
Abstract
Mantle cell lymphoma (MCL) is an aggressive B-cell neoplasm that follows a heterogeneous clinical course. Recurrent mutations have been described, but their applicability in the clinical setting is currently limited. The main reasons are challenges in the sequencing of DNA retrieved from formalin-fixed tissue commonly used for tissue collection in clinical biobanks. In this study, we sequenced 77 samples from a population-based de novo MCL cohort to investigate the utility of targeted sequencing in guiding personalized treatment approaches. Tumors were genetically variable, and a similar genetic landscape as previous studies using non-formalin fixed samples was identified, with recurrent mutations including ATM, KMT2D, and TP53. Novel alterations that can be considered actionable and/or indicative of treatment response were also identified. Our approach shows the potential benefits of using target sequencing of formalin fixed samples to facilitate treatment selection and individualized clinical decisions in MCL.
Introduction
The genetic hallmark of MCL is the t(11;14) (q13;q32), leading to cyclin D1 overexpression [Citation1]. MCL is an heterogeneous disease ranging from indolent to highly aggressive variants, the latter often characterized by blastoid morphology and high Ki-67 expression [Citation2]. Although substantial improvements in treatment have been achieved in the last decades, the median survival is only 3–5 years after diagnosis [Citation1]. To date, diagnosis is mostly based on the detection of cyclin D1 and SOX11 expression. However, the expression of these markers is insufficient to explain the pathogenesis of the disease [Citation2]. Secondary genetic events and disruption of other cellular pathways are needed for the development of MCL [Citation3]. Some molecular studies have pinpointed alterations in DNA damage pathways, cell survival, and cell-cycle regulation [Citation3–6]. Nevertheless, the current genomic knowledge of the disease cannot explain the high clinical heterogeneity and diverse responses to treatment of subsets of patients [Citation3].
Recent advances in sequencing technologies have provided tools for a deeper characterization of the genome and have brought insights into MCL recurrent single nucleotide variants (SNVs). ATM mutations are the most frequent (40–50%) aberration in the few studies that so far have evaluated the genomic landscape in MCL [Citation5–9]. Other recurrent mutations reported affect the CCND1, TP53, KMT2D, KMT2C, NOTCH1, NSD2, BIRC3, NOTCH2, and UBR5 genes [Citation5,Citation6,Citation8–11]. Although MCL is characterized by a widespread genetic heterogeneity [Citation12,Citation13], some recurrent SNVs might be relevant due to either their localization in targetable genes or correlation with prognostic characteristics. Of note, there is a lack of broader investigations in non-selected population-based cohorts. One reason has been the struggle with performing high-quality sequencing from formalin-fixed, paraffin-embedded (FFPE) tissue samples, which are the specimens of choice in routine pathology archives.
In the current study, we sequenced FFPE biopsies from patients with de novo MCL from a population-based cohort, with the intent to prove the utility of a targeted approach and sequencing technologies in tailoring treatment for MCL patients.
Material and methods
Patient cohort
The study encompasses 77 samples from patients diagnosed with MCL belonging to the BLISS (Biobank of Lymphomas in Southern Sweden). BLISS is a population-based cohort of FFPE lymphoma samples collected at the South Swedish Health region between 2000 and 2014. All cases were reviewed by a haematopathologist and classified as MCL. FFPE tumor tissue was available from a total of 191 patient biopsy samples collected at diagnosis from different sites, including lymph nodes, bone marrow, and the gastrointestinal tract (Figure S1). Information on morphological subtype of MCL [Citation1], Ki67 positivity, and p53 protein expression (Supplemental Material and Methods) were available for most patients included in the study. The study was approved by the Ethical Regional Committee in Lund (Dnr 2011/593).
DNA extraction, quality control, sequencing and analysis
DNA of FFPE samples was extracted with High Pure FFPET DNA isolation kit (Roche, Switzerland) according to the manufacturer instructions. DNA quality was assessed by quantitative PCR and samples with less than six delta quantification cycles (ΔCt) were sequenced.
Target sequencing data generation and sequencing pipeline
A customized lymphoma cancer panel (TWIST Bioscience, California, USA) was used to sequence 77 MCL samples. The panel covers the exonic parts of ∼200 genes with capture probes thought to be relevant in lymphomagenesis or other genes with known clinical significance (Table S1). Sequencing libraries were prepared from a maximum of 100 ng of DNA and sequenced on a NovaSeq (Illumina, California, USA). Burrow Wheels Alignment (BWA)-mem algorithm was used to align against the human genome version 37 to obtain sequence alignment. Variant calling was done using Sentieon TNscope (Sentieon Inc., California, USA) with the following parameters modified: –sv_mask_ext 10 –max_fisher_pv_active 0.05 –min_tumor_allele_frac 0.05 –assemble_mode 4. Additionally, dbSNP and cosmic variants were provided to TNscope to increase/reduce the germline risk factor of the variants found. After variant annotation with Ensemble Variant Effect Predictor (v. 93), the variants retained fulfilled the criteria: – FILTER = PASS, QUAL > 100, Allele frequency > 10%, ExAC (Exome Aggregation Consortium) population frequency < .1%.
Mutational data analysis
All mutations were manually validated with the Integrative Genomic Viewer (IGV) software [Citation14]. Only genes with non-synonymous mutations were considered in the study. To explore the landscape of mutations, Maftools package in R (v. 4.0.2 for Mac) was used. Fisher’s Exact test incorporated in the Maftools package was used to detect significant associations among pairs of genes and comparisons between cohorts to identify differentially mutated genes.
To determine pathways and cell functions of the mutated genes we used Reactome Webtool. The Cancer Genome Interpreter (CGI) webtool was used to help identify driver tumor alterations and detect the ones that can be therapeutically actionable [Citation15]. The Cancer Genome Interpreter webtool [Citation15] provides relevant information for each mutation, such as (i) gene mode of action; (ii) consequence of the mutation; (iii) cluster or hotspot mutation information; (iv) predicted functional impact; (vi) frequency among the human population. It takes into consideration mutation-centric measurements to stratify mutations in well-known oncogenic mutations, predicted cancer drivers, predicted passenger mutations, mutations that are likely to not have an impact in cancer development or polymorphisms. Information on genomic biomarkers of drug response was also available in the webtool.
Information about mutations classified as being actionable is available at the OncoKB website [Citation16]. OncoKB website is a platform to identify established actionable mutations and sub-divide them into four categories, according to their evidence-based level. Level 1 corresponds to alterations that are FDA-approved biomarkers for FDA-approved drugs in lymphoma. Level 2 includes standard care alterations recommended by expert panels to an FDA-approved drug in lymphoma. Level 3 alterations either have compelling clinical evidence that are drug response predictors in lymphoma or are standard care/investigational biomarkers on FDA-approved or investigational drugs in other disease. Level 4 alterations have compelling biological evidence to support them as being predictors of drug response.
Significance was considered when p-value < 0.05. A detailed description is found in Supplementary Material and Methods.
Results
Study design and clinical information
The population-based BLISS cohort included biopsies from 191 patients diagnosed with MCL. DNA extraction was performed for all 191 samples. From these, 28 samples (15%) did not yield sufficient DNA for library preparation, whereas DNA quality was considered too low for the remaining 86 samples (45%). In total, 77 samples were further processed for sequencing.
60 patients were male (78%). Average age at diagnosis was 70 years (range, 45 to 94 years). Table S2 shows the clinico-pathological characteristics of the original population-based cohort and the patients sequenced. No differences between the groups of patients was seen. Unfortunately, in a proportion of cases (39/77), information on the MCL international prognostic index (MIPI) was not available. For those where it could be calculated, 45% had high-risk MIPI. Ki-67 expression >30% was found in 14% of the cases and 11% had non-classical morphology. Eight patients (12%) had >30% strong nuclear p53 expression.
Patients were treated according to different protocols, the most frequently being R-Bendamustine (n = 21). The median overall survival time was 5.5 years.
FFPE DNA quality control for clinical setting application and sequencing performance
DNA was extracted from the same tissue used for diagnosis, which varied in tissue distribution for different patients. There is a difference in the distribution of sites where DNA quality was enough for sequencing (χ2 test, p-value = 0.03). There was a higher proportion of lymph node biopsies with high DNA quality and a lower proportion of biopsies taken from the bone marrow and the digestive system that passed DNA quality control (Table S3).
To determine the performance of the sequencing pipeline, we calculated the number of false positive mutations identified prior and after validation. Prior to validation, an average of 18 mutations per patient was detected (range, 0–210). After validation using IGV, an average of 5 mutations per patient remained (range, 0–11) (Figure S2(a)). Correlation analysis between the fraction of false positives identified and the ΔCt from the quality control analysis showed that low ΔCt, for a number of patients, could explain the high level of false positives in the samples prior to IGV validation (p-value < 0.001) (Figure S2(b)).
Samples had a median of 36 M reads (Inter Quartile Range 18–51) and a median depth of 361X across all samples. Most samples had the panel covered with at least 250X (Table S4).
Landscape of mutations in MCL accurately detected from FFPE samples
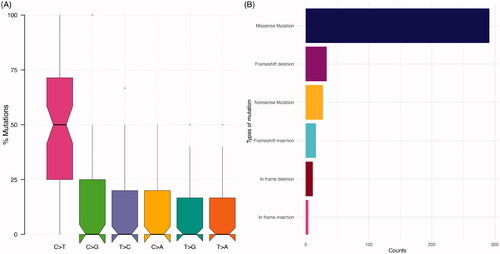
In total, 382 non-silent variants in 131 mutated genes were identified (). From the 77 patients analyzed, 75 (97%) presented with at least one variant. As expected [Citation9,Citation17], most of the variants were missense mutations (76%). Frameshift alterations corresponded to 13%, in-frame variations to 4%, and non-sense mutations to 7% of the 382 variants. Transitions were more common than transversions (). There was a predominance of C>T transitions.
Figure 1. Overview of genomic alterations identified in the cohort. (A) Distribution of variant classification in the cohort. (B) Distribution of the single-nucleotide variants (SNVs) in the cohort.
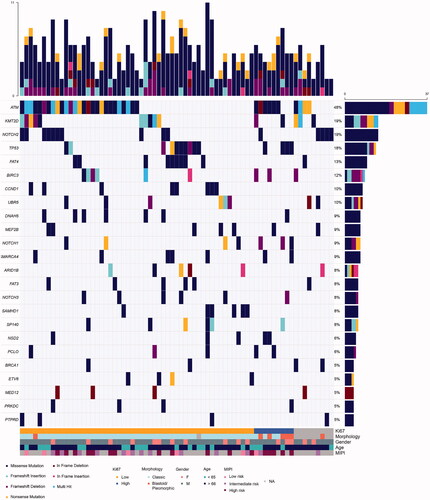
ATM was the most commonly mutated gene (48%), which is in line with previous reports () [Citation5,Citation6,Citation13]. It was followed by KMT2D (19%), NOTCH2 (19%), TP53 (18%) and FAT4 (13%). BIRC3 (12%), UBR5 (10%). Mutations in genes commonly studied in MCL, including CCND1 (Figure S3) and RB1 were less frequent than expected, with a mutation rate of 10% and 1.3%, respectively. Interestingly, ATM and TP53 mutations were mutually exclusive (p-value = 0.038), with only three patients harboring mutations in both genes simultaneously. The NOTCH family genes were mutated in 33% of the patients. NOTCH2, NOTCH1 and NOTCH3 mutated in 19%, 9% and 8%, respectively (Figure S4). Genes that code for proteins involved in the SWI-SNF chromatin remodeling complex were mutated in 20% of the tumors (Figure S5).
Figure 2. Landscape of mutations in the BLISS cohort. Oncoplot of the most commonly altered genes in the cohort. The upper bar plot represents counts of alterations per sample. The right panel bar plot refers to the total number of samples with alterations in that specific gene. Clinicopathological information is added on the lower region of the oncoplot.
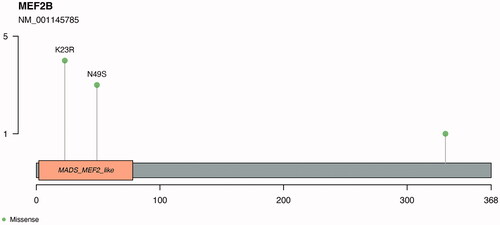
We found two recurrent mutations in the gene MEF2B, with four patients carrying a K23R alteration and three patients with an N49S mutation. According to SIFT and Polyphen analysis, both mutations are predicted to be deleterious/damaging ().
Figure 3. Recurrent mutations identified in the MEF2B gene. Identified in the image are the recurrent mutations K23R and N49S. The myocyte enhancer factor-2 binding domain is represented in orange in the plot.
We compared the mutation patterns among the different risk groups used in the clinical setting (Figure S6). In most of the groups, too few patients were included in the high-risk groups (non-classical morphology or Ki-67 expression >30%) to reach statistical significance. However, we observed that the UBR5 gene mutation was enriched among elderly MCL patients (age at diagnosis >66 years, p-value = 0.05). Mutations in FAT4, CCND1, SAMHD1, UBR5, and FAT3 were identified in patients with classic morphology and low Ki-67 expression only. Conversely, all patients with highly proliferative tumors had mutations either in ATM (70%) or in TP53 (40%) genes. Out of the seven patients classified with blastoid/pleomorphic variants, six harbored mutations in either ATM or TP53 genes (three each). Overall, the morphological groups were heterogeneous in the spectrum of mutated genes, but the majority of the mutations appeared in only a small fraction of patients.
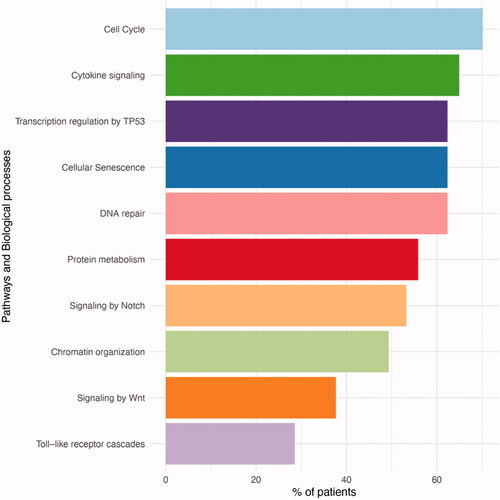
The genetic alterations commonly found in our cohort could be grouped into different categories, namely cell cycle, cytokine signaling and DNA repair (). Most patients (70%) had at least one mutation that affected cell cycle. Among the common oncogenic pathways, TP53 transcription pathway was highly represented, with 62% of patients having a mutation in a gene involved in the pathway, either ATM or TP53 genes.
Figure 4. Pathways and biological processes that were affected by mutations. The most commonly mutated pathways included cell cycle and TP53.
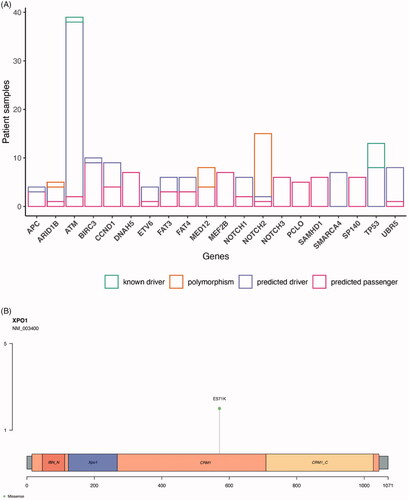
Our study indicates that there is a low number of mutated genes in each patient. Additionally, there is considerable variability in which genes are affected in each individual patient (Table S5). These circumstances made it less feasible to identify driver genes in our MCL cohort, although drivers appeared mutated at a similar rate as reported previously [Citation5,Citation6,Citation18]. However, when resorting to the CGI, we identified 64/75 patients (84%) that had mutations in either one established driver gene variant in cancer or a predicted driver mutation by OncodriveMUT (Table S6), which correspond to 138/382 identified variants. As expected, most mutations were predicted to be passenger mutations. The most commonly mutated gene that harbored variants with driver potential was the ATM gene (). TP53 variants were all considered disrupting or deleterious by the algorithms (except for the frameshift insertion that has not been previously reported, detailed information is published [Citation19]) and classified as predicted drivers. SMARCA4 gene variants and UBR5 mutations (6/7) were also among the predicted drivers in the cohort. One mutation classified as a predicted driver was the E571K mutation in the exportin 1 (XPO1) gene. This mutation is a known hotspot mutation and was identified in two patients ().
Figure 5. Prediction of driver genes in MCL. (A) Prediction of driver properties in recurrently altered genes. Known drivers refer to genes that have been functionally proven to drive cancer cells. Predicted drivers are genes that are hypothesized to drive cancer. Predicted passengers are mutations in genes without known driver role in carcinogenesis. Information gathered from the CGI analysis. Genes with alterations in at least four patients are shown grouped by their predicted driver properties. (B) E571K XPO1 mutations localization. IBN_N: Improtin-beta N-terminal domain; XPO1: exportin 1-like protein; CRM1: Chromosomal regional maintenance 1; CRM1_C: CRM1 C-terminal.
Targeted approach identifies mutations with potential clinical-decision impact
To assess the clinical impact of the identified mutations, we categorized them as potentially actionable according to the OncoKB classification (Table S7). A total of 34 variants were identified in 24 genes. The gene with more variants was ATM (five variants classified as targetable), followed by CCDC15, DNAH5, KMT2D, NOTCH1 and SF3B1, each with two targetable variants. 21% of the patients had at least one potentially actionable mutation and 10% had two or more actionable mutations ().
Figure 6. Target approach identifies mutations with potential impact on clinical decision making. (A) Number of actionable mutations in the cohort per patient sample addressed today by OncoKB platform, which provides information according to established guidelines. (B) Genes with druggable mutations and the number of drugs (y axis) for which they predict response according to literature. (C) Number of druggable mutations in the cohort per patient sample according to CGI database, that includes future addressable mutations according to scientific publication of both clinical and pre-clinical studies.
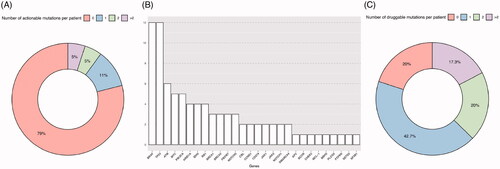
Although the number of mutations classified as potentially actionable was low in our cohort, many of the mutations have previously been associated to response to cancer therapy (, Table S8). A total of 60/75 analyzed samples with identified non-synonymous variants, had at least one mutation that has been proven, either in clinical trials or pre-clinical research, to predict response to a specific therapy ().
Discussion
The genetic landscape of MCL has been investigated in previous studies, although the total number of sequenced samples remains low. To our knowledge, previous larger targeting panel/global sequencing studies (studies with more than ∼50 studied genes) [Citation4–6,Citation17,Citation18,Citation20–22] include only approximately 300 de novo MCL patients in total. Thus, our data on 77 patients is a significant addition to the general knowledge base of the frequency of genetic alterations in MCL.
Most previous studies analyzed only a small set of genes, focusing on frequently mutated genes in MCL. In this study we adopt a more broad approach and sequence more than 200 genes, to enable identification of less frequent alterations. The top ten most frequent alterations in MCL include mutations in ATM, CCND1, TP53, KMT2D, KMT2C, NOTCH1, NSD2, BIRC3, NOTCH2, and UBR5 [Citation4–6,Citation23]. Among these alterations, seven were also identified in the present study as frequently mutated genes. Two genes were mutated at a lower frequency than expected and one, KMT2C, failed to be analyzed due to low coverage.
In this study, a population-based material of diagnostic biopsies was used. Thus, it is of interest to compare mutational rate to other studies with more selected cohorts of patients, either based on type of tissue or risk factors, such as histology and/or proliferation. ATM was the most commonly mutated gene, with 48% of the tumors harboring at least one mutation in this gene. The ATM gene has been reported as the most recurrently mutated gene in MCL (43.5%), with an increase of mutation rate at disease relapse/progression [Citation13]. The TP53 gene was mutated at a similar rate to what has been reported (18%). KMT2D was altered in 19% and has been suggested as an early event in the development of MCL [Citation6], with some controversy in relation to its impact on clinical outcome [Citation23,Citation24]. The frequency of patients with mutated CCND1 was lower than previously reported. Studies that reported higher number of CCND1 mutations analyzed a large proportion of peripherial blood samples and had a much higher number of cases classified as high-risk MIPI than our study (60–91% vs. 45%, respectively) [Citation6,Citation18]. We also found that NSD2 was mutated at a lower frequently than expected [Citation13].
NOTCH genes are frequently mutated in MCL and in our study, with the three genes showing alterations in 32.5% patients, most mutually exclusive. This highlights the relevance of the Notch signaling pathway in MCL pathogenesis [Citation11]. We report a higher frequency of NOTCH2 mutations compared to previous studies (6%) [Citation4,Citation13,Citation17,Citation18,Citation25], which is due to the presence of the SNP rs2603926 in 13 patients. The identified frequency of alterations in KMT2D, BIRC3 and UBR5 were similar to previous studies [Citation4–6,Citation11,Citation18].
In addition to recurrent mutations previously reported, we identified mutations that have not been commonly described in MCL lymphomagenesis. FAT4 was mutated in 13% of studied patients. This gene has, to our best knowledge, only been reported in one previous study of MCL [Citation5]. FAT4 is considered a tumor suppressor gene and it is commonly mutated in cancer [Citation26]. MEF2B was also commonly mutated in our cohort (9%). MEF2B encodes the MEF2B protein, which is proposed to be involved in the formation of germinal centers and a driver oncogene in B cell lymphomagenesis [Citation27]. Recently, Pararajalingam et al. [Citation18] reported that the pattern of MEF2B mutation hotspots in MCL is characteristic of this disease, including K23R and N49. Our results corroborate this finding. Bèa et al. [Citation6] also reported these specific mutations in MCL, albeit at lower frequency. The impact of this mutation is currently unknown, although it is suggested that it may limit the DNA-binding ability of the MEF2B protein, affecting the expression of down-stream transcriptional targets [Citation28].
Currently, the main prognostic factors for MCL patients include age, MIPI, histology, TP53 aberrations and tumor proliferation. When comparing cases with blastoid/pleomorphic vs. classic morphology and high vs. low proliferation rate measured by Ki-67 expression, we observed that TP53 and ATM were the most frequently altered genes in aggressive morphologic variants, being mutually exclusive, which has been shown previously [Citation20]. FAT4, CCND1, SAMHD1, UBR,5 and FAT3 genes were mutated only in non-aggressive cases, i.e. low Ki-67 and classic morphology. These results are in contrast to a previous study, where 39 MCL aggressive tumors were sequenced [Citation21], and UBR5 and CCND1 mutations were characteristic of aggressive histology cases. Our population-based cohort only contains seven cases classified as aggressive morphologic variants, which limited the possibility to perform sub-type analysis.
Only 21% of the patient samples analyzed harbored at least one mutation with potential to be actionable according to OncoKB [Citation16], which includes information curated from multiple unstructured resources, such as FDA recommendations and NCCN guidelines. Since new information is continuously being published and evaluated, we expanded the search with the CGI webtool, that also englobes curated information from pre-clinical studies [Citation15]. With this less strict analysis, most patients (60/77) had a mutation that had been associated with some type of treatment response in cancer. Lymphoid tumors with ATM alterations have been hypothesized to be efficiently treated with PARP inhibitors [Citation29], and as ATM is one of the most frequency altered genes this may constitute an attractive option.
To our knowledge, mutations in the XPO1 gene have not been shown previously in MCL. The affected specific mutation, E571K, was classified as a predicted driver in two patients with aggressive morphology and very short overall survival. XPO1 mediates the translocation from the nucleus of RNAs and proteins, namely tumor suppressor proteins [Citation30] and is highly expressed in MCL, where it is believed to be associated with disease pathogenesis and poor prognosis [Citation31]. Analysis suggests that the mutation is deleterious, and was reported to promote malignant B-cell growth and lymphomagenesis driven by BCL-2 and c-myc [Citation32]. This mutation should be exploited therapeutically in MCL, as XPO1 inhibition has shown promising results in other B-cell malignancies [Citation30,Citation32].
Also in the relapse or refractory setting, information on genetic alterations may help to identify targeted treatment options. A recent example showed that genes that coded for proteins of the SWI-SNF chromatic-remodeling complex confered resistance to treatment with venetoclax and ibrutinib in MCL [Citation33]. Here, 20% of the studied patients had one mutation in one gene involved in this complex. Additionally, the mutations in CCND1 have been shown to deregulate protein turnover and to be involved in ibrutinib resistance [Citation34]. It can be hypothesized that patients with SMARCA4 and CCND1 will not benefit from treatment with ibrutinib.
To our knowledge, this is one of the largest population-based studies using FFPE sample tissues in MCL. Jain et al. [Citation21] performed whole exome sequencing analysis in 81 samples, including ∼50% non-classic morphology cases and thus less clinically representative, as non-classic morphology tumors were rare [Citation35]. A large portion of samples could not be sequenced due to poor DNA quality but the selection did not alter the clinicopathological characteristics of the sequenced samples compared to the full population-based cohort. DNA QC analysis showed that samples with ΔCT value <6 robustly can be assessed. Most of the MCL samples that passed quality control derived from lymph nodes, and a likely explanation for lower representation from other tissue origins such as bone marrow is that they in general have fewer tumor cells and that the decalcification process hampers DNA quality. Our cohort comprises a high number of samples that have been processed more than 10 years prior to the study. The laboratory procedure might historically have varied between hospitals, and we hypothezise that fresh FFPE samples from standardized clinical routine would increase the success rate of DNA preparation in a prospective or clinical routine setting. As information about treatment was not availble for all patients, it limited the possibility to study the impact of mutations on outcome. Furthermore, a technical limitation of the pipeline was the lack of an algorithm for PCR duplicate exclusion, which resulted in a significant number of false positive mutations being identified that later were removed during IGV validation.
In summary, we show the utility of using FFPE samples in our comprehensive genomic study of a population-based cohort comprised of 77 MCL tumors, which pin-points the heterogeneity of this malignancy. We could identify genes that have been previously reported as commonly mutated in MCL and new rare alterations, which illustrates the possibilities for clinical decision-making based on specific mutations.
Author contributions
JMR was involved in the planning of the study, performed data and statistical analysis and wrote the manuscript. AP performed the pathology review of the BLISS cohort. MH performed experimental part and was involved in statistical analysis. SE was responsible for the planning of the study, analysis of the data and drafting the manuscript. MJ was the main responsible for the collection of the material and initiation of the study and was involved in the drafting of the manuscript. All authors approved the final version of the manuscript.
GLAL-2021-0298-File009.xlsx
Download MS Excel (49.8 KB)GLAL-2021-0298-File008.pdf
Download PDF (659.7 KB)Acknowledgments
We thank Center for Translational Genomics, Lund University and Clinical Genomics Lund, SciLifeLab for providing sequencing service; and Kristina Lövgren, Division of Oncology for handling tissue samples.
Disclosure statement
Mats Jerkeman received research support from Gilead, Abbvie, Roche, Celgene, Janssen. The other authors have no conflict of interests to disclose.
Additional information
Funding
References
- Swerdlow SC, Harris NL, Jaffe ES, et al. WHO classification of tumours of haematopoietic and lymphoid tissues. Revised 4th ed. Vol. 2. Lyon: IARC Publications; 2017.
- Klener P. Advances in molecular biology and targeted therapy of mantle cell lymphoma. Int J Mol Sci. 2019;20:18.
- Ahmed M, Zhang L, Nomie K, et al. Gene mutations and actionable genetic lesions in mantle cell lymphoma. Oncotarget. 2016;7(36):58638–58648.
- Yang P, Zhang W, Wang J, et al. Genomic landscape and prognostic analysis of mantle cell lymphoma. Cancer Gene Ther. 2018;25(5–6):129–140.
- Zhang J, Jima D, Moffitt AB, et al. The genomic landscape of mantle cell lymphoma is related to the epigenetically determined chromatin state of normal B cells. Blood. 2014;123(19):2988–2996.
- Bea S, Valdes-Mas R, Navarro A, et al. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci USA. 2013;110(45):18250–18255.
- Wiestner A, Tehrani M, Chiorazzi M, et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood. 2007;109(11):4599–4606.
- Rahal R, Frick M, Romero R, et al. Pharmacological and genomic profiling identifies NF-κB-targeted treatment strategies for mantle cell lymphoma. Nat Med. 2014;20(1):87–92.
- Meissner B, Kridel R, Lim RS, et al. The E3 ubiquitin ligase UBR5 is recurrently mutated in mantle cell lymphoma. Blood. 2013;121(16):3161–3164.
- Greiner TC, Dasgupta C, Ho VV, et al. Mutation and genomic deletion status of ataxia telangiectasia mutated (ATM) and p53 confer specific gene expression profiles in mantle cell lymphoma. Proc Natl Acad Sci USA. 2006;103(7):2352–2357.
- Kridel R, Meissner B, Rogic S, et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood. 2012;119(9):1963–1971.
- Ahmed M, Lorence E, Wang J, et al. Interrogating B cell signaling pathways: a quest for novel therapies for mantle cell lymphoma. Sci Signal. 2019;12:567.
- Hill HA, Qi X, Jain P, et al. Genetic mutations and features of mantle cell lymphoma: a systematic review and meta-analysis. Blood Adv. 2020;4(13):2927–2938.
- Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26.
- Tamborero D, Rubio-Perez C, Deu-Pons J, et al. Cancer genome interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018;10(1):25.
- Chakravarty D, Gao J, Phillips SM, et al. OncoKB: a precision oncology knowledge base. J Clin Oncol Precis Oncol. 2017;(1):1–16.
- Nadeu F, Martin-Garcia D, Clot G, et al. Genomic and epigenomic insights into the origin, pathogenesis, and clinical behavior of mantle cell lymphoma subtypes. Blood. 2020;136(12):1419–1432.
- Pararajalingam P, Coyle KM, Arthur SE, et al. Coding and noncoding drivers of mantle cell lymphoma identified through exome and genome sequencing. Blood. 2020;136(5):572–584.
- Rodrigues JM, Hassan M, Freiburghaus C, et al. p53 is associated with high-risk and pinpoints TP53 missense mutations in mantle cell lymphoma. Br J Haematol. 2020;191:796–805.
- Streich L, Sukhanova M, Lu X, et al. Aggressive morphologic variants of mantle cell lymphoma characterized with high genomic instability showing frequent chromothripsis, CDKN2A/B loss, and TP53 mutations: a multi-institutional study. Genes Chromosomes Cancer. 2020;59(8):484–494.
- Jain P, Zhang S, Kanagal-Shamanna R, et al. Genomic profiles and clinical outcomes of de novo blastoid/pleomorphic MCL are distinct from those of transformed MCL. Blood Adv. 2020;4(6):1038–1050.
- Wu C, de Miranda NF, Chen L, et al. Genetic heterogeneity in primary and relapsed mantle cell lymphomas: impact of recurrent CARD11 mutations. Oncotarget. 2016;7(25):38180–38190.
- Ferrero S, Rossi D, Rinaldi A, et al. KMT2D mutations and TP53 disruptions are poor prognostic biomarkers in mantle cell lymphoma receiving high-dose therapy: a FIL study. Haematologica. 2020;105(6):1604–1612.
- Eskelund CW, Dahl C, Hansen JW, et al. TP53 mutations identify younger mantle cell lymphoma patients who do not benefit from intensive chemoimmunotherapy. Blood. 2017;130(17):1903–1910.
- Sarkozy C, Ribrag V. Novel agents for mantle cell lymphoma: molecular rational and clinical data. Expert Opin Investig Drugs. 2020;29(6):555–566.
- Katoh M. Function and cancer genomics of FAT family genes (review). Int J Oncol. 2012;41(6):1913–1918.
- Brescia P, Schneider C, Holmes AB, et al. MEF2B instructs germinal center development and acts as an oncogene in B cell lymphomagenesis. Cancer Cell. 2018;34(3):453–465 e9.
- Dantas Machado AC, Cooper BH, Lei X, et al. Landscape of DNA binding signatures of myocyte enhancer factor-2B reveals a unique interplay of base and shape readout. Nucleic Acids Res. 2020;48(15):8529–8544.
- Kummar S, Ji J, Morgan R, et al. A phase I study of veliparib in combination with metronomic cyclophosphamide in adults with refractory solid tumors and lymphomas. Clin Cancer Res. 2012;18(6):1726–1734.
- Camus V, Miloudi H, Taly A, et al. XPO1 in B cell hematological malignancies: from recurrent somatic mutations to targeted therapy. J Hematol Oncol. 2017;10(1):47.
- Yoshimura M, Ishizawa J, Ruvolo V, et al. Induction of p53-mediated transcription and apoptosis by exportin-1 (XPO1) inhibition in mantle cell lymphoma. Cancer Sci. 2014;105(7):795–801.
- Taylor J, Sendino M, Gorelick AN, et al. Altered nuclear export signal recognition as a driver of oncogenesis. Cancer Discov. 2019;9(10):1452–1467.
- Agarwal R, Chan YC, Tam CS, et al. Dynamic molecular monitoring reveals that SWI-SNF mutations mediate resistance to ibrutinib plus venetoclax in mantle cell lymphoma. Nat Med. 2019;25(1):119–129.
- Mohanty A, Sandoval N, Das M, et al. CCND1 mutations increase protein stability and promote ibrutinib resistance in mantle cell lymphoma. Oncotarget. 2016;7(45):73558–73572.
- Jain P, Wang M. Blastoid Mantle Cell Lymphoma. Hematol Oncol Clin North Am. 2020;34(5):941–956.