
Abstract
Tyrosine kinases were first discovered as the protein products of viral oncogenes. We now know that this large family of metazoan enzymes includes nearly one hundred structurally diverse members. Tyrosine kinases are broadly classified into two groups: the transmembrane receptor tyrosine kinases, which sense extracellular stimuli, and the cytoplasmic tyrosine kinases, which contain modular ligand-binding domains and propagate intracellular signals. Several families of cytoplasmic tyrosine kinases have in common a core architecture, the “Src module,” composed of a Src-homology 3 (SH3) domain, a Src-homology 2 (SH2) domain, and a kinase domain. Each of these families is defined by additional elaborations on this core architecture. Structural, functional, and evolutionary studies have revealed a unifying set of principles underlying the activity and regulation of tyrosine kinases built on the Src module. The discovery of these conserved properties has shaped our knowledge of the workings of protein kinases in general, and it has had important implications for our understanding of kinase dysregulation in disease and the development of effective kinase-targeted therapies.
Graphical Abstract
Introduction
Tyrosine kinases play critical roles in metazoan signal transduction pathways, where they facilitate the conversion of external and internal stimuli into cellular responses that include cell proliferation, adhesion, motility, and the regulation of metabolism (Thomas and Brugge Citation1997; Hunter Citation2009; Lemmon and Schlessinger Citation2010). Tyrosine kinases can be divided into two major classes: (1) receptor tyrosine kinases, which have extracellular ligand-binding domains, a single-pass transmembrane domain, and an intracellular kinase domain, and (2) cytoplasmic tyrosine kinases, also referred to as nonreceptor tyrosine kinases, which are multidomain proteins that are often tethered to the inner leaflet of the plasma membrane by lipid anchors. There are ∼60 transmembrane receptors and ∼30 cytoplasmic tyrosine kinases in humans, and these kinases have ∼25 different domain architectures (Manning et al. Citation2002b).
Receptor tyrosine kinases are activated by the binding of cognate ligands to their extracellular domains. Ligand binding results in receptor dimerization/oligomerization, or a change in a preexisting oligomeric structure. These structural changes mediate activation of the intracellular kinase domain through a variety of mechanisms, including trans-autophosphorylation of kinase domains or the formation of allosterically activated kinase domain dimers (Lemmon and Schlessinger Citation2010). Individual receptor tyrosine kinases are often activated by several different ligands and are thus able to integrate information from a variety of extracellular stimuli into a conserved set of downstream signaling pathways ().
Figure 1. Architectures of eukaryotic protein tyrosine kinases. A. Activation of receptor tyrosine kinases through ligand-induced dimerization. B. Activation of cytoplasmic tyrosine kinases through phosphorylation or the engagement of modular ligand-binding domains. C. Domain architectures of the major families of metazoan cytoplasmic tyrosine kinases. Members of each family, found in humans, are listed. An asterisk for Srm and Txk denotes that these proteins differ slightly from other family members in their regulatory phosphosites or domain architecture. Numbering in panel C and throughout the text corresponds to the following sequences for representative members of each family: chicken c-Src and the human proteins Frk, c-Abl isoform 1b, Btk, Csk, Fes, Syk, Fak1, Jak1, and Ack. D. Schematic diagram of the key structural features of tyrosine kinase domains. Sidechains are numbered according to their position in chicken c-Src (see colour version of this figure at www.tandfonline.com/ibmg).
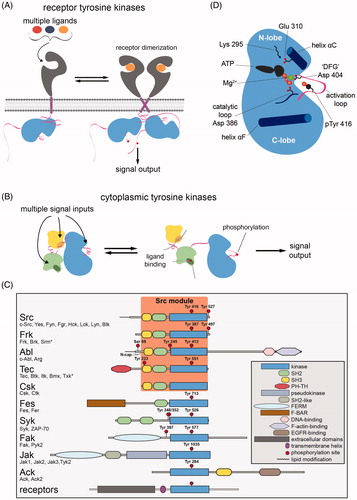
Receptor tyrosine kinases typically sit at the apex of signaling pathways and respond to specific biological cues. Cytoplasmic tyrosine kinases reside within the cores of these pathways and serve to amplify and propagate signals (Thomas and Brugge Citation1997; Takesono et al. Citation2002; Mócsai et al. Citation2010; Wang Citation2014). All cytoplasmic tyrosine kinases contain one or more ligand-binding domains, which allow them to integrate information from different inputs (). This multifunctionality is mediated by allosteric coupling between the activities of their ligand-binding domains and the catalytic domain (). Despite their structural diversity, cytoplasmic tyrosine kinases share the feature that they can adopt an auto-inhibited state, where intramolecular interactions with noncatalytic domains stabilize an inactive conformation of the kinase domain (Jura et al. Citation2011). The activation of cytoplasmic tyrosine kinases is mediated, in part, by engagement of these ligand-binding domains and the destabilization of the auto-inhibitory interactions to provide access to a catalytically competent state of the kinase domain. The discovery and characterization of the ligand-binding domains of cytoplasmic tyrosine kinases has been critical to our understanding of how metazoan signal transduction pathways are organized, and to our understanding of the structure and regulation of tyrosine kinases (Pawson and Kofler Citation2009).
Tyrosine kinases were first discovered by virtue of the catalytic activities of the proteins encoded by certain viral oncogenes, including the first oncogene to be identified, v-Src, which we now know is the viral variant of the cytoplasmic tyrosine kinase c-Src (Hunter and Sefton Citation1980; Witte et al. Citation1980; Martin Citation2004).1 An intensive investigation of the mechanism of regulation of Src and its relatives followed, leading to the identification of the Src-homology 2 (SH2) domain, which binds phosphotyrosine-containing peptide segments (Sadowski et al. Citation1986), and the Src-homology 3 (SH3) domain, which binds polyproline motifs (Mayer et al. Citation1988). These discoveries introduced the concept that signal-triggered change in the localization of proteins, governed by modular binding domains, is the most important consequence of the activation of tyrosine kinase pathways (Scott and Pawson Citation2000). This contrasts with the then-prevailing view of second-messenger signaling, where diffusible signaling molecules spread through the cell to transmit information. Signal-triggered protein–protein interactions, mediated by SH2, SH3, and other ligand-binding domains, such as phosphotyrosine-binding (PTB) and WW domains, are now understood to underlie all aspects of metazoan signal transduction.
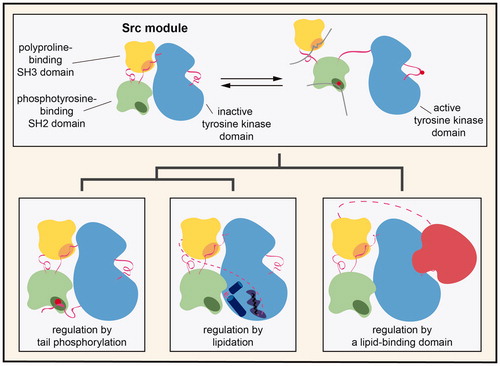
Half of the human cytoplasmic tyrosine kinases have the same core domain architecture, comprised of an SH3 domain, an SH2 domain, and a kinase domain, connected in that order. We refer to this conserved domain architecture as the “Src module” (). The Src module has been elaborated on numerous times over the course of evolution, through the addition of other modular binding domains, the incorporation of regulatory post-translational modification sites, and subtle alterations in conformational preferences. The result of this evolutionary tinkering on the same modular scaffold is the generation of distinct cytoplasmic tyrosine kinases that are controlled by different inputs and which participate in a wide range of signal transduction pathways.
Over the past three decades, substantial progress has been made in understanding the molecular basis for the regulation and activity of tyrosine kinases containing the Src module. Early progress was made through biochemical experiments and studies on oncogenic viruses containing mutant cytoplasmic tyrosine kinase alleles [see (Martin Citation2004) for a historical perspective]. Subsequent studies focused on characterizing the structure and sequence-specific ligand recognition of SH2 domains (Booker et al. Citation1992; Overduin et al. Citation1992; Waksman et al. Citation1992, Citation1993; Eck et al. Citation1993; Songyang et al. Citation1993, Citation1994) and SH3 domains (Musacchio et al. Citation1992; Yu et al. Citation1992, Citation1994; Feng et al. Citation1994; Lim et al. Citation1994). These investigations established the role of ligand-binding domains and modular protein–protein interactions in cellular signal transduction (Kuriyan and Cowburn Citation1997; Pawson and Kofler Citation2009). The determination of high-resolution crystal structures of the Src-family kinases c-Src and Hck in their auto-inhibited states revealed how the SH2 and SH3 domains stabilize the kinase domain in an inactive form (Sicheri et al. Citation1997; Williams et al. Citation1997; Xu et al. Citation1997, Citation1999; Schindler et al. Citation1999).
More recently, the determination of an increasing number of structures of cytoplasmic tyrosine kinases, coupled with biochemical, computational, and evolutionary analyses, have demonstrated how the Src module has been used in nature as a robust scaffold for the emergence of a variety of biologically important signaling switches. The principles gleaned from these studies have informed our understanding of the activity and regulation of all protein kinases, not just those bearing the Src module (Huse and Kuriyan Citation2002; Boggon and Eck Citation2004; Hubbard and Miller Citation2007; Kornev and Taylor Citation2015). These investigations have also revealed how the intrinsic conformational plasticity of kinases, which is essential for their function as signaling switches, can be a molecular weakness that is exploited by opportunistic diseases like cancers. Importantly, analysis of conformational plasticity in protein kinases has improved our understanding of drug binding and mechanisms of drug resistance (Noble et al. Citation2004). For example, structural analysis of the cytoplasmic tyrosine kinase Abl, bound to various small molecules including the cancer drug imatinib (Gleevec), have revealed how kinase inhibitors can synergize with the actions of regulatory domains of the Src module (Schindler et al. Citation2000; Nagar et al. Citation2002; Johnson Citation2009; Saleh et al. Citation2017). This finding has guided strategies to design selective kinase inhibitors (Wang et al. Citation2014), underscoring the broader importance of mechanistic studies of kinase regulation.
In this review, we describe the structural and biochemical elaborations on the Src module that have led to the functionally diverse cytoplasmic tyrosine kinases found in metazoans. We first survey the many mechanisms of regulation of kinases containing the Src module, highlighting recent structural, computational, and biochemical studies. We then describe a growing body of work that aims to understand the evolution of regulation in the Src module and examine how these investigations have influenced our view of human cytoplasmic tyrosine kinases. Finally, we discuss the impact that these basic mechanistic studies have had on our understanding of diseases driven by the dysregulation of protein kinases and on the development of efficacious targeted therapies to treat those diseases.
Conserved structural and regulatory features in the Src module
The nexus of regulatory control in all kinases is the kinase domain, which is biased toward states of greater or lesser activity by the actions of regulatory mechanisms. The fully active conformation of tyrosine kinases was first captured by a structure of the isolated kinase domain of the insulin receptor, phosphorylated on its activation loop (PDB code 1IR3)2 (Hubbard Citation1997). A general schematic of the active form of a tyrosine kinase domain, highlighting key structural elements, is shown in . In the insulin receptor structure, and in analogous structures of other active tyrosine kinases, ATP is bound in a productive conformation. A Mg2+ ion that coordinates the phosphate groups of ATP is, in turn, coordinated by the sidechain of the aspartate residue (Asp 404 in c-Src)3 found in the conserved Asp-Phe-Gly (DFG) motif at the base of the activation loop (). This conformation of the DFG motif, where the aspartate sidechain is pointed toward the ATP-binding pocket and the phenylalanine sidechain is pointed away, is referred to as the “DFG-in” state. In the active conformation of tyrosine kinases, helix αC, a key regulatory element in the N-lobe of the kinase domain, is rotated inwards with respect to its conformation in the inactive states of the kinase. The sidechain of Glu 310, presented by the helix αC, forms an ion pair with the sidechain of Lys 295, which in turn positions the phosphate groups of ATP appropriately for catalysis.
Figure 2. Activation of the Src module. A. Crystal structure of an active conformation of the c-Src kinase domain, bearing the activating T341I mutation and bound to the nonhydrolyzable ATP analog ATPγS (PDB code 3DQW). B. Crystal structure of c-Src in its auto-inhibited Cdk/Src inactive conformation, highlighting only the kinase domain, bound to the nonhydrolyzable ATP analog AMP-PNP (PDB code 2SRC). In panels A and B, the conserved catalytic and regulatory spines are highlighted in transparent surface representation. C. Crystal structure of the Src module of the Src-family kinase Hck in the assembled auto-inhibited conformation, bound to the ATP-competitive inhibitor PP1 (PDB code 1QCF). D. Geometry of the activation loop and active site residues in activated c-Src, highlighting the role of activation loop phosphorylation (PDB code 3DQW). E. Geometry of the activation loop and active site residues in auto-inhibited c-Src (PDB code 2SRC). F. Geometry of the activation loop and active site residues in auto-inhibited c-Abl, highlighting a flip of the “DFG” motif aspartate relative to its orientation in c-Src (PDB code 1OPL). G. A model for activation loop trans-autophosphorylation of Src-family kinases in which the activation loop of one Lck molecule is presented into the active site of another Lck molecule (Shah et al. Citation2016) (see colour version of this figure at www.tandfonline.com/ibmg).
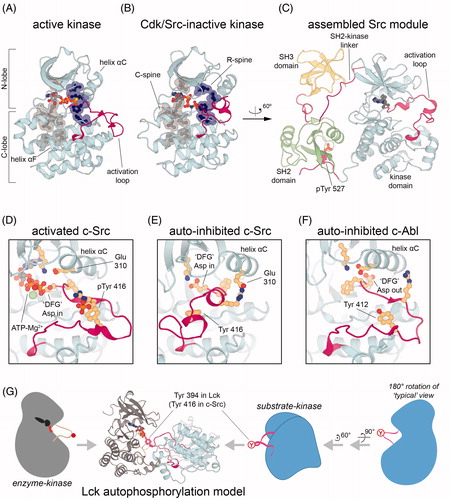
The active conformations of all kinases resemble each other, as demanded by the dictates of catalysis, but the inactive conformations can be structurally divergent because they are released from those constraints (Huse and Kuriyan Citation2002). As more structures of tyrosine kinases were determined, it became clear that inactive kinases, including those with the Src module, commonly adopt a small set of distinct conformations. In one such conformation, helix αC is rotated away from the N-lobe such that Glu 310 makes an alternative ion pair relative to the active state, and the activation loop is folded into the mouth of the kinase domain, blocking peptide substrate access (, PDB codes 2SRC and 1QCF). This conformation is referred to as the “Cdk/Src inactive conformation” because it was first observed in cyclin-dependent kinases and in Src-family kinases (De Bondt et al. Citation1993; Sicheri et al. Citation1997; Xu et al. Citation1997).
In another important set of inactive conformations, the DFG motif is flipped with respect to its orientation in active kinases, such that the aspartate sidechain (Asp 404) points away from the ATP-binding site (, PDB codes 1M52 and 1OPL) (Nagar et al. Citation2002). The “DFG-out” conformation is sometimes accompanied by a refolding of the activation loop so that the segment bearing the key regulatory tyrosine docks on the rest of the activation loop in a substrate-mimicking auto-inhibitory conformation (PDB codes 1FPU and 1IEP). Computational analysis suggests that the various inactive conformations are intermediates in a cycle connecting the active DFG-in conformation to the inactive DFG-out conformation (Levinson et al. Citation2006; Shan et al. Citation2009; Shukla et al. Citation2014). Particular interest in this conformational transition arises from the fact that kinase inhibitors often bind selectively to particular conformations in this cycle. The cancer drug, imatinib, for example, requires the DFG-out conformation for high affinity binding to the Abl kinase domain (Seeliger et al. Citation2007; Lin et al. Citation2013; Agafonov et al. Citation2014). All of the structural and regulatory mechanisms that we focus on below can be understood as acting on the kinase domain, to either stabilize one or the other of the common inactive kinase conformations, or to destabilize them and stabilize the active conformation instead.
The assembled, auto-inhibited state of Src-module-containing tyrosine kinases
The Src, Frk, Abl, Tec, and Csk families of cytoplasmic tyrosine kinases all contain the same core domain architecture, consisting of an SH3, SH2, and kinase domain, readily identified by comparison of their sequences (). The linkers connecting these globular domains, however, are different across the families. Despite the divergence in linker sequences, crystallographic studies of members from each family revealed that these proteins, except for the Csk-family kinases discussed later (Ogawa et al. Citation2002), can adopt roughly the same assembled and inactive structure () (Sicheri et al. Citation1997; Williams et al. Citation1997; Xu et al. Citation1997, Citation1999; Schindler et al. Citation1999; Nagar et al. Citation2003; Wang et al. Citation2015). This assembled state has two important features: (1) a compact configuration of the SH3-SH2-kinase module, in which the SH2-kinase linker provides an internal docking site for the SH3 domain, and (2) an inactive conformation of the activation loop in the kinase domain, with an unphosphorylated regulatory tyrosine. These structural features serve to suppress the activity of the kinase domain by stabilizing it in a catalytically incompetent conformation. The conservation of this auto-inhibited structure and regulatory mechanism across the Src-, Frk-, Abl-, and Tec kinases is the defining characteristic of the Src module.
The auto-inhibited conformation of the Src module was first revealed in structures of the Src-family kinases c-Src and Hck (, PDB codes 2SRC and 1QCF) (Sicheri et al. Citation1997; Xu et al. Citation1997). At the core of this structure is the SH2-kinase linker, which adopts a polyproline type II helix and is coordinated on one face by the canonical ligand-binding pocket of the SH3 domain. The other face of this linker is pressed up against the N-lobe of the kinase domain, holding the kinase in an inactive conformation. Indeed, displacement of the SH3 domain by an external ligand can activate Src-family kinases potently () (Moarefi et al. Citation1997). The assembled configuration is further stabilized by contacts between the SH2 domain and the C-lobe of the kinase domain, although the extent of this interaction varies across kinase families. Interaction of the SH2-kinase linker with the noncatalytic face of the kinase domain reduces kinase activity by allosterically perturbing the geometry of the active site (). This assembled structure is further stabilized by features that are unique to each kinase family, such as a phosphorylated tail that binds the SH2 domain, observed in the structures of Src-family kinases () (Sicheri et al. Citation1997; Williams et al. Citation1997; Xu et al. Citation1997). Despite general conservation of the auto-inhibitory conformation of Src-module-containing kinases, the structural details underlying allosteric control of the active site differ between each subfamily of kinases, as discussed in later sections.
The active conformation of Src-module-containing tyrosine kinases
Activation of the Src module is achieved by two principal mechanisms: (1) activation loop phosphorylation on Tyr 416, which stabilizes a catalytically competent active site geometry () and (2) displacement of the SH3 and SH2 domain to relieve constraints on helix αC (Moarefi et al. Citation1997). Since activation loop phosphorylation occurs in trans, this creates a molecular switch that is responsive to the enzymatic activity of another kinase molecule, thus allowing for signal relay or amplification. Activation by displacement of the SH2 and SH3 domains allows for switching of enzymatic activity in response to protein–protein interactions ().
Activation loop phosphorylation occurs on a conserved tyrosine found in all cytoplasmic tyrosine kinases (Tyr 416 in c-Src, ), except Csk and its paralog Ctk. The effect of phosphorylation on this residue is analogous to the effect of phosphorylating a corresponding serine or threonine residue in Ser/Thr kinases (De Bondt et al. Citation1993; Zheng et al. Citation1993; Jeffrey et al. Citation1995; Yamaguchi and Hendrickson Citation1996; Hubbard Citation1997), reviewed in (Huse and Kuriyan Citation2002; Nolen et al. Citation2004). In all cases, phosphorylation of a residue on the highly flexible activation loop results in the formation of one or more salt bridges. These salt bridges anchor the activation loop in a conformation that promotes substrate docking and aligns key residues involved in the coordination of ATP-Mg2+, most notably the Asp residue in the conserved DFG motif () (Ozkirimli and Post Citation2006; Banavali and Roux Citation2009; Shan et al. Citation2009). This activating modification often requires trans-autophosphorylation, where one active molecule of a tyrosine kinase phosphorylates the activation loop of another inactive molecule of the same kinase, thereby generating a positive feedback loop that may aid in signal amplification (Oliver et al. Citation2006; Pike et al. Citation2008; Wu et al. Citation2008; Zorba et al. Citation2014; Xu et al. Citation2015; Shah et al. Citation2016).
Src-family kinases robustly carry out trans-autophosphorylation of their activation loops in a manner that only requires their kinase domains (Shah et al. Citation2016). Some tyrosine kinase domains, such as that of c-Abl and the Tec family members Btk and Itk, have intrinsically inefficient autophosphorylation capabilities (Heyeck et al. Citation1997; Joseph et al. Citation2013; Lamontanara et al. Citation2014; Wang et al. Citation2015). In these cases, activation loop phosphorylation is enhanced by allosteric interdomain contacts, mediated by colocalization of two kinase molecules, or carried out through trans-phosphorylation by other kinases. These differences in trans-autophosphorylation capabilities suggest a specific structural mechanism for efficient activation loop phosphorylation. Recently, through analysis of crystallographic dimers of kinase domains in the Protein Data Bank, plausible structural models that explain the fast activation loop phosphorylation rates of Src-family kinases have been described (Xu et al. Citation2015; Shah et al. Citation2016) ().
Displacement of the SH2 and SH3 domains can be achieved by the competitive binding of ligands to these domains (). This mechanism not only facilitates activation of the kinase, but it also couples kinase activity to subcellular localization and/or colocalization with signaling partners, providing an extra layer of control over signal transduction pathways. The ability of the Src module to be activated in this way requires that the auto-inhibitory conformation is sufficiently stable to inactivate the kinase in the absence of stimulatory signals, but dynamic enough to provide access to ligands that competitively bind in trans. Consistent with this notion, increasing the flexibility of the SH3-SH2 linkers of both c-Src and c-Abl via glycine mutations has been shown to increase the activities of these kinases (Young et al. Citation2001; Hantschel et al. Citation2003).
Structural features that impact activation of the Src module
The transition from an inactive to active state is also controlled by the intrinsic dynamics and stability of the kinase domain. To understand how different protein kinases might vary in this respect while still relying on a conserved set of structural features, a bioinformatic technique known as “Local Spatial Patterns Alignment” was used to identify conserved patterns formed by residues of kinases in three-dimensional space, independent of protein sequence or backbone geometry (Kornev et al. Citation2006, Citation2008). This led to the discovery of two “spines,” denoted the R- and C-spines, that lie at the core of all protein kinase domains ().
The regulatory “R-spine” consists of four hydrophobic residues that connect the two lobes of the kinase domain and are anchored by a hydrogen bond and hydrophobic contacts to helix αF, a core structural element of the C-terminal lobe of the kinase domain (). Importantly, the R-spine contains the phenylalanine residue (Phe 405) in the highly conserved DFG motif at the base of the activation loop, a hydrophobic residue (Met 314) on helix αC, and a highly conserved histidine residue (His 384) on the catalytic loop (). The catalytic “C-spine” consists of a larger column of hydrophobic residues that span both lobes and are also anchored to the helix αF through hydrophobic interactions. Notably, the C-spine is completed by the adenine ring of ATP or ADP. The identities of the specific residues on or near the R- and C-spines are critical for the stability and activity of the kinase domain (Meharena et al. Citation2013; Meng et al. Citation2018). This is particularly apparent when considering that the R-spine is disassembled in the inactive states of most kinases () (Kornev et al. Citation2006). This has implications for our understanding of oncogenic and drug resistance mutations in many tyrosine kinases, which occasionally lie within or contact the spines (Azam et al. Citation2008).
Another important set of hydrophobic contacts in the Src module is illustrated by a recent study that examined the connection between nucleotide affinity and the transition from the inactive to active states of c-Src and the Tec-family kinase, Itk (von Rauszendorf et al. Citation2017). This study revealed that the auto-inhibited forms of c-Src and Itk bind ADP more than one order of magnitude tighter than ATP. Disassembly of the auto-inhibited state increases the affinity for ATP, making it comparable to that for ADP. This allosteric coupling between nucleotide binding and formation of the assembled Src module structure was shown to be controlled by the formation of a hydrophobic stack of residues from the N-lobe of the kinase domain (Trp 286 and Tyr 326) and the SH2-kinase linker (Leu 255 and Trp 260). Mutation of this hydrophobic stack, which is distinct from the R- and C-spines, facilitates disassembly of the auto-inhibited conformation and allows for inward rotation of helix αC, similar to what happens with SH3 domain displacement, and this disassembly increases ATP binding affinity. This allosteric affinity switch was shown to regulate nucleotide exchange in both c-Src and Itk, thereby controlling autophosphorylation and activation (von Rauszendorf et al. Citation2017). This critical hydrophobic stack is conserved across the Src-, Abl-, and Tec-family kinases.
Individual kinases containing the Src module also have unique structural features that differentiate their inactive states or provide distinct means of regulation in response to incoming signals. Some of these divergent features are subtle. For example, sequence heterogeneity across SH2-kinase linkers in Src-family kinases has been shown to correlate with the degree of allosteric coupling of the SH3 and SH2 domains to the ATP binding site (Register et al. Citation2014). Across the Src, Abl, and Tec families, kinase function and regulation has diversified in more dramatic ways. Each of these families bears a unique set of regulatory tails, ligand-binding domains, or post-translational modifications, in addition to divergent conformational landscapes of their inactive states. These differences, which enable the diverse signaling capabilities encoded within each protein family, are discussed in the next section.
Unique features of each Src-module-containing family
Src-family kinases
The eight Src-family kinases in humans (c-Src, Yes, Fyn, Fgr, Hck, Lck, Lyn, and Blk)4 are defined by three regulatory features that distinguish them from other kinases with Src modules: (1) a C-terminal tail bearing a tyrosine residue (Tyr 527 in c-Src) that can be phosphorylated to negatively regulate kinase activity, (2) an N-terminal myristoylation site, along with a palmitoylation site in most family members, that impacts subcellular localization, and (3) a unique region that varies in sequence and function between each family member (). Although not discussed here, two Src-related kinases, Frk and Brk, contain Src modules and are negatively regulated by tail phosphorylation, but they lack the canonical N-terminal lipidation motifs (Qiu and Miller Citation2002).
Regulation of Src-family kinases by tail phosphorylation
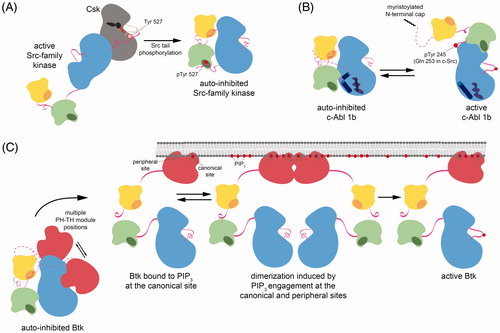
The principle mode of regulation in Src-family kinases is the interplay between two tyrosine phosphorylation sites: the conserved positive regulatory phosphorylation site in the activation loop and a negative regulatory site on a short tail at the C-terminus of the kinase domain, which is unique to Src-family kinases. High-resolution structures of the Src-family kinases c-Src and Hck, with their tails phosphorylated on Tyr 527 but activation loops unphosphorylated on Tyr 416, have revealed the mechanism of negative regulation by tail phosphorylation (PDB codes 2SRC and 1QCF). In these structures, the Src modules of both kinases adopt the assembled state described above, and this conformation is stabilized by the docking of the tyrosine-phosphorylated tail on the SH2 domain () (Sicheri et al. Citation1997; Williams et al. Citation1997; Xu et al. Citation1997, Citation1999; Schindler et al. Citation1999).
The tail sequences of Src-family kinases are not optimized for SH2 binding. For example, mutation of the Hck tail sequence from pYQQP to pYEEIP, an optimal motif for binding to the Hck SH2 domain (Songyang et al. Citation1993), results in a kinase that cannot be readily activated by an SH2 ligand in trans (Porter et al. Citation2000; Schindler et al. Citation1999). Thus, the wild-type tail sequences provide a sufficiently strong interaction to stabilize the inactive state, but not so strong that auto-inhibition cannot be relieved when a competitive SH2- or SH3-binding ligand is present. Although this SH2-tail interaction is a defining feature of Src-family kinases, it is likely a more recent innovation than the interaction between the SH2-kinase linker and the SH3 domain, as that interaction is conserved in all Src modules (except Csk).
The C-terminal tails of Src-family kinases are phosphorylated by the specialized C-terminal Src kinase, Csk, and its paralog Ctk (Okada et al. Citation1991; Okada and Nakagawa Citation1989). Csk is a poor kinase against peptide substrates. Csk is highly selective for Src-family tails, and efficient phosphorylation by Csk requires that Src-family tails be presented for phosphorylation by the substrate-kinase domain (Sondhi et al. Citation1998; Lee et al. Citation2003, Citation2006). In contrast to other tyrosine kinase domains, which phosphorylate many proteins promiscuously when overexpressed, over-expression Csk does not dramatically change overall phosphotyrosine levels in cells (Bergman et al. Citation1995).
Crystal structures of Csk bound to the c-Src kinase domain revealed the mechanism of tail phosphorylation and the strict specificity of Csk for the tails of Src-family kinases (PDB code 3D7T) (Levinson et al. Citation2008). In the co-crystal structures, the Csk and c-Src kinase domains interact through specific electrostatic interactions between their C-lobes, precisely positioning the c-Src tail in proximity to the Csk active site, as anticipated partly by mutagenesis studies () (Lee et al. Citation2006). The activation loop of Csk is unusually short and cannot efficiently dock peptide substrates as other protein kinases do, thereby explaining the inefficient peptide phosphorylation by this kinase (Lin et al. Citation2003; Levinson et al. Citation2008). Rather, specific recognition of the C-lobe of the c-Src kinase domain provides a high degree of substrate specificity and allows Csk to overcome its deficiencies as a tyrosine kinase that cannot anchor polypeptides on its activation loop.
Figure 3. Diverse mechanisms of regulation in Src-module-containing kinases. A. Inhibitory tail phosphorylation of Src-family kinases by Csk. Phosphorylation of the conserved C-terminal tail tyrosine in Src-family kinases occurs through specific recognition of Src-family kinase domains by Csk. This docking of tertiary structures positions the tail into the Csk active site for phosphorylation. B. A myristoyl/phosphotyrosine switch in c-Abl. Isoform 1b of c-Abl is auto-inhibited by docking of an N-terminal myristoyl moiety into the C-lobe of the kinase domain. Disruption of this interaction and phosphorylation of a tyrosine residue in the SH2-kinase linker activates the kinase. C. Regulation of Tec-family kinases by the PH-TH module. In Btk, the PH-TH module stabilizes the auto-inhibited configuration of the Src module by binding to the N-lobe of the kinase domain, however, the precise geometry of this binding is not yet known. Engagement of the PH-TH module by binding to soluble inositol phosphates or PIP lipids results in release of auto-inhibitory contacts and facilitates full activation by trans-autophosphorylation of the activation loop (see colour version of this figure at www.tandfonline.com/ibmg).
The mechanisms underlying activation by tail dephosphorylation are less well understood. Two recent studies of Lck, the primary Src-family kinase in T cells, and the phosphatase CD45, which is implicated in Lck tail dephosphorylation, have shed some light on this process. In one investigation, Lck activity was measured in the presence and absence of CD45, on liposomes, thereby mimicking the native membrane-tethered state of these proteins. These measurements demonstrated that Lck activity is enhanced 10-fold by CD45-mediated dephosphorylation of the Lck tail, and that tail dephosphorylation by CD45 is required for efficient trans-autophosphorylation of the activation loop (Hui and Vale Citation2014). The second study showed that dephosphorylation of the Lck tail by CD45 is facilitated by interactions between CD45 and the Lck SH2 domain (Courtney et al. Citation2017). Phosphorylation of a conserved tyrosine residue on the SH2 domain surface (Tyr 192 and Tyr 213 in Lck and c-Src, respectively) decreases the efficiency of dephosphorylation of the Lck tail by CD45 (Courtney et al. Citation2017). The phosphorylation of Tyr 192 has also been implicated in weakening the binding affinity of the Lck SH2 domain for phosphotyrosine-containing peptides (Couture et al. Citation1996). Phosphorylation of Tyr 192 on Lck is part of a negative feedback loop controlling Lck activity via CD45 (Goodfellow et al. Citation2015; Courtney et al. Citation2017), but the structural details of the CD45-Lck complex are still lacking.
N-terminal regulatory domains in Src-family kinases
The N-terminal segments of all Src-family kinases contain a myristoylation site, and all but c-Src also contain a palmitoylation site. These lipids, along with positively charged residues in the N-terminal segment, anchor the kinases to the plasma membrane, and control subcellular trafficking (Resh Citation1994). These lipidation sites are followed by a unique region in all Src-family kinases. High-resolution structural details of most Src-family unique regions are lacking, aside from that of Lck, which specifically binds to the CD4 and CD8 immune co-receptors through the formation of a zinc clasp (PDB codes 1Q68 and 1Q69) (Kim et al. Citation2003). Recently, a co-evolutionary analysis of Src-family unique domains, coupled with NMR data, revealed that although the sequences of individual unique regions vary, allowing for family-member-specific interactions, they all contain conserved aromatic motifs that drive the collapse of this partially disordered domain (Arbesú et al. Citation2017). This collapsed unique region structure can interact with SH3 domain of the Src-module and may regulate its activity.
The conformational and energetic landscapes of Src-family kinases
The Src-family kinases are regulated by phosphorylation on two sites (Tyr 416 and Tyr 527) with opposing effects on activity. Phosphorylation of Tyr 416 in the activation loop increases activity, whereas phosphorylation of Tyr 527 in the tail decreases activity. The basis for these opposing effects has been studied by molecular dynamics simulations, and this work has been reviewed in detail recently (Meng et al. Citation2017). Briefly, one investigation revealed an optimal pathway for the conversion of the isolated c-Src kinase domain from its inactive to active states, which entails first the opening of the activation loop, followed by rotation of helix αC (Gan et al. Citation2009). Further simulations, starting from several intermediate states along this pathway and facilitated by massively distributed computing, allowed for the construction of a free energy landscape for this activation transition (Shukla et al. Citation2014). These analyses and additional studies suggest that the unphosphorylated c-Src kinase domain can transiently adopt the active state, and that phosphorylation of the activation loop effectively locks the kinase in the active state (Meng and Roux Citation2014).
Similar analyses have also been carried out on the Src module of c-Src, both in an assembled inactive state with a phosphorylated C-terminal tail, and in a tail-unphosphorylated open state (Meng et al. Citation2016). Along with simulations of the isolated kinase domain of c-Src, these simulations show that the interdomain contacts in the assembled and inactive state of the Src module impose a significant thermodynamic penalty for the kinase domain to access its active conformation. A separate study, which also conducted simulations of the Src module of c-Src, led to the discovery of an allosteric network of residues spanning 40 Å across the kinase domain (Foda et al. Citation2015). This network includes the substrate binding site, contacts the regulatory SH2-kinase linker, and is perturbed dynamically by protonation of the DFG aspartate residue, an event that is thought to be coupled to the catalytic cycle of the kinase (Shan et al. Citation2009). Notably, the observed allosteric network mediates negative co-operativity between ATP binding and substrate docking, and may explain why some mutations that confer resistance to ATP-competitive inhibitors weaken ATP binding but enhance kinase activity (Foda et al. Citation2015). There is a growing body of computational work on Src-module-containing kinases aimed at revealing the thermodynamic and structural details underlying kinase activation (Meng et al. Citation2017). These investigations not only provide basic mechanistic insights into individual tyrosine kinases but they also provide a framework to understand how closely related kinases may differ, as in the case of Src- and Abl-family kinases.
Abl-family kinases
The Abl family of cytoplasmic tyrosine kinases consists of two members, Abl1, which is more commonly referred to as c-Abl, and Abl2, which is also known as Arg (Wang Citation2014). The N-terminal segments of Abl-family kinases not only vary between both family members, but also between isoforms of each individual family member, as a result of alternative promoters and variable mRNA splicing patterns. Immediately after this variable N-terminal “cap” is the canonical Src module, followed by a long sequence containing DNA- and actin-binding elements, as well as nuclear localization signals (). Here, we will focus our discussion of Abl-family kinases on the structural features of the N-terminal cap and Src module. The structure of the actin-binding element has been determined separately (PDB code 1ZZP) (Hantschel et al. Citation2005).
Regulation of c-Abl isoform 1b by a myristoyl/phosphotyrosine switch
Unlike Src-family kinases, which are inhibited by phosphorylation of the C-terminal tail, the most ubiquitous isoform of c-Abl, isoform 1b, is regulated by a unique myristoyl/phosphotyrosine switch (Hantschel et al. Citation2003; Nagar et al. Citation2003). The N-terminal cap region, which contains a canonical myristoylation site, is required for auto-inhibition but has little effect on subcellular localization, unlike the equivalent modification in Src-family kinases (Hantschel et al. Citation2003). The mechanism by which the N-terminal myristoyl group suppresses c-Abl activity was discovered through crystallographic analysis of the inactive kinase (PDB code 1OPL) (Nagar et al. Citation2003). These structures revealed that the N-terminal myristoyl group directly binds the C-lobe of the kinase domain in a hydrophobic pocket that is unique to c-Abl and does not appear to be present in Src-family kinases (). Binding of the myristoyl group causes a sharp bend in a C-terminal α helix in the kinase domain, allowing the SH2 domain to dock on the C-lobe of the kinase domain. The resulting binding surface of the SH2-kinase interaction is approximately twice as large as that of the analogous surfaces in Src-family kinases, and this interdomain interaction partially occludes the SH2-peptide binding cleft. Notably, this mechanism of Abl auto-inhibition by lipid binding to the kinase domain has been exploited for the development of a new class of inhibitors that modulate kinase activity allosterically by binding to the myristoyl-binding pocket, discussed later (Adrián et al. Citation2006; Wylie et al. Citation2017).
In addition to the myristoyl-binding pocket and increased SH2-kinase interaction, the assembled inactive state of the Src module in c-Abl also differs subtly from that of Src-family kinases at the SH3 interface with the SH2-kinase linker. In c-Abl, the SH3 recognition sequence in this linker contains a PxxY motif, as opposed to PxxP in Hck or PxxQ in c-Src. This unique tyrosine residue, Tyr 245 in c-Abl isoform 1b (Gln 253 in c-Src), is critical for auto-inhibition. In the auto-inhibited state, Tyr 245 is sequestered within a hydrophobic pocket on the kinase domain and stabilizes its inactive state (Nagar et al. Citation2003). Phosphorylation of this residue leads to activation (Brasher and Van Etten Citation2000), as does activation loop phosphorylation and destabilization of the myristoyl-kinase domain interaction by displacing the SH2 domain with phosphotyrosine-containing ligands (Hantschel et al. Citation2003).
Additional residues in the N-terminal cap of c-Abl, beyond the myristoylation site, are also important for the auto-inhibition of kinase activity. Crystal structures and small angle X-ray scattering data showed that a phosphorylated serine residue, Ser 69, in the cap region interacts with and helps rigidify the SH3-SH2 clamp in the auto-inhibited structure (Nagar et al. Citation2006). Biochemical studies on a construct of c-Abl lacking the first 45 residues, consistent with the naturally occurring isoform 1a, show that peptides derived from the natural c-Abl inhibitor Abi1 can allosterically inhibit c-Abl kinase activity by engaging its SH3 and SH2 domains (Xiong et al. Citation2008). This observation points to alternative mechanisms of c-Abl regulation in the absence of myristoylation.
Alternative conformations of the Src module in c-Abl
A recent NMR analysis of segments of c-Abl bearing all of the N-terminal regulatory domains indicates that these domains can adopt at least two distinct configurations in solution (Saleh et al. Citation2017). One configuration is consistent with the assembled state of the Src module, where the SH3 domain binds to the polyproline type II helix on the SH2-kinase linker. In the second stable structure, a PxxP motif in the N-terminal cap displaces the SH2-kinase linker and binds to the canonical ligand-binding pocket on the SH3 domain, indicating a novel activating role for the N-terminal cap in c-Abl.
An extended structure has also been observed in crystals of the c-Abl Src module and the isolated SH2-kinase construct, where the SH2 domain interacts directly with the N-lobe of the kinase domain (, PDB codes 1OPL, chain B, and 4XEY, chain B) (Nagar et al. Citation2003; Lorenz et al. Citation2015). This structure, which likely represents an active form of the kinase, is similar in general terms to the active form of the cytoplasmic tyrosine kinase Fes (Nagar et al. Citation2006; Filippakopoulos et al. Citation2008). For both c-Abl and Fes, this interdomain interaction is enhanced by engagement of the SH2 domain with a phosphotyrosine-containing ligand peptide, which stabilizes loops on the SH2 domain required for kinase domain binding. In the presence of a phosphopeptide ligand, SH2 domain docking on the kinase domain locks the helix αC in an active state. Interestingly, the Fes crystal structure where this SH2-kinase interaction is observed, the kinase has a well-ordered activation loop in the active conformation, despite being unphosphorylated (PDB code 3BKB), suggesting that the SH2-kinase interaction is a major determinant of kinase activity.
As for Fes and c-Abl, a structure of full length Csk revealed that the SH2 domain also docks onto the N-lobe of the kinase domain (PDB code 1K9A) (Ogawa et al. Citation2002). In this case, binding of an effector protein, Csk-binding protein, to the SH2 domain promotes the formation of this interface (Ogawa et al. Citation2002; Wong et al. Citation2005). The precise nature of the SH2 and kinase domain interactions are not conserved between Fes, c-Abl, and Csk, and it is not clear at present if the Src-family kinases are activated by a similar interaction between the SH2 domain and the kinase domain. A crystal structure of the Src module of c-Src with an unphosphorylated tail showed that the SH3 domain and the SH2-kinase linker can dock on the N-lobe of the kinase domain and potentially stabilize it in an active conformation (PDB code 1Y57), suggesting that the regulation of Src-family kinases may involve docking of the noncatalytic domains in alternative ways relative to c-Abl (Cowan-Jacob et al. Citation2005). Notably, biochemical measurements on the Tec-family kinase Btk, comparing an SH2-kinase construct to the isolated kinase domain, suggest that interactions between the SH2 domain and the kinase domain are also important for activation of Btk (Wang et al. Citation2015). These observations indicate that interactions between noncatalytic domains and the N-lobe of the kinase domain that promote activity might be an ancient feature of cytoplasmic tyrosine kinases.
Tec-family kinases
Building on the core Src module, the Tec-family kinases have evolved to respond to biochemical cues in a distinctive way, through the incorporation of a lipid-binding module. The Tec family consists of four kinases, Tec, Btk, Itk, and Bmx, all of which have an N-terminal PH-TH module, containing a lipid-binding pleckstrin homology (PH) domain fused to a Tec homology (TH) domain () (Mano Citation1999). A closely related fifth kinase, Txk (also known as Rlk), is missing the PH-TH module, and instead contains a cysteine rich sequence at its N-terminus that is palmitoylated and allows for constitutive membrane localization in vivo (Debnath et al. Citation1999). Txk appears to have emerged as a result of gene duplication of the Tec gene (Mano Citation1999).
Auto-inhibition of Tec-family kinases
There is, at present, no structure available for a full-length Tec-family kinase in an auto-inhibited state. Considerable insight regarding this state has been obtained, however, by integrating information from crystal structures of two constructs of Btk. One construct consists of the Src module of Btk, and the crystal structure shows that it adopts a conformation very similar to that of auto-inhibited Src-family kinases and c-Abl (PDB code 4XI2) (Wang et al. Citation2015). Consistent with the importance of this assembled state, mutations in the SH2-kinase linker in Btk are associated with an immunodeficiency known as X-linked agammaglobulinemia (XLA), discussed later.
The other construct is one in which the SH3 and SH2 domains have been deleted, leaving the PH-TH module connected to the kinase domain by a shortened version of the linker normally found between the PH-TH module and the SH3 domain. This engineered construct was based on the finding that a Btk construct in which the SH3 and SH2 domains are deleted has lower kinase activity than the isolated kinase domain, indicating that the PH-TH module is capable of inhibiting the kinase domain. The linker connecting the PH-TH module to the kinase domain, bypassing the SH3 and SH2 domains, was chosen to have the minimal length that is consistent with suppression of activity. The structure of this PH-TH-kinase construct has the PH-TH module docked on helix αC of the kinase domain, which is in the Cdk/Src inactive conformation (PDB code 4Y93) (Wang et al. Citation2015). The PH-TH module forms a dimer in the crystal. Mutational analysis showed that interactions made by the PH-TH module and those made by the SH2 and SH3 domains are both consistent with suppression of kinase activity. The two structures are compatible with each other, allowing an integrated model to be constructed, in which the SH2 and SH3 domains are docked on the distal face of the kinase domain, and the PH-TH module is docked above the kinase active site ( and , left panel) (Wang et al. Citation2015).
The nature of the Btk auto-inhibited conformation has also been probed by NMR (Joseph et al. Citation2017). This analysis confirmed the docking of the SH2 and SH3 domains as in the canonical structures of other auto-inhibited Src modules. This analysis also showed that although the Tec-family kinases lack the C-terminal tail observed in Src-family kinases, a conserved acidic latch at the C-terminus appears to play a similar role in the absence of a phosphorylation site. The acidic residue Asp 656 in Btk (Thr 521 in c-Src) forms a salt bridge with Arg 307 of the SH2 domain (Arg 175 in c-Src), a residue that engages the C-terminal tail phosphotyrosine in auto-inhibited Src-family kinases (Joseph et al. Citation2017).
The NMR analysis indicates that, in solution, the PH-TH module can dock in an alternative conformation that directly blocks the kinase active site () (Joseph et al. Citation2017). This configuration was first observed by NMR for Itk (Devkota et al. Citation2017), and it has a general resemblance to the auto-inhibited structure of the Ser/Thr kinase Akt, in which the PH domain of Akt is docked onto the catalytic face of the kinase domain (PDB code 3O96) (Wu et al. Citation2010). It is possible that there are two states of auto-inhibited Btk. A monomeric form, captured by NMR, and a dimeric form, which might be promoted at the membrane before activation. A detailed description of our current understanding of how Btk is activated at the membrane is provided below.
Regulation of Tec-family kinases by the PH-TH module
The defining feature of the Tec-family kinases, the PH-TH module, not only plays a role in auto-inhibition, but also facilitates the recruitment of these kinases to the plasma membrane (). PH domains recognize phosphatidylinositol lipids, and in the case of the Tec family members, the PH domain selectively binds phosphatidylinositol-(3,4,5)-trisphosphate (PIP3) (Salim et al. Citation1996). Phosphoinositide-3 kinase (PI3K), activated downstream of many transmembrane receptors, converts PIP2 into PIP3, thus, resulting in PI3K-activity-dependent recruitment of Tec-family kinases to the membrane (Mano Citation1999; Grasis and Tsoukas Citation2011).
The PH domain is present in many other proteins outside the Tec-family kinases (Lemmon and Ferguson Citation2000). It consists of a 7-stranded β sandwich structure with an α helix laying between the antiparallel β sheets. The phosphatidylinositol is recognized between the β1 and β2 loops, which contain lysine and arginine residues to coordinate phosphate groups on the ligand, while specificity is determined by other sidechains in these loops that determine the shape and charge of the binding pocket (Salim et al. Citation1996; Lemmon Citation2008). In Tec-family kinases, the TH domain forms a continuous fold with the PH domain, packs against the C-terminal half of the PH β sheet, and coordinates a zinc ion through three conserved cysteine residues and a histidine residue (Hyvönen and Saraste Citation1997).
The original crystal structure of the Btk PH-TH module by Matti Saraste and colleagues pointed out a specific homodimer of the module (PDB code 1BTK) (Hyvönen and Saraste Citation1997). Based on mutations found in XLA, it was hypothesized that this dimerization may be important for activation of Btk in vivo (Hyvönen and Saraste Citation1997). A secondary, peripheral inositol phosphate binding site in the Btk PH domain has been identified recently, and shown to play a role in dimerization and subsequent trans-autophosphorylation of the kinase domain activation loop (, top right panel, PDB code 4Y94) (Wang et al. Citation2015). Subsequent studies have shown that the Btk PH-TH module is capable of dimerizing on membranes, and that this dimerization depends on PIP3 binding at both the canonical and peripheral sites () (Chung et al. Citation2018). This work also suggests that dimerization allows for a switch-like response to PIP3 concentrations in the membrane. In the absence of PIP3, the PH-TH module plays an auto-inhibitory role by coordinating the kinase domain in solution, as discussed above. With sufficient PIP3 concentration in the plasma membrane, Btk is recruited to the membrane, where its auto-inhibition is partly relieved and it can be fully activated by PH-TH-mediated dimerization and trans-autophosphorylation of the activation loop ().
Additional regulatory features in the Src module of Tec-family kinases
All five domains of the Tec-family kinases (PH-TH-SH3-SH2-kinase) participate in auto-inhibitory interactions to reduce kinase activity in the cytosol, as described above () (Wang et al. Citation2015; Devkota et al. Citation2017; Joseph et al. Citation2017; Andreotti et al. Citation2018). Unlike Src-family kinases, however, the kinase domains of Btk and Itk alone have very little intrinsic activity. In line with this observation, it was found that Ile 432 in Btk, a residue that is conserved across the Tec family but divergent in the Src family (Leu 297 in c-Src), buttresses key regulatory elements in the kinase domain and seems to hamper the transition of the kinase to the active conformation (Boyken et al. Citation2014). Mutation of this isoleucine residue in Btk to the corresponding leucine residue in Src-family kinases increased the intrinsic activity of the Btk kinase domain. This isoleucine and the regulatory spine appear to be conserved across the Tec family, however, distinct differences in the activation loops are apparent within the Tec family. Biochemical studies comparing Btk and Itk suggest that Itk exhibits a lower intrinsic kinase activity that can be rescued to the level of intrinsic Btk activity by exchanging key residues in the activation loop with those of Btk (Joseph et al. Citation2013).
The SH3 domain of Btk also has regulatory features that distinguish it from other kinases with Src modules. The proline-rich region adjacent to the PH-TH module is capable of binding the SH3 domain in solution (Joseph et al. Citation2017). Mutational studies suggest that this interaction may help to shift Btk toward its active form, although there is no direct structural information about this interaction. In addition, Tyr 223 in the SH3 domain of Btk (Tyr 90 in c-Src) is autophosphorylated after activation loop phosphorylation at Tyr 551 (Tyr 416 in c-Src) (Park et al. Citation1996). Currently, the role of this phosphorylation site, if any, is not understood. The analogous site in Itk (Tyr 180) is also autophosphorylated and alters the ligand-binding properties of the Itk SH3 domain (Joseph et al. Citation2007). In the assembled state of the Src module, this tyrosine residue in the SH3 domain packs against the SH2-kinase linker, suggesting that its phosphorylation may impact the stability of the auto-inhibited state, although this effect has not been established experimentally.
Substrate specificity as a mechanism of signaling control
Kinase specificity guided by SH2 and SH3 domains
A consistent theme in the regulatory mechanisms of all cytoplasmic tyrosine kinases is that their noncatalytic domains play a direct role in stabilizing the inactive or active states of the kinase domain. Typically, these auto-inhibitory or activating interactions are modulated by post-translational modifications or by ligand binding to the noncatalytic domains. This coupling of noncatalytic domain function and kinase activation provides a way for these molecular switches to be responsive to diverse input signals, and it also has implications for the biochemical outputs of these signaling enzymes. Specifically, in the case of kinases bearing the Src module, the binding preferences of the SH2 and SH3 domains that are attached to the tyrosine kinase domain dictate when and where that kinase is activated. In this way, these ligand-binding domains play a critical role in defining the substrate specificities of tyrosine kinases (Pawson and Nash Citation2000).
SH2 domains are the most common noncatalytic domains found in cytoplasmic tyrosine kinases. Pioneering work using degenerate peptide libraries showed that individual SH2 domains recognize specific sequences containing phosphotyrosine (Songyang et al. Citation1993, Citation1994), and many subsequent structural studies have revealed the molecular basis for these sequence preferences (Kuriyan and Cowburn Citation1997; Waksman et al. Citation2004). SH3 domains have their own sequence specificities (Lim et al. Citation1994; Weng et al. Citation1995), and SH3-polyproline interactions have been implicated in guiding Src-family kinases to their substrates (Pellicena and Miller Citation2001). Likewise, Tec-family kinases are recruited to the membrane via the specific interaction of their PH-TH module with PIP3, where they encounter many of their substrates (Takesono et al. Citation2002).
The specific recognition of phosphoproteins by SH2 domains not only facilitates the activation of tyrosine kinases, but also enhances their ability to phosphorylate other spatially proximal tyrosines, as seen for Src-family kinases phosphorylating substrates with multiple tyrosine residues () (Mayer et al. Citation1995; Pellicena et al. Citation1998). Similarly, the engagement of the Abl and Fes SH2 domains can facilitate processive phosphorylation of substrates containing more than one tyrosine residue (Filippakopoulos et al. Citation2008), Syk-family kinases are recruited to transmembrane antigen receptors upon ligand binding to their tandem-SH2 domains () and phosphorylate substrates at the membrane (Katz et al. Citation2017), and Fak autophosphorylation creates a docking site for c-Src, thereby localizing c-Src to substrates at focal adhesions (Thomas et al. Citation1998). This sequence-specific enzyme-substrate colocalization mechanism is thought to be the dominant factor in defining the substrate specificities of cytoplasmic tyrosine kinases (Miller Citation2003; Ubersax and Ferrell Citation2007).
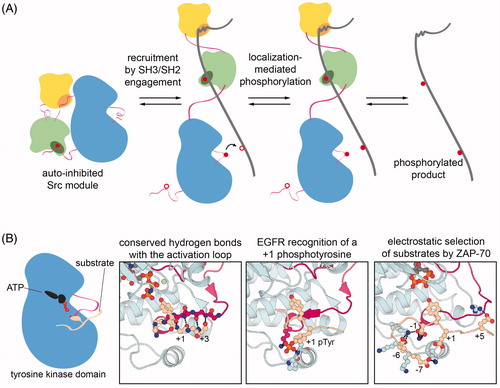
Figure 4. Mechanisms of substrate selection in tyrosine kinases. A. Localization-mediated specificity. Src-module-containing tyrosine kinases typically colocalize with their substrates through interactions mediated by their noncatalytic domains, and this localization is often coupled to activation of the kinase. Once localized, the kinase will phosphorylate proximal tyrosine residues. B. Structural features of tyrosine kinase-substrate interactions that impact sequence specificity. far-left: Schematic depiction of a substrate docked in the active site of a tyrosine kinase. middle-left: Structure of the insulin receptor kinase bound to a substrate, highlighting typical backbone hydrogen bonds between the kinase activation loop and residues downstream of the target tyrosine (PDB code 1IR3). middle-right: Preferred recognition of a substrate peptide bearing a phosphotyrosine residue at the +1 position by the EGFR kinase domain (PDB code 5CZH). far-right: Model based on molecular dynamics simulations of the extensive electrostatic interactions between ZAP-70 and a preferred substrate derived from the protein LAT (Shah et al. Citation2016) (see colour version of this figure at www.tandfonline.com/ibmg).
Intrinsic sequence preferences of the tyrosine kinase domain
The important role of SH2- and SH3-mediated localization in dictating tyrosine kinase substrate specificity raises the question of whether or not intrinsic substrate sequence preferences encoded within the catalytic domains of tyrosine kinases also play a role in determining biological function. Several studies using large peptide library screens and in vitro peptide phosphorylation measurements have clearly demonstrated that individual tyrosine kinase domains have distinct sequence preferences (Songyang et al. Citation1995; Schmitz et al. Citation1996; Till et al. Citation1999; Deng et al. Citation2014; Shah et al. Citation2016, Citation2018). The oncogenic forms of some tyrosine kinases, bearing point mutations within their kinase domains, have been shown to have altered substrate specificity relative to their wild-type counterparts, suggesting a connection between kinase domain specificity and biological activity (Songyang et al. Citation1995; Piao et al. Citation1996; Bocciardi et al. Citation1997; Bardelli et al. Citation1998; Griswold et al. Citation2006; Skaggs et al. Citation2006). Contrary to these observations, however, the sequences of true in vivo substrates of tyrosine kinases frequently do not fit the optimal sequence motifs determined using biochemical techniques (Miller Citation2003). Additionally, co-crystal structures of tyrosine kinases bound to substrate peptides reveal nonspecific hydrophobic interactions between the few residues downstream of the substrate tyrosine with a hydrophobic pocket on the kinase C-lobe (Hubbard Citation1997; Bose et al. Citation2006). The dominant feature of these interactions is the antiparallel β sheet formed by the substrate backbone and the activation loop (, left panels, PDB code 1IR3), which is sequence-independent and consistent with a nominal role for substrate sequence specificity encoded within the kinase domain.
Despite these contrasting observations, recent studies assessing the interplay between Src-family kinases and other tyrosine kinases suggest that the sequence specificities of tyrosine kinase domains can play a role in shaping the architecture of signaling pathways. In one study, using oriented peptide libraries, the receptor tyrosine kinase EGFR was found to strongly prefer phosphorylating substrates in which the target tyrosine was immediately followed by a phosphotyrosine (, middle-right panel, PDB code 5CZH) (Begley et al. Citation2015). A Y-Y motif is found in a number of relevant signaling molecules, most notably the adaptor protein Shc1. The authors found that c-Src preferentially phosphorylates the second tyrosine in the Y-Y motif in Shc1 and primes Shc1 for phosphorylation on the first tyrosine by EGFR. Notably, this doubly phosphorylated protein, which is the product of the cooperation between two kinases with distinct sequence specificities, has enhanced affinity for downstream signaling proteins, demonstrating how kinase specificities might be exploited to integrate biological signals.
In a second recent study, the interplay between the Src-family kinase, Lck, and Syk-family kinase, ZAP-70, was investigated (Shah et al. Citation2016). These kinases act sequentially to activate T cells upon antigen receptor stimulation, and they appear to have orthogonal substrate preferences in cells, despite the close spatial proximity of all proteins involved. Using an approach that coupled bacterial surface-display of peptide libraries with cell sorting and deep sequencing, the ZAP-70 kinase domain was found to strongly prefer substrates with an aspartic acid immediately preceding the target tyrosine residue. Preferred substrates also lacked positively charged residues and were enriched in negatively charged residues (, far-right panel). The Lck kinase domain, by contrast, prefers large aliphatic residues at the –1 position and positively charged residues distributed around the tyrosine. Notably, these are the defining features of ZAP-70 and Lck substrates in T cells, suggesting that the sequence preferences of these kinase domains play a critical role in defining the topology of this signaling pathway.
These two examples do not undermine the important role that SH2 and SH3 domains play in guiding tyrosine kinase specificity. Rather, they demonstrate that tyrosine kinase domains are fine-tuned enzymatic modules in their own right. In accordance with this idea, a recent comparison of the Src-family kinases c-Src and Lck revealed a subtle distinction in the sequence preferences of their catalytic domains, consistent with the specialization of Lck to function in concert with ZAP-70 in T cell signaling (Shah et al. Citation2018).
It is likely that when the various multidomain architectures of tyrosine kinases first evolved through the fusion of separate functional domains, these ancient proteins did not have the intricate mechanisms of interdomain allostery that we see in extant cytoplasmic tyrosine kinases. Before these regulatory mechanisms evolved, perhaps the intrinsic specificities of tyrosine kinase domains played a dominant role in controlling signal transduction. In the following section, we discuss recent efforts to explore how allosteric regulation may have emerged in various Src-module-containing kinases over the course of evolution.
Evolution of regulation in the Src module
Tyrosine kinase signaling networks, mediated by eukaryotic tyrosine kinases, SH2 and PTB domains, and protein tyrosine phosphatases, were once thought to be an exclusive feature of metazoans and thus an essential innovation that allowed for the emergence of one form of multicellularity (Pincus et al. Citation2008; Lim and Pawson Citation2010; Richter and King Citation2013). Although tyrosine phosphatases and SH2 domains had been observed in the genomes of unicellular slime molds and fungi, canonical tyrosine kinases had not been noted outside of the animal lineage (King and Carroll Citation2001). Over the past two decades, however, tyrosine kinases have been found in a number of organisms that diverged prior to the origin of the metazoan lineage and the emergence of multicellularity () (Manning et al. Citation2002a; Bradham et al. Citation2006; Srivastava et al. Citation2010; Suga et al. Citation2014). Initially, tyrosine kinases were thought to have emerged just before the divergence between metazoans and their closest nonmetazoan relatives, choanoflagellates (King and Carroll Citation2001; King et al. Citation2003). They have since been found in even more distantly related unicellular organisms, such as filasterea (Suga et al. Citation2012, Citation2014).
Figure 5. Evolution of cytoplasmic tyrosine kinases. A. Presence of cytoplasmic tyrosine kinase families across the metazoan lineage and in closely related nonmetazoan eukaryotes. left: A phylogenetic tree of eukaryotes highlighting specific metazoan species and several nonmetazoan clades. right: Incidence of tyrosine kinase genes across the represented eukaryotic phyla that are likely orthologs of tyrosine kinase genes from the indicated human families. A red circle indicates that a kinase from that family is present in that specific organism/clade, and a white circle indicates that no kinase from that family has been reported on that branch of the evolutionary tree. The yellow circle represents a kinase with domain architecture similar to a Jak-family kinase, but with a kinase domain sequence more homologous to a Syk-family kinase. The blue circles represent the presence of Syk-like tyrosine kinases known as SHARK tyrosine kinases, in which the two SH2 domains are separated by ankyrin repeats. Data for this figure were compiled from Bradham et al. Citation2006; Manning et al. Citation2002a; Srivastava et al. Citation2010; and Suga et al. Citation2014. B. Evolution of interdomain allostery in the Src module (conceptually adapted from Kuriyan and Eisenberg Citation2007). In an ancient organism, SH3, SH2, and tyrosine kinase domains likely existed as separte genes, which had no appreciable affinity for one another. It is possible that these individual domains retained some capacity for allosteric modulation, dictated by their intrinsic conformational dynamics. Multiple steps of gene fusion led to the assembly of a gene and its protein product with a domain architecture resembling the Src module. In the earliest iterations of this architecture, the domains interacted weakly and there was no allosteric modulation of kinase activity. Over time, through mutations, noncovent interactions between the domains strengthened, allowing for allosteric regulation of kinase activity through interdomain contacts (see colour version of this figure at www.tandfonline.com/ibmg).
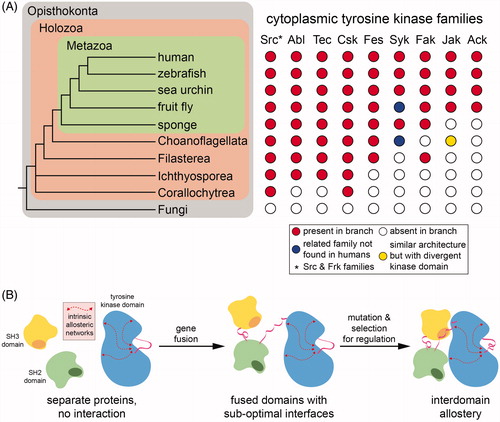
One intriguing feature of these anciently diverged nonmetazoan tyrosine kinomes is that they often contain an enormous repertoire of receptor tyrosine kinases (there are at least 88 receptor tyrosine kinases in the choanoflagellate Monosiga brevicollis), but the extracellular ligand-binding domains of these receptors do not resemble those found in animals (King et al. Citation2003; Manning et al. Citation2008). By contrast, several of the major cytoplasmic tyrosine kinases families found in metazoans are also present in the filasterea Capsaspora owczarzaki and Ministeria vibrans (Suga et al. Citation2012). These organisms contain at least one Src-, Abl-, Tec-, Csk-, and Fak-family kinase (), as well as one or more cytoplasmic tyrosine kinase types not found in metazoans. The fact that the Src module is present in filasterea with the same elaborations found in animal kinases indicates that this protein module has ancient origins. The lasting conservation of this core domain architecture strongly suggests its robustness and versatility as a scaffold for the evolution of signaling switches.
Evolution of allosteric regulation in the Src module
Although the SH3-SH2-kinase domain architecture of the Src module is ancient (), it is not clear when the assembled state and corresponding allosteric regulation of the Src module emerged. Multidomain proteins are most likely created as a result of the fusion of genes encoding distinct globular functional domains, and intramolecular interactions between the resulting tethered domains are unlikely to be optimized, initially, to exploit any intrinsic allosteric capabilities of the individual domains. Rather, the connections between the allosteric networks in these individual domains most likely evolve slowly after gene fusion () (Kuriyan and Eisenberg Citation2007). The fact that the Src-, Abl-, and Tec-family kinases can all adopt the same assembled, auto-inhibited state suggests that this structure pre-dates the separation of these kinase families.
The observation that Csk is present in our closest unicellular relatives suggests that tail phosphorylation of Src-family kinases also occurs in these organisms (). Studies on choanoflagellate Src and Csk orthologs have clearly shown that tail phosphorylation is conserved in those lineages (Li et al. Citation2008; Segawa et al. Citation2006; Taskinen et al. Citation2017). Analyses of Src and Csk from more divergent organisms show conflicting results. Early experiments with filasterean kinases suggested that although the Src-family kinase was active, Csk from these organisms was inactive (Schultheiss et al. Citation2012, Citation2014). This lack of activity was attributed to a poorly formed SH2-kinase interface, which is important for activation of human Csk, as described above (Ogawa et al. Citation2002; Wong et al. Citation2005).
In a more recent report, the activities of Csk orthologs were assessed in vitro using different protein constructs, and using a yeast viability assay, which exploits the fact that Src activity is toxic to yeast but suppressed by co-expression of Csk (Taskinen et al. Citation2017). These new experiments indicated that Csk orthologs are active in all distantly diverged lineages that are known to contain both Src and Csk. Notably, these studies also differ in their conclusions regarding allosteric regulation by tail phosphorylation. Previous work indicated that only metazoan Src kinases, but not choanoflagellate Src kinases, are negatively regulated by tail phosphorylation (Segawa et al. Citation2006; Li et al. Citation2008). The SH3 and SH2 domains of choanoflagellate Src kinases appear fully functional, and this regulatory defect was localized to the kinase domain (Li et al. Citation2008), but not specific residues, by the analysis of choanoflagellate-metazoan chimeric Src molecules (Segawa et al. Citation2006). The more recent study demonstrated that even nonmetazoan Src activity could be suppressed by Csk-mediated tail phosphorylation (Taskinen et al. Citation2017). The growing number of nonmetazoan genome sequences containing Csk- and Src-family kinases should allow for a more comprehensive biochemical and bioinformatic analysis of the function, regulation, and co-evolution of these families in the near future.
This general line of investigation has also provided insights into Src-family kinase regulation prior to the co-existence of Src and Csk. A distantly related ichthyosporean (), Creolimax fragrantissima, has a highly reduced kinome containing only one cytoplasmic tyrosine kinase, a Src-family kinase (Suga and Miller Citation2018). The lack of Csk in this kinome raises the question of how Src is negatively regulated in this organism. Biochemical analysis of the Src-family kinase from Creolimax revealed that its activity is enhanced by activation loop auto-phosphorylation, and that Csk molecules from other organisms cannot robustly phosphorylate its C-terminal tail (Suga and Miller Citation2018). Further analysis of the Creolimax genome revealed seven protein tyrosine phosphatases, of which one could dephosphorylate Creolimax Src and inhibit its activity in an in vitro biochemical assay. Notably, this phosphatase could also regulate Src in vivo in Creolimax and suppress a growth defect observed upon Src overexpression. This study suggests that, early in the evolution of phosphotyrosine signaling, tyrosine phosphatases likely played the major role in regulating tyrosine kinase activity. Indeed, tyrosine phosphatase genes appear to have evolved prior to the emergence of tyrosine kinases and may have served to suppress aberrant phosphorylation of tyrosine residues in the proteomes of ancient organisms (Pincus et al. Citation2008; Lim and Pawson Citation2010).
A related biochemical and evolutionary analysis has also been carried out on an Abl-family kinase from the choanoflagellate Monosiga brevicollis. In this organism, there are two Abl-like kinases, MbAbl1 and MbAbl2. MbAbl1 has a putative N-terminal myristoylation site, although it is not clear if this site is lipidated in vivo. MbAbl2 lacks the critical auto-inhibitory myristoylation site found in the N-terminal cap region in human c-Abl isoform 1b. Consistent with this lack of the negative-regulatory lipidation, biochemical and cell-based analysis of MbAbl2 showed that it has high constitutive activity relative to its mammalian ortholog (Aleem et al. Citation2015). Notably, the substrate specificity of MbAbl2 is similar to that of human c-Abl, suggesting that the sequence preferences of the catalytic domain were established early in the evolution of Abl-family kinases.
Dissecting divergent features of Src and Abl through evolutionary analysis
Current genomic data and bioinformatic analyses strongly support the notion that all Src-module-containing proteins have a common origin (Manning et al. Citation2002a, Citation2002b). This relationship affords an interesting opportunity to use evolutionary principles to dissect key molecular differences between each of these kinase lineages that have important functional consequences. This fact was highlighted by a recent study on the differential properties of the Abl and Src kinases (Wilson et al. Citation2015). In this study, the authors employed ancestral protein reconstruction, whereby multiple sequence alignments and inferred phylogenetic trees of extant orthologs and paralogs (leaves on the trees) are used, in combination with a statistical algorithm and evolutionary model, to infer the sequences of internal nodes in those phylogenetic trees (Harms and Thornton Citation2013). These predicted internal node sequences represent putative common ancestors of the extant sequences. By experimentally characterizing proteins that connect a path on the tree between two extant proteins, one can identify specific residue changes and epistatic interactions that dictate the divergent functions of those proteins.
The cancer drug imatinib binds tightly to the Abl kinase domain and inhibits it, but imatinib is a poor inhibitor of Src-family kinases (Zimmermann et al. Citation1997). The crystal structures of imatinib bound to the Abl kinase domain (PDB codes 1FPU and 1IEP) (Schindler et al. Citation2000; Nagar et al. Citation2002), and to the kinase domain of the receptor tyrosine kinase c-Kit, which imatinib also inhibits (PDB code 1T46) (Mol et al. Citation2004), showed that binding of the drug requires the DFG motif to flip into the DFG-out conformation, in which the aspartate sidechain points away from the ATP binding site. The initial inference was that the Src-family kinases could not readily adopt this DFG-out conformation (Nagar et al. Citation2002; Seeliger et al. Citation2007). This was shown to not be the case by studies on high-affinity Src inhibitors that also recognize the DFG-out state (Dar et al. Citation2008; Seeliger et al. Citation2009). The ancestral sequence reconstruction of the common ancestor of Abl and Src led to important insights concerning the differential ability of imatinib to inhibit Abl but not the Src-family kinases (Wilson et al. Citation2015). Using this approach, key residues were identified that confer a 3000-fold tighter binding of imatinib to Abl versus Src. Drug binding measurements and crystal structures of predicted ancestral proteins revealed a hydrogen bond network in the N-lobe of Src, lacking in Abl, that prohibits additional contacts with imatinib and allows for relatively fast dissociation of imatinib from Src but not Abl. This evolutionary analysis helped dissect the molecular basis for selective drug binding, a functional property that was not selected for in nature.
Structural mechanisms of dysregulation in the Src module
As essential enzymes in metazoan signal transduction, tyrosine kinases are often dysregulated in diseases, particularly cancer (Blume-Jensen and Hunter Citation2001). Indeed, the first ever oncogene to be discovered was v-Src, a variant of c-Src kinase found in the Rous sarcoma virus that causes cancer in chickens (Stehelin et al. Citation1976; Spector et al. Citation1978; Oppermann et al. Citation1979). The biological consequences of many of the sequence changes in oncogenic v-Src relative to c-Src can now be understood, in light of the large body of biochemical and structural work on Src-family kinases. We now know that most disease-causing mutations in Src and other tyrosine kinases act by disrupting the regulatory mechanisms controlling kinase function. Mutations in these dysregulated kinases can also confer resistance to targeted therapies by tapping into the same regulatory mechanisms (Azam et al. Citation2003).
Disease-associated mutations in the Src module
The most important mutation in v-Src relative to c-Src is an alteration of the C-terminal tail, resulting in loss of the negative-regulatory phosphorylation site. This change clearly causes a gain-of-function by removing a critical interaction that stabilizes the assembled, auto-inhibited state of the Src module. v-Src and c-Src also differ by several amino acid substitutions within the core domains of the Src module (). Several of these substitutions, along with other mutations not naturally found in Rous sarcoma viruses, are sufficient individually to induce the cell transforming ability of Src, even in the presence of the C-terminal tail (Kato et al. Citation1986; Levy et al. Citation1986), underscoring the distributed contributions of the entire Src module to the regulation of kinase activity. The Rous sarcoma virus is an avian pathogen, and surprisingly, oncogenic mutations in c-Src are rarely found in human cancers. Instead, cancer cells often unleash the oncogenicity of c-Src and other Src-family kinases through over-expression or hyper-activation of the wild-type proteins (Blume-Jensen and Hunter Citation2001). A small subset of patients with advanced human colon cancers contain a truncating mutation in their c-Src gene immediately adjacent to the regulatory C-terminal phosphorylation site at Tyr 527 () (Irby et al. Citation1999).
Figure 6. Disease-related and drug resistance mutations in in the Src module. A. Oncogenic mutations in c-Src, mapped onto a crystal structure of the auto-inhibited form of c-Src (PDB code 2SRC). Most of the mutation sites indicated by a black sphere are reported activating mutations in the v-Src gene of the Rous sarcoma virus, given in chicken c-Src numbering with the human c-Src numbering in parentheses: R95(98)W, T96(99)I, D117(120)N, R318(321)Q, T338(341)I. The sites at which the C-terminal tail of c-Src starts to differ from v-Src, and where a C-terminal truncation is observed a small subset of human colon cancers, are also marked with a black sphere. Note that the T338(341)I mutation is at the canonical ‘gatekeeper’ residue and also causes resistance to the drug dasatinib. Orange spheres mark the point mutations E378(381)G and I441(444)F, which were identified by passaging a virus encoding c-Src in fibroblast cells and selecting for cell transformation. These mutations are independently sufficient to hyperactivate c-Src (Kato et al. Citation1986; Levy et al. Citation1986). B. Sites of drug resistance mutations in Bcr-Abl, mapped onto a crystal structure of the auto-inhibited form of c-Abl (PDB code 1OPL). Mutation sites marked by a black sphere were identified in the first major survey of clinically observed resistance mutations to imatinib (Shah et al. Citation2002), and are given in human c-Abl isoform 1b numbering with the isoform 1a numbering in parentheses: M263(244)V, G269(250)E, Q271(252)H/R, Y272(253)F/H, E274(255)K, T334(315)I, F336(317)L, M370(351)T, E374(355)G, F378(359)V, L406(387)M, H415(396)R. Mutation sites marked by an orange sphere are examples of mutations distal to the imatinib binding site that confer resistance in a cell-based assay for oncogenic transformation (Azam et al. Citation2003): E298(279)K, V357(338)G, V358(339)A/G, A363(344)V, G391(372)R, G482(463)D, M491(472)I, F505(486)S, E513(494)A, I521(502)M, E528(509)D. C. Disease-related mutations in Btk, mapped onto a model of full-length Btk (left) from (Wang et al. Citation2015) (also see structures with PDB codes 4Y93 and 4XI2), a crystal structure of the Saraste dimer of the PH-TH module (top right, PDB code 4Y94), and a crystal structure of the Btk kinase domain bound to ibrutinib (bottom right, PDB code 5P9J). Residue numbering corresponds to that of the human Btk sequence. Sites of some inactivating mutations in XLA are shown as black spheres (R307G/K/T, L369F, R372G, G414R, R525K/G) or black ball and stick model in the case of R28H. The site of N-terminal truncation in an oncogenic form of Btk (Met 89), the critical cysteine (Cys 481) that reacts with the drug ibrutinib, and other sites of resistance mutations (T474I and L528W) are shown in orange (see colour version of this figure at www.tandfonline.com/ibmg).
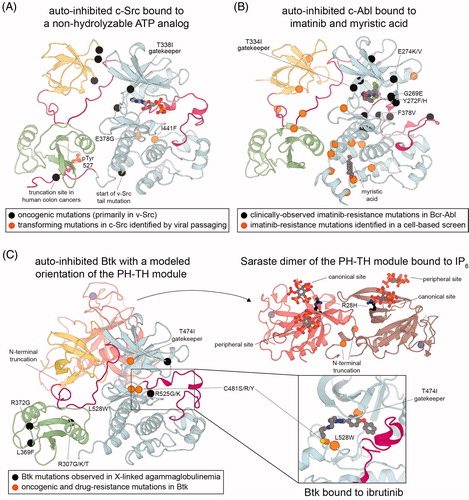
Like Src, Abl was also first discovered as a viral oncogene (Goff et al. Citation1980). The most common disease-causing variants of Abl in humans are the Bcr-Abl fusion proteins (Ben-Neriah et al. Citation1986). Bcr-Abl proteins are the result of DNA translocations found in certain patients with chronic myelogenous leukemia (CML) and acute lymphoblastic leukemia (ALL), which yield a hybrid of chromosomes 9 and 22 known as the Philadelphia chromosome (Nowell and Hungerford Citation1960). The fusion proteins contain part of the breakpoint cluster region of chromosome 9 and the majority of the c-Abl-coding gene, lacking part of the N-terminal cap and thus the myristoylation site in isoform 1b. In addition to dysregulation through the lack of the negative-regulatory myristoylation site, kinase activity and transforming potential is enhanced by oligomerization of Abl kinases through a coiled-coil domain in the Bcr protein, which most likely facilitates trans-autophosphorylation of the activation loop (McWhirter et al. Citation1993; Zhao et al. Citation2002). Interestingly, an oligomerization-deficient version of the Bcr-Abl oncogene can be partially inhibited by re-introducing the N-terminal cap of c-Abl isoform 1b, suggesting that this fusion protein is still subject to the same mechanisms of regulation as wild-type Abl (Pluk et al. Citation2002). Several other oncogenic fusion protein variants of both c-Abl and Arg have also been observed (Hantschel Citation2012). These fusion proteins lack N-terminal regulatory elements, including the myristoylation site and occasionally a functional SH3 domain, and they often bear an oligomerization motif, as in the Bcr-Abl proteins.
Although Bcr-Abl is the primary oncogenic form of Abl, a recent study employing a random mutagenesis screen on intact c-Abl isoform 1b yielded several point mutations that render the kinase constitutively active and able to transform a model cell line (Lee and Shah Citation2017). Most of these mutations were distributed throughout the Abl functional domains at key regulatory hotspots, including the N-terminal cap, SH3 ligand-binding groove, and myristoyl-binding pocket. Two of these mutations have been observed in clinical isolates from cancer patients, but their functional impact and roles as potential driver mutations had not been established previously. As discussed in the next section, many of these activating mutations in c-Abl isoform 1b also confer resistance to ATP-competitive inhibitors of Abl kinases, further supporting notion that there is allosteric coupling between the active site and regions dispersed throughout the Src module () (Azam et al. Citation2003).
In the Tec-family kinases, Btk was first discovered as the critical gene mutated in X-linked agammaglobulinemia (Vetrie et al. Citation1993). Mutations have been observed across all five domains of Btk in cases of XLA in humans and a related condition, X-linked immunodeficiency (Xid), in mice () (Rawlings et al. Citation1993; Thomas et al. Citation1993; Tsukada et al. Citation1993; Vetrie et al. Citation1993). These alterations include missense mutations, nonsense mutations, insertions, and deletions, which occur at nearly 100 positions across the gene (Smith Citation2017). Many of the Btk missense mutations in XLA clearly disrupt known functions of each domain, including inositol phosphate binding by the PH domain (such as R28H in Btk), phosphotyrosine binding by the SH2 domain (R307G/K/T, corresponding to Arg 175 in c-Src), and ATP-Mg2+ coordination by the kinase domain (G414R and R525G/K in Btk, corresponding to Gly 279 and Arg 388 in c-Src) () (Väliaho et al. Citation2006). Two such mutations in XLA patients, L369F and R372G (corresponding to Leu 237 and Arg 240 in c-Src), lie just upstream of the SH2-kinase linker and result in a loss of Btk-mediated calcium response, without greatly affecting protein stability or autophosphorylation rates (Guo et al. Citation2006). The large variety of mutations observed in XLA across all domains in Btk supports the idea that each module of Btk is integral to its ability to be activated and carry out downstream signaling.
Btk plays an important role in normal B cell homeostasis and proliferation. As a result, it has been the target of drug development efforts to treat B cell leukemias and lymphomas, where its wild-type form is often over-expressed and constitutively active (Hendriks et al. Citation2014). Despite the presumed tissue-specific expression of Btk in B lymphocytes, an alternatively spliced isoform of Btk was recently discovered in a subset of colon cancers and shown to be required for oncogenic transformation (Grassilli et al. Citation2016). This isoform bears an N-terminal truncation and is missing a large portion of the PH domain (). Consistent with our understanding of the role of the PH-TH module in Btk auto-inhibition (Wang et al. Citation2015; Joseph et al. Citation2017), this variant appears to have high activity in cells and acts as an oncogene in this setting.
Mechanisms of drug resistance in the Src module
Given their dysregulation in numerous human diseases, kinases containing the Src module have been the targets of many drug development efforts. Indeed, there are several dozen clinically approved protein kinase inhibitors with more than one hundred additional drugs in the pipeline (Wang et al. Citation2014), and several of these inhibitors are specifically designed to treat cancers driven by aberrations in kinases bearing a Src module. Although the development of targeted kinase inhibitors to treat cancers has been successful, treatment with these inhibitors almost inevitably leads to drug resistance and patient relapse (Sawyers Citation2003). This resistance may arise due to compensatory signaling mechanisms or over-expression of the target kinase, as well as point mutations in the target kinases.
The most intuitive mechanisms of resistance by point mutation are direct steric occlusion of drug binding or loss of specific contacts with the drug. Such mutations were reported in the first comprehensive survey of clinically observed resistance mutations in the Bcr-Abl kinase domain in response to imatinib treatment () (Shah et al. Citation2002). One of these, the T334I mutation at the “gatekeeper” position in kinases (T315I in c-Abl isoform 1b, residue Thr 338 in c-Src), was the first clinically observed mutation that caused imatinib resistance (Gorre et al. Citation2001), and it remains the most common resistance mutation to imatinib and related drugs (see PDB code 1EIP for imatinib-bound c-Abl structure). A similar “direct contact” mechanism of resistance has been seen for the Btk inhibitor, ibrutinib, which inactivates Btk by covalently reacting with Cys 481 (Ser 345 in c-Src) in the active site (PDB code 5P9J) (Bender et al. Citation2017; Pan et al. Citation2007). Patients treated with ibrutinib who have relapsed often have a C481S mutation in Btk (), rendering the drug substantially less effective (Woyach et al. Citation2014).
Many of the mutations that cause resistance to imatinib are not at positions that directly contact the drug (). It was proposed that these mutations most likely act by altering the ability of the Abl kinase domain to adopt the specific inactive conformation required for drug binding (Shah et al. Citation2002). This hypothesis was validated in an important study, where randomly mutated BCR-ABL genes were screened in a cell-based assay for their ability to confer resistance to imatinib and transform cells (Azam et al. Citation2003). This screen yielded 112 distinct amino acid substitutions at 90 different residues in Bcr-Abl, at least 10 of which had already been observed in patients. Many of the observed mutations were outside of the drug binding pocket and even outside of the kinase domain (). In light of our structural understanding of c-Abl auto-inhibition, these distal mutations can clearly be interpreted as impacting the stability of the auto-inhibited state, which should destabilize the imatinib-competent conformation of the kinase domain. In a recent study of c-Abl conformational energetics and dynamics using NMR spectroscopy, it was found that many of these distal resistance mutations have no effect on imatinib binding thermodynamics, but rather they perturbed the relative populations of the active and inactive states (Saleh et al. Citation2017). Even the gatekeeper T334I mutations in Abl, and analogous mutations in other kinases, act, in part, by stabilizing the active conformation of kinases (Azam et al. Citation2008).
Leveraging fundamental mechanistic insights to overcome drug resistance
To combat the emergence of imatinib-resistant mutant forms of Abl kinase, a series of second generation drugs have been developed that target the Abl active site. These largely overcome the effects of canonical resistance mutations, except for the gatekeeper T334I mutation, and third generation inhibitors have been developed to specifically target this mutant form (Lamontanara et al. Citation2013). In all cases, either through single or compound mutations, resistance has been observed in a clinical setting. Similar situations have been reported for virtually all other targeted kinase inhibitors to treat cancer, irrespective of the kinase (Krishnamurty and Maly Citation2010). This issue underscores the essential role that basic mechanistic studies can play in improving clinical outcomes, as exemplified below.
In an unbiased phenotypic screen for inhibitors of Bcr-Abl activity, a small molecule named GNF-2 was discovered that suppresses Bcr-Abl-mediated cell proliferation (Adrián et al. Citation2006). This molecule binds to the myristoyl binding pocket on the C-lobe of the Abl kinase domain and presumably acts allosterically to suppress Abl activity in cells through the same mechanism as the natural N-terminal myristoylation of c-Abl isoform 1b (Hantschel et al. Citation2003; Nagar et al. Citation2003). GNF-2 has limited potency and is not efficacious against the T334I mutant form of Abl. Building on these findings, a fragment-based screen was used, relying on an NMR signature of conformational changes near the myristoyl binding pocket (Jahnke et al. Citation2010), to identify small molecules that bound in that region in a manner that would robustly inhibit kinase activity (Wylie et al. Citation2017). The hits were further optimized by in silico docking to yield a molecule named ABL001, which binds tightly in the myristoyl binding pocket and potently allosterically inhibits Abl kinase activity. Structural analysis suggests that the myristoyl binding pocket is unique to Abl and not even present in the other closely related kinases with a Src module (Nagar et al. Citation2003). Thus, ABL001 is a highly selective kinase inhibitor. In an ABL001-treated xenograft mouse model, the mice gained resistance to the drug on a similar time-scale to those treated with the imatinib-like drug nilotinib, however, the resistance mutation profiles upon treating with these drugs were nonoverlapping. This observation, and the fact that both drugs can simultaneously bind the same kinase molecule (PDB code 5MO4), inspired the investigators to co-administer ABL001 and nilotinib. This combination therapy yielded a durable, apparently irreversible response in their mouse models (Wylie et al. Citation2017). While determination of the efficacy of such combination therapies in humans awaits the completion of rigorous clinical trials, the development of ABL001 highlights the value in understanding the structure and regulation of tyrosine kinases described throughout this review.
Concluding remarks
The assembled state of the Src module underlies an ancient structural mechanism for the regulation of many cytoplasmic tyrosine kinases. The determination of this structure revealed the critical role that interdomain allostery plays in the regulation of protein kinases in general, and foreshadowed the structural basis for the remarkable modularity that we now know is characteristic of phosphotyrosine signaling systems. A critical feature of all protein kinases is that their catalytic activity is dynamically regulated. In proteins bearing the Src module, this is achieved through the appendage of SH3 and SH2 domains, which can engage and release the catalytic domain in a signal-responsive manner to control kinase activity. While other cytoplasmic tyrosine kinases contain different combinations and arrangements of ligand-binding domains, the structural and regulatory principles gleaned from studies of the Src module can be generally applied to those other kinases.
The most notable feature of the Src module is that it provides an evolvable scaffold for the generation of new input–output relationships in kinase signaling. As discussed throughout this review, the introduction of unique post-translational modification sites and additional domains onto the Src module over the course of evolution has produced the distinct Src-, Abl-, and Tec-family kinase lineages. Each of these appendages facilitates control over catalytic activity by regulating the kinase domain in response to different upstream signaling events. Diverse downstream signaling is achieved not just through the differential specificities catalytic domains, but also through differences in the preferences of the ligand-binding domains.
The Src module was first discovered in the context of oncogenic signaling. The discovery and characterization of v-Src, the first oncogene to be identified, and of related kinases, demonstrated the importance of allosteric regulation and dysregulation in oncogenic signaling pathways and established tyrosine kinases as drug targets. The numerous structural and biochemical studies that have ensued since then have established an intimate connection between the activation cycle of protein kinases and their conformational flexibility. We are now beginning to understand how kinases traverse this activation cycle through the stabilization or destabilization of certain conformational states, and this information has proven useful for the design of selective, conformation-specific inhibitors of protein kinases. To date, most inhibitor design strategies have focused on targeting the ATP-binding pocket of the kinase domain. As evidenced by the interdomain allosteric regulation of the Src module, sites distal to the active site can modulate kinase activity. Future drug design efforts are likely to focus on the exploitation of these allosteric sites for the development of a next generation of effective kinase-targeted therapies.
| Abbreviations | ||
| SH2 | = | Src-homology 2 |
| SH3 | = | Src-homology 3 |
| PTB | = | phosphotyrosine binding |
| PH | = | pleckstrin homology |
| TH | = | Tec homology |
| Bcr | = | breakpoint cluster region |
| ATP | = | adenosine triphosphate |
| ADP | = | adenosine diphosphate |
| AMP-PNP | = | adenylyl-imidodiphosphate |
| PIP3 | = | phosphatidylinositol-(3,4,5)-trisphosphate |
| PIP2 | = | phosphatidylinositol-(4,5)-bisphosphate |
| XLA | = | X-linked agammaglobulinemia |
| Xid | = | X-linked immunodeficiency |
| CML | = | chronic myelogenous leukemia |
| ALL | = | acute lymphoblastic leukemia |
Disclosure statement
No potential conflict of interest was reported by the authors.
Notes
Additional information
Funding
Notes
1 The prefixes “v-“ and “c-“ are used to distinguish viral oncogenic forms and endogenous cellular forms of tyrosine kinases, respectively.
2 Throughout the review, we provide Protein Data Bank (PDB) accession numbers for protein structures discussed in detail.
3 In this review, we identify sidechains using the sequence of the historically important chicken c-Src. Residue numbers for specific tyrosine kinases are provided where appropriate. Numbering for the Src module of human c-Src is shifted by 3 additional residues (i.e. the regulatory activation loop tyrosine is Tyr 416 in chicken c-Src and Tyr 419 in human c-Src).
4 A ninth Src-family kinase, Yrk, is found in some vertebrates, such as chickens, but is not present in humans. Often, the Src family is said to be composed of nine kinases, when Yrk is included.
Related Research Data
References
- Adrián FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, Zhang G, Hur W, Ding S, Manley P, et al. 2006. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol. 2:95–102.
- Agafonov RV, Wilson C, Otten R, Buosi V, Kern D. 2014. Energetic dissection of Gleevec's selectivity toward human tyrosine kinases. Nat Struct Mol Biol. 21:848–853.
- Aleem SU, Craddock BP, Miller WT. 2015. Constitutive activity in an ancestral form of abl tyrosine kinase. PLoS One. 10:e0131062.
- Andreotti AH, Joseph RE, Conley JM, Iwasa J, Berg LJ. 2018. Multidomain control over TEC kinase activation state tunes the T cell response. Annu Rev Immunol. 36:549–578.
- Arbesú M, Maffei M, Cordeiro TN, Teixeira JMC, Pérez Y, Bernadó P, Roche S, Pons M. 2017. The unique domain forms a fuzzy intramolecular complex in Src family kinases. Structure. 25:630–640.e4.
- Azam M, Latek RR, Daley GQ. 2003. Mechanisms of autoinhibition and STI-571/imatinib resistance revealed by mutagenesis of BCR-ABL. Cell. 112:831–843.
- Azam M, Seeliger MA, Gray NS, Kuriyan J, Daley GQ. 2008. Activation of tyrosine kinases by mutation of the gatekeeper threonine. Nat Struct Mol Biol. 15:1109–1118.
- Banavali NK, Roux B. 2009. Flexibility and charge asymmetry in the activation loop of Src tyrosine kinases. Proteins. 74:378–389.
- Bardelli A, Longati P, Gramaglia D, Basilico C, Tamagnone L, Giordano S, Ballinari D, Michieli P, Comoglio PM. 1998. Uncoupling signal transducers from oncogenic MET mutants abrogates cell transformation and inhibits invasive growth. Proc Natl Acad Sci USA. 95:14379–14383.
- Begley MJ, Yun C, Gewinner CA, Asara JM, Johnson JL, Coyle AJ, Eck MJ, Apostolou I, Cantley LC. 2015. EGF-receptor specificity for phosphotyrosine-primed substrates provides signal integration with Src. Nat Struct Mol Biol. 22:983–990.
- Bender AT, Gardberg A, Pereira A, Johnson T, Wu Y, Grenningloh R, Head J, Morandi F, Haselmayer P, Liu-Bujalski L. 2017. Ability of Bruton’s tyrosine kinase inhibitors to sequester Y551 and prevent phosphorylation determines potency for inhibition of fc receptor but not B-cell receptor signaling. Mol Pharmacol. 91:208–219.
- Ben-Neriah Y, Daley GQ, Mes-Masson AM, Witte ON, Baltimore D. 1986. The chronic myelogenous leukemia-specific P210 protein is the product of the bcr/abl hybrid gene. Science. 233:212–214.
- Bergman M, Joukov V, Virtanen I, Alitalo K. 1995. Overexpressed Csk tyrosine kinase is localized in focal adhesions, causes reorganization of alpha v beta 5 integrin, and interferes with HeLa cell spreading. Mol Cell Biol. 15:711–722.
- Blume-Jensen P, Hunter T. 2001. Oncogenic kinase signalling. Nature. 411:355–365.
- Bocciardi R, Mograbi B, Pasini B, Borrello MG, Pierotti MA, Bourget I, Fischer S, Romeo G, Rossi B. 1997. The multiple endocrine neoplasia type 2B point mutation switches the specificity of the Ret tyrosine kinase towards cellular substrates that are susceptible to interact with Crk and Nck. Oncogene. 15:2257–2265.
- Boggon TJ, Eck MJ. 2004. Structure and regulation of Src family kinases. Oncogene. 23:7918–7927.
- Booker GW, Breeze AL, Downing AK, Panayotou G, Gout I, Waterfield MD, Campbell ID. 1992. Structure of an SH2 domain of the p85 alpha subunit of phosphatidylinositol-3-OH kinase. Nature. 358:684–687.
- Bose R, Holbert MA, Pickin KA, Cole PA. 2006. Protein tyrosine kinase-substrate interactions. Curr Opin Struct Biol. 16:668–675.
- Boyken SE, Chopra N, Xie Q, Joseph RE, Wales TE, Fulton DB, Engen JR, Jernigan RL, Andreotti AH. 2014. A conserved isoleucine maintains the inactive state of Bruton’s tyrosine kinase. J Mol Biol. 426:3656–3669.
- Bradham CA, Foltz KR, Beane WS, Arnone MI, Rizzo F, Coffman JA, Mushegian A, Goel M, Morales J, Geneviere A-M, et al. 2006. The sea urchin kinome: a first look. Dev Biol. 300:180–193.
- Brasher BB, Van Etten RA. 2000. c-Abl has high intrinsic tyrosine kinase activity that is stimulated by mutation of the Src homology 3 domain and by autophosphorylation at two distinct regulatory tyrosines. J Biol Chem. 275:35631–35637.
- Chung JK, Nocka LM, Wang Q, Kadlecek TA, Weiss A, Kuriyan J, Groves JT. 2018. Switch-like activation of Bruton’s tyrosine kinase by membrane-mediated dimerization. BioRxiv DOI:10.1101/284000.
- Courtney AH, Amacher JF, Kadlecek TA, Mollenauer MN, Au-Yeung BB, Kuriyan J, Weiss A. 2017. A phosphosite within the SH2 domain of Lck regulates its activation by CD45. Mol Cell. 67:498–511.e6.
- Couture C, Songyang Z, Jascur T, Williams S, Tailor P, Cantley LC, Mustelin T. 1996. Regulation of the Lck SH2 domain by tyrosine phosphorylation. J Biol Chem. 271:24880–24884.
- Cowan-Jacob SW, Fendrich G, Manley PW, Jahnke W, Fabbro D, Liebetanz J, Meyer T. 2005. The crystal structure of a c-Src complex in an active conformation suggests possible steps in c-Src activation. Structure. 13:861–871.
- Dar AC, Lopez MS, Shokat KM. 2008. Small molecule recognition of c-Src via the Imatinib-binding conformation. Chem Biol. 15:1015–1022.
- De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgan DO, Kim SH. 1993. Crystal structure of cyclin-dependent kinase 2. Nature. 363:595–602.
- Debnath J, Chamorro M, Czar MJ, Schaeffer EM, Lenardo MJ, Varmus HE, Schwartzberg PL. 1999. rlk/TXK encodes two forms of a novel cysteine string tyrosine kinase activated by Src family kinases. Mol Cell Biol. 19:1498–1507.
- Deng Y, Alicea-Velázquez NL, Bannwarth L, Lehtonen SI, Boggon TJ, Cheng H-C, Hytönen VP, Turk BE. 2014. Global analysis of human nonreceptor tyrosine kinase specificity using high-density peptide microarrays. J Proteome Res. 13:4339–4346.
- Devkota S, Joseph RE, Boyken SE, Fulton DB, Andreotti AH. 2017. An autoinhibitory role for the pleckstrin homology domain of interleukin-2-inducible tyrosine kinase and its interplay with canonical phospholipid recognition. Biochemistry. 56:2938–2949.
- Eck MJ, Shoelson SE, Harrison SC. 1993. Recognition of a high-affinity phosphotyrosyl peptide by the Src homology-2 domain of p56lck. Nature. 362:87–91.
- Feng S, Chen JK, Yu H, Simon JA, Schreiber SL. 1994. Two binding orientations for peptides to the Src SH3 domain: development of a general model for SH3-ligand interactions. Science. 266:1241–1247.
- Filippakopoulos P, Kofler M, Hantschel O, Gish GD, Grebien F, Salah E, Neudecker P, Kay LE, Turk BE, Superti-Furga G, et al. 2008. Structural coupling of SH2-kinase domains links Fes and Abl substrate recognition and kinase activation. Cell. 134:793–803.
- Foda ZH, Shan Y, Kim ET, Shaw DE, Seeliger MA. 2015. A dynamically coupled allosteric network underlies binding cooperativity in Src kinase. Nat Commun. 6:5939.
- Gan W, Yang S, Roux B. 2009. Atomistic view of the conformational activation of Src kinase using the string method with swarms-of-trajectories. Biophys J. 97:L8–L10.
- Goff SP, Gilboa E, Witte ON, Baltimore D. 1980. Structure of the Abelson murine leukemia virus genome and the homologous cellular gene: studies with cloned viral DNA. Cell. 22:777–785.
- Goodfellow HS, Frushicheva MP, Ji Q, Cheng DA, Kadlecek TA, Cantor AJ, Kuriyan J, Chakraborty AK, Salomon A, Weiss A. 2015. The catalytic activity of the kinase ZAP-70 mediates basal signaling and negative feedback of the T cell receptor pathway. Sci Signal. 8:ra49.
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, Sawyers CL. 2001. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 293:876–880.
- Grasis JA, Tsoukas CD. 2011. Itk: the rheostat of the T cell response. J Signal Transduct. 2011:297868.
- Grassilli E, Pisano F, Cialdella A, Bonomo S, Missaglia C, Cerrito MG, Masiero L, Ianzano L, Giordano F, Cicirelli V, et al. 2016. A novel oncogenic BTK isoform is overexpressed in colon cancers and required for RAS-mediated transformation. Oncogene. 35:4368–4378.
- Griswold IJ, MacPartlin M, Bumm T, Goss VL, O'Hare T, Lee KA, Corbin AS, Stoffregen EP, Smith C, Johnson K, et al. 2006. Kinase domain mutants of Bcr-Abl exhibit altered transformation potency, kinase activity, and substrate utilization, irrespective of sensitivity to imatinib. Mol Cell Biol. 26:6082–6093.
- Guo S, Wahl MI, Witte ON. 2006. Mutational analysis of the SH2-kinase linker region of Bruton’s tyrosine kinase defines alternative modes of regulation for cytoplasmic tyrosine kinase families. Int Immunol. 18:79–87.
- Hantschel O. 2012. Structure, regulation, signaling, and targeting of abl kinases in cancer. Genes Cancer. 3:436–446.
- Hantschel O, Nagar B, Guettler S, Kretzschmar J, Dorey K, Kuriyan J, Superti-Furga G. 2003. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell. 112:845–857.
- Hantschel O, Wiesner S, Güttler T, Mackereth CD, Rix LLR, Mikes Z, Dehne J, Görlich D, Sattler M, Superti-Furga G. 2005. Structural basis for the cytoskeletal association of Bcr-Abl/c-Abl. Mol Cell. 19:461–473.
- Harms MJ, Thornton JW. 2013. Evolutionary biochemistry: revealing the historical and physical causes of protein properties. Nat Rev Genet. 14:559–571.
- Hendriks RW, Yuvaraj S, Kil LP. 2014. Targeting Bruton's tyrosine kinase in B cell malignancies. Nat Rev Cancer. 14:219–232.
- Heyeck SD, Wilcox HM, Bunnell SC, Berg LJ. 1997. Lck phosphorylates the activation loop tyrosine of the Itk kinase domain and activates Itk kinase activity. J Biol Chem. 272:25401–25408.
- Hubbard SR. 1997. Crystal structure of the activated insulin receptor tyrosine kinase in complex with peptide substrate and ATP analog. EMBO J. 16:5572–5581.
- Hubbard SR, Miller WT. 2007. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr Opin Cell Biol. 19:117–123.
- Hui E, Vale RD. 2014. In vitro membrane reconstitution of the T-cell receptor proximal signaling network. Nat Struct Mol Biol. 21:133–142.
- Hunter T. 2009. Tyrosine phosphorylation: thirty years and counting. Curr Opin Cell Biol. 21:140–146.
- Hunter T, Sefton BM. 1980. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci USA. 77:1311–1315.
- Huse M, Kuriyan J. 2002. The conformational plasticity of protein kinases. Cell. 109:275–282.
- Hyvönen M, Saraste M. 1997. Structure of the PH domain and Btk motif from Bruton's tyrosine kinase: molecular explanations for X-linked agammaglobulinaemia. EMBO J. 16:3396–3404.
- Irby RB, Mao W, Coppola D, Kang J, Loubeau JM, Trudeau W, Karl R, Fujita DJ, Jove R, Yeatman TJ. 1999. Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet. 21:187–190.
- Jahnke W, Grotzfeld RM, Pellé X, Strauss A, Fendrich G, Cowan-Jacob SW, Cotesta S, Fabbro D, Furet P, Mestan J, et al. 2010. Binding or bending: distinction of allosteric Abl kinase agonists from antagonists by an NMR-based conformational assay. J Am Chem Soc. 132:7043–7048.
- Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massagué J, Pavletich NP. 1995. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature. 376:313–320.
- Johnson LN. 2009. Protein kinase inhibitors: contributions from structure to clinical compounds. Q Rev Biophys. 42:1–40.
- Joseph RE, Fulton DB, Andreotti AH. 2007. Mechanism and functional significance of Itk autophosphorylation. J Mol Biol. 373:1281–1292.
- Joseph RE, Kleino I, Wales TE, Xie Q, Fulton DB, Engen JR, Berg LJ, Andreotti AH. 2013. Activation loop dynamics determine the different catalytic efficiencies of B cell- and T cell-specific tec kinases. Sci Signal. 6:ra76.
- Joseph RE, Wales TE, Fulton DB, Engen JR, Andreotti AH. 2017. Achieving a graded immune response: BTK adopts a range of active/inactive conformations dictated by multiple interdomain contacts. Structure. 25:1481–1494.e4.
- Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. 2011. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell. 42:9–22.
- Kato JY, Takeya T, Grandori C, Iba H, Levy JB, Hanafusa H. 1986. Amino acid substitutions sufficient to convert the nontransforming p60c-Src protein to a transforming protein. Mol Cell Biol. 6:4155–4160.
- Katz ZB, Novotná L, Blount A, Lillemeier BF. 2017. A cycle of Zap70 kinase activation and release from the TCR amplifies and disperses antigenic stimuli. Nat Immunol. 18:86–95.
- Kim PW, Sun Z-YJ, Blacklow SC, Wagner G, Eck MJ. 2003. A zinc clasp structure tethers Lck to T cell coreceptors CD4 and CD8. Science. 301:1725–1728.
- King N, Carroll SB. 2001. A receptor tyrosine kinase from choanoflagellates: molecular insights into early animal evolution. Proc Natl Acad Sci USA. 98:15032–15037.
- King N, Hittinger CT, Carroll SB. 2003. Evolution of key cell signaling and adhesion protein families predates animal origins. Science. 301:361–363.
- Kornev AP, Haste NM, Taylor SS, Eyck LFT. 2006. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc Natl Acad Sci USA 103:17783–17788.
- Kornev AP, Taylor SS. 2015. Dynamics-Driven Allostery in Protein Kinases. Trends Biochem Sci. 40:628–647.
- Kornev AP, Taylor SS, Ten Eyck LF. 2008. A helix scaffold for the assembly of active protein kinases. Proc Natl Acad Sci USA. 105:14377–14382.
- Krishnamurty R, Maly DJ. 2010. Biochemical mechanisms of resistance to small-molecule protein kinase inhibitors. ACS Chem Biol. 5:121–138.
- Kuriyan J, Cowburn D. 1997. Modular peptide recognition domains in eukaryotic signaling. Annu Rev Biophys Biomol Struct. 26:259–288.
- Kuriyan J, Eisenberg D. 2007. The origin of protein interactions and allostery in colocalization. Nature. 450:983–990.
- Lamontanara AJ, Gencer EB, Kuzyk O, Hantschel O. 2013. Mechanisms of resistance to BCR-ABL and other kinase inhibitors. Biochim Biophys Acta. 1834:1449–1459.
- Lamontanara AJ, Georgeon S, Tria G, Svergun DI, Hantschel O. 2014. The SH2 domain of Abl kinases regulates kinase autophosphorylation by controlling activation loop accessibility. Nat Commun. 5:5470.
- Lee BJ, Shah NP. 2017. Identification and characterization of activating ABL1 1b kinase mutations: impact on sensitivity to ATP-competitive and allosteric ABL1 inhibitors. Leukemia. 31:1096–1107.
- Lee S, Ayrapetov MK, Kemble DJ, Parang K, Sun G. 2006. Docking-based substrate recognition by the catalytic domain of a protein tyrosine kinase, C-terminal Src kinase (Csk). J Biol Chem. 281:8183–8189.
- Lee S, Lin X, Nam NH, Parang K, Sun G. 2003. Determination of the substrate-docking site of protein tyrosine kinase C-terminal Src kinase. Proc Natl Acad Sci USA. 100:14707–14712.
- Lemmon MA. 2008. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 9:99–111.
- Lemmon MA, Ferguson KM. 2000. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J. 350Pt 1:1–18.
- Lemmon MA, Schlessinger J. 2010. Cell signaling by receptor tyrosine kinases. Cell. 141:1117–1134.
- Levinson NM, Kuchment O, Shen K, Young MA, Koldobskiy M, Karplus M, Cole PA, Kuriyan J. 2006. A Src-like inactive conformation in the abl tyrosine kinase domain. PLoS Biol. 4:e144.
- Levinson NM, Seeliger MA, Cole PA, Kuriyan J. 2008. Structural basis for the recognition of c-Src by its inactivator Csk. Cell. 134:124–134.
- Levy JB, Iba H, Hanafusa H. 1986. Activation of the transforming potential of p60c-Src by a single amino acid change. Proc Natl Acad Sci USA. 83:4228–4232.
- Li W, Young SL, King N, Miller WT. 2008. Signaling properties of a non-metazoan Src kinase and the evolutionary history of Src negative regulation. J Biol Chem. 283:15491–15501.
- Lim WA, Pawson T. 2010. Phosphotyrosine signaling: evolving a new cellular communication system. Cell. 142:661–667.
- Lim WA, Richards FM, Fox RO. 1994. Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature. 372:375–379.
- Lin X, Lee S, Sun G. 2003. Functions of the activation loop in Csk protein-tyrosine kinase. J Biol Chem. 278:24072–24077.
- Lin Y-L, Meng Y, Jiang W, Roux B. 2013. Explaining why Gleevec is a specific and potent inhibitor of Abl kinase. Proc Natl Acad Sci USA. 110:1664–1669.
- Lorenz S, Deng P, Hantschel O, Superti-Furga G, Kuriyan J. 2015. Crystal structure of an SH2-kinase construct of c-Abl and effect of the SH2 domain on kinase activity. Biochem J. 468:283–291.
- Manning G, Plowman GD, Hunter T, Sudarsanam S. 2002a. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci. 27:514–520.
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. 2002b. The protein kinase complement of the human genome. Science. 298:1912–1934.
- Manning G, Young SL, Miller WT, Zhai Y. 2008. The protist, Monosiga brevicollis, has a tyrosine kinase signaling network more elaborate and diverse than found in any known metazoan. Proc Natl Acad Sci USA 105:9674–9679.
- Mano H. 1999. Tec family of protein-tyrosine kinases: an overview of their structure and function. Cytokine Growth Factor Rev. 10:267–280.
- Martin GS. 2004. The road to Src. Oncogene. 23:7910–7917.
- Mayer BJ, Hamaguchi M, Hanafusa H. 1988. Characterization of p47gag-crk, a novel oncogene product with sequence similarity to a putative modulatory domain of protein-tyrosine kinases and phospholipase C. Cold Spring Harb Symp Quant Biol. 53:907–914.
- Mayer BJ, Hirai H, Sakai R. 1995. Evidence that SH2 domains promote processive phosphorylation by protein-tyrosine kinases. Curr Biol. 5:296–305.
- McWhirter JR, Galasso DL, Wang JY. 1993. A coiled-coil oligomerization domain of Bcr is essential for the transforming function of Bcr-Abl oncoproteins. Mol Cell Biol. 13:7587–7595.
- Meharena HS, Chang P, Keshwani MM, Oruganty K, Nene AK, Kannan N, Taylor SS, Kornev AP. 2013. Deciphering the structural basis of eukaryotic protein kinase regulation. PLoS Biol. 11:e1001680.
- Meng Y, Ahuja LG, Kornev AP, Taylor SS, Roux B. 2018. A catalytically disabled double mutant of Src tyrosine kinase can be stabilized into an active-like conformation. J Mol Biol. 430:881–889.
- Meng Y, Pond MP, Roux B. 2017. Tyrosine Kinase Activation and Conformational Flexibility: Lessons from Src-Family Tyrosine Kinases. Acc Chem Res. 50:1193–1201.
- Meng Y, Roux B. 2014. Locking the active conformation of c-Src kinase through the phosphorylation of the activation loop. J Mol Biol. 426:423–435.
- Meng Y, Shukla D, Pande VS, Roux B. 2016. Transition path theory analysis of c-Src kinase activation. Proc Natl Acad Sci USA. 113:9193–9198.
- Miller WT. 2003. Determinants of substrate recognition in nonreceptor tyrosine kinases. Acc Chem Res. 36:393–400.
- Moarefi I, LaFevre-Bernt M, Sicheri F, Huse M, Lee CH, Kuriyan J, Miller WT. 1997. Activation of the Src-family tyrosine kinase Hck by SH3 domain displacement. Nature. 385:650–653.
- Mócsai A, Ruland J, Tybulewicz VLJ. 2010. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 10:387–402.
- Mol CD, Dougan DR, Schneider TR, Skene RJ, Kraus ML, Scheibe DN, Snell GP, Zou H, Sang B-C, Wilson KP. 2004. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J Biol Chem. 279:31655–31663.
- Musacchio A, Noble M, Pauptit R, Wierenga R, Saraste M. 1992. Crystal structure of a Src-homology 3 (SH3) domain. Nature. 359:851–855.
- Nagar B, Bornmann WG, Pellicena P, Schindler T, Veach DR, Miller WT, Clarkson B, Kuriyan J. 2002. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 62:4236–4243.
- Nagar B, Hantschel O, Seeliger M, Davies JM, Weis WI, Superti-Furga G, Kuriyan J. 2006. Organization of the SH3-SH2 unit in active and inactive forms of the c-Abl tyrosine kinase. Mol Cell. 21:787–798.
- Nagar B, Hantschel O, Young MA, Scheffzek K, Veach D, Bornmann W, Clarkson B, Superti-Furga G, Kuriyan J. 2003. Structural basis for the autoinhibition of c-Abl tyrosine kinase. Cell. 112:859–871.
- Noble MEM, Endicott JA, Johnson LN. 2004. Protein kinase inhibitors: insights into drug design from structure. Science. 303:1800–1805.
- Nolen B, Taylor S, Ghosh G. 2004. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 15:661–675.
- Nowell P, Hungerford D. 1960. A minute chromosome in human chronic granulocytic leukemia. Science. 132:1497–1501.
- Ogawa A, Takayama Y, Sakai H, Chong KT, Takeuchi S, Nakagawa A, Nada S, Okada M, Tsukihara T. 2002. Structure of the carboxyl-terminal Src kinase, Csk. J Biol Chem. 277:14351–14354.
- Okada M, Nada S, Yamanashi Y, Yamamoto T, Nakagawa H. 1991. CSK: a protein-tyrosine kinase involved in regulation of Src family kinases. J Biol Chem. 266:24249–24252.
- Okada M, Nakagawa H. 1989. A protein tyrosine kinase involved in regulation of pp60c-Src function. J Biol Chem. 264:20886–20893.
- Oliver AW, Paul A, Boxall KJ, Barrie SE, Aherne GW, Garrett MD, Mittnacht S, Pearl LH. 2006. Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. EMBO J. 25:3179–3190.
- Oppermann H, Levinson AD, Varmus HE, Levintow L, Bishop JM. 1979. Uninfected vertebrate cells contain a protein that is closely related to the product of the avian sarcoma virus transforming gene (Src). Proc Natl Acad Sci USA 76:1804–1808.
- Overduin M, Rios CB, Mayer BJ, Baltimore D, Cowburn D. 1992. Three-dimensional solution structure of the Src homology 2 domain of c-abl. Cell. 70:697–704.
- Ozkirimli E, Post CB. 2006. Src kinase activation: A switched electrostatic network. Protein Sci. 15:1051–1062.
- Pan Z, Scheerens H, Li S-J, Schultz BE, Sprengeler PA, Burrill LC, Mendonca RV, Sweeney MD, Scott KCK, Grothaus PG, et al. 2007. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem. 2:58–61.
- Park H, Wahl MI, Afar DE, Turck CW, Rawlings DJ, Tam C, Scharenberg AM, Kinet JP, Witte ON. 1996. Regulation of Btk function by a major autophosphorylation site within the SH3 domain. Immunity. 4:515–525.
- Pawson T, Kofler M. 2009. Kinome signaling through regulated protein-protein interactions in normal and cancer cells. Curr Opin Cell Biol. 21:147–153.
- Pawson T, Nash P. 2000. Protein-protein interactions define specificity in signal transduction. Genes Dev. 14:1027–1047.
- Pellicena P, Miller WT. 2001. Processive phosphorylation of p130Cas by Src depends on SH3-polyproline interactions. J Biol Chem. 276:28190–28196.
- Pellicena P, Stowell KR, Miller WT. 1998. Enhanced phosphorylation of Src family kinase substrates containing SH2 domain binding sites. J Biol Chem. 273:15325–15328.
- Piao X, Paulson R, van der Geer P, Pawson T, Bernstein A. 1996. Oncogenic mutation in the Kit receptor tyrosine kinase alters substrate specificity and induces degradation of the protein tyrosine phosphatase SHP-1. Proc Natl Acad Sci USA 93:14665–14669.
- Pike ACW, Rellos P, Niesen FH, Turnbull A, Oliver AW, Parker SA, Turk BE, Pearl LH, Knapp S. 2008. Activation segment dimerization: a mechanism for kinase autophosphorylation of non-consensus sites. EMBO J. 27:704–714.
- Pincus D, Letunic I, Bork P, Lim WA. 2008. Evolution of the phospho-tyrosine signaling machinery in premetazoan lineages. Proc Natl Acad Sci USA 105:9680–9684.
- Pluk H, Dorey K, Superti-Furga G. 2002. Autoinhibition of c-Abl. Cell. 108:247–259.
- Porter M, Schindler T, Kuriyan J, Miller WT. 2000. Reciprocal regulation of Hck activity by phosphorylation of Tyr(527) and Tyr(416). Effect of introducing a high affinity intramolecular SH2 ligand. J Biol Chem. 275:2721–2726.
- Qiu H, Miller WT. 2002. Regulation of the nonreceptor tyrosine kinase Brk by autophosphorylation and by autoinhibition. J Biol Chem. 277:34634–34641.
- Rawlings DJ, Saffran DC, Tsukada S, Largaespada DA, Grimaldi JC, Cohen L, Mohr RN, Bazan JF, Howard M, Copeland NG. 1993. Mutation of unique region of Bruton's tyrosine kinase in immunodeficient XID mice. Science. 261:358–361.
- Register AC, Leonard SE, Maly DJ. 2014. SH2-catalytic domain linker heterogeneity influences allosteric coupling across the SFK family. Biochemistry. 53:6910–6923.
- Resh MD. 1994. Myristylation and palmitylation of Src family members: the fats of the matter. Cell. 76:411–413.
- Richter DJ, King N. 2013. The genomic and cellular foundations of animal origins. Annu Rev Genet. 47:509–537.
- Sadowski I, Stone JC, Pawson T. 1986. A noncatalytic domain conserved among cytoplasmic protein-tyrosine kinases modifies the kinase function and transforming activity of Fujinami sarcoma virus P130gag-fps. Mol Cell Biol. 6:4396–4408.
- Saleh T, Rossi P, Kalodimos CG. 2017. Atomic view of the energy landscape in the allosteric regulation of Abl kinase. Nat Struct Mol Biol. 24:893–901.
- Salim K, Bottomley MJ, Querfurth E, Zvelebil MJ, Gout I, Scaife R, Margolis RL, Gigg R, Smith CI, Driscoll PC, et al. 1996. Distinct specificity in the recognition of phosphoinositides by the pleckstrin homology domains of dynamin and Bruton’s tyrosine kinase. EMBO J. 15:6241–6250.
- Sawyers CL. 2003. Opportunities and challenges in the development of kinase inhibitor therapy for cancer. Genes Dev. 17:2998–3010.
- Schindler T, Bornmann W, Pellicena P, Miller WT, Clarkson B, Kuriyan J. 2000. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science. 289:1938–1942.
- Schindler T, Sicheri F, Pico A, Gazit A, Levitzki A, Kuriyan J. 1999. Crystal structure of Hck in complex with a Src family-selective tyrosine kinase inhibitor. Mol Cell. 3:639–648.
- Schmitz R, Baumann G, Gram H. 1996. Catalytic specificity of phosphotyrosine kinases Blk, Lyn, c-Src and Syk as assessed by phage display. J Mol Biol. 260:664–677.
- Schultheiss KP, Craddock BP, Suga H, Miller WT. 2014. Regulation of Src and Csk nonreceptor tyrosine kinases in the filasterean Ministeria vibrans. Biochemistry. 53:1320–1329.
- Schultheiss KP, Suga H, Ruiz-Trillo I, Miller WT. 2012. Lack of Csk-mediated negative regulation in a unicellular SRC kinase. Biochemistry. 51:8267–8277.
- Scott JD, Pawson T. 2000. Cell communication: the inside story. Sci Am. 282:72–79.
- Seeliger MA, Nagar B, Frank F, Cao X, Henderson MN, Kuriyan J. 2007. c-Src binds to the cancer drug imatinib with an inactive Abl/c-Kit conformation and a distributed thermodynamic penalty. Structure. 15:299–311.
- Seeliger MA, Ranjitkar P, Kasap C, Shan Y, Shaw DE, Shah NP, Kuriyan J, Maly DJ. 2009. Equally potent inhibition of c-Src and Abl by compounds that recognize inactive kinase conformations. Cancer Res. 69:2384–2392.
- Segawa Y, Suga H, Iwabe N, Oneyama C, Akagi T, Miyata T, Okada M. 2006. Functional development of Src tyrosine kinases during evolution from a unicellular ancestor to multicellular animals. Proc Natl Acad Sci USA 103:12021–12026.
- Shah NH, Löbel M, Weiss A, Kuriyan J. 2018. Fine-tuning of substrate preferences of the Src-family kinase Lck revealed through a high-throughput specificity screen. Elife. 7:e35190.
- Shah NH, Wang Q, Yan Q, Karandur D, Kadlecek TA, Fallahee IR, Russ WP, Ranganathan R, Weiss A, Kuriyan J. 2016. An electrostatic selection mechanism controls sequential kinase signaling downstream of the T cell receptor. Elife.5:e20105.
- Shah NP, Nicoll JM, Nagar B, Gorre ME, Paquette RL, Kuriyan J, Sawyers CL. 2002. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2:117–125.
- Shan Y, Seeliger MA, Eastwood MP, Frank F, Xu H, Jensen MØ, Dror RO, Kuriyan J, Shaw DE. 2009. A conserved protonation-dependent switch controls drug binding in the Abl kinase. Proc Natl Acad Sci USA 106:139–144.
- Shukla D, Meng Y, Roux B, Pande VS. 2014. Activation pathway of Src kinase reveals intermediate states as targets for drug design. Nat Commun. 5:3397
- Sicheri F, Moarefi I, Kuriyan J. 1997. Crystal structure of the Src family tyrosine kinase Hck. Nature. 385:602–609.
- Skaggs BJ, Gorre ME, Ryvkin A, Burgess MR, Xie Y, Han Y, Komisopoulou E, Brown LM, Loo JA, Landaw EM, et al. 2006. Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant BCR-ABL mutants. Proc Natl Acad Sci USA 103:19466–19471.
- Smith CIE. 2017. From identification of the BTK kinase to effective management of leukemia. Oncogene. 36:2045–2053.
- Sondhi D, Xu W, Songyang Z, Eck MJ, Cole PA. 1998. Peptide and protein phosphorylation by protein tyrosine kinase Csk: insights into specificity and mechanism. Biochemistry. 37:165–172.
- Songyang Z, Carraway KL, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng C. 1995. Catalytic specificity of protein-tyrosine kinases is critical for selective signalling. Nature. 373:536–539.
- Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ. 1993. SH2 domains recognize specific phosphopeptide sequences. Cell. 72:767–778.
- Songyang Z, Shoelson SE, McGlade J, Olivier P, Pawson T, Bustelo XR, Barbacid M, Sabe H, Hanafusa H, Yi T. 1994. Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Mol Cell Biol. 14:2777–2785.
- Spector DH, Varmus HE, Bishop JM. 1978. Nucleotide sequences related to the transforming gene of avian sarcoma virus are present in DNA of uninfected vertebrates. Proc Natl Acad Sci USA 75:4102–4106.
- Srivastava M, Simakov O, Chapman J, Fahey B, Gauthier MEA, Mitros T, Richards GS, Conaco C, Dacre M, Hellsten U, et al. 2010. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature. 466:720–726.
- Stehelin D, Varmus HE, Bishop JM, Vogt PK. 1976. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 260:170–173.
- Suga H, Dacre M, de Mendoza A, Shalchian-Tabrizi K, Manning G, Ruiz-Trillo I. 2012. Genomic survey of premetazoans shows deep conservation of cytoplasmic tyrosine kinases and multiple radiations of receptor tyrosine kinases. Sci Signal. 5:ra35.
- Suga H, Miller WT. 2018. Src signaling in a low-complexity unicellular kinome. Sci Rep. 8:5362.
- Suga H, Torruella G, Burger G, Brown MW, Ruiz-Trillo I. 2014. Earliest Holozoan expansion of phosphotyrosine signaling. Mol Biol Evol. 31:517–528.
- Takesono A, Finkelstein LD, Schwartzberg PL. 2002. Beyond calcium: new signaling pathways for Tec family kinases. J Cell Sci. 115:3039–3048.
- Taskinen B, Ferrada E, Fowler DM. 2017. Early emergence of negative regulation of the tyrosine kinase Src by the C-terminal Src kinase. J Biol Chem. 292:18518–18529.
- Thomas JD, Sideras P, Smith CI, Vorechovský I, Chapman V, Paul WE. 1993. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science. 261:355–358.
- Thomas JW, Ellis B, Boerner RJ, Knight WB, White GC, Schaller MD. 1998. SH2- and SH3-mediated interactions between focal adhesion kinase and Src. J Biol Chem. 273:577–583.
- Thomas SM, Brugge JS. 1997. Cellular functions regulated by Src family kinases. Annu Rev Cell Dev Biol. 13:513–609.
- Till JH, Chan PM, Miller WT. 1999. Engineering the substrate specificity of the Abl tyrosine kinase. J Biol Chem. 274:4995–5003.
- Tsukada S, Saffran DC, Rawlings DJ, Parolini O, Allen RC, Klisak I, Sparkes RS, Kubagawa H, Mohandas T, Quan S, et al. 1993. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell. 72:279–290.
- Ubersax JA, Ferrell JE. 2007. Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 8:530–541.
- Väliaho J, Smith CIE, Vihinen M. 2006. BTKbase: the mutation database for X-linked agammaglobulinemia. Hum Mutat. 27:1209–1217.
- Vetrie D, Vorechovský I, Sideras P, Holland J, Davies A, Flinter F, Hammarström L, Kinnon C, Levinsky R, Bobrow M. 1993. The gene involved in X-linked agammaglobulinaemia is a member of the Src family of protein-tyrosine kinases. Nature. 361:226–233.
- von Rauszendorf F, de Ruiter A, Leonard TA. 2017. A switch in nucleotide affinity governs activation of the Src and Tec family kinases. Sci Rep.Rep. 7:17405.
- Waksman G, Kominos D, Robertson SC, Pant N, Baltimore D, Birge RB, Cowburn D, Hanafusa H, Mayer BJ, Overduin M, et al. 1992. Crystal structure of the phosphotyrosine recognition domain SH2 of v-Src complexed with tyrosine-phosphorylated peptides. Nature. 358:646–653.
- Waksman G, Kumaran S, Lubman O. 2004. SH2 domains: role, structure and implications for molecular medicine. Expert Rev Mol Med. 6:1–18.
- Waksman G, Shoelson SE, Pant N, Cowburn D, Kuriyan J. 1993. Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell. 72:779–790.
- Wang JYJ. 2014. The capable ABL: what is its biological function? Mol Cell Biol. 34:1188–1197.
- Wang Q, Vogan EM, Nocka LM, Rosen CE, Zorn JA, Harrison SC, Kuriyan J. 2015. Autoinhibition of Bruton’s tyrosine kinase (Btk) and activation by soluble inositol hexakisphosphate. Elife.4:e06074.
- Wang Q, Zorn JA, Kuriyan J. 2014. A structural atlas of kinases inhibited by clinically approved drugs. Meth Enzymol. 548:23–67.
- Weng Z, Rickles RJ, Feng S, Richard S, Shaw AS, Schreiber SL, Brugge JS. 1995. Structure-function analysis of SH3 domains: SH3 binding specificity altered by single amino acid substitutions. Mol Cell Biol. 15:5627–5634.
- Williams JC, Weijland A, Gonfloni S, Thompson A, Courtneidge SA, Superti-Furga G, Wierenga RK. 1997. The 2.35 A crystal structure of the inactivated form of chicken Src: a dynamic molecule with multiple regulatory interactions. J Mol Biol. 274:757–775.
- Wilson C, Agafonov RV, Hoemberger M, Kutter S, Zorba A, Halpin J, Buosi V, Otten R, Waterman D, Theobald DL, Kern D. 2015. Kinase dynamics. Using ancient protein kinases to unravel a modern cancer drug’s mechanism. Science. 347:882–886.
- Witte ON, Dasgupta A, Baltimore D. 1980. Abelson murine leukaemia virus protein is phosphorylated in vitro to form phosphotyrosine. Nature. 283:826–831.
- Wong L, Lieser SA, Miyashita O, Miller M, Tasken K, Onuchic JN, Adams JA, Woods VL, Jennings PA. 2005. Coupled motions in the SH2 and kinase domains of Csk control Src phosphorylation. J Mol Biol. 351:131–143.
- Woyach JA, Furman RR, Liu T-M, Ozer HG, Zapatka M, Ruppert AS, Xue L, Li DH-H, Steggerda SM, Versele M, et al. 2014. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med. 370:2286–2294.
- Wu J, Li W, Craddock BP, Foreman KW, Mulvihill MJ, Ji Q, Miller WT, Hubbard SR. 2008. Small-molecule inhibition and activation-loop trans-phosphorylation of the IGF1 receptor. EMBO J. 27:1985–1994.
- Wu W-I, Voegtli WC, Sturgis HL, Dizon FP, Vigers GPA, Brandhuber BJ. 2010. Crystal structure of human AKT1 with an allosteric inhibitor reveals a new mode of kinase inhibition. PLoS One.5:e12913.
- Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, Marzinzik AL, Pelle X, Donovan J, Zhu W, et al. 2017. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature. 543:733–737.
- Xiong X, Cui P, Hossain S, Xu R, Warner B, Guo X, An X, Debnath AK, Cowburn D, Kotula L. 2008. Allosteric inhibition of the nonMyristoylated c-Abl tyrosine kinase by phosphopeptides derived from Abi1/Hssh3bp1. Biochim Biophys Acta. 1783:737–747.
- Xu Q, Malecka KL, Fink L, Jordan EJ, Duffy E, Kolander S, Peterson JR, Dunbrack RL. 2015. Identifying three-dimensional structures of autophosphorylation complexes in crystals of protein kinases. Sci Signal. 8:rs13.
- Xu W, Doshi A, Lei M, Eck MJ, Harrison SC. 1999. Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol Cell. 3:629–638.
- Xu W, Harrison SC, Eck MJ. 1997. Three-dimensional structure of the tyrosine kinase c-Src. Nature. 385:595–602.
- Yamaguchi H, Hendrickson WA. 1996. Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature. 384:484–489.
- Young MA, Gonfloni S, Superti-Furga G, Roux B, Kuriyan J. 2001. Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell. 105:115–126.
- Yu H, Chen JK, Feng S, Dalgarno DC, Brauer AW, Schreiber SL. 1994. Structural basis for the binding of proline-rich peptides to SH3 domains. Cell. 76:933–945.
- Yu H, Rosen MK, Shin TB, Seidel-Dugan C, Brugge JS, Schreiber SL. 1992. Solution structure of the SH3 domain of Src and identification of its ligand-binding site. Science. 258:1665–1668.
- Zhao X, Ghaffari S, Lodish H, Malashkevich VN, Kim PS. 2002. Structure of the Bcr-Abl oncoprotein oligomerization domain. Nat Struct Biol. 9:117–120.
- Zheng J, Trafny EA, Knighton DR, Xuong NH, Taylor SS, Ten Eyck LF, Sowadski JM. 1993. 2.2 A refined crystal structure of the catalytic subunit of cAMP-dependent protein kinase complexed with MnATP and a peptide inhibitor. Acta Crystallogr D Biol Crystallogr. 49:362–365.
- Zimmermann J, Buchdunger E, Mett H, Meyer T, Lydon NB. 1997. Potent and selective inhibitors of the Abl-kinase: phenylamino-pyrimidine (PAP) derivatives. Bioorg Med Chem Lett. 7:187–192.
- Zorba A, Buosi V, Kutter S, Kern N, Pontiggia F, Cho Y-J, Kern D. 2014. Molecular mechanism of Aurora A kinase autophosphorylation and its allosteric activation by TPX2. Elife. 3:e02667.