
Deregulation of normal epigenetic regulatory mechanisms, including histone acetylation, is increasingly recognized as a major driver in lymphoma, particularly germinal center origin non-Hodgkin lymphomas like follicular lymphoma (FL) and the germinal center subtype of diffuse large B-cell lymphoma (DLBCL) [Citation1]. Many studies have aimed to target these derangements using histone deacetylase (HDAC) inhibitors. However, prior studies of HDAC inhibitors in FL and DLBCL, including mocetinostat [Citation2], vorinostat [Citation3–5], panobinostat [Citation6], and abexinostat [Citation7], have shown heterogeneous response rates, from 11% to 64% in FL and 6% to 28% in DLBCL, and limited durations of response.
Two of the most common mutations observed in FL and GCB DLBCL are inactivating mutations of CREBBP and EP300. Both CREBBP and EP300 serve as transcriptional coactivators to other proteins, including key tumor suppressors like TP53, while inhibiting expression of the oncogenic BCL6 [Citation8–15]. By inhibiting HDACs that remove acetyl groups from histones, HDAC inhibitors may reestablish physiologic levels of acetylation and result in tumor death.
We hypothesized that HDAC inhibition with mocetinostat would be particularly effective in lymphomas with CREBBP or EP300 mutations, and designed a biomarker-driven clinical trial evaluating the efficacy of mocetinostat in patients with R/R FL or DLBCL and CREBBP or EP300 mutations.
We performed a phase II, open-label, single-arm clinical trial evaluating the efficacy of mocetinostat in patients with relapsed or refractory DLBCL or FL harboring mutations in CREBBP or EP300. The study was approved by the Institutional Review Board of Memorial Sloan Kettering Cancer Center, and registered at www.clinicaltrials.gov (NCT02282358). Patients with FL or DLBCL which was relapsed or refractory to two prior lines of treatment, good performance status, and adequate bone marrow and organ function were eligible for enrollment. Genetic testing was performed using any CLIA approved next-generation sequencing (NGS) platform, which was most commonly Foundation One Heme or MSK IMPACT based testing, with the latter utilizing normal tissue (nails or saliva) used as a germline comparison [Citation16,Citation17]. To be eligible, sequencing had to confirm the presence of CREBBP or EP300 mutation (deletion, frameshift, nonsense, or missense), known to affect the histone acetyltransferase function of CREBBP or EP300, based on previously published data and/or the COSMIC database [Citation1].
Patients were treated with mocetinostat 70 mg orally three times per week on a 28-day schedule in cycle 1. The dose was escalated in cycle 2 to 90 mg orally three times per week in the absence of grade 3 or higher drug related toxicities. Treatment was continued until disease progression, intolerable toxicity, or death.
The primary outcome of interest was efficacy as defined by overall response rate (ORR) at one year. Secondary objectives included assessing the event-free survival (EFS), duration of response (DOR), and safety and tolerability of mocetinostat. Exploratory assessments included correlation of Myc and Bcl-2 positivity with treatment response, assessment of mechanisms of resistance, and evaluation of T cell activation and exhaustion in peripheral blood.
From 29 October 2014 to 5 April 2017, we screened 102 patients with FL and DLBCL using NGS of tumor samples. Of these, 27 patients harbored mutations in CREBBP and/or EP300. Of the 27 patients harboring a CREBBP or EP300 mutation, seven patients were enrolled and treated.
Five patients had FL grade 1–3A, and two patients had transformed DLBCL from FL (). Six of seven patients had CREBBP mutations, and one patient with FL had an EP300 mutation (Supplementary Figure 1). Of five evaluable patients, four had BCL2 translocations. Median age was 67 (range 49–83), and five (71%) were male. Six (86%) were white and one (14%) was Asian. ECOG performance status was 0 in three patients and one in four patients. In patients with FL, FLIPI scores were 0–1 in two patients, 2 (moderate risk) in one patient, and 3–5 (high risk) in two patients. Both patients with DLBCL had IPI scores of 3. Patients had received a median of 4 prior lines of therapy (range 3–9); all had received prior anthracycline-based chemotherapy and anti-CD20 monoclonal antibody therapy, and three (43%) had received prior radiation. None had received prior autologous stem cell transplant.
Table 1. Patient characteristics and outcomes.
Of seven patients treated with mocetinostat, one achieved a partial remission and remained on treatment for 12.9 months, with a 1-year ORR of 14% (). Two patients had stable disease as best response and remained on treatment for 17.5 and 7.3 months; the remaining three response-evaluable patients had disease progression. One patient withdrew therapy for toxicity prior to response assessment. Median PFS was 4.6 months, and median EFS was 4.6 months.
Figure 1. Swimmer plot of all seven treated patients. DLBCL: diffuse large B-cell lymphoma; FL: follicular lymphoma; CR: complete response; PR: partial response; SD: stable disease; POD: progression of disease.
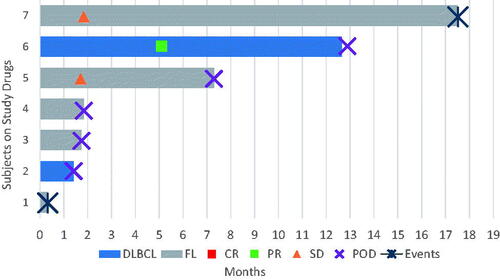
The most common adverse events were diarrhea (grade 1–2 in 57%, grade 3 in 14%), fatigue (grade 1 in 57%), rash (grade 1 in 57%), and nausea (grade 1 in 43%). Grade 3–4 hematologic events were grade 3 lymphopenia in one patient (14%) and grade 4 neutropenia in one patient (14%) without any episodes of neutropenic fever. One patient suffered grade 4 enterocolitis complicated by septic shock, hemolytic uremic syndrome, acute renal failure, and respiratory failure, reported as unrelated to study drug, 14 days after initiating therapy; treatment was discontinued due to ongoing comorbidities precluding participation. One patient with stable disease discontinued therapy for persistent grade 2 diarrhea and abdominal discomfort 17.4 months after starting treatment. The other five patients (71%) discontinued treatment for progression of disease.
The median time to onset of treatment-related adverse events (TRAEs) was 26 days (interquartile range, 2–53 days). The median time to onset of the most common TRAE, diarrhea, was 24 days (range, 0–53 days). No dose reductions of mocetinostat occurred; two patients had dose interruptions, one for the previously mentioned grade 4 enterocolitis, and another for grade 4 neutropenia.
Enrollment was limited due to withdrawal of drug support by the sponsor.
In summary, we performed a biomarker-driven clinical trial evaluating the anti-tumor activity of mocetinostat in patients with DLBCL or FL with mutations in CREBBP or EP300, which we hypothesized would have increased sensitivity to HDAC inhibitors. Study enrollment was limited and lymphoma activity was modest, with one patient achieving response to treatment, and response rates similar to those reported in an unselected cohort of patients with R/R NHL [Citation2].
Despite strong mechanistic rationale for the use of HDAC inhibitors in CREBBP and EP300-mutant lymphomas, limited clinical evidence to date has not clearly matched this expectation. Along with our study, subset analyses of a clinical trial of panobinostat in DLBCL showed that only 15% of CREBBP or EP300-mutant lymphomas responded to treatment, which was below the total ORR of 28% [Citation6]. Likewise, in a trial of vorinostat in FL, CREBBP and EP300 mutations were not associated with higher ORR or longer PFS [Citation4]. It should be noted that the EZH2 inhibitor tazemetostat, another epigenetic therapy, was associated with greater response rates in FL with gain of function mutations in EZH2, but responses were also seen in FL without these gain of function mutations, with similar DOR in mutant and wild-type disease, suggesting that the activity of epigenetic therapies is not restricted to diseases with single aberrations [Citation18].
Higher response rates have been reported in other HDAC inhibitor studies. A recent trial of abexinostat in 37 patients with FL showed a response rate of 70% [Citation19]. Differences may be due to patient population or the therapy itself; abexinostat is a pan-HDAC inhibitor, which may have different anti-lymphoma activity compared to the HDAC1-selective activity of mocetinostat. Molecular analysis, including CREBBP and EP300 mutations, were not reported in this study.
Despite modest response rates, this study demonstrates the feasibility of a molecularly targeted therapeutic approach for patients with lymphoma. Patients successfully completed clinical and molecular screening to determine eligibility and were enrolled in a timely manner. Molecular screening methods were also employed in the multicenter phase 2 trial of tazemetostat, with cohort assignment based on EZH2 mutation status [Citation18]. As our understanding of molecular pathogenesis and heterogeneity of FL and DLBCL advance, the ability to develop prospective trials using molecular testing to select patients who may be most likely to benefit will be essential. This study provides a potential platform by which to develop molecularly driven targeted and epigenetic trials in the future.
Author contributions
D.Q. and C.B. collected and analyzed data and wrote the manuscript. C.B. and A.Z. designed the study. G.S. contributed to data analysis, and reviewed and edited the manuscript.
GLAL-2022-0990-File003.pdf
Download PDF (79.4 KB)Acknowledgements
The authors would like to thank the patients for their participation and contribution. We would also like to thank the Mirati research team, including James Christensen and Chuck Baum, for their discussions regarding the clinical trial concept and guidance during the study.
Disclosure statement
Kumar: research support from AbbVie, Adaptive Biotechnologies, Celgene, Pharmacyclics, and Seattle Genetics; and has an advisory role with Celgene. A. Noy: research support from Abbvie, Cornerstone, Jannsen; honoraria from Abbvie; consultancy role Epizyme, Janssen, Morphosys, AstraZeneca, EUSA, TG therapeutics, and ADC therapeutics. M. Matasar: research support from Genentech, Roche, GlaxoSmithKline, Bayer, Pharmacyclics, Janssen, Rocket Medical, and Seattle Genetics; has received honoraria from Genentech, Roche, Bayer, Pharmacyclics, Janssen, Seattle Genetics, and GlaxoSmithKline; and has a consultancy role with Genentech, Bayer, Merck, Juno, Roche, Teva, Rocket Medical, and Seattle Genetics. G. Salles: consultancy role with AbbVie, Bayer, Beigene, BMS/Celgene, Debiopharm, Epizyme, Genentech/Roche, Genmab, Incyte, Ipsen, Janssen, Kite/Gilead, Loxo, Milteniy, Morphosys, Novartis, Rapt, Regeneron, Takeda, and Velosbio. A. Younes: employment at AstraZeneca. C.L. Batlevi: research support from Janssen, Novartis, Epizyme, Autolus, Roche, and Bayer; has received honoraria from Dava Oncology; and has a consultancy role with Skipta, Kite Pharma, MorphoSys, Bristol-Myers Squibb, Karyopharm Therapeutics, Genentech, and TG Therapeutics. D. Qualls, H.Y. Lam, K. Whiting, C. Owens, C. Nichols, J. Espleleta, S. Subzwari, E. Biggar, and V. Seshan have no conflicts of interest to disclose.
Data availability statement
The data that support the findings of this study are available from the corresponding author, C.B., upon reasonable request.
Additional information
Funding
References
- Pasqualucci L, Dominguez-Sola D, Chiarenza A, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471(7337):189–195.
- Batlevi CL, Crump M, Andreadis C, et al. A phase 2 study of mocetinostat, a histone deacetylase inhibitor, in relapsed or refractory lymphoma. Br J Haematol. 2017;178(3):434–441.
- Crump M, Coiffier B, Jacobsen ED, et al. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann Oncol. 2008;19(5):964–969.
- Ogura M, Ando K, Suzuki T, et al. A multicentre phase II study of vorinostat in patients with relapsed or refractory indolent B-cell non-Hodgkin lymphoma and mantle cell lymphoma. Br J Haematol. 2014;165(6):768–776.
- Kirschbaum M, Frankel P, Popplewell L, et al. Phase II study of vorinostat for treatment of relapsed or refractory indolent non-Hodgkin’s lymphoma and mantle cell lymphoma. J Clin Oncol. 2011;29(9):1198–1203.
- Assouline SE, Nielsen TH, Yu S, et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood. 2016;128(2):185–194.
- Ribrag V, Kim WS, Bouabdallah R, et al. Safety and efficacy of abexinostat, a pan-histone deacetylase inhibitor, in non-Hodgkin lymphoma and chronic lymphocytic leukemia: results of a phase II study. Haematologica. 2017;102(5):903–909.
- Kalkhoven E. CBP and p300: HATs for different occasions. Biochem Pharmacol. 2004;68(6):1145–1155.
- Goodman RH, Smolik S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 2000;14(13):1553–1577.
- Ogryzko VV, Schiltz RL, Russanova V, et al. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87(5):953–959.
- Bannister AJ, Kouzarides T. The CBP co-activator is a histone acetyltransferase. Nature. 1996;384(6610):641–643.
- Lill NL, Grossman SR, Ginsberg D, et al. Binding and modulation of p53 by p300/CBP coactivators. Nature. 1997;387(6635):823–827.
- Gu W, Shi XL, Roeder RG. Synergistic activation of transcription by CBP and p53. Nature. 1997;387(6635):819–823.
- Bereshchenko OR, Gu W, Dalla-Favera R. Acetylation inactivates the transcriptional repressor BCL6. Nat Genet. 2002;32(4):606–613.
- Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432(7017):635–639.
- Cheng DT, Mitchell TN, Zehir A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17(3):251–264.
- Ptashkin RN, Ewalt MD, Jayakumaran G, et al. Enhanced clinical assessment of hematologic malignancies through routine paired tumor:normal sequencing. medRxiv; 2022.
- Morschhauser F, Tilly H, Chaidos A, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol. 2020;21(11):1433–1442.
- Gui L, Cheng Y, Wang H, et al. Interim results of a phase II multicenter study with the oral histone deacetylase inhibitor abexinostat in patients with relapsed/refractory follicular lymphoma. J Clin Oncol. 2022;40(16_Suppl.):7513.