
Abstract
The BCR-ABL1 fusion gene, which causes aberrant kinase activity and uncontrolled cell proliferation, is the hallmark of chronic myeloid leukemia (CML). The development of tyrosine kinase inhibitors (TKI) that target the BCR-ABL oncoprotein has led to dramatic improvement in CML management. However, some challenges remain to be addressed in the TKI era, including patient stratification and the selection of frontline TKIs and CML progression. Additionally, with the emerging goal of treatment-free remission (TFR) in CML management, biomarkers that predict the outcomes of stopping TKI remain to be identified. Notably, recent reports have revealed the power of genome screening in understanding the role of genome aberrations other than BCR-ABL1 in CML pathogenesis. These studies have discovered the presence of disease-phase specific mutations and linked certain mutations to inferior responses to TKI treatment and CML progression. A personalized approach that incorporates genetic data in tailoring treatment strategies has been successfully implemented in acute leukemia, and it represents a promising approach for the management of high-risk CML patients. In this article, we will review current knowledge about the mutational profile in different phases of CML as well as patterns of mutational dynamics in patients having different outcomes. We highlight the effects of somatic mutations involving certain genes (e.g. epigenetic modifiers) on the outcomes of TKI treatment. We also discuss the potential value of incorporating genetic data in treatment decisions and the routine care of CML patients as a future direction for optimizing CML management.
Introduction
Chronic myeloid leukemia (CML) is a clonal myeloproliferative disorder that accounts for about 15% of adulthood leukemia with a median age at onset of 57 years [Citation1,Citation2]. The hallmark of CML is the Philadelphia chromosome (Ph), which emerges in the reciprocal translocation between chromosomes 9 and 22. This translocation results in the formation of the BCR-ABL1 hybrid gene, which encodes a constitutively active oncokinase protein [Citation3]. Tyrosine kinase inhibitors (TKI) have been developed as a targeted therapy inhibiting BCR-ABL1 kinase activity [Citation4]. The implementation of TKIs in CML management has led to a dramatic improvement in the disease’s outcomes and the nearly normal life expectancy of CML patients [Citation5,Citation6]. Furthermore, some patients with sustained deep molecular remission on TKI treatment have suspended therapy successfully without the incidence of disease relapse and have achieved treatment-free remission (TFR). Because of the improved survival rates, the achievement of TFR has been adopted as a new goal in CML management [Citation7–9]. Understanding the biological factors that contribute to successful TFR would enable the optimization of CML management.
Historically, since the discovery of the BCR-ABL1 fusion gene, CML has been considered a prototype of cancer evolution in which a single oncogene, BCR-ABL1, is capable of initiating and maintaining the cancer phenotype [Citation10]. Additionally, the high response rates of TKIs in most CML patients have added to the evidence showing the principal role of BCR-ABL1 in CML pathogenesis. Furthermore, the retroviral transduction of BCR-ABL1 in murine stem cells successfully induces a CML-like phenotype [Citation11,Citation12]. However, the heterogeneity of the clinical outcomes of CML in TKI treatment suggests the contribution of BCR-ABL1-independent mechanisms to CML pathogenesis. A proportion of CML patients have shown either primary (5–10%) or secondary resistance (20–30%) to TKI treatment, even with the use of more potent second- and third-generation TKIs [Citation2,Citation13]. Acquired Abl-kinase domain (Abl-KD) mutations are the most common cause of secondary resistance where they are detectable in about 60% of cases; however, the majority of primary resistance mechanisms remain elusive [Citation14,Citation15]. Moreover, many patients with secondary resistance lack Abl-KD mutations and have been suggested to have BCR-ABL1-independent mechanisms [Citation16,Citation17]. Furthermore, about 5% of CML patients progress from the chronic phase (CP) to the aggressive blast phase (BP) with very poor outcomes and few treatment options [Citation18,Citation19]. Additionally, leukemic stem cells (LSC) are the main culprit in CML relapse in about half of patients who attempt TKI discontinuation [Citation20,Citation21]. CML LSCs have been suggested to have additional survival pathways in addition to BCR-ABL1-induced signaling [Citation22,Citation23]. These mechanisms contribute to the persistence of CML LSCs despite efficient TKI treatment and the achievement of durable deep molecular responses [Citation24,Citation25].
The evidence accumulated with the advances made in sequencing technologies suggests the contribution of additional genetic events to CML pathogenesis. BCR-ABL1 has been detected in healthy individuals [Citation26], which suggests that additional genetic events may be required for leukemia transformation [Citation27]. Moreover, the genomic instability state induced by BCR-ABL1 leads to the acquisition of additional genetic aberrations ranging from point mutations to chromosomal abnormalities [Citation28–30]. Mechanisms that include enhanced DNA damage via induced reactive oxygen species (ROS) activity and the inhibition of DNA repair mechanisms [Citation31,Citation32] contribute to BCR-ABL1-induced mutagenesis. Furthermore, the acquisition of genetic aberrations involving known cancer genes has been linked to CML progression [Citation33,Citation34]. In elderly-onset leukemia, mutations related to clonal hematopoiesis of intermediate potential (CHIP) [Citation35] have been suggested to play a role in CML pathogenesis [Citation36]; however, this role remains elusive. Although genetic data have been incorporated in diagnosis, risk stratification, and treatment strategies for acute leukemia patients [Citation37], they are still lacking in CML. Currently, the risk stratification of CML patients involves mainly clinically based scoring systems (e.g. Sokal score) and molecular monitoring of BCR-ABL1 levels [Citation1,Citation2]. Systematic studies that investigate the mutational landscape of CML patients in different phases are highly warranted. The analysis of serial samples would also enable the in-depth examination of clonal evolution and mutational dynamics in CML patients under treatment. In addition, the role of genetic data in tailoring treatment strategies in high-risk CML patients remains to be investigated.
In this review, we discuss recent knowledge about the mutational landscape in different phases of CML, focusing on somatic single nucleotide variants (SNV), small insertions and deletions (indels), as well as focal deletions. We also highlight a potential association between non-BCR-ABL1 mutations and poor outcomes, including treatment resistance and CML progression. The current evidence emphasizes the significance of genetic data in CML management and supports the incorporation of genetic investigations in routine CML work-ups.
Genetic events in CP-CML
Initial studies that investigated genetic events beyond BCR-ABL1 focused on BP-CML rather than CP-CML, which has long been considered a genetically uniform disease. Early studies employed Sanger sequencing to analyze a limited number of selected genes with known cancer associations. One of the earliest studies reported a missense RUNX1 mutation in the diagnostic sample of a CP-CML patient with trisomy 21, secondary resistance to imatinib, and later progression to BP-CML [Citation38]. Another study screened the RUNX1 gene in 14 CML patients with trisomy 21 and identified RUNX1 mutations in six patients (one CP-CML patient and five myeloid BP-CML patients) [Citation39]. In the following studies [Citation40–42], ASXL1 was the most frequently mutated gene in CP-CML patients, while mutations in other leukemia-associated genes that were selected for screening, including the TET2 and IDH1/2 genes, have rarely been identified [Citation40,Citation43].
With the application of high-throughput sequencing techniques, a greater number of CML diagnosis samples have been sequenced using a targeted sequencing panel [Citation44–48], whole genome sequencing (WGS), or whole exome sequencing (WES) [Citation48–54] (). Data in high-throughput sequencing studies have suggested that CP-CML is a genetically heterogenic leukemia although it shows less heterogeneity compared with acute leukemia at diagnosis [Citation55]. Non-silent mutations affecting cancer-associated genes were detected in about 35% of CP-CML patients (range: 29%–50%) by either targeted sequencing or WES. In the above-mentioned studies, ASXL1 was the most frequently mutated gene in about 10% of CP-CML patients (). Other frequently mutated genes included IKZF1 mutations and deletions (4%), RUNX1 (2%), TET2 (2%), and DNMT3A (2%). Other mutated genes reported by more than one study included the KMT2D, TP53, KIA1594, CREBBP, and EP300 genes. Interestingly, typical AML-related mutations were seldom identified in CP-CML, including IDH1/2, FLT3, EZH2, and NRAS mutations.
Figure 1. Frequency of mutations in cancer-associated genes at diagnosis (CP) and BP-CML. The data used to build the figure were derived from 27 studies of CP and/or BP [Citation38–54,Citation61,Citation63–71] and included genes that were reported to be mutated in more than one patient and in more than one study. *The frequency of patients with mutated genes was calculated in relation to the number of patients screened for each individual gene, which was highly variable between different genes.
![Figure 1. Frequency of mutations in cancer-associated genes at diagnosis (CP) and BP-CML. The data used to build the figure were derived from 27 studies of CP and/or BP [Citation38–54,Citation61,Citation63–71] and included genes that were reported to be mutated in more than one patient and in more than one study. *The frequency of patients with mutated genes was calculated in relation to the number of patients screened for each individual gene, which was highly variable between different genes.](/cms/asset/2ac299af-647b-4611-bcde-6b28afcede7f/ilal_a_1894652_f0001_b.jpg)
Table 1. Studies of the genetic events at different phases of CML in the TKI era.
Preleukemic and CHIP-related mutations contribute to the complexity of CP-CML genetics. CHIP refers to the acquisition of somatic mutations in hematopoietic stem cells (HSC) in healthy individuals as a part of the aging process, which is associated with an increased risk of hematological malignancies, including AML, MDS, and MPN [Citation56,Citation57]; however, its role in CML remains unclear [Citation36]. Many of the genes reported to be frequently mutated in CP-CML are also known to be CHIP-associated genes, including the ASXL1, TET2, and DNMT3A genes. Two CML studies [Citation50,Citation51] reported a weak correlation between age and the number of somatic mutations, suggesting that some mutations may be passenger age-related mutations. Sequencing Ph-negative (Ph-neg) remission samples [Citation44] or T-cell samples [Citation45,Citation46,Citation48] from CP-CML patients enabled the identification of preleukemic mutations in many CHIP-related genes, including DNMT3A, TP53, TET2, ASXL1, BCOR, and CREBBP, which were detected in both leukemic and non-leukemic cells. However, somatic ASXL1 mutations were identified in 6/21 (29%) of children and young adult CML patients [Citation47], suggesting that the high frequency of ASXL1 mutations in CP-CML is not an age-related phenomenon. Another interesting observation is the low frequency of DNMT3A mutations in CML patients compared with CHIP and other myeloid leukemias (), which suggests that DNMT3A mutations might not greatly contribute to CML pathogenesis [Citation58]. The contribution of CHIP-related mutations to the mutational landscape of CP-CML remains to be addressed.
Figure 2. Frequency of the most common CHIP mutations in elderly healthy individuals and different leukemias. The frequency of DNMT3A, TET2, and ASXL1 mutations were calculated based on pivotal studies of elderly healthy individuals (>55 years) [Citation143], AML [Citation144,Citation145], and MDS [Citation146].
![Figure 2. Frequency of the most common CHIP mutations in elderly healthy individuals and different leukemias. The frequency of DNMT3A, TET2, and ASXL1 mutations were calculated based on pivotal studies of elderly healthy individuals (>55 years) [Citation143], AML [Citation144,Citation145], and MDS [Citation146].](/cms/asset/f7946e53-77a6-49af-9cc8-4b94d90601b7/ilal_a_1894652_f0002_b.jpg)
It is noteworthy that the above-mentioned studies showed considerable variations regarding (i) the selection of patients as random, consecutive, or response-based, (ii) the phenotype of the sample as diagnostic unsorted MNCs or sorted CD34+ cells, (iii) the availability and source of germline control as T-lymphocytes, mesenchymal cells, buccal swabs, or skin biopsy, and (iv) the number of screened genes in targeted sequencing panel studies. Such variations, in addition to the small sizes of the studied cohorts, preclude definitive conclusions and warrant further standardized screenings of larger cohorts.
Genetic events at advanced phase CML (BP-CML)
CML progression has been attributed to BCR-ABL1-induced genomic instability and the acquisition of genetic aberrations. However, the exact molecular mechanisms underlying CML progression are still not well characterized. Illegitimate activity of the RAG enzyme has been reported to contribute to the acquisition of structural aberrations, especially in lymphoid BP patients [Citation48,Citation59]. Recent studies have also suggested that a unique CML mutational signature is implicated in the mutagenesis process [Citation53]. Furthermore, mutations enriched for signatures of deficient DNA double-strand repair by homologous recombination and DNA mismatch repair system were identified in BP patients [Citation48]. Because of the prognostic and therapeutic implications of genetic events in acute leukemia [Citation60], further research is required to gain a comprehensive understanding of the mutational landscape of BP-CML, which would provide insights into BP-CML pathogenesis and enable the better management of this aggressive disease.
Genetic studies have shifted from using the Sanger sequencing of selected genes [Citation41,Citation43,Citation61–65] to more extensive high-throughput sequencing approaches that have revealed the genetic heterogeneity of BP-CML patients [Citation45,Citation48,Citation52–54,Citation66–70] (). Aggregated data have demonstrated that mutations involving several known leukemia-associated genes are frequently encountered in BP-CML patients. In the TKI era, Abl-KD mutations are the most common mutations, and they are detected in about 44% of BP-CML patients (). In addition to Abl-KD mutations, RUNX1 and ASXL1 are the most frequently mutated genes in 19% and 17% of BP-CML patients, respectively. IKZF1 deletions are commonly encountered in 20% of BP-CML patients. Two WES studies have provided insights into the mutation profile in different BP phenotypes [Citation48,Citation52] and showed that IKZF1 deletions are associated with a lymphoid phenotype, while ASXL1 mutations are more frequently encountered in patients with a myeloid phenotype. Other mutations that have been reported in BP-CML involve the BCORL1, BCOR, SETD1B, SETD2, TP53, IDH1/2, GATA2, TET2, EZH2, WT1, PHF6, SETBP1, CBL, PTPN11, and NRAS genes ().
The frequency of mutations in certain genes has shown considerable variation among BP-CML studies. This discrepancy could be explained by methodological and technical differences as well as the small cohort sizes used in most previous studies. WES-based studies [Citation48,Citation52–54,Citation70] have reported a higher frequency of IKZF1 deletions than earlier SNP array-based studies reported [Citation65,Citation67,Citation71] (27% and 11%, respectively). IKZF1 deletions are also common events in acute lymphoblastic leukemia (ALL), where they are associated with an inferior prognosis [Citation72,Citation73]. Another example is WT1, a recurrently mutated gene in AML [Citation74]. Two studies have reported high mutation frequencies (10% and 15%) in BP-CML [Citation53,Citation67], whereas other studies have shown much lower frequencies [Citation48,Citation52]. TP53 mutations were reported at higher frequencies in very early pre-TKI studies [Citation75,Citation76] compared with the low frequency (4%) reported in high-throughput sequencing studies. Recurrent mutations of BRCA2 in solid tumors [Citation77] were reported in 4/39 BP patients in a single recent study [Citation53]. Similarly, JAK3 and BRD3 mutations were reported to be recurrent in BP patients in another recent study [Citation48].
In addition to recurrent mutations in established leukemia-associated genes, recent genomic studies on BP-CML have identified recurrent novel mutations with a potential pathogenic role in CML progression. SETD2 mutations, which are frequent in solid tumors and, to a lesser degree, in acute leukemia [Citation78,Citation79], have been detected in recent studies [Citation48,Citation52,Citation53]. One study also reported SETD2 loss of function by post-translational mechanism was recurrent in BP-CML patients [Citation80]. Another epigenetic modifier, SETD1B [Citation81], was reported in several studies to be mutated in about 5% of patients [Citation48,Citation52,Citation53,Citation82]. BCOR/BCORL1 mutations common in MDS and MDS/MPN [Citation83–85] have been reported in about 13% of BP patients [Citation48,Citation52,Citation53]. Mutations in the ubiquitin-related gene, UBE2A [Citation86], were described by Magistroni et al. [Citation66] in 17% of BP-CML patients and at lower frequencies in other studies [Citation48,Citation52,Citation82]. CDKN2A deletions, known as leukemia-initiating events [Citation87], were identified in 5% of BP-CML patients and typically associated with lymphoid phenotype [Citation52,Citation53]. Because of the increasing number of WES studies in BP-CML, the synchronization and combination of the data are warranted to enable a better overview of the mutational landscape of BP-CML and overcome the limiting factor of small cohort sizes in individual studies.
Fusion genes represent another class of somatic mutations that have an established driver role in leukemia [Citation88]. Early cytogenetic studies described translocations, including cryptic translocations, that resulted in fusion genes involving known leukemia-associated genes, including CBFB-MYH11 [Citation89], as well as RUNX1 [Citation39,Citation90–93] and MLL [Citation94] fusions with various partners. RNA sequencing is a powerful tool for identifying clinically relevant fusion genes [Citation52,Citation95]. Recent studies that applied RNA sequencing have highlighted the potential role of fusion genes in BP-CML [Citation48,Citation52]. Branford et al. [Citation52] reported fusion genes in 14/33 BP-CML patients, including KMT2A (MLL) rearrangements (five patients) and CBFB-MYH11 (two patients). Known leukemia-associated genes, including RUNX1, MECOM, PAX5, and IKZF1, were involved in many fusions reported in the literature. Fusion genes occurred as the sole genetic event in 15% of BP-CML patients. Similarly, Adnan-Awad et al. [Citation48] reported fusion genes in 5/7 BP-CML patients, including CBFB-MYH11 and RUNX1 fusion. However, further RNA sequencing studies are needed to investigate the clinical value of fusion genes in CML progression.
Mutational dynamics and clonal evolution in CML
The analysis of serial samples of CML patients provides insights into the dynamics of the mutational profile in CP patients under TKI treatment, as well as during CML progression. Only a few studies have performed longitudinal analyses of CP-CML under TKI treatment. Mitani et al. [Citation50] analyzed matched diagnosis-remission samples from 20 CP patients and found that successful TKI treatment was associated with the elimination of almost all mutations identified at diagnosis. A handful of mutations, including a TET2 mutation, were identified in remission samples from 30% of patients. Interestingly, mutations were also detected at much lower variant allele frequency (VAF) in respective diagnosis samples, suggesting the expansion of Ph-neg clones. Similarly, Nteliopoulos et al. [Citation46] reported the clearance of mutations detected at diagnosis in remission samples drawn from TKI-treated CP patients. This finding was associated with the emergence of a few low-VAF mutations, including a DNMT3A mutation.
Kim et al. [Citation45] systematically analyzed matched diagnosis-follow-up (FU) samples from 100 patients and their sorted T-cell fractions. The results provided novel insights into the mutational profiles of TKI-treated CP patients, identifying five patterns of mutational dynamics associated with TKI treatment. In pattern 1, the mutations were presented at a stable VAF in both diagnosis and follow-up samples, despite a significant decline in BCR-ABL1 levels, which suggested the preleukemic nature of mutations. Pattern 2 demonstrated the acquisition of mutations in genes, including ABL1 and TP53, that were associated with poor responses and treatment resistance. Pattern 3 showed the elimination of diagnosis mutations in the FU samples, and it was associated with mixed outcomes. Patterns 4 and 5 included a few mutations that were detected in T-cell fractions, which suggested their preleukemic/Ph-neg origin. Notably, some of these patterns, such as the acquisition and clearance of diagnosis mutations on TKI treatment, were also described by Branford et al. [Citation52]. In a recent study by Adnan-Awad et al. [Citation48], a longitudinal analysis was performed on matched samples drawn from 28 CP patients. Similar to Kim et al. [Citation45], the persistence of truncal mutations and/or the acquisition of leukemia-associated mutations were associated with poor responses [Citation48]. Preleukemic and Ph-neg mutations involving TET2 and DNMT3A genes were identified and associated with mixed outcomes.
The analysis of matched diagnosis progression samples enables the identification of BP-specific mutations with a potential role in CML progression. Early studies performed WES on matched samples of individual cases [Citation41,Citation68,Citation69], which precluded the identification of recurrent mutations. One of the most comprehensive studies that addressed genetic events in CML progression was by Branford et al. [Citation52], which matched diagnosis progression samples from 25 patients. Abl-KD mutations were the most frequently acquired event associated with disease progression. IKZF1 deletions, in addition to RUNX1 and BCORL1 mutations, were frequently acquired during disease progression. Other progression-associated mutations involved BCOR, SETD1B, IDH1, and UBE2A genes, which were detected in individual cases. ASXL1 mutations showed variable progression-related patterns, which were acquired during progression in 3 of 25 patients, persistent in CP samples from 3 of 25 patients and lost during progression in 4 of 25 patients. Similar progression-associated mutational profiles were reported in abstract form [Citation82,Citation96], where Abl-KD and RUNX1 mutations were frequent progression-associated events. Magistroni et al. [Citation66] analyzed matched samples from 10 patients and found acquired mutations in the ABL1 (30%), UBE2A (20%), RUNX1 (10%), ASXL1 (10%), and NRAS (10%) genes. In a recent study by Ko et al. [Citation53] WES was performed on matched samples from 13 patients. In agreement with previous data, Abl-KD mutations were the most common, affecting 6 of 13 patients. Other progression-associated events were involved in RUNX1 (2/13), EZH2 (1/13) mutations, and IKZF1 deletion (1/13). Similarly, a study by Adnan-Awad et al. [Citation48] highlighted progression-specific mutations, including Abl-KD (2/3 patients), RUNX1, and ASXL1 (in each 1/3 patients) mutations. The study also investigated patterns of clonal dynamics in two BP-CML patients with matched BP-relapse samples using WES. A lymphoid BP patient showed clonal drift with the eradication, in treatment, of the diagnosed dominant clone with ABL1 (T315I) and RUNX1 mutations and the emergence of a new clone with an EZH2 mutation. Data on another patient demonstrated a linear evolution pattern in which the diagnosed DOT1L mutated clone expanded despite treatment after the acquisition of ABL1, MSH6, and SETD1B mutations.
Somatic mutations and TKI treatment in CP-CML
Several clinical scoring systems have been used for the risk assessment of CML patients at diagnosis, including the Sokal [Citation97], Hasford [Citation98], EUTOS [Citation99], and ELTS [Citation100] scoring systems. Despite their wide clinical use, these scoring systems have limited specificity and sensitivity [Citation101,Citation102]. Additional risk factors, including marrow fibrosis and high-risk additional chromosomal abnormalities (ACA), can predict inferior responses to TKI and a higher risk of progression [Citation103–105]. The integration of genetic data in risk stratification has been successfully implemented in the management of acute leukemia patients [Citation106,Citation107]. However, similar efforts are still pending in CML despite the increasing number of investigations of the prognostic value of genetic data in CML management. For example, germline mutations of the BIM gene have been reported to be associated with TKI resistance [Citation108], and they constitute an independent risk factor for inferior imatinib responses that can complement clinical risk scores in CP-CML patients [Citation109,Citation110]. Moreover, the polymorphism of the HMGCLL1 gene was suggested as a novel biomarker for predicting the achievement of deep molecular response in imatinib-treated patients [Citation111].
Recent high-throughput sequencing studies have suggested that somatic mutations at diagnosis are potential biomarkers for predicting TKI treatment outcomes. In a recent study [Citation48], the burden of somatic mutations was identified as an independent prognostic marker in CP-CML patients. Patients with poor responses showed a higher mutational burden, especially when the calculation was restricted to mutations in cancer-associated genes. Another study applied a scoring system to evaluate the oncogenic potential of the variants and reported a significant association between a high mutational burden and imatinib resistance [Citation51]. Mutational burden has been shown to correlate weakly with age in some studies [Citation49–51], suggesting that some variants are passenger mutations, which warrants further refinement of the data.
Mutations in the epigenetic modifiers are common events in leukemia, and they have been suggested to play a role in pathogenesis and treatment [Citation112,Citation113]. Recent studies have reported frequent mutations of epigenetic modifiers in 20–30% of CP-CML patients, and they have been associated with inferior responses to TKI treatment [Citation45,Citation48], especially imatinib [Citation46]. Kim et al. [Citation45] reported that patients carrying mutations in epigenetic modifiers had significantly inferior outcomes at the 12-, 24-, and 36-month milestones. Similarly, another study [Citation48] demonstrated an increased frequency of epigenetic mutation in poor responders compared with suboptimal and optimal responders. A study of 124 CP-CML patients [Citation46] reported that mutations in epigenetic modifiers at diagnosis could efficiently predict the achievement of major molecular responses and survival rates in imatinib-treated patients but not in the second-generation TKI cohort. Moreover, ASXL1 germline mutations were also reported to be strong biomarkers of imatinib responses in CP-CML [Citation114]. ASXL1 mutations were associated with TKI resistance and the increased risk of disease progression in a recent study [Citation115].
The potential effects of CHIP on treatment outcomes in CML patients remain elusive. CHIP-related mutations (i.e. ASXL1, PTPN11, ATM, and DNMT3A) have been reported at a high frequency in remission samples drawn from patients with Ph-neg clonal abnormalities and associated with reduced survival rates [Citation116]. Another interesting aspect is the association of CHIP mutations with the risk of cardiovascular disease [Citation117]. Cardiovascular events are also known as adverse effects of some TKIs [Citation118]. The frequency of CHIP mutations (especially DNMT3A, TET2, and ASXL1) was significantly higher in remission samples of patients who developed arterial occlusive disease (AOD) compared with patients with no AOD (65% vs. 32%, respectively) in a recent study involving 36 nilotinib-treated patients [Citation119].
In conclusion, there is an increasing amount of evidence of the predictive value of the mutational status of CP-CML patients at diagnosis regarding TKI treatment responses. However, further systematic studies involving larger patient cohorts are warranted to overcome the limiting factors of small cohort sizes and response-based patient selection in previous studies.
Genetic data in BP-CML management
The use of TKIs has dramatically improved survival rates in CP-CML patients [Citation120], whereas TKIs have only modestly improved the survival rates of BP-CML patients, even with the use of the more potent second-and third-generation TKIs [Citation33]. The current treatment of choice in BP-CML is a combination of TKIs with chemotherapy followed by allogenic stem cell transplantation [Citation19]. BP-CML remains the main clinical challenge in CML management in the TKI era, and there is still a major need to identify better treatment options for BP-CML patients.
Regarding genetic data, ex vivo high-throughput drug testing represents a promising complementary method in the personalized treatment of leukemia [Citation121–125]. High-throughput drug sensitivity and resistance testing (DSRT) of BP-CML cell lines and patient samples have identified novel candidate drugs, including VEGFR-, MEK-, and NAMPT inhibitors [Citation126]. Interestingly, ex vivo drug testing has led to the groundbreaking discovery of the selective and efficient inhibitory activity of axitinib against primary ABL1-T315I mutated cells, which highlights the potential benefits of repurposing approved targeted drugs in BP-CML management [Citation127]. A recent study emphasized the role of a personalized approach in tailoring treatment for BP-CML patients [Citation48]. The integrated approach of genetic, transcriptional, and drug sensitivity profiling was used to guide the treatment of two BP-CML patients with DSRT-based axitinib treatment, inducing significant clinical responses in both cases. Additionally, genetic data could indicate druggable targets and activated transcriptional pathways that underlie CML progression and relapse.
Many of the recurrently mutated genes in BP-CML are potential targets for targeted therapy. RUNX1, the most frequently mutated gene in BP-CML, is a transcription factor that is commonly mutated in other leukemias, including AML. Several studies have investigated potential targeted therapies for RUNX1-mutated AML, reporting specific sensitivity to glucocorticoids [Citation128], mTOR- [Citation129], VEGFR- [Citation130], and, recently, BET inhibitors [Citation131]. A recent study integrated genetic and DSRT profiling to characterize RUNX1-mutated BP-CML patients and identify a targeted therapy [Citation70]. The study identified distinct phenotypic and transcriptional criteria in RUNX1-mutated BP-CML patients, including the frequent expression of lymphoid markers in myeloid BP patients, enhanced off-target activity of the mutagenic AID/RAG axis, as well as the dysregulation of stem cell, lymphoid, and immune-related pathways. These genomic findings were translated into the sensitivity of RUNX1-mutated blasts to glucocorticoid, mTOR- and BCL2-inhibitor targeted therapy, suggesting the presence of a common RUNX1 signature in CML and AML. Furthermore, RUNX1-mutated blasts were sensitive to CD19-CAR T-cell immunotherapy not only in lymphoid BP patients but also in a myeloid patient with aberrant CD19 expression, which was in line with recent reports on RUNX1-mutated AML [Citation132].
IKZF1 is another potential target for targeted therapy in BP-CML. IKZF1 is a tumor suppressor that is commonly affected by focal deletions in ALL and lymphoid BP-CML [Citation72,Citation133]. In Ph + ALL, retinoids were reported to enhance dasatinib activity in IKZF1-mutated patients [Citation134]. Furthermore, the targeting of truncating mutations of the ASXL1 gene, which is another recurrently mutated gene in BP-CML, by BET inhibitors was recently reported [Citation135]. The known cancer-related genes, TP53 and EZH2, have been targeted by many specific drugs [Citation136–139]; however, they were mutated in only a minority of BP-CML. Rare BP-CML cases with IDH1/2 mutations could benefit from clinically approved drugs for IDH-mutated AML [Citation140]. In addition to the RUNX1-associated aberrant expression of CD19 in myeloid patients, WT1 represents another attractive target for immunotherapy [Citation141,Citation142].
In summary, the integration of genetic and drug sensitivity data provides an intriguing means of personalizing and improving BP-CML management. Only a very few of all recurrently mutated genes in BP-CML have been investigated in a limited number of cases. Thus, further systematic high-throughput drug testing of a greater number of BP-CML samples is required. This approach may allow for the identification of new potential biomarkers and treatment modalities for BP-CML.
Conclusion and further considerations
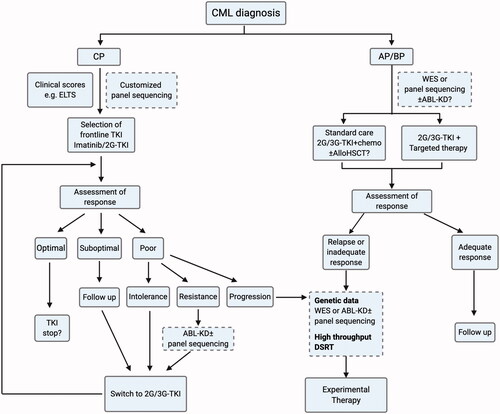
The concept of CML as a genetically uniform disease has changed as the number of studies has increased, which suggests the genetic heterogeneity of CML. Although BCR-ABL1 is the principal event in CML pathogenesis, mutations involving other genes play important roles in different phases of CML. Somatic mutations, especially those affecting epigenetic modifiers, have been suggested as affecting TKI treatment outcomes in CP-CML. Additionally, because somatic mutations have particularly pronounced effects in BP-CML, the integration of genetic and drug sensitivity data in a personalized approach represents a promising strategy for disease management. However, further studies are needed to reach definite conclusions. Additionally, the role of CHIP and preleukemic mutations in CML pathogenesis remains to be addressed. Clinically-based scores are the only scoring system in current CML practice, however; screening for additional cancer-associated mutations can provide clinically relevant information and may be incorporated in CML patients’ routine care in the future. Mutation profiling may be needed both at diagnosis and in the case of resistance or progression, as proposed in the algorithm shown in . Notably, there are still many open questions to be addressed, such as the selection of the sequencing method (i.e. WGS, WES, or pre-designed targeted panel sequencing), required sequencing depth (i.e. the detection of subclonal mutations), sample type (i.e. sorted vs. whole blood), and germline control samples. Nevertheless, we believe that further studies of CML genetics will enable the adoption of better personalized treatment strategies that will significantly improve the management of high-risk CML patients and provide a means of enhancing TFR rates in CML.
Figure 3. Algorithm of suggested future directions of CML management integrating genetic screening in risk stratification and drug selection. In BP, in case of non-fit patients ineligible for chemotherapy or allogenic hematopoietic stem cell transplantation (allo-HSCT), targeted therapy based on mutation profile can be considered.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Jabbour E, Kantarjian H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am J Hematol. 2020;95(6):691–709.
- Hochhaus A, Baccarani M, Silver RT, et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia. 2020;34(4):966–984.
- Cilloni D, Saglio G. Molecular pathways: BCR-ABL. Clin Cancer Res. 2012;18(4):930–937.
- Mughal TI, Goldman JM. Targeting cancers with tyrosine kinase inhibitors: lessons learned from chronic myeloid leukaemia. Clin Med (Lond). 2006;6(6):526–528.
- Bower H, Björkholm M, Dickman PW, et al. Life expectancy of patients with chronic myeloid leukemia approaches the life expectancy of the general population. J Clin Oncol. 2016;34(24):2851–2857.
- Russo D, Garcia-Gutierrez JV, Soverini S, et al. Chronic myeloid leukemia prognosis and therapy: criticisms and perspectives. J Clin Med. 2020;9(6):1709.
- Mahon F-X. Treatment-free remission in CML: who, how, and why? Hematology Am Soc Hematol Educ Program. 2017;2017(1):102–109.
- Molica M, Naqvi K, Cortes JE, et al. Treatment-free remission in chronic myeloid leukemia. Clin Adv Hematol Oncol. 2019;17(12):686–696.
- Baccarani M, Abruzzese E, Accurso V, et al. Managing chronic myeloid leukemia for treatment-free remission: a proposal from the GIMEMA CML WP. Blood Adv. 2019;3(24):4280–4290.
- Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7(6):441–453.
- Daley GQ, Van Etten RA, Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247(4944):824–830.
- Zhang X, Ren R. BCR-ABL efficiently induces a myeloproliferative disease and production of excess interleukin-3 and granulocyte-macrophage colony-stimulating factor in mice: a novel model for chronic myelogenous leukemia. Blood. 1998;92(10):3829–3840.
- Hochhaus A, La Rosée P. Imatinib therapy in chronic myelogenous leukemia: strategies to avoid and overcome resistance. Leukemia. 2004;18(8):1321–1331.
- Soverini S, de Benedittis C, Mancini M, et al. Mutations in the BCR-ABL1 kinase domain and elsewhere in chronic myeloid leukemia. Clin Lymphoma Myeloma Leuk. 2015;15:S120–S128.
- Braun TP, Eide CA, Druker BJ. Response and resistance to BCR-ABL1-targeted therapies. Cancer Cell. 2020;37(4):530–542.
- Bavaro L, Martelli M, Cavo M, et al. Mechanisms of disease progression and resistance to tyrosine kinase inhibitor therapy in chronic myeloid leukemia: an update. IJMS. 2019;20(24):6141.
- Patel AB, O'Hare T, Deininger MW. Mechanisms of resistance to ABL kinase inhibition in chronic myeloid leukemia and the development of next generation ABL kinase inhibitors. Hematol Oncol Clin North Am. 2017;31(4):589–612.
- Hehlmann R, Lauseker M, Jung-Munkwitz S, et al. Tolerability-adapted imatinib 800 mg/d versus 400 mg/d versus 400 mg/d plus interferon-α in newly diagnosed chronic myeloid leukemia. J Clin Oncol. 2011;29(12):1634–1642.
- Jain P, Kantarjian HM, Ghorab A, et al. Prognostic factors and survival outcomes in patients with chronic myeloid leukemia in blast phase in the tyrosine kinase inhibitor era: cohort study of 477 patients. Cancer. 2017;123(22):4391–4402.
- Campiotti L, Suter MB, Guasti L, et al. Imatinib discontinuation in chronic myeloid leukaemia patients with undetectable BCR-ABL transcript level: a systematic review and a meta-analysis. Eur J Cancer. 2017;77:48–56.
- Chomel JC, Bonnet ML, Sorel N, et al. Leukemic stem cell persistence in chronic myeloid leukemia patients in deep molecular response induced by tyrosine kinase inhibitors and the impact of therapy discontinuation. Oncotarget. 2016;7(23):35293–35301.
- Hamilton A, Helgason GV, Schemionek M, et al. Chronic myeloid leukemia stem cells are not dependent on BCR-ABL kinase activity for their survival. Blood. 2012;119(6):1501–1510.
- Graham SM, Jørgensen HG, Allan E, et al. Primitive, quiescent, Philadelphia-positive stem cells from patients with chronic myeloid leukemia are insensitive to STI571 in vitro. Blood. 2002;99(1):319–325.
- Houshmand M, Simonetti G, Circosta P, et al. Chronic myeloid leukemia stem cells. Leukemia. 2019;33(7):1543–1556.
- Muselli F, Peyron J-F, Mary D. Druggable biochemical pathways and potential therapeutic alternatives to target leukemic stem cells and eliminate the residual disease in chronic myeloid leukemia. IJMS. 2019;20(22):5616.
- Boquett JA, Alves JRP, de Oliveira CEC. Analysis of BCR/ABL transcripts in healthy individuals. Genet Mol Res. 2013;12(4):4967–4971.
- Fialkow PJ, Martin PJ, Najfeld V, et al. Evidence for a multistep pathogenesis of chronic myelogenous leukemia. Blood. 1981;58(1):158–163.
- Burke B, Carroll M. BCR-ABL: a multi-faceted promoter of DNA mutation in chronic myelogeneous leukemia. Leukemia. 2010;24(6):1105–1112.
- Soverini S, Gnani A, Colarossi S, et al. Philadelphia-positive patients who already harbor imatinib-resistant BCR-ABL kinase domain mutations have a higher likelihood of developing additional mutations associated with resistance to second- or third-line tyrosine kinase inhibitors. Blood. 2009;114(10):2168–2171.
- Koptyra M, Cramer K, Slupianek A, et al. BCR/ABL promotes accumulation of chromosomal aberrations induced by oxidative and genotoxic stress. Leukemia. 2008;22(10):1969–1972.
- Koptyra M, Falinski R, Nowicki MO, et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108(1):319–327.
- Slupianek A, Falinski R, Znojek P, et al. BCR-ABL1 kinase inhibits uracil DNA glycosylase UNG2 to enhance oxidative DNA damage and stimulate genomic instability. Leukemia. 2013;27(3):629–634.
- Hehlmann R, Saußele S, Voskanyan A, et al. Management of CML-blast crisis. Best Pract Res Clin Haematol. 2016;29(3):295–307.
- Jabbour EJ, Hughes TP, Cortés JE, et al. Potential mechanisms of disease progression and management of advanced-phase chronic myeloid leukemia. Leuk Lymphoma. 2014;55(7):1451–1462.
- Xie M, Lu C, Wang J, et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat Med. 2014;20(12):1472–1478.
- Walter MJ. Antecedent CHIP in CML? Blood. 2017;129(1):3–4.
- Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447.
- Corm S, Biggio V, Roche-Lestienne C, et al. Coexistence of AML1/RUNX1 and BCR-ABL point mutations in an imatinib-resistant form of CML. Leukemia. 2005;19(11):1991–1992.
- Roche-Lestienne C, Deluche L, Corm S, et al. RUNX1 DNA-binding mutations and RUNX1-PRDM16 cryptic fusions in BCR-ABL + leukemias are frequently associated with secondary trisomy 21 and may contribute to clonal evolution and imatinib resistance. Blood. 2008;111(7):3735–3741.
- Roche-Lestienne C, Marceau A, Labis E, et al. Mutation analysis of TET2, IDH1, IDH2 and ASXL1 in chronic myeloid leukemia. Leukemia. 2011;25(10):1661–1664.
- Menezes J, Salgado RN, Acquadro F, et al. ASXL1, TP53 and IKZF3 mutations are present in the chronic phase and blast crisis of chronic myeloid leukemia. Blood Cancer J. 2013;3:e157.
- Valikhani A, Poopak B, Ferdowsi S, et al. ASXL1 and JAK2V617F gene mutation screening in Iranian patients with chronic myeloid leukemia. Asia Pac J Clin Oncol. 2017;13(2):e41–e47.
- Soverini S, Score J, Iacobucci I, et al. IDH2 somatic mutations in chronic myeloid leukemia patients in blast crisis. Leukemia. 2011;25(1):178–181.
- Schmidt M, Rinke J, Schäfer V, et al. Molecular-defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR-ABL status. Leukemia. 2014;28(12):2292–2299.
- Kim T, Tyndel MS, Kim HJ, et al. Spectrum of somatic mutation dynamics in chronic myeloid leukemia following tyrosine kinase inhibitor therapy. Blood. 2017;129(1):38–47.
- Nteliopoulos G, Bazeos A, Claudiani S, et al. Somatic variants in epigenetic modifiers can predict failure of response to imatinib but not to second-generation tyrosine kinase inhibitors. Haematologica. 2019;104(12):2400–2409.
- Ernst T, Busch M, Rinke J, et al. Frequent ASXL1 mutations in children and young adults with chronic myeloid leukemia. Leukemia. 2018;32(9):2046–2049.
- Adnan Awad S, Kankainen M, Ojala T, et al. Mutation accumulation in cancer genes relates to nonoptimal outcome in chronic myeloid leukemia. Blood Adv. 2020;4(3):546–559.
- Togasaki E, Takeda J, Yoshida K, et al. Frequent somatic mutations in epigenetic regulators in newly diagnosed chronic myeloid leukemia. Blood Cancer J. 2017;7(4):e559.
- Mitani K, Nagata Y, Sasaki K, et al. Somatic mosaicism in chronic myeloid leukemia in remission. Blood. 2016;128(24):2863–2866.
- Mologni L, Piazza R, Khandelwal P, et al. Somatic mutations identified at diagnosis by exome sequencing can predict response to imatinib in chronic phase chronic myeloid leukemia (CML) patients. Am J Hematol. 2017;92(10):E623–E625.
- Branford S, Wang P, Yeung DT, et al. Integrative genomic analysis reveals cancer-associated mutations at diagnosis of CML in patients with high-risk disease. Blood. 2018;132(9):948–961.
- Ko TK, Javed A, Lee KL, et al. An integrative model of pathway convergence in genetically heterogeneous blast crisis chronic myeloid leukemia. Blood. 2020;135(26):2337–2353.
- Kim T, Tyndel MS, Zhang Z, et al. Exome sequencing reveals DNMT3A and ASXL1 variants associate with progression of chronic myeloid leukemia after tyrosine kinase inhibitor therapy. Leuk Res. 2017;59:142–148.
- Li S, Mason CE, Melnick A. Genetic and epigenetic heterogeneity in acute myeloid leukemia. Curr Opin Genet Dev. 2016;36:100–106.
- Genovese G, Kähler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med. 2014;371(26):2477–2487.
- Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16.
- Li X, Cen J, Wang Q, et al. Absence of DNMT3A gene mutation in chronic myeloid leukemia patients in blast crisis. Eur J Haematol. 2012;88(5):455–457.
- Thomson DW, Shahrin NH, Wang PPS, et al. Aberrant RAG-mediated recombination contributes to multiple structural rearrangements in lymphoid blast crisis of chronic myeloid leukemia. Leukemia. 2020;34(8):2051–2063.
- DiNardo CD, Cortes JE. Mutations in AML: prognostic and therapeutic implications. Hematology Am Soc Hematol Educ Program. 2016;2016(1):348–355.
- Makishima H, Jankowska AM, McDevitt MA, et al. CBL, CBLB, TET2, ASXL1, and IDH1/2 mutations and additional chromosomal aberrations constitute molecular events in chronic myelogenous leukemia. Blood. 2011;117(21):e198–e206.
- Zhao L-J, Wang Y-Y, Li G, et al. Functional features of RUNX1 mutants in acute transformation of chronic myeloid leukemia and their contribution to inducing murine full-blown leukemia. Blood. 2012;119(12):2873–2882.
- Zhang S-J, Ma L-Y, Huang Q-H, et al. Gain-of-function mutation of GATA-2 in acute myeloid transformation of chronic myeloid leukemia. Proc Natl Acad Sci USA. 2008;105(6):2076–2081.
- Yamamoto K, Tsuzuki S, Minami Y, et al. Functionally deregulated AML1/RUNX1 cooperates with BCR-ABL to induce a blastic phase-like phenotype of chronic myelogenous leukemia in mice. PLoS One. 2013;8(9):e74864.
- Boultwood J, Perry J, Zaman R, et al. High-density single nucleotide polymorphism array analysis and ASXL1 gene mutation screening in chronic myeloid leukemia during disease progression. Leukemia. 2010;24(6):1139–1145.
- Magistroni V, Mauri M, D'Aliberti D, et al. De novo UBE2A mutations are recurrently acquired during chronic myeloid leukemia progression and interfere with myeloid differentiation pathways. Haematologica. 2019;104(9):1789–1797.
- Grossmann V, Kohlmann A, Zenger M, et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76.9% of cases. Leukemia. 2011;25(3):557–560.
- Sklarz L-M, Wittke C, Krohn S, et al. Genetic mutations in a patient with chronic myeloid leukemia showing blast crisis 10 years after presentation. Anticancer Res. 2018;38(7):3961–3966.
- Huang Y, Zheng J, Hu JD, et al. Discovery of somatic mutations in the progression of chronic myeloid leukemia by whole-exome sequencing. Genet Mol Res. 2014;13(1):945–953.
- Adnan AS, Dufva O, Ianevski A, et al. RUNX1 mutations in blast-phase chronic myeloid leukemia associate with distinct phenotypes, transcriptional profiles, and drug responses. Leukemia. 2020:1–13. doi:10.1038/s41375-020-01011-5
- Mullighan CG, Miller CB, Radtke I, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008;453(7191):110–114.
- Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360(5):470–480.
- Marke R, Leeuwen FN, van, Scheijen B. The many faces of IKZF1 in B-cell precursor acute lymphoblastic leukemia. 1. Haematologica. 2018;103(4):565–574.
- Rampal R, Figueroa ME. Wilms tumor 1 mutations in the pathogenesis of acute myeloid leukemia. Haematologica. 2016;101(6):672–679.
- Ahuja H, Bar-Eli M, Arlin Z, et al. The spectrum of molecular alterations in the evolution of chronic myelocytic leukemia. J Clin Invest. 1991;87(6):2042–2047.
- Nakai H, Misawa S, Toguchida J, et al. Frequent p53 gene mutations in blast crisis of chronic myelogenous leukemia, especially in myeloid crisis harboring loss of a chromosome 17p. Cancer Res. 1992;52(23):6588–6593.
- Mersch J, Jackson M, Park M, et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. 2015;121(2):269–275.
- Dong Y, Zhao X, Feng X, et al. SETD2 mutations confer chemoresistance in acute myeloid leukemia partly through altered cell cycle checkpoints. Leukemia. 2019;33(11):2585–2598.
- Li J, Duns G, Westers H, et al. SETD2: an epigenetic modifier with tumor suppressor functionality. Oncotarget. 2016;7(31):50719–50734.
- Mancini M, Santis SD, Monaldi C, et al. SETD2 loss of function is a recurrent event in advanced-phase chronic myeloid leukemia. Blood. 2017;130:43.
- Krzyzewska IM, Maas SM, Henneman P, et al. A genome-wide DNA methylation signature for SETD1B-related syndrome. Clin Epigenetics. 2019;11(1):156.
- Ochi Y, Yoshida K, Huang Y-J, et al. Molecular profiling of blastic transformation in chronic myeloid leukemia. Blood. 2018;132(Supplement 1):1725.
- Tiacci E, Grossmann V, Martelli MP, et al. The corepressors BCOR and BCORL1: two novel players in acute myeloid leukemia. Haematologica. 2012;97(1):3–5.
- Damm F, Chesnais V, Nagata Y, et al. BCOR and BCORL1 mutations in myelodysplastic syndromes and related disorders. Blood. 2013;122(18):3169–3177.
- Cao Q, Gearhart MD, Gery S, et al. BCOR regulates myeloid cell proliferation and differentiation. Leukemia. 2016;30(5):1155–1165.
- Bergink S, Jentsch S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009;458(7237):461–467.
- Williams RT, Sherr CJ. BCR–ABL and CDKN2A: a dropped connection. Nat Rev Cancer. 2008;8(7):563.
- Wang Y, Wu N, Liu D, et al. Recurrent fusion genes in leukemia: an attractive target for diagnosis and treatment. Curr Genomics. 2017;18(5):378–384.
- Salem A, Loghavi S, Tang G, et al. Myeloid neoplasms with concurrent BCR-ABL1 and CBFB rearrangements: a series of 10 cases of a clinically aggressive neoplasm. Am J Hematol. 2017;92(6):520–528.
- Mitani K, Ogawa S, Tanaka T, et al. Generation of the AML1-EVI-1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukemia. Embo J. 1994;13(3):504–510.
- Paquette RL, Nicoll J, Chalukya M, et al. Frequent EVI1 translocations in myeloid blast crisis CML that evolves through tyrosine kinase inhibitors. Cancer Genet. 2011;204(7):392–397.
- Yin CC, Cortes J, Barkoh B, et al. t(3;21)(q26;q22) in myeloid leukemia: an aggressive syndrome of blast transformation associated with hydroxyurea or antimetabolite therapy. Cancer. 2006;106(8):1730–1738.
- Najfeld V, Wisch N, Mascarenhas J, et al. Development of t(8;21) and RUNX1-RUNX1T1 in the Philadelphia-positive clone of a patient with chronic myelogenous leukemia: additional evidence for multiple steps involved in disease progression. Cancer Genet. 2011;204(3):165–170.
- Wang W, Tang G, Cortes JE, et al. Chromosomal rearrangement involving 11q23 locus in chronic myelogenous leukemia: a rare phenomenon frequently associated with disease progression and poor prognosis. J Hematol Oncol. 2015;8:32.
- Lilljebjörn H, Ågerstam H, Orsmark-Pietras C, et al. RNA-seq identifies clinically relevant fusion genes in leukemia including a novel MEF2D/CSF1R fusion responsive to imatinib. Leukemia. 2014;28(4):977–979.
- Jain P, Kanagal-Shamanna R, Kantarjian HM, et al. Pattern of mutational changes in patients with chronic phase CML who are treated with frontline TKI and transform to blast phase CML. Blood. 2017;130:4172–4172.
- Sokal JE, Cox EB, Baccarani M, et al. Prognostic discrimination in “good-risk” chronic granulocytic leukemia. Blood. 1984;63(4):789–799.
- Hasford J, Pfirrmann M, Hehlmann R, et al. A new prognostic score for survival of patients with chronic myeloid leukemia treated with interferon alfa. Writing Committee for the Collaborative CML Prognostic Factors Project Group. J Natl Cancer Inst. 1998;90(11):850–858.
- Hasford J, Baccarani M, Hoffmann V, et al. Predicting complete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score. Blood. 2011;118(3):686–692.
- Pfirrmann M, Baccarani M, Saussele S, et al. Prognosis of long-term survival considering disease-specific death in patients with chronic myeloid leukemia. Leukemia. 2016;30(1):48–56.
- Hu B, Savani BN. Impact of risk score calculations in choosing front-line tyrosine kinase inhibitors for patients with newly diagnosed chronic myeloid leukemia in the chronic phase. Eur J Haematol. 2014;93(3):179–186.
- Suttorp M, Glauche I, Salas DG, et al. Scoring systems for predicting outcome of chronic myeloid leukemia in adults are poorly informative in pediatric patients treated with imatinib. Blood. 2013;122(21):2725–2725.
- Buesche G, Ganser A, Schlegelberger B, et al. Marrow fibrosis and its relevance during imatinib treatment of chronic myeloid leukemia. Leukemia. 2007;21(12):2420–2427.
- Fabarius A, Kalmanti L, Dietz CT, et al. Impact of unbalanced minor route versus major route karyotypes at diagnosis on prognosis of CML. Ann Hematol. 2015;94(12):2015–2024.
- Hehlmann R, Voskanyan A, Lauseker M, et al. High-risk additional chromosomal abnormalities at low blast counts herald death by CML. Leukemia. 2020;34(8):2074–2086.
- Estey EH. Acute myeloid leukemia: 2019 update on risk-stratification and management. Am J Hematol. 2018;93(10):1267–1291.
- Herold T, Rothenberg-Thurley M, Grunwald VV, et al. Validation and refinement of the revised 2017 European LeukemiaNet genetic risk stratification of acute myeloid leukemia. Leukemia. 2020;34(12):3161–3172.
- Ng KP, Hillmer AM, Chuah CTH, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012;18(4):521–528.
- Augis V, Airiau K, Josselin M, et al. A single nucleotide polymorphism in cBIM is associated with a slower achievement of major molecular response in chronic myeloid leukaemia treated with imatinib. PLoS One. 2013;8(11):e78582.
- Than H, Lye WK, Sng C, et al. BIM deletion polymorphism profiling complements prognostic values of risk scores in imatinib-treated Asian chronic myeloid leukemia patients. Leuk Lymphoma. 2019;60(1):234–237.
- Park J-H, Woo YM, Youm EM, et al. HMGCLL1 is a predictive biomarker for deep molecular response to imatinib therapy in chronic myeloid leukemia. Leukemia. 2019;33(6):1439–1450.
- Abdel-Wahab O, Levine RL. Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood. 2013;121(18):3563–3572.
- Fathi AT, Abdel-Wahab O. Mutations in epigenetic modifiers in myeloid malignancies and the prospect of novel epigenetic-targeted therapy. Adv Hematol. 2012;2012:469592.
- Marum JE, Yeung DT, Purins L, et al. ASXL1 and BIM germ line variants predict response and identify CML patients with the greatest risk of imatinib failure. Blood Adv. 2017;1(18):1369–1381.
- Wu W, Xu N, Zhou X, et al. Integrative genomic analysis reveals cancer-associated gene mutations in chronic myeloid leukemia patients with resistance or intolerance to tyrosine kinase inhibitor. Onco Targets Ther. 2020;13:8581–8591.
- Issa GC, Kantarjian HM, Gonzalez GN, et al. Clinical and molecular characterization of clonal chromosomal abnormalities appearing in philadelphia-negative metaphases of chronic phase CML. Blood. 2017;130(Supplement 1):47.
- Jaiswal S, Natarajan P, Silver AJ, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377(2):111–121.
- Jain P, Kantarjian H, Boddu PC, et al. Analysis of cardiovascular and arteriothrombotic adverse events in chronic-phase CML patients after frontline TKIs. Blood Adv. 2019;3(6):851–861.
- Hadzijusufovic E, Hoermann G, Herndlhofer S, et al. Cardiovascular scoring and age-related mutations predict the occurrence of adverse vascular events in CML patients during nilotinib therapy. Blood. 2017;130:1619.
- Radivoyevitch T, Weaver D, Hobbs B, et al. Do persons with chronic myeloid leukaemia have normal or near normal survival? Leukemia. 2020;34(2):333–335.
- Pemovska T, Kontro M, Yadav B, et al. Individualized systems medicine strategy to tailor treatments for patients with chemorefractory acute myeloid leukemia. Cancer Discov. 2013;3(12):1416–1429.
- Swords RT, Azzam D, Al-Ali H, et al. Ex-vivo sensitivity profiling to guide clinical decision making in acute myeloid leukemia: a pilot study. Leuk Res. 2018;64:34–41.
- Andersson EI, Pützer S, Yadav B, et al. Discovery of novel drug sensitivities in T-PLL by high-throughput ex vivo drug testing and mutation profiling. Leukemia. 2018;32(3):774–787.
- Dietrich S, Oleś M, Lu J, et al. Drug-perturbation-based stratification of blood cancer. J Clin Invest. 2018;128(1):427–445.
- Zhang H, Nakauchi Y, Köhnke T, et al. Integrated analysis of patient samples identifies biomarkers for venetoclax efficacy and combination strategies in acute myeloid leukemia. Nat Cancer. 2020;1(8):826–839.
- Pietarinen PO, Pemovska T, Kontro M, et al. Novel drug candidates for blast phase chronic myeloid leukemia from high-throughput drug sensitivity and resistance testing. Blood Cancer J. 2015;5:e309.
- Pemovska T, Johnson E, Kontro M, et al. Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation. Nature. 2015;519(7541):102–105.
- Simon L, Lavallée V-P, Bordeleau M-E, et al. Chemogenomic landscape of RUNX1-mutated AML reveals importance of RUNX1 allele dosage in genetics and glucocorticoid sensitivity. Clin Cancer Res. 2017;23(22):6969–6981.
- Fuka G, Kantner H-P, Grausenburger R, et al. Silencing of ETV6/RUNX1 abrogates PI3K/AKT/mTOR signaling and impairs reconstitution of leukemia in xenografts. Leukemia. 2012;26(5):927–933.
- Imai N, Shikami M, Miwa H, et al. t(8;21) acute myeloid leukaemia cells are dependent on vascular endothelial growth factor (VEGF)/VEGF receptor type2 pathway and phosphorylation of Akt. Br J Haematol. 2006;135(5):673–682.
- Mill CP, Fiskus W, DiNardo CD, et al. RUNX1-targeted therapy for AML expressing somatic or germline mutation in RUNX1. Blood. 2019;134(1):59–73.
- Danylesko I, Jacoby E, Yerushalmi R, et al. Remission of acute myeloid leukemia with t(8;21) following CD19 CAR T-cells. Leukemia. 2020;34(7):1939–1942.
- Tauer J, Kronnie G. t, Ekici A, et al. Copy number variations and IKZF1 mutations in pediatric CML. Blood. 2013;122(21):1473.
- Churchman ML, Low J, Qu C, et al. Efficacy of retinoids in IKZF1-mutated BCR-ABL1 acute lymphoblastic leukemia. Cancer Cell. 2015;28(3):343–356.
- Yang H, Kurtenbach S, Guo Y, et al. Gain of function of ASXL1 truncating protein in the pathogenesis of myeloid malignancies. Blood. 2018;131(3):328–341.
- Bykov VJN, Eriksson SE, Bianchi J, et al. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer. 2018;18(2):89–102.
- Sanz G, Singh M, Peuget S, et al. Inhibition of p53 inhibitors: progress, challenges and perspectives. J Mol Cell Biol. 2019;11(7):586–599.
- Li CH, Chen Y. Targeting EZH2 for cancer therapy: progress and perspective. Curr Protein Pept Sci. 2015;16(6):559–570.
- Jones BA, Varambally S, Arend RC. Histone methyltransferase EZH2: a therapeutic target for ovarian cancer. Mol Cancer Ther. 2018;17(3):591–602.
- Liu X, Gong Y. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomark Res. 2019;7:22.
- Chapuis AG, Egan DN, Bar M, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med. 2019;25(7):1064–1072.
- Dao T, Yan S, Veomett N, et al. Targeting the intracellular WT1 oncogene product with a therapeutic human antibody. Sci Transl Med. 2013;5(176):176ra33.
- Buscarlet M, Provost S, Zada YF, et al. DNMT3A and TET2 dominate clonal hematopoiesis and demonstrate benign phenotypes and different genetic predispositions. Blood. 2017;130(6):753–762.
- Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. New Eng J Med. 2013;368:2059–2074.
- Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221.
- Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–247.