Abstract
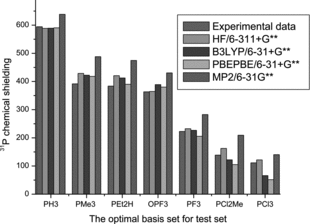
The performance of the ab initio and density functional theory (DFT) functional, B3LYP and PBEPBE, in conjunction with selected basis sets for the prediction of 31P shielding constants for small phosphorous-containing molecules is assessed. The effects of applied factors are discussed. By including the electron correlation treatment, B3LYP and PBEPBE are the most accurate methods to compute the chemical shielding for a set of small phosphines that was studied. Also, additional improvements of DFT were obtained by empirical scaling of basis sets in calculation of chemical shifts. For molecules containing only phosphorous and carbon atoms with sp3 hybridization, the PBEPBE/6-311G(d,p) level of theory has been shown to provide reliable 31P chemical shifts.
Supplemental materials are available for this article. Go to the publisher's online edition of Phosphorus, Sulfur, and Silicon and the Related Elements to view the free supplemental file.
GRAPHICAL ABSTRACT