
Abstract
Mutations in NPM1 can be used for minimal residual disease (MRD) analysis in acute myeloid leukemia (AML). We here applied a newly introduced method, deep sequencing, allowing for simultaneous analysis of all recurrent NPM1 insertions and thus constituting an attractive alternative to multiple PCRs for the clinical laboratory. We retrospectively used deep sequencing for measurement of MRD pre- and post-allogeneic hematopoietic stem cell transplantation (alloHCT). For 29 patients in morphological remission at the time of alloHCT, the effect of deep sequencing MRD on outcome was assessed. MRD positivity was defined as variant allele frequency ≥0.02%. Post-transplant MRD status was significantly and independently associated with clinical outcome; 3-year relapse-free survival 20% vs 85% (p < .001), HR 45 (95% CI 2–1260), and overall survival 20% vs 89% (p < .001), HR 49 (95% CI 2–1253). Thus, the new methodology deep sequencing is an applicable and predictive tool for MRD assessment in AML.
Introduction
Allogeneic hematopoietic stem cell transplantation (alloHCT) is the preferred treatment for patients with acute myeloid leukemia (AML) with high or intermediate risk of relapse. However, 30–40% of allografted patients eventually relapse, and after relapse survival is poor [Citation1,Citation2]. Several factors impact the risk of relapse including age, karyotype, presence of FLT3-ITD, refractory disease, and intensity of the conditioning regimen [Citation3–5]. Small amounts of remaining leukemic cells (minimal residual disease; MRD) pre- and post-transplant also herald an increased relapse risk after alloHCT [Citation6–10]. Post-transplant analyses of MRD may aid in treatment decisions including preemptive intervention with donor lymphocyte infusion (DLI), withdrawal of immunosuppressive drugs or addition of hypomethylating agents [Citation2,Citation11,Citation12]. Thus, assessment of MRD is becoming an increasingly important tool for treatment decisions in AML.
MRD assessed with multiparameter flow cytometry (MFC) pre- and post-transplant independently predict relapse [Citation6,Citation7,Citation13]. However, identification and quantification of MRD by MFC are afflicted by limitations including analytical difficulties such as heterogeneous leukemic cell immunophenotypes, and shifts in immunophenotype over time [Citation14]. Also, often only a subpopulation of the leukemic clone expresses the leukemia associated immunophenotype (LAIP), which renders the majority of leukemic cells unidentified and hampers the usefulness of MFC for monitoring purposes [Citation14]. Further, the routine usage of MFC MRD in clinical routine is hindered by technical drawbacks and requires standardization and highly skilled operators.
An alternative way to analyze MRD is to quantify the presence of leukemia specific genetic aberrations. In adult AML, the most common genetic aberration is a heterozygous mutation in exon 12 of the NPM1 gene, seen in ∼30% of all AML cases and in 50–60% of adult AML with normal karyotype (CN-AML) [Citation15]. The reported stability of mutations in NPM1 makes these ideal markers for MRD, as shown previously [Citation16–19]. Several studies have established the role of transcript-based analysis using reverse transcription quantitative polymerase chain reaction (RT-qPCR) for MRD analysis of NPM1 mutation load [Citation8,Citation9,Citation20]. A complicating factor in RT-qPCR for MRD analysis is the heterogeneity of described frameshift mutations in exon 12 of the NPM1 gene [Citation21–29]. To enable MRD analysis in all AML patients with NPM1 mutation, the clinical laboratory therefore needs to validate numerous specific RT-qPCR assays. We have recently shown the potential of targeted deep sequencing of NPM1 for MRD detection [Citation30]. The deep sequencing assay for NPM1 covers all insertions described to be recurrent (≥2) in exon 12 of the NPM1 gene according to the COSMIC database. The basis of the assay is to sequence the area of the gene containing the mutations without the need for specific primer and probe design for individual mutations.
Before introduction of any new method into clinical practice, it needs to be validated for its clinical value. The aim of this study was therefore to retrospectively determine the prognostic impact of pre- and post-transplant MRD as assessed by targeted deep sequencing, with relapse-free and overall survival as primary endpoints. As secondary endpoints, the prognostic impact of post-transplant chimerism on relapse-free and overall survival was determined.
Materials and methods
Patients and samples
This retrospective cohort study involving two centers 32 adult patients with AML (23 female and 9 male) with a mutation in exon 12 of the NPM1 gene (assessed in diagnostic samples by fragment analysis [Citation31]). All patients were allografted between 2005 and 2015 at Sahlgrenska University Hospital, Gothenburg, or Skåne University Hospital, Lund, Sweden. Patients in morphological remission before alloHCT were included in analyses of prognostic impact of MRD (n = 29). Of these, 9 patients had incomplete recovery before alloHCT (CRi) [Citation32]. Three patients not in morphological remission before alloHCT were described but excluded from analyses of prognostic impact of MRD. Clinical data, including results from MFC MRD (with 4–8 colors) performed in clinical routine, was collected from patient charts. The study was performed according to the Declaration of Helsinki. All patients had consented to biobanking of samples for research purposes. The study was approved by the Regional Ethical Review Board, Gothenburg.
DNA sequencing: library preparation, sequencing and data processing
Bone marrow aspirate slides were collected at diagnosis, at pre-transplant work-up, and post-transplant. For patients in morphological remission (CR/CRi) at the time of transplant, the following samples were available: at diagnosis (n = 25), at the pre-transplant work-up median 18 (8–92) days before alloHCT (n = 25) and post-transplant median 92 (68–115) days after alloHCT (n = 27). For 23 of these patients, samples were available at both time points. In addition, six peripheral blood smears (three pre-transplant and three post-transplant samples) taken at the same time as the bone marrow samples, were used for comparison with bone marrow. DNA was prepared from bone marrow aspirate/peripheral blood slides using QIAamp DNA Micro Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. The median DNA amount extracted from the bone marrow aspirate slides was 1660 ng (range 80—30,700 ng) and from blood smears 210 ng (range 120–240 ng). When possible, 100 ng DNA was used for the pre-sequencing amplification step. For targeted deep sequencing of exon 12 of the NPM1 gene, library preparation and sequencing were performed as previously described [Citation30]. The assay covers all recurrent (≥2) insertion mutations in exon 12 of the NPM1 gene reported in the COSMIC database (Supplementary Table S1). Primers were designed for the Illumina Truseq-library preparation system with unique eight base pair (bp) indexes in both the forward and reverse direction (Trugrade processing, Integrated DNA Technologies, Coralville, IA). The sequences for the specific primers targeting the NPM1 gene were intron 11 forward 5'-TGTCTATGAAGTGTTGTGGTTCC-3' and exon 12 reverse 5'-TCTGGCTGTCCTTTTTATAATGC-3' (Supplementary Figure S1). Paired-end sequencing (2 × 150 bp) was performed on a MiSeq (Illumina, San Diego, CA), with six samples run in multiplex together with a positive control (DNA from sorted leukemic cells with a NPM1 type A mutation diluted with normal cells) and a negative control (normal DNA). The Illumina PhiX bacteriophage genome was added to the pools to increase diversity. In the demultiplexing procedure no mismatches were allowed. Reads were first quality filtered and trimmed using the FASTX toolkit (http://hannonlab.cshl.edu/fastx_toolkit/). They were then merged using the Paired-End Read Merger (PEAR) [Citation33]. An in-house script was finally used to distinguish between wild-type and mutant NPM1 as previously described [Citation30]. MRD positivity was defined as NPM1mut variant allele frequency (VAF) > 0.02%, based on previously shown linearity of the assay [Citation30]. For Sanger sequencing for typing of the NPM1 mutation in diagnostic samples, the primers were as follows: intron 11 forward 5'-GATGTTGAACTATGCAAAGAGACA-3' and exon 12 reverse 5'- TGTGTTGTCCAAAATGCCTG -3' and results were analyzed in Lasergene SeqMan Pro (DNASTAR, Madison, WI) [Citation34].
Quantitative polymerase chain reaction of NPM1
Quantification of NPM1 type A mutation was performed using qPCR as previously described [Citation30,Citation35]. The limit of quantification (LOQ) in the assay was 0.039% (NPM1 type A mutation positive DNA/total concentration of DNA (albumin)).
Chimerism analysis
Chimerism analyses were performed as part of clinical routine on bone marrow samples taken at the same time point post-transplant as aspirates used for deep sequencing. Analyses were performed using polymerase chain reaction (PCR) of short tandem repeat (STR) sequences (n = 18) or FISH analysis (with Vysis X & Y Enumeration Probe, n = 3) on separated T cells (CD3+) and remaining cells (hereafter referred to as “non-T cells”). Complete donor chimerism was defined as ≥95/95% (T cells/non-T cells). Chimerism level of <95/95% (T cells/non-T cells) were considered mixed chimerism.
Statistical analyses
Comparisons between groups were performed with Mann-Whitney U test for continuous variables and Fisher’s exact test for categorical variables. Analyses of correlations between variables were performed with Spearman’s Rank correlation. Overall survival (OS) and relapse-free survival (RFS) were estimated with the Kaplan–Meier method and survival differences were assessed with the log-rank test. RFS was defined as the time from alloHCT to relapse or last follow-up. OS was defined as time between alloHCT and death or last follow up. Estimates of RFS and OS are given as percentage ± standard error. For the assessment of independent effects of continuous and categorical variables on OS and RFS, Cox regression analysis was used. Statistical analyses were performed using SPSS Statistics 22.0 (SPSS, Chicago, IL). All tests were two-sided and p < .05 was considered statistically significant. Graphs were created using GraphPad Prism version 7 (GraphPad Software, San Diego, CA).
Results
Patient characteristics
The clinical characteristics of the 29 patients in morphological remission before alloHCT are shown in . Of the patients, 25 had de novo AML and 4 had a history of previous myeloid disease. Median follow-up time after alloHCT was 43 (5–137) months, and 21 patients were alive at study termination. Follow-up time of non-relapsed patients was median 64 (15.5–137) months. Eight patients relapsed at a median of 7.3 (3.5–32) months after transplantation, and 7 of these died within 0.5–4 months after relapse. One patient died in remission 24 months after alloHCT.
Table 1. Patient and disease characteristics.
Deep sequencing identifies MRD positivity in a subset of patients
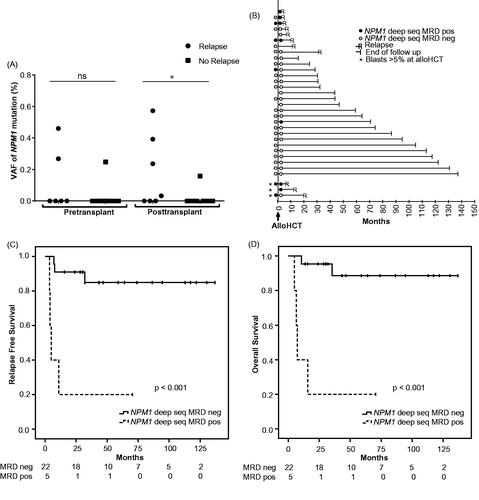
Levels of NPM1 MRD were assessed in bone marrow with targeted deep sequencing at ∼1 month before alloHCT (n = 25) and 3 months after alloHCT (n = 27). The targeted deep sequencing assay covered all recurrently occurring insertions in exon 12 of the NPM1 gene (Supplementary Table S1) and MRD was determined positive at VAF ≥0.02%. Three (12%) patients were MRD+ pre-transplant and five (19%) patients were MRD+ post-transplant. One patient converted from MRD+ pre-transplant to MRD− post-transplant and two patients converted from MRD− pre-transplant to MRD+ post-transplant. The mutation load of NPM1 ranged between 0 and 0.46% before alloHCT and 0–0.57% after alloHCT (). For confirmation with another method for NPM1 mutation detection, samples from patients with NPM1 type A mutation and available DNA were also analyzed with qPCR. Of these seven samples, six were determined positive also with this assay and one was below the LOQ (0.039%).
Figure 1. Outcome for AML patients with pre- and/or post-transplant deep sequencing MRD measurements. (A) Mutation load at 1-month pre-transplant (n = 25), and 3 months post-transplant (n = 27) in patients in morphological remission at the time of alloHCT; divided into patients with or without subsequent relapse. Negative MRD was set to 0. (B) Individual patient relapse-free survival in relation to pre- and/or post-transplant deep sequencing MRD status (n = 32). For patients in morphological remission with NPM1 MRD positivity and negativity 3 months after alloHCT, Kaplan–Meier curves for RFS are shown in (C) and OS in (D). Cutoff for MRD positivity with deep sequencing was VAF 0.02%, as described in Methods. *p < .05.
To test the applicability of the deep sequencing method in blood, we analyzed peripheral blood smears from MRD+ patients where such were available. In total six corresponding blood samples (from four patients) were analyzed (one before alloHCT only, one after alloHCT only, and two at both time points). Of the three pre-transplant blood samples where bone marrow samples were MRD+, one was MRD+, one showed NPM1 mutation VAF below the linearity of the assay (VAF 0.014%) and one had no detectable NPM1 mutation. The three patients that were MRD+ in bone marrow post-transplant showed MRD+ also in the blood samples and subsequently all relapsed.
We also assessed NPM1 mutation load in bone marrow samples from three patients with de novo AML who were not in morphological remission pre-transplant and all subsequently relapsed. Deep sequencing confirmed presence of NPM1 mutation in available pre-transplant samples and the only refractory patient who became deep sequencing MRD− after alloHCT relapsed late, after 21 months. Deep sequencing MRD status in bone marrow and relapse status for all patients are depicted in .
Pre-transplant deep sequencing MRD positivity is associated with worse outcome
Three patients in morphological remission at alloHCT were MRD+ pre-transplant as assessed with deep sequencing. Of these, 2 relapsed after alloHCT, whereas of 22 MRD− patients, 4 relapsed (p = .13; ). The resulting 2-year RFS and OS (±SE) was 33.3% ± 27.2% for MRD+ patients and 86.4% ±7.3% for MRD− patients (p = .019 and p = .031; Supplementary Figure 2(A,B). The result was confirmed when the analysis was performed for only patients analyzed within 30 days before alloHCT (n = 19, p = .04). Of note, the only pre-transplant MRD+ patient that did not relapse (follow-up time 28.3 months) was MRD− at 3 months post-transplant.
There were no significant differences between the pre-transplant deep sequencing MRD+ and MRD− groups regarding age, sex, FLT3-ITD status, cytogenetic risk category at diagnosis, conditioning intensity, T cell depletion, or disease status at transplantation (transplantation in first or second remission or whether in CR or CRi) (). Analysis of MRD using MFC as part of the work-up before transplant had been performed in 8/25 patients. Five patients were MRD− with both deep sequencing and MFC before alloHCT and of these, one relapsed. One patient was MRD+ with both methods and relapsed. One patient was MRD+ with deep sequencing and MRD− with MFC and relapsed. The last patient was MRD− with deep sequencing and MRD+ with MFC and remained in remission.
Table 2. Patient characteristics by MRD group before and after alloHCT, only patients in CR included.
Post-transplant deep sequencing MRD independently predicts outcome
Post-transplant, 5 patients were MRD+ as assessed with deep sequencing and of these 4 relapsed, whereas of 22 MRD− patients 3 relapsed (p = .009). For relapsing patients, median times from alloHCT to relapse were 4.5 (3.5–11) months for MRD+ patients and 7.7 (7–32) months for MRD− patients (p = .23). The MRD levels measured 3 months after alloHCT were significantly higher in patients who relapsed compared to those who did not (p = .036) (). The 3-year RFS was 20% ±17.9% for post-transplant MRD+ patients and 84.8% ±8.2% for MRD− patients (p < .001; ). The 3-year OS for MRD+ patients was 20% ±17.9% and for MRD− patients 88.6% ±7.8% (p < .001; ). Cox regression analysis including age, disease status at transplantation, conditioning intensity, cytogenetic risk stratification and FLT3-ITD status showed that MRD positivity 3 months post-transplant independently predicted both RFS and OS ().
Table 3. Multivariable Cox regression for RFS and OS after alloHCT.
There were no significant differences between the post-transplant deep sequencing MRD+ and MRD− groups regarding age, sex, FLT3-ITD status, cytogenetic risk category at diagnosis, conditioning intensity, T cell depletion, or disease status at transplantation (transplantation in first or second remission or whether in CR or CRi) (). Results from MFC MRD analyses performed as part of clinical routine ∼3 months post-transplant were available for 12/27 patients. Nine patients were MRD− with both deep sequencing and MFC, of which 2 relapsed. One patient was MRD+ with both methods and relapsed. Two patients were MRD+ with deep sequencing and MRD− with MFC and both relapsed. None of the investigated patients were MRD+ with MFC but MRD− with deep sequencing.
Bone marrow chimerism analysis did not predict relapse
Chimerism analyses of bone marrow were performed 3 months after alloHCT as part of clinical routine and data were available for 21 patients in morphological remission pre-transplant. Chimerism of T cells was complete donor in 16 patients and mixed in 5 patients. Non-T cell chimerism analyses were complete donor in all patients. Among the 5 patients with NPM1 deep sequencing MRD positivity and available chimerism data, only one displayed T cell mixed chimerism. As expected, levels of bone marrow recipient T and non-T cells correlated (rs = 0.52, p = .017). There was no correlation between chimerism levels and level of NPM1 deep sequencing MRD (rs = −0.16, p = .49 and rs = .038, p = .87 for T and non-T cells, respectively). Neither were there any significant associations between T cell chimerism status and RFS or OS. In patients with complete T cell chimerism, the 3-year RFS was 75% ± 10.8% and 3-year OS 74.5% ± 11% versus RFS 75% ± 21.7% and OS 75% ± 21.7% in patients with mixed T cell chimerism (p = .72 and p = .68, respectively).
Discussion
We here report that results from MRD analysis using targeted deep sequencing 3 months after alloHCT predict post-transplant relapse risk in adult AML with NPM1 mutation. The prognostic power of post-transplant NPM1 deep sequencing MRD positivity was independent of age, cytogenetic risk, FLT3-ITD status, conditioning intensity and disease status at transplantation. Our results indicate that the new methodology targeted deep sequencing can be a valuable tool for monitoring of patients with AML with mutated NPM1, and confirm the prognostic impact of MRD positivity before and after alloHCT.
Pre-transplant MRD− patients in CR had higher 2-year RFS and OS than MRD+ patients. Although the number of patients in our study was small, the results are in line with those reported by Kayser et al. who investigated the prognostic impact of pre-transplant MRD using RT-qPCR in 67 adult patients with AML with mutated NPM1. With this method, a ratio NPM1mut/ABL1 > 1% was shown to be an independent prognostic factor for poor outcome [Citation8]. Post-transplant NPM1 deep sequencing MRD status was a strong independent predictor for both relapse and overall survival. Thus, results using this new technology were in line with previous findings on the impact of post-transplant MRD status [Citation6,Citation8,Citation9,Citation13,Citation20]. Results that were available from MFC MRD analyses before and after alloHCT showed good concordance to deep sequencing of NPM1, but the results suggest that deep sequencing might be a more sensitive method. For MRD analysis, MFC has the advantage of being applicable to a majority of AML patients. However, it has a rather low sensitivity for relapse and can therefore not reliably identify true low-risk patients [Citation36,Citation37]. In some patients, MFC cannot even be applied due to major similarities between leukemic and normal myeloid cells. Also, analysis of MFC MRD is affected by immunophenotypic shifts often occurring during the progression to relapse, thus limiting its usefulness for monitoring [Citation38]. RT-qPCR is on the other hand a very sensitive method for quantification of mutated NPM1 transcript around the time of transplantation. Bacher et al. examined 13 patients with AML with mutated NPM1 with RT-qPCR for MRD following alloHCT, and demonstrated that relapse could be predicted earlier with RT-qPCR compared with morphology or chimerism analysis [Citation20]. Shayegi et al. reported that an increase in the ratio NPM1mut/ABL1 > 10% was prognostic for relapse after alloHCT [Citation9]. The drawback with RT-qPCR is that the assay needs to be mutation-specific, which in the accredited clinical laboratory setting makes it less feasible for cases with NPM1 mutations of less common types.
Monitoring of patients after alloHCT is today standard care, most often with chimerism analysis using STR-PCR. Several studies have shown association between increasing mixed chimerism and relapse [Citation39,Citation40]. Recent studies have explored digital PCR for chimerism analysis, reporting higher sensitivity than for STR-PCR [Citation41,Citation42]. The predictive value of lower levels of mixed chimerism remains to be determined. In this study, we analyzed bone marrow T cell and non-T cell chimerism from a single time point 3 months post-transplant and could not detect any association with outcome or correlation with MRD levels measured with deep sequencing. Thus, mixed chimerism as assessed at 3 months post-transplant is not necessarily associated with relapse. Our results hence support post-transplant monitoring based on MRD for relapse prediction as a complement to analysis of chimerism during engraftment.
The main advantage of deep sequencing is that it allows for simultaneous analysis of all recurrent insertions occurring in exon 12 of NPM1 in a single assay [Citation30,Citation43,Citation44]. With PCR of mutated NPM1, performed either with RT-qPCR, qPCR or droplet digital PCR, each NPM1 mutation requires a specific assay which should be validated and maintained in the clinical laboratory. Multiplex qPCR has been proposed as a solution for MRD detection of NPM1 mutations in the absence of sequence information. However, only a limited number of mutations can be analyzed in multiplex [Citation45]. The three most common NPM1 mutations, type A, type B and type D, constitute approximately 90% of NPM1 mutations in adult AML but there seems to be a larger diversity in childhood AML. Cazzaniga et al. investigated childhood AML for mutations in NPM1 and identified mutations in 7 of 107 cases. Only four of the mutations corresponded to type A, B or D [Citation46]. Further, in a recent abstract, data from the COG AML and SWOG young adult trial showed that non-type A NPM1 mutations are more frequent in childhood AML than in adult AML [Citation47]. The turnaround time for library preparation, multiplex targeted deep sequencing and bioinformatic analyses is 3–4 working days. In a clinical lab with sufficient throughput this would correspond to approximately 2 working weeks with a reagent cost per sample of 200$. Hence, the deep sequencing method constitutes a realistic option for MRD detection in clinical routine. The technique can also be used for patient specific mutations other than NPM1 [Citation30].
In conclusion, the results from this study suggest that deep sequencing is an applicable tool for MRD analysis and risk stratification in AML. Limitations of this study are its relatively small size, retrospective nature and limited comparisons with other methods. Therefore, the predictive value of deep sequencing needs to be validated in larger trials. Because the results despite these limitations confirm previous findings regarding residual disease after alloHCT, they emphasize the need for future interventional studies in MRD+ patients.
Potential conflict of interest
Disclosure forms provided by the authors are available with the full text of this article online at https://doi.org/10.1080/10428194.2018.1485910.
GLAL-2018-0177-File007.pdf
Download PDF (1.2 MB)GLAL-2018-0177-File006.docx
Download MS Word (121.3 KB)Acknowledgments
The authors thank the staff at the Department of Clinical Chemistry, Sahlgrenska University Hospital, Gothenburg, the Department of Hematology, Oncology and Radiation Physics Skåne University Hospital and the Department of Clinical Pathology, Lund, Laboratory Medicine Skåne and Louise Pettersson, Department of Pathology, Hallands Hospital Halmstad, Halmstad for assistance with routine diagnostics and/or biobanked samples. This study had financial support from Sahlgrenska University Hospital (grants ALFGBG-623831 and ALFGBG-720681), Sahlgrenska Academy at University of Gothenburg (salary for E.D.M.), Lion’s Cancer Foundation West (grant 2017:19), and Assar Gabrielsson’s Foundation (grant FB 15-23).
Related Research Data
References
- Devillier R, Crocchiolo R, Etienne A, et al. Outcome of relapse after allogeneic stem cell transplant in patients with acute myeloid leukemia. Leuk Lymphoma. 2013;54:1228–1234.
- Bejanyan N, Weisdorf DJ, Logan BR, et al. Survival of patients with acute myeloid leukemia relapsing after allogeneic hematopoietic cell transplantation: a center for international blood and marrow transplant research study. Biol Blood Marrow Transplant. 2015;21:454–459.
- Ogawa H, Ikegame K, Kawakami M, et al. Impact of cytogenetics on outcome of stem cell transplantation for acute myeloid leukemia in first remission: a large-scale retrospective analysis of data from the Japan society for hematopoietic cell transplantation. Int J Hematol. 2004;79:495–500.
- Schmid C, Labopin M, Socie G, et al. Outcome of patients with distinct molecular genotypes and cytogenetically normal AML after allogeneic transplantation. Blood. 2015;126:2062–2069.
- Walter RB, Sandmaier BM, Storer BE, et al. Number of courses of induction therapy independently predicts outcome after allogeneic transplantation for acute myeloid leukemia in first morphological remission. Biol Blood Marrow Transplant. 2015;21:373–378.
- Zhou Y, Othus M, Araki D, et al. Pre- and post-transplant quantification of measurable (“minimal”) residual disease via multiparameter flow cytometry in adult acute myeloid leukemia. Leukemia. 2016;30:1456–1464.
- Walter RB, Gooley TA, Wood BL, et al. Impact of pretransplantation minimal residual disease, as detected by multiparametric flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute myeloid leukemia. JCO. 2011;29:1190–1197.
- Kayser S, Benner A, Thiede C, et al. Pretransplant NPM1 MRD levels predict outcome after allogeneic hematopoietic stem cell transplantation in patients with acute myeloid leukemia. Blood Cancer J. 2016;6:e449.
- Shayegi N, Kramer M, Bornhauser M, et al. The level of residual disease based on mutant NPM1 is an independent prognostic factor for relapse and survival in AML. Blood. 2013;122:83–92.
- Wang Y, Wu DP, Liu QF, et al. In adults with t(8;21)AML, posttransplant RUNX1/RUNX1T1-based MRD monitoring, rather than c-KIT mutations, allows further risk stratification. Blood. 2014;124:1880–1886.
- Schmid C, Labopin M, Nagler A, et al. Treatment, risk factors, and outcome of adults with relapsed AML after reduced intensity conditioning for allogeneic stem cell transplantation. Blood. 2012;119:1599–1606.
- Sockel K, Wermke M, Radke J, et al. Minimal residual disease-directed preemptive treatment with azacitidine in patients with NPM1-mutant acute myeloid leukemia and molecular relapse. Haematologica. 2011;96:1568–1570.
- Bastos-Oreiro M, Perez-Corral A, Martinez-Laperche C, et al. Prognostic impact of minimal residual disease analysis by flow cytometry in patients with acute myeloid leukemia before and after allogeneic hemopoietic stem cell transplantation. Eur J Haematol. 2014;93:239–246.
- Kern W, Voskova D, Schoch C, et al. Determination of relapse risk based on assessment of minimal residual disease during complete remission by multiparameter flow cytometry in unselected patients with acute myeloid leukemia. Blood. 2004;104:3078–3085.
- Falini B, Nicoletti I, Martelli MF, et al. Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc + AML): biologic and clinical features. Blood. 2007;109:874–885.
- Kronke J, Bullinger L, Teleanu V, et al. Clonal evolution in relapsed NPM1-mutated acute myeloid leukemia. Blood. 2013;122:100–108.
- Kristensen T, Moller MB, Friis L, et al. NPM1 mutation is a stable marker for minimal residual disease monitoring in acute myeloid leukaemia patients with increased sensitivity compared to WT1 expression. Eur J Haematol. 2011;87:400–408.
- Kern W, Haferlach C, Haferlach T, et al. Monitoring of minimal residual disease in acute myeloid leukemia. Cancer. 2008;112:4–16.
- Jain P, Kantarjian H, Patel K, et al. Mutated NPM1 in patients with acute myeloid leukemia in remission and relapse. Leuk Lymphoma. 2014;55:1337–1344.
- Bacher U, Badbaran A, Fehse B, et al. Quantitative monitoring of NPM1 mutations provides a valid minimal residual disease parameter following allogeneic stem cell transplantation. Exp Hematol. 2009;37:135–142.
- Falini B, Mecucci C, Tiacci E, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266.
- Falini B, Martelli MP, Bolli N, et al. Immunohistochemistry predicts nucleophosmin (NPM) mutations in acute myeloid leukemia. Blood. 2006;108:1999–2005.
- Ivey A, Hills RK, Simpson MA, et al. Assessment of minimal residual disease in standard-risk AML. N Engl J Med. 2016;374:422–433.
- Thiede C, Koch S, Creutzig E, et al. Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood. 2006;107:4011–4020.
- Schnittger S, Schoch C, Kern W, et al. Nucleophosmin gene mutations are predictors of favorable prognosis in acute myelogenous leukemia with a normal karyotype. Blood. 2005;106:3733–3739.
- Verhaak RG, Goudswaard CS, van Putten W, et al. Mutations in nucleophosmin (NPM1) in acute myeloid leukemia (AML): association with other gene abnormalities and previously established gene expression signatures and their favorable prognostic significance. Blood. 2005;106:3747–3754.
- Dohner K, Schlenk RF, Habdank M, et al. Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood. 2005;106:3740–3746.
- Suzuki T, Kiyoi H, Ozeki K, et al. Clinical characteristics and prognostic implications of NPM1 mutations in acute myeloid leukemia. Blood. 2005;106:2854–2861.
- Jeon Y, Seo SW, Park S, et al. Identification of two novel NPM1 mutations in patients with acute myeloid leukemia. Ann Lab Med. 2013;33:60–64.
- Malmberg EB, Ståhlman S, Rehammar A, et al. Patient‐tailored analysis of minimal residual disease in acute myeloid leukemia using next‐generation sequencing. Eur J Haematol. 2017;98:26–37.
- Lin LI, Lin TC, Chou WC, et al. A novel fluorescence-based multiplex PCR assay for rapid simultaneous detection of CEBPA mutations and NPM mutations in patients with acute myeloid leukemias. Leukemia. 2006;20:1899–1903.
- Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–447.
- Zhang J, Kobert K, Flouri T, et al. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics. 2014;30:614–620.
- Fogelstrand L, Staffas A, Wasslavik C, et al. Prognostic implications of mutations in NOTCH1 and FBXW7 in childhood T-ALL treated according to the NOPHO ALL-1992 and ALL-2000 protocols. Pediatr Blood Cancer. 2014;61:424–430.
- Pettersson L, Leveen P, Axler O, et al. Improved minimal residual disease detection by targeted quantitative polymerase chain reaction in Nucleophosmin 1 type a mutated acute myeloid leukemia. Genes Chromosomes Cancer. 2016;55:750–766.
- Tierens A, Bjorklund E, Siitonen S, et al. Residual disease detected by flow cytometry is an independent predictor of survival in childhood acute myeloid leukaemia: results of the NOPHO-AML 2004 study. Br J Haematol. 2016;174:600–609.
- San Miguel JF, Martinez A, Macedo A, et al. Immunophenotyping investigation of minimal residual disease is a useful approach for predicting relapse in acute myeloid leukemia patients. Blood. 1997;90:2465–2470.
- Baer MR, Stewart CC, Dodge RK, et al. High frequency of immunophenotype changes in acute myeloid leukemia at relapse: implications for residual disease detection (Cancer and Leukemia Group B Study 8361). Blood. 2001;97:3574–3580.
- Tang X, Alatrash G, Ning J, et al. Increasing chimerism after allogeneic stem cell transplantation is associated with longer survival time. Biol Blood Marrow Transplant. 2014;20:1139–1144.
- Bader P, Kreyenberg H, Hoelle W, et al. Increasing mixed chimerism defines a high-risk group of childhood acute myelogenous leukemia patients after allogeneic stem cell transplantation where pre-emptive immunotherapy may be effective. Bone Marrow Transplant. 2004;33:815–821.
- Stahl T, Bohme MU, Kroger N, et al. Digital PCR to assess hematopoietic chimerism after allogeneic stem cell transplantation. Exp Hematol. 2015;43:462–468.e461.
- Waterhouse M, Pfeifer D, Follo M, et al. Early mixed hematopoietic chimerism detection by digital droplet PCR in patients undergoing gender-mismatched hematopoietic stem cell transplantation. Clin Chem Lab Med. 2017;55:1115–1121.
- Thol F, Kolking B, Damm F, et al. Next-generation sequencing for minimal residual disease monitoring in acute myeloid leukemia patients with FLT3-ITD or NPM1 mutations. Genes Chromosomes Cancer. 2012;51:689–695.
- Salipante SJ, Fromm JR, Shendure J, et al. Detection of minimal residual disease in NPM1-mutated acute myeloid leukemia by next-generation sequencing. Mod Pathol. 2014;27:1438–1446.
- Mencia-Trinchant N, Hu Y, Alas MA, et al. Minimal residual disease monitoring of acute myeloid leukemia by massively multiplex digital PCR in patients with NPM1 mutations. J Mol Diagn. 2017;19:537–548.
- Cazzaniga G, Dell'Oro MG, Mecucci C, et al. Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood. 2005;106:1419–1422.
- Selim D, Alonzo TA, Othus M, et al. Genomic subtypes of nucleophosmin (NPM1) mutations are associated with clinical outcome in AML – a COG and SWOG intergroup collaboration. Blood. 2016;128:285.