
Abstract
There is increasing evidence that therapy-related acute lymphoblastic leukemia (trALL) resulting from chemo- and/or radiotherapy represents a distinct entity. However, apart from KMT2A rearrangements, which have been repeatedly reported in this subgroup, the relevance of other aberrations remains controversial due to divergent study results and sparse molecular analyses. Within our ALL patient cohort, 15% (n = 19/131) met the criteria for trALL with a high proportion of Ph + and KMT2A rearrangements. On the molecular level, the most frequently observed mutation was KMT2D, followed by CDKN2A, KRAS and DNMT3A. No TP53 mutation was detected. Outcome was particularly poor in Ph + trALL compared to Ph+ de novo ALL, which seemed to be mitigated by allogeneic stem cell transplantation. Our findings further define trALL as a distinct entity but highlight the need for further molecular genome sequencing of somatic and germline variants to advance our understanding of trALL.
Introduction
Myeloid neoplasms arising after cytotoxic chemotherapy and/or radiation therapy (t-MN) for an unrelated condition have long been recognized as a specific entity in the WHO classification of hematolymphoid tumors [Citation1]. However, for lymphoid malignancies the association is less evident and therapy-related acute lymphoblastic leukemia (trALL) is still in its nascent stages of acknowledgment.
The association of alkylating agents and topoisomerase II inhibitors with large chromosomal deletions, such as del(5q), del(7q), and rearrangements of the KMT2A gene at chromosome 11q23 or RUNX1 gene at 21q22.1 are well described in the literature [Citation2,Citation3]. At the molecular level, a high incidence of mutations in TP53, IDH1/2, WT1, KIT, PTPN11, and EZH2 genes are reported for t-MN [Citation4]. Additionally, preexisting clonal molecular abnormalities, namely clonal hematopoiesis of indeterminate potential (CHIP), are thought to be involved in the development of t-MN, as it has been shown that CHIP increases the risk of developing t-MN with accelerated evolution of TP53-mutated t-MN after cytotoxic therapies [Citation5,Citation6].
In contrast, in trALL the understanding of the molecular profile is still very limited and primarily relies on small-scale studies that reported varying profile patterns as older studies commonly used the less specific term ‘secondary ALL’ instead of trALL, which limits comparability [Citation7–9]. Still, similarly to t-MN, rearrangement of the KMT2A gene was found more frequently in trALL than in de novo ALL (dnALL) [Citation10] and some studies reported a significantly higher presence of the Philadelphia chromosome (Ph+) in trALL [Citation11,Citation12]. However, due to the rarity of the disease with an incidence of 3% to 9% of all adult ALL patients, defining trALL as a distinct entity continues to pose a significant challenge.
With the present study, we aim to extend our understanding of the molecular events driving the development of trALL by using next-generation sequencing in our cohort of trALL.
Material and methods
Patient population and study design
Data collection included all patients, which were diagnosed with ALL between 2009 and 2019 and underwent treatment at the Department of Medical Oncology and Hematology of the University Hospital Zurich, Switzerland. Patients younger than 18 years or with a refusal to provide general research consent were excluded. The study was conducted according to the regulations of the local ethics committee (BASEC number 2020-00882 and 2018-01618) and the Declaration of Helsinki.
Patients diagnosed with ALL according to the WHO criteria for lymphoblastic leukemia and presenting after treatment with alkylating agents, topoisomerase inhibitors, radioactive iodine ablation (RAI) for thyroid cancer and/or irradiation due to a previous neoplasm were classified as trALL. Patients with germline mutations indicating genetic predisposition to develop hematological malignancies (e.g. Li-Fraumeni, GATA2 mutations, Fanconi anemia) were not considered for trALL diagnosis nor were patients with malignancies not requiring chemo- or radiotherapy.
Cytogenetic and molecular analyses
Cytogenetic analyses using the G-banding method were performed following standard protocol as part of the routine diagnostic work-up for each patient. At least 10 metaphase cells were examined and chromosomal abnormalities were described according to the International System of Human Cytogenomic Nomenclature [Citation13]. Three or more chromosomal abnormalities were defined as a complex karyotype. Presence of less than 46 chromosomes was considered a hypodiploid karyotype. FISH analyses for t(9;22)/BCR::ABL1 fusion, KMT2A (MLL) and TCF3 (E2A) rearrangement were performed using a dual fusion tricolor probe for BCR::ABL1 fusion and break-apart probes for KMT2A and TCF3 rearrangements. A total of 200 interphase cells were evaluated independently by two technicians.
Next-generation sequencing (NGS)
DNA and RNA were isolated from tissue sections (40 µm) of formalin-fixed paraffin-embedded (FFPE) bone marrow samples. DNA and RNA were isolated using the 16 LEV DNA FFPE and the 16 LEV RNA FFPE purification kits, respectively (Promega Corporation). No DNAse treatment was performed during RNA isolation. DNA and RNA were quantified using the Qubit 2.0 Fluorimeter (Thermo Fisher Scientific).
Analysis of somatic gene mutations was performed by NGS on the Illumina platform (NextSeq) using three gene sequencing panels. We used the FusionPlex ® ALL panel (ArcherDx) to identify fusions in 81 genes with an input of 250ng of RNA and our custom lymphoma panel (ArcherDx) which identifies mutations in 59 lymphoma-associated genes with an input of 100 ng of DNA. Coverage had to be above 200 reads. Mutations were reported at first diagnosis with a variant allele frequency (VAF) of 5%.
Statistics
Comparison of categorical data between groups was performed with the Fisher’s exact test and the Student’s t-test was used for continuous variables. Overall survival (OS) was calculated from the time of diagnosis until death from any cause or last follow-up using the Kaplan-Meier method and differences were compared with the Log-rank test. A p-value of <0.05 was considered significant. Data were compiled using Microsoft Excel (Microsoft Corporation, Redmond, WA, USA) and statistical analyses were performed using JMP version 17.1.0 (SAS Institute, Cary, NC, USA) and SPSS version 27 (IBM, Armonk, USA). Survival curves were plotted on Prism GraphPad version 10.0 (GraphPad Software Inc., La Jolla, CA, USA) and visualization of somatic clonal mutations was done with R (version 4.0.3, R Foundation for Statistical Computing, Vienna, Austria) and the package maftools [Citation14].
Results
Clinical characteristics of patients with trALL
From 131 patients with the diagnosis of ALL, 19 patients (15%) had been previously exposed to chemo- and/or radiotherapy for neoplastic disease (). Unsurprisingly, patients with trALL were significantly older than dnALL patients (median age 60 vs 40 years, p < 0.001) with no sex predilection (female prevalence in trALL 47% vs dnALL 46%). When gender-specific cancers (breast cancer and seminoma) were not taken into account, a higher proportion of men could be found in the trALL group (n = 8/9, 88%). Leukemia phenotypes (B or T cell precursor) were balanced between the two groups (p = 0.751). Clinically, around half of the patients in each group presented with splenomegaly (42% in the trALL vs 53% in the dnALL group, p = 0.302), but lymphadenopathy showed to be significantly less prevalent in trALL patients compared to those with dnALL (16% vs 38% respectively, p = 0.036). Only 1 trALL patient was found to have central nervous system involvement compared to 12 patients (11%) in the dnALL group (p = 0.687). No other primary extramedullary manifestations were noted in either group. Blood counts, peripheral and bone marrow blast counts as well as prevalence of comorbidities did not differ significantly between the two groups (except for the defining previous oncological disease in trALL).
Table 1. Patient characteristics in trALL and dnALL.
Breast cancer was the most frequent type of neoplasm (7/19, 37%), followed by testicular seminoma and thyroid cancer (each 3/19, 16%). Prior malignant hematological (lymphatic) disease was present in 3 patients (16%, one Hodgkin’s lymphoma, one diffuse large B-cell lymphoma (DLBCL) and one chronic lymphocytic leukemia). The combination of radiation and chemotherapy was given to 21% (4 patients), while 47% (9 patients) were treated with radiation and 26% (5 patients) with chemotherapy only (). In our cohort, Ph + trALL was significantly (p < 0.01) associated with radiation therapy but did not show a significant association with a specific primary malignancy or chemotherapy type. Overall median latency between genotoxic therapy and the diagnosis of trALL was 6.5 years (range 5–17 years) with a longer latency interval after radiotherapy (median 7 years) than chemotherapy (median 5 years), which is in line with published data [Citation15]. Latency was also affected by the type of chemotherapy with longer intervals after alkylating therapy than topoisomerase II inhibitors (median 9 vs 4 years).
Figure 1. Outline of primary cancer treatment, latency to trALL, and cytogenetic abnormalities. Gray shading indicates failed karyotype. * T-lymphoblastic phenotype.

Cytogenetic presentation
FISH analysis was available for all patients, while karyotyping failed in 1 patient in the trALL and 7 patients in the dnALL group. Cytogenetic analyses demonstrated a high proportion of Ph + patients in the trALL (7/19, 37%) compared to the dnALL group (20/112, 18%, p = 0.075). When looking more specifically at B-cell phenotype, the Ph + chromosome was significantly more common in trALL (7/15, 46%, p < 0.01). Ph + trALL BCR::ABL1 break point was mainly identified for mbcr (p190) with one exception with Mbcr (p210). The KMT2A rearrangement was present in 16% (3/19) of trALL compared to 6% of dn-ALL patients (7/112, p = 0.149). All KMT2A rearrangements involved the 11q23 locus. Deletions in chromosome 7 were found in 16% of trALL (3/19) vs 9% in dn-ALL (10/112, p = 0.404), but no deletions of chromosome 5 were identified in the trALL group. The number of patients with a complex karyotype (21% trALL vs 41% dnALL) or deletions of 17p were not significantly different between the two groups. Twenty-one percent of trALL patients had a normal karyotype (mainly with T-lymphoblastic subtype) comparable to patients with dnALL (19%, p = 0.532).
In , the most frequently reported cytogenetic changes are listed for each patient with trALL in relation to prior cancer treatment and latency period until the onset of leukemia. In line with previous results we found patients with KMT2A rearrangement to have a shorter interval to trALL (median 2 years) than those with deletion or monosomy 7 (median 4 years), most of which were exposed to prior topoisomerase II inhibitor treatment [Citation7,Citation16], whereas in Ph + trALL, the median latency period was 5 years with a broad range (2–17 years).
Mutation profiling
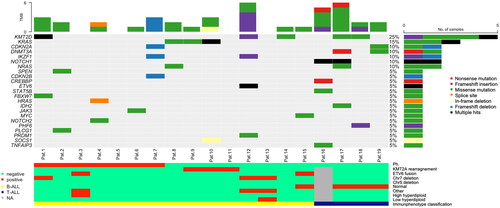
All patients with trALL had bone marrow material obtained from initial diagnosis, which was available for retrospective molecular genetic testing. Yet, one sample did not have the required quality for analysis. A 81 gene panel was able to detect at least one genetic mutation in 15 out of 18 trALL patients. The mutation profile is depicted in . The average number of somatic mutations per patient was 2 (range 0-6). On average, T-cell phenotype displayed a tendency for more mutations than B-cell phenotype (3.8 vs 1.8, p = 0.06). Missense mutations were most commonly detected, as frame shift deletions were found only in one patient with the history of prior DLBCL. This was also the only patient with del7p showing the highest mutational burden within the B-cell phenotype trALL group. The overall medium VAF was 36.2% (range 6.7% to 89%). The most frequently mutated gene was KMT2D in 26% (5/19) of the samples, followed by KRAS (16%, 3/19). The ALL-associated mutations IKZF1 and CDKN2A were present at 11%, all with a VAF below 40%. NOTCH1 was found in 2 of the 4 trALL patients with T-cell phenotype. Furthermore, mutations associated with myeloid neoplasms, such as DNMT3A and NRAS were found in 11% and HRAS in 5%. In our cohort, no mutations of the suppressor gene TP53 could be identified and only two patients showed alterations in the classical tumor suppressor genes CDKN2A and CDKN2B with VAF levels below 55%. Looking at the cytogenetic subgroups, KRAS was not found in Ph + trALL but appeared to be associated with KMT2A rearrangement. IKZF1 with a VAF of 31% was found in one patient with BCR::ABL1 translocation.
Figure 2. Oncoplot for visualization of mutations in trALL each column represents a patient sample and each row a different gene. The bottom barplot shows the corresponding cytogenetic and immunophenotypic profile. Percentages are rounded to 5% increments. Chr chromosome NA not available Ph Philadelphia chromosome TMB total mutation burden.
Outcome
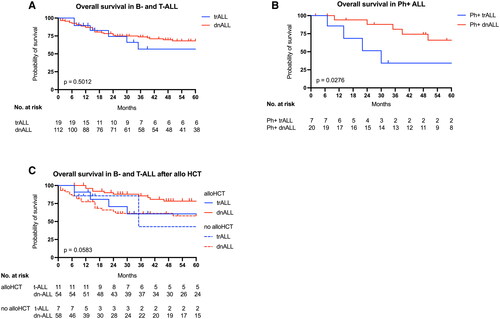
No significant difference in overall survival could be demonstrated between patients with trALL and dnALL (). However, survival for the high proportion of Ph + trALL patients was significantly worse than for Ph + dnALL patients (, p = 0.02). Outcome after allogeneic stem cell transplantation (allo HCT) did not differ significantly between trALL and dnALL when stratified by allo HCT (), although subgroup analysis of dnALL demonstrated superior survival in transplanted patients.
Figure 3. Overall survival in trALL shown are Kaplan–Meier plots for overall survival in trALL and dnALL (A) and in the subgroups of Philadelphia chromosome-positive trALL and dnALL (B). Overall survival in patients with and without allogeneic stem cell transplantation is depicted in C. Plots are calculated as the time to death or lost to follow-up, and p values are calculated by the log-rank test. Tick marks indicate censored data.
Discussion
KMT2A rearrangement, a prototypical cytogenetic finding in t-MN which is generally associated with a poor prognosis, was present with increased frequency in this retrospective study of molecular patterns in our trALL cohort. This is in line with several other studies [Citation10,Citation11,Citation17,Citation18]. Additionally, we identified a second group of patients with trALL that showed a strong association with Ph + as previously described [Citation7,Citation10,Citation19]. Other high-risk cytogenetic changes seen in t-AML, such as chromosome 5 and/or 7 abnormalities and complex karyotype that are not consistently associated with trALL were not substantially enriched in our cohort.
Comparing our data with other reports on the molecular landscape in trALL, it is worth mentioning that despite being relatively frequent, literature reports only include 13 adult patients with complete cytogenetic results for Ph + trALL (including our 7 Ph + cases). The majority of the reported cases had at least one additional cytogenetic abnormality [Citation9,Citation20,Citation21]. The BCR::ABL1 minor break point was observed most commonly but not exclusively, as BCR::ABL1 with Mbcr were also observed. Yet, the association of Ph + with trALL in general is debated, as the primary cytogenetic findings in trALL patient cohorts vary across published studies [Citation18]. While Ph + is a commonly observed genetic abnormality in adult ALL with increasing age, analysis of our Ph + trALL patients revealed a median age of 55 years, which is below the average age reported for trALL patients [Citation22]. Also, it is noteworthy that Ph + cases have also been found with higher frequencies in pediatric trALL cases, which is a very uncommon finding in this age group [Citation17]. Furthermore, most trALL cohorts comprise trALL patients with T-cell phenotype and only few studies focus specifically on B-ALL results within their trALL groups [Citation7,Citation8,Citation23]. Finally, the potential inclusion (or lack of clear exclusion) of germline-associated leukemias in several studies further contributes to a blurring effect for refined results in trALL [Citation8,Citation16], which is potentially also a confounding factor for consistent reports on TP53 mutations in trALL, that lead to contradictory reports on its association with trALL. Pourhassan et al. [Citation16] reported a high prevalence of TP53 gene mutations (12 patients, 40%) in their 30 trALL patients, a finding that was also reported in a small cohort of 8 trALL patients by Kook et al. [Citation9]. On the other hand, Saygin et al. detected more myeloid malignancy and probably CHIP associated profiles with a molecular analysis of 20 trALL patients such as mutations in DNMT3A, IDH2, RUNX1, ASXL, WT1, PHF, NRA, CUX1 and PRPF8 but only found two patients with a TP53 mutation [Citation7]. Together with cytogenomic studies of TP53 alterations, these results led to the assumption that higher frequencies of TP53 mutations/deletions are to be expected in trALL [Citation8, Citation24]. However, using a very strict definition of trALL, supported by Riazat et al. in their comprehensive review on trALL with a clear separation of trALL from secondary ALL, post malignancy ALL, and dnALL we did not observe any TP53 mutations in our cohort [Citation22]. Our results therefore align with the findings of Saygin et al. in which TP53 locus abnormalities do not play a major role in trALL [Citation7].
Despite the limitations of this study concerning its retrospective single-center design and the small patient numbers, this study adds substantial data to the increasing understanding of molecular changes in trALL. Focusing on the subgroup of Ph + trALL, we present the largest cohort with molecular analysis as of to date, as the publications of Saygin and Pourhassan did not allow for molecular subgroup analysis of Ph + patients. Additional molecular profiling for Ph + trALL indicates that it is not uncommon to find no additional mutations and that TP53, KRAS, NRAS, and probably other mutations associated with t-MN are mutually exclusive in this subgroup, while KMT2A showed an association with the presence of KRAS mutations as reported for KMT2A-rearranged acute myeloid leukemias [Citation25]. These findings confirm molecular genetic results on 8 trALL patients from the cohort of Kook et al. [Citation9]. Contrary to other studies that reported inferior survival in trALL, our data suggest that inferior outcomes are confined to the subgroup of Ph + trALL, which might be overcome by allo HCT; an interpretation that must be approached with caution due to the limited sample size and use of different induction therapies. Interestingly, the group of patients with trALL had a much higher median age, but still had a comparable survival rate to patients with dnALL, emphasizing the finding that a diagnosis of trALL is not inherently associated with inferior outcomes. On the other hand, patients with Ph + trALL, who were shown to perform worse, were not significantly older than patients with Ph + dnALL (p = 0.22), which is a further indicator that age-independent factors appear to influence long-term survival. In terms of treatment, patients in both groups were uniformly treated with tyrosine kinase inhibitors (mostly imatinib or dasatinib) in combination with polychemotherapy, while a higher proportion of Ph + trALL than Ph + dnALL patients received allo HCT (85%, 6/7 vs 70%, 14/20; p = 0.41).
In conclusion, our data confirm that trALL is associated with high-risk cytogenetics that share similarities with t-MN. Besides KMT2A rearrangements, a substantial part of patients with trALL harbor the Ph chromosome, that has a distinguished molecular profile. Establishing trALL as a coherent genetic entity with defined molecular differences continues to be challenging as sufficient data to compare mutated genes in trALL with those of dnALL are still lacking. Further study is warranted to validate and extend molecular findings in trALL with special attention to potential germline associated mutations.
Supplemental Material
Download MS Excel (18.4 KB)Acknowledgements
We thank Christine Fritz for her support in obtaining NGS results and Cyrill Rütsche for the independent review of mutation analysis.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
A dataset on all trALL patients is provided in the supplementary material. Further requests for data sharing may be submitted to Corinne C. Widmer ([email protected]).
Additional information
Funding
References
- Khoury JD, Solary E, Abla O, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36(7):1703–1719. doi:10.1038/s41375-022-01613-1
- McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer. 2017;17(9):513–527. doi:10.1038/nrc.2017.60
- Churpek JE, Larson RA. The evolving challenge of therapy-related myeloid neoplasms. Best Pract Res Clin Haematol. 2013;26(4):309–317. doi:10.1016/j.beha.2013.09.001
- Ok CY, Patel KP, Garcia-Manero G, et al. TP53 mutation characteristics in therapy-related myelodysplastic syndromes and acute myeloid leukemia is similar to de novo diseases. J Hematol Oncol. 2015;8(1):45. doi:10.1186/s13045-015-0139-z
- Gibson CJ, Lindsley RC, Tchekmedyian V, et al. Clonal hematopoiesis associated With adverse outcomes After autologous stem-cell transplantation for lymphoma. J Clin Oncol. 2017;35(14):1598–1605. doi:10.1200/JCO.2016.71.6712
- Kuzmanovic T, Patel BJ, Srinivasa R, et al. Genomics of therapy-related myeloid neoplasms. Haematologica. 2020;105(3):e98–e101. doi:10.3324/haematol.2019.219352
- Saygin C, Kishtagari A, Cassaday RD, et al. Therapy-related acute lymphoblastic leukemia is a distinct entity with adverse genetic features and clinical outcomes. Blood Adv. 2019;3(24):4228–4237. doi:10.1182/bloodadvances.2019000925
- Barnea Slonim L, Gao J, Burkart M, et al. Therapy-related B-cell acute lymphoblastic leukemia in adults has unique genetic profile with frequent loss of TP53 and inferior outcome. Leukemia. 2021;35(7):2097–2101. doi:10.1038/s41375-020-01061-9
- Kook HW, Kim JJ, Park MR, et al. Therapy-related acute lymphoblastic leukaemia has a unique genetic profile compared to de novo acute lymphoblastic leukaemia. J Cancer. 2022;13(12):3326–3332. doi:10.7150/jca.76719
- Aldoss I, Stiller T, Tsai N-C, et al. Therapy-related acute lymphoblastic leukemia has distinct clinical and cytogenetic features compared to de novo acute lymphoblastic leukemia, but outcomes are comparable in transplanted patients. Haematologica. 2018;103(10):1662–1668. doi:10.3324/haematol.2018.193599
- Abdulwahab A, Sykes J, Kamel-Reid S, et al. Therapy-related acute lymphoblastic leukemia is more frequent than previously recognized and has a poor prognosis. Cancer. 2012;118(16):3962–3967. doi:10.1002/cncr.26735
- Aldoss I, Stiller T, Song J, et al. Philadelphia chromosome as a recurrent event among therapy-related acute leukemia. American J Hematol. 2017;92(2):E18-E19. doi:10.1002/ajh.24604
- Liehr T. International system for human cytogenetic or cytogenomic nomenclature (ISCN): some thoughts. Cytogenet Genome Res. 2021;161(5):223–224. doi:10.1159/000516654
- Mayakonda A, Lin D-C, Assenov Y, et al. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28(11):1747–1756. doi:10.1101/gr.239244.118
- Ferraro F, Gao F, Stockerl-Goldstein K, et al. Secondary acute lymphoblastic leukemia, a retrospective analysis from Washington university and meta-analysis of published data. Leuk Res. 2018;72:86–91. doi:10.1016/j.leukres.2018.07.024
- Pourhassan H, Yang D, Afkhami M, et al. High prevalence and inferior long-term outcomes for TP53 mutations in therapy-related acute lymphoblastic leukemia. American J Hematol. 2022;97(5):E171-E173. doi:10.1002/ajh.26490
- Shivakumar R, Tan W, Wilding GE, et al. Biologic features and treatment outcome of secondary acute lymphoblastic leukemia—a review of 101 cases. Ann Oncol. 2008;19(9):1634–1638. doi:10.1093/annonc/mdn182
- Vasudevan Nampoothiri R, Viswabandya A. Allogeneic hematopoietic stem cell transplantation in therapy related acute leukemia. Indian J Hematol Blood Transfus. 2021;37(4):521–527. doi:10.1007/s12288-020-01334-4
- Ganzel C, Devlin S, Douer D, et al. Secondary acute lymphoblastic leukaemia is constitutional and probably not related to prior therapy. Br J Haematol. 2015;170(1):50–55. doi:10.1111/bjh.13386
- Matnani R, Parekh V, Borate U, et al. Therapy-related B-lymphoblastic leukemia associated with philadelphia chromosome and MLL rearrangement: single institution experience and the review of the literature. Pathol Int. 2015;65(10):536–540. doi:10.1111/pin.12337
- Lee S-G, Choi JR, Kim JS, et al. Therapy-related acute lymphoblastic leukemia with t(9;22)(q34;q11.2):a case study and review of the literature. Cancer Genet Cytogenet. 2009;191(1):51–54. doi:10.1016/j.cancergencyto.2009.02.002
- Riazat-Kesh YJRA, Mascarenhas J, Bar-Natan M. ‘Secondary’ acute lymphoblastic/lymphocytic leukemia - done playing second fiddle? Blood Rev. 2023;60:101070. doi:10.1016/j.blre.2023.101070
- Tang G, Zuo Z, Thomas DA, et al. Precursor B-acute lymphoblastic leukemia occurring in patients with a history of prior malignancies: is it therapy-related? Haematologica. 2012;97(6):919–925. doi:10.3324/haematol.2011.057752
- Aldoss I, Douer D, Pullarkat V. Therapy-related acute lymphoblastic leukemia: where do we stand with regards to its definition and characterization? Blood Rev. 2019;37:100584. doi:10.1016/j.blre.2019.06.001
- Bill M, Mrózek K, Kohlschmidt J, et al. Mutational landscape and clinical outcome of patients with de novo acute myeloid leukemia and rearrangements involving 11q23/KMT2A. Proc Natl Acad Sci USA. 2020;117(42):26340–26346. doi:10.1073/pnas.2014732117