
Abstract
The present study aims to identify genomic variants through a whole genome sequencing (WGS) approach and uncover biological pathways associated with adaptation and fitness in Indian yak populations. A total of 30 samples (10 from each population) were included from Arunachali, Himachali and Ladakhi yak populations. WGS analysis revealed a total of 32171644, 27260825, and 32632460 SNPs and 4865254, 4429941, and 4847513 Indels in the Arunachali, Himachali, and Ladakhi yaks, respectively. Genes such as RYR2, SYNE2, BOLA, HF1, and the novel transcript ENSBGRG00000011079 were found to have the maximum number of high impact variants in all three yak populations, and might play a major role in local adaptation. Functional enrichment analysis of genes harboring high impact SNPs revealed overrepresented pathways related to response to stress, immune system regulation, and high-altitude adaptation. This study provides comprehensive information about genomic variants and their annotation in Indian yak populations, thus would serve as a data resource for researchers working on the yaks. Furthermore, it could be well exploited for better yak conservation strategies by estimating population genetics parameters viz., effective population size, inbreeding, and observed and expected heterozygosity.
Introduction
The yaks (Poephagus grunniens) are ruminants that are distributed in two eco-environments, viz., the northwestern part of India (Himachal Pradesh and Union Territory of Ladakh) with arid dry climate and the eastern part (Arunachal Pradesh and Sikkim) with wet cold environment. Local farmers employ these animals for multiple purposes, including milk, meat, wool, fuel, and these also serve as pack animals for high-altitude transportation. Yaks can graze at an extremely high altitude and withstand extremely cold temperatures, down to −40 °C.Citation1 Yaks have certain special characteristics that enable them to thrive in high-altitude environments, such as heightened environmental sensing, relatively large size of lungs, and heart.Citation2
According to Indian Livestock Census (2019), the total Indian yak population is estimated to be around 58 thousands and has shown a significant decrease of 24.08 percent from the previous Livestock Census (2012).Citation3 Indian yaks are divided into four groups based on their area of inhabitation; Arunachali yaks, Himachali yaks, Ladakhi yaks, and Sikkimi yaks. However, only one yak breed, that is, the Arunachali yak is registered till to date which necessitates the characterization and conservation of other yak populations. It is widely acknowledged that comprehensive molecular data on within-breed and between-breed diversity is required for successful animal genetic resource management.Citation4 Additionally, deciphering the genomic variation would also serve as a valuable resource for studying complex traits in Indian yak populations.
Single nucleotide polymorphisms (SNPs) are the most common type of genetic markersCitation5 and are preferred over microsatellite markers for various genomic studies. SNP maps are denser and provide more detailed information than microsatellite maps. Recent technological developments and reductions in the cost of next-generation sequencing technology provide rapid and accurate identification of SNP markers for a large number of individuals. Sivalingam et al.Citation6 used a double digestion restriction-associated DNA (ddRAD) sequencing technique to find SNPs in Indian yak populations. Although ddRAD sequencing is less expensive than whole genome sequencing, it has the limitation of smaller genome analysis. Presently, whole genome sequencing (WGS) is increasingly used to uncover genetic variation,Citation7 and entails deciphering the order of bases in an individual’s entire genome, aided by automated DNA sequencing procedures and computer systems to put together the massive amounts of sequence data. Additionally, WGS has an edge over SNP chips in terms of providing details on rare variants. Various researchers have used the WGS technique in yak populations around the world to explore domestication signatures,Citation8 understand adaptation to high altitude,Citation9 evolution and divergence,Citation10 and in the development of de novo assembly.Citation11 Moreover, the role of whole genome resequencing methods in conservation biology has also been summarized.Citation12
To the best of our knowledge, this is the first study reporting genomic variant identification, annotation, and molecular pathways associated with local adaptation and fitness in Indian yak populations (Arunachali, Himachali, and Ladakhi yak) using whole genome sequencing. This study would not only provide genomic data resources for researchers working on the yaks but also help to steer future research into genetic mechanisms underlying various traits in Indian yak populations.
Materials and methods
Sample collection
A total of 30 yaks belonging to three different populations (10 samples from each population, that is, Himachali, Ladakhi, and Arunachali) were used for this study. The genomic DNA (gDNA) of these yaks were obtained from the DNA repository of various labs at ICAR-National Bureau of Animal Genetic Resources, Karnal, India. The quality and quantity of isolated gDNA were checked using Qubit ds DNA estimation and agarose gel electrophoresis.
Whole genome sequencing shotgun library preparation
DNASeq libraries for all samples were prepared using the NEBNext UltraII DNA library preparation kit for Illumina; Cat No. E7770 (New England Biolabs), according to manufacturer’s recommended protocol, and sequencing was done on the S4 flowcell of the NOVASEQ 6000 using 150 bp paired-end chemistry. A commercial service provider having adequate experience in NGS data generation was selected for the library preparation and sequencing of all the DNA samples.
Briefly, 1 µg of gDNA was taken for downstream processing, and it was end-repaired and A-tailed after fragmentation followed by ligation of indexed adapters. To make the final gDNA library, the products were purified and enriched using PCR. A Qubit Fluorometer (Invitrogen, Life Technologies, Grand Island, NY, USA) was used to quantify the library. Library fragment distribution was checked on the HSDNA kit using Tapestation (Agilent Technologies, USA). The tagged gDNA libraries were mixed in equal proportions and put into the c-bot automated system for cluster generation. Post cluster generation, the libraries were put into the Illumina S4 Flow Cell of Illumina Novaseq 6000 sequencing system and 2 × 150 bp paired end chemistry was used for sequencing. Further, the sequenced samples were demultiplexed and the CASAVA v1.8.2 (Illumina Inc.) programme was used to trim the indexed adapter sequences. FastQC, a quality control tool for high throughput sequence data was then used to assess the quality of raw sequencing data.
Read alignment, variant calling, and filtering
The Standard Broad Genomics Platform was followed for further analysis to call SNPs and indels. First, indexing of the reference genome (GCA_005887515.2) was done by BWACitation13 followed by thirty paired-end Illumina yak DNA sequenced fastq files into a unaligned BAM (Binary Alignment Map) files and marking of Illumina adapters using Picard tools. Further, SamToFastq was used to convert uBAM into fastq file and discount adapter sequences. The generated fastq files were aligned with the yak reference genome (GCA_005887515.2) using BWA-MEM to generate Sequence Alignment Map (SAM) files. MarkDuplicates from Picard was used to tag duplicate reads and output files were used to create the index “.bai” file. HaplotypeCaller of GATK version 4.2 was used to call germline SNPs and InDels with the -ERC GVCF, -G Standard and -G AS_Standard parameters. Further, GATK’s CombineGVCFs was used to combine GVCFs files of each population, followed by GenotypeGVCFs of GATK to perform joint genotyping on each group of samples pre-called with HaplotypeCaller. Hard-filtering was done with specific thresholds to remove variants that had annotation value lesser or greater than set threshold values using the -VariantFiltration command. QualByDepth (QD < 2.0), FisherStrand (FS > 60.0), StrandOddsRatio (SOR > 3.0), RMSMappingQuality (MQ < 40.0), MappingQualityRankSumTest (MQRankSum < −12.5), and ReadPosRankSumTest (ReadPosRankSum < −8.0) were used for variant filtration. Finally, generated .vcf file were used for further variant annotation study.
Variant annotation using SnpEff
Filtered SNPs and Indels were annotated functionally by SnpEff version 5.0e.Citation14 SnpEff is a toolbox that aids in the annotation of genetic variants and the prediction of functional effects. The SnpEff database LU_Bosgru_v3.0.99 was downloaded for variant annotation. SNPs were categorized based on number of effects by impact (high, low, moderate, and modifier); functional classes (missense, nonsense, and silence); type and region (downstream, exon, intergenic, intron, splice site acceptor, splice site donor, splice site region, transcript, upstream, UTR_3_prime, and UTR_5_prime).
Gene ontology analysis
Gene ontology (GO) analysis has been a commonly used approach for functional studies of large scale genomic or transcriptomic data. GO analysis has been widely used in functional analysis and allows the identification of important categories associated with functions of interests. GO terms can provide useful information to elucidate the regulatory mechanism. Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8Citation15 was used to identify over-represented biological processes for genes with high impact variants. P-value or EASE score along with fold enrichment parameter statistical method have been used by DAVID v6.8. which could rank the enriched terms in a more comprehensive way.
Results
Whole genome sequencing and read alignment
Whole genome of 30 yaks from three different native populations- Arunachali, Himachali and Ladakhi were sequenced on an average ∼10× coverage. These three different yak populations belong to different agroecological climates of India. An average of 19.9 billion reads were generated for each sample. Alignment of the reads with the yak reference genome using the Burrows-Wheeler AlignerCitation13 resulted into 97.6% mapping rate and 9.5 read depth on average ().
Table 1. Summary of whole genome sequencing data.
Variant identification
In the present investigation, a total of 32171644, 27260825, and 32632460 SNPs and, 4865254, 4429941, and 4847513 Indels were identified in the Arunachali, Himachali, and Ladakhi yak, respectively. The highest number of SNPs were found on chromosome 1, while the lowest were observed on chromosome Y. The highest number of InDels were found on chromosome 12, while the lowest number were found on chromosome Y. The chromosome-wise SNPs and InDels of these three populations have been summarized in . The differences between the number of SNPs and InDels, among the populations, provide us with raw idea related to population diversity.
Table 2. Chromosome-wise distribution of SNPs and InDels across the genome in Arunachali, Himachali, and Ladakhi yaks.
Variant annotation
The filtered variants (SNPs and InDels) were further annotated using SnpEff.Citation14 Annotation of SNPs and InDels provides detailed information regarding their position in the genome which includes intronic, untranslated region, upstream, downstream, splice site, or intergenic regions. Coding effects such as synonymous or non-synonymous amino acid replacement, start codon gains or losses, stop codon gains or losses, or frame shifts can also be predicted. The results revealed that a maximum number of SNPs and InDels were located in the intronic region in all three yak populations followed by the intergenic region. Region-specific distribution of SNPs and InDels have been mentioned in . After categorizing SNPs based on their impact, the majority fell into the modifier category (98.6%) followed by moderate, low and high (). However, based on the functional classes, the maximum SNPs were included under missense, followed by silent and nonsense. It is worth mentioning that RYR2, SYNE2, BOLA, and HF1 genes and the novel transcript ENSBGRG00000011079 were found to have a maximum number of high impact variants (>10) in all three yak populations.
Figure 1. Categorization of identified SNPs in Arunachali, Himachali and Ladakhi yaks (SNPs were classified: number of effects by impact (high, low, and moderate); number of effects by functional classes (Missense, Nonsense and Silence)).
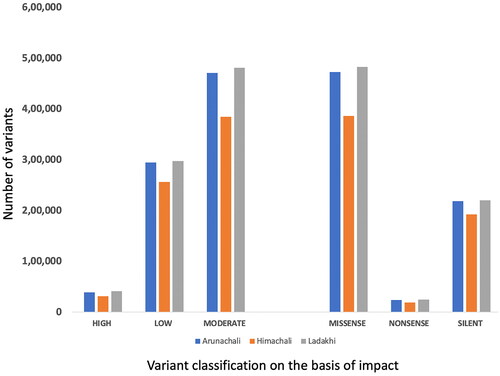
Table 3. Annotation of SNPs and InDels detected in Arunachali, Himachali, and Ladakhi yaks.
Gene ontology analysis
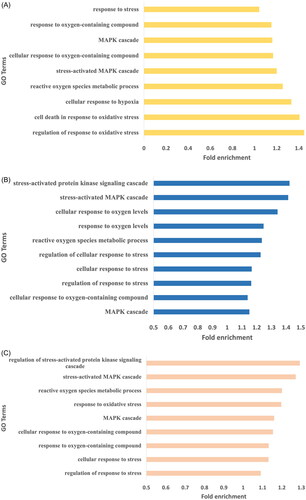
Genes harboring high impact SNPs were selected for gene ontology analysis. A total of 948, 862, and 761 biological pathways were identified in Arunachali, Himachali, and Ladakhi yaks, respectively (Supplementary Table 1; P < 0.05). All the biological pathways were further screened for the identification of immune-related pathways for a better understanding of fitness under local environmental conditions. Out of these three populations, the highest number of immune-related GO terms were found in Himachali yak as compared to Ladakhi and Arunachali yak populations (P < 0.05), including activation of immune response, positive regulation of innate immune response, activation of innate immune response, RIG-I signaling pathways, transport of virus, interleukin-1 beta secretion, and so on. Genes NOD2, SYK, C3, STAP1, CARD10, EIF2AK2, RC3H2, HDAC7, ADGRG3, RASSF2, AGO1, RPS3, CHI3L1, and SASH1 were found to be common in all three populations. Further, the mitogen-activated protein kinase (MAPK) cascade-related pathways were significant and highly enriched in all three yak populations. Each living body cell has the basic ability to cope up and respond to changing environmental conditions. The MAPK module, a three-tiered cascade of protein kinases, is a frequently used molecular tool for inducing such responses.Citation16 The identified immune-related pathways in all three populations have been summarized in . Additionally, generated data on biological pathways was also screened to identify pathways related to local adaptation and stress. Biological pathways, response to oxygen-containing compound, cellular response to oxygen-containing compound and reactive oxygen species metabolic process were found to be enriched in all three yak populations while cellular response to hypoxia was enriched in Arunachali yak and cellular response to oxygen levels was enriched in Himachali yak. The fold enrichment of GO terms for all these yak populations has been illustrated in .
Figure 2. The immune related enriched GO terms observed for genes harboring high impact variants. (A) Arunachali yaks. (B) Himachali yaks. (C) Ladakhi yaks.
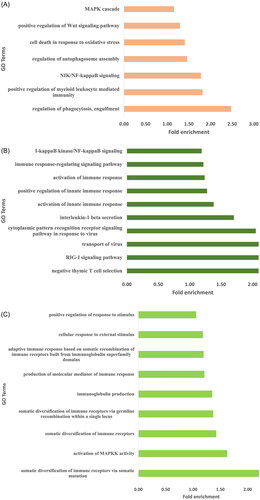
Figure 3. The hypoxia and stress related enriched GO terms observed for genes harboring high impact variants. (A) Arunachali yaks. (B) Himachali yaks. (C) Ladakhi yaks.
Discussion
In this study, a whole genome sequencing approach was used for the first time to decipher genomic variant information in Indian yak populations. The highest number of SNPs was found in Ladakhi yak (32632460), while the lowest was in Himachali yak (27260825). The number of InDels was approximately equal in the Arunachali and Ladakhi yaks, but slightly higher than the Himachali yak. Chai et al.Citation10 have reported approximately 44.3 million high quality SNPs in 108 yaks using whole genome sequencing analysis (average depth of 7.2×). Similarly, Qiu et al.Citation8 have identified 14.56 million SNPs across the genomic data (average depth of 6.7×) of 72 yaks. Similarly, 7.7 million quality SNPs were reported in six female Jinchuan yaks by whole genome sequencing (average depth of 25×, then adjusted to the sequencing depth to 6.77) approach.Citation17 Our findings revealed higher numbers of SNPs as compared to previous studies in terms of numbers of animals and populations, which might be due to the lesser selection intervention, a sizable base population, and diverse populations inhabiting different climatic conditions. A previous study based on ddRAD sequencing of Indian yak populations also revealed high molecular variation within all yak populations along with considerable effective population size.Citation6 Whole genome resequencing approach was used in the present study which presents an edge over previous studies related to variants information covering whole genome. This whole genome resequencing data could further be utilized for the detection of selection sweeps and copy number variations in indigenous yak populations. In our study, chromosome 1 had the maximum number of SNPs while chromosome Y had the least. However, the highest numbers of InDels were found in chromosome 12 in all the three populations. The highest and lowest numbers of SNPs on these chromosomes could be correlated with the chromosome sizes. The maximum number of SNPs and InDels were found in intronic and the intergenic region and this could be corroborated well with the previous reports.Citation8,Citation18
Annotation of identified SNPs was done which involved the categorization of variants based on their relationship with genomic sequences, and the way it may affect the gene product. To achieve this, SnpEff,Citation14 predicted the effects of genetic variants by categorizing the SNPs based on putative impact (high, moderate, low, and modifier) and function (missense, non-sense, and silent). Annotation study revealed that all the three yak populations had highest numbers of SNPs in the modifier and missense categories, while the least number of SNPs were detected in the high impact and nonsense categories. Mishra et al. have also reported similar results using ddRAD approach in buffaloes.Citation19 The identification of variants under the high impact and missense categories might be useful for studying their impact on complex traits. Further, the RYR2, SYNE2, BOLA, and HF1 genes, and novel transcript ENSBGRG00000011079 were observed with the highest number of high impact variants in all the three yak populations. The RYR2 gene product has been reported to regulate intracellular calcium concentration in heart,Citation20 SYNE2 maintains the structural integrity of the nucleus and BOLA is involved in immune responses. Previous investigations have found indications of positive selection on RYR2 in the Tibetan wolf, chicken, and goat, probably because of its potential function in hypoxia adaption. Inadequate oxygen delivery to the tissues induces release of reactive oxygen and an increase of intracellular calcium which ultimately induce cell contraction, a crucial response to hypoxia.Citation21 Further, the immune response is significantly influenced by major histocompatibility complex (MHC), while the presence of high impact variants in BOLA genes are thought to play a substantial role in the diversity of antigen presenting molecules in yaks. Overall, the presence of the highest number of variants in these genes might be crucial for high altitude adaptation and associated with local fitness in Indian yaks.
Further, gene ontology analysis was performed using DAVID to identify highly enriched pathways containing genes with high impact SNPs in protein coding region. The latest version, DAVID v6.8, includes a comprehensive set of functional annotation tools to understand various biological pathways behind a large number of genes. Out of total biological pathways, Himachali yak showed the highest number of immune related significant (P < 0.05) biological pathways followed by Ladakhi and Arunachali yak. The fold enrichment of immune related pathways varied from 1.15–2.1, 1.06–2.21, and 1.16–2.47 in Himachali, Ladakhi, and Arunachali yak, respectively. The fold enrichment ratio measures the magnitude of enrichment, and the ratio 1.5 and above are suggested to be considered as the threshold for significant results.Citation15
Genes were extracted from all immune related pathways to get more comprehensive information. Genes NOD2, C3, CARD10, SYK, STAP1, EIF2AK2, RC3H2, HDAC7, ADGRG3, RASSF2, AGO1, RPS3, CHI3L1, and SASH1 were found to be common in all three yak populations. NOD2 (Nucleotide-binding oligomerization domain 2) encodes a protein that acts as a sensor for muramyl-peptides generated from the bacterial cell wall. It is reported to be involved in innate immune regulation by activating type-I interferon and autophagy.Citation22 NOD2 was found to harbor 3, 2, and 1 high impact SNPs in Ladakhi, Himachali, and Arunachali yaks, respectively. C3 (complement C3) plays crucial role in triggering both classical and alternative complement activation pathways and was found to contain three high impact SNPs in Ladakhi and Himachali yak while 2 SNPs in Arunachali yak. Gene, CARD10 (caspase recruitment domain family member 10) is recognized as apoptosis signaling gene which activates NF-kappa-B through B-cell lymphoma/leukemia 10. This gene was found to harbor 1 high impact SNP in Arunachali and Himachali yak and 4 in Ladakhi yaks. However, the RASSF2 gene involved in Ras signaling and the AGO1gene involved in RNA silencing were found to harbor more high impact variants in Arunachali yaks as compared to Himachali and Ladakhi yaks. SYK protein is widely expressed in hematopoietic cells and involved in coupling activated immunoreceptors to downstream signaling events that mediate diverse cellular responses, including proliferation, differentiation, and phagocytosis. The SYK gene was found to have one high impact variant in each Arunachali and Himachali yaks, while two in Ladakhi yaks. Similarly, Ladakhi yak population was found to harbor more number of high impact variants in genes STAP-1 (2) and HDAC7 (6) as compared to Arunachali and Himachali yaks. STAP-1 is primarily expressed in the lymph nodes and spleen and regulates invariant natural killer T (iNKT) cells maintenance and activation,Citation23 and HDAC7 is highly expressed in CD4+/CD8+ thymocytes and functions as a signal-dependent repressor of gene transcription during T-cell development.Citation24
Arunachali yak population was found to harbor more number of high impact variants in genes such as CHI3L1 (2), RPS3 (3), and ADGRG3 (2) as compared to Himachali and Ladakhi yaks populations. CHI3L1 is a chitinase-like protein that is produced in innate immune cells and extensively expressed in a variety of inflammatory disorders with infectious and noninfectious aetiologies.Citation25 Similarly, RPS3 is a multifunctional protein that also plays central role in regulating numerous aspects of host–pathogen interactions.Citation26 Furthermore, Himachali yak population was found to harbor more number of high impact variants in RC3H2 (3) gene whereas, Arunachali and Himachali yaks were found to have one in each population. RC3H2 also defined as RING Finger And CCCH-Type Zinc Finger Domain-Containing Protein 2 is a protein coding gene that enables nucleic acid binding activity and ubiquitin protein ligase activity. Another gene EIF2AK2, also defined as eukaryotic translation initiation factor 2-alpha kinase 2 was found to have one high impact variant in each yak population. It is a serine/threonine-protein kinase that is IFN-induced and dsRNA-dependent and is essential for the innate immune response to viral infection.Citation27
More number of significant immune-related enriched biological pathways in Arunachali and Ladakhi yaks could suggest greater genetic diversity in the set of genes behind these pathways. This could be correlated with greater fitness to combat many viral infections and better innate immunity. However, this statement needs further validation by measuring a large number of diseased phenotypes and analyzing the genomic data associated with them.
Further, biological pathways were screened in all three populations to get a better understanding of local adaptation. These populations may vary in their adaptation tolerance for lesser oxygen or hypoxic conditions and evolve with specific abilities to cope with this condition. Previous studies on animals and humans have suggested that responses to hypoxic conditions are regulated by oxygen sensing and signal transduction pathways.Citation28 Ladakhi yaks are found at the highest altitude, while Arunachali yaks are found at the lowest altitude among three yak populations. Biological pathways viz., response to oxygen-containing compound, cellular response to oxygen-containing compound, reactive oxygen species metabolic process were found to be enriched in all three populations. However, GO terms including cellular response to hypoxia and cellular response to oxygen levels were found to be exclusively enriched in Arunachali yaks and Himachali yaks, respectively. Genes, HIF1A (Hypoxia-inducible factor-1alpha) and HIF3A were found to harbor one high impact polymorphism in protein coding region in Himachali and Arunachali yaks but were not detected in Ladakhi yaks. HIF-1α plays a key role in oxygen homeostasis, cellular responses to hypoxia and its abnormal expression has been reported in hypoxia related diseases.Citation29 HIF1A helps to maintain oxygen homeostasis by stimulating glycolysis, erythropoiesis, and angiogenesis.Citation30 Further, the PDK1 (Pyruvate Dehydrogenase Kinase 1) gene associated with hypoxia and Chronic Mountain Sickness was found to contain one high impact variant in both of their transcripts (ENSBGRT00000021747 and ENSBGRT00000021884) in Arunachali and Himachali yaks, while no high impact variant was found in Ladakhi yaks. The HIF-1 transcription factor directs the expression of genes involved in adaptation to low oxygen environments. It stimulates glycolysis and represses oxygen consumption by inducing PDK1, which results in increased availability of oxygen and reduced cell death.Citation31 Additionally, HIF-1 also targets angiogenic agents, which induces increased blood vessels formation for the supply of more oxygenated blood to hypoxic tissue. Genes, HMOX1 (Heme Oxygenase-1) and VEGF (Vascular endothelial growth factor) are downstream targets of HIF-1α, and both are reported to play pivotal role during hypoxic condition.Citation32 Furthermore, Arunachali yaks harbor 1 and Himachali yaks harbor two high impact variants in each transcript (ENSBGRT00000021970 and ENSBGRT00000022054) of the HMOX1 gene while no high impact variant was found in Ladakhi yaks. The gene VEGF is a potent angiogenic factorCitation33 that stimulates vascular endothelial cells and act as main mediator of hypoxia-induced-neovascularizationCitation34 and vascular permeability.Citation35 The gene EDN1 (endothelin 1) was also identified as a candidate gene for positive selection in the high-altitude populations.Citation36 EDN1 (Endothelins 1) was found to harbor three high impact variations in Arunachali yaks and one in Himachali yaks. However, no high impact variants were found in Ladakhi yaks. EDN1 are endothelium-derived vasoconstrictor peptides involved in a range of functions, including biological function, cellular response to hypoxia. Here, the disparity of high impact variants in the genes (HIF1A, HIF3A, PDK1, HMOX1, and EDN1) of these yak populations could be associated with positive selection or their adaptation against hypoxic conditions during evolution.
Biological pathways related to the regulation of stress were found to be enriched in all three yak populations. These pathways include regulation of response to stress, cellular response to stress, response to stress, regulation of response to oxidative stress, and pathways related to the MAPK cascade. MAPK signaling pathways are known to elicit the cellular response to environmental stress. Recently, a transcriptomic study on high altitude cattle revealed MAPK signaling pathway as a highly impacted pathway.Citation37 Some previous studies also established the correlation of this pathway related to high-altitude adaptations and hypoxic conditions.Citation38,Citation39 Therefore, enrichment of the above-mentioned pathways might be associated with high-altitude adaptations in Indian yaks.
Conclusion
In conclusion, whole genome analysis revealed that Ladakhi yaks were found to possess higher SNPs than Himachali and Arunachali yaks. In all the populations considered for this study, highest numbers of SNPs were found in chromosome 1, while the lowest in on chromosome Y. The highest numbers of InDels were found in on chromosome 12, while the lowest numbers were found in on chromosome Y. In addition to the numbers, annotation of SNPs and InDels provided detailed information regarding their position in the genome which includes intronic, untranslated region, upstream, downstream, splice site, or intergenic regions. Further, the disparity of high impact variants in HIF1A, HIF3A, PDK1, HMOX1, and EDN1 genes in these yak populations could be associated with positive selection or their high-altitude adaptation against hypoxic conditions during evolution. Genes RYR2, SYNE2, BOLA, and HF1, novel transcripts ENSBGRG00000011079, and HF1 were observed with the highest number of high impact variants in all yak populations, underlining the significance of these genes and their enriched biological pathways in adaptation and fitness for their native environment. Moreover, the information generated could be well exploited for better yak conservation strategies by estimating population genetics parameters viz., effective population size, inbreeding, and, observed and expected heterozygosity.
Author contributions
R.K.V. planned and designed the study with inputs from A.K. and M.D. S.K.N and S.A. provided yak DNA for WGS. A.K. and M.D. generated the WGS data. A.K. analyzed the WGS data and drafted the manuscript. S.K.N., R.A., A.Kour, and MA revised the manuscript with inputs from all authors.
Supplemental Material
Download MS Excel (2.2 MB)Acknowledgements
The authors thank Indian Council of Agricultural Research, New Delhi and Director, National Bureau of Animal Genetic Resources, Karnal for providing the financial assistance for this study. Special thanks are due to Dr. PJ Das, ICAR-NRC Pig for providing yak samples.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
The raw data and .vcf file have been submitted to Sequence Read Archive (PRJNA803425) and European Variant Archive (PRJEB50815), respectively.
References
- Pal RN. Yak (Poephagus grunniens L) of India. Anim. Genet. Resour. Inf. 1993;12:59–67.
- Guan J, Long K, Ma J, et al. Comparative analysis of the microRNA transcriptome between yak and cattle provides insight into high-altitude adaptation. PeerJ. 2017;5(11):e3959.
- BAHS. Basic Animal Husbandry Statistics-2019 | Department of Animal Husbandry & Dairying. Published, 2019. https://dahd.nic.in/circulars/basic-animal-husbandry-statistics-2019. Accessed August 26, 2021.
- Weitzman ML. What to preserve? An application of diversity theory to crane conservation. Quart. J. Econ. 1993;108(1):157–183.
- Xia W, Luo T, Zhang W, et al. Development of high-density snp markers and their application in evaluating genetic diversity and population structure in Elaeis guineensis. Front. Plant Sci. 2019;10(February):130.
- Sivalingam J, Vineeth MR, Surya T, et al. Genomic divergence reveals unique populations among Indian Yaks. Sci. Rep. 2020;10(1):3636.
- Yao RA, Akinrinade O, Chaix M, Mital S. Quality of whole genome sequencing from blood versus saliva derived DNA in cardiac patients. BMC Med Genomics. 2020;13(1):11.
- Qiu Q, Wang L, Wang K, et al. Yak whole-genome resequencing reveals domestication signatures and prehistoric population expansions. Nat. Commun. 2015;6(1):10283.
- Qiu Q, Zhang G, Ma T, et al. The yak genome and adaptation to life at high altitude. Nat Genet. 2012;44(8):946–949.
- Chai ZX, Xin JW, Zhang CF, Dawayangla , Luosang , Zhang Q, et al. Whole-genome resequencing provides insights into the evolution and divergence of the native domestic yaks of the Qinghai–Tibet Plateau. BMC Evol. Biol. 2020;20(1):137.
- Liu Y, Luo J, Dou J, et al. The sequence and de novo assembly of the wild yak genome. Sci. Data. 2020;7(1):66.
- Fuentes-Pardo AP, Ruzzante DE. Whole-genome sequencing approaches for conservation biology: advantages, limitations and practical recommendations. Mol. Ecol. 2017;26(20):5369–5406.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760.
- Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6(2):80–92.
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 1999;79(1):143–180.
- Lan D, Xiong X, Mipam TD, et al. Genetic diversity, molecular phylogeny, and selection evidence of Jinchuan Yak revealed by whole-genome resequencing. G3 (Bethesda). 2018;8(3):945–952.
- Dogan H, Can H, Otu HH. Whole genome sequence of a Turkish individual. PLoS One. 2014;9(1):e85233.
- Mishra DC, Sikka P, Yadav S, et al. Identification and characterization of trait-specific SNPs using ddRAD sequencing in water buffalo. Genomics. 2020;112(5):3571–3578.
- Ma MG, Liu XR, Wu Y, et al. RYR2 mutations are associated with benign epilepsy of childhood with centrotemporal spikes with or without arrhythmia. Front. Neurosci. 2021;15(April):629610.
- Wu DD, Yang CP, Wang MS, et al. Convergent genomic signatures of high-altitude adaptation among domestic mammals. Natl. Sci. Rev. 2020;7(6):952–963.
- Strober W, Watanabe T. NOD2, an intracellular innate immune sensor involved in host defense and Crohn’s disease. Mucosal Immunol. 2011;4(5):484–495.
- Kashiwakura JI, Saitoh K, Ihara T, et al. Expression of signal-transducing adaptor protein-1 attenuates experimental autoimmune hepatitis via down-regulating activation and homeostasis of invariant natural killer T cells. PLoS One. 2020;15(11):e0241440.
- Kasler HG, Verdin E. Histone deacetylase 7 functions as a key regulator of genes involved in both positive and negative selection of thymocytes. Mol. Cell Biol. 2007;27(14):5184–5200.
- Di Rosa M, Malaguarnera L. Chitinase 3 Like-1: an emerging molecule involved in diabetes and diabetic complications. Pathobiology. 2016;83(5):228–242.
- Gao X, Hardwidge PR. Ribosomal protein s3: a multifunctional target of attaching/effacing bacterial pathogens. Front Microbiol. 2011;2:137.
- Sadler AJ, Latchoumanin O, Hawkes D, Mak J, Williams BR. An antiviral response directed by PKR phosphorylation of the RNA helicase A. PLoS Pathog. 2009;5(2):e1000311.
- Rhodes J. Comparative physiology of hypoxic pulmonary hypertension: Historical clues from brisket disease. J. Appl. Physiol. (1985). 2005;98(3):1092–1100.
- Covello KL, Simon MC. HIFs, hypoxia, and vascular development. Curr. Top. Dev. Biol. 2004;62:37–54.
- Carmeliet P, Dor Y, Herber JM, et al. Role of HIF-1α in hypoxiamediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394(6692):485–490.
- Kim JW, Tchernyshyov I, Semenza GL, Dang C. v. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–185.
- Rashid I, Baisvar VS, Singh M, et al. Isolation and characterization of hypoxia inducible heme oxygenase 1 (HMOX1) gene in Labeo rohita. Genomics. 2020;112(3):2327–2333.
- Dw L GC, Wj K, Dv G NF. Vascular endothelial growth factor is a secreted angiogenic mitogen. Science. 1989;246(4935):1306–1309.
- Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J. Cell Sci. 2001;114(Pt 5):853–865.
- Senger DR. Vascular endothelial growth factor: much more than an angiogenesis factor. Mol. Biol. Cell. 2010;21(3):377–379.
- Scheinfeldt LB, Tishkoff SA. Living the high life: high-altitude adaptation. Genome Biol. 2010;11:133.
- Verma P, Sharma A, Sodhi M, et al. Transcriptome analysis of circulating PBMCs to understand mechanism of high altitude adaptation in native cattle of Ladakh region. Sci. Rep. 2018;8(1):7681.
- De B, Huajun X, Cuihong Z, Jun Z, Xiaoyan D, Xiaopeng L. Systems biology approach to study the high altitude adaptation in Tibetans. Braz. Arch. Biol. Technol. 2013;56(1):53–60.
- Ryter SW, Xi S, Hartsfield CL, Choi AM. Mitogen activated protein kinase (MAPK) pathway regulates heme oxygenase-1 gene expression by hypoxia in vascular cells. Antioxid Redox Signal. 2002;4(4):587–592.