000 mg/kg body weight, or the pesticide is corrosive to the skin.
Data from methods described in OECD test guidelines for acute toxicity testing are accepted (). Information provided to regulatory authorities should include LD50, or the range of lethal doses, clinical observations, gross necropsy, and changes in body weight. Currently, no alternatives to in vivo testing are accepted for acute systemic toxicity endpoints.
United States
EPA requires acute oral and inhalation toxicity data for all pesticide active ingredients and formulated products. These data are used to categorize the toxicity of pesticides using its own classification system (). These categories in turn determine the appropriate hazard labeling and level of personal protective equipment (clothing and respiratory protection) for pesticide handlers (EPA Citation2016b). Data are also used to determine engineering controls that should be used in handling end-use products as well as when it is safe for workers to enter fields after pesticide treatment. In addition, EPA uses acute oral, dermal, and inhalation LD50 and LC50 values to determine whether child-resistant packaging is needed for pesticides with residential applications. Such packaging is required for products with oral LD50 of 1500 mg/kg or less, dermal LD50 of 2000 mg/kg or less, or inhalation LC50 of 2 mg/L or less (40 CFR 157.22).
EPA recommends that acute lethality data be generated using EPA test guidelines (EPA Citation1998a, Citation1998b, Citation2002); although, data generated using methods described in OECD test guidelines () are accepted per the Mutual Acceptance of Data agreement. EPA has identified acute lethality testing as an area where alternative approaches may be applied and recommends that registrants wishing to submit alternative test method data consult with EPA prior to submission (EPA Citation2016d).
EPA has issued guidance for bridging to other data or for waiving animal studies to avoid testing and reduce animal use (EPA Citation2012; OECD Citation2016). Bridging may be supported for new pesticide formulations that are similar to currently registered end-use products. Waivers are considered when the data requirement is not relevant to assessment of the chemical. For example, an acute oral toxicity study may be waived for a material that is a gas because the oral exposure route is not relevant. EPA has also issued guidance for waiving all acute dermal acute LD50 studies for active ingredients and formulations (EPA Citation2016a, Citation2020). To explore an additional opportunity for waiving tests, EPA began a pilot program in December 2016 to evaluate the usefulness and acceptability of the GHS Mixtures Equation as an alternative to animal oral and inhalation toxicity studies for pesticide formulations (EPA Citation2016c). By using the GHS Mixtures Equation, end-use product toxicity can be estimated by adding together the individual ingredient toxicities without conducting new tests (Hamm et al. Citation2021).
Pharmaceuticals
The substances in this chemical sector may be referred to as pharmaceuticals, drugs, or medicinal products, depending upon the regulatory authority, and are typically those substances intended to diagnose, treat, mitigate, or prevent perceived adverse symptoms or diseases in humans. The pharmaceutical sector also includes vaccines, blood derived products, products produced through biotechnology (e.g. tissues and organs), disinfectants, and radiopharmaceuticals. Pharmaceuticals may be prescribed or available without a prescription. Some non-prescription products, as is the case for some cosmetics in Japan and South Korea, are classified as quasi-drugs, although these are generally not regulated under drug-related legislation. Herbs or dietary supplements are regulated as foods in most jurisdictions; however, in South Korea traditional herbal products (that are not simple herbs) qualify as pharmaceuticals.
The most frequent uses of acute systemic toxicity data for pharmaceuticals are for risk assessment and hazard assessment or classification (). The pharmaceutical sector does not use GHS hazard classifications. Except for Brazil, the regulatory authorities surveyed recognize Guideline M3(R2) of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) (ICH Citation2009) with respect to nonclinical data requirements for pharmaceuticals, although application of the guideline may vary among various authorities. In the context of this review, “nonclinical” refers to studies that are conducted in non-human test systems, usually before clinical trials in humans. The intent of Guideline M3(R2) is to provide information needed to support human trials and reduce the use of animals and drug development resources. Although lethality is not a recommended endpoint, the results of acute toxicity studies for the clinical route of exposure should be available to provide dose-limiting toxicity information for phase 3 clinical trials that intend to demonstrate that the drug is a safe and effective treatment in humans. An assessment of acute toxicity may be needed in earlier stages of pharmaceutical development for patient populations at higher risk for overdosing (e.g. patients diagnosed with depression, pain, and dementia) in out-patient clinical trials.
Stand-alone single-dose acute systemic toxicity studies are not required because acute toxicity information can be obtained from dose escalation studies or short-duration dose-ranging studies that define a maximum tolerated dose in the general toxicity test species using both a clinical and a parenteral route of administration (ICH Citation2009). Non-animal approaches for acute toxicity are not mentioned in the M3(R2) guidance except to state that validated methods accepted by all ICH regulatory authorities should be considered.
Brazil
Acute systemic toxicity data are required in Brazil on a case-by-case basis for new pharmaceuticals. Data are used for defining hazardous properties, setting exposure levels, risk assessment, dose setting for longer-term studies, WoE assessments, and defining mechanism of toxic action. Acceptable methods include internationally recognized methods such as those described in OECD test guidelines for acute systemic toxicity. The information needed from these tests include clinical observations and characterization of effects, along with mortality and LD50 or LC50.
Regulatory authorities in Brazil do not currently accept any non-animal methods to generate acute toxicity data for regulatory purposes but would consider any such methods that are validated in the future and recognized internationally for regulatory purposes by OECD. Toxicological studies can be waived with a technical justification that documents the impracticality of carrying out an in vivo study or provides information about the toxicological outcomes evaluated in the proposed study that are sufficient for a regulatory decision on product safety.
Canada
In Canada, prescription and non-prescription pharmaceuticals and disinfectants used on environmental surfaces and inanimate objects are regulated under the Food and Drugs Act, single-dose toxicity studies of prescription pharmaceuticals may be used in Canada to predict the consequences of human overdose (i.e. risk assessment) or to set doses for clinical trials. ICH Guideline M3(R2) is followed with respect to nonclinical data requirements to support use in clinical trials (Health Canada Citation2016). Health Canada’s guidance does not recommend non-animal approaches but indicates that stand-alone acute toxicity studies are generally not needed because acute toxicity information can be obtained from repeat-dose toxicity studies that define a maximum tolerated dose (Health Canada Citation2016).
For hard surface disinfectants, such as those used for countertops, acute toxicity information is used for hazard labeling such as that used for consumer products () (Health Canada Citation2018a) and for non-prescription drugs, acute toxicity information is used for safety assessment. For older non-prescription drugs that already have a known nonclinical safety profile, there are no requirements for acute systemic toxicity information; however, acute systemic toxicity studies must be submitted for new drugs that have never been approved for over-the-counter use. In most cases, it may only be necessary to submit toxicity data for the intended route of administration; however, data may also be needed for possible routes of unintended exposure (e.g. inhalation toxicity information may be needed for an aerosol sunscreen applied to the skin). Acute systemic toxicity tests of non-prescription and disinfectant drugs should be conducted according to OECD test guidelines. The nonclinical safety data requirements for acute systemic toxicity include LD50, gross pathology, clinical effects, and dose–response assessment. No non-animal tests are currently accepted; however, waivers may be accepted on a case-by-case basis with a sound scientific rationale. For example, corrosive disinfectants need not be tested systemically as long as these substances are labeled as corrosive.
China
Acute oral toxicity information for pharmaceuticals is required in China to identify mechanism of action, document lethality potential, conduct human health risk assessments, and to set doses for longer-term studies. Information needed from acute oral tests includes LD50, gross pathology, clinical effects, and dose response. Only finished products must be tested. Guidance for performing acute oral testing is provided in Technical Guidelines for General Pharmacology Research of Chemical Drugs, which are similar to ICH Guideline M3(R2) (National Medical Products Administration Citation2005). No non-animal approaches are accepted for acute oral testing.
European Union
Within the EU, acute toxicity information for pharmaceuticals is used to characterize their potential toxic effects or to support drug development in certain therapeutic fields such as oncology. The applicable guideline for acute toxicity studies for pharmaceuticals is ICH Guideline M3(R2) (EMA Citation2009). According to this guideline, lethality should not be an intended endpoint in studies assessing acute toxicity for pharmaceuticals.
When acute toxicity information is available from any other studies, separate single-dose studies are not needed. Studies providing acute toxicity information can be limited to the clinical route only under non-GLP conditions if the clinical administration route is supported by appropriate GLP repeated dose toxicity studies (EMA Citation2009).
Japan
Acute toxicity information for domestically manufactured medicines and imported chemicals is used for hazard classification under the Poisonous and Deleterious medicine designation criteria ( and ). These classify a substance as “poison,” “deleterious medicine,” or “none of the above” for product labeling purposes. Information is also used for risk assessment to identify the initial safe doses in humans and to provide information about the effect of drug overdoses.
Domestically manufactured and imported medicines must be tested for acute toxicity by the oral, dermal, and intraperitoneal injection routes. Pure medicines, as well as dilutions containing the active ingredients, must be tested, but testing is not required for mixtures or final formulations. Methods described in OECD acute toxicity test guidelines, which are currently in vivo tests, are acceptable (). No non-animal toxicity tests are accepted for acute systemic toxicity. However, dermal tests may be waived for substances that are positive for in vitro skin corrosivity, unless there is compelling evidence to suggest that the substance would be absorbed through the skin or exhibit toxicity. In such cases, the test substance may potentially be assessed as either a poisonous or deleterious substance based on toxic potency per the oral or inhalation routes.
For medicines that are not poisonous or deleterious, the Pharmaceuticals and Medical Devices Agency uses ICH Guideline M3(R2) on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals (ICH Citation2009). In addition to oral, dermal, and inhalation routes, acute toxicity information for intravenous and subcutaneous routes may be needed by the Pharmaceuticals and Medical Devices Agency. Lethality data are not required but are used if submitted. These data, along with the dose–response relationships of clinical effects, are needed to estimate systemic toxicity, select doses for other studies, identify target organ toxicity, and characterize mechanism of toxicity. Acute toxicity data may be waived because acute toxicity information can be obtained after initial dosing in repeated dose toxicity studies; however, no non-animal approaches are accepted.
South Korea
In South Korea, single-dose studies of pharmaceuticals intended for human use to determine acute systemic toxicity are used for hazard assessment. For single-dose toxicity, the approximate lethal dose, clinical effects, body weight change, gross pathology, and histopathology of organ effects are of interest.
The Standard for Toxicity Study of Pharmaceuticals, which is consistent with the ICH M3(R2) guidance (ICH Citation2009), provides guidance for pharmaceutical toxicity evaluations (Ministry of Food and Drug Safety Citation2022). The exposure route for single-dose toxicity studies must match the clinical route. Any method described in an OECD test guideline for acute systemic toxicity may be used to generate data (). Both active ingredients and drug formulations must be tested. For combination drugs, which contain more than one active ingredient, repeat-dose studies may substitute for the single-dose studies when the active ingredients have already been approved. Also, the requirement for single-dose studies for combination drugs may be waived if concomitant therapy of the active ingredients has already been approved or if the clinical literature shows that concomitant therapy causes no toxicological concerns. No non-animal approaches are accepted as substitutes for the single-dose studies.
Taiwan
Acute toxicity information is needed for hazard assessment of pharmaceuticals. The single-dose toxicity information is also used for dose-setting in repeat-dose studies. The route of administration must match the clinical route. The applicable guideline for acute toxicity studies for pharmaceuticals is the ICH M3(R2) guideline. The information needed from acute systemic toxicity studies, however, includes lethal dose range, mortality and/or signs of toxicity, clinical observations, gross necropsy findings, and body weight. No non-animal methods are accepted.
United States
In the USA, safety oversight of most pharmaceuticals is provided by the FDA Center for Drug Evaluation and Research, which relies on the ICH M3(R2) guidance (ICH Citation2009). However, acute toxicity assessment of cellular and gene therapy products is overseen by the FDA Center for Biologics Evaluation and Research. Their Guidance for Industry: Preclinical Assessment of Investigational Cellular and Gene Therapy Products (FDA Citation2013) notes that the nonclinical program for each investigational cellular and gene therapy product should be individualized with respect to scope, complexity, and design. Furthermore, safety assessment of such products should be sufficiently comprehensive to permit identification, characterization, and quantification of potential local and systemic toxicities, including whether their onset is acute or delayed. The submission of "classical" LD50 test data is discouraged (FDA Citation1988).
For the purposes of setting a single dose for human clinical trials, FDA does not recommend conducting acute nonclinical studies when acute toxicity information is or will be available from multidose studies (Strickland et al. Citation2018). Dose-setting information may also be available from prior human experience (e.g. data from other countries or regions) or toxicity studies in animals. Such studies must include complete clinical chemistry, hematology, histopathology, and toxicokinetic analyses for at least one dose. Usually, multidose animal studies rather than single-dose studies are submitted because multidose studies can be used to support both multidose clinical trials and marketing of products intended for a single dose in humans, per ICH M3(R2).
An alternative method can be used for testing products, if sufficient evidence is available to show that it can ascertain product safety in humans at least as well as the standard animal method (FDA Citation2005). FDA encourages sponsors to explore opportunities for reducing, refining, and replacing animal use in their nonclinical programs, and recommends that sponsors discuss any proposal for application of alternative methods with FDA prior to data submission as part of the routine consultation process.
Discussion
We have undertaken this review to clarify acute systemic toxicity testing needs, requirements, and uses for the purpose of identifying targets for development of non-animal methods and strategies for meeting regulatory needs. The review encompasses six chemical sectors within eight countries and regions that participate in ICATM. Compilation of this information was recommended by the workshop on Alternative Approaches for Identifying Acute Systemic Toxicity: Moving from Research to Regulatory Testing (Hamm et al. Citation2017). Our aim was to provide information needed to support development and international harmonization of non-animal test methods and strategies for these endpoints.
The information for this review was collected from the regulatory authorities in their respective regions by ICATM partners or participants, who are experts in chemical regulatory matters. In compiling this review, we found it difficult to obtain unambiguous information on whether and how regulatory agencies use acute systemic toxicity data and whether and what types of non-animal alternatives are accepted. Regulatory authorities often use terms such as “should” and “may” with respect to data that must be provided and “may be considered” with respect to the data that will be accepted. Thus, the requirements and needs for acute systemic toxicity data can be somewhat ambiguous. The ambiguity of requirements and needs likely increases animal use because when requirements are uncertain, chemical sponsors are likely to default to animal methods that have been acceptable in the past.
provides a summary of the preferred in vivo test methods for acute systemic toxicity and the acceptance of non-animal approaches by chemical sector. Although eight regions or countries and six chemical sectors were surveyed, 52 entries are shown because some countries subdivide specific chemical sectors. Brazil has separate consumer product categories for antimicrobials/disinfectants and toys/school supplies, the EU has separate pesticide categories for biocides and plant protection products, and Japan and Canada each have two pharmaceutical categories, with Japan distinguishing between pharmaceuticals and toxic medicines and Canada having separate categories for prescription and non-prescription pharmaceuticals. No acute systemic toxicity information is typically required for consumer products in Japan; for industrial chemicals in Brazil and the U.S.; or for cosmetics the EU, South Korea, Taiwan, and the U.S. For all the sectors where acute toxicity information is not required, some authorities accept such data if they are submitted. For most chemical sectors, authorities accept data generated using acute systemic toxicity test guidelines from OECD or other animal reduction and refinement methods. However, all countries/regions identified ISO guidelines, or similar methods, as approaches of choice for testing medical devices. Similarly, most countries/regions preferred ICH guidelines for pharmaceuticals.
Table 4. Acute systemic toxicity test methods accepted for regulatory use as of 2022.
The use of non-animal approaches for acute systemic toxicity is limited (see ) because there are few standardized and accepted test methods or approaches to replace animals for these endpoints. No non-animal approaches are accepted for the majority of the chemical sectors in China and Taiwan. Internationally, the most frequently used approach to reduce the use of animals is for the sponsor to obtain a test waiver that obviates the need for certain tests altogether. We suggest that the relatively frequent use of waivers, sometimes referred to as “non-testing” alternatives, is enhanced by the available guidance documents that provide rationale and direction for their use. For pharmaceuticals, which are typically evaluated according to ICH guidelines and without a lethality endpoint, waivers are accepted for stand-alone toxicity tests with adequate technical justification in all countries surveyed except China and Taiwan. ICH suggests that acute toxicity information can be obtained early in the course of repeated-dose studies. Another chemical sector for which regulatory authorities commonly accept test waivers is pesticides. Test waivers for certain exposure routes may be granted on the basis of physicochemical characteristics that limit or prevent exposure, or on similarity to formulations previously evaluated with animal tests. OECD has issued guidance on waiving and bridging principles as have other agencies for their specific needs (EPA Citation2012; Health Canada Citation2013; OECD Citation2016). Pesticide agencies in the U.S. and Canada have also indicated that the acute dermal systemic toxicity test may be waived and have produced guidance documents accordingly (EPA Citation2016a, Citation2020; PMRA Health Canada Citation2017). In South Korea, dermal testing of pesticides is performed only if absolutely necessary based on the potential for dermal toxicity.
Understanding regulatory requirements and decision contexts is critical to designing appropriate non-animal alternatives. A workshop in 2018 brought together 32 international groups from government, industry, and academic sectors to discuss regulatory needs and the potential utility of predictive models (Kleinstreuer et al. Citation2018). One specific non-animal test method that has achieved limited acceptance for the replacement of acute oral toxicity testing is the 3T3 neutral red uptake test, which is recommended by the EU Reference Laboratory for Alternatives to Animal Testing to classify nontoxic substances when used with additional supporting information in a WoE approach (JRC Citation2013). In this context, this test is currently accepted for medical device testing in Canada and cosmetics testing in Europe. The Regulatory Acceptance Board of the Japanese Center for the Validation of Alternative Methods has recommended use of the 3T3 neutral red uptake test in a WoE approach to classify nontoxic substances during the safety assessment of industrial substances, cosmetics, and quasi-drugs (JaCVAM Citation2019). This method would probably be used more widely if it were described in an OECD test guideline since the countries surveyed rely heavily on OECD test guidelines. In fact, regulatory authorities in Brazil, South Korea, and Taiwan indicate that non-animal methods adopted as OECD test guidelines would be readily accepted if they existed. In vitro cytotoxicity tests are also accepted by some regulatory authorities as a reduction alternative for setting starting doses for in vivo acute oral toxicity tests (OECD Citation2010). For example, such an approach is accepted in the USA by CPSC and internationally for medical devices in accordance with the ISO guidelines.
Regulatory authorities for some sectors will consider a WoE approach that uses multiple non-animal methods such as read-across, in vitro test methods, and in silico information, such as QSAR models, to assess acute systemic toxicity. WoE approaches are accepted for industrial chemical assessments in Canada, the EU, and South Korea; consumer product assessments in the EU and South Korea; cosmetics in the EU, Japan (additives for quasi-drugs), and South Korea; and pesticides in Canada and the EU. A few regulatory authorities consider in vitro and in silico methods outside the context of WoE approaches. Canada may consider data from in vitro and in silico methods for when animal data are unavailable, South Korea encourages the use of in vitro methods for pesticide testing, and the EU considers data from in vitro and in silico methods in the risk evaluation of plant protection products. Regulatory authorities for cosmetics in Brazil and for consumer products and medical devices in the U.S. will accept non-animal test methods on a case-by-case basis. One recently published in silico method, the Collaborative Acute Toxicity Modeling Suite (CATMoS; Mansouri et al. Citation2021), was the result of a large international collaboration among computational chemistry groups to build consensus models to predict acute oral systemic toxicity, including regulatory classification schemes highlighted here.
The information collected for this review suggests that the applications for which non-animal alternatives would have the most impact for replacing animals used for acute systemic toxicity testing would be hazard assessment, classification, and/or labeling because it is most frequent use of acute systemic toxicity data. The next most frequent application for acute systemic toxicity is risk assessment. The exposure route that would have the most impact regarding replacement of animals is the oral route since that is the route most often required across chemical sectors. Test method developers wanting to have the most impact on reducing animal use should target their efforts to these applications. Further, there may already be viable non-animal alternatives such as the CATMoS that could fulfill regulatory needs, and these could be included in regulatory submission packages for assessment by various agencies.
We noted that some regulatory authorities are unclear about whether non-animal methods or approaches are accepted. Statements such as non-alternative methods “are” or “may” be “considered” or “accepted on a case-by-case” leave doubt as to whether such methods are accepted. Sponsors may be discouraged from using non-animal alternatives when their regulatory acceptance is unclear. When acceptance of non-animal methods is uncertain, chemical sponsors are likely to default to animal methods that have been acceptable in the past, thereby unnecessarily using animals in situations where non-animal methods are acceptable. Transparency from regulatory agencies with respect to the acceptance of non-animal methods will prevent the unnecessary use of animals. Clear information about the needs of regulatory agencies also helps target the development of non-animal replacements.
While a number of regulatory authorities accept some non-animal alternatives for acute systemic toxicity, few communicate the specific acceptable methods. Guidance that provides clarity on the specific non-animal methods accepted by regulatory authorities is needed to improve the implementation of these methods. The guidance provided by the European Chemicals Agency (ECHA) for testing required for REACH (ECHA Citation2017b) serves as an example. This guidance provides a list of information sources that can be combined in a WoE approach to assess acute oral toxicity. Sources may include in silico and in vitro data, as well as in vivo data from 28-day studies. Our scope did not include a survey of the available non-animal methods for acute systemic toxicity; however, other international groups have reviewed available non-animal methods for acute systemic toxicity (Hamm et al. Citation2017; Clippinger et al. Citation2018; Sullivan et al. Citation2021). The current review provides the applications with which these methods could be matched to evaluate potential suitability and fitness for purpose.
An understanding of national and regional regulatory requirements for acute systemic toxicity testing is critical to achieve ICATM’s goal of acceptance and implementation of non-animal alternatives for meeting regulatory testing requirements. This information will be used to align the acute systemic toxicity needs and applications for the various chemical sectors and regulatory authorities with acceptable non-animal test methods and strategies. We expect that ICATM will work with other stakeholders through organizations such as OECD, ICH, and ISO, which produce test guidelines that are frequently relied upon by the countries surveyed to advance non-animal test guidelines for acute systemic toxicity. Such internationally harmonized test guidelines can go a long way toward reducing animal use for acute systemic toxicity testing because regulatory authorities regard them highly.
Disclaimer
This article may be the work product of an employee or group of employees of NIEHS or other organizations; however, the statements, opinions, or conclusions contained therein do not necessarily represent the statements, opinions, or conclusions of NTP, NIEHS, the United States government, or other organizations. Inotiv staff provide technical support for the NTP Interagency Center for the Evaluation of Alternative Toxicological Methods, but do not represent NIEHS, NTP, or the official positions of any federal agency.
| Abbreviations | ||
| CDRH | = | FDA Center for Devices and Radiological Health |
| CLP | = | Classification, Labelling, and Packaging (EU) |
| CONCEA | = | National Council for the Control of Animal Experimentation (Brazil) |
| CPSC | = | U.S. Consumer Product Safety Commission |
| EPA | = | U.S. Environmental Protection Agency |
| EU | = | European Union |
| FDA | = | U.S. Food and Drug Administration |
| GHS | = | United Nations Globally Harmonized System of Classification and Labelling of Chemicals |
| GLP | = | Good Laboratory Practices |
| ICATM | = | International Cooperation on Alternative Test Methods |
| ICH | = | International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use |
| ISO | = | International Organization for Standardization |
| NIEHS | = | National Institute of Environmental Health Sciences |
| NTP | = | National Toxicology Program |
| OECD | = | Organisation for Economic Co-operation and Development |
| QSAR | = | quantitative structure–activity relationship |
| REACH | = | Registration, Evaluation, Authorization and Restriction of Chemicals (EU) |
| SCCS | = | Scientific Committee on Consumer Safety (EU) |
| WoE | = | weight-of-evidence |
Acknowledgements
The authors thank Dr. Helena Hogberg and Dr. Pei-Li Yao for thoughtful critical reviews of this manuscript; and Ms. Catherine Sprankle for editorial review. This project was funded in part with federal funds from the National Institute of Environmental Health Sciences (NIEHS), National Institutes of Health under Contract No. HHSN273201500010C to Inotiv, Inc., in support of the National Toxicology Program (NTP) Interagency Center for the Evaluation of Alternative Toxicological Methods, and with support from the Carroll Petrie Foundation. The authors gratefully acknowledge the useful comments offered by the external reviewers selected by the editor whose identity was not known to the authors.
Declaration of interest
This work has been conceived, planned, and executed by the Physicians Committee for Responsible Medicine (Esther Haugabrooks and Kristie Sullivan) and the NIH/NIEHS/DTT/NICEATM (Nicole Kleinstreuer, Warren Casey), supported by Inotiv, Inc. (Judy Strickland, Dave Allen). Inotiv, Inc., staff were supported by NIEHS, the National Institutes of Health, under Contract No. HHSN273201500010C and Physicians Committee for Responsible Medicine staff were supported by the Carroll Petrie Foundation. Staff at government agencies that participate in the International Cooperation on Alternative Test Methods (ICATM) were instrumental in the execution of this work.
Notes
1 Although “systemic” is not used in the titles of the test guidelines referenced, we use “systemic” to distinguish these methods from those used to assess toxic effects that occur at the site of administration, such as skin and eye irritation.
References
- ANVISA. 2019. Resolution RDC N° 294/2019 on criteria for toxicological assessment and classification of 31 July 2019. Diário Oficial da União. 146(1):78–85.
- Brazil Ministry of Labor and Employment. Secretary of Labor Inspection. 2011. Ordinance No. 229 of May 24, 2011. Amends Regulatory Norm No. 26. Federal Official Gazette: Section 1, Brasília, DF, n. 101 [accessed 2011 May 27]; p. 140. http://www.normaslegais.com.br/legislacao/portariasit229_2011.htm.
- Buesen R, Oberholz U, Sauer U, Landsiedel R. 2018. Comment on “Alternative acute oral toxicity assessment under REACH based on sub-acute toxicity values”. Ludwigshfen, Germany/Neubiberg, Germany: BASF SE, Experimental Toxicology/Scientific Consultancy-Animal Welfare. doi: 10.14573/altex.1710111.
- Clippinger AJ, Allen D, Behrsing H, BéruBé KA, Bolger MB, Casey W, DeLorme M, Gaça M, Gehen SC, Glover K, et al. 2018. Pathway-based predictive approaches for non-animal assessment of acute inhalation toxicity. Toxicol In Vitro. 52:131–145. doi: 10.1016/j.tiv.2018.06.009.
- CONCEA. 2014. Normative resolution no. 18/2014 on alternative methods of 24 September 2014. Diário Oficial da União. 185(1):9.
- Corvaro M, Gehen S, Andrews K, Chatfield R, Arasti C, Mehta J. 2016. GHS additivity formula: a true replacement method for acute systemic toxicity testing of agrochemical formulations. Regul Toxicol Pharmacol. 82:99–110. doi: 10.1016/j.yrtph.2016.10.007.
- CPSC. 2012. Recommended procedures regarding the CPSC’s policy on animal testing. https://www.cpsc.gov/business–manufacturing/testing-certification/recommended-procedures-regarding-the-cpscs-policy-on-animal-testing.
- CPSC. 2015. Statement on animal testing policy. 16 CFR part 1500.232. www.gpo.gov/fdsys/pkg/CFR-2015-title16-vol2/pdf/CFR-2015-title16-vol2-sec1500-232.pdf.
- CPSC. 2019. Definitions. 16 CFR Part 1500.3. https://ecfr.io/Title-16/pt16.2.1500#se16.2.1500_13.
- CPSC. 2021. Notice of availability: proposed guidance on alternative test methods and integrated testing approaches. Fed Regist. 86(60):16704–16705.
- EC. 2006. Regulation (EC) No. 1907/2006 of the European Parliament and of the Council of 18 December 2006 concerning the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH), establishing a European Chemicals Agency, amending Directive 1999/45/EC and repealing Council Regulation (EEC) No. 793/93 and Commission Regulation (EC) No. 1488/94 as well as Council Directive 76/769/EEC and Commission Directives 91/155/EEC, 93/67/EEC, 93/105/EC and 2000/21/EC. Off J Eur Union. 396:1–520.
- EC. 2008. Council Regulation (EC) No. 440/2008 of 30 May 2008 laying down test methods pursuant to Regulation (EC) No. 1907/2006 of the European Parliament and of the Council on the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH). Off J Eur Union. L142:1–739.
- EC. 2012. Regulation (EU) No. 528/2012 of the European Parliament and of the Council of 22 May 2012 concerning the making available on the market and use of biocidal products. Off J Eur Union. L167:1–123.
- EC. 2013a. Commission Regulation (EU) No. 283/2013 of 1 March 2013 setting out the data requirements for active substances, in accordance with Regulation (EC) No. 1107/2009 of the European Parliament and of the Council concerning the placing of plant protection products on the market. Off J Eur Union. L93:1–84.
- EC. 2013b. Commission Regulation (EU) No. 284/2013 of 1 March 2013 setting out the data requirements for plant protection products, in accordance with Regulation (EC) No. 1107/2009 of the European Parliament and of the Council concerning the placing of plant protection products on the market. Off J Eur Union. L93:85–152.
- EC. 2013c. Impact assessment on the animal testing provisions in regulation (EC) 1223/2009 on Cosmetics. Commission Staff Working Document. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:52013SC0066&from=EN.
- ECHA. 2012. Guidance on information requirements and chemical safety assessment. Chapter R.8: characterisation of dose [concentration]-response for human health. Helsinki: European Chemicals Agency (ECHA).
- ECHA. 2017a. Guidance on the application of the CLP criteria. Helsinki: European Chemicals Agency (ECHA).
- ECHA. 2017b. Guidance on information requirements and chemical safety assessment. Chapter R.7a: endpoint specific guidance. Helsinki: European Chemicals Agency (ECHA).
- ECHA. 2017c. Guidance on the biocidal products regulation. Volume III: human health – assessment & evaluation (parts B + C). Helsinki: European Chemicals Agency (ECHA).
- ECHA. 2022. Guidance on the biocidal products regulation. Volume III: human health. Part A: information requirements. Helsinki: European Chemicals Agency (ECHA).
- EMA. 2009. ICH Guideline M3(R2) on non-clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals step 5. Local Tolerance Testing of Medicinal Products. https://www.ema.europa.eu/en/documents/scientific-guideline/ich-guideline-m3r2-non-clinical-safety-studies-conduct-human-clinical-trials-marketing-authorisation_en.pdf.
- EPA. 1998a. Health effects test guidelines OPPTS 870.1200 acute dermal toxicity. 712-C-98-192. https://www.regulations.gov/document?D=EPA-HQ-OPPT-2009-0156-0004.
- EPA. 1998b. Health effects test guidelines OPPTS 870.1300 acute inhalation toxicity. 712-C-98-193. https://www.regulations.gov/document?D=EPA-HQ-OPPT-2009-0156-0005.
- EPA. 2002. Health effects test guidelines OPPTS 870.1100 acute oral toxicity. 712-C-02-190. https://www.regulations.gov/document?D=EPA-HQ-OPPT-2009-0156-0003.
- EPA. 2012. Guidance for waiving or bridging of mammalian acute toxicity tests for pesticides and pesticide products (acute oral, acute dermal, acute inhalation, primary eye, primary dermal, and dermal sensitization). http://www.epa.gov/sites/production/files/documents/acute-data-waiver-guidance.pdf.
- EPA. 2016a. Guidance for waiving acute dermal toxicity tests for pesticide formulations & supporting retrospective analysis. https://www.epa.gov/sites/production/files/2016-11/documents/acute-dermal-toxicity-pesticide-formulations_0.pdf.
- EPA. 2016b. Label review manual. https://www.epa.gov/sites/production/files/2016-12/documents/lrmcomplete.pdf.
- EPA. 2016c. Mixtures equation pilot program to reduce animal testing. https://www.epa.gov/pesticide-registration/mixtures-equation-pilot-program-reduce-animal-testing.
- EPA. 2016d. Process for establishing and implementing alternative approaches to traditional in vivo acute toxicity studies for FIFRA regulatory use. https://www.epa.gov/pesticide-science-and-assessing-pesticide-risks/process-establishing-implementing-alternative.
- EPA. 2020. Guidance for waiving acute dermal toxicity tests for pesticide technical chemicals & supporting retrospective analysis. https://www.epa.gov/sites/default/files/2021-01/documents/guidance-for-waiving-acute-dermal-toxicity.pdf.
- FDA. 1975. Cosmetic products: warning statements/package labels. Fed Reg. 40(42):8912–8916.
- FDA. 1988. LD 50 test policy [Docket No. 86P-0224]. https://cdn.loc.gov/service/ll/fedreg/fr053/fr053196/fr053196.pdf.
- FDA. 2005. General biological products standards: equivalent methods and processes. 21 CFR 610.9. http://www.ecfr.gov/cgi-bin/text-idx?SID=0fe7ae5ae5d0af1a60a09e9b4a770a4b&mc=true&node=se21.7.610_19&rgn=div8.
- FDA. 2013. Guidance for industry: preclinical assessment of investigational cellular and gene therapy products. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/preclinical-assessment-investigational-cellular-and-gene-therapy-products.
- FDA. 2016. Cosmetic product warning statements, 21 CFR 740. http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=740.10&SearchTerm=cosmetic.
- FDA. 2020. Use of International Standard ISO 10993-1, “Biological evaluation of medical devices – part 1: evaluation and testing within a risk management process". https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-international-standard-iso-10993-1-biological-evaluation-medical-devices-part-1-evaluation-and.
- General Administration of Quality Supervision Inspection and Quarantine of the People’s Republic of China. 2008. General principles for risk assessment of consumer product safety (GB/T 227602008). Beijing.
- Gissi A, Louekari K, Hoffstadt L, Bornatowicz N, Aparicio AM. 2017. Alternative acute oral toxicity assessment under REACH based on sub-acute toxicity values. ALTEX. 34(3):353–361. doi: 10.14573/altex.1609121.
- Gissi A, Louekari K, Hoffstadt L, Bornatowicz N, Aparicio AM. 2018. Reply to comment on “Alternative acute oral toxicity assessment under REACH based on sub-acute toxicity values. ALTEX. 35(1):121–122. doi: 10.14573/altex.1712011.
- Government of Canada. 2005. Guidelines for the notification and testing of new substances: chemicals and polymers, pursuant to Section 69 of the Canadian Environmental Protection Act, 1999. http://publications.gc.ca/collections/Collection/En84-25-2005E.pdf.
- Ha S, Seidle T, Lim K-M. 2016. Act of the registration and evaluation of chemicals (K-REACH) and replacement, reduction or refinement best practices. Environ Health Toxicol. 31:e2016026. doi: 10.5620/eht.e2016026.
- Hamm J, Allen D, Ceger P, Flint T, Lowit A, O’Dell L, Tao J, Kleinstreuer N. 2021. Performance of the GHS mixtures equation for predicting acute oral toxicity. Regul Toxicol Pharmacol. 125:105007. doi: 10.1016/j.yrtph.2021.105007.
- Hamm J, Sullivan K, Clippinger AJ, Strickland J, Bell S, Bhhatarai B, Blaauboer B, Casey W, Dorman D, Forsby A, et al. 2017. Alternative approaches for identifying acute systemic toxicity: moving from research to regulatory testing. Toxicol In Vitro. 41:245–259. doi: 10.1016/j.tiv.2017.01.004.
- Health Canada. 2007. Reference manual for the Consumer Chemicals and Containers Regulations, 2001 of the Hazardous Products Act. Minister of Health. https://www.canada.ca/en/health-canada/services/consumer-product-safety/reports-publications/industry-professionals/reference-manual-consumer-chemicals-containers-regulations/updates.html.
- Health Canada. 2013. Guidance for waiving or bridging of mammalian acute toxicity tests for pesticides. https://www.canada.ca/en/health-canada/services/consumer-product-safety/reports-publications/pesticides-pest-management/policies-guidelines/guidance-waiving-bridging-mammalian-acute-toxicity-tests-pesticides.html#a2.6.
- Health Canada. 2016. Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals, ICH Topic M3(R2). https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/international-conference-harmonisation/multidisciplinary/guidance-nonclinical-safety-studies-conduct-human-clinical-trials-marketing-authorization-pharmaceuticals.html#a13.
- Health Canada. 2017. Acute dermal toxicity study waiver. Science Policy Note SPN2017-03. Pest Management Regulatory Agency. https://www.canada.ca/en/health-canada/services/consumer-product-safety/reportspublications/pesticides-pest-management/policies-guidelines/science-policy-notes/2017/acute-dermal-toxicity-waiver-spn2017-03.html.
- Health Canada. 2018a. Guidance document: disinfectant drugs. https://www.canada.ca/content/dam/hc-sc/documents/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/disinfectants/disinfectant-drugs/disinfectant-drug-eng.pdf.
- Health Canada. 2018b. Guidance for developing datasets for conventional pest control product applications. https://www.canada.ca/en/health-canada/services/consumer-product-safety/reports-publications/pesticides-pest-management/policies-guidelines/guidance-developing-applications-data-codes-parts-1-2-3-4-5-6-7-10.html.
- ICCVAM. 2018. A strategic roadmap for establishing new approaches to evaluate the safety of chemicals and medical products in the United States. Fed Regist. 83(35):7487.
- ICH. 2009. Guidance on nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals M3(R2). www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Multidisciplinary/M3_R2/Step4/M3_R2__Guideline.pdf.
- ISO. 2006. ISO 10993-11: biological evaluation of medical devices – part 11: tests for systemic toxicity. https://www.iso.org/standard/35977.html.
- ISO. 2009. ISO 10993–1: biological evaluation of medical devices – part 1: evaluation and testing within a risk management process. https://www.iso.org/standard/44908.html.
- ISO. 2017. ISO 10993-11: biological evaluation of medical devices – part 11: tests for systemic toxicity. https://www.iso.org/standard/68426.html.
- ISO. 2018. ISO 10993-1: biological evaluation of medical devices – part 1: evaluation and testing within a risk management process. https://www.iso.org/standard/68936.html.
- JaCVAM. 2019. JaCVAM statement on in vitro cytotoxicity assay for estimating acute oral toxicity. http://jacvam.jp/files/list/07/07_02_Aen1.pdf.
- JRC. 2013. EURL ECVAM recommendation on the 3T3 neutral red uptake cytotoxicity assay for acute oral toxicity testing. https://publications.jrc.ec.europa.eu/repository/bitstream/JRC79556/lbna25946enn.pdf.
- Keene AT. 2020. Biological evaluation and regulation of medical devices in the European Union. In: Boutrand J-P, editor. Biocompatibility and performance of medical devices. 2nd ed. Duxford (UK): Woodhead Publishing; p. 413–440.
- Kleinstreuer NC, Karmaus AL, Mansouri K, Allen DG, Fitzpatrick JM, Patlewicz G. 2018. Predictive models for acute oral systemic toxicity: a workshop to bridge the gap from research to regulation. Comput Toxicol. 8(11):21–24. doi: 10.1016/j.comtox.2018.08.002.
- Kojima K, Sakaguchi K. 2020. Biological evaluation and regulation of medical devices in Japan. In: Boutrand J-P, editor. Biocompatibility and performance of medical devices. 2nd ed. Duxford (UK): Woodhead Publishing; p. 441–474.
- Mansouri K, Karmaus AL, Fitzpatrick J, Patlewicz G, Pradeep P, Alberga D, Alepee N, Allen TEH, Allen D, Alves VM, et al. 2021. CATMoS: collaborative acute toxicity modeling suite. Environ Health Perspect. 129(4):47013. doi: 10.1289/EHP8495.
- MAPA, ANVISA, IBAMA. 2013. Joint Normative Instruction N°1/2013 on changing pesticide formulation of 22 April 2013. Diário Oficial da União. 76(1):4–14.
- MHLW. 2017. Guidebook on the manufacture and sales of cosmetics and quasi-drugs. Tokyo, Japan: Yakuji Nippo Ltd.
- Mielke H, Strickland J, Jacobs MN, Mehta JM. 2017. Biometrical evaluation of the performance of the revised OECD Test Guideline 402 for assessing acute dermal toxicity. Regul Toxicol Pharmacol. 89:26–39. doi: 10.1016/j.yrtph.2017.07.007.
- Minister of Justice Canada. 2019. Consumer Chemicals and Containers Regulations, 2001 (SOR/2001-269).
- Ministry of Agriculture. 2017. Toxicological test methods for pesticides registration (GB/T 15670-2017).
- Ministry of Food and Drug Safety. 2014. Common standards and specifications on biological safety of medical devices (Public Notification No. 2014-115).
- Ministry of Food and Drug Safety. 2021. Regulation on the examination of functional cosmetics (Cosmetics Policy Division, 043-719-3400).
- Ministry of Food and Drug Safety. 2022. Standard for toxicity study of pharmaceuticals. https://www.mfds.go.kr/eng/brd/m_18/view.do?seq=71459&srchFr=&srchTo=&srchWord=&srchTp=&itm_seq_1=0&itm_seq_2=0&multi_itm_seq=0&company_cd=&company_nm=&page=1.
- Ministry of Health and Welfare. 2000. Notification No. 331. Standards for cosmetics. https://www.mhlw.go.jp/english/dl/cosmetics.pdf.
- Ministry of Health and Welfare. 2019. Regulations governing the application of animal testing for the safety assessment of cosmetics or cosmetic ingredients. https://law.moj.gov.tw/ENG/LawClass/LawAll.aspx?pcode=L0030085.
- National Medical Products Administration. 2005. Technical guidelines for general pharmacology research of chemical drugs. [H]GPT3-1. Beijing.
- National Medical Products Administration. 2017. Biological evaluation of medical devices. GB/T 16886. Beijing.
- National Medical Products Administration. 2021. Safety and technical standards for cosmetics. https://www.nmpa.gov.cn/xxgk/ggtg/qtggtg/20210409160436155.html.
- OECD. 2000. No. 19: guidance document on the recognition, assessment and use of clinical signs as humane endpoints for experimental animals used in safety evaluation. OECD Series on Testing and Assessment. https://www.oecd-ilibrary.org/environment/guidance-document-on-the-recognition-assessment-and-use-of-clinical-signs-as-human-endpoints-for-experimental-animals-used-in-safety-evaluation_9789264078376-en.
- OECD. 2002a. Test No. 420. Acute oral toxicity – fixed dose procedure. In: OECD guidelines for the testing of chemicals, section 4: health effects. Paris: OECD Publishing. http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2001)4&doclanguage=en.
- OECD. 2002b. Test No. 423. Acute oral toxicity – acute toxic class method. In: OECD guidelines for the testing of chemicals, section 4: health effects. Paris: OECD Publishing. http://www.oecd-ilibrary.org/environment/test-no-423-acute-oral-toxicity-acute-toxic-class-method_9789264071001-en.
- OECD. 2008. Test No. 425. Acute oral toxicity – up-and-down procedure. In: OECD guidelines for the testing of chemicals, section 4: health effects. Paris: OECD Publishing. http://www.oecd-ilibrary.org/environment/test-no-425-acute-oral-toxicity-up-and-down-procedure_9789264071049-en.
- OECD. 2009a. Test No. 403. Acute inhalation toxicity. In: OECD guidelines for the testing of chemicals, section 4: health effects. Paris: OECD Publishing. http://www.oecd-ilibrary.org/environment/test-no-403-acute-inhalation-toxicity_9789264070608-en.
- OECD. 2009b. Test No. 436. Acute inhalation toxicity – acute toxic class method. In: OECD guidelines for the testing of chemicals, section 4: health effects. Paris: OECD Publishing. http://www.oecd-ilibrary.org/environment/test-no-436-acute-inhalation-toxicity-acute-toxic-class-method_9789264076037-en.
- OECD. 2010. No. 129: guidance document on using cytotoxicity tests to estimate starting doses for acute oral systemic toxicity tests. OECD Series on Testing and Assessment. www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2010)20&doclanguage=en.
- OECD. 2016. No. 237: guidance document on considerations for waiving or bridging of mammalian acute toxicity tests. OECD guidelines for the testing of chemicals, section 4. http://www.oecd.org/env/ehs/testing/mono%202016%2032.pdf.
- OECD. 2017. Test No. 402. Acute dermal toxicity. In: OECD guidelines for the testing of chemicals, section 4: health effects. Paris: OECD Publishing. https://www.oecd-ilibrary.org/environment/test-no-402-acute-dermal-toxicity_9789264070585-en.
- OECD. 2018. Test No. 433. Acute inhalation toxicity – fixed concentration procedure. In: OECD guidelines for the testing of chemicals, section 4: health effects. Paris: OECD Publishing. https://www.oecd-ilibrary.org/environment/test-no-433-acute-inhalation-toxicity-fixed-concentration-procedure_9789264284166-en.
- Pistollato F, Madia F, Corvi R, Munn S, Grignard E, Paini A, Worth A, Bal-Price A, Prieto P, Casati S, et al. 2021. Current EU regulatory requirements for the assessment of chemicals and cosmetic products: challenges and opportunities for introducing new approach methodologies. Arch Toxicol. 95(6):1867–1897. doi: 10.1007/s00204-021-03034-y.
- PMRA Health Canada. 2017. Science Policy Note SPN2017-03, acute dermal toxicity study waiver. https://www.canada.ca/en/health-canada/services/consumer-product-safety/reports-publications/pesticides-pest-management/policies-guidelines/science-policy-notes/2017/acute-dermal-toxicity-waiver-spn2017-03.html.
- Prieto P, Cole T, Curren R, Gibson RM, Liebsch M, Raabe H, Tuomainen AM, Whelan M, Kinsner-Ovaskainen A. 2013. Assessment of the predictive capacity of the 3T3 neutral red uptake cytotoxicity test method to identify substances not classified for acute oral toxicity (LD50 > 2000 mg/kg): results of an ECVAM validation study. Regul Toxicol Pharmacol. 65(3):344–365. doi: 10.1016/j.yrtph.2012.11.013.
- Scarano L. 2015. Requirements and evaluation of toxicity testing in the TSCA New Chemicals Program (presentation at Alternative approaches for identifying acute systemic toxicity: moving from research to regulatory setting).
- SCCS. 2021. The SCCS notes of guidance for the testing of cosmetic ingredients and their safety evaluation. 11th revision. doi: 10.1016/j.yrtph.2021.105052.
- Sewell F, Ragan I, Indans I, Marczylo T, Stallard N, Griffiths D, Holmes T, Smith P, Horgan G. 2018. An evaluation of the fixed concentration procedure for assessment of acute inhalation toxicity. Regul Toxicol Pharmacol. 94:22–32. doi: 10.1016/j.yrtph.2018.01.001.
- Shan C, Liu M. 2020. Medical device regulations in China. In: Boutrand J-P, editor. Biocompatibility and performance of medical devices. 2nd ed. Duxford (UK): Woodhead Publishing; p. 475–488.
- Standardization Administration of China. 2008a. Chemicals – test method of acute oral toxicity – fixed dosed procedure (GB/T 21826-2008). Beijing.
- Standardization Administration of China. 2008b. Chemicals – test method of acute oral toxicity for chemicals – acute toxic class method (GB/T 21757-2008). Beijing.
- Standardization Administration of China. 2008c. Test method of acute inhalation toxicity for chemicals (GB/T 21605-2008). Beijing.
- Standardization Administration of China. 2011. Chemicals – test method of acute inhalation toxicity – fixed concentration procedure (GB/T 27824-2011). Beijing.
- Standardization Administration of China. 2012a. Chemicals – acute inhalation toxicity testing – acute toxic class method (GB/T 28648-2012). Beijing.
- Standardization Administration of China. 2012b. Chemicals – test method of acute oral toxicity – up and down procedure (UDP) (GB/T 28648-2012). Beijing.
- Strickland J, Clippinger AJ, Brown J, Allen D, Jacobs A, Matheson J, Lowit A, Reinke EN, Johnson MS, Quinn MJ, et al. 2018. Status of acute toxicity testing requirements and data uses by U.S. Regulatory Agencies. Regul Toxicol Pharmacol. 94:183–196. doi: 10.1016/j.yrtph.2018.01.022.
- Sullivan K, Allen DG, Clippinger AJ, Wilson DM, Edwards SW, Glover K, Mansouri K, Settivari R, Wijeyesakere S, Casey W. 2021. Mind the gaps: prioritizing activities to meet regulatory needs for acute systemic lethality. ALTEX. 38(2):327–335. doi: 10.14573/altex.2012121.
- UN. 2021a. Globally harmonised system of classification and labelling of chemicals (GHS). Rev9. New York: United Nations.
- UN. 2021b. GHS implementation by country. GHS Sub-Committee. https://unece.org/sites/default/files/2021-10/GHS%20implementation%20by%20country_2021-10-19.pdf.