
Abstract
Dieldrin is an organochlorine insecticide that was widely used until 1970 when its use was banned because of its liver carcinogenicity in mice. Several long-term rodent bioassays have reported dieldrin to induce liver tumors in in several strains of mice, but not in rats. This article reviews the available information on dieldrin liver effects and performs an analysis of mode of action (MOA) and human relevance of these liver findings. Scientific evidence strongly supports a MOA based on CAR activation, leading to alterations in gene expression, which result in increased hepatocellular proliferation, clonal expansion leading to altered hepatic foci, and ultimately the formation of hepatocellular adenomas and carcinomas. Associative events include increased liver weight, centrilobular hypertrophy, increased expression of Cyp2b10 and its resulting increased enzymatic activity. Other associative events include alterations of intercellular gap junction communication and oxidative stress. Alternative MOAs are evaluated and shown not to be related to dieldrin administration. Weight of evidence shows that dieldrin is not DNA reactive, it is not mutagenic, and it is not genotoxic in general. Furthermore, activation of other pertinent nuclear receptors, including PXR, PPARα, AhR, and estrogen are not related to dieldrin-induced liver tumors nor is there liver cytotoxicity. In previous studies, rats, dogs, and non-human primates did not show increased cell proliferation or production of pre-neoplastic or neoplastic lesions following dieldrin treatment. Thus, the evidence strongly indicates that dieldrin-induced mouse liver tumors are due to CAR activation and are specific to the mouse, which are qualitatively not relevant to human hepatocarcinogenesis. Thus, there is no carcinogenic risk to humans. This conclusion is also supported by a lack of positive epidemiologic findings for evidence of liver carcinogenicity. Based on current understanding of the mode of action of dieldrin-induced liver tumors in mice, the appropriate conclusion is that dieldrin is a mouse specific liver carcinogen and it does not pose a cancer risk to humans.
1. Introduction
Dieldrin (CAS no. 60-57-1) is an organochlorine insecticide that was widely used in the United States in the 1950s until 1970, at which time its use was canceled by the U.S. Department of Agriculture based primarily on the rodent carcinogenicity bioassays reported by Walker et al. (Citation1973) and Thorpe and Walker (Citation1973). These studies showed an increase in liver tumors in mice following chronic administration of dieldrin in the diet. While additional studies examining the carcinogenicity of dieldrin have been performed, the Walker et al. (Citation1973) and Thorpe and Walker (Citation1973) studies remain the primary ones on which the evidence for hepatocarcinogenicity of dieldrin in mice is based. In contrast to the results in mice, no increase in tumors have been identified in rats administered dieldrin in the diet (National Cancer Institute (NCI) Citation1978; IARC Citation2019).
Dieldrin was banned for use in the US in 1970. Although no longer used or manufactured, dieldrin is still found in hazardous waste sites in the US according to the Agency for Toxic Substances and Disease Registry (ATSDR) (Citation2022) and has been identified in at least 312 of the 1,867 hazardous waste sites that have been proposed for inclusion on the EPA National Priorities List (NPL) (Agency for Toxic Substances and Disease Registry (ATSDR) Citation2022, page 111). The reported induction of hepatic cancer in mice led to a concern about the potential human cancer risk to humans. While experimental studies have been reported on some of the hepatic toxic effects of dieldrin, the mode of action for the observed cancer induction in the mouse had not been defined. Thus, a scientifically based risk assessment of liver cancer from dieldrin in humans utilizing more recent mechanistic investigations is needed for a more up to date evaluation of human cancer risk. Dieldrin is one of a group of organochlorine pesticides (including DDT and toxaphene) that exhibit mouse liver tumor induction following chronic treatment. Like dieldrin, these other organochlorine pesticides are still found in the environment. Understanding the mechanisms by which these chemicals produce the liver tumors is important in defining the potential liver cancer risk in humans.
The United States Environmental Protection Agency (USEPA) classified dieldrin as a Group B2 (probable human carcinogen) based on sufficient evidence of carcinogenicity (liver tumors) in mice (USEPA Citation1987; Citation2012). The USEPA considered the human carcinogenicity data for dieldrin to be inadequate. The International Agency for Research on Cancer (IARC) determined that there was limited evidence of dieldrin carcinogenicity in humans but sufficient evidence in experimental animals for the carcinogenicity of dieldrin (IARC Citation2019). Therefore, the overall IARC classification was that dieldrin is probably carcinogenic to humans (Group 2 A). Based on current understanding of the mode of action (MOA) of dieldrin-induced liver tumors in mice, a more appropriate conclusion is that dieldrin is a mouse specific liver carcinogen and that it does not pose a cancer risk to humans. The basis for this conclusion is the purpose of this manuscript.
The original regulatory decision regarding dieldrin was made in 1970 at a time when all chemicals that produced tumors (carcinogens) were considered to involve the same mechanism of action which involved DNA reactivity and genetic mutation. The role of DNA reactivity and mutagenicity in carcinogenesis was evolving, culminating in the publication of the development and application of the Salmonella bacterial mutagenicity assay (Ames assay) in 1975 (Ames et al. Citation1975). The concept of two major classes of carcinogens, mutagenic and non-mutagenic (also referred to as epigenetic) was not distinguished until 1981 (Weisburger and Williams Citation1981). The role of increased cell proliferation as the basis for non-mutagenic carcinogenesis was published in 1981 (Moolgavkar and Knudson Citation1981; Greenfield et al. Citation1984; Cohen and Ellwein Citation1990, Citation1991). As will be described in this article, the scientific evidence supports that dieldrin is non-mutagenic, and the mode of action for the mouse liver tumors induced by dieldrin is via activation of the constitutive androstane receptor (CAR) leading to increased hepatocellular proliferation. This mode of action (CAR) is not relevant to human carcinogenesis (Elcombe et al. Citation2014; Yamada Citation2021a; Yamada et al. Citation2021b; Shizu et al. Citation2024). An updated and revised assessment of dieldrin’s possible cancer risk to humans is needed given the extensive new experimental findings as well as our better understanding of the carcinogenesis process since the previous human risk evaluations were performed.
Recent experimental evidence has been reported regarding mode of action analyses, indicating that constitutive androstane receptor (CAR) activation is the process through which dieldrin-induced liver changes occur. In this review, we evaluate the evidence for carcinogenicity of dieldrin in mice, discuss the evidence supporting CAR activation as the MOA, evaluate alternative MOAs, and evaluate possible relevance to human cancer risk. A discussion regarding epidemiology studies evaluating the risk of liver toxicity and cancer in humans is then presented.
We present this case study as an example of applying current knowledge to a MOA analysis for a substance that had been evaluated at a previous time when no mechanistic information was available. Current MOA knowledge has regulatory implications.
A literature search was conducted using four sources, including the US National Library of Medicine database (PubMed), the Elsevier citation database (Scopus), the regulatory agencies reviews of dieldrin (USEPA, ATSDR, and IARC), and references cited in the above three sources. Specific search terms used in the literature search strategy are listed in Supplemental Material.
2. Carcinogenicity studies of dieldrin in animals
Several long-term feeding studies of dieldrin in rats and mice have been performed in various strains, including Osborne-Mendel rats (Deichmann et al. Citation1970; Fitzhugh et al. Citation1964; National Cancer Institute (NCI) Citation1978), Carworth farm “E” strain rats (Walker et al. Citation1969; Stevenson et al. Citation1976), rats with the strain not reported (Cleveland Citation1966), C3Heb/Fe/J mice (Davis and Fitzhugh Citation1962), and B6C3F1 mice (National Cancer Institute (NCI) Citation1978). In these studies, no evidence of liver carcinogenicity of dieldrin was reported in rats. In contrast, dieldrin in these studies and subsequent reported studies consistently induced liver tumors in mice. Positive results in the mouse liver were also reported in the neonatal mouse model using B6C3F1 mice (Vesselinovitch et al. Citation1979). However, all of these studies had significant shortcomings in their experimental design (such as outdated histopathology classification system, poorly defined histopathology criteria, too few numbers of animals, inappropriate controls, and others) or did not show statistically significant results (IARC Working Group Citation1974). This included the study in rats and mice performed by the National Cancer Institute (National Cancer Institute (NCI) Citation1978), a study which showed no evidence of tumorigenesis in rats but showed an increased incidence of liver tumors in mice. The NCI study had several flaws. It was performed before a standardized protocol was established for the long-term cancer bioassay by the National Cancer Institute (subsequently the National Toxicology Program). In the NCI study (1978), the number of animals used in the concurrent control group was small, therefore the investigators pooled controls from several other ongoing studies performed at about the same time. This is an unacceptable approach for both experimental design and statistical analysis. In addition, the histopathology of the resulting liver tumors was all classified as carcinomas. Reevaluation by us based on the descriptions and photographs in these publications and based on current criteria, most of the carcinomas would be classified as adenomas (Thoolen et al. Citation2010). The use of the term hepatocellular carcinoma in the NCI study infers a more aggressive tumor type. Thus, in the National Cancer Institute (NCI) (Citation1978) study, a better descriptor would have been total liver tumors instead of hepatocellular carcinoma. Nevertheless, even though these studies had numerous deficiencies, they provided some evidence of the selective hepatocarcinogenicity of dieldrin in mice and the lack of liver or other carcinogenicity in rats. Although the studies in rats had deficiencies in these and in shorter term studies described below, there was no evidence of increased incidences of liver tumors or precursor lesions (foci) or evidence of increased cell proliferation in the rat.
The studies reported in Walker et al. (Citation1973) and Thorpe and Walker (Citation1973) are considered the definitive studies for the hepatocarcinogenicity of dieldrin in mice. In these reports, several experiments were performed in mice, with a thorough analysis of the diets for presence of various organochlorine contaminants. In the study by Walker et al. (Citation1973), the Carworth Farm No. 1 (CFI) strain of mice was used. Because of the large number of mice in that study, starting times were staggered. Different numbers of mice were used in the various groups, with the largest number in the control groups (). Male and female mice were fed doses of 0, 0.1, 1.0, and 10 ppm dieldrin in the diet for up to 132 weeks. Survival was similar to the controls in the low dose groups, but survival was markedly decreased in the high dose group, both in males and females, primarily due to the development of liver tumors, many of which were actually palpable masses during the course of the experiment. Palpable masses were not detected in the lower dose groups or in the controls.
Table 1. Incidence of liver tumors in mice fed dieldrin for 132 weeks (from Walker et al. Citation1973).
The histology of liver tumors was described as being of two types, both of which were classified as hepatocellular carcinomas. The first type (referred to as Walker A) included tumors that today (Thoolen et al. Citation2010) would be classified either as adenomas or carcinomas with little anaplasia, expansive growth pattern with pushing borders, and without the presence of invasion into the adjacent liver parenchyma. The second type (Walker B) involved papilliform and trabecular formations of liver cells, with marked nuclear pleomorphism (anaplasia) and invasion of adjacent hepatic parenchyma. Today, the Walker B tumors would be classified as carcinomas. However, since there is a continuum of histopathologic changes between adenomas and carcinomas, it has been concluded that it is best to combine these tumors for incidence comparisons (Quist et al. Citation2019). Therefore, in , the total number of animals with liver tumors is indicated. Based on this initial experiment, there was a significant increase in liver tumor incidences in both the male and the female mice at the dose of 10 ppm of the diet but not at the lower doses.
In a follow up study (Walker et al. Citation1973) involving administration of dieldrin to male and female CF1 mice for 128 weeks at doses of 0, 1.25, 2.5, 5.0, 10.0 and 20.0 ppm in the diet, a more detailed dose response evaluation was performed (). In this experiment, the incidence of liver tumors was increased at doses of 2.5 ppm and higher. Survival was also decreased at the 2 highest doses, resulting in smaller numbers of animals being considered in the results.
Table 2. Incidence of liver tumors in mice fed dieldrin for 128 weeks (dose-response experiment) (from Walker et al. Citation1973).
Additional experiments were performed to evaluate the effect of sterilizing the diet with ethylene oxide or irradiation compared to unsterilized diet (Walker et al. Citation1973). Sterilization did not affect the overall results. Likewise, coadministration of dieldrin (5 ppm) with dichlorodiphenyltrichloroethane (DDT) (50 ppm) for 2 years did not show any impact of dieldrin on the overall incidence of hepatic tumors induced by DDT or vice versa. In the livers in the mice administered dieldrin with DDT, the histopathologic appearances were essentially similar to that of DDT alone.
Studies on rats and dogs by the same group of investigators (Walker et al. Citation1969) showed no evidence of an increased incidence of liver tumors or hepatocellular toxicity. Nevertheless, examination of the livers by transmission electron microscopy showed alterations of the smooth endoplasmic reticulum in response to dieldrin administration in the mouse and in the dog and the rat. These electron microscopic changes are consistent with later experiments demonstrating an effect of dieldrin on induction of microsomal enzymes, some of which are linked to CAR activation.
In a subsequent experiment, Thorpe and Walker (Citation1973) also evaluated dieldrin and other chlorinated hydrocarbons for their effect on mouse liver that were considered enzyme inducers. This included dieldrin administered at a dose of 10 ppm (equivalent to a dose of approximately 1.5 mg/kg bw/day based on the conversion ratio proposed by Lehman Citation1959). Thirty males and 30 females were administered dieldrin at 10 ppm of the diet compared to 45 and 44 males and females, respectively, in the control groups. All of the male mice administered dieldrin developed liver tumors whereas 87% of the female mice developed liver tumors. The histologic appearance was similar to that in the earlier study reported by Walker et al. (Citation1973).
In the NCI chronic study (1978) performed in rats, it was reported that there was an increased incidence of adrenal cortical adenomas and carcinomas in the high dose dieldrin treated females. However, compared to the concurrent controls, the incidence was not statistically significantly increased. There was some indication of an increase when compared to pooled controls, but as indicated above, such comparison is statistically inappropriate. Based on the review of all of the dieldrin rodent carcinogenicity data, it can be concluded that the hepatic tumorigenic effect of dieldrin is limited to mice and carcinogenicity is limited to hepatocellular tumors.
Examination of other models, including dieldrin used as a “liver tumor promoter” in mice have been reviewed, with the majority of these studies showing a positive response for increased hepatocellular tumors. Using this model, no tumorigenic effect has been observed in rats by dieldrin. Such experiments involve administration of the test chemical after a loading dose of a DNA reactive carcinogen, usually diethylnitrosamine (DEN). Tumor promotors are actually non-DNA reactive carcinogens (Cohen and Ellwein Citation1990, Citation1991), causing an increase in tumor incidences by increasing cell proliferation (see discussion below).
Kolaja et al. (Citation1996a) evaluated the promoting effect of dieldrin in F344 male rats and in B6C3F1 male mice injected with DEN prior to the administration of dieldrin in the diet. The regimen that was used produced preneoplastic foci in mice at 10 ppm of dieldrin after 30 and 60 days, but not in the rats. A lower concentration (≤ 1 ppm) did not produce a tumorigenic effect in the liver in mice, similar to what was observed in the long term bioassay of dieldrin by itself (Walker et al. Citation1973). This protocol has been frequently used by Kolaja et al. (Citation1996a, Citation1996b) and by numerous other investigators (Farber and Sarma Citation1987; Ito et al. Citation1989; Pitot et al. Citation2000; Pitot Citation2007) to show an increased tumorigenic effect of various non-genotoxic carcinogens in the livers of mice pretreated with DEN or other carcinogens. Kolaja et al. (Citation1996b) were also able to show that the hepatocellular foci induced by dieldrin following DEN pretreatment showed some evidence of reversibility, particularly the eosinophilic lesions. These are the types of responses that one expects for non-DNA reactive hepatocarcinogens, such as dieldrin, including ones known to be related to CAR activation (Elcombe et al. Citation2014).
Reuber (Citation1976) evaluated the histopathology of the liver tumors in the mouse study of dieldrin by Fitzhugh and Davis (Davis and Fitzhugh Citation1962; Davis Citation1965). The diagnoses were confirmed as hepatocellular tumors, mostly carcinomas, ranging from well to poorly differentiated. This did not change the overall assessment of the tumorigenicity. Reuber also suggested that there were increased incidences of lung tumors, but this was not confirmed in any other studies. A search of the literature after 2020 showed no additional long-term bioassays, with references identified before that time based on previous reviews (Agency for Toxic Substances and Disease Registry (ATSDR) Citation2022; IARC Working Group Citation1974; IARC Citation2019; USEPA Citation1987; Citation2012; Stevenson et al. Citation1999; Hooker et al. Citation2014).
3. Mode of action evaluation
Hepatocellular tumors have been produced by a variety of agents in animals as well as in humans. Fundamentally, all of the causes are related to either an increase in DNA reactivity (direct genotoxicity, mutagenicity) or secondary to increased cell proliferation, either following evidence of cytotoxicity secondary to either chemical damage, viral damage, inherited disorders, or other causes, with regenerative increased cell proliferation, or by direct mitogenesis () (Cohen Citation2010).
Table 3. Modes of action for hepatocarcinogens in rodents.
A consistent finding in animal studies with dieldrin has been the presence of increased liver weight, a common occurrence with CAR activation. In the mouse, increased smooth endoplasmic reticulum is present as detected by transmission electron microscopy, a marker of CAR activation (Walker et al. Citation1969). Centrilobular hypertrophy is also common for these agents, as exemplified by the prototype CAR activator, phenobarbital (PB).
An analysis of the experimental data (described in more detail below) strongly supports the observed induction of mouse liver tumors by dieldrin by a CAR activating MOA. Activation of CAR altered gene expression specific to CAR activation, increased cell proliferation, clonal expansion leading to altered hepatic foci, and ultimately liver tumor formation are considered to be key events, as they constitute necessary steps in the MOA (Elcombe et al. Citation2014). In addition, induction of hepatic CYP2B enzymes and liver hepatocellular centrilobular or panlobular hypertrophy (as evidenced by morphological changes and increases in liver weight) are considered associative events and as such represent reliable markers of CAR activation (Elcombe et al. Citation2014; Yamada et al. Citation2021b). While other associative events and/or modulating factors (e.g. decreased apoptosis, inhibition of gap junctional intercellular communication, induction of oxidative stress) may also be involved in tumor formation, data on such endpoints are not specific to CAR activation and are not required to establish a CAR-dependent MOA for rodent liver tumor formation (Elcombe et al. Citation2014; Lake Citation2018). Similarly, while much MOA data can be obtained from short-term studies, data on clonal expansion to altered hepatic foci and formation of liver tumors (adenomas and carcinomas) can only be obtained in long-term studies, and, depending on the time points selected, such increases may not be observed. The accepted evolution of hepatic tumors in mice is development of hepatocellular foci, evolving to hepatocellular adenomas which evolve to carcinomas (Farber and Sarma Citation1987; Maronpot et al. Citation1987; Pitot Citation2007; Thoolen et al. Citation2010). Some studies will not have evidence of foci at the end of the experiment as most will have evolved to actual tumors by that time. The presence of adenomas and/or carcinomas implies the presence of hepatocellular foci at earlier time points. The earlier times at which the foci are present might not have been examined in a given experiment but had to have been present for the ultimate development of the actual tumors.
As noted above, dieldrin has been shown to induce liver tumors selectively in mice after chronic exposure. Multiple studies that have examined the mechanism of dieldrin-induced mouse liver tumors have been reported. As discussed above, the accepted MOAs for liver tumor induction by chemicals fall into three general categories of MOA: genotoxicity, receptor mediated mitogenicity, and cytotoxicity with consequent regenerative proliferation. Our comprehensive review of the published studies concluded that dieldrin is working through a nongenotoxic MOA most likely through a receptor mediated process. As shown in several nuclear receptors have been shown to be targets for non-genotoxic liver carcinogens in rodents. These include CAR (constitutive androstane receptor), AhR (aryl hydrocarbon receptor), PPARα (peroxisome proliferator activated receptor alpha), PXR (pregnane X receptor), and estrogen (although there is some evidence that estrogen-induced liver tumors involve DNA reactivity, Yager and Davidson Citation2006). Recently, Wang et al. (Citation2020) examined the activation of these nuclear receptors in the liver of dieldrin-treated mice. Their results definitively showed that CAR activation in the liver of mice following dieldrin treatment was the MOA for dieldrin-induced liver tumors in mice and that these other receptors were not activated.
We apply the International Programme on Chemical Safety (IPCS) MOA/human relevance framework to the analysis of the dieldrin-induced liver tumors (Meek et al. Citation2003; Sonich-Mullin et al. Citation2001; Seed et al. Citation2005; Boobis et al. Citation2006, Citation2008). This framework was developed by a committee sponsored by USEPA and Health Canada (Meek et al. Citation2003, Citation2014; Seed et al. Citation2005), and formed the basis for the MOA recommendations in the 2005 USEPA cancer guidelines (USEPA Citation2005). The MOA is described by a sequence of key events. A “Key Event” is an observable necessary precursor in development of the tumor (Sonich-Mullin et al. Citation2001; Cohen et al. Citation2004). The MOA analysis begins with the identification of a specific neoplasm (usually in rodents) that is linked to exposure of a specific chemical. Based on the pharmacology, toxicology, and pathology of the chemical and the neoplasm induced, a hypothesized rodent cancer MOA using defined Key Events is developed that explains how exposure to the chemical leads to the observed neoplasm. Besides the Key Events, other important biological processes linked to the chemical exposure may be observed. While not required for the cancer MOA, these other endpoints can be used as an indicator or biomarker for a Key Event (Associate Events). In addition, various factors, including external or internal (host) factors, can modulate the dose–response relationship of one or more of the Key Events, thereby changing the probability and/or magnitude of the end result (Cohen et al. Citation2004).
4. Postulated key events
From the published studies on dieldrin liver carcinogenesis, we hypothesize that the MOA for Dieldrin-induced mouse liver tumors is through the well establish CAR MOA for rodent liver tumors. The key events of the Dieldrin – CAR mode of action include:
Key event 1: Activation of CAR;
Key event 2: Altered gene expression;
Key event 3: Increased hepatocellular proliferation;
Key event 4: Clonal expansion leading to altered hepatic foci;
Key event 5: Formation of hepatocellular adenomas/carcinomas.
A description of each Key event and the scientific support for the event are described below:
Key Event 1. CAR activation
Wang et al. (Citation2020) treated male C57BL/6 mice with dieldrin in diet (10 ppm) (the carcinogenic dose) (equivalent to approximately 1.5 mg/kg bw/day) for 7, 14, and 28 days and examined the activation of the liver nuclear receptors CAR, AhR, PPARα, and PXR in the treated mouse livers compared to untreated mouse liver. The results showed that the expression level of Cyp2b10 (a specific CAR responsive gene) was significantly induced in the C57BL/6 mice following 7, 14, and 28 days of treatment with dieldrin. The increase in Cyp2b10 gene expression showed an exposure dose pattern with a 430-fold increased expression at 7 days, a 2200-fold increase at 14 days of treatment and a 2700-fold increase after 21 days of dieldrin exposure compared to untreated control mice. In this same study, mice treated with PB (500 ppm in the diet), an established liver tumorigen in rodents that functions through the CAR MOA, was also examined for receptor activation. The PB-treated mice showed a similar increase in Cyp2b10 gene expression, demonstrating a 2200-fold, 400-fold, and 1400-fold increase in Cyp2b10 gene expression relative to untreated control levels at 7, 14 and 21 days of treatment, respectively. Dieldrin also induced a significant increase in PXR activation (measured by Cyp3a11, up to 5- to 11-fold) over untreated controls. The PXR receptor activation often is linked with the CAR receptor activation, and frequently one sees an increase in expression in both CAR and PXR (Lake Citation2018; Yamada et al. Citation2021b). The other nuclear receptors examined in this study, AhR and PPARα, showed little to no activation by the dieldrin treatment.
A follow up experiment (Wang et al. Citation2020) confirmed the specific activation of CAR by dieldrin treatment using genetically modified KO mice in C57B1/6 (one of the parent strains of the B6C3F1 mice used in the chronic bioassays). In this study, male CAR KO mice (CAR−/−) and wild type mice (CAR+/+) were treated with dieldrin (10 ppm; 1.5 mg/kg bw/day), or vehicle control for 14 days. In the dieldrin treated CAR knock out mice, no increase in Cyp2b10 gene expression. This was in contrast to dieldrin-treated wild type mice that showed a 2000-fold increase in Cyp2b10 over the untreated wild type control mice. Cyp2b10 expression levels in the untreated and dieldrin-treated CAR KO mouse livers were not measurable. This confirms the CAR KO efficacy. In addition, owing to the observed activation of PXR in the initial study, Wang et al. (Citation2020) looked at the potential contribution of PXR with dieldrin treatment using PXR−/− (PXR knock out) and PXR−/−/CAR−/− (PXR and CAR knock out) dieldrin-treated KO mice in their analysis. In the CAR and CAR/PXR KO mice no gene activation was seen following dieldrin treatment. In contrast, dieldrin treated PXR KO mice showed activation of CAR as measured by Cyp2b10 expression. To summarize, these two studies by Wang et al. (Citation2020) showed that the dieldrin treatment at the tumorigenic dose used in chronic mouse studies resulted in an increase in Cyp2b10 expression (the cytochrome p450 specific to CAR activation) and using KO mice for CAR and PXR showed that the activation of the gene expression was strictly a CAR mediated process. In addition the CAR and PXR knock out studies showed that the induction of DNA synthesis and increase in relative liver weight following dieldrin treatment were dependent on a functional CAR.
Key Event 2. Altered gene expression
The activation of the CAR receptor produces several biologic changes in the liver. The induction of hepatic CYP2B enzymes is a specific marker of CAR altered gene expression. Other less specific hepatic changes that have been related to CAR activation in the liver include hepatocellular centrilobular or panlobular hypertrophy identified by histopathologic examination and increased relative liver weight. These are considered associative events and as such represent reliable but nonspecific markers of CAR activation (Elcombe et al. Citation2014). Kolaja et al. (Citation1996c) showed that acute dietary exposure of rats or mice to either dieldrin or PB produced several liver changes, including centrilobular hypertrophy, induction of hepatic cytochrome P450, increased DNA synthesis and increased liver weight. Additionally, Wang et al. (Citation2020) reported an increase in pentoxyresorufin-O-dealkylase (PROD) (which assesses Cyp2b10 activity; Paolini et al. Citation1995) as well as relative liver weight and DNA synthesis in mice treated with dieldrin for 7, 14, and 28 days continuously with 10 ppm dieldrin in the diet. In addition, at terminal sacrifice, liver weights were measured and dieldrin-treated mice had a slight but statistically significant increase in relative liver weights compared to control mice after 14 and 28 days of treatment (Wang et al. Citation2020). No liver toxicity was seen in the dieldrin-treated mice based on no increase in liver serum enzymes (alanine amino transferase, ALT; aspartate aminotransferase, AST; alkaline phosphatase, ALP; gamma glutamyl transpeptidase, GGT) and no evidence of liver cytotoxicity by histopathologic evaluation.
Wang et al. (Citation2020) also reported a significant induction of PROD activity in C57BL/6 mice following treatment with dieldrin in the diet for 7, 14, or 28 days compared with untreated control mice. The expression level of Cyp2b10 (CAR responsive gene) was induced statistically significantly in wild-type C57BL/6 mice following 7, 14, and 28 days of treatment with dieldrin (up to 430-, 2200-, and 2700-fold, respectively), comparable to the positive control CAR chemical, PB (2200-, 400-, and 1400-fold, respectively). The expression level of the PXR target gene Cyp3a11, which is also regulated by CAR, was increased after treatment with dieldrin (4- to 6-fold) and with PB (10- to 14-fold) for 7, 14, and 28 days. Both PROD and Cyp2b10 were not increased in CAR KO mice treated with dieldrin, further supporting a CAR mediated mode of action for dieldrin.
Wright et al. (Citation1972) reported that after a 1-week dietary treatment of rats (200 ppm: 8.0 mg/kg bw/day) and mice (10 ppm: 1.6 mg/kg bw/day), increases in hepatocyte smooth endoplasmic reticulum, microsomal protein, and mixed function oxidase system activity were detected in the liver. Similar hepatic changes were reported in dogs after dieldrin treatment (via a daily capsule) for 4 weeks at 2 mg/kg bw/day of dieldrin. Exposure of monkeys to concentrations as high as 0.1 mg/kg bw/day of dieldrin for up to 6 years (not lifetime) also produced an increase in cytochrome P-450 content in the liver cells (Wright et al. Citation1972, Citation1978). While the monkeys and rats showed liver changes consistent with CAR (or other receptor mediated) activation, it is important to note that neither of these species exhibited increased hepatocellular proliferation or hepatic tumors following dieldrin chronic treatment. In rats, this is likely due to inadequate stimulation of proliferation whereas in the dogs there is no evidence of increased cell proliferation with CAR activators. Differential responses in proliferation and tumorigenesis to CAR activators in mice compared to rats has been reported with various chemicals, not just dieldrin (Yamada et al. Citation2021b).
Key Event 3. Increased hepatocellular proliferation
The induction of cell proliferation by a non-genotoxic carcinogenic compound is essential in the formation of neoplasia (Cohen and Ellwein Citation1990). In the case of dieldrin, several studies have been reported that demonstrate the induction of increased DNA synthesis in a dose dependent manner in mouse liver following dieldrin treatment (Kolaja and Klaunig Citation1997; Klaunig et al. Citation1998a; Citation1998b). Kolaja et al. (Citation1995a, Citation1996c) examined the acute and subchronic treatment (7, 14, 21, 28, and 90 days) with dieldrin (0.1, 1.0, 3.0, 10.0 mg/kg diet) and PB (10, 50, 100, 500 mg/kg diet) on the induction of hepatic DNA synthesis and hepatocyte lethality in male B6C3F1 mice and male F344 rats. Mice and rats were evaluated for hepatic DNA synthesis after 7, 14, 21, 28, and 90 days of continuous treatment with dieldrin or PB. The maximum increase in hepatic DNA synthesis in mice was seen with dieldrin only at 10 ppm at the 14-, 21-, and 28-day sampling times. In rats, no increase in hepatic DNA synthesis was observed at any dieldrin dose. In contrast, PB treatment increased hepatic DNA synthesis in both rat and mouse liver following 7 days of treatment as has been reported previously (Yamada et al. Citation2021b). In the rat, the PB induction of liver DNA synthesis rate was transient, with the labeling index returning to control rates by the 14-day sampling time, whereas mice treated with PB showed a dose related increase in hepatic DNA synthesis rate at the 7, 14, 21, 28 days sampling times. In mice, dieldrin induced hepatic DNA synthesis selectively in the centrilobular region of the hepatic lobule. No increase in liver serum enzymes indicative of hepatic damage or necrosis was detected in mice or rats fed dieldrin or PB. The authors concluded that the induction of DNA synthesis was not mediated by a cytolethal, compensatory hyperplastic response (Kolaja et al. (Citation1995a, Citation1996c) but through a mitogenic mechanism. The mouse specific induction of hepatic DNA synthesis by either dieldrin or PB correlated with the previously observed species-specific induction of hepatic cancer by these two compounds.
Kolaja et al. (Citation1996c) confirmed that exposure to 10 mg dieldrin/kg diet results in a transient increase in the DNA synthesis rate in the mouse liver, but not rat liver. In addition, doses of 0.1 and 1.0 mg dieldrin/kg diet did not increase DNA synthesis in either species. This lack of induction of DNA synthesis correlated with previously observed non-carcinogenic doses of dieldrin. It has been suggested that increased DNA synthesis in naïve liver may be an indicator of non-genotoxic hepatocarcinogenic compounds that also have so-called tumor promoting activity. A dose of 10 mg dieldrin/kg diet to mice led to a rapidly increased rate of DNA synthesis, followed by a steadily decreasing rate of DNA synthesis. However, although the rate of DNA synthesis decreases, the number of hepatocytes has increased as indicated by the increased liver weight, so that the total number of hepatocyte DNA replications is actually continuously increased. Although not examined specifically for dieldrin, the increased liver weight has been shown with a prototypic CAR activator, phenobarbital, to be due to hypertrophy and increased hepatocyte number (Carthew et al. Citation1998). As recently reported by Wang et al. (Citation2020), using CAR KO mice, dieldrin-induced relative liver weight and hepatocyte cell proliferation was CAR dependent. The key parameter is number of DNA replications, not rate, although the two frequently occur together (Wood et al. Citation2015; Strupp et al. Citation2023).
The effect of dieldrin on hepatic nuclear ploidy has also been investigated by Kamendulis et al. (Citation2001) and by van Ravenzwaay and Kunz (Citation1988). Kamendulis et al. (Citation2001) examined hepatocyte DNA synthesis, mitosis, apoptosis, and ploidy in mouse liver and rat liver after treatment with dieldrin for 7, 14, 28, or 90 days dosed at 0, 1, 3 and 10 ppm dieldrin in the diet (equivalent to 0.0, 0.15, 0.45, or 1.5 mg/kg bw/day). Rates of DNA synthesis and mitoses were increased at 14, 28, and 90 days at 3 and 10 ppm in the mouse. No increase in DNA synthesis was seen in dieldrin-treated rats. The apoptotic index in liver of dieldrin treated mice and rats did not change with treatment from controls. Mice treated with 10 ppm dieldrin showed a significant increase in octaploid (8 N) hepatocytes and a relative decrease in diploid (2 N) hepatocytes in the liver that was primarily seen in centrilobular hepatocytes. Hepatocytes in the portal region of the liver in dieldrin-treated mice showed little to no change in ploidy compared to untreated control mice. No change in hepatocyte nuclear ploidy was observed in the rat following dieldrin treatment. The observed increase in hepatocyte ploidy may reflect an adaptive response of non-lesion hepatocytes to dieldrin exposure. Given that the majority of mouse liver tumors and preneoplastic lesions are diploid (2 N), the fact that most of the hepatocytes seen in this study were octoploid suggest that a subpopulation of hepatocytes after dieldrin treatment maintained the diploid state and were selectively destined to become preneoplastic lesions and potentially tumors.
More recently (Wang et al. Citation2020), the hepatic DNA synthesis rate in the dieldrin-treated mouse liver was determined using BrdU immunohistochemical detection (Wang et al. Citation2015). C57BL/6 male mice treated with 10 ppm dieldrin (1.5 mg/kg bw/day) for 7, 14 and 28 days showed a duration dependent increase in the DNA synthesis rate over untreated mouse liver. This observed increase in DNA synthesis was specifically CAR activation dependent (not PXR) as CAR KO mice (CAR -/-) treated with dieldrin failed to show an increase in the induction of cell proliferation. In contrast, similarly treated PXR KO mice showed a 2-fold increase in DNA synthesis while a CAR/PXR combination KO failed to show an increase in DNA synthesis in the liver following dieldrin treatment. These findings confirm the role CAR activation in modulating the dieldrin- induced liver DNA synthesis.
Key Event 4. Clonal expansion leading to altered hepatic foci
Hepatocarcinogenesis is a multistep process with defined, demonstrable pathologic and operational stages (Farber and Sarma Citation1987; Pitot Citation2007). For modeling purposes, these stages have been operationally labeled initiation, promotion, and progression. Initiation is the irreversible alteration of a cell’s genomic DNA. Promotion in this model is defined as the selective reversible clonal expansion of these initiated cells, and progression as the final irreversible stage where additional genetic alterations occur resulting in the formation of a neoplasm. While dieldrin has been shown to cause some similar acute and subchronic hepatic effects in both the mouse and the rat liver, including increased relative liver weight, centrilobular hypertrophy, and p450 induction, only the mouse responds with liver neoplasms after chronic exposure. These results raise questions about the mechanism involved in the different species susceptibility, although different responses to CAR activators in mice and rats has been reported with other chemicals (Lake Citation2018; Yamada et al. Citation2021b). As noted above (Key Event 3), besides the selective liver tumorigenicity, only the mouse (not the rat) responds to acute and subchronic dieldrin exposure with increased cell proliferation, a characteristic of non-genotoxic carcinogens.
Kolaja et al. (Citation1995b, Citation1996a) examined the dose-response effect of dieldrin on focal lesion growth, hepatocyte apoptosis, and DNA synthesis in rat and mouse liver. Preneoplastic focal hepatic lesions were produced by a DEN protocol (Klaunig et al. Citation1988). After the lesions developed, mice and rats were placed into one of the following dose groups: control (NIH-07 diet) or 0.1, 1.0, or 10.0 mg dieldrin/kg diet (0.015, 0.15, or 1.5 mg/kg bw/day). Dieldrin at dietary concentrations of 0.1, 1.0, and 10 mg/kg diet produced an increase in preneoplastic focal volume (0.1 mg/kg diet at 7 and 30 days, 1.0 mg/kg diet at 30 days; 10 mg/kg diet at all sampling times), but only the high dose (10 mg/kg diet produced and increased focal DNA labeling index.) This increased labeling index at the 10 mg/kg diet treatment was seen in both eosinophilic and basophilic lesions. Apoptosis was not modified in foci by any concentration of dieldrin in either rats or mice. The increase in foci volume seen with the 0.1 and 1.0 mg/kg dose was transitory and may reflect an increase in P450 enzyme induction. At dietary concentrations of 0.1, 1.0, and 10 mg dieldrin/kg diet, no change in lesion volume, number, or preneoplastic lesion DNA labeling index was seen in treated rats. The authors concluded that these results showed that dieldrin functions as a mouse-specific tumor promoter through expansion of preneoplastic focal lesions.
Kolaja et al. (Citation1996b) also examined the reversibility of dieldrin and PB-induced focal lesion growth in mice. Using the liver foci induction model noted above (Klaunig et al. Citation1988), the effect of dieldrin and PB treatment on focal liver growth and the effect of cessation of PB and dieldrin treatment on hepatic focal lesion growth was assessed. Mice received dieldrin (10.0 mg/kg diet) or PB (500 mg/kg diet) for 30 days followed by a return to control diet for 30 days. Dieldrin and PB increased the number of focal lesions, focal lesion volume, and DNA synthesis. This study demonstrated that feeding of dieldrin or PB to B6C3F1 mice stimulated the growth of DEN-induced hepatic foci. Eosinophilic lesions in particular showed an increase in lesion number and volume in mice fed either dieldrin or PB. Cessation of either dieldrin or PB exposure was accompanied by a decrease in the number and the volume of altered hepatic foci when compared to that seen in mice continuously fed the chemical. The regression of focal lesions following removal of either dieldrin or PB was primarily attributed to the loss of eosinophilic foci. This decrease in focal hepatocytes was directly related to an increased incidence of apoptosis, however, reversion of altered hepatocytes to a phenotype not demonstrable by hematoxylin and eosin staining cannot be precluded. This study indicates that dieldrin and PB promote hepatic focal lesions and that these lesions (eosinophilic in particular) are dependent on continuous exposure to the chemical agent. These data indicated that induction and maintenance of the growth of some preneoplastic lesions in the mouse is dependent upon the continuous treatment with the chemical.
Key Event 5. Hepatocellular adenomas/carcinomas
Progression from preneoplastic foci to adenomas to carcinomas in liver tumorigenesis has been well-documented (Farber and Sarma Citation1987; Pitot et al. Citation2000; Pitot Citation2007; Thoolen et al. Citation2010). In the case of dieldrin, multiple chronic studies using multiple strains of mice (BALB/c, CF1, B6C3F1, C3HeB/Fe, C3H/He, and C57BL/6J mice) have reported the induction of liver tumors following dietary dieldrin administration. Unlike the mouse, no liver tumor induction in rats, hamsters, dogs, or monkeys (Walker et al. Citation1969) have been seen following chronic treatment. The induction of liver tumors in mice is dose dependent, and as shown above, occurs by a CAR MOA. NCI (Citation1978) reported a significantly increased incidence of hepatocellular carcinoma in male B6C3F1 mice exposed to 0.86 mg/kg/day dieldrin in the diet for 80 weeks; but not in males at 0.43 mg/kg/day. Additional chronic exposure studies (Davis and Fitzhugh Citation1962; Thorpe and Walker Citation1973; Walker et al. Citation1973; Epstein et al. Citation1972; NCI, Citation1978; Tennekes et al. Citation1979, Citation1981; Meierhenry et al. Citation1983; Ruebner et al. Citation1984; Lipsky et al. Citation1989) have reported increased incidences of hepatocellular tumors in male C3H/He, B6C3F1, CF1, Balb/c and C57BL/6J mice treated with dieldrin, confirming the moue specific effect with dieldrin without regard to strain.
4.1. Evaluation of modified Bradford Hill criteria applied to mode of action in animal models
The evaluation of the MOA by which a chemical produces an adverse effect such as cancer utilizes the Bradford Hill (Citation1965) criteria originally developed for an evaluation of causality in epidemiology studies but modified for use in evaluating MOA in animal models (Sonich-Mullin et al. Citation2001). These criteria include temporality, dose response, strength, consistency and specificity, biologic plausibility and coherence, followed by an overall assessment of the strength of the evidence for the postulated mode of action (Sonich-Mullin et al. Citation2001; Meek et al. Citation2003; Seed et al. Citation2005; USEPA Citation2005; Boobis et al. Citation2006, Citation2008; Meek et al. Citation2014; Frank and Meek Citation2024).
4.1.1. Temporality
The sequence of the key events is well established for CAR activation in general and for dieldrin specifically. Activation of the CAR (Key Event 1) occurs essentially immediately after absorption of dieldrin from the gastrointestinal tract and its arrival in the liver. Associative events related to CAR activation, including Cyp2b10 activation, increased liver weight, and centrilobular and panlobular hepatocellular hypertrophy, have been identified by 7 days (earliest time evaluated) after administration of dieldrin in the diet, the earliest time at which it has been examined. For PB, the prototypical CAR activator, these changes have been identified earlier and it is presumed that they occur earlier than 7 days for dieldrin. Associated changes in gene expression (Key Event 2) and increased cell proliferation (Key Event 3) are also identified by 7 days, again the earliest time evaluated. Evolution of increased cell proliferation to the clonal expansion of foci (Key Event 4) and the subsequent development of hepatocellular tumors (Key Event 5) is a well-established sequence of events in rodent hepatocellular carcinogenesis (Pitot et al. Citation2000; Pitot Citation2007; Thoolen et al. Citation2010). Thus, the criterion of temporality is clearly met.
4.1.2. Dose response
Hepatocellular tumors were increased at dietary doses of 2.5, 5.0, and 10 ppm (equivalent to 0.375, 0.75, or 1.5 mg/kg bw/day based on the EPA IRIS dose conversion, Lehman Citation1959) in mice (Walker et al. Citation1973) but not at 1 ppm (0.15 mg/kg bw/day). The various key events have been evaluated in experimental studies predominantly at the 10 ppm dieldrin dose (liver carcinogenic dose), a dose at which all of the key events occurred. Increased cell proliferation was also reported in two studies, albeit transitory at 3.0 ppm in dieldrin-treated mice. Thus, there was concordance in dose response for the key events and the formation of tumors, but detailed dose response information of Key events 2, 3, and 4 has not been evaluated at dieldrin doses lower than 10 ppm to the same extent. Although this is a limitation of this criterion, the data overall supports a concordance for the key events with ultimate tumor formation (see ).
Table 4. Concordance table for dose response of key events in dieldrin induced liver tumors.
4.1.3. Strength, consistency, and specificity
The strength of the evidence for CAR activation as the mode of action for dieldrin-induced liver tumors is high, based on the multiple studies showing the relationship between dieldrin administration and CAR activation, including changes in liver weight, histopathology (hepatocellular hypertrophy), activation of Cyp2b10, and increased PROD enzyme activity (Kolaja et al. Citation1995a, Citation1996a, Citation1996b; Kolaja and Klaunig Citation1997; Klaunig et al. Citation1998; Wang et al. Citation2020). The findings have been consistent from study to study, and pertain specifically to liver changes related to CAR. The studies in the various KO mouse models described above clearly show that the effect of dieldrin is on CAR and not on other nuclear receptors such as PXR, AhR, PPARα, or estrogen (Wang et al. Citation2020). Also, other possible modes of action have been excluded (see below).
4.1.4. Biologic plausibility and coherence
CAR activation is a well-known MOA for rodent hepatocellular tumors in mice, and has been demonstrated for a variety of chemicals, including the prototypical CAR activator PB (Elcombe et al. Citation2014; Yamada et al. Citation2021b). The same sequence of key events has been demonstrated for PB and for many of the other chemicals shown to be CAR activators. The key to this MOA in rodents is the induction of increased cell proliferation (Key Event 3). This key event occurs in mice but not in other species, such as the rat. The reason for this difference between mice and rats is not entirely clear (see below), but induction of increased cell proliferation is clearly the critical step in the MOA that distinguishes whether a tumorigenic effect will occur or not. The fact that CAR activation in general does not lead to increased hepatocellular proliferation in humans indicates that humans will not be susceptible to tumorigenic effects of dieldrin, similar to a lack of a tumorigenic effect in humans by PB (see below). Thus, the criteria of biologic plausibility and coherence are met.
4.1.5. Overall assessment and data gaps
Bioassay and mechanistic studies involved in assessing the hepatocellular carcinogenicity of dieldrin in mice have been performed over the past 50 plus years. It is clear that chronic feeding of dieldrin to rodents results in liver tumors only in mice. Recent mechanistic studies definitively point to the activation of CAR as the MOA for mouse liver tumors. The support for this MOA is strong. One data gap is the lack of detailed dose response analysis of the key events leading to tumors. Nevertheless, with the data available there is clearly a strong correlation between the doses at which the key events are observed compared to the dose response for the tumors. A remaining data gap is determining why rats do not also develop liver tumors secondary to dieldrin administration. Although we have leads into the differential liver tumor response in mice versus rats, a definitive mechanism has not been determined. Different tumorigenic responses in mice and rats to various CAR activators has been reported (Lake Citation2018; Yamada et al. Citation2021b). Several additional studies could be performed to further define the reason for the species difference in response, including evaluation of blood levels between species. Specifically, the metabolism of dieldrin in mouse and rats requires more attention. In particular, is there a metabolite that forms only in mouse that is responsible for the observed different response in rats. Second, additional studies could be performed to definitively show that the hypertrophy and P450 induction in rats treated with dieldrin is CAR dependent. And third, the major difference between rat and mouse response of tumor induction and early cellular markers appears to be the disparate induction of cell proliferation seen only in the mouse. Regardless of these data gaps, the evidence for CAR activation as the MOA for dieldrin-induced liver tumors in mice is strong. This is particularly true based on the specific studies utilizing various KO strains of mice to distinguish effects on CAR vs. PXR as well as the specific studies evaluating other nuclear receptors and other possible MOAs (see below).
4.2. Potential mechanisms for the selective induction of liver tumors in mice
Other hepatic carcinogenic compounds, particularly chlorinated pesticides, have also shown mouse specific effects (Newberne Citation1983; Maronpot et al. Citation1987; Wang et al. Citation2015). It is of critical importance to further understand the mechanism of nongenotoxic carcinogenicity in general and in the mouse liver specifically. Because the B6C3Fl mouse is associated with a high spontaneous hepatic tumor incidence compared to the Fisher 344 rat (Maronpot et al. Citation1987), an initial suggestion was that dieldrin functions as a selective promoter of hepatocarcinogenesis in the mouse because of the substantially greater spontaneously initiated cell population in the mouse compared to the rat. If dieldrin is selectively carcinogenic in the mouse liver because of a higher rate of spontaneous initiation than the rat, then DEN-induced hepatic lesions should show expansion in growth in both rat and mouse liver after dieldrin treatment since the fewer spontaneous foci in the rat will be compensated with the use of DEN to induce the foci. This was not the case (Kolaja et al. Citation1996a, Citation1996b), as the DEN-induced rat foci failed to show a response to dieldrin treatment. In contrast, the highest dose of dieldrin examined (10 mg/kg diet) (equivalent to 1.5 mg/kg bw /day) resulted in significant increases in the number, volume, and labeling index of mouse preneoplastic lesions (Kolaja et al. Citation1996a, Citation1996b). This dose enhanced the growth of both basophilic and eosinophilic foci. Kolaja et al. also showed that the lower doses of dieldrin (0.1 and 1.0 mg/kg diet) did not produce an observably consistent tumor-enhancing effects, in particular a lack of cell proliferation induction. A dose of 1.0 mg dieldrin/kg diet only increased the growth of eosinophilic lesions, as measured by an increased labeling index of eosinophilic focal lesions after 60 days of treatment and an increased eosinophilic lesion volume after 30 and 60 days. Rather than an inherent difference in number of potential preneoplastic cells in the rat versus the mouse, the results suggest that there is a difference in the manner in which dieldrin (intrinsic difference) is handled by the mouse hepatocytes, specifically the preneoplastic focal hepatocytes that may account for the species dependent carcinogenicity. Studies by Baldwin et al. (Citation1972) and Hutson (Citation1976) have shown that the rat metabolizes dieldrin quicker than the mouse, producing mainly pentachloroketone, while the mouse produces only very small amounts of pentachloroketone. Dieldrin metabolism studies, however, in the rat and more so in the mouse are limited and incomplete, and therefore conclusions concerning metabolism difference in the two species as the basis for the difference in tumorigenicity of dieldrin must be interpreted with caution. Both rats and mice responded to dieldrin treatment with increased P450 activity and liver hypertrophy, but only the mouse showed increased DNA synthesis in response to dieldrin treatment. CAR activation is definitely seen in the liver of mice treated with dieldrin and appears responsible for the tumorigenicity. CAR activation has not been specifically studied in the rat although the hypertrophy and increased P450 enzyme activity seen following dieldrin treatment support an activation of CAR in the rat by dieldrin. The earlier results with Gap Junctional Intercellular Communication (GJIC) and oxidative stress, while participatory but not causal for dieldrin hepatic carcinogenicity, showed a correlation along with induction of cell proliferation with the species difference in dieldrin susceptibility.
4.3. Modifying/associative events
In the 1970s it became clear that many rodent liver carcinogens were not acting thorough DNA reactive mechanisms. This led to the classification of these agents as nongenotoxic or epigenetic (Weisburger and Williams Citation1981). The exact mechanisms by which these agents produced liver cancer was not defined. A review of the experimental evidence pointed to three cellular mechanisms related to the activity of the nongenotoxic compounds, increased cell proliferation, modification of GJIC, and induction of oxidative stress. In the case of the former, the role of cell proliferation is obvious given the need for neoplastic growth. Increased cell proliferation leads to an increase in the opportunities for the spontaneous errors to occur that accompany DNA replication. The other two mechanisms are associated with the increased cell proliferation process, and multiple studies have examined these processes in more detail. In addition, an application of these two processes were also seen as a much-needed screen for nongenotoxic chemicals.
4.3.1. Gap junctional intercellular communication
GJIC is considered to play a key role in the maintenance of tissue independence and homeostasis by controlling the growth of GJIC-connected cells (Fitzgerald et al. Citation1989). Gap junction channels are composed of connexin molecules. Most neoplastic cells display modified GJIC either between cells within the neoplasm and/or between the normal surrounding cells and the neoplastic cells. A role for GJIC in growth control was initially shown after partial hepatectomy, where a significant change in GJIC and reduction of cx32 protein within 3 h after partial hepatectomy correlated with cell proliferation suggesting that blocking of GJIC precedes S-phase induction. The level of cx32 protein in the liver continues to decrease until 24 h. after partial hepatectomy confirms that partial hepatectomy induces a decrease of GJIC (Tsuda et al. Citation1995). Specific connexins are necessary to control growth of specific cell types. In liver tumorigenesis, Janssen-Timmen et al. (Citation1986) found a 70% decrease in the number of gap junctional (cx32) immunofluorescent spots in rat hepatocellular carcinomas. More recently, using connexin knockout mice, Willecke and coworkers (Ott et al. Citation2006) have shown that ablation of liver connexins Cx26 and Cx32 did not lead to increased spontaneous liver tumor formation but enhanced the carcinogenic effect of DEN administration and resulted in a higher susceptibility to hepatocellular neoplasms, but did not appear to initiate hepatic tumor development. These studies and others have therefore linked the blockage of GJIC to the tumor promotion stage of the cancer processes.
With regard to dieldrin carcinogenesis, several studies have been reported that have examined the effect of dieldrin on in vitro hepatocyte GJIC. Trosko and coworkers used a metabolic cooperation assay in V79 cells and showed that dieldrin, aldrin, and toxaphene inhibited GJIC in a dose dependent manner (Trosko et al. Citation1987). Using primary cultured mouse hepatocytes, Ruch and Klaunig (Citation1986) reported that non-cytotoxic concentrations of PB (20-500 micrograms/ml) and dieldrin (1-10 micrograms/ml) inhibited GJIC. In a follow up study, Klaunig and Ruch (Citation1987) examined the effect of dieldrin and PB on GJIC in primary cultured hepatocytes from four strains of male mice (B6C3F1, C3H, C57BL, and Balb/c strains) and male F344 male rat. Dieldrin blocked GJIC in the mouse hepatocytes in all mouse strains at concentrations greater than 1 μg/ml. There was no inhibition of rat hepatocyte GJIC by dieldrin. PB in contrast blocked hepatocyte GJIC in both mouse (all strains examined) and rat hepatocytes. Thus, the inhibition of GJIC by dieldrin and PB showed a correlation with the in vivo liver tumorigenicity of these compounds.
Baker et al. (Citation1995) further examined the role of dieldrin on GJIC inhibition. They reported a dose dependent inhibition of GJIC in mouse hepatocytes but a lack of GJIC inhibition by dieldrin in primary cultured rat, monkey, and human hepatocytes. Given these results as well as the linkage between GJIC and cell proliferation (in tumors and following partial hepatectomy) led to the hypothesis by Stevenson et al. (Citation1999) that GJIC modification by nongenotoxic compounds may play a role in regulating cellular proliferation and therefore has tumor promotion activity. Jones et al. (Citation1987) examined the effect of dieldrin treatment on a rat cultured hepatocyte line using a metabolic corporation assay. Jones et al. (Citation1987) found that dieldrin (as well as aldrin) at a concentration of greater than 3 mg/ml inhibited GJIC. This cell line was derived from rat liver. These results are in contrast to previous studies by the Klaunig group (Klaunig and Ruch Citation1987) that showed the effect of dieldrin on liver GJIC was specific for the mouse. Two concerns with the Jones et al. (Citation1987) study revolve around the use of an immortalized cell line and the method of GJIC evaluation.
In the context of the MOA framework (Sonich-Mullin et al. Citation2001; Meek et al. Citation2003; Seed et al. Citation2005; USEPA Citation2005), modification of GJIC should be categorized as an associative or modifying event.
4.3.2. Oxidative stress
Oxidative stress results in an imbalance between reactive oxygen species (ROS) or reactive nitrogen species (RNS) and biological antioxidant systems can lead to functional and structural modification of cellular macromolecules including DNA, lipids, and proteins (Klaunig et al. Citation1998; Citation2011; Klaunig Citation2018). Because the redox status (oxidizing/reducing conditions) of cells is involved in regulating various transcription factors/activators (e.g. AP-1, NF-κB and p53), thereby influencing cellular target gene expression and modulating cellular signaling pathways, appropriate ROS and RNS levels are necessary for normal physiological function of the living organisms (Klaunig Citation2018). Interest in a role for oxidative stress in hepatic carcinogenesis increased as a possible mechanism in general for nongenotoxic hepatic carcinogenesis. Oxidative stress is defined as an increase in the imbalance between ROS and antioxidants in the cell. This can occur through either a decrease in antioxidants and/or an increase in ROS. Oxidative stress is frequently measured by oxidative DNA adducts (8-hydroxydeoxyguanosine, 8 OH-dG) or lipid peroxidation. Increased ROS due to high activity of P450 enzymes resulting in futile cell cycling has been postulated as a possible mechanism for liver carcinogens (Klaunig et al. Citation2011). Reddy and coworkers (Reddy and Rao Citation1989; Rao and Reddy Citation1991) proposed a role for hydrogen peroxide generation in the hepatocyte peroxisome as a cause for liver tumors by PPARα activating compounds. Similarly, Takagi et al. (Citation1990) showed an increase in 8-OH-dG in liver DNA of rats following short-term exposure to the peroxisome proliferator phthalates suggesting an involvement of oxidative DNA damage in hepatocarcinogenesis by peroxisome proliferators. Kinoshita et al. (Citation2003) after PB treatment of rats for 4 days showed an increased in hydroxyl radical levels and an accumulation of 8-OH-dG in genomic DNA. The levels of P-450 isoenzymes CYP2B1/2 and CYP3A2 were also increased with PB treatment 8-OH-dG levels decreased with an increase in mRNA expression for the 8-OH-dG repair enzyme, DNA glycosylase 1 (Ogg1).
In line with the above investigations, a number of studies have examined the effect of dieldrin on oxidative stress. Stevenson et al. (Citation1995) fed male B6C3F1 mice (10 per dose group), diets containing dieldrin for 7 or 28 days and examined their livers, including the hepatocytes. Separate groups of mice were simultaneously dosed with the antioxidants, vitamins C or vitamin E, in addition to dieldrin. No evidence of liver cytotoxicity was seen based on serum levels of liver enzymes and histopathology. Dieldrin-induced DNA synthesis was partially inhibited by co-treatment with vitamin E, suggesting a role for oxidative stress in the induction of cell proliferation by dieldrin. Suppression of DNA replication by vitamin E was independent of the increase in relative liver weight and hepatocyte hypertrophy observed with dieldrin treatment.
Bachowski et al. (Citation1998) fed mice and rats (20 per dose group) with diets containing 0, 0.1, 1, or 10 ppm dieldrin (equivalent to 0.0, 0.015, 0.15, or 1.5 mg/kg bw/day) for up to 90 days. Consistent with the observations in Stevenson et al. (Citation1995), DNA labeling increased with dieldrin dose in mice. In mice, DNA labeling was significantly increased above controls for the 1 and 10 ppm dieldrin diets and reached a maximum plateau at 14 and 28 days, returning to the 7-day level at 90 days. After 7 days of the 10 ppm diets, liver vitamin E concentrations in mice decreased by about 50%. Malonaldehyde (MDA), a lipid oxidation product, increased transiently in mouse liver peaking at 14 days and then declining to control levels. In this study, liver DNA 8-OH-dG did not increase with dieldrin treatment, but 8-OH-dG repair products were detected in the urine of dieldrin-treated mice. In this study, in contrast to the mice, similarly treated rats did not show an increase in DNA labeling in response to dieldrin. As with the mice, after 7 days of the 10 ppm dieldrin treatment, vitamin E concentrations in rats decreased, but unlike the mice, the decrease was uniform at all dieldrin doses. Vitamin E concentrations in control liver tissue were 4 times larger in rats than in mice and remained about 2–3 times higher than mouse control values even with dieldrin exposure. Unlike the mice, MDA levels in rats did not increase with dieldrin treatment.
Kolaja and Klaunig (Citation1997), using a preneoplastic formation protocol, examined the effect of vitamin E on preneoplastic mouse liver lesions, focal lesion growth, and DNA synthesis. Treatment consisted of 10 ppm dieldrin (1.5.mg/kg bw/day), 10 ppm dieldrin plus 450 ppm vitamin E, 450 ppm vitamin E alone, and control diet (with a standard 50 ppm vitamin E) for either 30 or 60 days. Dieldrin increased hepatic DNA labeling within focal hepatic lesions as well as surrounding non-lesion liver. Dieldrin plus vitamin E exposure decreased DNA labeling index to control levels in normal non-lesion hepatocytes. Dieldrin treatment resulted in a significant increase in the volume of hepatic focal lesions while treatment with vitamin E reduced the focal lesion volume from that seen with dieldrin treatment only. Vitamin E co-treatment also decreased the DNA synthesis rate seen in dieldrin-treated mice. Overall, these results appear to confirm a role of oxidative stress in lesion growth. One issue addressed after publication was that the dieldrin and vitamin E were mixed together in the diet. It is unknown if the inclusion of vitamin E reduced the available dieldrin and thus the effects seen may be from the lack of available dieldrin in the diet.
Klaunig et al. (Citation1995) reviewed the role of chemically induced oxidative stress in carcinogenesis generally, and specifically with respect to dieldrin. Several possible mechanisms for oxidative stress participation resulting from the production of ROS can participate in the formation of tumors. These mechanism included: 1) direct modification of DNA through the production of hydroxylated bases (8-OH-dG) that results in modified gene expression or mutation; 2) lipid and protein oxidation by ROS resulting in loss of cellular homeostasis; 3) production of DNA-MDA adducts secondary to lipid peroxidation; 4) changes in membranes leading to changes in calcium and potassium signaling pathways; 5) changes in receptor proteins and gap junction proteins leading to changes in signaling and growth responses; 6) activation of transcription factors resulting in changes in cell growth; and 7) promotion of initiated cells through one or more of the above mechanisms. How the treatment with dieldrin results in ROS in mouse hepatocytes is not known. Parke and Sapota (Citation1996) have suggested that futile cycling of P450 enzymes after induction by a xenobiotic result in the production of ROS.
Wang et al. (Citation2020) examined the effect of dieldrin liver gene expression in male C57bl mice after treatment with dieldrin at 10 ppm (1.5 mg/kg bw/day). With regard to oxidative stress, the gene expression of two enzymes, aldehyde oxidase 1 (AOX1) and superoxide dismutase 1 (SOD1), that are involved in oxidative stress induction (AOX1)and detoxification (SOD1) were examined. AOX1 is a cytosolic enzyme highly expressed in liver that with xenobiotic metabolism produces ROS, specifically hydrogen peroxide and superoxide. SOD1 is found on the outer mitochondrial membrane where superoxide anions are generated. There is a suggestion that SOD1 may inhibit apoptosis by interacting with BCL-2. Gene expression of AOX1 was upregulated in mouse liver after treatment with toxaphene or PB. In that study it was considered that activation of AOX1 was dependent on CAR activation.
Dieldrin treatment of C57Bl mice resulted in increased AOX1 expression at levels comparable to PB treatment after 28 days of treatment (Wang et al. Citation2020). SOD1 gene expression was not affected by dieldrin or PB. In another study (Wang et al. Citation2020), KO mice, PXR−/, CAR−/− and PXR−/−/CAR−/−,were treated with dieldrin for 14 days. In the CAR−/− and PXR−/−/CAR−/− KO mice no increase in AOX1 gene expression was seen. However, in PXR−/− mice, dieldrin treatment induced AOX1 gene expression. The authors concluded that dieldrin treatment-related induction of AOX1s was secondary to the activation of CAR and therefore the induction of oxidative stress by dieldrin in the mouse liver is dependent on CAR activation. The authors further concluded that the oxidative stress and damage previously described in dieldrin-treated mice may be viewed as an associative event or modulating factor to the process of dieldrin-induced hepatocarcinogenesis and is not causal.
4.3.3. Metabolism
Studies on the metabolism of dieldrin in the liver have been limited and predominantly directed to the rat liver. Proposed metabolic pathways for dieldrin are shown in . Dieldrin and its metabolites are mainly excreted in the feces (via bile) and to a lesser extent in the urine. Following oral dosing with radiolabeled aldrin or dieldrin, high levels of radioactivity were detected in the liver, blood, stomach, and duodenum of dosed rats within 1–5 h (Heath and Vandekar Citation1964). Twenty-four hours following a single oral administration of dieldrin (10 mg/kg) to rats, half of the dose was detected in adipose tissue (Hayes Citation1974). The liver accumulation of orally administered dieldrin occurs through the gastrointestinal tract via the hepatic portal vein (Heath and Vandekar Citation1964). Hutson (Citation1976) noted species differences in tissue distribution of dieldrin in rodents following an injection of a single dose of 14C labeled dieldrin (3 mg/kg). Higher levels of dieldrin were seen in liver and fat in mice compared to rats 8 days post treatment. The liver concentration in mice (0.94 mg/kg) was nine times higher than in rats (0.11 mg/kg). Dieldrin levels in adipose tissue were twice as high in mice compared to similarly treated rats after 8 days (11.6 mg/kg verses 5.6 mg/kg, Hutson Citation1976). In rats, dieldrin is hydroxylated to 9-hydroxydieldrin by liver microsomal monooxygenases (Matthews et al. Citation1971). Metabolism of dieldrin is 3–4 times more rapid in male rats than in female rats (Matthews et al. Citation1971). The difference is attributed to the greater ability of males to metabolize dieldrin to its more polar metabolites, primarily 9-hydroxydieldrin. Species differences in rates of metabolism have been reported in rats and mice. The hydroxylation reaction occurs more rapidly in rats than it does in mice (Hutson Citation1976). Species differences for the excretion of dieldrin and/or its metabolites have been reported (Baldwin et al. Citation1972; Hutson Citation1976). Excretion, which occurs predominantly via the feces (80 to 90%), was more rapid in the rat than in the mouse (Hutson Citation1976). Pentachloroketone is the major component in rat urine after dieldrin treatment (Matthews et al. Citation1971; Baldwin et al. Citation1972; Hutson Citation1976). The mouse, unlike the rat, does not appear to excrete pentachloroketone as a urinary metabolite. The mouse produces an urinary metabolite that has not been identified. This unidentified metabolite is not a precursor of the pentachloroketone found in the rat, but it may be produced as a metabolite of the pentachloroketone in the mouse (Baldwin et al. Citation1972). Pretreatment of rats with dieldrin increased excretion of the pentachloroketone in the urine. Pretreatment of mice with dieldrin had no effect on the pattern of excretion of urinary metabolites in the CF1 mouse (Baldwin et al. Citation1972). The observed species difference in liver tumor induction by dieldrin (rats are refractory, mice are sensitive) may be related to differences in metabolism and specific metabolite generation. Unfortunately, experimental support for a specific metabolite in mice that may be responsible for the selective tumor response to dieldrin is lacking. Therefore, it is unclear whether the parent compound or a specific metabolite of dieldrin is responsible for the tumor induction in mice remains unresolved.
Figure 1. The proposed metabolic pathway for dieldrin metabolism (taken from ATSDR Citation2022).
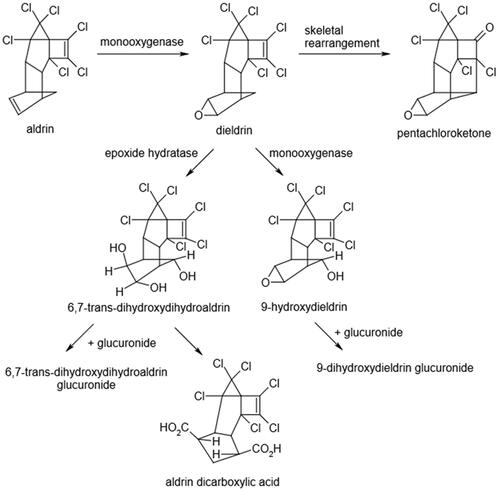
In a study by Dail et al. (Citation2007), Sprague Dawley male rats were treated with either PB (ip; 80 mg/kg/day) for 5 days or dieldrin (2.5 mg/kg/day for 13 days via oral gavage). Liver CYP2B1/2 mRNA levels were compared using laser capture microdissection followed by real-time reverse transcriptase PCR. Liver mRNA samples exhibited a significant induction in CYP2B1/2 mRNA levels over control of approximately 6 times for PB and 2,200 times for dieldrin. The dieldrin induction of CYP2B1/2 mRNA levels followed the direction of lobular blood flow from periportal (300-fold over control) to midzonal (600-fold over control) and to centrilobular (1700-fold over control).
4.4. Evaluation of alternative modes of action
Although the experimental evidence strongly points to CAR activation as the MOA for dieldrin-induced liver tumors, the MOA framework requires that alternative MOAs be evaluated.
4.4.1. Genotoxicity
The DNA reactivity and mutagenicity of dieldrin has been previously reviewed (USEPA Citation1987; Citation2012; Agency for Toxic Substances and Disease Registry (ATSDR) Citation2022; Stevenson et al. Citation1999; Ashwood-Smith Citation1981; Stern Citation2014), the most recent being the Agency for Toxic Substances and Disease Registry (ATSDR) review (ATSDR, Citation2022). A number of mutagenesis studies have been reported for dieldrin, with a majority giving negative results in the Ames assay with or without S9 metabolic activating system present (Bidwell et al. Citation1975; McCann et al. Citation1975; Marshall et al. Citation1976; Shirasu et al. Citation1976; Anderson and Styles Citation1978; Wade et al. Citation1979; Probst et al. Citation1981; Nishimura et al. Citation1982; Glatt et al. Citation1983; Haworth et al. Citation1983). Only one study (Majumdar et al. Citation1977) reported dieldrin as mutagenic for S. typhimurium, but this study was deemed unacceptable because of toxicity and lack of positive controls. In addition, dieldrin was negative in E. coli reverse mutation assays (Ashwood-Smith et al. Citation1972; Probst et al. Citation1981) and in two forward mutation assay systems (Gal Rz2 and streptomycin resistance) (Fahrig Citation1974). Dieldrin did not induce mitotic gene conversion in two strains of Saccharomyces cerevisiae (Fahrig Citation1974; Dean et al. Citation1975), and dieldrin did not induce mutagenicity in the fungus Aspergillus nidulans (Crebelli et al. Citation1986). In contrast, positive results were reported for gene mutation in mouse L5178Y lymphoma cells (McGregor et al. Citation1991) and in Chinese hamster V79 lung fibroblasts (Ahmed et al. Citation1977) in the absence of exogenous metabolic activation. However, negative results were obtained from in vivo tests of dieldrin-induced gene mutations in livers or liver tumors from treated mice (Bauer-Hofmann et al. Citation1990; Citation1992) or dieldrin exposed Drosophila melanogaster (Osaba et al. Citation1999). The overall weight of the evidence approach (Gooderham et al. Citation2020) given negative results in most mutation assays in vitro and negative results in vivo indicates that dieldrin does not have mutagenic activity.
Klaunig et al. and Probst et al. examined unscheduled DNA synthesis (UDS) in primary cultured rat and mouse hepatocytes (Probst et al. Citation1981; Klaunig et al. Citation1984) and found it to be negative. Although negative in the UDS assay, this assay is no longer recognized as a valid evaluation of genotoxicity by the Organization for Economic Cooperation and Development (OECD) (OECD Citation2016). An important consideration for mutagenicity evaluation is whether a chemical can produce mutagenic DNA adducts. Using calf thymus DNA, dieldrin did not induce DNA adduct formation (Decloitre et al. Citation1975).
Dieldrin’s effects on chromosomal aberrations and micronuclei have also been investigated for indirect genotoxicity. Dieldrin did not induce chromosomal aberrations in bone marrow from Chinese hamsters (Dean et al. Citation1975). Similarly, dieldrin did not induce dominant lethality in mice exposed via oral or intraperitoneal injection routes (Epstein et al. Citation1972; Dean et al. Citation1975). Dieldrin was also negative in chromosome aberration induction in CHO-W-B1 cells (Galloway et al. Citation1987). In contrast, Majumdar et al. (Citation1976) reported induction of chromosomal aberrations by dieldrin in the bone marrow of treated mice in vivo and in vitro in W138 lung cells, but evaluation for cytotoxicity was not adequate. Also, Cicchetti et al. (Citation1999) detected an increase in micronuclei in bone marrow from mice following in vivo treatment with dieldrin. Cicchetti and Argentin (Citation2003) also showed micronuclei formation in mouse lung fibroblasts following in vitro treatment with dieldrin. Although Stern (Citation2014) recently suggested that these positive findings may be due to an increase in dieldrin-induced oxidative stress, the studies were not adequately controlled for cytotoxicity as a confounding factor and were not performed to guideline standards. Again, based on an overall weight of the evidence evaluation (Gooderham et al. Citation2020), dieldrin is considered negative for indirect genotoxicity similar to the conclusion regarding mutagenicity. Further supporting the conclusion that there is not a genotoxic risk to humans are the findings by Dean et al. (Citation1975). They compared the frequencies of both chromatid- and chromosome-type aberrations in lymphocytes isolated from workers exposed to dieldrin to an unexposed cohort and found no statistically significant differences in the frequencies.
4.4.2. Oncogenes
Bauer-Hofmann et al. (Citation1992) analyzed c-Ha-ras protooncogene mutations at codon 61 in hepatic lesions (glucose-6-phosphatase deficient (G6P-) foci) in male C3H/He mice. Mice received either 10 ppm dieldrin, 500 ppm PB, or no compound in their diet for 52 weeks. At the end of the treatment period, the incidences of G6P- hepatic lesions was reported to increase from 41% (15/37) in the control group to 67% (10/15) and 63% (10/16) in the dieldrin and PB treated groups, respectively. c-Ha-ras mutations were found in 57% (12/21) of the liver lesions from untreated control mice but only in 22% (5/23) and 25% (4/16) lesions from dieldrin and PB-treated mice, respectively. The authors concluded that the c-Ha-ras mutations occurred spontaneously rather than as a result of dieldrin treatment. No differences in the mutation spectra were noted between control and dieldrin-treated mice. It is important to note that these oncogene mutation studies were performed on hepatic lesions using a specific marker G6P- to identify the hepatic lesions and were not performed on the non-neoplastic hepatocytes. Also, a specific histochemical marker (G6P-) was used to identify the lesions. In mouse liver carcinogenesis, the G6P- lesions represent a subtype of foci and neoplasms, but other lesion phenotypes are also found. More importantly, the mutation spectrum is being evaluated in the neoplastic lesions, not in the non-neoplastic hepatocytes. Such changes do not indicate an actual MOA since similar genetic alterations occur in the tumors regardless of the MOA (Riva et al. Citation2020).
4.4.3. Receptor mediated modes of action
As noted above, a number of nuclear receptors have been shown to be involved in the MOA of hepatic carcinogens. These include CAR, PPARα, PXR, AhR. Wang et al. (Citation2015) examined activation of these receptors by dieldrin treatment by assessing specific genes associated with their activation using real time PCR in wild-type C57BL/6 mice after treatment with dieldrin (10 ppm in the diet) (1.5 mg/kg bw/day equivalent) for 7, 14, or 28 days: CAR/PXR (Cyp2b10, Cyp3a11), AhR (Cyp1a1, Cyp1a2), PPARα (Acox1, Cyp4a10). As described above, compared with untreated control mice, the expression level of Cyp2b10 (CAR responsive gene) was induced significantly in wild-type C57BL/6 mice in a time dependent manner. Similarly, activation expression of PXR (measured by target gene Cyp3a11) was detected. It is important to note that PXR and CAR activation are frequently seen together with CAR activators. In the dieldrin-treated mice, the expression levels of AhR target genes Cyp1a1 and Cyp1a2 (indicative of AHR activation) and of PPARα responsive genes Acox1 and Cyp4a10 (indicative of PPAR activation) were not significantly increased after treatment with dieldrin. These results suggest that dieldrin, like PB, activates primarily CAR/PXR nuclear receptors.
A follow-up experiment (Wang et al. Citation2020) showed that hepatic expression levels of Cyp2b10 genes were not increased in CAR deficient mice or in PXR−/−/CAR−/− double KO mice following treatment with dieldrin for 14 days at 10 ppm, whereas Cyp2b10 expression (CAR activation) was significantly induced in dieldrin-treated PXR−/− mice. Cyp3a11 gene expression (PXR) level was not significantly increased in any of the three KO mice exposed to dieldrin for 14 days. These studies confirm that the dieldrin MOA is through CAR activation and other common nuclear receptors (PXR, Ahr, and PPARα) are not activated.
4.4.4. Cytotoxicity
The criteria for the cytotoxicity MOA classification has been previously reviewed (Holsapple et al. Citation2006; Cohen Citation2010; Wood et al. Citation2015; Strupp et al. Citation2023). Multiple studies have assessed liver cytotoxicity in mice and rats following dieldrin administration through serum liver enzyme evaluation and liver histopathology, although mild changes in liver serum enzymes at high doses does not necessarily mean cytotoxicity if there are no corresponding histopathologic changes. At tumorigenic doses of dieldrin, no liver cytotoxicity has been reported that would fulfill the requirements for a cytotoxic mode of action as seen with carbon tetrachloride (Cohen et al. Citation2023). Kolaja et al. (Citation1996a) examined the acute and subchronic treatment (7, 14, 21, 28, and 90 days with dieldrin; 0.1, 1.0, 3.0, 10.0 mg/kg diet (0.015, 0.15, 0.45, or 1.5 mg/kg bw/day equivalent)). At these sampling times, no increase in liver serum enzyme or histopathology demonstration of necrosis was seen. More recently, Wang et al. (Citation2020) examined dieldrin-treated (10 ppm via diet) (1.5 mg/kg bw/day equivalent) mice at 7, 14 and 28 days of treatment. The liver of these mice, while showing increases in cell proliferation and CAR mediated gene expression, displayed no increase in liver serum enzymes (ALT, AST, ALP, GGT) or liver necrosis or increased apoptosis. In conclusion, dieldrin does not function through a cytotoxic MOA in the mouse studies nor in other species. In the various Toxcast assays, (https://comptox.epa.gov/dashboard/chemical/invitrodb/DTXSID9020453?list=EDSPUOC), there was evidence of cytotoxicity but only at concentrations of 20 μm and higher, which would not be relevant to in vivo concentrations. In addition, there have been no reports of increased iron accumulation in the livers of dieldrin-treated animals as a possible mechanism of inducting cytotoxicity.
Besides the rodent studies, several human studies have also examined for possible liver cytotoxicity of dieldrin. A slight increase in serum hepatic enzymes (ALT and AST) was reported in one epidemiology study of pesticide workers (Morgan and Lin Citation1978). In contrast, other studies of workers involved in the manufacture of dieldrin showed no evidence of any hepatic effects of dieldrin exposure (Hoogendam et al. Citation1965; Jager Citation1970; Hunter et al. Citation1972; Warnick and Carter Citation1972; Morgan and Roan Citation1974; van Sittert and de Jong Citation1987; de Jong Citation1991). The liver endpoints that were examined in these workers included serum hepatic enzyme activity (Hoogendam et al. Citation1965; Jager Citation1970; Warnick and Carter Citation1972; Morgan and Roan Citation1974; van Sittert and de Jong Citation1987), hepatic enlargement (Jager Citation1970), and microsomal enzyme induction (Jager Citation1970; Hunter et al. Citation1972; Morgan and Roan Citation1974; van Sittert and de Jong Citation1987). All of these endpoints showed no modification from dieldrin exposure in humans. In a controlled study with healthy male subjects who were given 0.003 mg/kg/day (equivalent to 0.02 ppm) of dieldrin for 18 months, no general clinical changes or signs were reported nor hepatic effects as measured by changes in liver serum enzymes (ALT, AST, ALP) (Hunter and Robinson Citation1967).
4.4.5. Immunosuppression
There is no evidence for immunosuppression playing a role in dieldrin liver carcinogenesis in the mouse. Further, liver tumors are not increased in immunosuppressed rodents or humans (Penn Citation1988; Krynitz et al. Citation2013; Lebrec et al. Citation2016). Examination of immunosuppression by dieldrin has been reported in several studies in mice. These studies have used viral or parasitic infection in the mice and determined if dieldrin treatment enhanced the lethality of the infection. Mice infected with mouse hepatitis virus 3 showed a decrease in the antigenic response to the virus after the mice were treated with a single oral dose of dieldrin (18 mg/kg) (Krzystyniak et al. Citation1985). An increase in lethality of parasitic infections (malaria, Plasmodium berghei, or Leishmania tropica) in mice was seen after treatment with dieldrin in the diet (0.18 mg/kg/day) for 10 weeks (Loose Citation1982). In another study, Loose et al. (Citation1981) reported a decrease in macrophage tumor cell killing activity in dieldrin (0.13 mg/kg/day) treated mice for 3–18 weeks (Loose et al. Citation1981). However, the doses used in these studies were high, and the relevance of these studies to the carcinogenicity of dieldrin is doubtful. There has been no reported abnormality in the mouse studies indicating an abnormality in immune-related tissues such as the spleen, thymus, bone marrow, or lymph nodes. However, as far as we can ascertain there have been no immunotoxicity guideline studies reported.
4.4.6. Estrogenic activity
Several reviews have addressed the effect of dieldrin on estrogen binding. (Agency for Toxic Substances and Disease Registry (ATSDR) Citation2022; Snedeker Citation2011; Soto et al. Citation1995). These reviews noted that dieldrin displays only very weak binding to ER-α, the estrogen receptor. Thus, neither aldrin nor dieldrin have shown sufficient evidence of estrogenicity. Tully et al. (Citation2000) examined the effect on estrogen-responsive reporter genes that had been transfected into HeLa cells. Dieldrin treatment produced no induction of transcriptional activation. Syversen et al. (Citation2000) examined a possible synergistic estrogenic effect of dieldrin and toxaphene on bone mass density in rats. Dieldrin alone showed no estrogenic activity but when co-administered with toxaphene, a slight increase in activity was noted. Similarly, Ramamoorthy et al. (Citation1997) examined the estrogenic activity of a dieldrin/toxaphene mixture in the mouse uterus, MCF-7 human breast cancer cells, and a yeast-based estrogen receptor assay. No apparent synergism between dieldrin and toxaphene was seen in these assays (Ramamoorthy et al. Citation1997). The conclusions from these studies are that dieldrin does not have estrogenic properties and the dieldrin-induced mouse liver tumors are not due to an estrogenic MOA.
5. Relevance of CAR activation to human hepatocarcinogenesis
5.1. Mechanistic studies
CAR activation as a MOA for hepatocellular carcinogenesis in mice and/or rats has been demonstrated for a number of agents, the prototype chemical being PB (Elcombe et al. Citation2014; Lake Citation2018; Yamada et al. Citation2021b). The evidence that dieldrin is acting by this MOA is strong, and other possible MOAs have been excluded. Fundamentally, the question then remains is whether such a MOA is relevant to human carcinogenesis. This has been reviewed extensively in Elcombe et al. (Citation2014) and in Yamada et al. (Citation2021b), and most recently in a seminal publication by Shizu et al. (Citation2024). Human relevance of the animal findings can be evaluated using the USEPA and Health Canada (Meek et al. Citation2003; Seed et al. Citation2005; USEPA Citation2005) and the IPCS (Sonich-Mullin et al. Citation2001; Boobis et al. Citation2006, Citation2008; Meek et al. Citation2014) MOA/human relevance framework. This framework poses three questions. First is whether the MOA for the adverse effect in animals is known. The answer to this question for dieldrin-induced mouse liver tumors is yes, based on the above discussion. The second question asks whether the key events in the MOA can qualitatively occur in humans. Since the second key event following CAR activation is increased cell proliferation and does not occur in humans, the answer to this question is no. Thus, the MOA is not relevant to humans, and the third question, are the key events likely to occur in humans based on a quantitative assessment, does not need to be addressed.
Fundamentally, CAR activation leading to down-stream induction of various cytochrome P450 metabolizing enzymes is well known in rodents and has been demonstrated in humans at high exposure levels of various agents, such as PB (Elcombe et al. Citation2014; Yamada et al. Citation2021b). Thus, this second key event (following CAR Activation) can occur in humans. However, it is the third key event, increased cell proliferation, that clearly differs between rodents and humans. Although the metabolic effects of CAR activation are well known to occur in humans, there is no evidence for an increase in hepatocellular proliferation following administration of such agents (Elcombe et al. Citation2014; Yamada et al. Citation2021b; Shizu et al. Citation2024). The lack of a proliferative response in human cells has been demonstrated in vitro utilizing primary human hepatocytes and also using immortalized human cell lines. However, the most convincing evidence that there is a lack of a proliferative effect in human hepatocytes following administration of CAR activators has come from studies using chimeric mice (Yamada Citation2021a; Yamada et al. Citation2021b). These mice have human hepatocytes replacing mouse hepatocytes. Administration of CAR activators, such as PB, even at doses as high as those utilized in animal carcinogenesis experiments and human therapeutic doses, shows no evidence of an increase in cell proliferation in the human hepatocytes in these chimeric mice. There are the expected metabolic effects from CAR activation, but not the proliferative effects that occur in rodents. This model retains the potential for liver cell proliferation, as has been demonstrated utilizing chimeric mice with rat hepatocytes instead of the human hepatocytes. In such cases, CAR activation produces both the metabolic and proliferative effects in the rat hepatocytes and in the mouse hepatocytes, in contrast to the lack of a proliferative effect in human cells. This provides strong evidence that human cells do not have the proliferative response to CAR activation, and since this is a necessary key event in the carcinogenic process for CAR activators in rodents, this provides strong evidence that hepatocarcinogenicity will not occur in humans. The basis for the lack of a proliferative response in humans has recently been demonstrated by Shizu et al. (Citation2024). They showed that the proliferative response in rodents is due to down-stream interaction with yes-associated protein (YAP). In contrast, this interaction did not occur with the human YAP protein in the human liver, precluding CAR-dependent hepatocyte proliferation. The lack of a proliferative and tumorigenic response is supported by extensive epidemiology studies in individuals exposed to PB or permethrin, with the PB studies involving exposures as high as the levels in the rodent studies leading to liver tumors (Elcombe et al. Citation2014; Yamada et al. Citation2021b). Numerous chemicals have shown liver tumorigenesis by a CAR activating MOA, including pharmaceuticals (PB, oxazepam), pesticides (benfluralin, cyproconazole, DDT, epoxiconazole, fluopyram, metazachlor, metofluthrin, momfluthrin, piperonyl butoxide, prinomide, propiconazole, sedaxane, sulfoxaflor, toxaphene, triadimefon), solvents (methylisobutyl ketone, tetrahydrofuran), and nitrogen stabilizer (nitrapyrin) (see Yamada et al. Citation2021b). Based on such studies, it is concluded that dieldrin, like other CAR hepatocarcinogens in rodents, will not be carcinogenic to humans.
IARC in its assessment of dieldrin did not conclude that the MOA of the liver tumors in mice was due to CAR activation, but rather they focused on the ten key characteristics approach for evaluating mechanism. This approach is not appropriate (Becker et al. Citation2017). To begin with, the key characteristics were based on an assessment only of known chemical carcinogens. There was no evaluation of these characteristics in non-carcinogens, thus, no assessment of specificity is possible. Furthermore, the key characteristics approach was designed as a method for organizing the literature regarding a chemical, not for mechanistic evaluation. This approach essentially becomes a checkbox exercise, but does not evaluate MOA and more importantly doesn’t evaluate human relevance. The mode of action/human relevance is designed specifically for such an evaluation.
An example of the deficiencies of the key characteristics approach is seen with examples such as the statins. Statins have seven of the ten key characteristics of carcinogens. Statins produce liver tumors in rats and mice, but based on mode of action analysis the effect is not relevant to humans (MacDonald and Halleck Citation2004). A lack of cancer risk in humans by statins, whether cancer overall, liver tumors specifically, or any other type of tumor, has been demonstrated in several very large epidemiology studies involving hundreds of thousands of people (Frils et al. Citation2005; Dale et al. Citation2006; Jacobs et al. Citation2006; Coogan et al. Citation2007; Alsheikh-Ali et al. Citation2008; Farwell et al. Citation2008; Chiu et al. Citation2011). If the IARC approach to mechanistic evaluation of rodent carcinogens based on the key characteristics was used, these valuable pharmaceuticals which have saved thousands if not millions of lives would not be available. There are numerous classes of pharmaceuticals which produce cancer in animal bioassays which are widely used by humans without evidence of a cancer risk (Friedrich and Olejniczak Citation2011; Brambilla et al. Citation2012). The evaluation must take into account an assessment of the key events and an evaluation of their human relevance.
5.2. Studies in humans
A study involving ingestion of dieldrin in gelatin capsules to men at doses up to 211 µg/day, much higher than occupational or environmental exposure, extended over a period of 18 months with a follow-up period of six months during which dieldrin was not administered (Hunter and Robinson Citation1967; Hunter et al. Citation1968; Citation1969). Several tests were performed including routine hematology and blood chemistry examinations. No clinical adverse events occurred, and there were no alterations in the hematologic and blood chemistry parameters examined, including alkaline phosphatase. The lack of an effect on alkaline phosphatase supports a conclusion of no adverse liver effects. In a study of factory workers (Hunter et al. Citation1972), there was an increase in d-glucaric acid excretion, suggesting increased enzyme induction, but the changes were not statistically significant. Workers were exposed to aldrin and/or dieldrin during manufacture and/or use of these pesticides. Slightly increased serum ALT and AST levels were reported in one study of pesticide-exposed workers (Morgan and Lin Citation1978). However, there was no evidence of exposure-related hepatic effects in other studies (Hoogendam et al. Citation1965; Jager Citation1970; Hunter et al. Citation1972; Warnick and Carter Citation1972; Morgan and Roan Citation1974; van Sittert and de Jong Citation1987; de Jong Citation1991). No adverse hepatic effects were observed in a study of healthy male subjects who consumed dieldrin at up to 0.0003 mg/kg/day (equivalent to 0.02 ppm) for 18 months in a study designed to evaluate dieldrin kinetics (Hunter and Robinson Citation1967). Liver dysfunction has been reported in cases of inadvertent or intentional ingestion of relatively large amounts of dieldrin (Garrettson and Curley Citation1969; Black Citation1974).
Aldrin and dieldrin have not been manufactured in the United States since 1974 and all uses were banned by 1987. In the environment, aldrin is readily converted to dieldrin. It is not likely that the general population is exposed to dieldrin today, and certainly not at levels high enough to cause adverse hepatic effects. The hepatotoxicity of aldrin or dieldrin has been reported in a number of experimental animal species with the common finding of effects on the liver (Kitselman Citation1953; Fitzhugh et al. Citation1964; Harr et al. Citation1970; Thorpe and Walker Citation1973; Kohli et al. Citation1977; Shakoori et al. Citation1982; Ahmed et al. Citation1986; Goel et al. Citation1988), except in monkeys. In a monkey study covering administration for 6 years, there was no evidence of increased liver weight, histopathologic changes, or changes in total DNA (indicative of a lack of increased cell proliferation), although there was an increase in cytochrome P450 (Wright et al. Citation1978).
Exposures in the case reports were predominantly via oral exposure, whereas exposures in the epidemiological studies were mainly inhalation and dermal. Given that the strongest data are in workers, finding additional exposed populations for future studies is unlikely given that aldrin and dieldrin have not been manufactured in the United States since 1974 and all uses were banned by 1987. Regarding cancer risk to dieldrin, epidemiologic reports showed no association between dieldrin exposure and human cancer risk (Stevenson et al. Citation1999; Hooker et al. Citation2014). Dose response models for carcinogens generally do not incorporate the possibility of a threshold. In mice, the dose response for the liver tumors induced by dieldrin involves a threshold. However, based on the above discussion, the mouse liver tumors induced by dieldrin occur by a MOA that is not qualitatively relevant to humans, so no cancer risk is anticipated in humans and no dose response modeling for cancer is appropriate. The cancer mortality data on male workers indicated that low-dose exposure to aldrin and dieldrin did not significantly increase human cancer risk. The suggested decrease in cancer risk at low doses of aldrin and dieldrin was opposite to the USEPA cancer potency based on mouse liver tumors. The USEPA's upper bound calculation suggests a lifetime average daily doses of 0.0000625 and 0.00625 microgram/kg body weight/day (0.0000000625 and 0.00000625 mg/kg body weight/day) would result in an increased risk of cancer 0.000001 and 0.0001. Using a non-threshold dose response model is inappropriate for the mouse liver tumor data, and completely ignores the lack of human relevance of these tumors. In assessing cancer hazard characterization, it is critical to evaluate the human relevance of the MOA. If not relevant to humans, it is inappropriate to characterize the animal findings as a cancer hazard for humans, as has been concluded for many types of MOA involving various tissues (IARC Working Group Citation1999; Meek et al. Citation2003; Holsapple et al. Citation2006; Cohen Citation2010; Cohen et al. Citation2019).
5.3. Epidemiological studies
The database of information regarding the potential human carcinogenicity of aldrin and dieldrin includes evaluation of cohorts involved in aldrin and dieldrin production, or case-control studies involving pesticide applicators. A major limitation common to the epidemiological studies for aldrin and dieldrin is the lack of quantitative measures of exposure. Various investigations have been reported in several previous reviews (IARC Working Group Citation1974; USEPA Citation1987; Citation2012; Stevenson et al. Citation1999; Hooker et al. Citation2014; IARC Citation2019; ATSDR, Citation2022), and a search of the literature since 2020 produced no additional epidemiology studies of dieldrin.
The potential carcinogenicity of aldrin and dieldrin in workers was examined at an organochlorine manufacturing plant in Colorado (Ditraglia et al. Citation1981). No increased risk of death from all malignant neoplasms, or cancers of the esophagus, stomach, intestines, rectum, liver, pancreas, respiratory system, bladder and urinary system, or lymphatic and hematopoietic system was reported. Follow up studies of this cohort (Amoateng-Adjepong et al. Citation1995) continued to show no evidence of an increased risk of all cancers or specific cancers. Evaluation of workers in the manufacture of aldrin, dieldrin and endrin in the Netherlands was reported. No increased risk for stomach, intestinal, liver, pancreas, lung, prostate, bladder, multiple myeloma, leukemia, or kidney cancer was observed (de Jong Citation1991; Swaen et al. Citation2002; Van Amelsvoort et al. Citation2009). Although earlier estimates suggested an increased risk for rectal cancer (de Jong Citation1991) in dieldrin workers, a follow-up review of the data (Van Amelsvoort et al. Citation2009) concluded no statistical increase in rectal cancer. A number of cohort or case-control studies have used data collected as part of the Agricultural Health Study of pesticide applicators in Iowa and North Carolina to evaluate possible associations between self-reported use of aldrin or dieldrin and risk (Flower et al. Citation2004; Purdue et al. Citation2007). Studies found no association between self-reported aldrin or dieldrin use and risk of selected cancer types (Hooker et al. Citation2014; ATSDR, Citation2022). A variety of studies have been reported evaluating specific tumor types, but no statistically significant increased risk has been detected, including for any cancer (Swaen et al. Citation2002; Van Amelsvoort et al. Citation2009) or specific cancers of the lung (Purdue et al. Citation2007), non-Hodgkin’s lymphoma (McDuffie et al. Citation2001; Schroeder et al. Citation2001; Cantor et al. Citation2003; Quintana et al. Citation2004; Cocco et al. Citation2008), breast (Ward et al. Citation2000; Hoyer et al. Citation2001, Citation2002; Gammon et al. Citation2002; Mathur et al. Citation2002; Ibarluzea et al. Citation2004; Engel et al. Citation2005; Xu et al. Citation2010), prostate (Ritchie et al. Citation2003; Belpomme et al. Citation2009; Xu et al. Citation2010), pancreas (Clary and Ritz Citation2003), or gallbladder (Shukla et al. Citation2001), and for childhood cancers (Flower et al. Citation2004). In the first Hoyer et al. (Citation2001) study there appeared to be an increase in estrogen receptor negative breast cancers at the highest quartile, but the authors concluded that this should be interpreted with caution due to the small number of cases, the lack of an increase in estrogen receptor positive cases, significant differences in tumor size at diagnosis, and possible confounding by other variables known to influence cancer risk such as body weight, hormone replacement therapy, and other estrogen-modifying factors. In addition, in a follow-up study (Hoyer et al. Citation2002), these investigators did not detect an increased risk of breast cancer with dieldrin exposure based on p53 analyses.
6. Conclusions
Dieldrin administered in the diet induces liver tumors in mice but not in rats. The MOA for the liver tumor induction in mice is CAR activation, with key events defined as CAR activation leading to altered gene expression and increased hepatocellular proliferation. This leads to the usual sequence of events for hepatocellular tumors in rodents of formation of hepatocellular foci ultimately evolving into adenomas and carcinomas. Alternative MOAs for these mouse liver tumors have been excluded including genotoxicity, cytotoxicity with consequent regenerative cell proliferation, and other cell receptor-based mechanisms of mitogenicity. The conclusion for CAR activation as the MOA for the dieldrin-induced liver tumors is strong. A role for CAR activation in the mouse liver tumor MOA of other chlorinated insecticides (DDT and toxaphene) has also been reported (Wang et al. Citation2015; Harada et al. Citation2016: Wang et al. Citation2017). These compounds, like dieldrin, selectively produce liver tumors in the mouse. Thus, CAR activation appears to be a common liver tumor MOA for these compounds and like dieldrin the CAR MOA has important implications for human liver tumor risk.
The strength of the evidence is that CAR activation as a MOA for liver carcinogenicity is rodent specific and not relevant to human cancer risk, as has been shown for the prototypical CAR activator PB. Thus, the overall conclusion regarding dieldrin is that it does not pose a cancer risk to humans. Rodent specificity is based on CAR activation leading to increased cell proliferation. The enzyme inductive effect of CAR activation occurs in other species, including humans, but not the hepatocellular proliferative response. The reason for this species difference in the induction of cell proliferation has recently been identified as being due the interaction of CAR with YAP in rodents. The interaction with YAP does not occur in humans and thus does not lead to increased cell proliferation. Without this increase in hepatocellular proliferation, tumors will not be produced. USEPA applied a q1* quantitative risk assessment model for the dose response for dieldrin-induced tumors since at that time MOA data were insufficient so they defaulted to a no-threshold dose response model as described in the 2005 Cancer Guidelines (USEPA Citation2005). That approach is not appropriate today given the current state of the scientific literature taking into account the MOA of CAR activation. Based on this understanding of the MOA of dieldrin-induced liver tumors in mice, a more appropriate conclusion is that dieldrin is a mouse specific liver carcinogen, it does not pose a cancer risk to humans and the q1* dose response model is not appropriate.
Most studies on dieldrin metabolism have been directed to the rat liver. This limits the application of any differences in metabolism to clarify the mouse-specific induction of liver tumors. The pesticide aldrin is rapidly metabolized to dieldrin. There are species differences in the distribution of dieldrin in the liver (Hutson (Citation1976). After a single injection of dieldrin, greater levels of dieldrin (9 times higher) were seen in the liver of mice compared to rats. In rats, dieldrin is hydroxylated to 9-hydroxydieldrin by liver microsomal monooxygenases (Matthews et al. Citation1971). Hydroxylation occurs more rapidly in rats than it does in mice (Hutson Citation1976), and the fecal excretion of dieldrin and its metabolites was much more rapid in the rat. Pentachloroketone is the major urinary metabolite in the rat but has not been detected in the mouse (Baldwin et al. Citation1972). Although there are species differences in the metabolism of dieldrin, the exact metabolite responsible for the selective mouse liver tumor induction is not known (taken in part from ATSDR Citation2022).
PubMed search terms
Dieldrin; dieldrin and liver; dieldrin and liver and cancer; dieldrin and liver and cancer and mode of action; dieldrin and liver and rodent cancer; liver and rodent cancer; mouse and liver and cancer; mouse and liver and cancer and dieldrin; mouse and liver and dieldrin; mouse and liver and mode of action; dieldrin and genotoxicity; dieldrin and oxidative stress; oxidative stress and cancer; oxidative stress and cancer and liver; dieldrin and metabolism; dieldrin and liver and metabolism; dieldrin and liver and metabolism and mouse; dieldrin and mutagenesis; dieldrin and nongenotoxic dieldrin and human; dieldrin and human and cancer; dieldrin and human and liver and cancer; rodent and liver and cancer and mode of action; gap junction and dieldrin; dieldrin and cytotoxicity; dieldrin and cytotoxicity and liver: dieldrin and receptor; dieldrin and receptor and liver; dieldrin and receptor and liver and CAR; liver and CAR.
SCOPUS search terms
Dieldrin; dieldrin AND liver; dieldrin AND liver AND mouse; dieldrin AND liver AND mouse AND cancer; dieldrin AND liver AND cancer; dieldrin AND liver AND cancer AND mode of action; mouse AND liver AND cancer; dieldrin AND metabolism; dieldrin AND metabolism AND mouse; dieldrin AND metabolism AND mouse AND liver; dieldrin AND cytotoxicity; dieldrin AND cytotoxicity AND liver; dieldrin AND cancer; dieldrin AND cancer AND human; Dieldrin AND genotoxicity; dieldrin AND mutation; dieldrin AND oxidative AND stress; dieldrin AND gap AND junction; dieldrin AND gap AND junction AND liver; dieldrin AND liver AND oxidative AND stress; dieldrin AND liver AND mouse AND oxidative AND stress; dieldrin AND human; dieldrin AND human AND cancer; dieldrin AND human AND cancer AND liver; dieldrin AND receptor; dieldrin AND receptor AND liver; dieldrin AND receptor AND liver AND CAR.
A similar search of references in the regulatory reviews did not yield any additional references to those found in the PubMed and Scopus searches or in the authors’ personal reference database.
| Abbreviations | ||
| 8-OH-dG | = | 8-hydroxydeoxyguanosine |
| AhR | = | aryl hydrocarbon receptor |
| ALP | = | alkaline phosphatase |
| ALT | = | alanine aminotransferase |
| AOX1 | = | aldehyde oxidase 1 |
| AST | = | aspartate aminotransferase |
| ATSDR | = | Agency for Toxic Substances and Disease Registry |
| CAR | = | constitutive androstane receptor |
| DDT | = | dichlorodiphenyltrichloroethane |
| DEN | = | diethylnitrosamine |
| G6P | = | glucose-6-phosphatase |
| GGT | = | gamma-glutamyl transpeptidase |
| GJIC | = | gap junctional intercellular communication |
| IARC | = | International Agency for Research on Cancer |
| IPCS | = | International Programme on Chemical Safety |
| KO | = | knockout |
| MDA | = | malondialdehyde |
| MOA | = | mode of action |
| NCI | = | National Cancer Institute |
| OECD | = | Organization for Economic Cooperation and Development |
| PB | = | phenobarbital |
| PPARα | = | peroxisome proliferator activated receptor alpha |
| PROD | = | pentoxyresorufin-O-dealkylase |
| PXR | = | pregnane X receptor |
| RNS | = | reactive nitrogen species |
| ROS | = | reactive oxygen species |
| USEPA | = | United States Environmental Protection Agency |
| YAP | = | yes-associated protein |
FINAL DIELDRIN MANUSCRIPT6-28-24 clean.docx
Download MS Word (352.1 KB)Acknowledgments
The manuscript was reviewed by six reviewers (anonymous to the authors) provided by the editor of the journal and also Drs. Satinder S. Sarang and Troy Carrington of Shell and Dr. Gary Krieger of NewFields. The latter provided suggestions on organization of the document, syntax, and clarification of tests. These suggestions were considered by the authors and incorporated where appropriate. Similarly, the comments from the anonymous reviewers were considered and changes incorporated where appropriate in the manuscript. The final content and conclusions of the manuscript are strictly those of the two authors.
We also gratefully acknowledge the expert assistance of Lisa Allen, University of Nebraska Medical Center, in the preparation of this manuscript.
Supplemental material
Supplemental material for this article is available online here.
An extensive and comprehensive literature search was conducted. Four major sources of literature were examined: 1) the US National Library of Medicine database (PubMED); 2) the Elsevier citation database (Scopus); 3) the regulatory agencies’ reviews of dieldrin (USEPA Citation1987, Citation2012; IARC Working Group Citation1974, IARC Citation2019; ATSDR Citation2022); and 4) pertinent references cited in the selected publications. In addition, each of the authors have an extensive personal database of publications related to dieldrin, liver carcinogenesis, mode of action, and human risk assessment. The overall objective of the literature search strategy was to identify relevant publications and reviews that addressed the overall topic of dieldrin-induced mouse liver tumors, mode of action, and human relevance.
Declaration of interests
This review was supported by a contract to Drs. Klaunig and Cohen from NewFields (an environmental, engineering, and construction management consulting firm (https://ww.newfields.com)). It is the authors’ understanding that Shell (a former manufacturer of dieldrin (http://www.shell.com) and NewFields have a working relationship on ag-chem impacted sites. Dieldrin, although no longer used or manufactured, is still found in hazardous sites (ATSDR, Citation2022). The reported induction of hepatic tumors in mice had been a concern in part because the mechanism of liver cancer induction was not defined, and as such, the relative risk of liver to humans by dieldrin was unresolved. With regard to this document, it is the authors’ understanding that Shell identified Drs. Klaunig and Cohen as experts in the area of chemically induced carcinogenesis and in particular in liver cancer mode of action analysis and as such requested NewFields (working through their established contract with Shell) to contract with Drs. Klaunig and Cohen to perform an independent, scientifically based mode of action assessment of potential human liver cancer risk. Drs. Klaunig and Cohen used the established mode of action framework that both were instrumental in developing. The review of the scientific literature and the conclusions reached in this manuscript are solely those of Drs. Klaunig and Cohen using their scientific expertise. Drs. Klaunig and Cohen required independence in the contract for the approach and conclusions reached on the topic.
An extensive literature search on this topic was performed by both authors. Of note, a review of the literature revealed significant omissions in regulatory documents on the mechanism of liver cancer induction by dieldrin and particularly the topic of liver tumor mode of action. The present manuscript addressed those deficiencies using current scientific approaches. The authors are not aware of any current litigation or reregistration activities with dieldrin. The results presented here are important in understanding the mode of action for dieldrin-induced liver tumors, and coupled with other studies on organochlorinated pesticides (Wang et al. Citation2015; Harada et al. Citation2016; Wang et al. Citation2017) provide scientific basis for understanding the rodent liver tumor response and the application of these findings to human liver cancer risk. Dr. Klaunig had been invited and served as an external peer reviewer in 2020 and for the 2022 ATSDR Toxicological Profile for Aldrin and Dieldrin. (https://wwwe.atsdr.cdc.gov/toxprofiles/tp1.pdf).
Additional information
Funding
References
- Agency for Toxic Substances and Disease Registry (ATSDR). 2022. Toxicological Profile for Aldrin and Dieldrin. Atlanta, GA: U.S. Department of Health and Human Services, Public Health Service. PMID: 37040456.
- Ahmed FE, Lewis NJ, Hart RW. 1977. Pesticide induced ouabain resistant mutants in Chinese hamster V79 cells. Chem. Biol. Interact. 19(3):369–374. doi: 10.1016/0009-2797(77)90059-x.
- Ahmed NA, Rawi SM, el-Behary MH. 1986. Effect of dieldrin injection on the level of certain amino acids and some enzymes in rat brain. Comp Biochem Physiol C Comp Pharmacol Toxicol. 85(2):437–442. doi: 10.1016/0742-8413(86)90222-7.
- Alsheikh-Ali AA, Trikalinos TA, Kent DM, Karas RH. 2008 Statins, low-density lipoprotein cholesterol, and risk of cancer. J. Am. Coll. Carciol. 52(14):1141–1147. doi: 10.1016/j.jacc.2008.06.037.
- Ames BN, McCann J, Yamasaki E. 1975. Methods for detecting carcinogens and mutagens with the salmonella/mammalian-microsome mutagenicity test. Mutat Res. 31(6):347–364. doi: 10.1016/0165-1161(75)90046-1.
- Amoateng-Adjepong Y, Sathiakumar N, Delzell E, Cole P. 1995. Mortality among workers at a pesticide manufacturing plant. J Occup Environ Med. 37(4):471–478. doi: 10.1097/00043764-199504000-00020.
- Anderson D, Styles JA. 1978. The bacterial mutation test. Six tests for carcinogenicity. Br J Cancer. 37(6):924–930. doi: 10.1038/bjc.1978.134.
- Ashwood-Smith MJ. 1981. The genetic toxicology of aldrin and dieldrin. Mutat Res. 86(2):137–154. doi: 10.1016/0165-1110(81)90022-1.
- Ashwood-Smith MJ, Trevino J, Ring R. 1972. Mutagenicity of dichlorvos. Nature. 240(5381):418–420. doi: 10.1038/240418a0.
- Bachowski S, Xu Y, Stevenson DE, Walborg EF, Klaunig JE. 1998. Role of oxidative stress in the selective toxicity of dieldrin in the mouse liver. Toxicol Appl Pharmacol. 150(2):301–309. doi: 10.1006/taap.1998.8372.
- Baker TK, Bachowski S, Stevenson DE, Walborg EF, Klaunig JE. 1995. Modulation of gap junctional intercellular communication in rodent, monkey and human hepatocyte by nongenotoxic compounds. Prog Clin Biol Res. 391:71–80.
- Baldwin MK, Robinson J, Parke DV. 1972. A comparison of the metabolism of HEOD (dieldrin) in the CF1 mouse with that in the CFE rat. Food Cosmet Toxicol. 10(3):333–351. doi: 10.1016/s0015-6264(72)80252-9.
- Bauer-Hofmann R, Buchmann A, Mahr J, Kress S, Schwarz M. 1992. The tumour promoters dieldrin and phenobarbital increase the frequency of c-Ha-ras wild-type, but not of c-Ha-ras mutated focal liver lesions in male C3H/He mice. Carcinogenesis. 13(3):477–481. doi: 10.1093/carcin/13.3.477.
- Bauer-Hofmann R, Buchmann A, Wright AS, Schwarz M. 1990. Mutations in the Ha-ras proto-oncogene in spontaneous and chemically induced liver tumours of the CF1 mouse. Carcinogenesis. 11(10):1875–1877. doi: 10.1093/carcin/11.10.1875.
- Becker RA, Dreier DA, Manibusan MK, Cox LAT, Simon TW, Bus JS. 2017. How well can carcinogenicity be predicted by high throughput “characteristics of carcinogens” mechanistic data? Regul Toxicol Pharmacol. 90:185–196. doi: 10.1016/j.yrtph.2017.08.021.
- Belpomme D, Irigaray P, Ossondo M, Vacque D, Martin M. 2009. Prostate cancer as an environmental disease: an ecological study in the French Caribbean islands, Martinique and Guadeloupe. Int J Oncol. 34(4):1037–1044. doi: 10.3892/ijo_00000229.
- Bidwell K, Weber E, Nienhold I, Connor T, Legator MS. 1975. Comprehensive evaluation for mutagenic activity of dieldrin. Mutat. Res. 31(5):314. doi: 10.1016/0165-1161(75)90110-7.
- Black AM. 1974. Self poisoning with dieldrin: a case report and pharmacokinetic discussion. Anaesth Intensive Care. 2(4):369–374. doi: 10.1177/0310057X7400200413.
- Boobis AR, Cohen SM, Dellarco V, McGregor D, Meek ME, Vickers C, Willcocks D, Farland W. 2006. IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit Rev Toxicol. 36(10):781–792. doi: 10.1080/10408440600977677.
- Boobis AR, Doe JE, Heinrich-Hirsch B, Meek ME, Munn S, Ruchirawat M, Schlatter J, Seed J, Vickers C. 2008. IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit Rev Toxicol. 38(2):87–96. doi: 10.1080/10408440701749421.
- Brambilla G, Mattioli F, Robbiano L, Martelli A. 2012. Update of carcinogenicity studies in animals and humans of 535 marketed pharmaceuticals. Mutat Res. 750(1):1–51. doi: 10.1016/j.mrrev.2011.09.002.
- Cantor KP, Strickland PT, Brock JW, Bush D, Helzlouer K, Needham LL, Zahm SH, Comstock GW, Rothman N. 2003. Risk of non-Hodgkin’s lymphoma and prediagnostic serum organochlorine: beta-hexachlorocyclohexane, chlordane/heptachlor-related compounds, dieldrin, and hexachlrobenene. Environ. Health Perspect. 111(2):179–183. doi: 10.1289/ehp.4347.
- Carthew P, Edwards RE, Nolan BM. 1998. The quantitative distinction of hyperplasia from hypertrophy in hepatomegaly induced in the rat liver by phenobarbital. Toxicol Sci. 44(1):46–51. doi: 10.1006/toxs.1998.2473.
- Chiu H-F, Ho S-C, Chen C-C, Yang C-Y. 2011. Statin use and the risk of liver cancer: a population-based case-control study. Am J Gastroenterol. 106(5):894–898. doi: 10.1038/ajg.2010.475.
- Cicchetti R, Argentin G. 2003. The role of oxidative stress in the in vitro induction of micronuclei by pesticides in mouse lung fibroblasts. Mutagenesis. 18(2):127–132. doi: 10.1093/mutage/18.2.127.
- Cicchetti R, Bari M, Argentin G. 1999. Induction of micronuclei in bone marrow by two pesticides and their differentiation with CREST staining: an in vivo study in mice. Mutat Res. 439(2):239–248. doi: 10.1016/s1383-5718(98)00185-5.
- Clary T, Ritz B. 2003. Pancreatic cancer mortality and organochlorine pesticide exposure in California, 1989-1996. Am J Ind Med. 43(3):306–313. doi: 10.1002/ajim.10188.
- Cleveland FP. 1966. A summary of work on aldrin and dieldrin toxicity at the Kettering Laboratory. Arch Environ Health. 13(2):195–198. doi: 10.1080/00039896.1966.10664532.
- Cocco P, Brennan P, Ibba A, de Sanjosé Llongueras S, Maynadié M, Nieters A, Becker N, Ennas MG, Tocco MG, Boffetta P. 2008. Plasma polychlorobiphenyl and organochlorine pesticide level and risk of major lymphoma subtypes. Occup Environ Med. 65(2):132–140. doi: 10.1136/oem.2007.033548.
- Cohen SM. 2010. Evaluation of possible carcinogenic risk to humans based on liver tumors in rodent assays: the two-year bioassay is no longer necessary. Toxicol Pathol. 38(3):487–501. doi: 10.1177/0192623310363813.
- Cohen SM, Bevan C, Gollapudi B, Klaunig JE. 2023. Evaluation of the carcinogenicity of carbon tetrachloride. J Toxicol Environ Health B Crit Rev. 26(6):342–370. doi: 10.1080/10937404.2023.2220147.
- Cohen SM, Boobis AR, Dellarco VL, Doe JE, Fenner-Crisp PA, Moretto A, Pastoor TP, Schoeny RS, Seed JG, Wolf DC. 2019. Chemical carcinogenicity revisited 3: risk assessment of carcinogenic potential based on the current state of modern knowledge of carcinogenesis in humans. Reg. Toxicol. Pharmacol. 103:100–105. doi: 10.1016/j.yrtph.2019.01.017.
- Cohen SM, Ellwein LB. 1990. Cell proliferation in carcinogenesis. Science. 249(4972):1007–1011. doi: 10.1126/science.2204108.
- Cohen SM, Ellwein LB. 1991. Genetic errors, cell proliferation, and carcinogenesis. Cancer Res. 51(24):6493–6505.
- Cohen SM, Klaunig J, Meek ME, Hill RN, Pastoor T, Lehman-McKeeman L, Bucher J, Longfellow DG, Seed J, Dellarco V, et al. 2004. Evaluating the human relevance of chemically-induced animal tumors. Toxicol Sci. 78(2):181–186. doi: 10.1093/toxsci/kfh073.
- Coogan PF, Smith J, Rosenberg L. 2007. Statin use and risk of colorectal cancer. J Natl Cancer Inst. 99(1):32–40. doi: 10.1093/jnci.djk003.
- Crebelli R, Bellincampi D, Conti G, Conti L, Morpurgo G, Carere A. 1986. A comparative study on selected chemical carcinogens for chromosome malsegregation, mitotic crossing-over and forward mutation induction in Aspergillus nidulans. Mutat Res. 172(2):139–149. doi: 10.1016/0165-1218(86)90070-4.
- Dail MB, Burgess SC, Meek EC, Wagner J, Baravik J, Chambers JE. 2007. Spatial distribution of CYP2B1/2 messenger RNA within the rat liver acinus following exposure to the inducers phenobarbital and dieldrin. Toxicol. Sci. 99(1):35–42. doi: 10.1093/toxsci/kfm129.
- Dale KM, Coleman CI, Henyan NN, Kluger J, White CM. 2006. Statins and cancer risk: a meta-analysis. JAMA. 295(1):74–80. doi: 10.1001/jama.295.1.74.
- Davis KJ. 1965. Pathology report on mice fed aldrin, dieldrin, heptachlor or heptachlor epoxide for two years. International FDA memorandum to Dr. A.J. Lehman.
- Davis KJ, Fitzhugh OG. 1962. Tumorigenic potential of aldrin and dieldrin for mice. Toxicol Appl Pharmacol. 4(2):187–189. doi: 10.1016/0042/008x(62)90056-x.
- de Jong G. 1991. Long-term health effects of aldrin and dieldrin. A study of exposure, health effects and mortality of workers engaged in the manufacture and formulation of the insecticides aldrin and dieldrin. Toxicol Lett. Suppl1:1–206. doi: 10.1016/0378-4274(91)90002-N.
- Dean BJ, Doak SMA, Somerville H. 1975. The potential mutagenicity of dieldrin (HOED) in mammals. Food Cosmet. Toxicol. 13(3):317–323. doi: 10.1016/s0015-6264(75)80292-6.
- Decloitre F, Chauveau J, Benoit A, Martin M. 1975. Métabolisation et fixation in vitro par le DNA de thymus de veau et les protéines microsomiques de foie de rat de quelques pesticides organochlorés et organophosphorés [Metabolism and in vitro binding of several organochlorine and organophosphate pesticides to calf thymus DNA and rat liver microsomal proteins]. C. R. Acad. Hebd. Seances Acad. Sci. D. 280(8):1027–1030. French.
- Deichmann WB, MacDonald WE, Blum E, Bevilacqua M, Radomski J, Keplinger M, Balkus M. 1970. Tumorigenicity of aldrin, dieldrin and endrin in the albino rat. IMS Ind Med Surg. 39(10):426–434.
- Ditraglia D, Brown DP, Namekata T, Iverson N. 1981. Mortality study of workers employed at organochlorine pesticide manufacturing plants. Scand J Work Environ Health. 7(Suppl 4):140–146.
- Elcombe CR, Peffer RC, Wolf DC, Bailey J, Bars R, Bell D, Cattley RC, Ferguson SS, Geter D, Goetz A, et al. 2014. Mode of action and human relevance analysis for nuclear receptor-mediated liver toxicity: a case study with phenobarbital as a model constitutive androstane receptor (CAR) activator. Crit. Rev. Toxicol. 44(1):64–82. doi: 10.3109/10408444.2013.835786.
- Engel LS, Hill DA, Hoppin JA, Lubin JH, Lynch CF, Pierce J, Samanic C, Sandler DP, Blair A, Alavanja MC. 2005. Pesticide use and breast cancer risk among farmers’ wives in the agricultural health study. Am J Epidemiol. 161(2):121–135. doi: 10.1093/aje/kwi022.
- Epstein SS, Arnold E, Andrea J, Bass W, Bishop Y. 1972. Detection of chemical mutagens by the dominant lethal assay in the mouse. Toxicol Appl Pharmacol. 23(2):288–325. doi: 10.1016/0041-008x(72)90192-5.
- Fahrig R. 1974. Comparative mutagenicity with pesticides. IARC Publ. (U.N.). 10:161–181.
- Farber E, Sarma DSR. 1987. Hepatocarcinogenesis: a dynamic cellular perspective. Lab Invest. 56(1):4–22.
- Farwell WR, Scranton RE, Lawler EV, Lew RA, Brophy MT, Fiore LD, Gaziano JM. 2008. The association between statins and cancer incidence in a veterans population. J Natl Cancer Inst. 100(2):134–139. doi: 10.1093/jnci/djm286.
- Fitzgerald DJ, Mesnil M, Oyamada M, Tsuda H, Ito N, Yamasaki H. 1989. Changes in gap junction protein (connexin 32) gene expression during rat liver carcinogenesis. J Cell Biochem. 41(2):97–102. doi: 10.1002/jcb.240410206.
- Fitzhugh OG, Nelson AA, Quaife ML. 1964. Chronic oral toxicity of aldrin and dieldrin in rats and dogs. Food Cosmet Toxicol. 2:551–562. doi: 10.1016/s0015-6264(64)80354-0.
- Flower KB, Hoppin JA, Lynch CF, Blair A, Knott C, Shore DL, Sandler DP. 2004. Cancer risk and parental pesticide application in children of Agricultural Health Study participants. Environ Health Perspect. 112(5):631–635. doi: 10.1289/ehp.6586.
- Frank EA, Meek MEB. 2024. Procedural application of mode-of-action and human relevance analysis: styrene-induced lung tumors in mice. Crit Rev Toxicol. 54(2):134–151. doi: 10.1080/10408444.2024.2310600.
- Friedrich A, Olejniczak K. 2011. Evaluation of carcinogenicity studies of medicinal products for human use authorized via the European centralized procedure (1995-2009). Regul. Toxicol. Pharmacol. 60(2):225–248. doi: 10.1016/j.yrtph.2011.04.001.
- Frils S, Poulsen AH, Johnsen SP, McLaughlin JK, Fryzek JP, Dalton SO, Sorensen HT, Olsen JH. 2005. Cancer risk among statin users: a population-based cohort study. Int. J. Cancer. 114(4):643–647. doi: 10.1002/IJC.20758.
- Galloway SM, Armstrong MJ, Reuben C, Colman S, Brown B, Cannon C, Bloom AD, Nakamura F, Ahmed M, Duk S, et al. 1987. Chromosome aberrations and sister chromatid exchanges in Chinese hamster ovary cells: evaluations of 108 chemicals. Environ. Mol. Mutagen. 10:1–175. doi: 10.1002/em.2850100502.
- Gammon MD, Wolff MS, Neugut AI, Eng SM, Teitelbaum SL, Britton JA, Terry MB, Levin B, Stellman SD, Kabat GC, et al. 2002. Environmental toxins and breast cancer on Long Island. II. Organochlorine compound levels in blood. Cancer Epidemiol Biomarkers Prev. 11(8):686–697.
- Garrettson LK, Curley A. 1969. Dieldrein: studies in a poisoned child. Arch Environ Health. 19(6):814–822. doi: 10.1080/00039896.1969.10666935.
- Glatt H, Jung R, Oesch F. 1983. Bacterial mutagenicity investigation of epoxides: drugs, drug metabolites, steroids and pesticides. Mutat Res. 111(2):99–118. doi: 10.1016/0027-5107(83)90056-8.
- Goel MR, Shara MA, Stohs SJ. 1988. Induction of lipid peroxidation by hexachlorocyclohexane, dieldrin, TCDD, carbon tetrachloride, and hexachlorobenzene in rats. Bull Environ Contam Toxicol. 40(2):255–262. doi: 10.1007/bf01881048.
- Gooderham NJ, Cohen SM, Eisenbrand G, Fukushima S, Guengerich FP, Hecht SS, Rietjens IMCM, Rosol TJ, Bastaki M, Linman MJ, et al. 2020. The safety evaluation of food flavoring substances: the role of genotoxicity studies. Crit Rev Toxicol. 50(1):1–27. (PMID: 32162576) doi: 10.1080/10408444.2020.1712589.
- Greenfield RE, Ellwein LB, Cohen SM. 1984. A general probabilistic model of carcinogenesis: analysis of experimental urinary bladder cancer. Carcinogenesis. 5(4):437–445. doi: 10.1093/carcin/5.4.437.
- Harada T, Takeda M, Kojima S, Tomiyama N. 2016. Toxicity and carcinogenicity of dichlorodiphenyltrichloroethane (DDT). Toxicol Res. 32(1):21–33. doi: 10.5487/tr.2016.32.1.021.
- Harr JR, Claeys RR, Bone JF, McCorde TW. 1970. Dieldrin toxidosis: rat reproduction. Am. J. Vet. Res. 3(1):181–189. (PMID: 5414277)
- Haworth S, Lawlor T, Mortelmans K, Speck W, Zeiger E. 1983. Salmonella mutagenicity test results for 250 chemicals. Environ Mutagen. 5(S1):3–49. (PMID: 6365529) doi: 10.1002/em.2860050703.
- Hayes WJ. 1974. Distribution of dieldrin following a single oral dose. Toxicol Appl Pharmacol. 28(3):485–492. doi: 10.1016/0041-008x(74)90233-6.
- Heath DF, Vandekar M. 1964. Toxicity and metabolism of dieldrin in rats. Br J Ind Med. 21(4):269–279. doi: 10.1136/oem.21.4.269.
- Hill AB. 1965. The environment and disease: association or causation? Proc R Soc Med. 58(5):295–300. (PMID: 14283879)
- Holsapple MP, Pitot HC, Cohen SM, Boobis AR, Klaunig JE, Pastoor T, Dellarco V, Dragan YP. 2006. Mode of action in relevance of rodent liver tumors to human cancer risk. Toxicol Sci. 89(1):51–56. doi: 10.1093/toxsci/kfj001.
- Hoogendam I, Versteeg JP, de Vlieger M. 1965. Nine years’ toxicity control in insecticide plants. Arch Environ Health. 10(3):441–448. doi: 10.1080/00039896.1965.10664026.
- Hooker EP, Fulcher KG, Gibb HJ. 2014. Aldrin and dieldrin: a reevaluation of the cancer and noncancer dose-response assessments. Risk Anal. 34(5):865–878. (PMID: 24955469) doi: 10.1111/risa.12145.
- Hoyer AP, Gerdes A-M, Jørgensen T, Rank F, Bøggild Hartvig H. 2002. Organchlorines, p53 mutations in relation to breast cancer risk and survival. A Danish cohort-nested case-controls study. Breast Cancer Res Treat. 71(1):59–65. doi: 10.1023/A:1013340327099.
- Hoyer AP, Jørgensen T, Rank F, Grandjean P. 2001. Organochlorine exposures influence on breast cancer risk and survival according to estrogen receptor status: a Danish cohort-nested case-control study. BMC Cancer. 1(1):8. doi: 10.1186/1471-2407-1-8.
- Hunter J, Maxwell JD, Stewart DA, Williams R, Robinson J, Richardson A. 1972. Increased hepatic microsomal enzyme activity from occupational exposure to certain organochlorine pesticides. Nature. 237(5355):399–401. doi: 10.1038/237399a0.
- Hunter CG, Robinson J. 1967. Pharmacodynamics of dieldrin (HEOD). I. Ingestion by human subjects for 18 months. Arch Environ Health. 15(5):614–626. doi: 10.1080/00039896.1967.10664977.
- Hunter CG, Robinson, J, Aldrin. 1968. Dieldrin, and man. Food Cosmet. Toxicol. 6(2):253–560. doi: 10.1016/0015-6264(68)90206-x.
- Hunter CG, Robinson J, Roberts M. 1969. Pharmacodynamics of dieldrin (HEOD). Ingestion by human subjects for 18 to 24 months, and post exposure for eight months. Arch Environ Health. 18(1):12–21. doi: 10.1080/00039896.1969.10665367.
- Hutson DH. 1976. Comparative metabolism of dieldrin in the rat (CFE) and in two strains of mouse (CF1 and LACG). Food Cosmet Toxicol. 14(6):577–591. doi: 10.1016/s0015-6264(76)80012-0.
- IARC Working Group. 1974. International Agency for Research on Cancer (IARC) Monographs on the Evaluation of Carcinogenic Risk of Chemicals to Man: Some Organochlorine Pesticides. Volume 5.
- IARC Working Group. 1999. Consensus report. IARC Sci. Pub. 147:1–32.
- IARC. 2019. Pentachlorophenol and some related compounds. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Volume 117.
- Ibarluzea jm, Fernández MF, Santa-Marina L, Olea-Serrano MF, Rivas AM, Aurrekoetxea JJ, Expósito J, Lorenzo M, Torné P, Villalobos M, et al. 2004. Breast cancer risk and the combined effect of environmental estrogens. Cancer Causes Control. 15(6):591–600. doi: 10.1023/b:caco.0000036167.51236.86.
- Ito N, Imaida K, Hasegawa R, Tsuda H, Cohen SM. 1989. Rapid bioassay methods for carcinogens and modifiers of hepatocarcinogenesis. Crit Rev Toxicol. 19(4):385–415. doi: 10.3109/10408448909029328.
- Jacobs EJ, Rodriguez C, Brady KA, Connell CJ, Thun MJ, Calle EE. 2006. Cholesterol-lowering drugs and colorectal cancer incidence in a large United States cohort. J. Natl. Cancer Inst. 98(1):69–72. doi: 10.1093/jnci/djj006.
- Jager KW. 1970. Aldrin, dieldrin, endrin and telodrin: an epidemiological and toxicological study of long-term occupational exposure. Amsterdam, Netherlands: Elsevier Publishing Company.
- Janssen-Timmen U, Traub O, Dermietzel R, Rabes HM, Willecke K. 1986. Reduced number of gap junctions in rat hepatocarcinomas detected by monoclonal antibody. Carcinogenesis. 7(9):1475–1482. doi: 10.1093/carcin/7.9.1475.
- Jones C, Trosko JE, Chang CC. 1987. Characterization of a rat liver epithelial cell line to detect inhibitors of metabolic cooperation. In Vitro Cell Dev Biol. 23(3):214–220. (PMID: 3558255. doi: 10.1007/BF02623582.
- Kamendulis LM, Kolaja KL, Stevenson DE, Walborg EF, Jr, Klaunig JE. 2001. Comparative effects of dieldrin on hepatic ploidy, cell proliferation, and apoptosis in rodent liver. J. Toxicol. Environ. Health A. 62(2):127–141. doi: 10.1080/009841001455535.
- Kinoshita A, Wanibuchi H, Morimura K, Wei M, Shen J, Imaoka S, Funae Y, Fukushima S. 2003. Phenobarbital at low dose exerts hormesis in rat hepatocarcinogenesis by reducing oxidative DNA damage, altering cell proliferation, apoptosis and gene expression. Carcinogenesis. 24(8):1389–1399. doi: 10.1093/carcin/bgg079.
- Kitselman CD. 1953. Long term studies on dogs fed aldrin and dieldrin in sublethal dosages, with reference to the histopathological findings and reproduction. J. Am. Vet. Med. Assoc. 132:28–30.
- Klaunig JE. 2018. Oxidative stress and cancer. Curr Pharm Des. 24(40):4771–4778. doi: 10.2174/1381612825666190215121712.
- Klaunig JE, Goldblatt PJ, Hinton DE, Lipsky MM, Trump BF. 1984. Carcinogen induced unscheduled DNA synthesis in mouse hepatocytes. Toxicol Pathol. 12(2):119–125. doi: 10.1177/019262338401200202.
- Klaunig JE, Ruch RJ. 1987. Strain and species effects on the inhibition of hepatocyte intercellular communication by liver tumor promoters. Cancer Lett. 36(2):161–168. doi: 10.1016/0304-3835(87)90087-5.
- Klaunig JE, Wang Z, Pu X, Zhou S. 2011. Oxidative stress and oxidative damage in chemical carcinogenesis. Toxicol Appl Pharmacol. 254(2):86–99. doi: 10.1016/j.taap.2009.11.028.
- Klaunig JE, Weghorst CM, Pereira MA. 1988. Effect of phenobarbital on diethylnitrosamine and dimethylnitrosamine induced hepatocellular tumors in male B6C3F1 mice. Cancer Lett. 42(1-2):133–139. doi: 10.1016/0304-3835(88)90250-9.
- Klaunig JE, Xu Y, Bachowski S, Ketcham CA, Isenberg JS, Kolaja KL, Baker TK, Walborg EF, Stevenson, DE. 1995. Oxidative stress in nongenotoxic carcinogenesis. Toxicol Lett. 82-83:683–691. doi: 10.1016/0378-4274(95)03514-1.
- Klaunig JE, Xu Y, Isenberg JS, Bachowski S, Kolaja KL, Jiang J, Stevenson DE, Walborg EF. 1998. The role of oxidative stress in chemical carcinogens. Environ Health Perspect. 106(Suppl 1):289–295. doi: 10.1289/ehp.98106s1289.
- Kohli KK, Maggon KK, Venkitasubramanian TA. 1977. Induction of mixed function oxidases on oral administration of dieldrin. Chem Biol Interact. 17(3):249–255. doi: 10.1016/0009-2797(77)90089-8.
- Kolaja KL, Klaunig JE. 1997. Vitamin E modulation of hepatic focal lesion growth in mice. Toxicol Appl Pharmacol. 143(2):380–387. PMID: 9144454. doi: 10.1006/taap.1996.8089.
- Kolaja KL, Stevenson DE, Johnson JT, Walborg EF, Klaunig JE. 1995a. Hepatic effects of dieldrin and phenobarbital in male B6C3F1 mice and Fisher 344 rats: species selective induction of DNA synthesis. Prog Clin Biol Res. 391:397–408. PMID: 8532732
- Kolaja KL, Stevenson DE, Johnson JT, Walborg EF, Klaunig JE. 1996c. Subchronic effects of dieldrin and phenobarbital on hepatic DNA synthesis in mice and rats. Fundam Appl Toxicol. 29(2):219–228. doi: 10.1006/faat.1996.0025.
- Kolaja KL, Stevenson DE, Walborg EF, Jr., Klaunig JE. 1996a. Selective dieldrin promotion of hepatic focal lesions in mice. Carcinogenesis. 17(6):1243–1250. doi: 10.1093/carcin/17.6.1243.
- Kolaja KL, Stevenson DE, Walborg EF, Jr., Klaunig JE. 1996b. Reversibility of promoter induced hepatic focal lesion growth in mice. Carcinogenesis. 17(7):1403–1409. doi: 10.1093/carcin/17.7.1403.
- Kolaja KL, Stevenson DE, Walborg EF, Klaunig JE. 1995b. The effect of dieldrin and phenobarbital on preneoplastic hepatic lesion growth in male F344 rat and B6C3F1 mouse. Prog Clin Biol Res. 391:409–423. PMID: 8532733
- Krynitz B, Edgren G, Lindelöf B, Baecklund E, Brattström C, Wilczek H, Smedby KE. 2013. Risk of skin cancer and other malignancies in kidney, liver, heart and lung transplant recipients 1970 to 2008–a Swedish population-based study. Int J Cancer. 132(6):1429–1438. doi: 10.1002/ijc.27765.
- Krzystyniak K, Hugo P, Flipo D, Fournier M. 1985. Increased susceptibility to mouse hepatitis virus 3 of peritoneal macrophages exposed to dieldrin. Toxicol Appl Pharmacol. 80(3):397–408. doi: 10.1016/0041-008x(85)90384-9.
- Lake BG. 2018. Human relevance of rodent liver tumour formation by constitutive androstane receptor (CAR) activators. Toxicol Res (Camb). 7(4):697–717. doi: 10.1039/c8tx00008e.
- Lebrec H, Brennan FR, Haggerty H, Herzyk D, Kamperschroer C, Maier CC, Ponce R, Preston BD, Weinstock D, Mellon RD. 2016. HESI/FDA workshop on immunomodulators and cancer risk assessment: building blocks for a weight-of-evidence approach. Regul Toxicol Pharmacol. 75:72–80. doi: 10.1016/j.yrtph.2015.12.018.
- Lehman A. 1959. Appraisal of the safety of chemicals in foods, drugs, and cosmetics. Association of food and Drug Officials of the United States (as cited in USEPA, 1992 and USEPA, 1988).
- Lipsky MM, Trump BF, Hinton DE. 1989. Histogenesis of dieldrin and DDT-induced hepatocellular carcinoma in Balb/c mice. J Environ Pathol Toxicol Oncol. 9(1):79–93.
- Loose LD. 1982. Macrophage induction of T-suppressor cells in pesticide-exposed and protozoan-infected mice. Environ Health Perspect. 43:89–97. doi: 10.1289/ehp.824389.
- Loose LD, Silkworth JB, Charbonneau T, Blumenstock F. 1981. Environmental chemical-induced macrophage dysfunction. Environ Health Perspect. 39:79–92. doi: 10.1289/ehp.813979.
- MacDonald JS, Halleck MM. 2004. The toxicology of HMG-CoA reductase inhibitors: prediction of human risk. Toxicol Pathol. 32 Suppl 2(2_suppl):26–41. doi: 10.1080/01926230490462057.
- Majumdar SK, Kopelman HA, Schnitman MJ. 1976. Dieldrin-induced chromosome damage in mouse bone-marrow and WI-38 human lung cells. J. Hered. 67(5):303–307. doi: 10.1093/oxfordjournals.jhered.a108736.
- Majumdar SK, Maharam LG, Viglianti GA. 1977. Mutagenicity of dieldrin in the Salmonella-microsome test. J Hered. 68(3):184–185. doi: 10.1093/oxfordjournals.jhered.a108805.
- Maronpot RR, Haseman JK, Boorman GA, Eustis SE, Rao GN, Huff JE. 1987. Liver lesions in B6C3F1 mice: the National Toxicology Program, experience and position. Arch Toxicol Suppl. 10:10–26. doi: 10.1007/978-3-642-71617-1_2.
- Marshall TC, Dorough HW, Swim HE. 1976. Screening of pesticides for mutagenic potential using Salmonella typhimurium mutants. J Agric Food Chem. 24(3):560–563. doi: 10.1021/jf60205a013.
- Mathur V, Bhatnagar P, Sharma RG, Acharya V, Sexana R. 2002. Breast cancer incidence and exposure to pesticides among women originating from Jaipur. Environ Int. 28(5):331–336. doi: 10.1016/s0160-4120(02)00031-4.
- Matthews HB, McKinney JD, Lucier GW. 1971. Dieldrin metabolism, excretion, and storage in male and female rats. J Agric Food Chem. 19(6):1244–1248. doi: 10.1021/jf60178a035.
- McCann J, Choi E, Yamasaki E, Ames BN. 1975. Detection of carcinogens as mutagens in the Salmonella/microsome test: assay of 300 chemicals. Proc Natl Acad Sci U S A. 72(12):5135–5139. doi: 10.1073/pnas.72.12.5135.
- McDuffie HH, Pahwa P, McLaughlin JR, Spinelli JJ, Fincham S, Dosman JA, Robson D, Skinnider LF, Choi NW. 2001. Non-Hodgkin’s lymphoma and specific pesticide exposure in men: Cross-Canada study of pesticides and health. Cancer Epidemiol Biomarkers Prev. 10(11):1155–1163. (PMID: 11700263)
- McGregor DB, Brown AG, Howgate S, McBride D, Riach C, Caspary WJ. 1991. Responses of the L5178Y mouse lymphoma cell forward mutation assay. V: 27 coded chemicals. Environ Mol Mutagen. 17(3):196–219. doi: 10.1002/em.2850170309.
- Meek ME, Bucher JR, Cohen SM, Dellarco V, Hill RN, Lehman-McKeeman LD, Longfellow DG, Pastoor T, Seed J, Patton DE. 2003. A framework for human relevance analysis of information on carcinogenic modes of action. Crit. Rev. Toxicol. 33(6):591–653. doi: 10.1080/713608373.
- Meek MEB, Palermo CM, Bachman AN, North CM, Lewis RJ. 2014. Mode of action human relevance (species concordance) framework: evolution of the Bradford Hill considerations and comparative analysis of weight of evidence. J Appl Toxicol. 34(6):595–606. doi: 10.1002/jat.2984.
- Meierhenry EF, Ruebner BH, Gershwin ME, Hsieh LS, French SW. 1983. Dieldrin-induced Mallory bodies in hepatic tumors of mice of different strains. Hepatology. 3(1):90–95. doi: 10.1002/hep.1840030115.
- Moolgavkar SH, Knudson AG. 1981. Mutation and cancer: a model for human carcinogenesis. J Natl Cancer Inst. 66(6):1037–1052. doi: 10.1093/jnci/66.6.1037.
- Morgan DP, Lin LI. 1978. Blood organochlorine pesticide concentrations, clinical hematology and biochemistry in workers occupationally exposed to pesticides. Arch Environ Contam Toxicol. 7(4):423–447. doi: 10.1007/bf02332069.
- Morgan DP, Roan CC. 1974. Liver function in workers having high tissue stores of chlorinated hydrocarbon pesticides. Arch Environ Health. 29(1):14–17. doi: 10.1080/00039896.1974.10666519.
- National Cancer Institute (NCI). 1978. Bioassays of aldrin and dieldrin for possible carcinogenicity. Report number 309-00-2 and 60-57-1.
- Newberne PM. 1983. How relevant are mice hepatomas to human risk? Toxicol Pathol. 11(2):113–114. doi: 10.1177/019262338301100201.
- Nishimura N, Nishimura H, Oshima H. 1982. Survey on mutagenicity of pesticides by the Salmonella-microsome test. Aichi Ika Daig Igak Zass. 10:305–312.
- OECD. 2016. Overview of the set of OECD genetic toxicology test guidelines and updates performed in 2014-2015. Series on Testing and Assessment No. 238.
- Osaba L, Aguirre A, Alonso A, Graf U. 1999. Genotoxicity testing of six insecticides in two crosses of the Drosophila wing spot test. Mutat Res. 439(1):49–61. doi: 10.1016/s1383-5718(98)00173-9.
- Ott T, Jokwitz M, Lenhard D, Romualdi A, Dombrowski F, Ittrich C, Schwarz M, Willecke K. 2006. Ablation of gap junctional communication in hepatocytes of transgenic mice does not lead to disrupted cellular homeostasis or increased spontaneous tumourigenesis. Eur J Cell Biol. 85(8):717–728. doi: 10.1016/j.ejcb.2006.03.004.
- Paolini M, Mesirca R, Pozzetti L, Sapone A, Cantelli-Forti G. 1995. Induction of CYP2B1 mediated pentoxyresorufin O-dealkylase activity in different species, sex and tissue by prototype 2B1-inducers. Chem Biol Interact. 95(1-2):127–139. doi: 10.1016/0009-2797(94)03352-8.
- Parke DV, Sapota A. 1996. Chemical toxicity and reactive oxygen species. Int J Occup Med Environ Health. 9(4):331–340. (PMID: 9117192)
- Penn I. 1988. Tumors of the immunocompromised patient. Annu Rev Med. 39(1):63–73. doi: 10.1146/annurev.me.39.020188.000431.
- Pitot HC. 2007. Adventures in hepatocarcinogenesis. Annu Rev Pathol. 2(1):1–29. doi: 10.1146/annurev.pathol.2.010506.092027.
- Pitot HC, Hikita H, Dragan Y, Sargent L, Haas M. 2000. Review article: the stages of gastrointestinal carcinogenesis—application of rodent models to human disease. Aliment Pharmacol Ther. 14 Suppl 1(s1):153–160. doi: 10.1046/j.1365-2036.2000.014s1153.x.
- Probst GS, McMahon RE, Hill LE, Thompson CZ, Epp JK, Neal SB. 1981. Chemically-induced unscheduled DNA synthesis in primary rat hepatocyte cultures: a comparison with bacterial mutagenicity using 218 compounds. Environ Mutagen. 3(1):11–32. doi: 10.1002/em.2860030103.
- Purdue MP, Hoppin JA, Blair A, Dosemeci M, Alavanja MC. 2007. Occupational exposure to organochlorine insecticides and cancer incidence in the Agricultural Health Study. Int J Cancer. 120(3):642–649. (PMID: 17096337) doi: 10.1002/ijc22258.
- Quintana PJE, Delfino RJ, Korrick S, Zopgas A, Kutz FW, Jones EL, Laden F, Garshick E. 2004. Adipose tissue levels of organiochlorine pesticides and polychlorinated biphenlys and risk of non-Hodgkin’s lymphoma. Environ. Health Perspect. 112(8):854–861. (PMID: 15175172) doi: 10.1289/6726.
- Quist EM, Boorman GA, Cullen JM, Maronpot RR, Remick AK, Swenberg JA, Freshwater L, Hardisty JF. 2019. Reevaluation of hepatocellular neoplasms in CD-1 mice from a 2-year oral carcinogenicity study with permethrin. Toxicol Pathol. 47(1):11–17. doi: 10.1177/0192623318809304.
- Ramamoorthy K, Wang F, Chen IC, Norris JD, McDonnell DP, Leonard LS, Gaido KW, Bocchinfuso WP, Korach KS, Safe S. 1997. Estrogenic activity of a dieldrin/toxaphene mixture in the mouse uterus, MCF-7 human breast cancer cells, and yeast-based estrogen receptor assays: no apparent synergism. Endocrinology. 138(4):1520–1527. doi: 10.1210/endo.138.4.5056.
- Rao MS, Reddy JK. 1991. An overview of peroxisome proliferator-induced hepatocarcinogenesis. Environ Health Perspect. 93:205–209. doi: 10.1289/ehp.9193205.
- Reddy JK, Rao MS. 1989. Oxidative DNA damage caused by persistent peroxisome proliferation: its role in hepatocarcinogenesis. Mutat Res. 214(1):63–68. doi: 10.1016/0027-5107(89)90198-x.
- Reuber MD. 1976. Histopathology of carcinomas of the liver in mice ingesting dieldrin or aldrin. Tumori. 62(5):463–471. doi: 10.1177/030089167606200501.
- Ritchie JM, Vial SL, Fuortes LJ, Guo H, Reedy VE, Smith EM. 2003. Organochlorines and risk of prostate cancer. J Occup Environ Med. 45(7):692–702. doi: 10.1097/01.jom.0000071510.96740.0b.
- Riva L, Pandiri AR, Li YR, Droop A, Hewinson J, Quail MA, Iyer V, Shepherd R, Herbert RA, Campbell PJ, et al. 2020. The mutational signature profile of known and suspected human carcinogens in mice. Nat Genet. 52(11):1189–1197. doi: 10.1038/s41588-020-0692-4.
- Ruch RJ, Klaunig JE. 1986. Effects of tumor promoters, genotoxic carcinogens and hepatocytotoxins on mouse hepatocyte intercellular communication. Cell Biol Toxicol. 2(4):469–483. doi: 10.1007/BF00117849.
- Ruebner BH, Gershwin ME, Meierhenry EF, Hsieh LS, Dunn PL. 1984. Irreversibility of liver tumors in C3H mice. J Natl Cancer Inst. 73(2):493–498. doi: 10.1093/jnci/73.2.493.
- Schroeder JC, Olshan AF, Baric R, Dent GA, Weinberg CR, Yount B, Cerhan JR, Lynch CF, Schuman LM, Tolbert PE, et al. 2001. Agricultural risk factors for t(14;18) subtypes of non-Hodgkin’s lymphoma. Epidemiology. 12(6):701–709. doi: 10.1097/00001648-200111000-00020.
- Seed J, Carney EW, Corley RA, Crofton KM, DeSesso JM, Foster PM, Kavlock R, Kimmel G, Klaunig J, Meek ME, et al. 2005. Using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Crit Rev Toxicol. 35(8-9):664–672. doi: 10.1080/10408440591007133.
- Shakoori AR, Rasul YG, Ali, SS 1982. Effect of dieldrin feeding for six months on albino rats-biochemical and histological changes in liver. Pakistan J. Z. 14:191–204.
- Shirasu Y, Moriya M, Kato K, Furuhashi A, Kada T. 1976. Mutagenicity screening of pesticides in the microbial system. Mutat. Res. 40(1):19–30. doi: 10.1016/0165-1218(76)90018-5.
- Shizu R, Makida N, Sobe K, Ishimura M, Takeshita A, Hosaka T, Kanno Y, Sasaki T, Yoshinari K. 2024. Interaction with YAP underlies the species differences between humans and rodents in CAR-dependent hepatocyte proliferation. Toxicol. Sci. 198(1):101–112. doi: 10.1093/toxsci/kfad129.
- Shukla VK, Rastogi AN, Adukia TK, Raizada RB, Reddy DC, Singh S. 2001. Organchlorine pesticides in carcinoma of the gallbladder: a case-control study. Eur J Cancer Prev. 10(2):153–156. doi: 10.1097/000084669-200104000-00006.
- Snedeker SM. 2011. Pesticides and breast cancer risk: a review of DDT, DDE, and dieldrin. Environ. Health Perspect. 109(Suppl 1):35–47. doi: 10.1289/ehp.01109s135.
- Sonich-Mullin C, Fielder R, Wiltse J, Baetcke K, Dempsey J, Fenner-Crisp P, Grant D, Hartley M, Knaap A, Kroese D, et al. 2001. IPCS conceptual framework for evaluating a mode of action for chemical carcinogenesis. Regul Toxicol Pharmacol. 34(2):146–152. doi: 10.1006/rtph.2001.1493.
- Soto AM, Sonnenschein C, Chung KL, Fernandez MF, Olea N, Serrano FO. 1995. The E-SCREEN assay as a tool to identify estrogens: an update on estrogenic environmental pollutants. Environ Health Perspect. 103 Suppl 7(Suppl 7):113–122. doi: 10.1289/ehp.95103s7113.
- Stern AH. 2014. Hazard identification of the potential for dieldrin carcinogenicity to humans. Environ Res. 131:188–214. doi: 10.1016/j.envres.2014.02.007.
- Stevenson DE, Thorpe E, Hunt PF, Walker AI. 1976. The toxic effects of dieldrin in rats: a reevaluation of data obtained in a two-year feeding study. Toxicol Appl Pharmacol. 36(2):247–254. doi: 10.1016/0041-008x(76)90004-1.
- Stevenson DE, Walborg EF, Klaunig JE. 1995. The species specificity of dieldrin- or phenobarbital- induced hepatocarcinogenesis: case studies with implications for human health risk assessment. Prog. Clin. Biol. Res. 391:337–345. (PMID: 8532726)
- Stevenson DE, Walborg EF, North DW, Sielken RL, Ross CE, Wright AS, Xu Y, Kamendulis LM, Klaunig, JE 1999. Monograph: reassessment of human cancer risk of aldrin/dieldrin. Toxicol Lett. 109(3):123–186. doi: 10.1016/s0378-4274(99)00132-0.
- Strupp C, Corvaro M, Cohen SM, Corton JC, Ogawa K, Richert L, Jacobs MN. 2023. Increased cell proliferation as a key event in chemical carcinogenesis: application in an integrated approach for the testing and assessment of non-genotoxic carcinogenesis. IJMS. 24(17):13246. doi: 10.3390/ijms241713246.
- Swaen GM, de Jong G, Slangen JJ, van Amelsvoort LG. 2002. Cancer mortality in workers exposed to dieldrin and aldrin: an update. Toxicol Ind Health. 18(2):63–70. doi: 10.1191/0748233702th132oa.
- Syversen U, Waldum HL, Slordahl KW, Chen D. 2000. Synergistic effect of the pesticides toxaphene and dieldrin on bone mineral density in rats. J. Bone Miner. Res. 15(Suppl 1):s451.
- Takagi A, Sai K, Umemura T, Hasegawa R, Kurokawa Y. 1990. Significant increase of 8-hydroxydeoxyguanosine in liver DNA of rats following short-term exposure to the peroxisome proliferators di(2-ethylhexyl)phthalate and di(2-ethylhexyl)adipate. Jpn J Cancer Res. 81(3):213–215. doi: 10.1111/j.1349-7006.1990.tb02551.x.
- Tennekes HA, Wright AS, Dix KM. 1979. The effects of dieldrin, diet and other environmental components on enzyme function and tumour incidence in livers of CF-1 mice. In: Chambers PL, Günzel P, editors. Mechanism of Toxic Action on Some Target Organs. Archives of Toxicology, vol 2. Berlin: Springer. https://doi.org/10.1007/978-3-642-67265-1_17
- Tennekes HA, Wright AS, Dix KM, Koeman JH. 1981. Effects of dieldrin, diet and bedding on enzyme function and tumor incidence in livers of male CF-1 mice. Cancer Res. 41(9 Pt 1):3615–3620. PMID: 7260918.
- Thoolen B, Maronpot RR, Harada T, Nyska A, Rousseaux C, Nolte T, Malarkey DE, Kaufmann W, Küttler K, Deschl U, et al. 2010. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol Pathol. 38(7 Suppl):5S–81S. doi: 10.1177/0192623310386499.
- Thorpe E, Walker AI. 1973. The toxicology of dieldrin (HEOD). II. Comparative long-term oral toxicity studies in mice with dieldrin, DDT, phenobarbitone, β-BHC and γ-BHC. Food Cosmet Toxicol. 11(3):433–442. doi: 10.1016/0015-6264(73)90008-4.
- Trosko JE, Jone C, Chang, CC 1987. Inhibition of gap junctional-mediated intercellular communication in vitro by aldrin, dieldrin, and toxaphene: a possible cellular mechanism for their tumor-promoting and neurotoxic effects. Mol Toxicol. 1(1):83–93. (PMID: 3449752)
- Tsuda H, Asamoto M, Baba H, Iwahori Y, Matsumoto K, Iwase T, Nishida Y, Nagao S, Hakoi K, Yamaguchi S, et al. 1995. Cell proliferation and advancement of hepatocarcinogenesis in the rat are associated with a decrease in connexin 32 expression. Carcinogenesis. 16(1):101–105. doi: 10.1093/carcin/16.1.101.
- Tully DB, Cox VT, Mumtaz MM, Davis VL, Chapin RE. 2000. Six high-priority organochlorine pesticides, either singly or in combination, are nonestrogenic in transfected HeLa cells. Reprod Toxicol. 14(2):95–102. doi: 10.1016/s0890-6238(00)00060-5.
- USEPA (United States Environmental Protection Agency). 2012. Dieldrin. Integrated Risk Information System (IRIS) updated 3/14/12. Available at: https://iris.epa.gov/ChemicalLanding/&substance_nmbr=225.
- USEPA (United States Environmental Protection Agency). Carcinogenicity Assessment of Aldrin and Dieldrin (Appendix B). August 1987. Available at: https://cfpub.epa.gov/ncea/risk/recordisplay.cfm?deid=49651.
- USEPA. 2005. Guidelines for Carcinogen Risk Assessment. Report number 630-P-03-001F.
- Van Amelsvoort LGPM, Slangen JJM, Tsai SP, de Jong G, Kant I. 2009. Cancer mortality in workers exposed to dieldrin and aldrin: over 50 years of follow up. Int Arch Occup Environ Health. 82(2):217–225. doi: 10.1007/s00420-008-0325-1.
- van Ravenzwaay B, Kunz W. 1988. Quantitative aspects of accelerated nuclear polyploidization and tumour formation in dieldrin treated CF-1 mouse liver. Br J Cancer. 58(1):52–56. doi: 10.1038/bjc.1988.160.
- van Sittert NJ, de Jong G. 1987. Evaluation of liver function with biochemical tests of operators engaged in aldrin and dieldrin manufacturing. Shell Oil Company.
- Vesselinovitch SD, Rao KV, Mihailovich N. 1979. Neoplastic response of mouse tissues during perinatal age periods and its significance in chemical carcinogenesis. Natl Cancer Inst Monogr. (51):239–250.
- Wade MJ, Moyer JW, Hine CH. 1979. Mutagenic action of a series of epoxides. Mutat Res. 66(4):367–371. doi: 10.1016/0165-1218(79)90047-8.
- Walker AI, Stevenson DE, Robinson J, Thorpe E, Roberts M. 1969. The toxicoclogy and pharmacodynamics of dieldrin (HEOD): two-year oral exposures of rats and dogs. Toxicol Appl Pharmacol. 15(2):345–373. doi: 10.1016/0041-008x(69)90034-9.
- Walker AI, Thorpe E, Stevenson DE. 1973. The toxicology of dieldrin (HEOD). I. Long-term oral toxicity studies in mice. Food Cosmet Toxicol. 11(3):415–432. doi: 10.1016/0015-6264(73)90007-2.
- Wang Z, Li X, Wu Q, Lamb JC, Klaunig JE. 2017. Toxaphene-induced mouse liver tumorigenesis is mediated by the constitutive androstane receptor. J Appl Toxicol. 37(8):967–975. doi: 10.1002/jat.3445.
- Wang Z, Neal BH, Lamb JC, Klaunig JE. 2015. Mechanistic investigation of toxaphene induced mouse liver tumors. Toxicol Sci. 147(2):549–561. doi: 10.1093/toxsci/kfv151.
- Wang Z, Wu Q, Li X, Klaunig JE. 2020. Constitutive androstane receptor (CAR) mediates dieldrin-induced liver tumorigenesis in mouse. Arch Toxicol. 94(8):2873–2884. doi: 10.1007/s00204-020-02781-8.
- Ward EM, Schulte P, Grajewski B, Andersen A, Patterson DG, Turner W, Jellum E, Deddens JA, Friedland J, Roeleveld N, et al. 2000. Serum organochlorine levels and breast cancer: a nested case-control study of Norwegian women. Cancer Epidemiol Biomarkers Prev. 9(12):1357–1367. (PMID: 11142422)
- Warnick SL, Carter JE. 1972. Some findings in a study of workers occupationally exposed to pesticides. Arch Environ Health. 25(4):265–270. doi: 10.1080/00039896.1972.10666172.
- Weisburger JH, Williams GM. 1981. Carcinogen testing: current problems and new approaches. Science. 214(4519):401–407. (PMID: 7291981). DOI: 10/1126.science.7291981
- Wood CE, Hukkanen RR, Sura R, Boyce R, Jacobson-Kram D, Nolte T, Odin M, Cohen SM. 2015. Scientific and regulatory policy committee (SRPC) review: Interpretation and use of cell proliferation data in cancer risk assessment. Toxicol Pathol. 43(6):760–775. doi: 10.1177/0192623315576005.
- Wright AS, Donninger C, Greenland RD, Stemmer KL, Zavon MR. 1978. The effects of prolonged ingestion of dieldrin on the livers of male rhesus monkeys. Ecotoxicol Environ Saf. 1(4):477–502. doi: 10.1016/0147-6513(78)90016-7.
- Wright AS, Potter D, Wooder MF, Donninger C, Greenland RD. 1972. The effects of dieldrin on the subcellular structure and function of mammalian liver cells. Food Cosmet Toxicol. 10(3):311–332. doi: 10.1016/s0015-6264(72)80251-7.
- Xu X, Dailey AB, Talbott EO, Ilacqua VA, Kearney G, Asal NR. 2010. Associations of serum concentrations of organochlorine pesticides with breast cancer and prostate cancer in U.S. adults. Environ Health Perspect. 118(1):60–66. doi: 10.1289/ehp.0900919.
- Yager JD, Davidson NE 2006. Estrogen carcinogenesis in breast cancer. N Engl J Med. 354(3):270–282. doi: 10.1056/nejmra050776.
- Yamada T. 2021a. Application of humanized mice to toxicology studies: evaluation of the human relevance of the mode of action for rodent liver tumor formation by activators of the constitutive androstane receptor (CAR). J Toxicol Pathol. 34(4):283–297. doi: 10.1293/tox.20210027.
- Yamada T, Cohen SM, Lake BG. 2021b. Critical evaluation of the human relevance of the mode of action for rodent liver tumor formation by activators of the constitutive androstane receptor (CAR). Crit. Rev. Toxicol. 51(5):373–394. (PMID: 34264181) doi: 10.1080/10408444.2021.1939654.