
Abstract
Bisantrene (Bis), a topoisomerase-II inhibitor, is less cardiotoxic than the current anthracyclines. Its synergistic cytotoxicity with newly developed antineoplastic drugs has not been reported. We demonstrated the synergism of [Bis + ABT199/venetoclax] in combination with panobinostat (Pano), decitabine (DAC), or olaparib (Ola), known inhibitors of BCL2, histone deacetylase, DNA methyltransferase, and poly(ADP-ribose) polymerase, respectively, in AML cells. [Bis + ABT199] with Pano, DAC, or Ola synergistically inhibited cell proliferation with combination indices of 0.25–0.6, 0.2–0.35, and 0.2–0.4 (at 50% inhibition of proliferation), respectively. Increased γ-H2AX suggests enhanced DNA damage; cleavages of Caspase 3 and PARP1, DNA fragmentation, increased ROS, and decreased MMP indicate potent apoptosis activation. Similar results were observed using mononuclear cells from leukemia patients but not from healthy donors. The SAPK/JNK signaling pathway was strongly activated by the combination treatments, whereas the PI3K/mTOR and Wnt/β-catenin pro-survival pathways were inhibited. These drug combinations may be used in cytoreductive clinical trials for AML patients.
Introduction
Anthracycline drugs have been used to treat acute myeloid leukemia (AML) for the past 40 years, but despite being of proven efficacy their associated cardiotoxicity limits their utility. The enhanced production of reactive oxygen species (ROS) is a major contributor to anthracycline cardiotoxicity [Citation1]. Bisantrene is an anthracene derivative, which is much less cardiotoxic than the commonly used anthracyclines, including daunorubicin and doxorubicin [Citation2]. The antineoplastic activity of these agents, including bisantrene, is primarily attributed to their inhibition of topoisomerase II-mediated relaxation of the DNA supercoil torsion, which prevents DNA replication and cancer cell proliferation [Citation3]. Bisantrene inhibits the fat mass and obesity-associated protein (FTO), an RNA N6-methyladenosine (m6A) demethylase, and suppresses self-renewal of leukemia stem/initiating cells, further emphasizing its pharmacological and clinical value in therapy for AML as well as other cancers [Citation4]. Prior clinical studies on the efficacy of bisantrene as a salvage drug led to its approval for the treatment of AML in France in 1990 [Citation5,Citation6]. In a recent phase II study, bisantrene showed low toxicity in relapsed/refractory AML patients with an overall response rate of 40% [Citation7], consistent with earlier reports.
Since treatment of AML requires combinatory drug therapy to address the complexity of its genetic and epigenetic abnormalities, we hypothesized that combinations of bisantrene with established and newly developed agents may provide safer and more effective treatment options for patients with AML. We recently reported the synergistic cytotoxicity of bisantrene with ABT199/venetoclax in the presence of the nucleoside analogs cytarabine, cladribine, fludarabine and clofarabine in AML cell lines and patient-derived leukemic cells [Citation8]. The effects of bisantrene and ABT199/venetoclax when combined with panobinostat, decitabine and olaparib, drugs with different mechanisms of action, have not been explored. ABT199/venetoclax, a known inhibitor of the anti-apoptotic BCL2 protein, is effective as a single agent for the treatment of AML [Citation9]. Its specific binding to BCL2 releases its proapoptotic partner proteins and triggers activation of intrinsic apoptosis [Citation10].
Panobinostat, on the other hand, is a nonselective inhibitor of histone deacetylase (HDAC) which alters gene expression through epigenetic modifications and inhibition of protein metabolism [Citation11]. Pre-clinical and clinical studies showed the efficacy of a panobinostat plus anthracycline combination in AML cells [Citation12,Citation13]. Decitabine is another epigenetic modifier which inhibits DNA methylation and induces DNA damage by inactivating and trapping DNA methyltransferase (DNMT) on DNA [Citation14]. Sequential combination of decitabine and the anthracycline idarubicin synergistically inhibited AML cell proliferation by downregulation of the Wnt/β-catenin pathway [Citation15]. Epigenetic priming with decitabine followed by low dose idarubicin/cytarabine resulted in 67% complete remission in high-risk patients with myeloid neoplasms [Citation16].
Olaparib inhibits poly(ADP-ribose) polymerase (PARP), a critical enzyme in the repair of DNA damage, resulting in an increased level of DNA strand breaks, which activates the DNA-damage response and cell death [Citation17]. The ability of olaparib to trap PARP at damaged DNA sites is considered even more cytotoxic than unrepaired DNA damage caused by PARP inactivation [Citation18]. When combined with daunorubicin, olaparib showed an additive effect on the killing of IDH1/2MUT AML cells [Citation19], whereas strong synergistic cytotoxicity of these two drugs was observed in colorectal cancer cells [Citation20].
To evaluate the possibility of using bisantrene and ABT199/venetoclax with panobinostat, decitabine or olaparib as a salvage regimen for AML patients, we determined the synergistic cytotoxicity and mechanisms of action of these drug combinations in AML cell lines and patient-derived cell samples. The drug combinations were effective in inducing tumor cell death, which may be attributed to combined effects on the DNA damage response, apoptosis, SAPK/JNK, PI3K/mTOR and Wnt/β-catenin pathways.
Materials and methods
Cell lines and drugs
The AML cell lines OCI-AML3, MOLM13 and MV4-11 were obtained from Dr. Michael Andreeff (UTMDACC). Cells were cultured in RPMI 1640 (Mediatech) with 10% heat-inactivated fetal bovine serum (FBS: Gemini Bio Products), 100 U/ml penicillin and 100 μg/ml streptomycin (Mediatech) at 37 °C in a humidified atmosphere of 5% CO2. Bisantrene dihydrochloride was provided by Race Oncology Ltd. Other drugs were obtained from SelleckChem. Stock solutions of bisantrene were dissolved in water and the other drugs were prepared in DMSO.
Patient and healthy donor-derived cell samples
Peripheral blood samples from leukemia patients were collected with written informed consent. Mononuclear cells were purified using lymphocyte separation medium (Mediatech) and cryopreserved in liquid nitrogen. Mononuclear cells from healthy donors were purchased from ALLCELLS LLC (Almeda, CA, USA). Enriched normal CD34+ progenitor cells were not used in the study due to possible changes in their phenotype upon exposure to growth factors during stimulation and cell purification. Prior to drug exposure, frozen cells were thawed at 37 °C, washed with culture medium, and incubated overnight in RPMI 1640 medium with 10% FBS, 100 IU/mL penicillin, and 100 µg/mL streptomycin. Cells were exposed to drugs and analyzed after 48 h of incubation at 37 °C. Use of patient-derived cells was performed according to a protocol approved by the Institutional Review Board of the UTMDACC, in accordance with the Declaration of Helsinki.
Cytotoxicity assay
Cell suspensions (6 ml of 0.5 × 106 cells/ml) in T25 flasks were exposed to drugs or solvent alone for 48 h, aliquoted (100 μl) in triplicate into 96-well plates, and added 10 µl of the CCK8 (Cell counting kit 8) assay reagent (APExBIO) and incubating at 37 °C for 2 h. Cell viability was determined by measuring optical density at 450 nm using a Victor X3 (Perkin Elmer Life and Analytical Sciences) plate reader. Proliferation was determined relative to the control cells exposed to solvent alone. Graphical analyses, including calculations of IC10–IC20 values (the concentration of drug required for 10–20% growth inhibition), were done using Prism 5 (GraphPad Software). Drug combination effects were calculated based on the combination index (CI) values using the CalcuSyn software (Biosoft). This program was developed based on the median-effect method: CI < 1 indicates synergy, CI∼1 indicates additivity, and CI < 1 suggests antagonism.
Apoptosis assay
Cells were analyzed for apoptosis by flow-cytometric measurements of phosphatidylserine externalization with Annexin-V-FLUOS (Roche Diagnostics) and a fluorescent DNA-binding marker 7-aminoactinomycin D (BD Biosciences) using a Muse Cell Analyzer (EMD Millipore). The extent of cleavage of PARP-1 and Caspase 3, determined by western blotting, was also used as an indicator of apoptosis.
Western blot analysis
Cells were collected by centrifugation, washed with ice-cold phosphate-buffered saline (PBS) and lysed with cell lysis buffer (Cell Signaling Technology). Total protein concentrations were determined using a BCA Protein Assay kit (ThermoFisher Scientific). The protein extracts were combined with loading buffer, boiled for 5 min, and aliquots of equal amounts of protein were resolved on polyacrylamide-SDS gels and blotted onto nitrocellulose membranes (Bio-Rad). Western blot analyses were done by chemiluminescence using the Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore). The antibodies, their sources and other relevant information were previously described [Citation21,Citation22].
Analysis of reactive oxygen species (ROS)
Cells were analyzed for production of ROS using CM-H2DCFDA (5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein diacetate, acetyl ester), an ROS indicator that diffuses into cells where it is oxidized to a fluorescent product (Life Technologies). Briefly, cells were aliquoted (0.4 ml) into tubes and CM-H2DCFDA (2 μl of 0.12 mM solution in DMSO) was added. Cells were incubated at 37 °C for 1 h and immediately analyzed with a Gallios Flow Cytometer (Beckman Coulter, Inc.) using excitation/emission wavelengths of 492/520 nm. Arithmetic means of the fluorescence intensities were compared and the relative fold increase in ROS production was calculated.
Analysis of mitochondrial membrane potential (MMP)
An MMP kit (Cayman Chemical Co.) was used to determine changes in the MMP using the JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) reagent. Cells to be analyzed were aliquoted (0.4 ml) into tubes. Diluted (1:10 with cell growth medium, 40 μl) MMP-sensitive fluorescent dye JC-1 was added to each tube, incubated at 37 °C for 20 min, and analyzed by flow cytometry (λex = 488 nm) using the 530 nm (FL-1 channel, green) and 585 nm (FL-2 channel, red) band-pass filters. Healthy cells with functional mitochondria and high MMP exhibit red fluorescence (aggregated JC-1), whereas cells with disrupted mitochondria and low MMP show green fluorescence (monomeric JC-1).
Caspase 3 assay
Cells were exposed to drugs with or without the Caspase 3 inhibitor Z-VAD-FMK, harvested and washed with ice-cold PBS. Total cell extracts were prepared using the Caspase-3 Colorimetric Activity Assay kit (Chemicon International). Total protein concentration was determined as described above. Equal amounts of protein were analyzed for Caspase 3 activity using the same kit.
Statistical analysis
Results are presented as the mean ± standard deviation of at least three independent experiments.
Results
The combinations of bisantrene and ABT199/venetoclax with panobinostat (pan-HDAC inhibitor), decitabine (hypomethylating agent), or olaparib (PARP inhibitor) in AML cell lines are synergistic
We first determined the cytotoxicity and potential synergism of bisantrene when combined with various drugs. shows the cytotoxicity of bisantrene and ABT199 when combined with panobinostat (Pano), decitabine (DAC) or olaparib (Ola) in the OCI-AML3, MOLM13 and MV4-11 cell lines. Exposure of the three cell lines to individual drugs resulted in ∼3.2 − 31.5% inhibition of proliferation. [Bisantrene + ABT199] exposure resulted in ∼37.3% (OCI-AML3), ∼43% (MOLM13), and ∼41.2% (MV4-11) inhibition of proliferation. The three-drug exposures in which [Bisantrene + ABT199] were combined with Pano, DAC or Ola inhibited the proliferation of OCI-AML3 cells by ∼67%, ∼67.2, and ∼58.3%, respectively; of MOLM13 cells by ∼55.1%, ∼65.3, and ∼69%; and of MV4-11 cells by ∼86.2%, ∼70.7%, and ∼77.6% (, upper panel). These findings are consistent with the increase in Annexin V-positive cells, an indication of apoptosis; exposure to individual drugs resulted in ∼11–18% positivity in OCI-AML3 cells, whereas the three-drug combinations increased Annexin V positivity to ∼62.7% [Bisantrene + ABT199 + Pano], ∼55.6% [Bisantrene + ABT199 + DAC], and ∼56.3% [Bisantrene + ABT199 + Ola]. Similar results were obtained for MOLM13 and MV4-11 cells ().
Figure 1. Cytotoxicity and synergism. AML cells were exposed to the indicated concentrations of bisantrene (Bis), venetoclax/ABT199 (ABT), panobinostat (Pano), decitabine (DAC) and olaparib (Ola), alone or in combination, for 48 h prior to determination of relative cell proliferation by the CCK8 assay (bar graph) and apoptosis by the Annexin V (line graph) assay. The results are averages of at least three independent experiments. To determine drug synergism, cells were exposed to various drug combinations at constant ratio concentrations for 48 h prior to proliferation assay. The relationships between the calculated combination indexes (CI, Y-axis) and fraction affected (Fa, X-axis) are shown below the bar graphs. CI <1.0 indicates synergism. The graphs are representatives of two independent experiments.
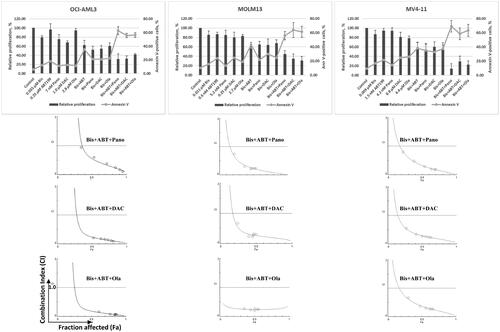
To quantitatively assess drug synergism, cells were exposed to different concentrations of individual drugs or to the three-drug combination at a constant concentration ratio and cell proliferation was analyzed after 48 h. Combination index (CI) values at increasing drug effects were graphically analyzed as shown in (below bar graphs). At 50% inhibition of cell proliferation, or 0.5 fraction affected (Fa), the calculated CI values for [Bisantrene + ABT199 + Pano] were ∼0.6 (OCI-AML3), ∼0.25 (MOLM13) and ∼0.3 (MV4-11); for [Bisantrene + ABT199 + DAC], the CI values were ∼0.2 (OCI-AML3), ∼0.35 (MOLM13), and ∼0.3 (MV4-11); and for [Bisantrene + ABT199 + Ola], the CI values were ∼0.2 (OCI-AML3), ∼0.25 (MOLM13), and ∼0.4 (MV4-11), indicating strong synergism (CI < 1) of the three-drug combinations in all three AML cell lines.
The three-drug combinations activate the apoptosis pathway
We initially assessed the biochemical activities of Pano, DAC and Ola by determining their effects on the acetylation of histone H3, DNMT1 protein levels, and poly(ADP)ribosylation of proteins by western blot analysis. At lower concentrations of Pano, there was no observed increase in the acetylation of histone H3; however, its combination with [Bisantrene + ABT199] markedly increased the level of AcH3K9 (). DAC is known to inhibit DNA methylation by triggering the degradation of DNMTs [Citation23]. Examination of DNMT1 showed a significant decrease in its protein level in cells exposed to [Bisantrene + ABT199 + DAC] (). Similarly, exposure of cells to [Bisantrene + ABT199 + Ola] significantly decreased the level of poly(ADP)ribosylated proteins, suggesting inhibition of PARP enzyme.
Figure 2. Determination of drug biochemical activities and activation of apoptosis. AML cells were exposed to the indicated concentrations of bisantrene (Bis or B), venetoclax/ABT199 (ABT or A), panobinostat (Pano or P), decitabine (DAC or D) and olaparib (Ola or O), alone or in combination, for 48 h. (A) Western blot analysis showing the effects of the histone deacetylase inhibitor Pano, the DNA methyl transferase inhibitor DAC, and the PARP inhibitor Ola on the level of AcH3K9, DNMT1 and PAR, respectively. (B) Changes in the level of modifications of protein markers of apoptosis. (C) Genomic DNA was isolated from drug-treated cells and resolved according to size using agarose gel electrophoresis in the presence of ethidium bromide. (D) Cells were exposed to drugs without or with 40 mM Caspase 3 inhibitor Z-VAD-FMK (Casp-i) for 48 h and analyzed for enzymatic activity and cleavage of Caspase 3 (Cleaved Casp 3).
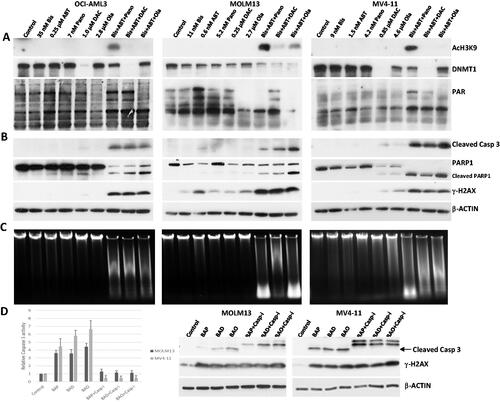
We next determined possible mechanisms of the enhanced cytotoxicity of the three-drug combinations. Each of the three-drug combinations increased the cleavage of Caspase 3 (), suggesting activation of the intrinsic apoptosis pathway. This cleavage of Caspase 3 was associated with the cleavage of PARP1, another molecular hallmark of apoptosis. The observed increase in the phosphorylation of histone 2AX (γ-H2AX) in cells exposed to the three-drug combinations () is typically indicative of drug-mediated DNA damage/chromatin remodeling as well as of apoptosis itself.
Activation of apoptosis is further indicated by increased genomic DNA fragmentation in cells exposed to the three-drug combinations as determined by agarose gel analysis (), suggesting activation of caspase-dependent DNase [Citation24]. To further determine the involvement of caspases in these effects, the enzymatic activity of Caspase 3 in cells exposed to the three-drug combinations was assayed in vitro. Caspase 3 activity increased 3.5–6.5-fold in drug-treated cells relative to the control cells; addition of the Caspase 3 inhibitor Z-VAD-FMK decreased Caspase 3 activity and prevented Caspase 3 cleavage without having a significant effect on the phosphorylation of histone H2AX (), indicating that the increased level of γ-H2AX is independent of Caspase 3-mediated DNA fragmentation. Collectively these results suggest activation of the intrinsic apoptosis pathway in AML cells exposed to the three-drug combinations.
The three-drug combinations have synergistic effects in patient-derived cells similar to their effects in cultured cells
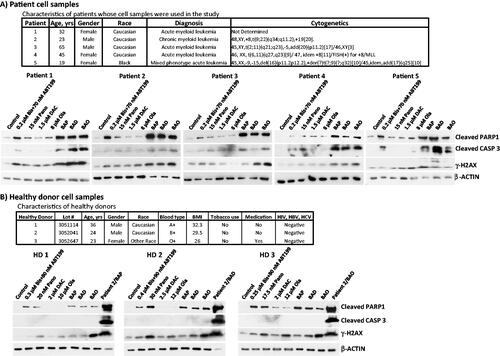
To assess the potential clinical implications of our observations, we isolated mononuclear cells (MNCs) from peripheral blood of patients with myeloid leukemia and exposed the cells to individual or combined drugs. (upper panel) shows the characteristics of the patients whose MNCs were used in this study. Exposure of the isolated MNCs to the three-drug combinations increased the cleavage of PARP1 and Caspase 3 (), consistent with what was observed in the three cultured cell lines (). In contrast, exposure of MNCs from healthy donors to higher concentrations of the drugs did not elicit similar responses (), suggesting greater sensitivity of MNCs from leukemia patients to the three-drug combinations. These observations suggest that these three-drug combinations are highly and selectively cytotoxic to patient-derived myeloid leukemia cells.
Figure 3. Drug effects on leukemia patient- and healthy donor-derived cells. Mononuclear cells were isolated from peripheral blood of (A) patients (characteristics shown in the upper panel) with hematological malignancies and (B) healthy donors, and exposed to the indicated drugs for 48 h prior to western blot analysis. Patient 2 cells exposed to triple drug combinations in panel B were used as positive control for western blot. Bis or B, bisantrene; ABT199 or A, venetoclax; Pano or P, panobinostat; DAC or D, decitabine; Ola or O, olaparib.
The three-drug combinations activate the production of ROS and decrease MMP
To better understand the cellular responses underlying drug-mediated cell death, we examined the production of ROS, elevated levels of which are known to mediate cell death. Exposure of the three cell lines to the three-drug combinations increased ROS production 2–3-fold (). These results suggest that the drug combinations have perturbed mitochondrial metabolism, resulting in enhanced generation of ROS.
Figure 4. Drug-mediated changes in the production of reactive oxygen species (ROS) and in mitochondrial membrane potential (MMP). AML cells were exposed to the indicated concentrations of bisantrene (Bis or B), venetoclax (ABT199 or A), panobinostat (Pano or P), decitabine (DAC or D) and olaparib (Ola or O), alone or in combination, for 48 h prior to flow cytometric analysis.
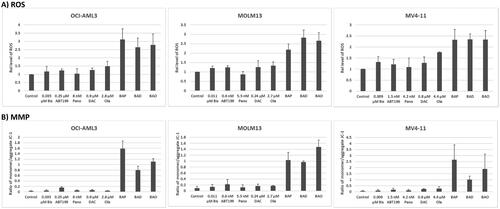
To substantiate the effects of the three-drug combinations on the integrity of the mitochondria, their impact on MMP was determined by flow cytometry using JC-1 reagent as a surrogate marker/tracer. A higher ratio of monomeric JC-1 to its aggregated form would indicate leakage of JC-1 from the mitochondria into the cytoplasm, suggesting a decreased MMP. As shown in , exposure of the three cell lines to [Bisantrene + ABT199 + Pano] resulted in monomer/aggregate ratios of ∼1.0–2.6 (compared with ∼0.03–0.09 in the control cells); exposure to [Bisantrene + ABT199 + DAC] similarly resulted in monomer/aggregate ratios of ∼0.8–1.0; and exposure to [Bisantrene + ABT199 + Ola] resulted in ratios of ∼1.1–1.9 (). These marked increases in comparison to the control cells are indicative of a major decrease in the cellular MMP.
The three-drug combinations activate the SAPK/JNK stress-signaling pathway
The observed production of ROS and perturbation of mitochondria () suggest a role for drug-mediated activation of stress pathways leading to apoptosis. We, therefore, sought to determine the effects of these drug combinations on the activation of the stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) signal transduction pathway, which is known to transmit and convert stress signaling into apoptosis signaling in various cell types [Citation25]. Increased phosphorylation of SAPK/JNK at threonine 183/tyrosine 185 was observed in MOLM13 and MV4-11 cells exposed to the three-drug combinations relative to the control or single drug-treated cells (). Analysis of one of the SAPK/JNK downstream target proteins, the ATF2 transcription factor, showed its increased phosphorylation in cells exposed to the three-drug combinations (). The increased level of P-SAPK/JNK (T183/Y185) might be due to activation by phosphorylation of the mitogen-activated protein kinase cascades [Citation26] as indicated by the observed SEK1/MKK4 phosphorylation at serine 257 (), which is upstream of SAPK/JNK in the stress-signaling pathway.
Figure 5. Effects of the three-drug combinations on the SAPK/JNK and PI3K signaling pathways in AML cells. Cells were exposed to drugs for 48 h and analyzed by western blotting. Drug abbreviations are as in .
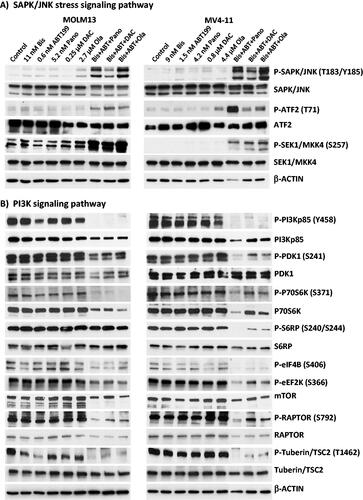
The three-drug combinations downregulate the PI3Kp85/P70S6K/mTOR signaling pathway
The phosphoinositide 3-kinase (PI3K) signaling pathway is important for cell survival and it is constitutively activated by phosphorylation in most cancer cells [Citation27]. We therefore sought to determine if the observed synergistic cytotoxicities were associated with the inhibition of phosphorylation of its regulatory subunit PI3Kp85. Combination of [Bis + ABT199] with panobinostat, decitabine or olaparib dramatically decreased the level of P-PI3Kp85 (Y458) in MOLM13 and MV4-11 cells (). The phosphorylations of its downstream targets, including PDK1, P70S6K, and S6RP, were significantly decreased by the three-drug combinations, suggesting that cell survival might indeed have been compromised through inhibition of the PI3K signaling pathway. The decreased phosphorylation of P70S6K is consistent with decreased phosphorylation of its target proteins including S6RP, eIF4B, and eEF2K, which may reflect an inhibition of protein translation. Furthermore, the observed drug-mediated decrease in mTOR and P-RAPTOR (S792; ) may also contribute to the inhibition of protein translation [Citation28].
Tuberin/TSC2 is a tumor suppressor protein which inhibits the mTOR complex resulting in a downregulation of protein translation [Citation29]. Activation of the PI3K pathway is known to cause inhibition of Tuberin/TSC2 by phosphorylation at threonine 1462 [Citation30]. The observed drug-mediated downregulation of PI3K might therefore have mediated the decrease in the level of P-Tuberin/TSC2 (T1462) seen in cells exposed to the three-drug combinations () and thus may have contributed to inhibition of cell proliferation by relieving the PI3K-mediated inhibition of Tuberin/TSC2.
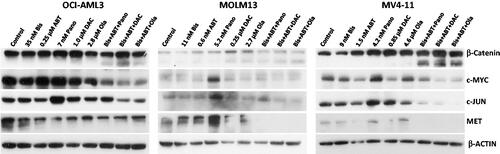
Effects of the three-drug combinations on the wnt/β-catenin pathway
The Wnt/β-catenin survival pathway can be constitutively activated in myeloid leukemia cells. We, therefore, examined whether the Wnt/β-catenin pathway was inhibited by the three-drug combinations in all three cell lines used in the study. shows a marked cleavage of β-catenin protein (a known substrate of activated Caspase 3) in cells exposed to the triple-drug combinations. These results correlate with a decrease in the expression of the β-catenin-target genes including c-MYC, c-JUN, and MET [Citation31], as shown by a decrease in their protein levels in cells exposed to the three-drug combinations (). These results suggest that the cytotoxicity of the three-drug combinations is partly due to inhibition of the pro-survival Wnt/β-catenin signal transduction pathway.
Figure 6. Drug-mediated inhibition of the Wnt/β-catenin pathway. AML cells were exposed to the indicated concentrations of bisantrene (Bis), venetoclax/ABT199 (ABT), panobinostat (Pano), decitabine (DAC) and olaparib (Ola), alone or in combination, for 48 h prior to Western blot analysis.
Discussion
We recently demonstrated the antineoplastic activity of [Bisantrene + ABT199 + nucleoside analog(s)] in AML cells [Citation8]. We now report strong synergistic cytotoxicity of [Bisantrene + ABT199] combined with the HDAC inhibitor Pano, the hypomethylating agent DAC, or the PARP inhibitor Ola in AML cell lines and patient-derived samples. This synergism might be attributed to activation of the DNA-damage response, intrinsic apoptosis and SAPK/JNK stress-signaling pathways, as well as downregulation of the pro-survival PI3K/mTOR and Wnt/β-catenin pathways.
Bisantrene may initiate apoptosis by inhibiting topoisomerase II that results in the formation of DNA strand breaks. DNA damage is exacerbated by increased acetylation of histones () mediated by the HDAC inhibitor panobinostat, which maintains chromatin in its relaxed conformation, likely making it more accessible to bisantrene. Furthermore, panobinostat has been shown to cause structural chromosomal and oxidative DNA damage as well as DNA hypomethylation, in addition to its alteration of the expression of genes involved in the DNA damage response [Citation32,Citation33].
These genomic insults send a stress signal to the mitochondria, resulting in depolarization of the mitochondrial membrane and increased production of ROS (), with a concomitant leakage of pro-apoptotic proteins into the cytoplasm. ABT199 is known to bind to the hydrophobic groove of the BCL2 protein on the outer membrane of the mitochondria [Citation34], which further facilitates this leakage resulting in enhanced activation of the apoptotic cascade, followed by PARP1 cleavage and DNA fragmentation (). This model is consistent with the increased Annexin V positivity () and activation of Caspase 3 (by cleavage) in AML cell lines and patient-derived cell samples exposed to [Bis + ABT199 + Pano] ( and ).
Similarly, decitabine epigenetically modifies the structure of chromosomes and gene expression. It causes DNMT1 depletion, DNA hypomethylation, and DNA damage induction in AML cell lines [Citation35], a mechanism which probably sends apoptotic signals to the mitochondria and synergistically induces cell death when combined with bisantrene and ABT199, similarly to [Bis + ABT199 + Pano].
Olaparib, on the other hand, is known to enhance DNA damage accumulation and apoptosis induction in acute leukemia [Citation36] by inhibiting the DNA repair enzyme PARP1 and trapping it at DNA damage sites [Citation18]. The combined trapping of PARP1 and topoisomerase II by olaparib and bisantrene, respectively, may cause gross chromosomal abnormalities leading to apoptosis induction, which is further enhanced by ABT199.
The effects of these three-drug combinations in the nucleus likely transmits signals to the mitochondria as evidenced by increased ROS production and decreased MMP (), both of which promote cell death. Such mitochondrial stress is further aggravated by the activation of the SAPK/JNK stress signaling pathway (), probably triggered by DNA damage [Citation37], which also induces apoptosis by phosphorylating mitochondrial proteins involved in the release of cytochrome c and AIF [Citation25].
An oncogenic pathway inhibited by the three-drug combinations in AML cells is the PI3K/mTOR axis. One major target of PI3K is PDK1, which affects the protein translation machinery through mTOR. These drugs decreased the phosphorylation of the p85 regulatory subunit of PI3K, which correlates with the observed decrease in the phosphorylation status of PDK1 (). As the level of pan-PDK1 was also decreased, it is possible that these drugs affected both the expression and phosphorylation status of PDK1, at least in MOLM13 cells. Downstream of PDK1 is mTOR, a kinase that phosphorylates the p70S6 kinase. Exposure of AML cells to the three-drug combinations resulted in an inhibition of the phosphorylation of p70S6 kinase that may invoke, through a cascade of events, an inhibition of protein translation.
Another pro-survival target of the three-drug combinations is β-catenin. When activated in the cytoplasm, β-catenin translocates to the nucleus and mediates transcription of pro-survival genes including c-MYC, c-JUN and MET [Citation30]. In the present study we observed drug-mediated cleavage of β-catenin and down-regulation of its downstream targets c-MYC, c-JUN and MET (). The activation of Caspase 3 probably mediates the cleavage of β-catenin [Citation38].
In summary, the combination of [Bis + ABT199] with panobinostat, decitabine or olaparib exerts synergistic cytotoxicity in AML cell lines and patient-derived cell samples through combined effects on the DNA damage response, apoptosis, SAPK/JNK, PI3K/mTOR and Wnt/β-catenin pathways. These preclinical findings provide a rationale to propose a clinical trial of these three-drug combinations as cytoreductive treatments for AML patients. Specifically, such drug combinations might be used as part of the reinduction regimen for relapsed/refractory AML patients to minimize their disease burden and thus improve their long-term prognosis if consolidated with allogeneic hematopoietic stem cell transplantation.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Papazoglou P, Peng L, Sachinidis A. Epigenetic mechanisms involved in the cardiovascular toxicity of anticancer drugs. Front Cardiovasc Med. 2021;8:658900.
- Spadea A, Petti MC, Aloespiriti MA, et al. Bisantrene in relapsed and refractory acute myelogenous leukemia. Leuk Lymphoma. 1993;9(3):217–220.
- Marinello J, Delcuratolo M, Capranico G. Anthracyclines as topoisomerase II poisons: from early studies to new perspectives. IJMS. 2018;19(11):pii:3480.
- Su R, Dong L, Li Y, et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 2020;38(1):79–96.
- Tosi P, Visani G, Colombini R, et al. Phase II study of bisantrene in relapsed/refractory acute non lymphoid leukemias (ANLL). Haematologica. 1989;74(6):555–558.
- Mills GM, Dahlberg S, Cowan J, et al. Phase II evaluation of bisantrene in acute leukemia. A southwest oncology group study. Am J Clin Oncol. 1989;12(6):507–510.
- Canaani J, Danylesko I, Shemtov N, et al. A phase II study of bisantrene in patients with relapsed/refractory acute myeloid leukemia. Eur J Haematol. 2021;106(2):260–266.
- Valdez BC, Murray D, Li Y, et al. Synergism of the anthracene-derivative anti-cancer agent bisantrene with nucleoside analogs and a BCL-2 inhibitor in acute myeloid leukemia cells. J Clin Exp Oncol. 2021;10:4.
- Naqvi K, Konopleva M, Ravandi F. Targeted therapies in acute myeloid leukemia: a focus on FLT-3 inhibitors and ABT199. Expert Rev Hematol. 2017;10(10):863–874.
- Mihalyova J, Jelinek T, Growkova K, et al. Venetoclax: a new wave in hematooncology. Exp Hematol. 2018;61:10–25.
- Laubach JP, Moreau P, San-Miguel JF, et al. Panobinostat for the treatment of multiple myeloma. Clin Cancer Res. 2015;21(21):4767–4773.
- Maiso P, Colado E, Ocio EM, et al. The synergy of panobinostat plus doxorubicinin acute myeloid leukemia suggests a role for HDAC inhibitors in the control of DNA repair. Leukemia. 2009;23(12):2265–2274.
- Wieduwilt MJ, Pawlowska N, Thomas S, et al. Histone deacetylase inhibition with panobinostat combined with intensive induction chemotherapy in older patients with acute myeloid leukemia: phase I study results. Clin Cancer Res. 2019;25(16):4917–4923.
- Jüttermann R, Li E, Jaenisch R. Toxicity of 5-aza-2'-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci U S A. 1994; 91(25):11797–11801.
- Li K, Hu C, Mei C, et al. Sequential combination of decitabine and idarubicin synergistically enhances anti-leukemia effect followed by demethylating WNT pathway inhibitor promoters and downregulating WNT pathway nuclear target. J Transl Med. 2014;12:167.
- Ye XN, Zhou XP, Wei JY, et al. Epigenetic priming with decitabine followed by low-dose idarubicin/cytarabine has an increased anti-leukemic effect compared to traditional chemotherapy in high-risk myeloid neoplasms. Leuk Lymphoma. 2016;57(6):1311–1318.
- Wei H, Yu X. Functions of PARylation in DNA damage repair pathways. Genomics Proteomics Bioinformatics. 2016;14(3):131–139.
- Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72(21):5588–5599.
- Molenaar RJ, Radivoyevitch T, Nagata Y, et al. IDH1/2 mutations sensitize acute myeloid leukemia to PARP inhibition and this is reversed by IDH1/2-mutant inhibitors. Clin Cancer Res. 2018;24(7):1705–1715.
- Tavares TS, Hofman J, Lekešová A, et al. Olaparib synergizes the anticancer activity of daunorubicin via interaction with AKR1C3. Cancers (Basel). 2020;12(11):3127.
- Valdez BC, Li Y, Murray D, et al. The synergistic cytotoxicity of clofarabine, fludarabine and busulfan in AML cells involves ATM pathway activation and chromatin remodeling. Biochem Pharmacol. 2011;81(2):222–232.
- Valdez BC, Li Y, Murray D, et al. Comparison of the cytotoxicity of cladribine and clofarabine when combined with fludarabine and busulfan in AML cells: enhancement of cytotoxicity with epigenetic modulators. Exp Hematol. 2015;43(6):448–461.
- Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci U S A. 1984;81(22):6993–6997.
- Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980;284(5756):555–556.
- Aoki H, Kang PM, Hampe J, et al. Direct activation of mitochondrial apoptosis machinery by c-Jun N-terminal kinase in adult cardiac myocytes. J Biol Chem. 2002;277(12):10244–10250.
- Ichijo H. From receptors to stress-activated MAP kinases. Oncogene. 1999;18(45):6087–6093.
- Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657.
- Morita M, Gravel SP, Hulea L, et al. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle. 2015;14(4):473–480.
- Sparagana SP, Roach ES. Tuberous sclerosis complex. Curr Opin Neurol. 2000;13(2):115–119.
- Manning BD, Tee AR, Logsdon MN, et al. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151–162.
- Kawano Y, Kypta R. Secreted antagonists of the WNT signalling pathway. J Cell Sci. 2003;116(Pt 13):2627–2634.
- Xie C, Drenberg C, Edwards H, et al. Panobinostat enhances cytarabine and daunorubicin sensitivities in AML cells through suppressing the expression of BRCA1, CHK1, and Rad51. PLoS ONE. 2013;8(11):e79106.
- Al-Hamamah MA, Alotaibi MR, Ahmad SF, et al. Genetic and epigenetic alterations induced by the small-molecule panobinostat: a mechanistic study at the chromosome and gene levels. DNA Repair (Amst). 2019;78:70–80.
- Souers AJ, Leverson JD, Boghaert ER, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202–208.
- Hollenbach PW, Nguyen AN, Brady H, et al. A comparison of azacitidine and decitabine activities in acute myeloid leukemia cell lines. PLoS One. 2010;5(2):e9001.
- Garcia TB, Snedeker JC, Baturin D, et al. A small-molecule inhibitor of WEE1, AZD1775, synergizes with olaparib by impairing homologous recombination and enhancing DNA damage and apoptosis in acute leukemia. Mol Cancer Ther. 2017;16(10):2058–2068.
- Hayakawa J, Depatie C, Ohmichi M, et al. The activation of c-Jun NH2-terminal kinase (JNK) by DNA-damaging agents serves to promote drug resistance via activating transcription factor 2 (ATF2)-dependent enhanced DNA repair. J Biol Chem. 2003;278(23):20582–20592.
- Steinhusen U, Badock V, Bauer A, et al. Apoptosis-induced cleavage of beta-catenin by caspase-3 results in proteolytic fragments with reduced transactivation potential. J Biol Chem. 2000;275(21):16345–16353.