
Abstract
Pelabresib (CPI-0610), a BET protein inhibitor, is in clinical development for hematologic malignancies, given its ability to target NF-κB gene expression. The MANIFEST phase 1 study assessed pelabresib in patients with acute leukemia, high-risk myelodysplastic (MDS) syndrome, or MDS/myeloproliferative neoplasms (MDS/MPNs) (NCT02158858). Forty-four patients received pelabresib orally once daily (QD) at various doses (24–400 mg capsule or 225–275 mg tablet) on cycles of 14 d on and 7 d off. The most frequent drug-related adverse events were nausea, decreased appetite, and fatigue. The maximum tolerated dose (MTD) was 225 mg tablet QD. One patient with chronic myelomonocytic leukemia (CMML) showed partial remission. In total, 25.8% of acute myeloid leukemia (AML) patients and 38.5% of high-risk MDS patients had stable disease. One AML patient and one CMML patient showed peripheral hematologic response. The favorable safety profile supports the ongoing pivotal study of pelabresib in patients with myelofibrosis using the recommended phase 2 dose of 125 mg tablet QD.
CLINICAL TRIAL REGISTRATION: NCT02158858
Introduction
Bromodomain and extra-terminal (BET) proteins, including BRD2, BRD3, BRD4, and BRDT, regulate the expression of an array of genes including NF-κB target gene expression [Citation1,Citation2], resulting in the overexpression of proinflammatory cytokines, abnormal cell development, and fibrosis in myeloproliferative neoplasms (MPNs) [Citation3–5]. NF-κB, a transcription factor essential for inflammatory responses, is constitutively activated in hematologic malignancies [Citation6,Citation7]. BET inhibition can reduce NF-κB kinase signaling and reverse the expression of specific genes linked to malignancy, including MYC, BCL-2, IL-6, and IL-10 [Citation3,Citation8]. Early preclinical trials have shown that BET inhibitors have anticancer effects in patients with acute myeloid leukemia [Citation9], and are emerging as a promising therapeutic target in myelofibrosis (MF) [Citation10,Citation11]. Phase 1 clinical trials in several BET inhibitors have also shown clinical activity as monotherapies and in combination with other drugs for acute myeloid leukemia (AML) and high-risk myelodysplastic syndromes (MDSs) [Citation12,Citation13].
Pelabresib is an investigational oral, small-molecule inhibitor of BET proteins that downregulates the expression of genes with the potential for disease-modifying effects in patients with hematologic malignancies [Citation8]. In a phase 1 study of patients with relapsed or refractory lymphoma, pelabresib treatment suppressed BET target gene expression (IL-8 and CCR1) in a dose-dependent manner within 2 h of post-dose application and with clinical activity well below the maximum tolerated dose (MTD) of 225 mg once daily (QD) [Citation14]. Pelabresib also decreased blood mRNA concentrations of the NF-kB target gene IL-8 by a median of 55% in patients with MF at 4 h post dose, with corresponding reductions in proinflammatory cytokines at day 14 and bone marrow megakaryocyte histotopography at week 24 [Citation15].
MANIFEST is a phase 1/2 clinical trial investigating dose escalation of pelabresib (CPI-0160) in patients with hematologic malignancies (phase 1) and dose expansion of pelabresib with and without ruxolitinib in patients with MPNs. Here we present results from the MANIFEST phase 1 dose-escalation study designed to determine the MTD of pelabresib.
Methods
Study conduct
This study was approved by the institutional review board at each participating site and was conducted in accordance with the principles of the Declaration of Helsinki. It was also overseen by an independent ethics review committee. Written informed consent was obtained from each patient prior to study entry. Data were collected by the study investigators and analyzed by the study sponsor, Constellation Pharmaceuticals, a MorphoySys Company. All the authors, including authors employed by the study sponsor, contributed to authoring the manuscript for publication.
Study design
Study 0610–02 (MANIFEST) was a phase 1, multicenter, open-label, sequential dose-escalation study of pelabresib (CPI-0610) in patients with acute leukemia, chronic myelomonocytic leukemia (CMML) (classified as an MDS/MPN [Citation16]), high-risk MDS or MDS/MPN (NCT02158858). Eligible patients were adults with indicated hematologic malignancies for whom an effective standard treatment was not available. Inclusion criteria included age ≥18 years with a histologically or cytologically confirmed diagnosis of AML, acute lymphoblastic leukemia, acute undifferentiated leukemia, acute biphenotypic leukemia, chronic myelogenous leukemia (CML) in blast crisis, high-risk MDS, or MDS/MPN.
Exclusion criteria included patients with newly diagnosed acute leukemia that had not been treated with a standard induction chemotherapeutic regimen or another antileukemic agent (although those with newly diagnosed, untreated AML and bone marrow containing myelodysplasia-related changes, and 20–30% blasts could be enrolled). Other exclusions included relapsed or refractory acute leukemia where additional induction chemotherapy was of potential clinical benefit, acute leukemia in relapse less than 6 months following allogeneic stem cell transplantation, CML in blast crisis previously treated with only one BCR-ABL tyrosine kinase inhibitor, very low- or low-risk MDS without previous treatment, or leukemia with central nervous system involvement (e.g. leptomeningeal).
Pelabresib was administered orally QD for 14 d, followed by a 7-day break, in continuous 21-day cycles. Successive cohorts of three to eight patients were sequentially enrolled with increasing doses of pelabresib (24, 48, 120, 170, 230, 300, and 400 mg with the capsule formulation, and 225 and 275 mg with the micronized tablet formulation) until the MTD was determined (the highest dose that caused a dose-limiting toxicity [DLT] in fewer than two of six patients) (Supplementary Figure 1). Adverse events (AEs) that were considered for the MTD determination were defined as DLTs occurring during cycle 1 and/or assessed as a DLT by both the investigator and the sponsor after confirmation of investigational medicinal product causality. In each cohort, at least three patients were required to be evaluated for one cycle before the next dose cohort was initiated. Pelabresib was administered under fasting conditions (no food intake 2 h before and 1 h after study drug administration).
The DLT population was defined as all patients who received at least 85% of their planned dose of pelabresib in cycle 1 (≥12 of 14 doses), unless interrupted by a DLT, and who had sufficient follow-up data to allow the investigators and sponsor to determine whether a DLT occurred (≥21 d after the first daily dose). Any patient who received 1–11 doses of pelabresib in cycle 1 and experienced a DLT in cycle 1 was also included.
If a DLT occurred, the patient was withdrawn from the study after appropriate follow-up was completed. However, if the patient’s disease was at least stable and per the investigator it is in the patient’s best interest to continue therapy with pelabresib, then consideration was given to resume treatment at a lower dose. Resumption of treatment at a lower dose was not allowed in the case of Common Terminology Criteria for Adverse Events (CTCAE) grade 4 nonhematologic toxicities. When a dose reduction of pelabresib was required, no re-escalation of dose was permitted.
To characterize the plasma pharmacokinetics (PK) of pelabresib and profile its potential metabolites for the first dose and at steady state for multiple dosing, blood samples were taken: before and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, and 24 h after the initial dose; before dosing on day 8; on days 8–13; before and at 0.5, 1, 1.5, 2, 3, 4, 6, and 8 h after dosing on day 14; anytime on day 15, 16, 17, 18, and 19; and prior to dosing on cycle 2 day 1.
Endpoints
The primary objective of the study was to determine the MTD of pelabresib and its DLTs. Secondary objectives were to characterize PK, safety (AEs were graded using the National Cancer Institute CTCAE version 4.03), and efficacy of pelabresib. Secondary PK parameters included maximum observed concentration (Cmax), area under the concentration–time curve from zero to last quantifiable concentration (AUClast), terminal elimination half-life (t1/2), volume of distribution, and apparent total body clearance.
Statistical analysis
The MTD of pelabresib was defined as the highest dose during cycle 1 that could be given without causing a DLT in 33% or more patients treated at that dose (capsules or tablets).
Dose escalation followed a traditional ‘3 + 3′ design based on the frequency of DLTs.
All patients who were treated with at least one dose of pelabresib were included in the safety population and used for all tabulations of demographic information, baseline characteristics, PK, safety, and efficacy analyses.
Results
Patient disposition and demographics
Forty-four patients were enrolled and treated with pelabresib. Thirty patients were treated with the capsule formulation of pelabresib at seven dose levels, ranging from 24 to 400 mg QD; 14 patients received the tablet formulation at 225 mg (n = 8) and 275 mg (n = 6).
The median age was 67 years (range 18–82 years), 30 (68.2%) were male, and 36 (81.8) were White/Caucasian. The most common diagnosis/type was AML (70.5%), followed by high-risk MDS (18.2%), and CMML (11.4%). Patients had received a median of 3 (range 1–11) prior therapies ().
Table 1. Demographic and disease characteristics.
All patients had discontinued pelabresib treatment at the time of data cutoff. The most common reasons for treatment discontinuation were the development of progressive disease in 18 patients (40.9%); AE or a lab abnormality in 10 patients (22.7%); patient withdrawal in 9 patients (20.5%); and principal investigator decision in 3 patients (6.8%). The remaining reasons for treatment discontinuation (‘death’ and ‘other’) were reported by two patients (4.5%) each.
Dose-limiting toxicities
Thirty-eight of the 44 treated patients met the criteria for the MTD determination population. Seven DLTs in six (18.4%) evaluable patients occurred; 1/6 (16.7%) patients in the 400 mg capsule cohort, 2/6 (33%) patients in the 225 mg tablet cohort, and 4/5 (80%) patients in the 275 mg tablet cohort (where one patient experienced grade 3 diarrhea and nausea). No DLTs were observed in the 21 evaluable patients treated at the 24, 48, 120, 170, 230, and 300 mg capsule cohorts.
Overall, four patients had DLTs that were considered for MTD determination. Of those included in the analysis, one patient in the 400 mg capsule cohort had grade 3 fatigue, and three patients in the 275 mg tablet group experienced one grade 3 DLT (one of vomiting, one of nausea, and one of diarrhea). The 225 mg tablet (QD for 14 d, followed by a 7-d break) was determined as the MTD for pelabresib monotherapy in this study ().
Table 2. Summary of DLTs.
Pharmacokinetics
Mean plasma concentration–time profiles of pelabresib for the groups of patients evaluated at each dose level are shown in for the initial dose and in for the day 14 dose. Pelabresib was rapidly absorbed, with mean peak plasma concentrations occurring between 1.5 and 4 h post dose at all dose levels on day 1, and between 1.5 and 3 h post dose at all dose levels on day 14. Geometric mean Cmax and AUClast values generally increased with increasing dose, with inter-subject variability being 11–35% and 20–79%, respectively, on cycle 1 day 14. Half-life ranged from 5 to 9 h on cycle 1 day 1 (representative of distribution t1/2) and 9–23 h on cycle 1 day 14 (representative of terminal elimination t1/2).
Figure 1. Mean pelabresib plasma profiles (semi-logarithmic scale). A) Mean pelabresib plasma profile over time at cycle 1 day 1. B) Mean pelabresib plasma profile over time at cycle 1 day 14.
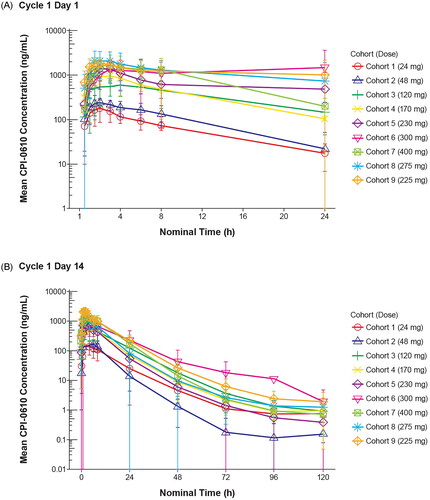
Safety
All 44 patients in the safety analysis set received at least one dose of pelabresib. A total of 14 patients (31.8%) completed at least one cycle of treatment. The mean treatment duration was 2.4 cycles, and the median treatment duration was 2.0 cycles (range 1–9). The median relative dose intensity was 100% (minimum 14% in the 225 mg tablet cohort; maximum 100% across all cohorts) and consistent across treatment cycles.
All 44 patients had at least one treatment-emergent adverse event (TEAE) during the study. The most frequently reported TEAEs were nausea (24 patients [54.5%]), fatigue (23 patients [52.3%]), diarrhea (20 patients [45.5%]), and decreased appetite (19 patients [43.2%]). TEAEs that led to treatment discontinuation were reported for 16 patients (36.4%): diarrhea (three patients), and pneumonia and AML (two patients each); all other TEAEs that led to discontinuation were reported in one patient (2.3%) each. Nine patients (20.5%) died due to a TEAE; eight patients (18.2%) died on study, and one death occurred >30 d after the last dose of pelabresib. None of these fatal TEAEs were considered to be related to the study drug. TEAEs of grade 3 or higher with an incidence of at least 5% of patients are provided in Supplementary Table 1. At least one TEAE of grade 3 or higher was reported in 39 patients (88.6%). Serious TEAEs were reported for 33 patients (75.0%); for five patients (11.4%), at least one serious AE was related to the study drug (Supplementary Table 2).
Study drug-related TEAEs occurring in ≥10% of patients are presented in . For 38 patients (86.4%), at least one TEAE was considered to be related to the study drug; for 15 patients (34.1%), at least one drug-related TEAE was of grade 3 or higher (Supplementary Table 2). The three most frequently reported drug-related TEAEs were nausea in 18 patients (40.9%); decreased appetite in 17 patients (38.6%); and fatigue in 14 patients (31.8%).
Table 3. Treatment-emergent adverse events by relationship to study drug (incidence ≥10% of patients overall; safety population).
Efficacy
Complete remission was not observed in any patient. One patient with CMML experienced partial remission (from 4% blasts on cycle 1 day 1, to 0% blasts) after two cycles of treatment. This patient was discontinued after four cycles of treatment to receive bone marrow transplant. 25.8% of AML patients and 38.5% of high-risk MDS/CMML patients had stable disease. Progressive disease was observed in 45.2% of AML patients and in 23.1% of high-risk MDS/CMML patients.
Two patients had a peripheral hematologic response: one AML patient in the 48 mg capsule group had erythroid, platelet, and neutrophil response, and one CMML patient in the 120 mg capsule group had erythroid and platelet response (Supplementary Table 3).
Discussion
In this phase 1 study of pelabresib in patients with acute leukemia, high-risk MDS, or MDS/MPN (NCT02158858), the MTD for pelabresib monotherapy was determined as the 225 mg tablet (administered daily for 14 consecutive days, followed by a 7-d break in 21-d cycles). Pelabresib administered at this dose was generally tolerable, and no new safety concerns were identified in this study. In a previous phase 1 study in patients with relapsed or refractory lymphoma, hematologic events were the primary cause of DLTs (thrombocytopenia and neutropenia) [Citation14]; however, in this study, gastrointestinal disorders were the most frequently reported TEAEs (vomiting, nausea, and diarrhea). All doses of pelabresib demonstrated a rapid absorption that supported daily dosing. While complete remission did not occur in the efficacy population, one patient (4.8%) with CMML experienced partial response, two patients (8.0%) achieved a hematologic response, and 13 patients (25.0%) had stable disease. Interpretation of efficacy findings is limited owing to small sample sizes per dose level and indication; a further study in a larger study population is required in patients with acute leukemia, high-risk MDS, or MDS/MPN to draw meaningful conclusions.
Although the MTD was identified as the 225 mg QD tablet, a lower dose of 125 mg QD was chosen for the ongoing, multi-arm MANIFEST phase 2 trial starting dose in patients with MF (as monotherapy and in combination with the Janus kinase inhibitor [JAKi] ruxolitinib). This was in light of emerging pharmacodynamic data from the CPI-0610–01 study of pelabresib in patients with relapsed or refractory lymphoma, where doses above 120 mg QD and 170 mg QD showed sustained suppression of IL8 and CCR1 mRNA, respectively, indicating NF-κB attenuation [Citation14]. Additionally, although in this study all patients experienced an AE and 75% experienced a serious AE, these were only considered related to pelabresib in <12% of patients, and no fatal AEs were considered related to pelabresib. There were no notable findings in vital signs, physical findings, or Eastern Cooperative Oncology Group performance status. AEs related to electrocardiogram findings occurred in two patients (4.5%), which were considered related to pelabresib and resolved within 24 h. Phase 2 will also investigate the 125 mg QD tablet dose in patients with high-risk essential thrombocythemia who are resistant or intolerant to hydroxyurea [Citation10,Citation17]. Treatment-related AEs reported with pelabresib are consistent with those reported for other BET inhibitors, which include fatigue, decreased appetite, gastrointestinal disorders, thrombocytopenia, and skin disorders [Citation18–20].
In phase 2, Arm 3 of MANIFEST, combining pelabresib at 125 mg QD starting dose given in combination with ruxolitinib in JAKi-naïve patients with MF was well tolerated and demonstrated clinically meaningful durable improvements in splenomegaly, symptoms, and hematologic responses at 24 weeks [Citation10]. Potential disease-modifying activity was also demonstrated by decreased bone marrow fibrosis and a reduction in the JAK2 V617F mutant allele fraction [Citation10]. These findings indicate the potential for enhancing the standard of care for patients with myeloid diseases through pelabresib treatment.
In this dose-escalation study in patients with acute leukemia, high-risk MDS, or MDS/MPN, all doses of pelabresib showed rapid absorption and a half-life that supported daily dosing. The MTD for pelabresib monotherapy was determined as the 225 mg tablet. Pelabresib was generally well tolerated, with no new safety concerns identified. The recommended phase 2 dosing of pelabresib in MF development and the ongoing phase 3 trial in MF (NCT04603495), which is investigating pelabresib in combination with ruxolitinib, is for cycles of 125 mg QD for 14 d with a 7-d break [Citation21].
Author contributions
ES and ATF helped with patient recruitment, enrollment, and conduct of the study. All authors contributed equally to the development of this manuscript.
Ethics approval
This study was approved by the institutional review board at each participating site and was conducted in accordance with the principles of the Declaration of Helsinki. It was also overseen by an independent ethics review committee.
Patient consent
Written informed consent was obtained from each patient prior to study entry.
Supplemental Material
Download MS Word (106.2 KB)Acknowledgments
The authors would like to express their gratitude to the study participants, investigators, and trial staff. Constellation Pharmaceuticals (subsequently acquired by MorphoSys AG) provided funding for this study. The development of pelabresib was funded in part by the Leukemia and Lymphoma Society. Medical writing support was provided by Laura Travers of LiNK Health Group and funded by Constellation Pharmaceuticals (subsequently acquired by MorphoSys AG).
Disclosure statement
EMS received research funding to his institution from MorphoSys. ATF participates in advisory board/consulting with AbbVie, Agios/Servier, Amgen, Astellas, Blueprint, Celgene/Bristol Myers Squibb, Daiichi Sankyo, EnClear, Foghorn, Genentech, Immunogen, Kite, Kura Oncology, Mablytics, Menarini, MorphoSys, Novartis, Orum, Pfizer, PureTech, Remix, Rigel, Seattle Genetics, Takeda, and Trillium; consulting for Daiichi Sankyo, Forma, Ipsen, Menarini, Remix, Gilead, and Rigel, and receives clinical research funding from AbbVie, Agios/Servier, and Celgene/Bristol Myers Squibb. WAH has nothing to disclose. GC is employed by Constellation Pharmaceuticals, Inc., a MorphoSys Company, and is a current holder of stock options in a privately held company and stock options at MorphoSys AG. AF is employed by Constellation Pharmaceuticals, Inc., a MorphoSys Company. JKM participated in advisory boards for Astellas, Kura Oncology, Adicet Bio, Gilead, and Rigel Pharma.
Data availability statement
Data sharing requests by qualified researchers pertaining to the MANIFEST phase 1 study will be considered only for noncommercial use on a case-by-case basis (to be approved by MorphoSys; [email protected]), starting 12 months from acceptance of the manuscript and until 36 months thereafter; approval may be subject to a data access agreement.
Additional information
Funding
References
- Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13(5):337–356. doi:10.1038/nrd4286
- Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014;54(5):728–736. doi:10.1016/j.molcel.2014.05.016
- Ceribelli M, Kelly PN, Shaffer AL, et al. Blockade of oncogenic IκB kinase activity in diffuse large B-cell lymphoma by bromodomain and extraterminal domain protein inhibitors. Proc Natl Acad Sci USA. 2014;111(31):11365–11370. doi:10.1073/pnas.1411701111
- Pemmaraju N, Verstovsek S, Mesa R, et al. Defining disease modification in myelofibrosis in the era of targeted therapy. Cancer. 2022;128(13):2420–2432. doi:10.1002/cncr.34205
- Naymagon L, Mascarenhas J. Myelofibrosis-related anemia: current and emerging therapeutic strategies. Hemasphere. 2017;1(1):e1. doi:10.1097/HS9.0000000000000001
- Di Francesco B, Verzella D, Capece D, et al. NF-κB: a druggable target in acute myeloid leukemia. Cancers (Basel). 2022;14(14):3557. doi:10.3390/cancers14143557
- Braun T, Carvalho G, Fabre C, et al. Targeting NF-kappaB in hematologic malignancies. Cell Death Differ. 2006;13(5):748–758. doi:10.1038/sj.cdd.4401874
- Albrecht BK, Gehling VS, Hewitt MC, et al. Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J Med Chem. 2016;59(4):1330–1339. doi:10.1021/acs.jmedchem.5b01882
- Zhang L, Cai T, Lin X, et al. Selective inhibition of the second bromodomain of BET family proteins results in robust antitumor activity in preclinical models of acute myeloid leukemia. Mol Cancer Ther. 2021;20(10):1809–1819. doi:10.1158/1535-7163.MCT-21-0029
- Mascarenhas J, Kremyanskaya M, Patriarca A, et al. MANIFEST: pelabresib in combination with ruxolitinib for Janus kinase inhibitor treatment-naïve myelofibrosis. J Clin Oncol. 2023;41(32):4993–5004. doi:10.1200/JCO.22.01972.
- Tremblay D, Mascarenhas J. Next generation therapeutics for the treatment of myelofibrosis. Cells. 2021;10(5):1034. doi:10.3390/cells10051034
- Mims AS, Solh M, Saultz JN, et al. Final results of a phase 1b study of BET inhibitor PLX2853 in patients with relapsed or refractory acute myeloid leukemia or high risk myelodysplastic syndrome. Blood. 2021;138(1):3420–3420. doi:10.1182/blood-2021-152040
- Borthakur G, Odenike O, Aldoss I, et al. A phase 1 study of the pan-bromodomain and extraterminal inhibitor mivebresib (ABBV-075) alone or in combination with venetoclax in patients with relapsed/refractory acute myeloid leukemia. Cancer. 2021;127(16):2943–2953. doi:10.1002/cncr.33590
- Blum KA, Supko JG, Maris MB, et al. A phase I study of pelabresib (CPI-0610), a small-molecule inhibitor of BET proteins, in patients with relapsed or refractory lymphoma. Cancer Res Commun. 2022;2(8):795–805. doi:10.1158/2767-9764.CRC-22-0060
- Keller P, Cui J, Mertz J, et al. BET inhibitor pelabresib decreases inflammatory cytokines, improves bone marrow fibrosis and function, and demonstrates clinical response irrespective of mutation status in myelofibrosis patients. Presented at the European Hematology Association; June 9–17, 2021; virtual congress.
- Kuendgen A, Kasprzak A, Germing U. Hybrid or mixed myelodysplastic/myeloproliferative disorders - epidemiological features and overview. Front Oncol. 2021;11:778741. doi:10.3389/fonc.2021.778741
- ClinicalTrials.gov. A phase 2 study of CPI-0610 with and without ruxolitinib in patients with myelofibrosis. NCT02158858. 2022. Available from: https://clinicaltrials.gov/study/NCT02158858
- Falchook G, Rosen S, LoRusso P, et al. Development of 2 bromodomain and extraterminal inhibitors with distinct pharmacokinetic and pharmacodynamic profiles for the treatment of advanced malignancies. Clin Cancer Res. 2020;26(6):1247–1257. doi:10.1158/1078-0432.CCR-18-4071
- Lewin J, Soria JC, Stathis A, et al. Phase Ib trial with birabresib, a small-molecule inhibitor of bromodomain and extraterminal proteins, in patients with selected advanced solid tumors. J Clin Oncol. 2018;36(30):3007–3014. doi:10.1200/JCO.2018.78.2292
- Shapiro GI, LoRusso P, Dowlati A, et al. A phase 1 study of RO6870810, a novel bromodomain and extra-terminal protein inhibitor, in patients with NUT carcinoma, other solid tumours, or diffuse large B-cell lymphoma. Br J Cancer. 2021;124(4):744–753. doi:10.1038/s41416-020-01180-1
- ClinicalTrials.gov. Phase 3 study of pelabresib (CPI-0610) in myelofibrosis (MF) (MANIFEST-2). NCT04603495. 2022. Available from: https://clinicaltrials.gov/study/NCT04603495