
Abstract
Electroporation is a widely used method for delivering CRISPR components into cells; however, it presents challenges when applied to difficult-to-transfect cells like adult buffalo fibroblasts. In this study, the ITGB2 gene (encoding the CD18 protein), plays vital for cellular adhesion and immune responses, was selected for editing experiments. To optimize electroporation conditions, we investigated parameters such as electric field strength, pulse duration, plasmid DNA amount, cuvette type, and cell type. The best transfection rates were obtained in a 4 mm gap cuvette with a single 20-millisecond pulse of 300 V using a 10 μg of all-in-one CRISPR plasmid for 106 cells in 100 μL of electroporation buffer. Increasing DNA quantity enhanced transfection rates but compromised cell viability. The 4 mm cuvette gap had high transfection rates than the 2 mm gap, and newborn cells exhibited higher transfection rates than adult cells. We achieved transfection rates of 10–12% with a cell viability of 25–30% for adult fibroblast cells. Subsequently, successfully edited the ITGB2 gene with a 30% editing efficiency, confirmed through various analysis methods, including T7E1 assay, TIDE and ICE analysis, and TA cloning. In conclusion, electroporation conditions reported here can edit buffalo gene(s) for various biotechnological research applications.
Introduction
Both viral and non-viral methods have been used for delivery of exogenous components into mammalian cells; however, non-viral methods are favored due to their advantages in biosafety, ease of application, and rapid results.Citation1 In farm animals, various non-viral techniques such as liposomes,Citation2,Citation3 nucleofection,Citation4,Citation5 and electroporation,Citation3,Citation6–9 have been utilized to introduce exogenous components into cultured cells. Electroporation involves the application of high-voltage electric pulses to transiently permeabilize cell membranes, enabling the entry of exogenous materials into the cells.Citation10 Several critical factors, such as electric field strength, pulse duration, exogenous material quantity, cell count, cell age, and cell type, influence the efficiency of electroporation.Citation9,Citation11,Citation12 The development of genome editing tools, mainly clustered regularly interspaced short palindromic repeats (CRISPR), provide precise control of gene insertion, deletion, or modification.Citation13 Electroporation methods have been used to deliver CRISPR components into various cell types with varying extent of gene editing success.Citation14–16 Each cell type necessitates specific electroporation conditions, which require extensive experimental optimization to establish the most effective transfection protocols. Among several cell types, adult fibroblasts are the preferred for genome editing studies due to their straightforward culture maintenance and biological significance; however, they are also considered to be difficult to transfect cells.Citation17 In recent years, CRISPR components have been successfully delivered in different type of buffalo cells such as fetal cells, newborn cells, stem cells, and corpus luteum cells;Citation18–24 however, there is no systematic study on adult buffalo fibroblast cells for editing of any gene locus using all-in-one CRISPR plasmids.
Buffalo is an economically important bovine animal species providing milk, meat, and work power,Citation25 and is increasingly used as a model species for biomedical and veterinary research.Citation18,Citation26–28 For CRISPR-based genome editing in adult buffalo cells, we chose to focus on the ITGB2 gene. This gene encodes the CD18 protein, which plays pivotal roles in cellular adhesion, cell surface signaling, and immune responses. Previous studies reported the severe disorders associated with mutations in this gene in livestock species.Citation29–31 Bovine Leukocyte Adhesion Deficiency (BLAD) is a fatal autosomal recessive disorder, characterized by a significant reduction in CD18 adhesion molecule expression on leukocytes.Citation29 This genetic anomaly compromises the immune response, rendering affected bovine highly susceptible to severe and recurrent bacterial infections.Citation29 BLAD's molecular basis lies in a single nucleotide mutation (A→G) at position 383 of the ITGB2 gene.Citation29 In the another recent study, researchers utilized zinc-finger nucleases (ZFN) gene editing tool to introduce a Q(–5)G substitution in signal peptide coding region of ITGB2 gene. This modification effectively eliminated the intact CD18 signal peptide of ruminant leukocytes. They also demonstrated that leukocytes from an edited bovine fetus were resistant to leukotoxin-induced cytolysis caused the Mannheimia haemolytica.Citation31 Keeping significance of ITGB2 gene and advances of CRISPR editing into consideration, this study objective was to optimize electroporation parameters for the successful delivery of an all-in-one CRISPR/Cas9 plasmids specifically targeting the ITGB2 gene in adult buffalo fibroblast cells.
Materials and methods
Ethics statements
All experiments were performed according to the ethical standards of the institute. Animal procedures including biopsy collection were approved by the Institute Animal Ethics Committee, ICAR-Central Institute for Research on Buffaloes, Hisar, India. The study is reported in accordance with ARRIVE guidelines.
Cell culture
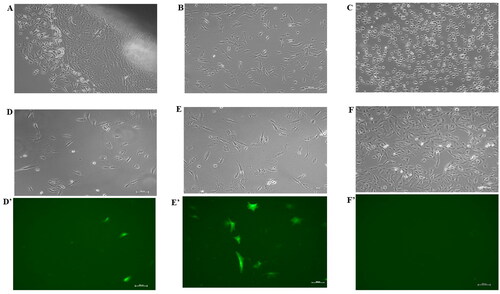
Isolation, culture, and cryopreservation of adult buffalo skin fibroblasts were performed as previously described by us.Citation32 Briefly, the tail skin tissue biopsy, from an adult buffalo, was collected aseptically and chopped with a sterile surgical blade in Dulbecco’s phosphate-buffered saline (DPBS) supplemented with antibiotics. Chopped tissue pieces (<1 mm3) were cultured in high glucose-Dulbecco’s Modified Eagle Medium (DMEM) (Sigma#D5796) supplemented with 10% fetal bovine serum (FBS) (Gibco#10270-106), 1% (v/v) non-essential amino acids (Himedia#ACL006), 1X vitamin mix (Himedia#VA001), and 1X antibiotic solution (Gibco#15240-062) under a humidified 5% CO2 in air at 38.5 °C. After 7–10 days of culture, fibroblast cells outgrowths were obtained (), and then routinely passaged using 0.25% trypsin EDTA (Himedia#T001) solution. The aliquots of early passage cells (below 2) were cryopreserved in DMEM supplemented with 10% FBS and 10% dimethyl sulfoxide (Sigma#D2650) using a standard slow freezing method. Aliquots were stored at −80 °C and were thawed as per need. Cells between three to six passages were used for electroporation experiments.
Figure 1. (A) Buffalo fibroblast cells outgrowth from a tissue explant in a T-25 culture flask. (B) Cells in a T-25 flask before electroporation experiments. (C) Cells immediately after electroporation. Fluorescence microscopic images showing transfection with best electroporation parameters in 2 mm cuvette (D’) with its corresponding bright field image (D), in 4 mm cuvette (E’) with its corresponding bright field image (E), and in electroporated control without plasmid (F’) with its corresponding bright field (F). Scale bar = 100 µm.
Design of CRISPR guides
In this study, we used two Addgene plasmids, namely the pSpCas9(BB)-2A-GFP (pX458) plasmid (#48138, 9.229 kb size) and the pSpCas9(BB)-2A-Puro (pX459) V2.0 plasmid (#62,988 9.174 kb size). The sgRNA (forward sequence, CCAGTCCGGTAAGTCCCACG; and reverse sequence, CGTGGGACTTACCGGACTGG) targeting exon 2 of the bovine ITGB-2gene was designed using the CHOPCHOP design tool website (https://chopchop.cbu.uib.no/). Ligation experiments were performed as described previously by Ran et al., 2013.Citation33 Briefly, both all-in-one CRISPR plasmids were linearized using the BbsI restriction enzyme (NEB# R0539S), and then annealed sgRNA was overnight ligated using the T4 DNA ligase (Sigma# 10481220001) at 4 °C. The ligated plasmid was amplified by transforming into E. coli SIG10 5α (Sigma#CMC0007), and the plasmid DNA was isolated using a geneJET plasmid miniprep kit (Thermo Scientific#K0503). The ligation of sgRNA sequences into CRISPR plasmids were confirmed by restriction enzyme analysis and by the Sanger sequencing. A successfully ligated plasmid was again transformed into E. coli SIG10 5α, and a large quantity of plasmid was isolated using a pure yield plasmid maxiprep kit (Promega#A2393). The isolated plasmids were stored at −20 °C and used as per need.
Optimization of electroporation parameters
An electrofusion machine (BTX-ECM 2001+) and two types of cuvettes (2 and 4 mm gap) were used to deliver square-wave electric pulses. Buffalo fibroblasts cultured in T-25 culture flasks (Thermo scientific#156367). When cells in the flask reached approximately 70–80% confluence (), they were trypsinized with a 0.25% trypsin EDTA solution, and then, adjusted the concentration to 1 × 106 cells/100 µL of transfection volume using Neubauer counting chamber. Adjusted cells in 100 µL of electroporation buffer (Bio-Rad#165-2676) were transferred to an electroporation cuvette (2 and 4‐mm gap, BTX#45-0125 and #45-0126, respectively). The different pX458 plasmid concentrations, 2.5, 5, 10 µg, were used in electroporation using the 4 different electric strength parameters (i) 10 milliseconds (ms), 1 square pulse (sqP), 300 voltage (V), (ii) 10 ms, 2 sqP, 300 V, (iii) 20 ms, 1 sqP, 300 V, (iv) 20 ms, 2 sqP, 300 V. Following electroporation (), the pulsed cells were resuspended in 2 mL of above-mentioned DMEM-based cell culture medium and cultured in 6‐well plates (Thermo Scientific#140675) under humidified 5% CO2 in air at 38.5 °C. We also analyzed the effect of cell types (adult vs. newborn) on transfection rates using best-optimized electroporation parameters.
Analysis of electroporated cells
Cells electroporated with different combinations of parameters were analyzed for transfection rates (percentage of GFP expression positive cells) and cell viability. The cells were observed 48 hr post-electroporation using an inverted microscope (Nikon Ti-E eclipse, Tokyo, Japan) equipped with fluorescence optics under appropriate excitation filters, and images were captured. Transfection percentage was calculated within each parameter group as a number of GFP positive cells/total numbers of cells. Cell viability was assessed by trypan blue stain exclusion test immediately after electroporation and viable cells counted by Neubauer chamber. The cell viability results of best electroporation parameters in 2 mm and 4 mm cuvette gap were further assessed by a flow cytometer (CytoFLEX, Beckman Coulter-Life Sciences) using Annexin V-FITC Kit (Sigma #APOAF-20TST). The transfection rates of adult and newborn fibroblasts at the best conditions were also further confirmed by flow cytometry, in which GFP expressing cells were analyzed in the FITC channel. All data were collected and analyzed using CytExpert software (v.2.3).
Electroporation of pX459 plasmid using best optimized parameters
Adult buffalo fibroblast cells were electroporated with the best electroporation parameters (electric strength, cuvette size, and amount of plasmid) achieved in the above optimization experiments. And, cells were cultured as described above. After 24 hr of electroporation, the cell culture media was changed to new culture media having 1.5 µg/mL puromycin (Sigma#P8833) and allowed cells to grow for 72 hr. Puromycin selection allows enriching the population of transfected cells. Puromycin was withdrawn after 72 hr, cells were continuously cultured for 8–10 days. When they reached 70–80% confluence, the cells were used for the analysis of genome editing efficiency.
Analysis of genome editing efficiency
We used different methods for the analysis of editing efficiency. These were T7E1 assay, TIDE (Tracking of indels by decomposition), ICE (interference of CRISPR edits), and PCR product Sanger sequencing following pX459 plasmid experiments. The genomic region surrounding the target site was amplified using one pair of primers (Forward-GCCTCCCAACTCTGTGTCTC, and reverse-GAGAGACACGGGCTGGTAAG), resulting in a 480-bp PCR product. Cells were lysed for DNA extraction by resuspending cell pellets in 50 µL of cell lysis buffer and 2 µL protein degrader mix (Invitrogen# A24372). Cell lysate preparation conditions were: 68 °C for 15 min, 95 °C for 10 min followed by hold at 4 °C. The PCR reaction was 3 µL of cell lysate,1 µL of 1 µM each of forward and reverse primer, 25 µL of AmpliTaq Gold™ Master mix (Applied Biosystems # 4398876), and then, added nuclease-free water to make a total volume of 50 µL reaction. PCR amplification conditions were as follows: 1 cycle at 95 °C for 5 min; 30 cycles at 95 °C for the 30 sec, 60 °C for 15 sec, and 72 °C for 30 sec; followed by a final extension at 72 °C for 5 min. The PCR products were used to determine the editing efficiency. The T7E1 assay was performed using a GeneArt genomic cleavage detection kit (Invitrogen# A24372) as per the manufacturer’s instructions and the editing efficiency was calculated by using the formula i.e., efficiency= [(sum of cleaved band intensities)/sum of cleaved and parental band intensities)] × 100. For TIDE and ICE analysis, PCR products were sent for Sanger sequencing. The Sanger sequencing files were then uploaded into the TIDE web tool (http://tide.nki.nl) and the ICE web tool (https://ice.synthego.com) for analysis. Both algorithms presented the percentage of indels. Further, the PCR products were cloned into the pJet1.2/blunt TA cloning vector using a CloneJET PCR cloning kit (Thermo Scientific#K1232). Cloned PCR products were then transformed into E.coli SIG10 5α, and plasmid was isolated from 10 E. coli colonies using a geneJET plasmid miniprep kit (Thermo Scientific#K0503). Isolated plasmids were sent for Sanger sequencing, and data were analyzed to determine the editing efficiency.
Statistical analysis
All data were expressed as mean ± SEM. The statistical significance analyses in percent cell viability and transfection rates in different cuvette sizes at different electroporation parameters were performed using the SPSS 16.0 statistical software package and One-way ANOVA was performed. For two-group comparison, means were compared with independent samples T-test. The minimum significant range of confidence was evaluated at 0.05 levels.
Results
Transfection rates at different electroporation parameters
To identify the optimal parameters for maximizing transfection rates in buffalo fibroblasts, a series of experiments were conducted using the all-in-one CRISPR plasmid (pX458) and different combinations of electroporation pulse conditions. These conditions included four settings: (i) 10 ms pulse duration, 1 square pulse (sqP), and 300 V, (ii) 10 ms pulse duration, 2 sqP, and 300 V, (iii) 20 ms pulse duration, 1 sqP, and 300 V, and (iv) 20 ms pulse duration, 2 sqP, and 300 V. Plasmid amounts of 2.5, 5, and 10 µg were tested using 2 and 4 mm gap size cuvettes. Transfection rates showed a positive correlation with increased plasmid amounts across all electroporation conditions. In the case of 2 mm cuvette gap size (), transfection rate was found significantly high at 10 ms pulse duration, 1 sqP, and 300 V, with a significant increase in transfection rates as plasmid amounts increased from 2.5 µg (5.0 ± 0.39) to 5 µg (6.97 ± 0.10) and 10 µg (7.22 ± 0.34). For 4 mm gap size cuvettes (), the highest transfection rates were achieved at 20 ms pulse duration, 1 sqP, and 300 V. However, increasing plasmid amounts from 2.5 µg (5.76 ± 0.41) to 5 µg (6.48 ± 0.38) did not significantly improve transfection rates, whereas it was higher with 10 µg (10.90 ± 0.13).
Figure 2. Transfection rates (A and B) and cell viability (C and D) of all-in-one CRISPR plasmid (pX458) at different electroporation conditions in 2 mm cuvette and 4 mm cuvette. Transfection rates (E) and cell viability (F) of pX458 plasmid for the best conditions in 2 and 4 mm cuvette. All values are shown as Mean ± SEM. Different superscripts denote statistically different values (p < 0.05). small alphabets indicate within groups and capital alphabets indicate between groups.
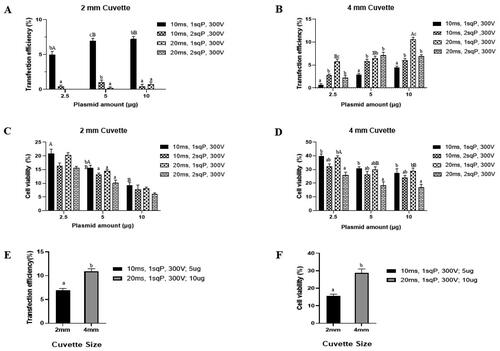
Consequently, the optimal electroporation parameters selected for subsequent experiments were 10 ms pulse duration, 1 sqP, and 300 V with 5 µg plasmid for 2 mm cuvette size, and 20 ms pulse duration, 1 sqP, 300 V with 10 µg plasmid for 4 mm cuvette size. Fluorescence images showing transfection with their optimal electroporation parameters in 2 mm cuvette (, 1D’), in 4 mm cuvette ( and 1E’) and in electroporated control ( and Citation1F’). We also noticed that using 4 mm cuvette with 2 sqP of 20 ms duration had higher transfection rates than 1 sqP of 10 ms duration, indicating potential for high transfection. However, extending pulse duration and increasing the number of pulses had compromised cell viability ().
Cell viability rates at different electroporation parameters
On comparing cell viability across different plasmid amount groups (2.5, 5, and 10 µg) under various electroporation conditions, it was found that cell viability decreased with higher plasmid amounts, with the lowest viability observed at 10 µg plasmid. Using 4 mm gap size cuvette and subjecting cells to 20 ms pulse duration, 2 sqP, and 300 V resulted in the lowest cell viability compared to other conditions (). In contrast, when using 2 mm cuvette size, the impact of electric pulse conditions on cell viability was less pronounced (). However, it’s important to note that cell viability in 2 mm cuvette was generally lower than that in 4 mm cuvette across all electroporation conditions (). This suggests that the cuvette size itself have a notable influence on cell viability, with smaller cuvette gap size potentially exerting greater stress on the cells during electroporation.
Effect of cuvette gap size on transfection rates and cell viability
Following the optimization process discussed above, the transfection rates () were higher when using the 4 mm cuvette (10.90 ± 0.13) compared to the 2 mm cuvette (6.97 ± 0.10). Additionally, cell viability () was significantly improved in the 4 mm cuvette (28.7 ± 0.90) compared to the 2 mm cuvette (15.6 ± 0.59). This improvement in cell viability was further confirmed by flow cytometry analysis, which revealed a higher viability percentage in the 4 mm cuvette (41.12 ± 0.79) in contrast to the 2 mm cuvette (20.59 ± 0.21). As a result, the best optimal electroporation parameters, specifically using a 4 mm cuvette with settings of 20 ms pulse duration, 1sqP, 300 V, and 10 µg plasmid, were identified as the most effective for delivering all-in-one CRISPR plasmids into adult buffalo fibroblast cells.
Effect of cell age on transfection rates
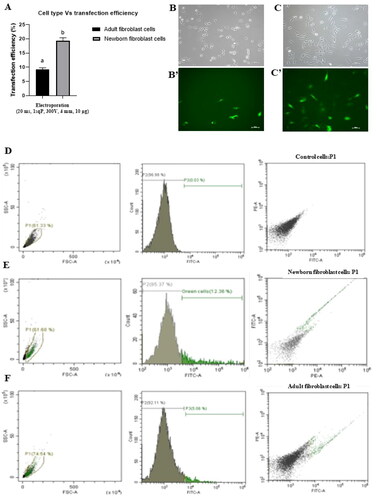
In addition to optimizing electroporation parameters, we investigated the influence of cell age, comparing adult and newborn fibroblast cells using the best parameters (20 ms pulse duration, 1sqP, 300 V, 4 mm cuvette size, and 10 µg plasmid). The results () revealed significantly higher transfection rates in newborn fibroblast cells (19.40 ± 0.98) compared to adult fibroblast cells (9.22 ± 0.59). Fluorescence images presented the expression of green fluorescence protein in both cell types, with newborn fibroblast cells displaying more robust fluorescence (') compared to adult cells ('). Further, flow cytometry analysis () supported these findings, showing higher transfection rates in newborn fibroblast cells (12.18 ± 0.22) than that of adult fibroblast cells (4.38 ± 0.23).This experiment indicates that age of cell is another important factor that greatly affects the efficiency of transfection by electroporation.
Figure 3. Transfection rates (A) in adult and newborn fibroblast cells following the electroporation of cells with best optimized conditions (4 mm cuvette gap, 20 ms, 1 sqP, 300 V, 10 µg plasmid DNA). Fluorescence images showing transfection efficiency in adult fibroblast cells (B’) with its corresponding bright field image (B) and in newborn fibroblast cells (C’) with its corresponding bright field (C).transfection efficiency was also determined using the flow cytometry, control cells (D) in which no plasmid DNA was used, newborn fibroblast cells (E) have 12.18 ± 0.22% transfection rates, and adult buffalo fibroblast cells (F) have 4.38 ± 0.23. Values are shown as Mean ± SEM. Different superscripts denote statistically different values (p < 0.05). Scale bar for microscopy images = 100 µm.
Evaluation of the gene editing efficiency
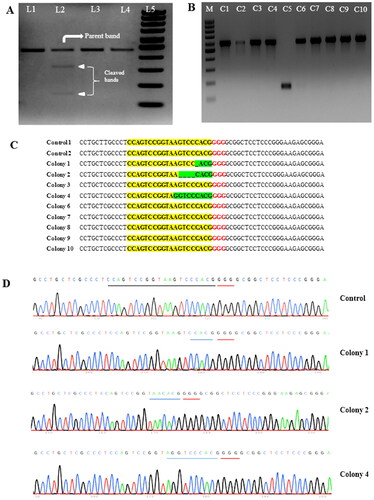
Adult buffalo fibroblast cells were subjected to electroporation using an all-in-one CRISPR plasmid (pX459), containing a puromycin antibiotic resistance marker, with the best optimized conditions (20 ms pulse duration, 1 sqP, 300 V, and 10 µg plasmid in a 4 mm cuvette). Subsequently, puromycin cell selection was performed, and then, genomic DNA was isolated for the T7E1 assay to detect mutations induced by the all-in-one CRISPR plasmid at the targeted ITGB2 gene locus. The T7E1 assay results demonstrated indel formation at the gene-specific locus (), with editing efficiency of 19.5%. Further, the Sanger sequencing data from electroporated mixed population cells and non-electroporated control cells were used in TIDE and ICE algorithms to determine editing efficiency. TIDE analysis resulted in 26.7% editing efficiency () and an aberrant sequence plot (’), whereas, ICE analysis resulted in 24% editing efficiency () and a discordance plot (’). To further validate the editing efficiency determined through these methods, we employed the TA cloning method. Plasmids from 10 distinct E. coli colonies were TA cloned and sent for Sanger sequencing. Among these, colony 5 exhibited self-ligation, which was excluded from data analysis (). Of the remaining 9 plasmid sequencing datasets, 3 colonies (colony 1, 2 and 4) exhibited indels at the target gene locus () that resulted in an editing efficiency of 33%. Chromatograms of the control (wild type) and the three edited colonies at the target site are shown in .
Figure 4. CRISPR editing efficiency determined using TIDE and ICE software. TIDE (Tracking of indels by decomposition) analyses showing indel spectra and aberrant PCR sequencing plot. The graph on the left shows indels frequencies within ±10 bp from theoretical gRNA breakpoints (a), and the graph on the right depict PCR sequence aberrations and theoretical gRNA cuts indicated by a blue line (A’), and total editing efficiency was 26.7%. ICE (inference of CRISPR edits) analyses show 24% editing efficiency (B) and discordance plots (B’). The gRNA region in cells showed a knockout score of 24.
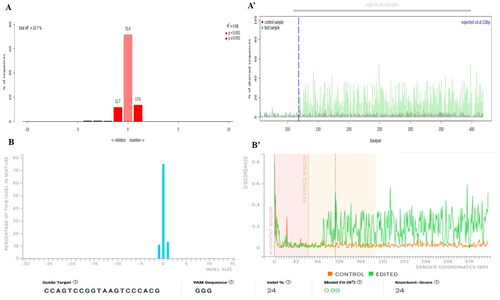
Figure 5. Gel image (A) of genomic cleavage detection assay (T7E1) performed in buffalo fibroblast cells electroporated with all-in-one CRISPR plasmid (pX459) targeting the ITGB2 gene. Lane 1, sample without T7E1 enzyme Digestion; Lane 2, sample, showing parent and both the cleaved bands, after addition of T7E1 enzyme. The negative control sample (with intact gene locus) without T7E1 enzyme (Lane 3) and with T7E1 enzyme (Lane 4); Lane 5, 100 bp DNA ladder. Gel image (B) shows the correct ligation of PCR products encompassing the targeted region of ITGB2 gene into the pJET1.2/blunt cloning vector (480 bpPCR product size + 94 bp vector backbone) in 9 E.coli colonies, except colony C5 that has self-ligated vector. Sanger sequencing results (C) at target site showing three colonies have indels such as colony 1 (−1bp), colony 2 (−4 bp), colony 4 (−1bp/+1bp). sequences highlighted yellow represents guide sequence, red represents PAM sequence, and green represents changes in the targeted sequence. Chromatogram of the control (wild type) and the three edited colonies at the target site (D).
Discussion
Electroporation is a commonly used non-viral transfection method, in which high voltage electric pulses are used to create temporary pores in cell membranes, to deliver CRISPR components in mammalian cells.Citation20,Citation34 It has been well established that if the electric strength parameters are appropriate, the electroporated cells have high transfection rates and cell viability.Citation35 Several studies reported that electroporation-based transfection efficiency differs with experimental conditions.Citation9,Citation36–38 Therefore, parameters such as pulse duration, number of pulses, electric strength, plasmid DNA amount, and cuvette type need to be optimized to initiate electroporation of any new cell type to achieve effective transfection outcomes. In the present study, we conducted the experiments to optimize the electroporation conditions for the delivery of all-in-one CRISPR plasmids into adult buffalo fibroblast cells. We found that for transfection of CRISPR plasmids in 100 µL of electroporation buffer having 10 µg of plasmid DNA and 106 cells, optimal conditions were a single square pulse for the 20 ms duration at 300 V in a 4 mm cuvette which resulted in an 11% transfection rate and successful editing of ITGB2 gene with 30% efficiency in adult buffalo fibroblast cells.
The amount of DNA added into the cell suspension during electroporation experiments have been identified as a critical factor in the successful delivery of CRISPR plasmids into mammalian cells. In this study, we investigated different DNA concentrations, such as 2.5, 5, and 10 μg per 100 μL electroporation volume. It was evident that increasing plasmid concentrations led to higher transfection rates, irrespective of the electroporation conditions. However, this improved transfection rates were counterbalanced by reduced cell viability. Similar observations were also previously reported,Citation9,Citation39,Citation40 validating the importance of plasmid amount in optimizing transfection protocols. In this study, the optimal DNA amount for all-in-one CRISPR plasmids for buffalo fibroblast transfection was determined to be 10 μg. In agreement with our results, this same DNA quantity (10 μg) was used for gene editing in bovine fibroblasts.Citation41
Another important parameter in electroporation is cuvette gap size. We observed that transfection rates and cell viability were higher in the 4 mm cuvette gap than in the 2 mm cuvette gap. This is possibly due to the more distance of cells from electrodes preventing direct contact of cells with electrodes that further protects the cells from harmful effects of non-physiological pH, toxic electrode products, and extensive heat generated near the electrodes during the electroporation protocol.Citation42 Several studies reported the use of 4 mm cuvette for efficient electroporation of fibroblast cells in farm animals.Citation3,Citation4,Citation7,Citation9,Citation16,Citation24 In buffalo species, electroporation has demonstrated varying efficiencies. For instance, Zhao et al. 2020Citation18 reported a 40% transfection rate, while Lu et al. 2018Citation3 reported a 35.5% transfection rate. In this study, we achieved an 11% transfection rate, which may be attributed to the relatively large plasmid size (9.2 kb) used; whereas, they used smaller plasmids of around 5 kb in size and fetal fibroblast cells.
We also investigated the influence of cell age by subjecting both adult and newborn buffalo fibroblast cells. We observed significantly higher transfection rates (20%) in newborn fibroblasts compared to adult fibroblasts (12%). This discrepancy can be attributed to high proliferative activity of newborn fibroblast cells, since, it has been reported that actively dividing cells have high transfection efficiency.Citation43 Varying transfection rates have been reported in different cell types. For examples, Hyder et al. 2020 achieved a transfection efficiency of 23% in bovine fetal fibroblast cells using T2-RMCE-Venus (6.3 kb) plasmid.Citation9 Wei et al. 2020 obtained a transfection efficiency of 17.80% in Guangdong small-ear spotted (GDSS) pig kidney fibroblast cells with a pEGFP-N1 vector (4.7 kb).Citation16 Ishino et al. 2018 reported a lower transfection efficiency of 0.35 ± 0.1% with a pSpCas9(BB)-2A-GFP plasmid (9.2 kb) in fibroblasts from the ears of adult Japanese Black cattle.Citation41 For buffalo fibroblasts, prior studies used the different pulse conditions, for examples, Kumar et al. 2018Citation8 used a 300 V single pulse for 10 ms in a 2 mm cuvette gap for water buffalo fetal fibroblasts, Su et al. 2018Citation20 used with 225 V single pulse for 10 ms in a 4 mm cuvette gap for swamp buffalo fetal fibroblasts, and Lu et al. 2018Citation3 applied a 300 V single pulse for 10 ms in a 4 mm cuvette gap for male fetus ear skin fibroblasts. However, in this study, we identified the 300 V single pulse for 20 ms in a 4 mm cuvette gap as optimal for the transfection of the 9.2 kb all-in-one CRISPR plasmid into adult buffalo fibroblast cells. It has been suggested that large plasmids can pose challenges in transfection, and an increase in pulse duration is often necessary to facilitate the uptake of such large plasmids.Citation12,Citation44
The optimized conditions discussed above were then used to edit the ITGB2 gene. This gene encodes the CD18 protein, which plays a pivotal role in cellular adhesion, cell surface signaling, and immune responses. Previous research has reported the severe disorders associated with mutations of this gene in livestock species, notably Bovine Leukocyte Adhesion Deficiency (BLAD) and pathology of Mannheimia haemolytica leukotoxin in bovines.Citation29,Citation30 In recent study, it has been demonstrated the potential of gene editing techniques in introducing specific modifications to the ITGB2 gene.Citation31 In this study, using optimized conditions, we edited the ITGB2 gene in buffalo fibroblasts. Based on T7E1 results, we achieved successful editing with 19.5% efficiency. We further validated the T7E1 assay results using the TIDE and ICE algorithms, which rely on Sanger sequencing data. TIDE analysis revealed indels frequency of 26.7%, while ICE analysis revealed indels frequency of 24%. Further validation involved cloning the PCR products from the targeted ITGB2 gene region into a TA cloning vector, followed by Sanger sequencing. This validation also resulted 33% editing efficiency. These validation methods collectively confirmed successful editing, ranging from 1 to 3 nucleotide deletion and A/G replacement, of ITGB2 gene in adult buffalo fibroblast cells. Previously, various editing efficiencies have been reported according to the experimental conditions, Wang et al., 2015Citation38 reported 21% editing efficiency in pig fibroblasts, Ishino et al., 2018Citation41 reported 11.3% gene editing in cattle fibroblasts, and Su et al., 2018Citation20 reported 75% editing in buffalo fibroblasts. These discrepancies may be due to differences in the type of targeted gene loci and experimental conditions. The optimized conditions reported in this study represent a valuable tool for efficiently introducing targeted gene modifications into somatic cells with desired edits of any gene(s), including ITGB2 gene. Therefore, future research aims to harness these optimized conditions to implement desired genetic modifications in buffalo cells.
In summary, electroporation-based transfection conditions allows successful CRISPR-based gene editing in difficult-to-transfect cells like adult buffalo primary fibroblasts. Key parameters include plasmid DNA amount, pulse conditions, cuvette gap size, and cell type. These parameters enable the generation of gene-edited buffalo cells for various research applications, including desired edits within the ITGB2 gene.
Author contributions
K.K.B. conducted experiments, analyzed data, and prepared figures. M.P. contributed to cell viability experiments and data analysis. D.K. and P.S.Y. provided chemicals, interpreted data, and assisted in cell culture establishment. C.R.L contributed in study design and execution. N.L.S. contributed to study design, provided chemicals, interpreted data, and drafted and reviewed the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets generated during the current study are available in the NCBI-Gen Bank repository, [Accession numbers: ON900160, ON900161, ON900162, ON900163, ON900164, ON900165, ON900166, ON900167, ON900168, ON900169, ON900170].
Additional information
Funding
References
- Chicaybam L, Barcelos C, Peixoto B, et al. An efficient electroporation protocol for the genetic modification of mammalian cells. Front Bioeng Biotechnol. 2016;4:99.
- Hyun S, Lee G, Kim D, et al. Production of nuclear transfer-derived piglets using porcine fetal fibroblasts transfected with the enhanced green fluorescent protein. Biol Reprod. 2003;69(3):1060–1068.
- Lu F, Luo C, Li N, et al. Efficient generation of transgenic buffalos (Bubalus bubalis) by nuclear transfer of fetal fibroblasts expressing enhanced green fluorescent protein. Sci Rep. 2018;8(1):6967.
- Nakayama A, Sato M, Shinohara M, et al. Efficient transfection of primarily cultured porcine embryonic fibroblasts using the Amaxa nucleofection system™. Cloning Stem Cells. 2007;9(4):523–534.
- Mehta P, Kaushik R, Singh KP, et al. Establishment, growth, proliferation, and gene expression of buffalo (Bubalus bubalis) transgenic fetal fibroblasts containing human insulin gene, and production of embryos by handmade cloning using these cells. Cell Reprogram. 2018;20(2):135–143.
- Watanabe S, Iwamoto M, Suzuki SI, et al. A novel method for the production of transgenic cloned pigs: electroporation-mediated gene transfer to non-cultured cells and subsequent selection with puromycin. Biol Reprod. 2005;72(2):309–315.
- Yekta AA, Dalman A, Sanati MH, et al. Optimization of the electroporation conditions for transfection of human factor IX into the goat fetal fibroblasts. Cell J. 2013;14(4):270.
- Kumar D, Sharma P, Vijayalakshmy K, et al. Generation of venus fluorochrome expressing transgenic handmade cloned buffalo embryos using sleeping beauty transposon. Tissue Cell. 2018;51:49–55.
- Hyder I, Eghbalsaied S, Kues WA. Systematic optimization of square-wave electroporation conditions for bovine primary fibroblasts. BMC Mol Cell Biol. 2020;21(1):1–8.
- Neumann E, Schaefer-Ridder M, Wang Y, et al. Gene transfer into mouse lyoma cells by electroporation in high electric fields. Embo J. 1982;1(7):841–845.
- Heiser WC. Optimizing electroporation conditions for the transformation of mammalian cells. Transcription Factor Protocols. 2000;130:117–134.
- Lesueur LL, Mir LM, Andre FM. Overcoming the specific toxicity of large plasmids electro transfer in primary cells in vitro. Mol Ther Nucleic Acids. 2016;5(3):e291.
- Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15(5):321–334.
- Liang X, Potter J, Kumar S, et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J Biotechnol. 2015;208:44–53.
- Qin W, Wang H. Delivery of CRISPR-Cas9 into mouse zygotes by electroporation. In: Liu C, Du Y, editors. Microinjection. Methods in Molecular Biology. New York, NY: Humana Press; 2019;1874:179–190.
- Wei YY, Zhan QM, Zhu XX, et al. Efficient CRISPR/Cas9-mediated gene editing in Guangdong small-ear spotted pig cells using an optimized electro transfection method. Biotechnol Lett. 2020;42(11):2091–2109.
- Jin L, Kim D, Roh S. Comparison of various transfection methods in human and bovine cultured cells. Intern J Oral Biol. 2014;39(4):177–185.
- Zhao X, Nie J, Tang Y, et al. Generation of transgenic cloned buffalo embryos harboring the EGFP gene in the Y chromosome using CRISPR/Cas9-mediated targeted integration. Front Vet Sci. 2020;7:199.
- Heo YT, Quan X, Xu YN, et al. CRISPR/Cas9 nuclease-mediated gene knock-in in bovine-induced pluripotent cells. Stem Cells Dev. 2015;24(3):393–402.
- Su X, Cui K, Du S, et al. Efficient genome editing in cultured cells and embryos of Debao pig and swamp buffalo using the CRISPR/Cas9 system. In Vitro Cell Dev Biol Anim. 2018;54(5):375–383.
- Punetha M, Chouhan VS, Sonwane A, et al. Early growth response gene mediates in VEGF and FGF signaling as dissected by CRISPR in corpus luteum of water buffalo. Sci Rep. 2020;10(1):6849.
- Punetha M, Kumar S, Paul A, et al. Deciphering the functional role of EGR1 in Prostaglandin F2 alpha induced luteal regression applying CRISPR in corpus luteum of buffalo. Biol Res. 2021;54(1):9.
- Paul A, Bharati J, Punetha M, et al. Transcriptional regulation of thrombospondins and its functional validation through CRISPR/Cas9 mediated gene editing in corpus luteum of water buffalo (Bubalus bubalis). Cell Physiol Biochem. 2019;52(3):532–552.
- Dua S, Bansal S, Gautam D, et al. Production of MSTN Gene-Edited Embryos of Buffalo Using the CRISPR/Cas9 System and SCNT. Cell Reprogram. 2023;25(3):121–127.
- Selokar NL, Saini M, Palta P, et al. Hope for restoration of dead valuable bulls through cloning using donor somatic cells isolated from cryopreserved semen. PLOS One. 2014;9(3):e90755.
- Deng Y, Liu Q, Luo C, et al. Generation of induced pluripotent stem cells from buffalo (Bubalus bubalis) fetal fibroblasts with buffalo defined factors. Stem Cells Dev. 2012;21(13):2485–2494.
- Meng F, Li H, Wang X, et al. Optimized production of transgenic buffalo embryos and offspring by cytoplasmic zygote injection. J Anim Sci Biotechnol. 2015;6(1):1–7.
- Rizk MA, Mostafa NY. Extraction and characterization of collagen from buffalo skin for biomedical applications. Orient J Chem. 2016;32(3):1601–1609.
- Gerardi AS. Bovine leukocyte adhesion deficiency: a brief overview of a modern disease and its implications. Acta Vet Hung. 1996;44(1):1–8.
- Shanthalingam S, Srikumaran S. Intact signal peptide of CD18, the β-subunit of β2-integrins, renders ruminants susceptible to Mannheimia haemolytica leukotoxin. Proc Natl Acad Sci USA. 2009;106(36):15448–15453.
- Shanthalingam S, Tibary A, Beever JE, et al. Precise gene editing paves the way for derivation of Mannheimia haemolytica leukotoxin-resistant cattle. Proc Natl Acad Sci USA. 2016;113(46):13186–13190.
- Selokar NL, Sharma P, Krishna A, et al. Establishment of a somatic cell bank for Indian buffalo breeds and assessing the suitability of the cryopreserved cells for somatic cell nuclear transfer. Cell Reprogram. 2018;20(3):157–163.
- Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308.
- Roth TL, Puig-Saus C, Yu R, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature. 2018;559(7714):405–409.
- Ovcharenko D, Jarvis R, Hunicke-Smith S, et al. High-throughput RNAi screening in vitro: from cell lines to primary cells. RNA. 2005;11(6):985–993.
- Somiari S, Glasspool-Malone J, Drabick JJ, et al. Theory and in vivo application of electroporative gene delivery. Mol Ther. 2000;2(3):178–187.
- Ross JW, Whyte JJ, Zhao J, et al. Optimization of square-wave electroporation for transfection of porcine fetal fibroblasts. Transgenic Res. 2010;19(4):611–620.
- Wang X, Yu H, Lei A, et al. Generation of gene-modified goats targeting MSTN and FGF5 via zygote injection of CRISPR/Cas9 system. Sci Rep. 2015;5(1):13878.
- Chopra S, Ruzgys P, Maciulevicius M, et al. Investigation of plasmid DNA delivery and cell viability dynamics for optimal cell electrotransfection in vitro. Appl Sci. 2020;10(17):6070.
- Yao S, Rana S, Liu D, et al. Electroporation optimization to deliver plasmid DNA into dental follicle cells. Biotechnol J. 2009;4(10):1488–1496.
- Ishino T, Hashimoto M, Amagasa M, et al. Establishment of protocol for preparation of gene-edited bovine ear-derived fibroblasts for somatic cell nuclear transplantation. Biomed Res. 2018;39(2):95–104.
- Grys M, Madeja Z, Korohoda W. Avoiding the side effects of electric current pulse application to electroporated cells in disposable small volume cuvettes assures good cell survival. Cell Mol Biol Lett. 2017;22(1):1–13.
- Anderson ML, Spandidos DA, Coggins JR. Electroporation of lymphoid cells: factors affecting the efficiency of transfection. J Biochem Biophys Methods. 1991;22(3):207–222.
- Lucas ML, Heller R. Immunomodulation by electrically enhanced delivery of plasmid DNA encoding IL-12 to murine skeletal muscle. Mol Ther. 2001;3(1):47–53.