
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Protein aggregation is implicated in multiple diseases, so-called proteinopathies, ranging from neurodegenerative disorders such as Alzheimer’s disease and Parkinson’s disease (PD) to type 2 diabetes mellitus and sickle cell disease (SCD). The structure of the protein aggregates and the kinetics and mechanisms of aggregation have been the object of intense research over the years toward the development of therapeutic routes, including the design of aggregation inhibitors. Nonetheless, the rational design of drugs targeting aggregation inhibition remains a challenging endeavor because of multiple, disease-specific factors, including an incomplete understanding of protein function, the multitude of toxic and non-toxic protein aggregates, the lack of specific drug binding targets, discrepant action mechanisms of aggregation inhibitors, or a low selectivity, specificity, and/or drug potency, reflected in the high concentrations required for some inhibitors to be effective. Herein, we provide a perspective of this therapeutic route with emphasis on small molecules and peptide-based drugs in two diverse diseases, PD and SCD, aiming at establishing links among proposed aggregation inhibitors. The small and large length-scale regimes of the hydrophobic effect are discussed in light of the importance of hydrophobic interactions in proteinopathies. Some simulation results are reported on model peptides, illustrating the impact of hydrophobic and hydrophilic groups in water’s hydrogen-bond network with an impact on drug binding. The seeming importance of aromatic rings and hydroxyl groups in protein-aggregation-inhibitor-drugs is emphasized along with the challenges associated with some inhibitors, limiting their development into effective therapeutic options, and questioning the potential of this therapeutic route.
Introduction
Protein aggregation is implicated in several human pathologies, ranging from sickle cell disease (SCD) (Pauling et al. Citation1949; Ingram Citation1956, Citation1957; Noguchi and Schechter Citation1985; Eaton and Hofrichter Citation1990), a red blood cell disorder, to various neurodegenerative diseases (NDs) (Aguzzi and O’Connor Citation2010; Sweeney et al. Citation2017), such as Alzheimer’s, Huntington’s, or Parkinson’s disease (PD), commonly known as proteinopathies or protein conformational diseases. Although the root cause of most NDs, including PD, is not exactly known, compelling evidence (Spillantini et al. Citation1997, Citation1998; Feany and Bender Citation2000; Masliah et al. Citation2000; Bucciantini et al. Citation2002; Soto Citation2003) posits a relationship with protein misfolding and aggregation, resulting in the formation of abnormal protein aggregates, generally referred to as amyloids (Trojanowski and Lee Citation2003; Aguzzi and O’Connor Citation2010; Sweeney et al. Citation2017). SCD, on the other hand, is probably the most well-known proteinopathy, coined the first molecular disease by Pauling et al. (Citation1949), being associated with the reversible aggregation of a mutated form of hemoglobin (i.e. sickle cell hemoglobin) most common in some parts of sub-Saharan Africa (Williams Citation2016) and known to confer protection against malaria (Ferreira et al. Citation2011).
Several therapeutic routes have been concomitantly explored both in SCD and NDs in general. A common route includes the reduction of the monomeric precursor protein. This is because the kinetics of aggregation of both amyloid and HbS fibers depends on the concentration of the precursor as well as on the cell environment. Hydroxyurea, for instance, the most widely used drug to treat SCD, increases the levels of fetal hemoglobin (HbF), which does not polymerize (Charache et al. Citation1995; Mehanna Citation2001; Vinjamur et al. Citation2018). The increase in HbF decreases the concentration of HbS, enhancing the delay time that precedes fiber growth, ultimately reducing the percentage of sickled erythrocytes.
Preventing or reducing protein aggregation in SCD (Eaton and Bunn Citation2017; Tisdale et al. Citation2020; Eaton Citation2022) and NDs through a direct disruption of the aggregation mechanism is another possible therapeutic strategy since it could potentially prevent the diseases’ progression without compromising the biological function of the protein. Ultimately, this route requires the rational design of drugs that can hinder or delay the aggregation process and, thus, a molecular knowledge of the aggregation mechanism of the proteins involved in the disease.
Herein, we provide an overview of the main features of two very different proteinopathies, PD (section “Parkinson’s disease”) and SCD (section “Sickle cell disease”), along with some of the molecules, including small peptides, that have been found to exhibit some aggregation inhibitory activity and their proposed action mechanisms (section “Protein aggregation inhibitors”).
Protein aggregation, much like protein folding, is deeply connected with hydrophobic interactions (Kauzmann Citation1959; Tanford Citation1980; Dill Citation1990), which in turn are influenced by the surrounding environment, driving the conformational search toward a thermodynamic stable or native state. Hence, hydrophobic interactions are also discussed (section “Hydrophobic effect and protein aggregation”) concerning protein aggregation and their influence on the design of aggregation inhibitors (section “Hydrophobic effect and drug design”).
The main goal of this review is twofold: (i) expose putative similarities among aggregation inhibitors proposed for distinct proteinopathies, including their seeming “universal” or non-specific action mechanism, and (ii) point out some of the main challenges concerning drug design associated with proteinopathies, with emphasis on the harnessing of the hydrophobic effect and the understanding of the action mechanism of some inhibitors and its relationship with specific chemical groups long reported to be pivotal to protein aggregation inhibition.
Parkinson’s disease
PD is the second most prevalent disease among NDs, next to Alzheimer’s disease, and the most common movement disorder. PD’s etiology has been associated with the formation of cytotoxic oligomers (Breydo et al. Citation2012; Cremades et al. Citation2017), of which α-synuclein (α-syn) (Maroteaux et al. Citation1988) is the primary component. These prefibrillar transient oligomers accumulate in intracellular inclusions called Lewy bodies and Lewy neurites (Spillantini et al. Citation1997, Citation1998), and are now (Goldberg and Lansbury Citation2000; Caughey and Lansbury Citation2003; Karpinar et al. Citation2009; Winner et al. Citation2011) thought to be the main culprit, as opposed to the misfolded monomer and mature (insoluble) fibrils, in the loss of nigral dopaminergic neurons (Feany and Bender Citation2000; Masliah et al. Citation2000). Despite the cell toxicity mechanism remains elusive, this is believed to be connected with perturbations in the neuronal membrane (Lashuel et al. Citation2002; Winner et al. Citation2011; Goedert et al. Citation2017). In addition, α-syn aggregates spread through a prion-like mechanism across brain cells, accelerating disease’s progression (Masuda-Suzukake et al. Citation2013; Ma et al. Citation2019; Cascella et al. Citation2022).
α-Synuclein aggregation
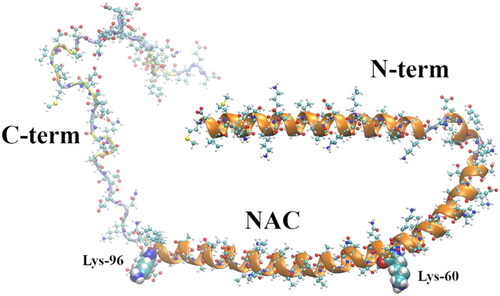
α-syn is a 140 amino acid intrinsically disordered protein (IDP) mainly expressed in the central nervous system and whose function is not completely understood, although several putative functions have been put forward (Fink Citation2006; Cheng et al. Citation2011; Breydo et al. Citation2012; Lashuel et al. Citation2013; Lee et al. Citation2008; Norris et al. Citation2004). α-syn is comprised of three distinct domains (see ), the N-terminal (N-term), a membrane-binding domain that tends to form α-helices, encompassing amino acids 1–60 (Davidson et al. Citation1998; Eliezer et al. Citation2001); the so-called non-amyloid-β component (NAC) (Uéda et al. Citation1993), a highly hydrophobic and amyloidogenic domain comprising amino acids 61–95; and the C-terminal (C-term) domain, a more disordered region comprised of the amino acids 96–140 (Breydo et al. Citation2012). Besides PD, α-syn aggregates are at the center of dementia with Lewy bodies (Baba et al. Citation1998) and multiple system atrophy (Spillantini et al. Citation1998; Tu et al. Citation1998), jointly known as synucleinopathies or Lewy body diseases (Trojanowski and Lee Citation2003; Uversky Citation2007; Goedert et al. Citation2017).
Figure 1. Membrane-bound α-syn structure (2kkw.pdb) (Rao et al. Citation2010) showing its three regions, N-term (positively charged), the NAC (highly hydrophobic), and the C-term (negatively charged). The last amino acid of the N-term domain (Lys-60) and the first amino acid of the C-term domain (Lys-96), encompassing the NAC, are shown as vdW spheres. The chain colors correspond to the following secondary structures: α-helix (orange), turn (yellow), and coil (ice blue).
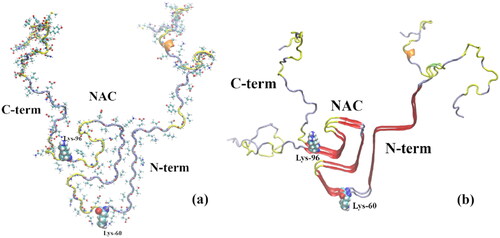
The structure of α-syn amyloid polymorph fibrils has been assessed by cryo-electron microscopy and NMR (Tuttle et al. Citation2016; Li, Ge, et al. Citation2018; Li, Zhao, et al. Citation2018; Ni et al. Citation2019) (see ) including membrane-bound α-syn (Ulmer et al. Citation2005; Rao et al. Citation2010) (). In addition, the structure of toxic oligomers (Cremades et al. Citation2012; Bengoa-Vergniory et al. Citation2017) has been assessed through multiple experimental techniques, including circular dichroism, small-angle X-ray scattering, and atomic force microscopy-infrared spectroscopy (Hong et al. Citation2008; Ruggeri et al. Citation2015; Zhou and Kurouski Citation2020).
Figure 2. (a) α-syn monomer extracted from the α-syn experimental (NMR spectroscopy) protofibril reported by Tuttle et al. (Citation2016) (2n0a.pdb). (b) α-syn dimer extracted from the same experimental protofibril; cartoon representation showing a β-sheet-rich region in the NAC (amino acids 61–95) domain; some β-sheet is also visible in the N-term region. The last amino acid of the N-term domain (Lys-60) and the first amino acid of the C-term domain (Lys-96), encompassing the NAC, are shown as vdW spheres. The chain colors correspond to the following secondary structures: 310 helix (orange), β-sheet (red), turn (yellow), and coil (ice blue).
The transient (Zurlo et al. Citation2021) and heterogeneous aggregational nature of α-syn poses a challenge to a comprehensive understanding of the aggregation mechanism(s) and kinetics (Arter et al. Citation2020), and their relationship with the onset of PD (Breydo et al. Citation2012; Cremades et al. Citation2017; Cremades and Dobson Citation2018). Nucleation–polymerization and nucleation–conversion–polymerization kinetic mechanisms have been proposed to describe the formation of soluble oligomeric species and the growth into insoluble mature fibrils (Cremades et al. Citation2017). The latter foresees a conversion stage where disordered oligomers with little or no stable β-sheet structure convert into more orderly and stable oligomers before they grow into fibrils (Cremades et al. Citation2017). Furthermore, the trigger behind the nucleation stage is thought to be associated with a conformational transformation (i.e. misfolding) of the natively unfolded protein into a more aggregation-prone partially folded intermediate (Uversky et al. Citation2001; Uversky and Eliezer Citation2009). The latter induces the formation of β-sheet-rich structures, a hallmark of the cytotoxic oligomers, nearly absent in the native α-syn. Further, the kinetics of aggregation is promoted by its binding with lipid membranes (Galvagnion et al. Citation2015, Citation2016), in addition to the temperature, pH, or osmolytes.
In addition to the wild type α-syn, several pathogenic missense mutations (Polymeropoulos et al. Citation1997; Krüger et al. Citation1998; Zarranz et al. Citation2004) (e.g. A53T, A30P, and E46K) have been implicated in early-onset familial PD, as opposed to the most common sporadic form of PD, with some mutated forms accelerating the kinetics of oligomerization (Conway et al. Citation1998; El-Agnaf et al. Citation1998; Li et al. Citation2001; Greenbaum et al. Citation2005; Ghosh et al. Citation2013).
Hydrophobic interactions (section “Hydrophobic effect and protein aggregation”), although insufficient to promote the formation of secondary and tertiary structures, are thought to be pivotal to IDP’s aggregation. Thus, several domains within the NAC region have been shown to be key to the aggregation process (Bodles et al. Citation2001; Giasson et al. Citation2001; Du et al. Citation2003; El-Agnaf et al. Citation2004; Bertoncini et al. Citation2005; Madine et al. Citation2008; Eugene et al. Citation2014; Rodriguez et al. Citation2015). That is to say that oligomerization decreases when specific changes in this region are promoted, opening the way for the development of drugs targeting these NAC sub-domains or stabilizing conformations of monomeric α-syn where intramolecular interactions shield the NAC region (Bertoncini et al. Citation2005; Dedmon et al. Citation2005; Esteban-Martín et al. Citation2013). In addition, small domains in the NAC flanking regions have also been associated with α-syn’s aggregation mechanism and function (Afitska et al. Citation2017; Doherty et al. Citation2020; Landeck et al. Citation2020; Stephens et al. Citation2020; Tripathi Citation2020; Farzadfard et al. Citation2022; Ulamec et al. Citation2022), including the domain comprised by amino acids 46–53 where the abovementioned missense mutations, implicated in familial PD, are found. The fact that these mutations are located in the N-term region suggests this segment could be equally important to the aggregation process. shows a β-sheet-rich domain in this region of the N-term. This is also consistent with molecular dynamics (MD) results which indicate that the dimerization process already involves regions beyond the NAC segment (Guzzo et al. Citation2022). Some of the abovementioned studies (Doherty et al. Citation2020; Ulamec et al. Citation2022) identified putative master-controller sequence motifs in the N-term region, namely, a segment formed by residues 36–42 coined “P1”, that may regulate α-syn aggregation. These segments, could, therefore, constitute key alternative (to NAC segments) targets to inhibit the aggregation of α-syn (Doherty et al. Citation2020; Tripathi Citation2020; Horsley et al. Citation2022).
Several other proteins, IDPs and globular proteins, are connected with the formation and deposition of aggregates implicated in different NDs. Well-known examples include aggregates of mutated forms of the Huntingtin protein, forming intranuclear inclusions in Huntington’s disease (Davies et al. Citation1997), an autosomal dominant inherited disease, and the amyloid-β peptide and the tau protein, which form, respectively, extracellular amyloid plaques and intracellular neurofibrillary tangles in the brain, implicated in Alzheimer’s disease (Aguzzi and O’Connor Citation2010; Sweeney et al. Citation2017). Although not discussed in this work, many of the drugs discussed in section “Protein aggregation inhibitors” (e.g. polyphenols) were found to inhibit aggregation of the proteins and peptides implicated in these diseases.
Sickle cell disease
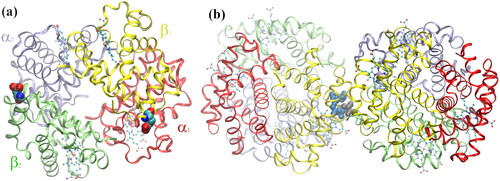
At the other end of proteinopathies’ spectrum is SCD, an autosomal recessive inherited disorder that affects hemoglobin, the protein responsible for the transport of O2 and CO2 in the red blood cells. Opposite to IDPs, hemoglobin (normal adult Hb; HbA) has well-defined secondary, tertiary, and quaternary structures (Perutz et al. Citation1960). HbA is an allosteric protein, existing in low-oxygen affinity and high-oxygen affinity quaternary conformational states, known as the T-state (tense or deoxyhemoglobin) and the R-state (relaxed or oxyhemoglobin), respectively. HbA is composed of four polypeptide chains, two α subunits (α-globin) and two β subunits (β-globin); each α-globin is formed by 141 amino acids and a Heme group whereas β-globin is formed by 146 amino acids and a Heme group (see ).
Figure 3. (a) Normal deoxygenated (T-state) hemoglobin, deoxy-HbA (2dn2.pdb) (Park et al. Citation2006), showing the four heme groups and the Glu-β6 amino acid in β1 and β2, replaced by Val-β6 in HbS; (b) sickle-cell deoxy-HbS dimer (2HBS.pdb) (Harrington et al. Citation1997) showing the Val-β6 (grey spheres) in 2β2, lodged in a hydrophobic cavity in 1β1 formed by Ala-β70, Phe-β85, and Leu-β88 (blue spheres) (Wishner et al. Citation1975; Dykes et al. Citation1979; Padlan and Love Citation1985a, Citation1985b).
HbS aggregation
SCD is caused by a monogenic mutation in the β-globin gene that results in the substitution of a surface glutamic acid (charge −1) for valine (neutral and hydrophobic) at the 6th position of the β-globins of HbA (Pauling et al. Citation1949; Ingram Citation1956; Citation1957; Noguchi and Schechter Citation1985; Eaton and Hofrichter Citation1990). This mutation, while not significantly changing the conformation of HbS (Perutz et al. Citation1951), relative to HbA, reduces the solubility (Eaton and Hofrichter Citation1990) of deoxygenated sickle cell hemoglobin (deoxy-HbS) from 7.0 g·cm−3 to 1.7 g·cm−3, inducing the (reversible) aggregation of deoxy-HbS into 14-stranded helical fibers (Eaton and Hofrichter Citation1990; Dykes et al. Citation1979; Harrington et al. Citation1977; Padlan and Love Citation1985b, Citation1985a; Eaton and Bunn Citation2017; Tisdale et al. Citation2020; Perutz Citation1970; Wishner et al. Citation1975; Edelstein Citation1980; Dykes et al. Citation1978). These fibers are ∼20 nm in diameter and distort the red blood cells into a stiff, non-deformable sickle-like shape, disrupting microcirculation and causing hemolysis (Kato et al. Citation2018). Vaso-occlusion is responsible for pain crisis and organ failure, which ultimately can lead to death (Piel et al. Citation2017).
HbS fibers involve a lateral contact where Val-β6 has its hydrophobic side chain lodged in a hydrophobic cavity of a neighbor HbS tetramer (see ). Further, the structure of the fibers has been shown to be similar to that of the deoxy-HbS crystal (Nagel et al. Citation1980). The kinetics of polymerization of HbS is characterized by a delay time and an exceedingly large dependence of the HbS intra-cellular concentration (Adachi and Asakura Citation1978; Eaton and Hofrichter Citation1990, Citation1987; Hofrichter et al. Citation1974). This delay time has been shown to be highly correlated with HbS supersaturation (Cellmer et al. Citation2016). The kinetics is thought to proceed via a double nucleation mechanism (Ferrone et al. Citation1985a, Citation1985b) involving a stochastic homogeneous nucleation stage (Galkin et al. Citation2007) in which the formation of HbS fibers occurs, followed by a heterogeneous nucleation stage involving the nucleation of additional polymers on the surface of the original fibers (Samuel et al. Citation1990). A kinetic model based on classic nucleation theory, which accounts for such a mechanism, aimed at probing the effectiveness of potential anti-sickling drug candidates was recently reported (Lu et al. Citation2019). Another recent kinetic study found the HbS polymerization process to be a rather rapid and inefficient process, namely, fiber growth, re-opening the window for drugs directly targeting aggregation (Castle et al. Citation2019).
Whereas in vitro polymerization is commonly studied in a high concentration phosphate buffer, allowing reducing the solubility of HbS (Itano Citation1953) and therefore the amount of HbS required to observe polymerization, a similar structure (Wang et al. Citation2000) and double nucleation mechanism is seemingly found. Thus, although some differences have been reported (see for instance Chen et al. Citation2004 and references therein) polymerization is characterized by a lag time, similar to that observed at physiological conditions (Adachi and Asakura Citation1978, Citation1979). A similar solubility decrease is found in 2,3-bisphosphoglycerate (2,3-DPG) solutions (Poillon et al. Citation1986, Citation1995; Poillon and Kim Citation1990); 2,3-DPG is an allosteric effector found in erythrocytes that enhances oxygen delivery, shifting the hemoglobin R-T equilibrium to the T conformational state, thus, enhancing aggregation by increasing the concentration of deoxy-HbS. In addition, 2,3-DPG stabilizes the fibers by decreasing the solubility of HbS (Eaton and Bunn Citation2017).
Upon oxygenation, the fibers disassemble without a delay period and most (5–10% erythrocytes sickle irreversibly, Dean and Schechter Citation1978a) erythrocytes recover their biconcave disk shape (Eaton and Hofrichter Citation1990). However, the rate of uptake of O2 of deoxygenated sickled erythrocytes (82 ± 4.7 ms) is slower than that of normal erythrocytes (135 ± 17.6 ms), reflecting the time of depolymerization, among other possible factors (Harrington et al. Citation1977; Dean and Schechter Citation1978a). These sickling–unsickling repeated cycles possibly damage the erythrocytes’ membrane leading ultimately to extra- and intravascular hemolysis (Eaton and Hofrichter Citation1990; Kato et al. Citation2017).
Concerning the molecular nature of the aggregation, this is believed to be primarily associated with hydrophobic interactions because of the nature of the abovementioned lateral contact (Eaton and Hofrichter Citation1990; Cao and Ferrone Citation1997; Wang and Ferrone Citation2013). A negative free energy around −3 kcal·mol−1 along with a nearly zero enthalpy was found for the polymerization (i.e. gelation) process, in a 0.15 M potassium phosphate solution at 37 °C (Ross et al. Citation1977). The solubility was found to decrease with increasing temperature, with a minimum (0.16 g cm−3) at 37 °C, increasing at higher temperatures. A monomer–polymer contact binding free energy of −7.5 kcal·mol−1 was also reported by Cao and Ferrone (Citation1997) from nucleation theory and kinetic rate measurements. Assuming each Val-β6 and hydrophobic cavity in each β-globin contributes (Nozaki and Tanford Citation1971) ∼1.5 kcal·mol−1, Cao and Ferrone (Citation1997) estimated hydrophobic interactions to contribute 80% of the monomer-polymer binding free energy (Cao and Ferrone Citation1997). This estimate, however, assumes the host pocket as well as the other Val-β6 not involved in the lateral contact, also contributes ∼1.5 kcal·mol−1, an assumption that can be questioned because the pocket also exists in HbA and should be largely dewetted; recent MD simulations support this view (Galamba Citation2019).
Wang and Ferrone (Citation2013) studied through light scattering experiments, the aggregation of deoxy-HbS and several nonpolymerizable species, including deoxy-HbA, below the solubility for polymer formation. A positive binding free energy was found at 1 mM and 302 K for deoxy-HbA (4.0 kcal·mol−1) and deoxy-HbS (1.8 kcal·mol−1); the dimerization of deoxy-HbA involves, however, the (weaker) axial contact, as opposed to the lateral contact, which involves the β6 mutation site. Furthermore, HbS association was found to be entropically favored and enthalpically disfavored, consistent with the view that deoxy-HbS aggregation is impelled by hydrophobic interactions.
Nonetheless, it has also been suggested that electrostatic interactions could be at least equally important (Kuczera et al. Citation1990; Harrington et al. Citation1997; Galamba and Pipolo Citation2018; Galamba Citation2019). Kuczera et al. (Citation1990) in fact have long suggested that “for a fiber-like dimer structure, it is not the stabilizing hydrophobic interaction of Val in HbS that is the dominant factor, but the loss of the destabilizing interaction of Glu in HbA.” Galamba and Pipolo (Citation2018) provided evidence, based on MD simulations, that aggregation could be triggered by the formation of hydrogen-bonded ion pairs (i.e. salt bridges) between several surface residues (Lys, Asp, Glu) as well as heme groups. A subsequent study (Galamba Citation2019) provided evidence that the absence of Glu-β6 was more important than the presence of Val-β6, consistent with Kuczera et al. (Citation1990). In particular, it was shown that the Glu-β6→Val-β6 mutation favors aggregation through the elimination of strong electrostatic repulsions involving Glu-β6 and several residues, noteworthy, Asp-β73 and Glu-β90, as well as heme, whereas a mild attractive potential energy was found to be connected with Val-β6 (Galamba Citation2019). Asp-β73 was found to be especially relevant, being associated with strong electrostatic repulsions with Glu-β6 in the deoxy-HbA dimer, while forming a major attractive residue pair with Val-β6 in deoxy-HbS (see ), supporting the view that damping of electrostatic repulsions involving Glu-β6 in deoxy-HbA could explain the polymerization of deoxy-HbA at high potassium phosphate concentrations (Eaton and Hofrichter Citation1990).
Figure 4. (a) Glu-β6 (HbA) and (b) Val-β6 (HbS) interactions with every residue from adjacent HbA and HbS tetramers, respectively. The residues forming the hydrophobic pocket in HbS-1, Ala-β70, Phe-β85, and Leu-β88, are shown in orange in (b). The most important residues in the 1β1 (blue) and Glu-β6 and Val-β6 in the 2β2 (red) polypeptides from a MD snapshot are shown below. The potential energy, and not the electrostatic and vdW energy, are plotted in (a) and (b), respectively. Reprinted from Galamba (Citation2019).
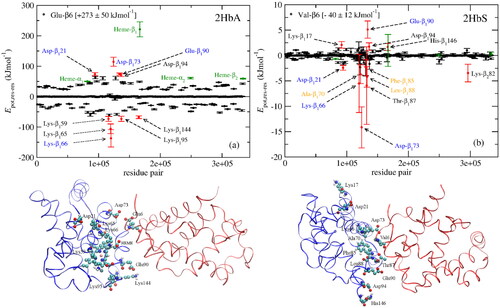
Interestingly, Asp-β73 is a point mutation in HbC-Harlem (Glu-β6→Val-β6; Asp-β73→Asn-β73) which differs from HbS in this single amino acid (i.e. Asp-β73→Asn-β73) (Bookchin et al. Citation1967). This mutation was shown to have a pronounced effect in the aggregation of HbC-Harlem, relative to HbS, resulting in the formation of crystals as opposed to polymers, seemingly through a similar mechanism, although with a much slower kinetics than HbS gelation (Adachi and Asakura Citation1980; Ivanova et al. Citation2001). Adachi et al. (Citation2003) related some of these differences with the formation of a hydrogen bond between Asp-β73 and Thr-β4, further influencing the hydrophobic interaction between Val-β6 and the hydrophobic pocket in deoxy-HbS.
A binding free energy of −14 kcal·mol−1 and −4 kcal·mol−1 was found, respectively, for the HbS and HbA lateral contact dimers in the abovementioned MD studies (Galamba and Pipolo Citation2018; Galamba Citation2019). These studies further suggested that possible aggregation inhibitors could target several salt bridges found in the HbS dimer, instead, or in addition to the hydrophobic contact.
MD simulations of sickle and normal hemoglobin and hemoglobin fibril models were also recently reported (Lu et al. Citation2016; Maity and Pal Citation2021; Olagunju et al. Citation2022). Maity and Pal (Citation2021) argued that the presence of hydrophobic residues without a bulky side chain at β6 in hemoglobin explained the stability of the fibrils, consistent with the experiments by Adachi et al. (Citation1993), which showed that some substituents in the β6 position, such as phenylalanine and tryptophan, polymerized less readily compared to deoxy-HbS and that when oversaturated polymerization occurred without the delay time observed for HbS. Adachi et al. argued that the difficulty of insertion of the bulky side chains of phenylalanine and tryptophan into the hydrophobic acceptor pocket on an adjacent tetramer could inhibit nuclei formation prior to polymerization. Interestingly, however, phenylalanine, tryptophan and derivatives, and analogues (Noguchi and Schechter Citation1977, Citation1978; Dean and Schechter Citation1978a; Poillon Citation1982) are themselves aggregation inhibitors, although it is not known whether these interact with the amino acids that form the hydrophobic pocket that lodges Val-β6.
Olagunju et al. (Citation2022)) found through MD simulations that both electrostatic and hydrophobic interactions involving the mutation site are important in the HbS aggregation. The authors suggested that a potential aggregation inhibitor could, in addition to target HbS–HbS interactions involving Val-β6, aim to interrupt an electrostatic contact involving Lys-β17 and the Glu-β90 of a neighbor tetramer. This same contact was identified as being among the lowest energy contacts in the deoxy-HbS and deoxy-HbA dimers (see SI of Galamba and Pipolo Citation2018; Galamba Citation2019).
While not an exhaustive road map through the drugs developed for NDs and SCD, in what follows we aim at providing a broad perspective on some of the early and most recent molecules shown to have aggregation inhibitory activity, and their putative action mechanism, in the context of SCD and NDs, especially PD. In addition, we aim to establish some contact between aggregation inhibitors found for such different diseases, involving archetypes of an IDP and an allosteric globular protein.
Protein aggregation inhibitors
Parkinson’s disease
A potential disease-modifying drug for PD, that is, one beyond the symptomatic treatment, should aim at reducing the α-syn expression, aggregation, membrane affinity and cytotoxicity, and/or propagation (Lashuel et al. Citation2013). In addition, reversing α-syn aggregation could be pivotal to treating several synucleinopathies because of delayed diagnosis. Despite several molecules being found to induce at least one of these responses in vitro and/or in vivo, these either have some major drawback, ruling out their therapeutic potential, or have not passed clinical trials. Hence, the most effective drug still used today against PD is l-3,4-dihydroxyphenylalanine (aka levopoda or l-dopa) (Abbott Citation2010), a dopamine precursor developed in the late 1960s. l-dopa increases the dopaminergic flux in the striatum, without, therefore, curing the disease nor influencing non-dopamine-associated symptoms (Jenner Citation2003; Huot et al. Citation2013; Stoker and Barker Citation2020).
Thus, several alternative therapeutic routes continue to be actively explored (Fields et al. Citation2019; Stoker and Barker Citation2020). These include, for instance, drugs that target non-dopaminergic neurotransmitters (Barone Citation2010; Blair and Dhillon Citation2017), associated with symptoms not alleviated by l-dopa and other related drugs, neuroprotective therapies (Rosenblad et al. Citation1999), aggregation inhibitors, gene therapies, or cell-based treatments (Cyranoski Citation2017). The latter, as discussed in the next section (section “Sickle cell disease”), were already successfully applied in the cure of SCD, although several challenges persist.
Concerning protein aggregation inhibitors, covered herein, although there are still no approved drugs that preclude or even delay the formation of α-syn oligomers (Stoker and Barker Citation2020), major advances have been achieved in recent years both in unraveling the structure of the multiple putative cytotoxic oligomers and in the design of new drugs targeting aggregation-inhibition. Molecules that showed aggregation-inhibition potential in PD and other proteinopathies encompass peptides (Madine et al. Citation2008; El-Agnaf et al. Citation2004; Horsley et al. Citation2022; Armiento et al. Citation2020; Cheruvara et al. Citation2015; Torpey et al. Citation2020; Kim et al. Citation2009), peptidomimetics and macrocycle peptides (Wrasidlo et al. Citation2016; Dougherty et al. Citation2017), antibodies (El-Turk et al. Citation2016), heat-shock proteins (Luk et al. Citation2008; Gao et al. Citation2015), and small organic molecules, including natural products (Stefani and Rigacci Citation2013; Doig and Derreumaux Citation2015; Bu et al. Citation2016; Young et al. Citation2017; Giorgetti et al. Citation2018; Javed et al. Citation2018). Here, we discuss various small molecules and peptide-based aggregation inhibitors developed through in silico, in vitro, and in vivo model studies.
Small molecule drugs
Several small organic molecules were found to exhibit anti-amyloid activity, although their action mechanism is not always completely understood. Some of the most explored small molecule anti-amyloid agents, including natural products (Khatoon et al. Citation2018), are catecholamines (Conway et al. Citation2001; Rochet et al. Citation2004; Li et al. Citation2005; Norris et al. Citation2005; Mazzulli et al. Citation2006, Citation2007; Herrera et al. Citation2008; Latawiec et al. Citation2010), phthalocyanines (Lee et al. Citation2004; Lamberto et al. Citation2009, Citation2011; Valiente-Gabioud et al. Citation2016), and polyphenols (Giorgetti et al. Citation2018; Borah et al. Citation2021; Mohammad-Beigi et al. Citation2019; Palazzi et al. Citation2018; Meng et al. Citation2009, Citation2010; Masuda et al. Citation2006; Caruana et al. Citation2011; Singh et al. Citation2013; Gautam et al. Citation2017; Ahmad and Lapidus Citation2012; Bieschke et al. Citation2010; Gautam et al. Citation2014; Rao et al. Citation2008; Ono and Yamada Citation2006; Arbo et al. Citation2020; Yang et al. Citation2023; Zhang et al. Citation2018), and among the latter, flavonoids (Hong et al. Citation2008; Jia et al. Citation2019; Meng et al. Citation2010, Citation2009; Tinku et al. Citation2021; Yang et al. Citation2023; Zhu et al. Citation2004). Many other small molecule drugs (Dong et al. Citation2019; Masuda et al. Citation2006; Mohankumar et al. Citation2020; Perni et al. Citation2017, Citation2018; Pujols et al. Citation2018; Saffari and Amininasab Citation2021; Tatenhorst et al. Citation2016; Tóth et al. Citation2014; Vittorio et al. Citation2020), were, however, reported to inhibit the aggregation of α-syn. In addition, many such anti-amyloid drugs are not protein specific, exhibiting anti-amyloid activity for different proteins involved in several proteinopathies (Masuda et al. Citation2006; Ehrnhoefer et al. Citation2008; Bieschke et al. Citation2010; Doig and Derreumaux Citation2015; Valiente-Gabioud et al. Citation2016; Giorgetti et al. Citation2018). Some of these small molecules stabilize the monomeric form, inducing the formation of disordered nontoxic oligomers, whereas others prevent aggregation into toxic and nontoxic oligomeric species either by interacting with specific domains or by displacing the protein away from the lipid membrane. In addition, several molecules were shown to disrupt preformed fibrils (Ehrnhoefer et al. Citation2008; Meng et al. Citation2010; Pujols et al. Citation2018; Saffari and Amininasab Citation2021; Zhu et al. Citation2004). The mechanism of several drugs (e.g. polyphenols) generally considered neuroprotective agents does not necessarily involve, however, protein aggregation inhibition alone, being also or exclusively associated with other PD pathogenic events such as increased oxidative stress and defective mitochondrial function (Moon and Paek Citation2015).
Among the most studied catecholamines is dopamine (), in addition to several oxidation derivatives and analogs (Li et al. Citation2005; Herrera et al. Citation2008; Latawiec et al. Citation2010), which have been shown to inhibit aggregation through different mechanism (Conway et al. Citation2001; Rochet et al. Citation2004; Norris et al. Citation2005; Mazzulli et al. Citation2006, Citation2007). A non-covalent aggregation-inhibition mechanism of dopamine, in particular, was associated with nonspecific interactions with the 125YEMPS129 sequence region in the C-term tail, and with long range electrostatic interactions involving E83 in the NAC domain (Mazzulli et al. Citation2007; Herrera et al. Citation2008).
Figure 5. Molecular structure of several α-syn aggregation-inhibitors: dopamine, OleA, EGCG, Curcumin, SynuClean-D, and Fasudil.
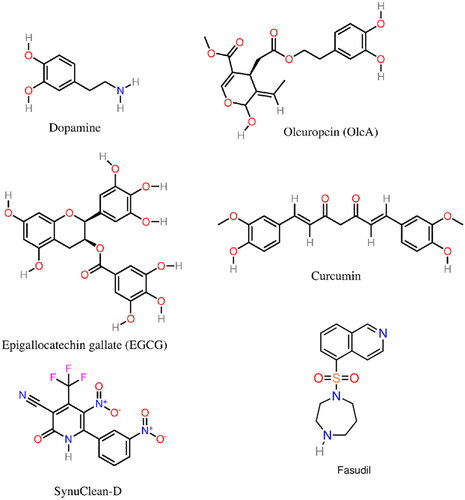
Oleuropein and derivatives (Mohammad-Beigi et al. Citation2019), including oleuropein aglycone (OleA) (Palazzi et al. Citation2018; Mohammad-Beigi et al. Citation2019; Borah et al. Citation2021) (), a phenolic compound found in olive oil, were shown to stabilize less aggregation-prone conformations of monomeric α-syn, favoring the growth of stable nontoxic aggregates (Palazzi et al. Citation2018; Mohammad-Beigi et al. Citation2019). A cytotoxicity reduction was also connected with a reduction of the propensity of oligomers to bind to cell membrane components by interacting with the membrane-binding N-term domain of α-syn (Palazzi et al. Citation2018; Borah et al. Citation2021). In addition, it was argued (Palazzi et al. Citation2018) that OleA stabilized the NAC and C-term regions of α-syn, preventing long-range and hydrophobic interactions between these sub-domains, which could favor aggregation.
Interestingly, however, long-range (tertiary) interactions involving the NAC and C-term regions were found in other studies to prevent, not promote, aggregation (Bertoncini et al. Citation2005; Dedmon et al. Citation2005; Esteban-Martín et al. Citation2013). Furthermore, truncation of the C-term region has been shown to induce an acceleration of fibril formation in vitro (Crowther et al. Citation1998; Breydo et al. Citation2012; Farzadfard et al. Citation2022). This illustrates the apparent wide spectrum of monomeric α-syn conformations which might be involved in aggregation pathways of toxic and nontoxic oligomers.
Epigallocatechin gallate (EGCG) (), a polyphenol within the group of flavonoids, and an aggregation inhibitor not specific to α-syn, was also suggested to induce the formation of less toxic disordered oligomers (Ehrnhoefer et al. Citation2008) and remodel mature α-syn fibrils into less toxic aggregates (Bieschke et al. Citation2010). Another study, however, reported that EGCG binds to the oligomers without changing either the secondary structure or its size distribution. In this study, the EGCG-induced toxicity reduction was linked with a decrease in the oligomers’ membrane affinity (Lorenzen et al. Citation2014).
The molecular mechanism underlying the aggregation-inhibition of α-syn by several flavonoids (e.g. baicalein) was shown to be associated with the restriction of conformational changes as well as the stabilization of α-syn’s monomeric and oligomeric species (Zhu et al. Citation2004; Hong et al. Citation2008; Meng et al. Citation2009, Citation2010). Furthermore, flavonoids with three vicinal hydroxyl groups exhibited enhanced inhibitory effects on α-syn aggregation (Meng et al. Citation2009); this was related with the flavonoids anti-oxidant activity, although limitations of this correlation were pointed out (Meng et al. Citation2009). In addition, the oxidized species (e.g. baicalein quinone) rather than the polyphenol (e.g. baicalein) were found to be the main aggregation inhibitors; aggregation inhibition of baicalein is significantly reduced under anaerobic conditions (Zhu et al. Citation2004; Meng et al. Citation2009). A complex mechanism encompassing the auto-oxidation of baicalein and other flavonoids and the subsequent covalent bonding to α-syn, through the formation of a Schiff base between the quinone of baicalein and a Lys of α-syn were, in fact, found to be the key factors for the inhibition of α-syn aggregation (Zhu et al. Citation2004; Meng et al. Citation2009). Caruana et al. (Citation2011) also suggested that the main factors underpinning α-syn self-assembly inhibition and destabilization are the existence of aromatic elements that bind to α-syn monomers/oligomers, and neighbor hydroxyl groups on a single phenyl ring.
A drawback of some potential drugs found in small molecule libraries, in particular several aggregation inhibition polyphenols (e.g. curcumin; see ), quinones, and catechols, is the fact that these might be pan-assay interference compounds (PAINS) (Baell and Holloway Citation2010; Baell and Walters Citation2014), that is, molecules that give false positives in high-throughput screening assays for several possible reasons. Since the action mechanism of these molecules is not always completely understood, neither as PAINS nor as aggregation inhibitors, and because of their nonspecificity, it is difficult to predict whether molecules such as curcumin can both be PAINS and effective aggregation inhibitors (Gautam et al. Citation2014, Citation2017).
In addition, polyphenols at dietary concentrations have been connected with the prevention and attenuation of PD through alternative mechanisms, including oxidative stress (i.e. a reduction of reactive oxygen species) and neuroinflammation reduction (de Andrade Teles et al. Citation2018; Carecho et al. Citation2022).
Another class of compounds explored, concerning anti-amyloid activity, are phthalocyanines, which suppress aggregation through the interaction of the aromatic rings with aromatic amino acids via π–π interactions (π-stacking) (Valiente-Gabioud et al. Citation2016). This mechanism led to the suggestion that aromatic interactions could be key players in the aggregation mechanism of α-syn (Lamberto et al. Citation2009, Citation2011), in spite of a relatively reduced number of aromatic amino acids. α-syn has only four tyrosine (Tyr39, Tyr125, Tyr133, Tyr136), two phenylalanine (Phe4, Phe94), and no tryptophan amino acids, of which only Phe94 is in the NAC segment. The anti-amyloid activity of these compounds also depends on the type of metal ion coordinated to the tetrapyrrolic system (Valiente-Gabioud et al. Citation2016).
A series of pyridinyl-triazole derivatives were also recently reported to inhibit α-syn aggregation from in vitro screening and docking studies (Vittorio et al. Citation2020). Fasudil (5-(1,4-diazepan-1-ylsulfonyl)isoquinoline) (), a small isoquinoline derivative, was shown to inhibit α-syn aggregation through direct binding to tyrosine residues Y133 and Y136 in the C-term region of α-Syn (Tatenhorst et al. Citation2016). SynuClean-D (2-hydroxy-5-nitro-6-(3-nitrophenyl)-4-(trifluoromethyl)nicotinonitrile) (), an aromatic compound, was shown to reduce the in vitro aggregation of wild-type α-syn and the A30P and H50Q variants in a sub-stoichiometric molar ratio (Pujols et al. Citation2018). In addition, this compound was found to disrupt mature amyloid fibrils and prevent fibril propagation.
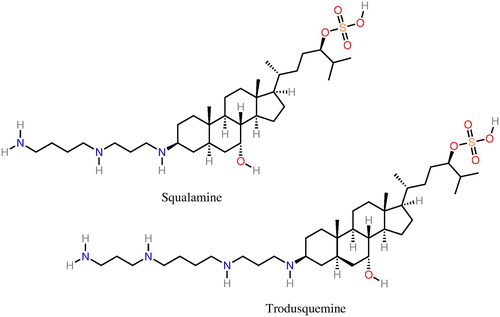
Squalamine (Perni et al. Citation2017) (), a natural product isolated from the dogfish shark was shown to inhibit the aggregation of α-syn in vitro and in vivo by blocking the nucleation of α-syn. The mechanism of action of squalamine is not linked with specific protein–drug interactions but instead with a competition with α-syn for binding the membrane. The latter stimulates nucleation (Galvagnion et al. Citation2015) and, thus, the displacement of α-syn from the membrane hampers the first steps of the aggregation process. A later study by Perni et al. (Citation2018) showed that the related compound, trodusquemine (), interferes not only with the nucleation of α-syn but also with fibril-dependent secondary pathways. In addition, trodusquemine was shown to suppress the toxicity of α-syn oligomers in neuronal cells (Perni et al. Citation2018). These molecules are already relatively long, potentially binding to larger protein/cell domains relative to most small molecules previously discussed. In this sense, these are more similar to peptides, discussed in the next section.
Figure 6. Molecular structure of the α-syn aggregation inhibitors squalamine and trodusquemine.
While small molecules remain appealing because they have a good metabolic stability and can, in principle, more easily cross the blood–brain barrier, in addition to other reasons, including economic, they suffer in general from poor selectivity, specificity, and potency, regarding protein aggregation inhibition. Thus, alternative aggregation-inhibitors have been explored. Among these, we focus on some small peptide-based drugs (El-Agnaf et al. Citation2004; Wrasidlo et al. Citation2016; Torpey et al. Citation2020; Liang et al. Citation2021; Meade et al. Citation2021).
Peptide drugs
Peptide-based drugs re-emerged as promising alternatives to small molecules concerning proteinopathies because of their specificity and potency (Neddenriep et al. Citation2011; Cunningham et al. Citation2017; Armiento et al. Citation2020). The enhanced potency of small peptides is directly related with their larger interaction surfaces, allowing, in principle, to interfere with extended protein domains linked to the aggregation process (Cunningham et al. Citation2017). Linear peptide drugs, however, suffer from other drawbacks such as bioavailability and proteolytic instability (Armiento et al. Citation2020). This has stimulated the development of macrocycle peptides and peptidomimetics to overcome some of these limitations.
Anti-amyloid designed peptides are often β-sheet breakers or blockers, disrupting or inhibiting the formation of cross-β structures, a structural hallmark of toxic oligomers (Young et al. Citation2017; Serpell et al. Citation2000; Bartolini and Andrisano Citation2010; Sciarretta et al. Citation2006; Kim et al. Citation2009). Furthermore, in addition to aggregation inhibitors, since peptides are often modified segments of the amyloid proteins, they can provide insight into pivotal aggregation-prone domains (Neddenriep et al. Citation2011). Peptide modification strategies include, for instance, peptide termini modifications, insertion of prolines, which are potent β-sheet breakers, backbone modifications, or peptide cyclization (Sciarretta et al. Citation2006). In addition, polar amino acids can be inserted to enhance the solubility (El-Agnaf et al. Citation2004).
El-Agnaf et al. (Citation2004) showed that modified peptides containing amino acid sequences 68GAVVT72 from the NAC inhibit aggregation into oligomers and mature amyloid fibrils. The peptides’ modifications included the insertion of RG and GR amino acids in the N-term and C-term. Aggregation inhibition was observed at (α-syn:peptide) 2:1, 1:1, and 1:2 molar ratios and the shortest peptide that inhibited α-syn aggregation had the central sequence 69AVVT72. These peptides were also observed to inhibit NAC aggregation suggesting that aggregation-inhibition is due to the binding of the peptides to their homologous sequence in α-syn.
Madine et al. (Citation2008) showed that a peptide from NAC (77VTGVTAVAQKTV82), N-methylated in the C-terminus, disrupted the aggregation of α-syn. This peptide is a sub-domain of the 72–84 region of α-syn, absent in β-syn, which in spite of sharing 78% of similarity with α-syn, does not aggregate (Biere et al. Citation2000).
Kim et al. (Citation2009) also proposed a small peptide (72PGVTAV77) able to block aggregation and to dissolve preassembled fibrils. This is a modified (T72 → P72) sub-domain of NAC (72TGVTAV77), part of the NACore (68GAVVTGVTAVA78) an 11 amino acid peptide later shown (Rodriguez et al. Citation2015) to play an important role in the aggregation and cytotoxicity of α-syn.
A 10 amino acid peptide (KDGIVNGVKA) was proposed by Cheruvara et al. (Citation2015) from intracellular screening of a peptide library based on α-syn residues 45–54, involved in several familial PD mutations (E46K, H50Q, and A53T). The peptide was shown to inhibit the aggregation of α-syn and the associated toxicity. Interestingly, Torpey et al. (Citation2020) found, through NMR, that although this peptide precludes oligomerization of the wild-type and several mutations associated with familial PD, it does not bind to the monomer neither to low-n (n < 4) oligomers. Thus, the aggregation-inhibition mechanism of this peptide is not completely understood.
A cyclic peptidomimetic (NPT100-18A) with a seemingly similar mechanism to squalamine was proposed by Wrasidlo et al. (Citation2016). This peptidomimetic has the ability to displace α-syn from the membrane by interacting with domains in the C-terminus of α-syn.
Rezaeian et al. (Citation2017) proposed two peptides, KISVRV and GQTYVLPG, which suppressed the aggregation of α-syn in vitro. These peptides were chosen based on their binding affinity to the amino acids 70–75 (VVTGVT) and 46–53 (EGVVHGVA) of α-syn. The first region is among the several regions of NAC found to be pivotal in the aggregation process. The second corresponds to the region where several missense mutated forms of α-syn (E46K, H50Q, G51D, A53T), implicated in familial PD, were found, and shown to induce an acceleration of the aggregation process, as previously discussed.
More recently, a small ubiquitin-related modifier 1 (SUMO1) derived peptide SUMO1 (15–55) (see ), which targets two SUMO-interacting motifs within the N-term region flanking the NAC was shown to inhibit α-syn aggregation (Liang et al. Citation2021). Another recent study (Santos et al. Citation2021) proposed the use of α-helical peptides with a low affinity toward the monomeric form, avoiding perturbing the natural function of α-syn, while interrupting aggregation by binding to toxic oligomers and fibers. The PSMα3 peptide was found to have a high affinity toward a large number of binding sites in the oligomers, inhibiting aggregation (Santos et al. Citation2021).
Table 1. Examples of α-syn aggregation inhibition peptides.
Popova et al. (Citation2021) also found two synthetic peptides, through a high-throughput screening study (), 25 and 19 residues-long, that suppress α-syn aggregation. While the action mechanism was not disclosed, the peptides were shown to significantly suppress the first steps of oligomerization. The peptides were also shown to be specific to α-syn. Furthermore, the larger and more potent peptide was shown to reduce α-syn aggregation in human cells.
A recent study (Horsley et al. Citation2022) compared the aggregation inhibitory activity of two peptides, GVLYVGS-Aib and VAQKTV-Aib (L and D isomers; Aib stands for aminobutyric acid, a β-breaker), targeting respectively, the “P1” region (residues 36–42) of the N-term domain (Doherty et al. Citation2020), previously discussed, and the segment comprised by the residues 77–82 within the NAC domain. Horsley et al. (Citation2022) showed that the D isomer of the peptide targeting the “P1” region was the most effective in preventing fibril formation, increasing the solubility of α-syn, and altering the conformation of the monomer into a less aggregation-prone state.
We now discuss the nature of some of the drugs, including amino acids and peptides, found to inhibit the aggregation of HbS, aiming at establishing some contact points between potential common drugs and aggregation inhibition mechanisms.
Sickle cell disease
Opposite to NDs, SCD can be potentially cured, either through hematopoietic stem cell transplantation (Eapen et al. Citation2019) or gene therapy (Ribeil et al. Citation2017), although several limitations and challenges, including economic, persist, preventing these treatments’ widespread (Eaton and Bunn Citation2017; Kapoor et al. Citation2018; Salinas Cisneros and Thein Citation2020; Tisdale et al. Citation2020). Thus, great interest remains in the development of drugs that can be used in the treatment of SCD.
In addition to hydroxyurea, long used in the treatment of SCD, other drugs became recently available. These include l-glutamine (Niihara et al. Citation1997, Citation1998; Ortiz de Montellano Citation2018), whose action mechanism, although thought to reduce the oxidative stress in the erythrocytes, remains largely unknown, and voxelotor (Vichinsky et al. Citation2019), an allosteric modulator (i.e. an oxygen affinity modifying drug) aimed at stabilizing the nonpolymerizing R quaternary structure of HbS. l-Glutamine was approved in 2017 by the Food and Drug Administration (FDA) for adult and pediatric patients 5 years and older. Voxelotor was approved by the FDA and the European Medicines Agency (EMA) and recently extended to the treatment of children of ages 4–11 years old. Nonetheless, in spite of representing important alternatives and/or potential co-adjuvants to hydroxyurea, these drugs have limitations (Tisdale et al. Citation2020; Henry et al. Citation2021). For instance, Henry et al. recently provided evidence that although voxelotor significantly reduces sickling, oxygen delivery to tissues is offset by increased hemoglobin O2 affinity (Henry et al. Citation2021). The latter is the biggest potential drawback of allosteric modulators, in general. Nonetheless, allosteric modulators are among the most studied anti-sickling agents, and several other drugs were recently reported (Abdulmalik et al. Citation2005; Nakagawa et al. Citation2014; Oder et al. Citation2016; Oksenberg et al. Citation2016; Strader et al. Citation2019; Gopalsamy et al. Citation2021; Pagare et al. Citation2022). Other anti-sickling drugs that neither bind to HbS nor change the HbS oxygen affinity were also recently reported; the antisickling mechanism of this compound (SCD-101) is unknown (Swift et al. Citation2016).
Various SCD therapeutic strategies have evolved over the years (see Eaton and Bunn Citation2017; Telen et al. Citation2019; Tisdale et al. Citation2020 for recent reviews). These include (Eaton and Bunn Citation2017; Tisdale et al. Citation2020): (i) increase of HbF or the increase of the erythrocyte volume to decrease the intra-cellular HbS concentration, (ii) decrease the concentration of the allosteric effector, 2,3-DPG, increasing the solubility and decreasing the fibers’ stability, (iii) shift the allosteric equilibrium toward the R-state, and (iv) block protein–protein contacts by binding to HbS. In addition, several drugs aiming to reduce adhesion of erythrocytes to the vascular endothelium, decreasing transit times, have been investigated (Ataga et al. Citation2017; Eaton and Bunn Citation2017; Telen et al. Citation2019). Although most of these approaches started being explored in the 1970s (see of Dean and Schechter Citation1978a), only recently there have been some important advances in the development of effective drugs falling within the purview of at least one of the above action mechanisms, as briefly discussed above.
Our main focus herein is on non-covalent stereospecific aggregation inhibitors that block protein–protein contacts. Again, our approach is not an exhaustive one, but rather to provide some examples of small molecules, including amino acids and peptides, that may share common features across the drugs studied in other proteinopathies.
Small molecule drugs
A foremost obstacle to the design of an effective anti-sickling molecule addressing the polymerization process, concerns the high concentration of HbS in the erythrocytes (Dean and Schechter Citation1978a) (∼5 mM) and therefore the putative high concentration required of any effective aggregation inhibition drug (Eaton and Bunn Citation2017). Nonetheless, the fact that a small change in the solubility of HbS can have a major impact in the lag period that characterizes the polymerization process, motivated the continuous exploration of this therapeutic strategy (Eaton and Bunn Citation2017). Furthermore, a recent study provided evidence that HbS fiber growth is a rather inefficient process (∼4% efficiency), as previously mentioned in the Introduction, where monomer addition and loss are nearly equal. This led to the suggestion that HbS fiber growth is a viable therapeutic target even at drug concentrations below the total hemoglobin concentration. Nonetheless, this concentration will depend further on drug selectivity, among other factors, since many drugs will bind to additional domains of HbS, not affecting or even enhancing polymerization.
Many small molecules (Abdulmalik et al. Citation2011; Abraham et al. Citation1983; Dampier Citation2015; Dash et al. Citation2013; Dean and Schechter Citation1978b; Iyamu et al. Citation2002; Mehanna Citation2001; Syed et al. Citation2019; Votano et al. Citation1984; Xu et al. Citation2017) including some amino acids (Noguchi and Schechter Citation1977; Dean and Schechter Citation1978a; Noguchi and Schechter Citation1978; Schechter Citation1980; Russu et al. Citation1986), were long found to decrease the deoxy-HbS aggregation in vitro by increasing the solubility of HbS. While these molecules, many reported in the 1970s and 1980s, failed their purpose as effective drugs for the treatment of SCD it is of interest to contrast some of these molecules with those proposed more recently to address SCD and other proteinopathies such as PD.
Examples of small molecules long shown to increase the HbS solubility include for instance (Dean and Schechter Citation1978a; Mehanna Citation2001): alkyl ureas (Elbaum et al. Citation1974, Citation1976, Citation1978), Hofmeister (lyotropic) salts (Poillon and Bertles Citation1979), aromatic compounds with a phenyl group and a pendant side chain terminating in a hydrogen bond donor/acceptor (e.g. NH3+, COO–, and OH) (Ross and Subramanian Citation1977), benzyl and phenoxy acids (Abraham et al. Citation1984), and clofibric acid and gemfibrozil (Abraham et al. Citation1983) (see ).
Figure 7. Molecular structure of several α-syn deoxy-HbS aggregation inhibitors: n-propyl urea, benzyl alcohol, clofibric acid, and gemfibrozil.
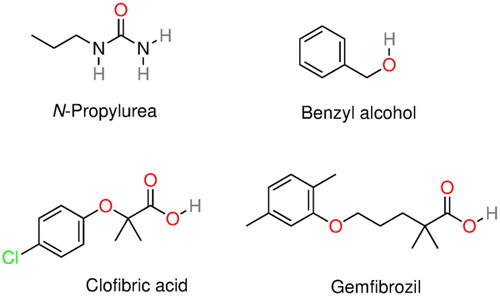
Ross and Subramanian (Citation1977) provided a comprehensive analysis of several small molecules and concluded that deoxy-HbS aggregation inhibition (i.e. solubility increase) was promoted by the combination of a hydrophobic and a (single) hydrophilic group. In this regard, aromatic rings were more potent than aliphatic chains, and the hydrophilic group should be located on an aliphatic side chain attached to the aromatic ring to provide proper flexibility and/or distance to interact with deoxy-HbS. The latter was based upon the fact that molecules such as phenol, aniline, and salicylic acid do not inhibit aggregation. According to Ross and Subramanian, a similar logic should apply to alkyl ureas. They proposed an aggregation inhibition mechanism where the deoxy-HbS lateral contact was blocked through the interaction of the aromatic ring with the hydrophobic pocket (i.e. Phe-β85 and Leu-β88) whereas the hydrophilic group formed a hydrogen bond with the Asp-β73.
This mechanism is at odds with the later study by Adachi et al. (Citation1993), which showed that phenylalanine and tryptophan in the Val-β6 position largely precluded aggregation, presumably due to stereo hindrance, as previously discussed. With regard to the requirement of an aromatic ring and a hydrophilic moiety at a given distance, this is common to many small molecules found to inhibit aggregation, although their specific action mechanism is not always understood.
Among amino acids, phenylalanine and tryptophan (see ) were the only amino acids found to exhibit aggregation inhibition activity in vitro (i.e. gelation inhibition) (Dean and Schechter Citation1978a; Schechter et al. Citation1978). Again, following Ross and Subramanian (Citation1977), this was consistent with their findings regarding aromatic small molecules. A study in 1975, nonetheless, showed that 3.8 mM homoserine, asparagine, and glutamine but no other amino acid, reversed the erythrocytes sickling (Rumen Citation1975). This suggested at the time that these amino acids should inhibit sickling through a completely different (unknown) path than that observed in in vitro antigelling experiments.
Figure 8. Molecular structure of several α-syn deoxy-HbS aggregation inhibitors: phenylalanine, tryptophan, alizarin, piperine, capsaicin, and cubebin molecular structures. Phenylalanine and tryptophan exhibit some aggregation inhibition activity and were used as building blocks in aggregation inhibition peptides (Dean and Schechter Citation1978a; Schechter et al. Citation1978). Alizarin (hydroxyl anthraquinone) is a bioactive compound from the plant Rubia cordifolia. Piperine, capsaicin, and cubebin have been pointed out (Ameh et al. Citation2012) as possible aggregation inhibitors present in Niprisan (Iyamu et al. Citation2002) (drug Nix-0699) a product of the extracts of four different plants.
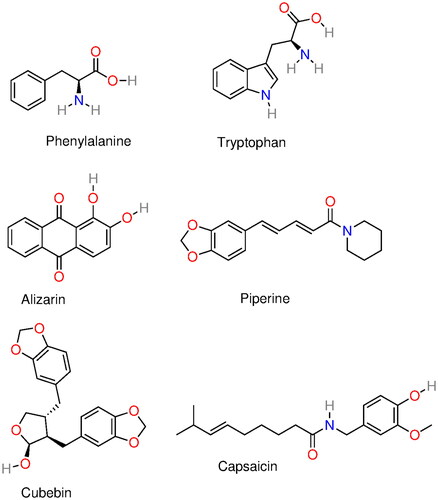
More recently, an experimental and simulation/docking study investigated alizarin (Syed et al. Citation2019), a hydroxyl anthraquinone (see ) found in the plant Rubia cordifolia (aka Indian madder). HbS polymerization was shown to decrease with the concentration of alizarin. The proposed mechanism involved the perturbation of the Val-β6 binding pocket through hydrogen bonding and hydrophobic interactions of HbS with alizarin. Many other natural products found in plants and long used in traditional medicine have also been shown to have some anti-sickling activity (Fall et al. Citation1999; Moody et al. Citation2003; Mpiana et al. Citation2007; Ameh et al. Citation2012; Imaga Citation2013). For instance, piperine, capsaicin, and cubebin (see ) have been pointed out (Ameh et al. Citation2012) as possible aggregation inhibitors present in Niprisan (Iyamu et al. Citation2002) (drug Nix-0699), a product of the extracts of four different plants.
A high-throughput assay encompassing 12,657 compounds of the Scripps ReFRAME drug repurposing library, where sickling times following deoxygenation to 0% oxygen of red cells from sickle trait individuals were assessed, was recently reported (Metaferia et al. Citation2022). summarizes 50 compounds, among the 106 with antisickling activity, reported in that study.
Table 2. Set of 50 compounds, among the 106, reported by Metaferia et al. (Citation2022) with antisickling activity (see Table 1 of Metaferia et al. Citation2022 for additional details).
Among the compounds depicted in , there are several polyphenols, including butein and curcumin, as well as some derivatives (e.g. cyclovalone) with reported neuroprotective effects, including for Parkinson, via an aggregation inhibitory mechanism (Tinku et al. Citation2021). Resveratrol derivatives were also recently studied concerning their potential as HbF inducers (Bosquesi et al. Citation2020). In addition, some of the compounds in were already studied for SCD and later abandoned whereas other are currently being investigated. Tucaresol, for instance, a substituted benzaldehyde, was originally developed for treating SCD as an aggregation inhibitor by enhancing oxygen affinity, reducing hemolysis (Rolan et al. Citation1993; Arya et al. Citation1996), but later discontinued. Mitapivat, recently approved in the United States of America and Europe to treat hemolytic anemia, is also being investigated concerning SCD (Quezado et al. Citation2022; van Dijk et al. Citation2022; Xu et al. Citation2022). Mitapivat increases adenosine triphosphate (ATP) levels and decreases the 2,3-diphosphoglycerate (2,3-DPG) concentration, thus potentially reducing sickling, although other (unknown) mechanisms may play a role (Quezado et al. Citation2022). The large number of antisickling compounds reported by Metaferia et al. (Citation2022) holds the promise that one or more compounds can be developed in a near future with one or more of the abovementioned action mechanisms.
Several potential drugs were also recently investigated through docking studies (Olubiyi et al. Citation2018; Das et al. Citation2020). A recent in silico drug repurposing study (Olubiyi et al. Citation2018) identified a series of compounds (praziquantel, losartan, ketoprofen, glipizide, rosuvastatin, atorvastatin, ergotamine, and risperidone) interfering in the Val-β6 lateral contact with the hydrophobic cavity in the neighbor HbS tetramer. This perturbation involved mostly the interaction of the drugs’ aromatic rings with the pocket formed by Ala-β70, Phe-β85, and Leu-β88. Unfortunately, assessing the aggregation inhibition activity of these and other compounds through docking or molecular simulations remains a challenging problem because of computational limitations.
Similar to other proteinopathies, the deoxy-HbS aggregation inhibitory effect of small peptides in SCD, now discussed, has long been probed.
Peptide drugs
Several peptides were long shown to exhibit anti-sickling activity (Dean and Schechter Citation1978b; Gorecki et al. Citation1980; Kubota et al. Citation1976; Schechter Citation1980; Schechter et al. Citation1978; Votano et al. Citation1977; Votano and Rich Citation1985). The latter were the object of a recent comprehensive review (Olubiyi et al. Citation2019). Herein, we highlight the main features that characterize the inhibition mechanism of peptides as opposed to small molecules, as well as putative resemblances with some of the peptide-drugs explored within the context of α-syn aggregation, discussed in section “Peptide drugs”.
Gorecki et al. (Citation1980) found, based on a study of over 30 peptides, that the hydrophobicity of the side chains was the most important feature with respect to the HbS aggregation inhibition (i.e. antigelling activity). Votano and Rich (Citation1985) later reported that “compounds containing bicyclic or multi-aromatic residues have a higher activity than those that carry a single aromatic or aliphatic side chain”. In addition, an increase in the apolar content of the aromatic residue and ring polarizability further enhanced the antigelling activity of such compounds. The peptides in these studies commonly comprised one to two Phe or Trp and a Gly, and were succinylated to enhance the solubility (Votano and Rich Citation1985); additional amino acids comprising the peptides included Arg, Nle, and Lys (Gorecki et al. Citation1980). Whereas the binding sites in HbS were not completely disclosed it was argued that peptide-HbS contacts should involve several amino acids near the hydrophobic cavity where Val-β6 is enclosed in the fibers. The fact that peptides containing several aromatic residues are more potent that those with a single Phe or Trp suggest that the aggregation inhibition mechanism should involve a larger domain than that comprised by the principal amino acids that form the Val-β6 binding pocket.
A pivotal advantage of peptide-based drugs concerning SCD is the possibility to enhance the protein–drug contact area, decreasing, in principle, the drug:HbS ratio. The low stereoselectivity of small molecules toward protein surfaces can translate into the interaction with several protein domains, which can disfavor, promote, or exert no significant effect on aggregation. In principle, larger peptides should depict a greater aggregation inhibitory activity, relative to single amino acid or di- and tri-peptides because of the increased contact area. However, some critical length should exist above which aggregation inhibition “saturates”. The latter, however, will depend on the contact area, which in turn depends on the chemical nature of the peptide. Several works (Kubota et al. Citation1976; Noguchi and Schechter Citation1978; Akbar et al. Citation2006) addressed this problem concluding that the aggregation inhibitory activity increased with the peptide size, although large concentrations were still required to observe effective antigelation effects (see also Olubiyi et al. Citation2019 for a broader discussion of this aspect).
A possible explanation for the large concentrations required for observing antigelation activity by small molecules and peptides involving aromatic rings (Phe and Trp) is the fact that these may not interact with the hydrophobic pocket where Val-β6 is inserted. The latter can be speculated based on the results of Adachi et al. (Citation1993), previously discussed. Thus, such a pocket could only be blocked at high drug concentrations when interactions with neighbor sites imped the entrance of Val-β6 from a neighbor tetramer. An alternative explanation is that some of these peptides and or small molecules are neither selective enough nor the aggregation inhibition mechanism is associated with the Val-β6 binding pocket. The abovementioned studies, point, nevertheless, to a common direction, that is, aromatic amino acids are key to the blockage of protein–protein contacts behind nucleation (increasing the lag time), and heterogeneous nucleation, preventing the formation of fibers. In the following sections, we attempt to rationalize some of the above information regarding the role of hydrophobic interfaces, aromatic rings, and hydroxyl groups, all seemingly playing a part in protein aggregation inhibition in SCD, PD, among other proteinopathies.
Hydrophobic effect and protein aggregation
The design of a drug to oppose or delay protein aggregation could, in principle, aim at increasing the solubility of the protein, in principle, by weakening the hydrophobic effect. The hydrophobic effect comprehends two related phenomena, hydrophobic hydration, associated with the low solubility of apolar molecules and groups in water, and hydrophobic interactions, the spontaneous association of apolar molecules in water. The most remarkable feature of hydrophobic hydration is, perhaps, its system size dependence, with the hydration of small (RS < ∼1 nm) and large radius (RS > ∼1 nm) spherical solutes being dominated, respectively, by entropy and enthalpy (Pratt and Chandler Citation1977; Hummer et al. Citation1996; Lum et al. Citation1999; Chandler Citation2005; Ashbaugh and Pratt Citation2006; Berne et al. Citation2009; Jamadagni et al. Citation2011).
Hydrophobic hydration
Small length hydration
Hydration of small apolar molecules is governed by the formation of a suitable cavity to lodge the solute. For hard spheres, this probability is related to the hydration free energy (i.e. excess chemical potential) by , where R is the solvent accessible radius, given by
, and RS and RW are the radius of the solute and water, modeled as hard spheres, whereas
is the probability that a sphere of radius R randomly inserted in water is devoid of water molecules (Widom Citation1982; Garde et al. Citation1996; Ashbaugh and Pratt Citation2006).
Whereas solute–water interactions are favorable, the hydration enthalpy is smaller in magnitude than the hydration entropy and the process is, therefore, entropic. Although displaying differences relative to bulk water, the water structure (and dynamics) and, therefore, water–water interactions, produce a small net effect on the hydration enthalpy and entropy. For instance, a tetrahedral enhancement (Davis et al. Citation2012; Galamba Citation2013, Citation2014; Conti Nibali et al. Citation2020) of a sub-population (e.g. ∼70% for methane) of water molecules in the hydration layer of small apolar solutes has been found, along with an enhancement of the hydrogen bond strength between those water molecules and the water molecules that comprise the vertices of the imperfect “tetrahedrons” (Galamba et al. Citation2019; Tamoliūnas and Galamba Citation2020; Galamba Citation2021). This water population illustrated in is denoted here W-4W. This is opposed by water molecules (e.g. ∼30% for methane) that are not tetrahedral (i.e. water molecules closer to the solute than to the four nearest water neighbors (W-3W; see ), which cannot, therefore, form up to four hydrogen bonds), where a weakening of the average water–water interactions with the third and fourth water neighbors is observed, making a positive contribution to the hydration enthalpy. These populations (i.e. tetrahedral and non-tetrahedral; see and the discussion in the next section) decrease and increase, respectively, with the solute size (Tamoliūnas and Galamba Citation2020). The excluded volume induced weakening of water–water interactions (i.e. hydrogen bond breaking), nonetheless, outweighs the tetrahedral enhancement, except for small solutes at low temperatures (Galamba Citation2021), making a small positive contribution to the enthalpy. Furthermore, notwithstanding these contributions to the hydration entropy and enthalpy, the water reorganization around the cavity is characterized by a nearly exact entropy–enthalpy compensation, thus, not contributing to the hydration free energy (Ben-Naim Citation1975, Citation1978; Lee Citation1991; Grunwald and Steel Citation1995; Graziano Citation2005).
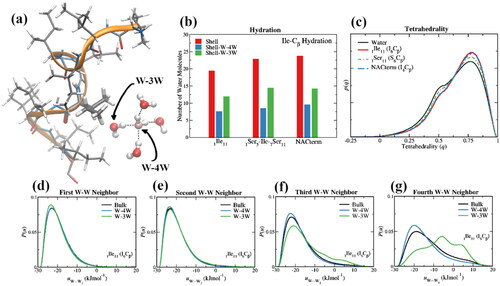
Figure 9. (a) A water pentamer next to Ile-6 in an 11-mer isoleucine peptide (1ILE11) displaying a water in the Cβ coordination sphere with at least four nearest water neighbors (W-4W) and a water with three or less nearest water neighbors (W-3W) because of the proximity of the solute; (b) number of water molecules in the first hydration layer (rmin ≤ 6.45 Å) of the Cβ of, Ile-6 in an 11-mer isoleucine peptide (1ILE11), Ile-6 in an 11-mer serine peptide with a single (middle) isoleucine (1SER5-ILE-7SER11), and Ile-4 in NAC-term (85AGSIAAATGFV95), an 11-mer peptide comprised of the last 11 amino acids of NAC; (c) tetrahedrality of the W-4W water populations in the first hydration shell of the Cβ of Ile and Ser, respectively, in isoleucine (1ILE11) and serine (1SER11) 11-mer peptides, compared with the tetrahedrality of bulk water; (d–g) water pair interaction energy distributions, P(W···Wn) for n = 1–4, for bulk water and W-4W and W-3W water populations in the first hydration shell of the Cβ of Ile in isoleucine (1ILE11).
For long linear hydrophobic solutes such as n-alkanes, a behavior similar to small solutes is observed as these can be accommodated in water almost as an ensemble of small apolar molecules and the hydration free energy is a linear function of the carbon number (nC) up to nC ∼20 (Chandler Citation2005; Galamba Citation2021; Singh and Sharma Citation2022). Folding in long alkanes (nC > ∼20) was recently shown to result in the violation of entropy and enthalpy convergence (Galamba Citation2021). This was demonstrated to result from the release of water molecules in the hydration layer of methylene groups upon folding, increasing the hydration entropy and reducing solute–water interactions, thus, lowering the enthalpy (Galamba Citation2021). Entropy convergence violation is observed for low curvature (RS > ∼1 nm) hard spheres (Huang and Chandler Citation2000; Ashbaugh and Pratt Citation2006) and for many globular proteins upon unfolding (Robertson and Murphy Citation1997), because of the heterogeneous nature of protein–water interactions (Sedlmeier et al. Citation2011) and probably folding/unfolding differences (Galamba Citation2021).
Large length hydration
For low curvature solutes (RS > ∼1 nm), hydration is governed instead by the formation of a solute–water interface, as opposed to density fluctuations and spontaneous cavity formation, resulting in a substantial loss of water–water HBs and, therefore, an enthalpy-dominated hydration free energy and a positive hydration entropy (Chandler Citation2005; Jamadagni et al. Citation2011). Hydrophobic hydration of such large surfaces is characterized by an increase of the local water fluctuations and isothermal compressibility, relative to bulk water (Chandler Citation2005; Jamadagni et al. Citation2011). Furthermore, a more favorable binding of hydrophobic molecules has been observed because of facilitated cavity formation next to the surface, favoring the binding of hydrophobic drug-groups (Jamadagni et al. Citation2011).
The above picture, nonetheless, applies to hydrophobic molecules or interfaces, as opposed to (amphiphilic) proteins and other biomolecules where chemical heterogeneities result in heterogeneous solvation, with water molecules next to hydrophilic groups sharing the hydration layer of hydrophobic side chains of neighbor amino acids, leading to quenching of density fluctuations (Chandler Citation2005; Jamadagni et al. Citation2011). In addition, topological (microscopic) irregularities influence protein hydration and density fluctuations. Hydrophobic hydration has, for this reason, been more difficult to characterize in biomolecules than in hard spheres, hydrocarbons, or flat planar self-assembled monolayers (Granick and Bae Citation2008; Jamadagni et al. Citation2011).
To illustrate some of the above points, displays some MD results on the size of the water populations next to hydrophobic and hydrophilic amino acids in 11-mer peptides along with the tetrahedrality (Errington and Debenedetti Citation2001) and energetics of water–water interactions. The peptides and water were described by the AMBER99sb (Hornak et al. Citation2006) and TIP4P-Ew (Horn et al. Citation2004) force fields, respectively. Further details about these simulations can be found elsewhere (Galamba Citation2022). shows a hydration increase by 15% of an Ile amino acid when sided by Ser amino acids in an 11-mer peptide compared to an Ile amino acid in an 11-mer Ile peptide. This increase is related both with the hydrophilic nature of Ser and with a reduction of the excluded volume. The latter is confirmed by an even larger hydration increase in a C-term segment of NAC (85AGSIAAATGFV95), denoted herein NACterm peptide (Bertoncini et al. Citation2005; Galamba Citation2022), where Ile is sided by a Ser and an Ala; although no significant folding is found for 11-mer peptides (Galamba Citation2022), hydration is also influenced by peptide structural fluctuations.
As previously discussed, excluded volume, either next to a hydrophobic or hydrophilic amino acid results in a tetrahedrality increase of a water population that retains four water neighbors (W-4W, ). This can be seen in for Ile in an 11-mer Ile and NACterm peptides, and Ser (Ser-6), in an 11-mer Ser peptide. also confirms the water HB enhancement next to Ile within the W-4W population, compared to bulk water, more pronounced for the 3rd and 4th potential HB partners. The W-3W population shows an enhancement of only the first HB, whereas solute exclusion volume hinders the formation of more than two HBs, in average, within this population. The fact that such a population dominates in low curvature solutes (), along with the weak nature of dispersion interactions involved in hydrophobic group–water interactions, is responsible for a mild dewetting not found around molecules and groups that form HBs with water.
However, hydrophobic segments in proteins are generally limited in length and may not favor hydrophobic drug binding over protein–protein aggregation at therapeutically relevant concentrations. The latter is among the main obstacles to the rational drug design targeting protein–protein hydrophobic contacts.
Hydrophobic interactions
Hydrophobic interactions, the other related manifestation of the hydrophobic effect, are indirect, solvent-induced interactions associated with the dewetting of apolar groups and the formation of a liquid–vapor-like interface that culminates in the so-called hydrophobic collapse for sufficiently large hydrophobic surfaces (Stillinger Citation1973; Chandler Citation2005; Berne et al. Citation2009). This microscopic dewetting, while not opposed by solute–water interactions in ideal hard (i.e. repulsive) flat surfaces is outweighed by van der Waals interactions in real hydrophobic systems bringing water closer to the hydrophobic surface (Granick and Bae Citation2008; Jamadagni et al. Citation2011), resulting in a small (i.e. less than the size of a water molecule, Godawat et al. Citation2009), although important, width depletion region (Chandler Citation2005; Chattopadhyay et al. Citation2010). A similar phenomenon is not observed for small solutes, unless an extended surface cluster can form (Chandler Citation2005); even relatively large (e.g. neopentane) and long (e.g. dodecane) alkanes show small tendency to aggregate (Xavier and Galamba Citation2021).
Hydrophobic interactions are the main driving “force” responsible for protein folding (Kauzmann Citation1959; Dill Citation1990) and IDPs such as wild-type α-syn are thought not to fold because of their high net charge and low hydrophobicity. Hydrophobic interactions are also thought to be the main driving force in protein aggregation, as previously discussed for HbS and α-syn.
In light of the above discussion, small and large length-scale hydrophobic effects should be important to protein hydration, (mis)folding, and aggregation. In addition, water-mediated electrostatic interactions should also play an important part. For instance, rationalization of the hydrophobic interaction of Val-β6 with the host pocket in the neighbor HbS within the small length hydrophobic picture, would suggest a low tendency for aggregation since the hydrophobic pocket, which also exists in HbA, should be largely dehydrated in the monomer, as previously discussed. Thus, the main hydrophobic driving force should be associated with the dewetting of the side chain (isopropyl) of Val-β6. However, following the above discussion, other hydrophobic and electrostatic interactions resulting both from the departure of Glu-β6 and the insertion of Val-β6 should be important in triggering the aggregation process and the formation of a nucleus. Thus, although undoubtedly linked with the interaction of Val-β6 with the acceptor hydrophobic pocket (see ) and neighbor hydrophilic and hydrophobic amino acids, a comprehensive understanding of the initiation of the homogeneous nucleation at deoxy-HbS saturation conditions, remains elusive.
Figure 10. (a) Deoxy-HbS monomer showing the hydrophobic pocket (yellow); (b) deoxy-HbS monomer showing Val-β6 (yellow); (c) α-syn. (blue) Hydrophobic amino acids with aliphatic side chains (Ala, Ile, Leu, Met, and Val); (red) hydrophobic amino acids with aromatic side chains (Phe, Trp, Tyr); (orange) remaining amino acids; (green) Hem in deoxy-HbS.
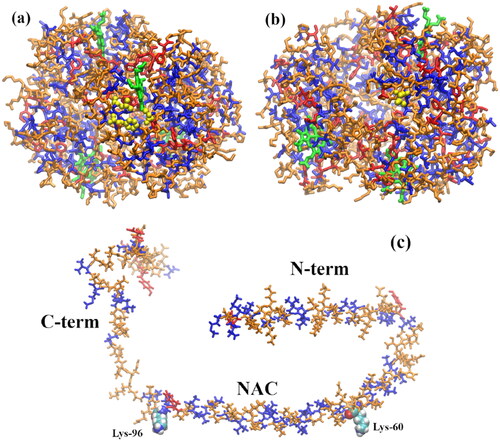
With respect to α-syn aggregation, again, in spite of copious differences relative to HbS, both hydrophobic length scales should be pivotal. For instance, NAC (35 amino acids) has 17 hydrophobic amino acids, mostly Val and Ala, but no more than four contiguous hydrophobic amino acids (88IAAA91), with the main hydrophobic domains being 88IAAA91, 69AVA71, 76AVA78, and 94FV95 (). The aggregation of the 11-mer domain (Bertoncini et al. Citation2005) NACterm (85AGSIAAATGFV95) of α-syn, which contains this longer hydrophobic domain (i.e. 88IAAA91) was recently studied through molecular simulations (Galamba Citation2022). A binding free energy of ∼−10 kJ mol−1 was found (Galamba Citation2022). Urea, a protein denaturant, that also induces protein disaggregation at high concentrations (∼6–8 M) was shown to strongly inhibit aggregation through the occupation of the dewetted interpeptide region precluding hydrophobic collapse. Whether some of the small molecules discussed in section “Protein aggregation inhibitors”, including some polyphenols, inhibit aggregation by directly interacting with the NAC or other domains of α-syn and/or reduce density fluctuations next to hydrophobic groups remains unknown. Furthermore, the relationship between polyphenols and other small molecules that stabilize specific oligomers and the hydrophobic effect, seemingly responsible for the oligomerization process, is also not clear. Nevertheless, some chemical groups such as aromatic rings and hydroxyl groups seem to play a key role in the aggregation and aggregation inhibition of distinct proteins. This is now briefly discussed, including some of the limitations and challenges associated with these ubiquitous groups and compound families, in drug design.
Hydrophobic effect and drug design
A small molecule or peptide aimed at inhibiting aggregation in vivo through some protein–protein contact impediment must (i) cross the cell membrane in intracellular aggregation-related diseases and the blood–brain barrier in NDs, (ii) selectively bind to some key protein aggregation site(s), and (iii) inhibit aggregation or induce the formation of non-toxic oligomers, and/or disrupt pathological oligomers or fibrils without compromising the protein’s function(s). Furthermore, the drug should be soluble, have high selectivity, specificity and potency, and low toxicity, along with good bioavailability and metabolic stability. We pause to note that in Alzheimer’s disease and PD, the diseases further induce changes in the blood–brain barrier, altering drug permeability, that, in spite of possible menaces, can also lead to novel drug therapies (Desai et al. Citation2007). In SCD, erythrocyte membrane alterations might also change drug permeability, in addition to known cation permeability perturbation, which results in erythrocyte dehydration (Brugnara Citation2018), favoring aggregation through the increase of deoxy-HbS concentration.
With regard to the selective binding to specific protein sites, knowledge of the aggregation mechanism is pivotal to define suitable targets to block aggregation and/or promote oligomer and fiber disassembly. For proteinopathies, the most common targets are hydrophobic regions such as the hydrophobic host pocket in deoxy-HbS and the NAC or the N-term region in α-syn because these are involved in the main contacts found in the respective fibers. However, structural changes affecting these regions, and thus, aggregation, can, in principle, also be driven by protein–drug interactions involving protein segments far-off from the main protein aggregation binding contacts.
A link between many protein aggregation inhibitors, including amino acids and peptides, is the existence of aromatic rings, which allow the formation of non-covalent stacked structures with aromatic amino acids and the interaction with aliphatic hydrophobic side chains. In addition, hydroxyl groups can interact with polar side chains and have been found to play a role in the aggregation inhibitory potency of small aromatic molecules (Ross and Subramanian Citation1977; Meng et al. Citation2009; Caruana et al. Citation2011). In this sense and considering the large collection of polyphenols that showed aggregation inhibitory activity in vitro for different proteins, although with reported diverse mechanisms, it could seem that finding a drug for therapeutic purposes would not be difficult. Nonetheless, although polyphenols occupy a relevant position among protein aggregation inhibitors, in addition to their long reported role as protective agents in NDs through various mechanisms (Freyssin et al. Citation2018; Henríquez et al. Citation2020), their general protective role remains elusive (Colizzi Citation2019). In particular, concerning their reputation as aggregation inhibitors, a more fundamental knowledge about the action mechanism of polyphenols and aromatic rings and hydroxyl groups, in general, seems important, namely, (i) their impact in protein solvation, including water density fluctuations and structural transformations, (ii) whether direct (i.e. protein-polyphenol) and/or indirect (i.e. polyphenol induced alterations of water–water and protein–water interactions) aggregation inhibition mechanisms are involved, (iii) their selectivity toward hydrophobic and hydrophilic domains, or (iv) their influence on the structure of the monomeric proteins. This could allow unraveling the fundamental intermolecular interactions through which some small molecules induce the formation of stable (not-toxic) oligomers, precluding oligomerization and even disrupt mature fibrils (Zhu et al. Citation2004; Hong et al. Citation2008; Bieschke et al. Citation2010; Lorenzen et al. Citation2014), whereas others may inhibit nucleation by stabilizing the monomer, although not disrupting preformed fibrils.
We stress that, the fact that some polyphenols were found to inhibit aggregation of distinct proteins (Henríquez et al. Citation2020), behaving almost as universal-aggregation-inhibitors, could mean these are not effective therapeutic drugs because of low selectivity, requiring concentrations too high to hamper key contacts. For instance, there is a common view that a drug, either a small molecule or a peptide-based drug (Armiento et al. Citation2020), that can shield specific domains of the NAC (Bertoncini et al. Citation2005; Dedmon et al. Citation2005) is of potential therapeutic interest in PD. The concentration necessary to block some pivotal sub-domain of the NAC, however, may not be therapeutically relevant because of a selectivity deficit. Indeed, looking at the hydrophobic–hydrophilic amino acid sequence in α-syn (), it is difficult to argue on the putative preferential binding of a polyphenol toward a specific domain, let alone its influence on the structure, solubility, and aggregation propensity of the monomer.
A similar rationale applies to peptide-based drugs. While the importance of aromatic rings was long reported in the context of deoxy-HbS aggregation, concentrations too high for therapeutic purposes were required to observe significant solubility increases (Olubiyi et al. Citation2019). This suggests that, despite peptides carrying aromatic rings can compete more effectively than small molecules with proteins for specific extended amphiphilic surfaces involved in fiber contacts, they still lack enough selectivity. In addition, some peptides may also aggregate, a characteristic possibly shared with some (low solubility) polyphenols.
Thus, although the rational design of aggregation inhibitors continues to hold the promise of developing effective therapeutics for several proteinopathies, the fact that many small molecules in drug libraries are soluble polyphenols or other amphiphilic molecules with aromatic rings (and OH groups), some of which have been considered PAINS, can taint drug screening assays (Baell and Nissink Citation2018), unless the aggregation inhibition mechanisms of some of these molecules, at a molecular-level, is unraveled. In this sense, it seems particularly promising to investigate the molecular origin of the seeming “universal” aggregation inhibitory activity of some small molecule drugs, either to improve drug design strategies based on these molecules or to rule them out as promising leads possibly for reasons that put them into the category of PAINS.
Conclusions
The failure of many potential drugs for NDs, especially Alzheimer’s disease, propelled the discussion (Makin Citation2018) on whether the amyloid hypothesis is, in fact, correct, and protein aggregation is the primary cause of NDs. However, unlike NDs, the molecular origin of SCD is well-known and there has also been limited progress in finding effective aggregation inhibitors. This naturally raises questions concerning the efficacy of this approach in SCD, NDs, and other proteinopathies. However, given the large number of diseases that involve some form of protein aggregate and the economic burden associated with future stem cell and gene therapies, the development of small molecule or peptide-based drugs remains a worth-exploring therapeutic route.
Herein, we provided an overview of the drugs developed to treat two completely different proteinopathies, namely, PD and SCD, aiming to establish some links associated with the importance of hydrophobic interactions in protein aggregation. The small and large length-scale regimes of the hydrophobic effect were discussed, along with the apparent relevance of aromatic rings and hydroxyl groups to aggregation inhibition. Although the ubiquity of polyphenols as potential small molecule drugs concerning a multitude of diseases, ranging from cardiovascular diseases to cancer and NDs, stimulates their study, their mechanisms as protein aggregation inhibitors remain poorly understood. As a consequence, the design of improved drugs that could dodge intrinsic drawbacks of polyphenols that showed aggregation inhibitory activity in vitro even at nontoxic concentrations, including low bioavailability and selectivity, has proven difficult.
Significant advances might emerge in a near future, nevertheless, linked with improved machine learning-assisted drug design methodologies (Vamathevan et al. Citation2019) and a more fundamental understanding of the action mechanism of potential drugs, polyphenols and others, concerning their influence on the impediment of intertwined hydrophobic and electrostatic interactions and protein solvation. In addition, the design of cyclic peptides represents a promising route toward the development of alternative drugs with enhanced selectivity while avoiding some of the limitations of linear peptides.
Disclosure statement
The authors report no declarations of interest.
Additional information
Funding
References
- Abbott A. 2010. Levodopa: the story so far. Nature. 466(7310):S6–S7.
- Abdulmalik O, Ghatge MS, Musayev FN, Parikh A, Chen Q, Yang J, Nnamani I, Danso-Danquah R, Eseonu DN, Asakura T, et al. 2011. Crystallographic analysis of human hemoglobin elucidates the structural basis of the potent and dual antisickling activity of pyridyl derivatives of vanillin. Acta Crystallogr D Biol Crystallogr. 67(Pt 11):920–928.
- Abdulmalik O, Safo MK, Chen Q, Yang J, Brugnara C, Ohene-Frempong K, Abraham DJ, Asakura T. 2005. 5-Hydroxymethyl-2-furfural modifies intracellular sickle haemoglobin and inhibits sickling of red blood cells. Br J Haematol. 128(4):552–561.
- Abraham DJ, Kennedy PE, Mehanna AS, Patwa DC, Williams FL. 1984. Design, synthesis, and testing of potential antisickling agents. 4. Structure–activity relationships of benzyloxy and phenoxy acids. J Med Chem. 27(8):967–978.
- Abraham DJ, Perutz MF, Phillips SE. 1983. Physiological and X-ray studies of potential antisickling agents. Proc Natl Acad Sci U S A. 80(2):324–328.
- Adachi K, Asakura T. 1978. Demonstration of a delay time during aggregation of diluted solutions of deoxyhemoglobin S and hemoglobin CHarlem in concentrated phosphate buffer. J Biol Chem. 253(19):6641–6643.
- Adachi K, Asakura T. 1979. Nucleation-controlled aggregation of deoxyhemoglobin S. Possible difference in the size of nuclei in different phosphate concentrations. J Biol Chem. 254(16):7765–7771.
- Adachi K, Asakura T. 1980. Polymerization of deoxyhemoglobin CHarlem (B6 Glu → Val, B73 Asp → Asn). J Mol Biol. 144(4):467–480.
- Adachi K, Ding M, Wehrli S, Reddy KS, Surrey S, Horiuchi K. 2003. Effects of different B73 amino acids on formation of 14-stranded fibers of Hb S versus double-stranded crystals of Hb C-Harlem. Biochemistry. 42(15):4476–4484.
- Adachi K, Konitzer P, Kim J, Welch N, Surrey S. 1993. Effects of beta 6 aromatic amino acids on polymerization and solubility of recombinant hemoglobins made in yeast. J Biol Chem. 268(29):21650–21656.
- Afitska K, Fucikova A, Shvadchak VV, Yushchenko DA. 2017. Modification of C terminus provides new insights into the mechanism of α-synuclein aggregation. Biophys J. 113(10):2182–2191.
- Aguzzi A, O’Connor T. 2010. Protein aggregation diseases: pathogenicity and therapeutic perspectives. Nat Rev Drug Discov. 9(3):237–248.
- Ahmad B, Lapidus LJ. 2012. Curcumin prevents aggregation in α-synuclein by increasing reconfiguration rate. J Biol Chem. 287(12):9193–9199.
- Akbar MGK, Tamura Y, Ding M, Ding H, Rosenblatt MM, Reddy KS, Surrey S, Adachi K. 2006. Inhibition of hemoglobin S polymerization in vitro by a novel 15-mer EF-helix β73 histidine-containing peptide. Biochemistry. 45(27):8358–8367.
- Ameh SJ, Tarfa FD, Ebeshi BU. 2012. Traditional herbal management of sickle cell anemia: lessons from Nigeria. Anemia. 2012:607436.
- Arbo BD, André-Miral C, Nasre-Nasser RG, Schimith LE, Santos MG, Costa-Silva D, Muccillo-Baisch AL, Hort MA. 2020. Resveratrol derivatives as potential treatments for Alzheimer’s and Parkinson’s disease. Front Aging Neurosci. 12:103.
- Armiento V, Spanopoulou A, Kapurniotu A. 2020. Peptide-based molecular strategies to interfere with protein misfolding, aggregation, and cell degeneration. Angew Chem Int Ed Engl. 59(9):3372–3384.
- Arter WE, Xu CK, Castellana-Cruz M, Herling TW, Krainer G, Saar KL, Kumita JR, Dobson CM, Knowles TPJ. 2020. Rapid structural, kinetic, and immunochemical analysis of alpha-synuclein oligomers in solution. Nano Lett. 20(11):8163–8169.
- Arya R, Rolan PE, Wootton R, Posner J, Bellingham AJ. 1996. Tucaresol increases oxygen affinity and reduces haemolysis in subjects with sickle cell anaemia. Br J Haematol. 93(4):817–821.
- Ashbaugh HS, Pratt LR. 2006. Colloquium: scaled particle theory and the length scales of hydrophobicity. Rev Mod Phys. 78(1):159–178.
- Ataga KI, Kutlar A, Kanter J, Liles D, Cancado R, Friedrisch J, Guthrie TH, Knight-Madden J, Alvarez OA, Gordeuk VR, et al. 2017. Crizanlizumab for the prevention of pain crises in sickle cell disease. N Engl J Med. 376(5):429–439.
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. 1998. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol. 152(4):879–884.
- Baell J, Walters MA. 2014. Chemistry: chemical con artists foil drug discovery. Nature. 513(7519):481–483.
- Baell JB, Holloway GA. 2010. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 53(7):2719–2740.
- Baell JB, Nissink JWM. 2018. Seven year itch: pan-assay interference compounds (PAINS) in 2017—utility and limitations. ACS Chem Biol. 13(1):36–44.
- Barone P. 2010. Neurotransmission in Parkinson’s disease: beyond dopamine: neurotransmission in Parkinson’s disease. Eur J Neurol. 17(3):364–376.
- Bartolini M, Andrisano V. 2010. Strategies for the inhibition of protein aggregation in human diseases. Chembiochem. 11(8):1018–1035.
- Bengoa-Vergniory N, Roberts RF, Wade-Martins R, Alegre-Abarrategui J. 2017. Alpha-synuclein oligomers: a new hope. Acta Neuropathol. 134(6):819–838.
- Ben-Naim A. 1975. Hydrophobic interaction and structural changes in the solvent. Biopolymers. 14(7):1337–1355.
- Ben-Naim A. 1978. A simple model for demonstrating the relation between solubility, hydrophobic interaction, and structural changes in the solvent. J Phys Chem. 82(8):874–885.
- Berne BJ, Weeks JD, Zhou R. 2009. Dewetting and hydrophobic interaction in physical and biological systems. Annu Rev Phys Chem. 60(1):85–103.
- Bertoncini CW, Jung Y-S, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M. 2005. From the cover: release of long-range tertiary interactions potentiates aggregation of natively unstructured -synuclein. Proc Natl Acad Sci U S A. 102(5):1430–1435.
- Biere AL, Wood SJ, Wypych J, Steavenson S, Jiang Y, Anafi D, Jacobsen FW, Jarosinski MA, Wu G-M, Louis J-C, et al. 2000. Parkinson’s disease-associated α-synuclein is more fibrillogenic than β- and γ-synuclein and cannot cross-seed its homologs. J Biol Chem. 275(44):34574–34579.
- Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE. 2010. EGCG remodels mature α-synuclein and amyloid-β fibrils and reduces cellular toxicity. Proc Natl Acad Sci U S A. 107(17):7710–7715.
- Blair HA, Dhillon S. 2017. Safinamide: a review in Parkinson’s disease. CNS Drugs. 31(2):169–176.
- Bodles AM, Guthrie DJS, Greer B, Irvine GB. 2001. Identification of the region of non-Aβ component (NAC) of Alzheimer’s disease amyloid responsible for its aggregation and toxicity: bioactive region of NAC. J Neurochem. 78(2):384–395.
- Bookchin RM, Nagel RL, Ranney HM. 1967. Structure and properties of hemoglobin C-Harlem, a human hemoglobin variant with amino acid substitutions in 2 residues of the beta-polypeptide chain. J Biol Chem. 242(2):248–255.
- Borah P, Sanjeev A, Mattaparthi VSK. 2021. Computational investigation on the effect of oleuropein aglycone on the α-synuclein aggregation. J Biomol Struct Dyn. 39(4):1259–1270.
- Bosquesi PL, Melchior ACB, Pavan AR, Lanaro C, de Souza CM, Rusinova R, Chelucci RC, Barbieri KP, Fernandes GFDS, Carlos IZ, et al. 2020. Synthesis and evaluation of resveratrol derivatives as fetal hemoglobin inducers. Bioorg Chem. 100:103948.
- Breydo L, Wu JW, Uversky VN. 2012. α-Synuclein misfolding and Parkinson’s disease. Biochim Biophys Acta. 1822(2):261–285.
- Brugnara C. 2018. Sickle cell dehydration: pathophysiology and therapeutic applications. Clin Hemorheol Microcirc. 68(2–3):187–204.
- Bu X-L, Rao PPN, Wang Y-J. 2016. Anti-amyloid aggregation activity of natural compounds: implications for Alzheimer’s drug discovery. Mol Neurobiol. 53(6):3565–3575.
- Bucciantini M, Giannoni E, Chiti F, Baroni F, Formigli L, Zurdo J, Taddei N, Ramponi G, Dobson CM, Stefani M. 2002. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 416(6880):507–511.
- Cao Z, Ferrone FA. 1997. Homogeneous nucleation in sickle hemoglobin: stochastic measurements with a parallel method. Biophys J. 72(1):343–352.
- Carecho R, Figueira I, Terrasso AP, Godinho-Pereira J, de Oliveira Sequeira C, Pereira SA, Milenkovic D, Leist M, Brito C, Nunes dos Santos C. 2022. Circulating (poly)phenol metabolites: neuroprotection in a 3D cell model of Parkinson’s disease. Mol Nutr Food Res. 66(21):2100959.
- Caruana M, Högen T, Levin J, Hillmer A, Giese A, Vassallo N. 2011. Inhibition and disaggregation of α-synuclein oligomers by natural polyphenolic compounds. FEBS Lett. 585(8):1113–1120.
- Cascella R, Bigi A, Cremades N, Cecchi C. 2022. Effects of oligomer toxicity, fibril toxicity and fibril spreading in synucleinopathies. Cell Mol Life Sci. 79(3):174.
- Castle BT, Odde DJ, Wood DK. 2019. Rapid and inefficient kinetics of sickle hemoglobin fiber growth. Sci Adv. 5(3):eaau1086.
- Caughey B, Lansbury PT. 2003. Separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 26(1):267–298.
- Cellmer T, Ferrone FA, Eaton WA. 2016. Universality of supersaturation in protein-fiber formation. Nat Struct Mol Biol. 23(5):459–461.
- Chandler D. 2005. Interfaces and the driving force of hydrophobic assembly. Nature. 437(7059):640–647.
- Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR. 1995. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. N Engl J Med. 332(20):1317–1322.
- Chattopadhyay S, Uysal A, Stripe B, Ha Y, Marks TJ, Karapetrova EA, Dutta P. 2010. How water meets a very hydrophobic surface. Phys Rev Lett. 105(3):037803.
- Chen K, Ballas SK, Hantgan RR, Kim-Shapiro DB. 2004. Aggregation of normal and sickle hemoglobin in high concentration phosphate buffer. Biophys J. 87(6):4113–4121.
- Cheng F, Vivacqua G, Yu S. 2011. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J Chem Neuroanat. 42(4):242–248.
- Cheruvara H, Allen-Baume VL, Kad NM, Mason JM. 2015. Intracellular screening of a peptide library to derive a potent peptide inhibitor of α-synuclein aggregation. J Biol Chem. 290(12):7426–7435.
- Colizzi C. 2019. The protective effects of polyphenols on Alzheimer’s disease: a systematic review. Alzheimers Dement. 5(1):184–196.
- Conti Nibali V, Pezzotti S, Sebastiani F, Galimberti DR, Schwaab G, Heyden M, Gaigeot M-P, Havenith M. 2020. Wrapping up hydrophobic hydration: locality matters. J Phys Chem Lett. 11(12):4809–4816.
- Conway KA, Harper JD, Lansbury PT. 1998. Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med. 4(11):1318–1320.
- Conway KA, Rochet J-C, Bieganski RM, Lansbury PT. 2001. Kinetic stabilization of the α-synuclein protofibril by a dopamine-α-synuclein adduct. Science. 294(5545):1346–1349.
- Cremades N, Chen SW, Dobson CM. 2017. Structural characteristics of α-synuclein oligomers. Int Rev Cell Mol Biol. 329:79–143.
- Cremades N, Cohen SIA, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, et al. 2012. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell. 149(5):1048–1059.
- Cremades N, Dobson CM. 2018. The contribution of biophysical and structural studies of protein self-assembly to the design of therapeutic strategies for amyloid diseases. Neurobiol Dis. 109(Pt B):178–190.
- Crowther RA, Jakes R, Spillantini MG, Goedert M. 1998. Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett. 436(3):309–312.
- Cunningham AD, Qvit N, Mochly-Rosen D. 2017. Peptides and peptidomimetics as regulators of protein–protein interactions. Curr Opin Struct Biol. 44:59–66.
- Cyranoski D. 2017. Trials of embryonic stem cells to launch in China. Nature. 546(7656):15–16.
- Dampier C. 2015. Orphan drugs for sickle vaso-occlusion: dawn of a new era of targeted treatment. Orphan Drugs Res Rev. 5:99.
- Das DR, Kumar D, Kumar P, Dash BP. 2020. Molecular docking and its application in search of antisickling agent from Carica papaya. J Appl Biol Biotechnol. 8(1):105–116.
- Dash B, Archana Y, Satapathy N, Naik S. 2013. Search for antisickling agents from plants. Pharmacogn Rev. 7(13):53–60.
- Davidson WS, Jonas A, Clayton DF, George JM. 1998. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem. 273(16):9443–9449.
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. 1997. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 90(3):537–548.
- Davis JG, Gierszal KP, Wang P, Ben-Amotz D. 2012. Water structural transformation at molecular hydrophobic interfaces. Nature. 491(7425):582–585.
- de Andrade Teles RB, Diniz TC, Costa Pinto TC, de Oliveira Júnior RG, Gama e Silva M, de Lavor ÉM, Fernandes AWC, de Oliveira AP, de Almeida Ribeiro FPR, da Silva AAM, et al. 2018. Flavonoids as therapeutic agents in Alzheimer’s and Parkinson’s diseases: a systematic review of preclinical evidences. Oxid Med Cell Longev. 2018:7043213.
- Dean J, Schechter AN. 1978a. Sickle-cell anemia: molecular and cellular bases of therapeutic approaches: (second of three parts). N Engl J Med. 299(15):804–811.
- Dean J, Schechter AN. 1978b. Sickle-cell anemia: molecular and cellular bases of therapeutic approaches: (third of three parts). N Engl J Med. 299(16):863–870.
- Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. 2005. Mapping long-range interactions in α-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc. 127(2):476–477.
- Desai BS, Monahan AJ, Carvey PM, Hendey B. 2007. Blood–brain barrier pathology in Alzheimer’s and Parkinson’s disease: implications for drug therapy. Cell Transplant. 16(3):285–299.
- Dill KA. 1990. Dominant forces in protein folding. Biochemistry. 29(31):7133–7155.
- Doherty CPA, Ulamec SM, Maya-Martinez R, Good SC, Makepeace J, Khan GN, van Oosten-Hawle P, Radford SE, Brockwell DJ. 2020. A short motif in the N-terminal region of α-synuclein is critical for both aggregation and function. Nat Struct Mol Biol. 27(3):249–259.
- Doig AJ, Derreumaux P. 2015. Inhibition of protein aggregation and amyloid formation by small molecules. Curr Opin Struct Biol. 30:50–56.
- Dong C, Garen CR, Mercier P, Petersen NO, Woodside MT. 2019. Characterizing the inhibition of α-synuclein oligomerization by a pharmacological chaperone that prevents prion formation by the protein PrP. Protein Sci. 28:1690–1702.
- Dougherty PG, Qian Z, Pei D. 2017. Macrocycles as protein–protein interaction inhibitors. Biochem J. 474(7):1109–1125.
- Du H-N, Tang L, Luo X-Y, Li H-T, Hu J, Zhou J-W, Hu H-Y. 2003. A peptide motif consisting of glycine, alanine, and valine is required for the fibrillization and cytotoxicity of human alpha-synuclein. Biochemistry. 42(29):8870–8878.
- Dykes G, Crepeau RH, Edelstein SJ. 1978. Three-dimensional reconstruction of the fibres of sickle cell haemoglobin. Nature. 272(5653):506–510.
- Dykes GW, Crepeau RH, Edelstein SJ. 1979. Three-dimensional reconstruction of the 14-filament fibers of hemoglobin S. J Mol Biol. 130(4):451–472.
- Eapen M, Brazauskas R, Walters MC, Bernaudin F, Bo-Subait K, Fitzhugh CD, Hankins JS, Kanter J, Meerpohl JJ, Bolaños-Meade J, et al. 2019. Effect of donor type and conditioning regimen intensity on allogeneic transplantation outcomes in patients with sickle cell disease: a retrospective multicentre, cohort study. Lancet Haematol. 6(11):e585–e596.
- Eaton WA, Bunn HF. 2017. Treating sickle cell disease by targeting HbS polymerization. Blood. 129(20):2719–2726.
- Eaton WA, Hofrichter J. 1987. Hemoglobin S gelation and sickle cell disease. Blood. 70(5):1245–1266.
- Eaton WA, Hofrichter J. 1990. Sickle cell hemoglobin polymerization. Adv Protein Chem. 40:63–279.
- Eaton WA. 2022. Impact of hemoglobin biophysical studies on molecular pathogenesis and drug therapy for sickle cell disease. Mol Aspects Med. 84:100971.
- Edelstein SJ. 1980. Structure of the fibers of hemoglobin S. Tex Rep Biol Med. 40:221–232.
- Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, Engemann S, Pastore A, Wanker EE. 2008. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol. 15(6):558–566.
- El-Agnaf OMA, Jakes R, Curran MD, Wallace A. 1998. Effects of the mutations Ala30 to Pro and Ala53 to Thr on the physical and morphological properties of α-synuclein protein implicated in Parkinson’s disease. FEBS Lett. 440(1–2):67–70.
- El-Agnaf OMA, Paleologou KE, Greer B, Abogrein AM, King JE, Salem SA, Fullwood NJ, Benson FE, Hewitt R, Ford KJ, et al. 2004. Strategy for designing inhibitors of α-synuclein aggregation and toxicity as a novel treatment for Parkinson’s disease and related disorders. FASEB J. 18(11):1315–1317.
- Elbaum D, Harrington JP, Bookchin RM, Nagel RL. 1978. Kinetics of Hb S gelation effect of alkylureas, ionic strength and other hemoglobins. Biochim Biophys Acta. 534(2):228–238.
- Elbaum D, Nagel RL, Bookchin RM, Herskovits TT. 1974. Effect of alkylureas on the polymerization of hemoglobin S. Proc Natl Acad Sci U S A. 71(12):4718–4722.
- Elbaum D, Roth EF, Neumann G, Jaffé ER, Bookchin RM, Nagel RL. 1976. Molecular and cellular effects of antisickling concentrations of alkylureas. Blood. 48(2):273–282.
- Eliezer D, Kutluay E, Bussell R, Browne G. 2001. Conformational properties of α-synuclein in its free and lipid-associated states. J Mol Biol. 307(4):1061–1073.
- El-Turk F, Newby FN, De Genst E, Guilliams T, Sprules T, Mittermaier A, Dobson CM, Vendruscolo M. 2016. Structural effects of two camelid nanobodies directed to distinct C-terminal epitopes on α-synuclein. Biochemistry. 55(22):3116–3122.
- Errington JR, Debenedetti PG. 2001. Relationship between structural order and the anomalies of liquid water. Nature. 409(6818):318–321.
- Esteban-Martín S, Silvestre-Ryan J, Bertoncini CW, Salvatella X. 2013. Identification of fibril-like tertiary contacts in soluble monomeric α-synuclein. Biophys J. 105(5):1192–1198.
- Eugene C, Laghaei R, Mousseau N. 2014. Early oligomerization stages for the non-amyloid component of α-synuclein amyloid. J Chem Phys. 141(13):135103.
- Fall AB, Vanhaelen-Fastré R, Vanhaelen M, Lo I, Toppet M, Ferster A, Fondu P. 1999. In vitro antisickling activity of a rearranged limonoid isolated from Khaya senegalensis. Planta Med. 65(3):209–212.
- Farzadfard A, Pedersen JN, Meisl G, Somavarapu AK, Alam P, Goksøyr L, Nielsen MA, Sander AF, Knowles TPJ, Pedersen JS, et al. 2022. The C-terminal tail of α-synuclein protects against aggregate replication but is critical for oligomerization. Commun Biol. 5(1):123.
- Feany MB, Bender WW. 2000. A Drosophila model of Parkinson’s disease. Nature. 404(6776):394–398.
- Ferreira A, Marguti I, Bechmann I, Jeney V, Chora Â, Palha NR, Rebelo S, Henri A, Beuzard Y, Soares MP. 2011. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell. 145(3):398–409.
- Ferrone FA, Hofrichter J, Eaton WA. 1985a. Kinetics of sickle hemoglobin polymerization. I. Studies using temperature-jump and laser photolysis techniques. J Mol Biol. 183(4):591–610.
- Ferrone FA, Hofrichter J, Eaton WA. 1985b. Kinetics of sickle hemoglobin polymerization. II. A double nucleation mechanism. J Mol Biol. 183(4):611–631.
- Fields CR, Bengoa-Vergniory N, Wade-Martins R. 2019. Targeting alpha-synuclein as a therapy for Parkinson’s disease. Front Mol Neurosci. 12:299.
- Fink AL. 2006. The aggregation and fibrillation of α-synuclein. Acc Chem Res. 39(9):628–634.
- Freyssin A, Page G, Fauconneau B, Rioux Bilan A. 2018. Natural polyphenols effects on protein aggregates in Alzheimer’s and Parkinson’s prion-like diseases. Neural Regen Res. 13(6):955–961.
- Galamba N, Paiva A, Barreiros S, Simões P. 2019. Solubility of polar and nonpolar aromatic molecules in subcritical water: the role of the dielectric constant. J Chem Theory Comput. 15(11):6277–6293.
- Galamba N, Pipolo S. 2018. On the binding free energy and molecular origin of sickle cell hemoglobin aggregation. J Phys Chem B. 122(30):7475–7483.
- Galamba N. 2013. Water’s structure around hydrophobic solutes and the iceberg model. J Phys Chem B. 117(7):2153–2159.
- Galamba N. 2014. Water tetrahedrons, hydrogen-bond dynamics, and the orientational mobility of water around hydrophobic solutes. J Phys Chem B. 118(15):4169–4176.
- Galamba N. 2019. On the nonaggregation of normal adult hemoglobin and the aggregation of sickle cell hemoglobin. J Phys Chem B. 123(50):10735–10745.
- Galamba N. 2021. Free energy convergence in short- and long-length hydrophobic hydration. J Mol Liq. 339:116699.
- Galamba N. 2022. Aggregation of a Parkinson’s disease-related peptide: when does urea weaken hydrophobic interactions? ACS Chem Neurosci. 13(12):1769–1781.
- Galkin O, Pan W, Filobelo L, Hirsch RE, Nagel RL, Vekilov PG. 2007. Two-step mechanism of homogeneous nucleation of sickle cell hemoglobin polymers. Biophys J. 93(3):902–913.
- Galvagnion C, Brown JWP, Ouberai MM, Flagmeier P, Vendruscolo M, Buell AK, Sparr E, Dobson CM. 2016. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of α-synuclein. Proc Natl Acad Sci U S A. 113(26):7065–7070.
- Galvagnion C, Buell AK, Meisl G, Michaels TCT, Vendruscolo M, Knowles TPJ, Dobson CM. 2015. Lipid vesicles trigger α-synuclein aggregation by stimulating primary nucleation. Nat Chem Biol. 11(3):229–234.
- Gao X, Carroni M, Nussbaum-Krammer C, Mogk A, Nillegoda NB, Szlachcic A, Guilbride DL, Saibil HR, Mayer MP, Bukau B. 2015. Human Hsp70 disaggregase reverses Parkinson’s-linked α-synuclein amyloid fibrils. Mol Cell. 59(5):781–793.
- Garde S, Hummer G, García AE, Paulaitis ME, Pratt LR. 1996. Origin of entropy convergence in hydrophobic hydration and protein folding. Phys Rev Lett. 77(24):4966–4968.
- Gautam S, Karmakar S, Batra R, Sharma P, Pradhan P, Singh J, Kundu B, Chowdhury PK. 2017. Polyphenols in combination with β-cyclodextrin can inhibit and disaggregate α-synuclein amyloids under cell mimicking conditions: a promising therapeutic alternative. Biochim Biophys Acta Proteins Proteom. 1865(5):589–603.
- Gautam S, Karmakar S, Bose A, Chowdhury PK. 2014. β-Cyclodextrin and curcumin, a potent cocktail for disaggregating and/or inhibiting amyloids: a case study with α-synuclein. Biochemistry. 53(25):4081–4083.
- Ghosh D, Mondal M, Mohite GM, Singh PK, Ranjan P, Anoop A, Ghosh S, Jha NN, Kumar A, Maji SK. 2013. The Parkinson’s disease-associated H50Q mutation accelerates α-synuclein aggregation in vitro. Biochemistry. 52(40):6925–6927.
- Giasson BI, Murray IVJ, Trojanowski JQ, Lee VM-Y. 2001. A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J Biol Chem. 276(4):2380–2386.
- Giorgetti S, Greco C, Tortora P, Aprile F. 2018. Targeting amyloid aggregation: an overview of strategies and mechanisms. Int J Mol Sci. 19(9):2677.
- Godawat R, Jamadagni SN, Garde S. 2009. Characterizing hydrophobicity of interfaces by using cavity formation, solute binding, and water correlations. Proc Natl Acad Sci U S A. 106(36):15119–15124.
- Goedert M, Jakes R, Spillantini MG. 2017. The synucleinopathies: twenty years on. J Parkinsons Dis. 7(S1):S51–S69.
- Goldberg MS, Lansbury PTJr. 2000. Is there a cause-and-effect relationship between α-synuclein fibrillization and Parkinson’s disease? Nat Cell Biol. 2(7):E115–E119.
- Gopalsamy A, Aulabaugh AE, Barakat A, Beaumont KC, Cabral S, Canterbury DP, Casimiro-Garcia A, Chang JS, Chen MZ, Choi C, et al. 2021. PF-07059013: a noncovalent modulator of hemoglobin for treatment of sickle cell disease. J Med Chem. 64(1):326–342.
- Gorecki M, Votano JR, Rich A. 1980. Peptide inhibitors of sickle hemoglobin aggregation: effect of hydrophobicity. Biochemistry. 19(8):1564–1568.
- Granick S, Bae SC. 2008. A curious antipathy for water. Science. 322(5907):1477–1478.
- Graziano G. 2005. Entropy convergence in the hydration thermodynamics of n-alcohols. J Phys Chem B. 109(24):12160–12166.
- Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. 2005. The E46K mutation in α-synuclein increases amyloid fibril formation. J Biol Chem. 280(9):7800–7807.
- Grunwald E, Steel C. 1995. Solvent reorganization and thermodynamic enthalpy–entropy compensation. J Am Chem Soc. 117(21):5687–5692.
- Guzzo A, Delarue P, Rojas A, Nicolaï A, Maisuradze GG, Senet P. 2022. Wild-type α-synuclein and variants occur in different disordered dimers and pre-fibrillar conformations in early stage of aggregation. Front Mol Biosci. 9:910104.
- Harrington DJ, Adachi K, Royer WE. 1997. The high resolution crystal structure of deoxyhemoglobin S. J Mol Biol. 272(3):398–407.
- Harrington JP, Elbaum D, Bookchin RM, Wittenberg JB, Nagel RL. 1977. Ligand kinetics of hemoglobin S containing erythrocytes. Proc Natl Acad Sci U S A. 74(1):203–206.
- Henríquez G, Gomez A, Guerrero E, Narayan M. 2020. Potential role of natural polyphenols against protein aggregation toxicity: in vitro, in vivo, and clinical studies. ACS Chem Neurosci. 11(19):2915–2934.
- Henry ER, Metaferia B, Li Q, Harper J, Best RB, Glass KE, Cellmer T, Dunkelberger EB, Conrey A, Thein SL, et al. 2021. Treatment of sickle cell disease by increasing oxygen affinity of hemoglobin. Blood. 138(13):1172–1181.
- Herrera FE, Chesi A, Paleologou KE, Schmid A, Munoz A, Vendruscolo M, Gustincich S, Lashuel HA, Carloni P. 2008. Inhibition of α-synuclein fibrillization by dopamine is mediated by interactions with five C-terminal residues and with E83 in the NAC region. PLOS One. 3(10):e3394.
- Hofrichter J, Ross PD, Eaton WA. 1974. Kinetics and mechanism of deoxyhemoglobin S gelation: a new approach to understanding sickle cell disease. Proc Natl Acad Sci U S A. 71(12):4864–4868.
- Hong D-P, Fink AL, Uversky VN. 2008. Structural characteristics of alpha-synuclein oligomers stabilized by the flavonoid baicalein. J Mol Biol. 383(1):214–223.
- Horn HW, Swope WC, Pitera JW, Madura JD, Dick TJ, Hura GL, Head-Gordon T. 2004. Development of an improved four-site water model for biomolecular simulations: TIP4P-Ew. J Chem Phys. 120(20):9665–9678.
- Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. 2006. Comparison of multiple amber force fields and development of improved protein backbone parameters. Proteins. 65(3):712–725.
- Horsley JR, Jovcevski B, Pukala TL, Abell AD. 2022. Designer D-peptides targeting the N-terminal region of α-synuclein to prevent Parkinsonian-associated fibrilization and cytotoxicity. Biochim Biophys Acta Proteins Proteom. 1870(10):140826.
- Huang DM, Chandler D. 2000. Temperature and length scale dependence of hydrophobic effects and their possible implications for protein folding. Proc Natl Acad Sci U S A. 97(15):8324–8327.
- Hummer G, Garde S, Garcia AE, Pohorille A, Pratt LR. 1996. An information theory model of hydrophobic interactions. Proc Natl Acad Sci U S A. 93(17):8951–8955.
- Huot P, Johnston TH, Koprich JB, Fox SH, Brotchie JM. 2013. The pharmacology of l-DOPA-induced dyskinesia in Parkinson’s disease. Pharmacol Rev. 65(1):171–222.
- Imaga NA. 2013. Phytomedicines and nutraceuticals: alternative therapeutics for sickle cell anemia. ScientificWorldJournal. 2013:269659.
- Ingram VM. 1956. A specific chemical difference between the globins of normal human and sickle-cell anaemia haemoglobin. Nature. 178(4537):792–794.
- Ingram VM. 1957. Gene mutations in human haemoglobin: the chemical difference between normal and sickle cell haemoglobin. Nature. 180(4581):326–328.
- Itano HA. 1953. Solubilities of naturally occurring mixtures of human hemoglobin. Arch Biochem Biophys. 47(1):148–159.
- Ivanova M, Jasuja R, Krasnosselskaia L, Josephs R, Wang Z, Ding M, Horiuchi K, Adachi K, Ferrone FA. 2001. Flexibility and nucleation in sickle hemoglobin. J Mol Biol. 314(4):851–861.
- Iyamu EW, Turner EA, Asakura T. 2002. In vitro effects of NIPRISAN (Nix-0699): a naturally occurring, potent antisickling agent: antisickling effects of Nix-0699. Br J Haematol. 118(1):337–343.
- Jamadagni SN, Godawat R, Garde S. 2011. Hydrophobicity of proteins and interfaces: insights from density fluctuations. Annu Rev Chem Biomol Eng. 2(1):147–171.
- Javed H, Nagoor Meeran MF, Azimullah S, Adem A, Sadek B, Ojha SK. 2018. Plant extracts and phytochemicals targeting α-synuclein aggregation in Parkinson’s disease models. Front Pharmacol. 9:1555.
- Jenner P. 2003. Dopamine agonists, receptor selectivity and dyskinesia induction in Parkinsonʼs disease. Curr Opin Neurol. 16:S3–S7.
- Jia L, Wang Y, Sang J, Cui W, Zhao W, Wei W, Chen B, Lu F, Liu F. 2019. Dihydromyricetin inhibits α-synuclein aggregation, disrupts preformed fibrils, and protects neuronal cells in culture against amyloid-induced cytotoxicity. J Agric Food Chem. 67(14):3946–3955.
- Kapoor S, Little JA, Pecker LH. 2018. Advances in the treatment of sickle cell disease. Mayo Clin Proc. 93(12):1810–1824.
- Karpinar DP, Balija MBG, Kügler S, Opazo F, Rezaei-Ghaleh N, Wender N, Kim H-Y, Taschenberger G, Falkenburger BH, Heise H, et al. 2009. Pre-fibrillar α-synuclein variants with impaired β-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 28(20):3256–3268.
- Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, Smith WR, Panepinto JA, Weatherall DJ, Costa FF, et al. 2018. Sickle cell disease. Nat Rev Dis Primers. 4(1):18010.
- Kato GJ, Steinberg MH, Gladwin MT. 2017. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. 127(3):750–760.
- Kauzmann W. 1959. Some factors in the interpretation of protein denaturation. Adv Protein Chem. 14:1–63.
- Khatoon SS, Rehman M, Rahman A. 2018. The role of natural products in Alzheimer’s and Parkinson’s disease. Stud Nat Prod Chem. 56:69–127.
- Kim YS, Lim D, Kim JY, Kang SJ, Kim Y-H, Im H. 2009. β-Sheet-breaking peptides inhibit the fibrillation of human α-synuclein. Biochem Biophys Res Commun. 387(4):682–687.
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kösel S, Przuntek H, Epplen JT, Schols L, Riess O. 1998. AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet. 18(2):106–108.
- Kubota S, Chang CT, Samejima T, Yang JT. 1976. Oligopeptides as potential antiaggregation agent for proteins: hemoglobin S gel and insulin dimer. J Am Chem Soc. 98(9):2677–2678.
- Kuczera K, Gao J, Tidor B, Karplus M. 1990. Free energy of sickling: a simulation analysis. Proc Natl Acad Sci U S A. 87(21):8481–8485.
- Lamberto GR, Binolfi A, Orcellet ML, Bertoncini CW, Zweckstetter M, Griesinger C, Fernández CO. 2009. Structural and mechanistic basis behind the inhibitory interaction of PcTS on α-synuclein amyloid fibril formation. Proc Natl Acad Sci U S A. 106(50):21057–21062.
- Lamberto GR, Torres-Monserrat V, Bertoncini CW, Salvatella X, Zweckstetter M, Griesinger C, Fernández CO. 2011. Toward the discovery of effective polycyclic inhibitors of α-synuclein amyloid assembly. J Biol Chem. 286(37):32036–32044.
- Landeck N, Strathearn KE, Ysselstein D, Buck K, Dutta S, Banerjee S, Lv Z, Hulleman JD, Hindupur J, Lin L-K, et al. 2020. Two C-terminal sequence variations determine differential neurotoxicity between human and mouse α-synuclein. Mol Neurodegener. 15(1):49.
- Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT. 2002. Amyloid pores from pathogenic mutations. Nature. 418(6895):291.
- Lashuel HA, Overk CR, Oueslati A, Masliah E. 2013. The many faces of α-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci. 14(1):38–48.
- Latawiec D, Herrera F, Bek A, Losasso V, Candotti M, Benetti F, Carlino E, Kranjc A, Lazzarino M, Gustincich S, et al. 2010. Modulation of alpha-synuclein aggregation by dopamine analogs. PLOS One. 5(2):e9234.
- Lee B. 1991. Solvent reorganization contribution to the transfer thermodynamics of small nonpolar molecules. Biopolymers. 31(8):993–1008.
- Lee E-N, Cho H-J, Lee C-H, Lee D, Chung KC, Paik SR. 2004. Phthalocyanine tetrasulfonates affect the amyloid formation and cytotoxicity of α-synuclein. Biochemistry. 43(12):3704–3715.
- Lee S-J, Jeon H, Kandror KV. 2008. Alpha-synuclein is localized in a subpopulation of rat brain synaptic vesicles. Acta Neurobiol Exp. 68(4):509–515.
- Li B, Ge P, Murray KA, Sheth P, Zhang M, Nair G, Sawaya MR, Shin WS, Boyer DR, Ye S, et al. 2018. Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun. 9(1):3609.
- Li H-T, Lin D-H, Luo X-Y, Zhang F, Ji L-N, Du H-N, Song G-Q, Hu J, Zhou J-W, Hu H-Y. 2005. Inhibition of alpha-synuclein fibrillization by dopamine analogs via reaction with the amino groups of alpha-synuclein. Implication for dopaminergic neurodegeneration. FEBS J. 272(14):3661–3672.
- Li J, Uversky VN, Fink AL. 2001. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry. 40(38):11604–11613.
- Li Y, Zhao C, Luo F, Liu Z, Gui X, Luo Z, Zhang X, Li D, Liu C, Li X. 2018. Amyloid fibril structure of α-synuclein determined by cryo-electron microscopy. Cell Res. 28(9):897–903.
- Liang Z, Chan HYE, Lee MM, Chan MK. 2021. A SUMO1-derived peptide targeting SUMO-interacting motif inhibits α-synuclein aggregation. Cell Chem Biol. 28(2):180–190.e6.
- Lorenzen N, Nielsen SB, Yoshimura Y, Vad BS, Andersen CB, Betzer C, Kaspersen JD, Christiansen G, Pedersen JS, Jensen PH, et al. 2014. How epigallocatechin gallate can inhibit α-synuclein oligomer toxicity in vitro. J Biol Chem. 289(31):21299–21310.
- Lu L, Li X, Vekilov PG, Karniadakis GE. 2016. Probing the twisted structure of sickle hemoglobin fibers via particle simulations. Biophys J. 110(9):2085–2093.
- Lu L, Li Z, Li H, Li X, Vekilov PG, Karniadakis GE. 2019. Quantitative prediction of erythrocyte sickling for the development of advanced sickle cell therapies. Sci Adv. 5(8):eaax3905.
- Luk KC, Mills IP, Trojanowski JQ, Lee VM-Y. 2008. Interactions between Hsp70 and the hydrophobic core of α-synuclein inhibit fibril assembly. Biochemistry. 47(47):12614–12625.
- Lum K, Chandler D, Weeks JD. 1999. Hydrophobicity at small and large length scales. J Phys Chem B. 103(22):4570–4577.
- Ma J, Gao J, Wang J, Xie A. 2019. Prion-like mechanisms in Parkinson’s disease. Front Neurosci. 13:552.
- Madine J, Doig AJ, Middleton DA. 2008. Design of an N-methylated peptide inhibitor of α-synuclein aggregation guided by solid-state NMR. J Am Chem Soc. 130(25):7873–7881.
- Maity D, Pal D. 2021. Molecular dynamics of hemoglobin reveals structural alterations and explains the interactions driving sickle cell fibrillation. J Phys Chem B. 125(35):9921–9933.
- Makin S. 2018. The amyloid hypothesis on trial. Nature. 559(7715):S4–S7.
- Maroteaux L, Campanelli J, Scheller R. 1988. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci. 8(8):2804–2815.
- Masliah E, Rockenstein E, Veinbergs I, Mallory M, Hashimoto M, Takeda A, Sagara Y, Sisk A, Mucke L. 2000. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science. 287(5456):1265–1269.
- Masuda M, Suzuki N, Taniguchi S, Oikawa T, Nonaka T, Iwatsubo T, Hisanaga S, Goedert M, Hasegawa M. 2006. Small molecule inhibitors of α-synuclein filament assembly. Biochemistry. 45(19):6085–6094.
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DMA, Hasegawa M. 2013. Prion-like spreading of pathological α-synuclein in brain. Brain. 136(Pt 4):1128–1138.
- Mazzulli JR, Armakola M, Dumoulin M, Parastatidis I, Ischiropoulos H. 2007. Cellular oligomerization of α-synuclein is determined by the interaction of oxidized catechols with a C-terminal sequence. J Biol Chem. 282(43):31621–31630.
- Mazzulli JR, Mishizen AJ, Giasson BI, Lynch DR, Thomas SA, Nakashima A, Nagatsu T, Ota A, Ischiropoulos H. 2006. Cytosolic catechols inhibit -synuclein aggregation and facilitate the formation of intracellular soluble oligomeric intermediates. J Neurosci. 26(39):10068–10078.
- Meade RM, Watt KJC, Williams RJ, Mason JM. 2021. A downsized and optimised intracellular library-derived peptide prevents alpha-synuclein primary nucleation and toxicity without impacting upon lipid binding. J Mol Biol. 433(24):167323.
- Mehanna A. 2001. Sickle cell anemia and antisickling agents then and now. Curr Med Chem. 8(2):79–88.
- Meng X, Munishkina LA, Fink AL, Uversky VN. 2009. Molecular mechanisms underlying the flavonoid-induced inhibition of α-synuclein fibrillation. Biochemistry. 48(34):8206–8224.
- Meng X, Munishkina LA, Fink AL, Uversky VN. 2010. Effects of various flavonoids on the α-synuclein fibrillation process. Parkinsons Dis. 2010:1–16.
- Metaferia B, Cellmer T, Dunkelberger EB, Li Q, Henry ER, Hofrichter J, Staton D, Hsieh MM, Conrey AK, Tisdale JF, et al. 2022. Phenotypic screening of the ReFRAME drug repurposing library to discover new drugs for treating sickle cell disease. Proc Natl Acad Sci U S A. 119(40):e2210779119.
- Mohammad-Beigi H, Aliakbari F, Sahin C, Lomax C, Tawfike A, Schafer NP, Amiri-Nowdijeh A, Eskandari H, Møller IM, Hosseini-Mazinani M, et al. 2019. Oleuropein derivatives from olive fruit extracts reduce α-synuclein fibrillation and oligomer toxicity. J Biol Chem. 294(11):4215–4232.
- Mohankumar T, Chandramohan V, Lalithamba HS, Jayaraj RL, Kumaradhas P, Sivanandam M, Hunday G, Vijayakumar R, Balakrishnan R, Manimaran D, et al. 2020. Design and molecular dynamic investigations of 7,8-dihydroxyflavone derivatives as potential neuroprotective agents against alpha-synuclein. Sci Rep. 10(1):599.
- Moody JO, Ojo OO, Omotade OO, Adeyemo AA, Olumese PE, Ogundipe OO. 2003. Anti-sickling potential of a Nigerian herbal formula (Ajawaron HF) and the major plant component (Cissus populnea L. CPK). Phytother Res. 17(10):1173–1176.
- Moon HE, Paek SH. 2015. Mitochondrial dysfunction in Parkinson’s disease. Exp Neurobiol. 24(2):103–116.
- Mpiana PT, Tshibangu DST, Shetonde OM, Ngbolua KN. 2007. In vitro antidrepanocytary activity (anti-sickle cell anemia) of some Congolese plants. Phytomedicine. 14(2–3):192–195.
- Nagel RL, Johnson J, Bookchin RM, Garel MC, Rosa J, Schiliro G, Wajcman H, Labie D, Moo-Penn W, Castro O. 1980. β-Chain contact sites in the haemoglobin S polymer. Nature. 283(5750):832–834.
- Nakagawa A, Lui FE, Wassaf D, Yefidoff-Freedman R, Casalena D, Palmer MA, Meadows J, Mozzarelli A, Ronda L, Abdulmalik O, et al. 2014. Identification of a small molecule that increases hemoglobin oxygen affinity and reduces SS erythrocyte sickling. ACS Chem Biol. 9(10):2318–2325.
- Neddenriep B, Calciano A, Conti D, Sauve E, Paterson M, Bruno EA, Moffet D. 2011. Short peptides as inhibitors of amyloid aggregation. Open Biotechnol J. 5(1):39–46.
- Ni X, McGlinchey RP, Jiang J, Lee JC. 2019. Structural insights into α-synuclein fibril polymorphism: effects of Parkinson’s disease-related C-terminal truncations. J Mol Biol. 431(19):3913–3919.
- Niihara Y, Zerez CR, Akiyama DS, Tanaka KR. 1998. Oral l-glutamine therapy for sickle cell anemia: I. Subjective clinical improvement and favorable change in red cell NAD redox potential. Am J Hematol. 58(2):117–121.
- Niihara Y, Zerez CR, Akiyama DS, Tanaka KR. 1997. Increased red cell glutamine availability in sickle cell anemia: demonstration of increased active transport, affinity, and increased glutamate level in intact red cells. J Lab Clin Med. 130(1):83–90.
- Noguchi CT, Schechter AN. 1977. Effects of amino acids on gelation kinetics and solubility of sickle hemoglobin. Biochem Biophys Res Commun. 74(2):637–642.
- Noguchi CT, Schechter AN. 1978. Inhibition of sickle hemoglobin gelation by amino acids and related compounds. Biochemistry. 17(25):5455–5459.
- Noguchi CT, Schechter AN. 1985. Sickle hemoglobin polymerization in solution and in cells. Annu Rev Biophys Biophys Chem. 14(1):239–263.
- Norris EH, Giasson BI, Hodara R, Xu S, Trojanowski JQ, Ischiropoulos H, Lee VM-Y. 2005. Reversible inhibition of α-synuclein fibrillization by dopaminochrome-mediated conformational alterations. J Biol Chem. 280(22):21212–21219.
- Norris EH, Giasson BI, Lee VM-Y. 2004. α-Synuclein: normal function and role in neurodegenerative diseases. Curr Top Dev Biol. 60:17–54.
- Nozaki Y, Tanford C. 1971. The solubility of amino acids and two glycine peptides in aqueous ethanol and dioxane solutions. J Biol Chem. 246(7):2211–2217.
- Oder E, Safo MK, Abdulmalik O, Kato GJ. 2016. New developments in anti-sickling agents: can drugs directly prevent the polymerization of sickle haemoglobin in vivo? Br J Haematol. 175(1):24–30.
- Oksenberg D, Dufu K, Patel MP, Chuang C, Li Z, Xu Q, Silva-Garcia A, Zhou C, Hutchaleelaha A, Patskovska L, et al. 2016. GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half-life in a murine model of sickle cell disease. Br J Haematol. 175(1):141–153.
- Olagunju MO, Loschwitz J, Olubiyi OO, Strodel B. 2022. Multiscale MD simulations of wild-type and sickle hemoglobin aggregation. Proteins. 90(11):1811–1824.
- Olubiyi OO, Olagunju MO, Oni JO, Olubiyi AO. 2018. Structural basis of antisickling effects of selected FDA approved drugs: a drug repurposing study. Curr Comput Aided Drug Des. 14(2):106–116.
- Olubiyi OO, Olagunju MO, Strodel B. 2019. Rational drug design of peptide-based therapies for sickle cell disease. Molecules. 24(24):4551.
- Ono K, Yamada M. 2006. Antioxidant compounds have potent anti-fibrillogenic and fibril-destabilizing effects for alpha-synuclein fibrils in vitro. J Neurochem. 97(1):105–115.
- Ortiz de Montellano PR. 2018. A new step in the treatment of sickle cell disease: published as part of the Biochemistry series “Biochemistry to Bedside”. Biochemistry. 57(5):470–471.
- Padlan EA, Love WA. 1985a. Refined crystal structure of deoxyhemoglobin S. II. Molecular interactions in the crystal. J Biol Chem. 260(14):8280–8291.
- Padlan EA, Love WE. 1985b. Refined crystal structure of deoxyhemoglobin S. I. Restrained least-squares refinement at 3.0-8 resolution. J Biol Chem. 260(14):8272–8279.
- Pagare PP, Rastegar A, Abdulmalik O, Omar AM, Zhang Y, Fleischman A, Safo MK. 2022. Modulating hemoglobin allostery for treatment of sickle cell disease: current progress and intellectual property. Expert Opin Ther Pat. 32(2):115–130.
- Palazzi L, Bruzzone E, Bisello G, Leri M, Stefani M, Bucciantini M, Polverino de Laureto P. 2018. Oleuropein aglycone stabilizes the monomeric α-synuclein and favours the growth of non-toxic aggregates. Sci Rep. 8(1):8337.
- Park S-Y, Yokoyama T, Shibayama N, Shiro Y, Tame JRH. 2006. 1.25 Å resolution crystal structures of human haemoglobin in the oxy, deoxy and carbonmonoxy forms. J Mol Biol. 360(3):690–701.
- Pauling LL, Itano HA, Singer SJ, Wells IC. 1949. Sickle cell anemia, a molecular disease. Science. 110(2865):543–548.
- Perni M, Flagmeier P, Limbocker R, Cascella R, Aprile FA, Galvagnion C, Heller GT, Meisl G, Chen SW, Kumita JR, et al. 2018. Multistep inhibition of α-synuclein aggregation and toxicity in vitro and in vivo by trodusquemine. ACS Chem Biol. 13(8):2308–2319.
- Perni M, Galvagnion C, Maltsev A, Meisl G, Müller MBD, Challa PK, Kirkegaard JB, Flagmeier P, Cohen SIA, Cascella R, et al. 2017. A natural product inhibits the initiation of α-synuclein aggregation and suppresses its toxicity. Proc Natl Acad Sci U S A. 114(6):E1009–E1017.
- Perutz MF, Liquori AM, Eirich F. 1951. X-ray and solubility studies of the haemoglobin of sickle-cell anaemia patients. Nature. 167(4258):929–931.
- Perutz MF, Rossmann MG, Cullis AF, Muirhead H, Will G, North ACT. 1960. Structure of haemoglobin: a three-dimensional Fourier synthesis at 5.5-Å. Resolution, obtained by X-ray analysis. Nature. 185(4711):416–422.
- Perutz MF. 1970. Stereochemistry of cooperative effects in haemoglobin: haem–haem interaction and the problem of allostery. Nature. 228(5273):726–739.
- Piel FB, Steinberg MH, Rees DC. 2017. Sickle cell disease. N Engl J Med. 376(16):1561–1573.
- Poillon W, Kim B. 1990. 2,3-Diphosphoglycerate and intracellular pH as interdependent determinants of the physiologic solubility of deoxyhemoglobin S. Blood. 76(5):1028–1036.
- Poillon WN, Bertles JF. 1979. Deoxygenated sickle hemoglobin. Effects of lyotropic salts on its solubility. J Biol Chem. 254(9):3462–3467.
- Poillon WN, Kim BC, Labotka RJ, Hicks CU, Kark JA. 1995. Antisickling effects of 2,3-diphosphoglycerate depletion. Blood. 85(11):3289–3296.
- Poillon WN, Kim BC, Welty EV, Walder JA. 1986. The effect of 2,3-diphosphoglycerate on the solubility of deoxyhemoglobin S. Arch Biochem Biophys. 249(2):301–305.
- Poillon WN. 1982. Noncovalent inhibitors of sickle hemoglobin gelation: effects of aryl-substituted alanines. Biochemistry. 21(6):1400–1406.
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. 1997. Mutation in the -synuclein gene identified in families with Parkinson’s disease. Science. 276(5321):2045–2047.
- Popova B, Wang D, Rajavel A, Dhamotharan K, Lázaro DF, Gerke J, Uhrig JF, Hoppert M, Outeiro TF, Braus GH. 2021. Identification of two novel peptides that inhibit α-synuclein toxicity and aggregation. Front Mol Neurosci. 14:659926.
- Pratt LR, Chandler D. 1977. Theory of the hydrophobic effect. J Chem Phys. 67(8):3683–3704.
- Pujols J, Peña-Díaz S, Lázaro DF, Peccati F, Pinheiro F, González D, Carija A, Navarro S, Conde-Giménez M, García J, et al. 2018. Small molecule inhibits α-synuclein aggregation, disrupts amyloid fibrils, and prevents degeneration of dopaminergic neurons. Proc Natl Acad Sci U S A. 115(41):10481–10486.
- Quezado ZMN, Kamimura S, Smith M, Wang X, Heaven MR, Jana S, Vogel S, Zerfas P, Combs CA, Almeida LEF, et al. 2022. Mitapivat increases ATP and decreases oxidative stress and erythrocyte mitochondria retention in a SCD mouse model. Blood Cells Mol Dis. 95:102660.
- Rao JN, Dua V, Ulmer TS. 2008. Characterization of α-synuclein interactions with selected aggregation-inhibiting small molecules. Biochemistry. 47(16):4651–4656.
- Rao JN, Jao CC, Hegde BG, Langen R, Ulmer TS. 2010. A combinatorial NMR and EPR approach for evaluating the structural ensemble of partially folded proteins. J Am Chem Soc. 132(25):8657–8668.
- Rezaeian N, Shirvanizadeh N, Mohammadi S, Nikkhah M, Arab SS. 2017. The inhibitory effects of biomimetically designed peptides on α-synuclein aggregation. Arch Biochem Biophys. 634:96–106.
- Ribeil J-A, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, Caccavelli L, Neven B, Bourget P, El Nemer W, et al. 2017. Gene therapy in a patient with sickle cell disease. N Engl J Med. 376(9):848–855.
- Robertson AD, Murphy KP. 1997. Protein structure and the energetics of protein stability. Chem Rev. 97(5):1251–1268.
- Rochet J-C, Outeiro TF, Conway KA, Ding TT, Volles MJ, Lashuel HA, Bieganski RM, Lindquist SL, Lansbury PT. 2004. Interactions among α-synuclein, dopamine, and biomembranes: some clues for understanding neurodegeneration in Parkinson’s disease. J Mol Neurosci. 23(1–2):23–34.
- Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, Sangwan S, Guenther EL, Johnson LM, Zhang M, et al. 2015. Structure of the toxic core of α-synuclein from invisible crystals. Nature. 525(7570):486–490.
- Rolan P, Parker J, Gray S, Weatherley B, Ingram J, Leavens W, Wootton R, Posner J. 1993. The pharmacokinetics, tolerability and pharmacodynamics of tucaresol (589C80; 4[2-formyl-3-hydroxyphenoxymethyl] benzoic acid), a potential anti-sickling agent, following oral administration to healthy subjects. Br J Clin Pharmacol. 35(4):419–425.
- Rosenblad C, Kirik D, Devaux B, Moffat B, Phillips HS, Björklund A. 1999. Protection and regeneration of nigral dopaminergic neurons by neurturin or GDNF in a partial lesion model of Parkinson’s disease after administration into the striatum or the lateral ventricle: NTN and GDNF protect nigrostriatal DA neurons. Eur J Neurosci. 11(5):1554–1566.
- Ross PD, Hofrichter J, Eaton WA. 1977. Thermodynamics of gelation of sickle cell deoxyhemoglobin. J Mol Biol. 115(2):111–134.
- Ross PD, Subramanian S. 1977. Inhibition of sickle cell hemoglobin gelation by some aromatic compounds. Biochem Biophys Res Commun. 77(4):1217–1223.
- Ruggeri FS, Longo G, Faggiano S, Lipiec E, Pastore A, Dietler G. 2015. Infrared nanospectroscopy characterization of oligomeric and fibrillar aggregates during amyloid formation. Nat Commun. 6(1):7831.
- Rumen N. 1975. Inhibition of sickling in erythrocytes by amino acids. Blood. 45(1):45–48.
- Russu IM, Lin AKLC, Yang CP, Ho C. 1986. Molecular basis for the antisickling activity of aromatic amino acids and related compounds: a proton nuclear magnetic resonance investigation. Biochemistry. 25(4):808–815.
- Saffari B, Amininasab M. 2021. Crocin inhibits the fibrillation of human α-synuclein and disassembles mature fibrils: experimental findings and mechanistic insights from molecular dynamics simulation. ACS Chem Neurosci. 12(21):4037–4057.
- Salinas Cisneros G, Thein SL. 2020. Recent advances in the treatment of sickle cell disease. Front Physiol. 11:435.
- Samuel RE, Salmon ED, Briehl RW. 1990. Nucleation and growth of fibres and gel formation in sickle cell haemoglobin. Nature. 345(6278):833–835.
- Santos J, Gracia P, Navarro S, Peña-Díaz S, Pujols J, Cremades N, Pallarès I, Ventura S. 2021. α-Helical peptidic scaffolds to target α-synuclein toxic species with nanomolar affinity. Nat Commun. 12(1):3752.
- Schechter AN, Noguchi CT, Schwartz WA. 1978. Amino acids and peptides as inhibitors of sickle hemoglobin gelation. In: Winslow Caughey, editor. Biochemical and clinical aspects of hemoglobin abnormalities. New York: Elsevier; p. 129–141.
- Schechter AN. 1980. Stereospecific inhibitors of the gelation of sickle hemoglobin. Hemoglobin. 4(3–4):335–345.
- Sciarretta KL, Gordon DJ, Meredith SC. 2006. Peptide-based inhibitors of amyloid assembly. In: Indu Kheterpal and Ronald Wetzel, editors. Methods in enzymology. Vol. 413. Amsterdam: Elsevier; p. 273–312.
- Sedlmeier F, Horinek D, Netz RR. 2011. Entropy and enthalpy convergence of hydrophobic solvation beyond the hard-sphere limit. J Chem Phys. 134(5):055105.
- Serpell LC, Berriman J, Jakes R, Goedert M, Crowther RA. 2000. Fiber diffraction of synthetic alpha-synuclein filaments shows amyloid-like cross-beta conformation. Proc Natl Acad Sci U S A. 97(9):4897–4902.
- Singh H, Sharma S. 2022. Hydration of linear alkanes is governed by the small length-scale hydrophobic effect. J Chem Theory Comput. 18(6):3805–3813.
- Singh PK, Kotia V, Ghosh D, Mohite GM, Kumar A, Maji SK. 2013. Curcumin modulates α-synuclein aggregation and toxicity. ACS Chem Neurosci. 4(3):393–407.
- Soto C. 2003. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. 4(1):49–60.
- Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M. 1998. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett. 251(3):205–208.
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. 1998. Alpha-synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A. 95(11):6469–6473.
- Spillantini MG, Schmidt ML, Lee VM-Y, Trojanowski JQ, Jakes R, Goedert M. 1997. α-Synuclein in Lewy bodies. Nature. 388(6645):839–840.
- Stefani M, Rigacci S. 2013. Protein folding and aggregation into amyloid: the interference by natural phenolic compounds. Int J Mol Sci. 14(6):12411–12457.
- Stephens AD, Zacharopoulou M, Moons R, Fusco G, Seetaloo N, Chiki A, Woodhams PJ, Mela I, Lashuel HA, Phillips JJ, et al. 2020. Extent of N-terminus exposure of monomeric alpha-synuclein determines its aggregation propensity. Nat Commun. 11(1):2820.
- Stillinger FH. 1973. Structure in aqueous solutions of nonpolar solutes from the standpoint of scaled-particle theory. J Solution Chem. 2(2–3):141–158.
- Stoker TB, Barker RA. 2020. Recent developments in the treatment of Parkinson’s disease. F1000Res. 9:862.
- Strader MB, Liang H, Meng F, Harper J, Ostrowski DA, Henry ER, Shet AS, Eaton WA, Thein SL, Alayash AI. 2019. Interactions of an anti-sickling drug with hemoglobin in red blood cells from a patient with sickle cell anemia. Bioconjug Chem. 30(3):568–571.
- Sweeney P, Park H, Baumann M, Dunlop J, Frydman J, Kopito R, McCampbell A, Leblanc G, Venkateswaran A, Nurmi A, et al. 2017. Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener. 6(1):6.
- Swift R, Abdulmalik O, Chen Q, Asakura T, Gustafson K, Simon JE, Zaman V, Quiusky KA, Hassell KL, Shapira I, et al. 2016. SCD-101: a new anti-sickling drug reduces pain and fatigue and improves red blood cell shape in peripheral blood of patients with sickle cell disease. Blood. 128(22):121.
- Syed MM, Doshi PJ, Kulkarni MV, Dhavale DD, Kadam NS, Kate SL, Doshi JB, Sharma N, Uppuladinne M, Sonavane U, et al. 2019. Alizarin interaction with sickle hemoglobin: elucidation of their anti-sickling properties by multi-spectroscopic and molecular modeling techniques. J Biomol Struct Dyn. 37(17):4614–4631.
- Tamoliunas K, Galamba N. 2020. Protein denaturation, zero entropy temperature, and the structure of water around hydrophobic and amphiphilic solutes. J Phys Chem B. 124(48):10994–11006.
- Tanford C. 1980. The hydrophobic effect: formation of micelles and biological membranes. 2nd ed. New York: Wiley.
- Tatenhorst L, Eckermann K, Dambeck V, Fonseca-Ornelas L, Walle H, Lopes da Fonseca T, Koch JC, Becker S, Tönges L, Bähr M, et al. 2016. Fasudil attenuates aggregation of α-synuclein in models of Parkinson’s disease. Acta Neuropathol Commun. 4(1):39.
- Telen MJ, Malik P, Vercellotti GM. 2019. Therapeutic strategies for sickle cell disease: towards a multi-agent approach. Nat Rev Drug Discov. 18(2):139–158.
- Tinku, Paithankar H, Rane AR, Hosur RV, Choudhary S. 2021. Mechanistic insights into chalcone butein-induced inhibition of α-synuclein fibrillation: biophysical and in silico studies. J Mol Liq. 334:116105.
- Tisdale JF, Thein SL, Eaton WA. 2020. Treating sickle cell anemia. Science. 367(6483):1198–1199.
- Torpey JH, Meade RM, Mistry R, Mason JM, Madine J. 2020. Insights into peptide inhibition of alpha-synuclein aggregation. Front Neurosci. 14:561462.
- Tóth G, Gardai SJ, Zago W, Bertoncini CW, Cremades N, Roy SL, Tambe MA, Rochet J-C, Galvagnion C, Skibinski G, et al. 2014. Targeting the intrinsically disordered structural ensemble of α-synuclein by small molecules as a potential therapeutic strategy for Parkinson’s disease. PLOS One. 9(2):e87133.
- Tripathi T. 2020. A Master Regulator of α-synuclein aggregation. ACS Chem Neurosci. 11(10):1376–1378.
- Trojanowski JQ, Lee VM-Y. 2003. Parkinson’s disease and related α-synucleinopathies are brain amyloidoses. Ann N Y Acad Sci. 991(1):107–110.
- Tu P, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, Iwatsubo T, Trojanowski JQ, Lee VM-Y. 1998. Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol. 44(3):415–422.
- Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney J, Kim M, Barclay JK, Kendall AM, et al. 2016. Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat Struct Mol Biol. 23(5):409–415.
- Uéda K, Fukushima H, Masliah E, Xia Y, Iwai A, Yoshimoto M, Otero DA, Kondo J, Ihara Y, Saitoh T. 1993. Molecular cloning of CDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 90(23):11282–11286.
- Ulamec SM, Maya-Martinez R, Byrd EJ, Dewison KM, Xu Y, Willis LF, Sobott F, Heath GR, van Oosten Hawle P, Buchman VL, et al. 2022. Single residue modulators of amyloid formation in the N-terminal P1-region of α-synuclein. Nat Commun. 13(1):4986.
- Ulmer TS, Bax A, Cole NB, Nussbaum RL. 2005. Structure and dynamics of micelle-bound human α-synuclein. J Biol Chem. 280(10):9595–9603.
- Uversky V, Eliezer D. 2009. Biophysics of Parkinsons disease: structure and aggregation of α-synuclein. Curr Protein Pept Sci. 10(5):483–499.
- Uversky VN, Li J, Fink AL. 2001. Evidence for a partially folded intermediate in α-synuclein fibril formation. J Biol Chem. 276(14):10737–10744.
- Uversky VN. 2007. Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J Neurochem. 103:17–37.
- Valiente-Gabioud AA, Miotto MC, Chesta ME, Lombardo V, Binolfi A, Fernández CO. 2016. Phthalocyanines as molecular scaffolds to block disease-associated protein aggregation. Acc Chem Res. 49(5):801–808.
- Vamathevan J, Clark D, Czodrowski P, Dunham I, Ferran E, Lee G, Li B, Madabhushi A, Shah P, Spitzer M, et al. 2019. Applications of machine learning in drug discovery and development. Nat Rev Drug Discov. 18(6):463–477.
- van Dijk MJ, Rab MAE, van Oirschot BA, Bos J, Derichs C, Rijneveld AW, Cnossen MH, Nur E, Biemond BJ, Bartels M, et al. 2022. Safety and efficacy of mitapivat, an oral pyruvate kinase activator, in sickle cell disease: a phase 2, open-label study. Am J Hematol. 97(7):E226–E229.
- Vichinsky E, Hoppe CC, Ataga KI, Ware RE, Nduba V, El-Beshlawy A, Hassab H, Achebe MM, Alkindi S, Brown RC, et al. 2019. A phase 3 randomized trial of voxelotor in sickle cell disease. N Engl J Med. 381(6):509–519.
- Vinjamur DS, Bauer DE, Orkin SH. 2018. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br J Haematol. 180(5):630–643.
- Vittorio S, Adornato I, Gitto R, Peña-Díaz S, Ventura S, De Luca L. 2020. Rational design of small molecules able to inhibit α-synuclein amyloid aggregation for the treatment of Parkinson’s disease. J Enzyme Inhib Med Chem. 35(1):1727–1735.
- Votano JR, Altman J, Wilchek M, Gorecki M, Rich A. 1984. Potential use of biaromatic l-phenylalanyl derivatives as therapeutic agents in the treatment of sickle cell disease. Proc Natl Acad Sci U S A. 81(10):3190–3194.
- Votano JR, Gorecki M, Rich A. 1977. Sickle hemoglobin aggregation: a new class of inhibitors. Science. 196(4295):1216–1219.
- Votano JR, Rich A. 1985. Inhibition of deoxyhemoglobin s polymerization by biaromatic peptides found to associate with the hemoglobin molecule at a preferred site. Biochemistry. 24(8):1966–1970.
- Wang Y, Ferrone FA. 2013. Dissecting the energies that stabilize sickle hemoglobin polymers. Biophys J. 105(9):2149–2156.
- Wang Z, Kishchenko G, Chen Y, Josephs R. 2000. Polymerization of deoxy-sickle cell hemoglobin in high-phosphate buffer. J Struct Biol. 131(3):197–209.
- Widom B. 1982. Potential-distribution theory and the statistical mechanics of fluids. J Phys Chem. 86(6):869–872.
- Williams TN. 2016. Sickle cell disease in sub-Saharan Africa. Hematol Oncol Clin North Am. 30(2):343–358.
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, et al. 2011. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A. 108(10):4194–4199.
- Wishner BC, Ward KB, Lattman EE, Love WE. 1975. Crystal structure of sickle-cell deoxyhemoglobin at 5 Å resolution. J Mol Biol. 98(1):179–194.
- Wrasidlo W, Tsigelny IF, Price DL, Dutta G, Rockenstein E, Schwarz TC, Ledolter K, Bonhaus D, Paulino A, Eleuteri S, et al. 2016. A de novo compound targeting α-synuclein improves deficits in models of Parkinson’s disease. Brain. 139(Pt 12):3217–3236.
- Xavier P, Galamba N. 2021. Effect of urea on the hydration and aggregation of hydrophobic and amphiphilic solute models: implications to protein aggregation. J Chem Phys. 155(14):144501.
- Xu GG, Pagare PP, Ghatge MS, Safo RP, Gazi A, Chen Q, David T, Alabbas AB, Musayev FN, Venitz J, et al. 2017. Design, synthesis, and biological evaluation of ester and ether derivatives of antisickling agent 5-HMF for the treatment of sickle cell disease. Mol Pharm. 14(10):3499–3511.
- Xu JZ, Conrey A, Frey I, Gwaabe E, Menapace LA, Tumburu L, Lundt M, Lequang T, Li Q, Glass K, et al. 2022. A phase 1 dose escalation study of the pyruvate kinase activator mitapivat (AG-348) in sickle cell disease. Blood. 140(19):2053–2062.
- Yang Z, Yao Y, Zhou Y, Li X, Tang Y, Wei G. 2023. EGCG attenuates α-synuclein protofibril-membrane interactions and disrupts the protofibril. Int J Biol Macromol. 230:123194.
- Young LM, Ashcroft AE, Radford SE. 2017. Small molecule probes of protein aggregation. Curr Opin Chem Biol. 39:90–99.
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, et al. 2004. The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia: new α-synuclein gene mutation. Ann Neurol. 55(2):164–173.
- Zhang L, Yu X, Ji M, Liu S, Wu X, Wang Y, Liu R. 2018. Resveratrol alleviates motor and cognitive deficits and neuropathology in the A53T α-synuclein mouse model of Parkinson’s disease. Food Funct. 9(12):6414–6426.
- Zhou L, Kurouski D. 2020. Structural characterization of individual α-synuclein oligomers formed at different stages of protein aggregation by atomic force microscopy-infrared spectroscopy. Anal Chem. 92(10):6806–6810.
- Zhu M, Rajamani S, Kaylor J, Han S, Zhou F, Fink AL. 2004. The flavonoid baicalein inhibits fibrillation of α-synuclein and disaggregates existing fibrils. J Biol Chem. 279(26):26846–26857.
- Zurlo E, Kumar P, Meisl G, Dear AJ, Mondal D, Claessens MMAE, Knowles TPJ, Huber M. 2021. In situ kinetic measurements of α-synuclein aggregation reveal large population of short-lived oligomers. PLOS One. 16(1):e0245548.