
Abstract
Diet is an important component to influence microbiota, there are less data available about the microbiome of Suffolk cross with Tibetan (SCT) animals with different fodders. The current study was conducted for comparing the fungi microbiota in SCT sheep fed with different forages. Sequencing of ileum samples from sheep groups of AH (alfalfa and oat grass), BH (mixture of grass and concentrated feeds), CH (concentrated feed I), DH (concentrated feed II) and EH (concentrated feed III) achieved 3,171,271 raw and 2,719,649 filtered sequences. Concentrated feeds changed fungi microbiota in SCT sheep with three phyla and 47 genera significantly different among the groups. Genera include positive genus of Scytalidium and negative fungi of Sarocladium, Kazachstania, Gibberella, Scytalidium, Candida, Wickerhamomyces. The findings of our study will contribute to efficient feeding of SCT sheep at cold plateau areas.
Introduction
Intestine microbiota are composed of numerous microbes which include fungi, viruses, bacteria and some protozoaCitation1 which are responsible for 98% of the organisms genetic activity.Citation2 Gut microbiota plays a significant role in functions related to the nutrition, metabolism, immune system development, nervous system development and behaviour of the host.Citation3 Fungi in gut biomes have not been studied as much as bacteria. However, it has been reported that gut fungi play an important role on health and diseases of animalsCitation4 as they affect intestinal bacteria through fungal–bacterial interaction.Citation5 In the human gut, there are over 400 fungi species which is approximately 0.1% of the total microbial DNA in the intestine.Citation6 The main fungi in the intestine include Candida, Cryptococcus and Cladosporium contributing greatly to the host’s immunity and gastrointestinal function.Citation7,Citation8 Studies have revealed that probiotic Saccharomyces cerevisiae var. boulardii is useful in treating gastroenteritis.Citation9
An increase in useful gut bacteria results in better growth performance however is also related to age, diet, management and environmental factors.Citation10,Citation11 Fungi dysbiosis is commonly associated with diverse diseases like non-alcoholic fatty liver diseaseCitation12 inflammatory bowel diseaseCitation13 and diarrhea.Citation14 Diet is another factor affecting microbiota.Citation15 To date, limited information is available about gut fungi of SCT (Suffolk cross with Tibetan) sheep reared on the cold plateau with different feeding regimes. Therefore, we conducted this study to compare the fungi flora in SCT sheep fed with different forages.
Materials and methods
Experiment design
A total of 50 SCT sheep with average age of 3 months were equally grouped (n = 10) into AH, BH, CH, DH and EH. Sheep in group AH, CH, DH and EH were given alfalfa and oat grass, concentrated feed #1 (corn 35%, wheat bran 23.5%, bean pulp 10%, alfalfa 20%, highland barley straw 10.5%, premix feed 0.5% and salt 0.5%), concentrated feed #2 (corn 45%, wheat bran 11.8%, soybean oil 2.5%, bean pulp 14.3%, alfalfa 17.4%, highland barley straw 8%, premix feed 0.5% and salt 0.5%) and concentrated feed #3 (corn 50%, wheat bran 5.9%, soybean oil 3.8%, bean pulp 16.2%, alfalfa 16.3%, highland barley straw 6.8%, premix feed 0.5% and salt 0.5%), respectively. Group BH was fed with equal amount of alfalfa and oat grass, and concentrated provender I, II and III. The SCT ruminates were reared for 120 days and weighed twice daily before slaughtering to collect ileum samples.
SCT sheep microbiota sequencing
Total DNA extraction from ileum samples of SCT sheep (n = 25) were carried out through MolPure® Stool DNA Kit (Yeasen, China) by following the manufacturer’s instructions. The quality and quantity of SCT sheep’s DNA extracts were checked by 1.5% agarose gel electrophoresis and NanoDrop One/OneC (Thermo Scientific, USA), respectively. Targeting gene amplification of SCT sheep products was performed by piloting ITS1 primers pairs as described previously,Citation16 and then, the amplified products were again checked for quality and quantity. Final results obtained from SCT sheep were used through MiSeq reagent kit v3 (Illumina, USA), whereas sequencing was done through platform of Illlumina MiSeq in Bioyi biotechnology company (Wuhan, China).
Fungal microbiota analysis
Amplicon sequence variants (ASVs) were generated by DADA2 after raw sequence quality control.Citation17 Then, ASVs were used to align ITS database for obtaining taxonomy table through vsearch software.Citation18 Differences of alpha (Shannon, observed OTUs, Faith’s phylogenetics and Chao1) and beta indexes (Qiime2β, unweighted pair-group method with arithmetic means, principal coordinate analysis and non-metric multidimensional scaling) were calculated via QIIME2.Citation19–22 The distinguished marker fungi species among SCT sheep in group AH compared to other groups were analysed through analysis of variance (ANOVA), LEfSe and DEseq2.Citation23,Citation24 The KEGG Ortholog function of SCT sheep gut fungi was calculated by PICRUSt.Citation25
Statistical analysis
Analysis of variance and Duncans multiple range test was selected for obtaining significance among SCT sheep by using SPSS (IBM, 26.0). Data are presented as means ± SD and differences were perceived as statistically significant at P < 0.05.
Results
Fungal microbiota analysis of SCT ruminant with different fodders
It was observed that the average raw sequences and filtered sequences in AH (108865, 93806) were lower than BH (134633, 114510), CH (136942, 116449), DH (128162, 110202) and EH (125651, 108961) (). A total of 3249 ASVs were aligned in SCT sheep (AH = 609, BH = 591, CH = 875, DH =473 and EH = 1145) with 111 shared ASVs in small ruminants (). The shared ASVs between AH and BH, AH and CH, AH and DH, and AH and EH was 202, 208, 171 and 268, respectively. There were 11 (AH), 12 (BH), 12 (CH), 11 (DH) and 13 (EH) phyla whereas, 181 (AH), 135 (BH), 197 (CH), 119 (DH) and 261 (EH) genera observed in SCT sheep (). Alpha diversity shows that no valid difference is examined among different SCT groups ( and ).
Figure 1. ASVs Aligning analysis. a: Venn map, b; annotation statistics.
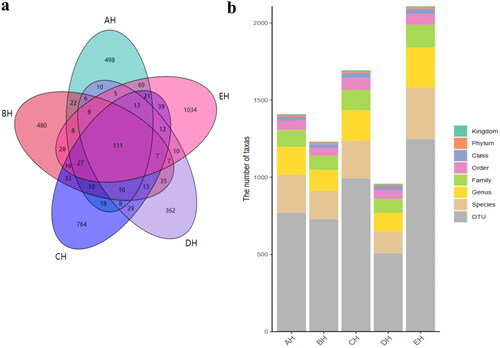
Figure 2. Comparing analysis of alpha diversity in SCT ruminants. a: chao1, b: faith_pd, c: observed_features, d: shannon_entropy, e: simpson.
Table 1. The obtained sequencing data in SCT ruminants.
Table 2. Comparing analysis of alpha diversity index in SCT ruminants.
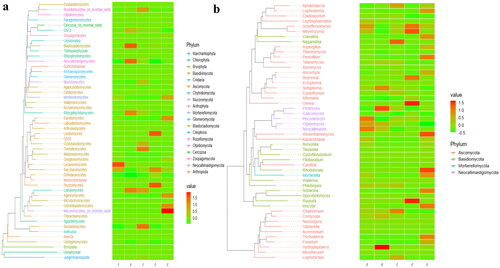
At the phylum level, Ascomycota, Neocallimastigomycota and Chytridiomycota were primary phyla in AH (44.64%, 25.56%, 7.14%) and BH (14.23%, 49.01%, 2.03%), whereas Ascomycota, Neocallimastigomycota and Basidiomycota were the dominating phyla in CH (29.83%, 35.81%, 11.43%), DH (54.29%, 23.12%, 5.20%) and EH (52.20%, 14.98%, 13.53%) (). At class levels, Neocallimastigomycetes, Saccharomycetes and Eurotiomycetes were mainly examined in AH (25.56%, 30.08%, 3.36%) and EH (14.98%, 31.21%, 10.59%). The principally classes in BH were Neocallimastigomycetes (49.01%), Saccharomycetes (6.12%) and Sordariomycetes (2.78%), in CH Neocallimastigomycetes (35.81%), Saccharomycetes (15.85%), Tremellomycetes (9.62%) and in DH were Neocallimastigomycetes (23.12%), Saccharomycetes 39.06%) and Agaricomycetes (3.36%), respectively (). At the order level, Neocallimastigales, Saccharomycetales and Eurotiales were the central orders in AH (25.56%, 30.08%, 3.26%), DH (23.12%, 39.06%, 2.48%) and EH (14.98%, 31.21%, 10.36%) while, Neocallimastigales (49.01%), Saccharomycetales (6.12%) and Hypocreales (2.00%) were found higher in BH and Neocallimastigales (35.81%), Saccharomycetales (15.85%) and Filobasidiales (9.53%) were prime orders in CH (). At family level, Neocallimastigaceae, Debaryomycetaceae and Aspergillaceae were mainly found in AH (25.56%, 29.39%, 2.84%), BH (49.01%, 5.77%, 1.23%) and DH (23.12%, 38.94%, 2.08%), while Neocallimastigaceae (23.12%), Debaryomycetaceae (38.94%) and Filobasidiaceae (2.08%) were the primary families in CH. Neocallimastigaceae (14.98%), Debaryomycetaceae (18.49%) and Phaffomycetaceae (12.01%) were dominating families in EH (). At genera levels, the main genus in different SCT sheep groups were Scheffersomyces (29.09%), Pecoramyces (6.21%) and Neocallimastix (5.22%) in AH, Piromyces (31.97%), Scheffersomyces (5.73%) and Orpinomyces (4.73%) in BH, Scheffersomyces (15.21%), Piromyces (13.47%) and Caecomyces (10.89%) in CH, Scheffersomyces (38.76%), Caecomyces (9.96%) and Piromyces (4.50%) in DH and Scheffersomyces (18.37%), Wickerhamomyces (12.00%) and Piromyces (7.91%) in EH, respectively (). Grouping and clustering heat map detected that Ascomycota was more abundant in AH, Neocallimastigomycota was higher in BH, Ascomycota was more abundant in DH and Ascomycota and Mortierellomycota were more abundant in EH (). At the class level, Saccharomycetes, Lecanoromycetes and Cystobasidiomycetes were more abundant in AH Neocallimastigomycetes was higher in BH, Tremellomycetes was higher in CH, Saccharomycetes, Leotiomycetes and Pezizomycetes were higher in DH, and Saccharomycetes, Eurotiomycetes, Agaricomycetes, Mortierellomycetes and Microbotryomycetes were more abundant in EH, respectively (). At the order level, higher abundance of Saccharomycetales in AH, Neocallimastigales in BH, Filobasidiales in CH, Saccharomycetales, Helotiales and Thelephorales in DH, and Saccharomycetales, Eurotiales, Sebacinales, Agaricales and Mortierellales in EH were detected, respectively, in SCT sheep groups (). Higher abundance families of Debaryomycetaceae in AH, Neocallimastigaceae in BH, Filobasidiaceae and Chaetomiaceae in CH, Debaryomycetaceae in DH and Aspergillaceae, Phaffomycetaceae, Sebacinaceae, Mortierellaceae, Inocybaceae and Hypocreaceae were revealed in EH (). At the genera level, abundance of Scheffersomyces, Neocallimastix and Pecoramyces in AH, Piromyces and Orpinomyces in BH, Naganishia and Chaetomium in CH, Scheffersomyces in DH, and Penicillium, Wickerhamomyces, Sebacina, Mortierella and Inocybe in EH were found (). Evolutionary tree of species with heat map analysis exhibited higher abundance of Saccharomycetes and Lecanoromycetes in AH, Lobulomycetes, Rhizophlyctidomycetes, Blastocladiomycetes and Neocallimastigomycetes in BH, Sordariomycetes, Lobulomycetes, Tremellomycetes and Rozellomycotina_cls_Incertae_sedis in CH, Pezizomycetes, Saccharomycetes and Leotiomycetes in DH and Agaricomycetes, Geminibasidiomycetes, Mucoromycotina_cls_Incertae_sedis, Eurotiomycetes and Mortierellomycetes in EH (). Higher genus of Scheffersomyces, Pecoramyces, Neocallimastix and Orpinomyces in AH, Hydropisphaera, Setophoma and Piromyces in BH, Chaetomium and Naganishia in CH, Russula, Genea, Meyerozyma, Scheffersomyces and Boeremia in DH, and Trichoderma, Sebacina, Inocybe, Clavulina, Lophiostoma, Penicillium, Mortierella, Rhodotorula and Wickerhamomyces in EH were also observed ().
Figure 3. Fungi microbiota structure of SCT sheep at different taxa presented in percentile pile bar chart. a: Phylum, b: Class, c: Order, d: Family, e: Genera.
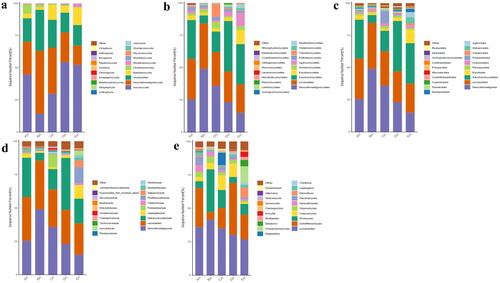
Figure 4. Comparing analysis of fungi microbiota structure of SCT sheep via grouping and clustering heat map. a: Phylum, b: Class, c: Order, d: Family, e: Genera.
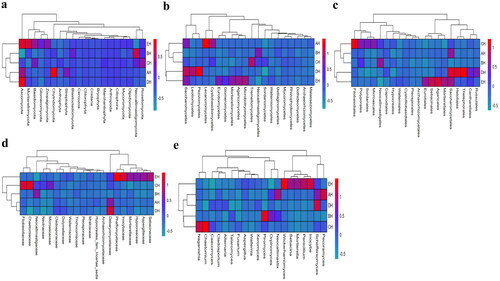
Figure 5. Evolutionary tree of species with heat map analysis of the fungi microbiota of SCT sheep in (a) class and (b) genera.
Examining the marker fungi species in SCT sheep
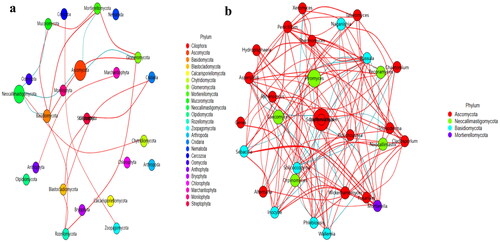
No strong difference regarding group distance among SCT groups was found by analysis of NMDS (), PCA (), PCoA () and Qiime2β (); however, effective difference was discovered in plots between AH and BH (p < 0.01) and CH (p < 0.01) respectively (). The LEfSe showed that p__Neocallimastigomycota (p < 0.05) and p__Ascomycota (p < 0.05) were expressively higher in BH and DH, respectively (). At the level of genera, g__Stephanonectria (p < 0.01) and f__Hypocreales_fam_Incertae_sedis (p < 0.05) in AH, g__Myrothecium (p < 0.05), f__Neocallimastigaceae (p < 0.05), f__Stachybotryaceae (p < 0.05), g__Phlebiopsis (p < 0.05), c__Ustilaginomycetes (p < 0.05), f__Ustilaginaceae (p < 0.05), c__Neocallimastigomycetes (p < 0.05), g__Sporisorium (p < 0.05), o__Polyporales (p < 0.05), o__Neocallimastigales (p < 0.05), p__Neocallimastigomycota (p < 0.05), f__Phanerochaetaceae (p < 0.05), g__Piromyces (p < 0.05) and o__Ustilaginales (p < 0.05) in BH, g__Simplicillium (p < 0.01), g__Lecanicillium (p < 0.05), g__Galerella (p < 0.01), o__Sordariomycetes_ord_Incertae_sedis (p < 0.05), g__Conioscypha (p < 0.01), f__Chaetomiaceae (p < 0.05), c__Sordariomycetes (p < 0.05), g__Lophotrichus (p < 0.01), g__Caecomyces (p < 0.05), o__Microascales (p < 0.05), g__Wongia (p < 0.05), g__Aphanoascus (p < 0.01), f__Onygenaceae (p < 0.01), o__Sordariales (p < 0.05), f__Nectriaceae (p < 0.05), g__Naganishia (p < 0.05), o__Filobasidiales (p < 0.05), f__Filobasidiaceae (p < 0.05), c__Tremellomycetes (p < 0.05), f__Bolbitiaceae (p < 0.05), f__Microascaceae (p < 0.05) and f__Papulosaceae (p < 0.05) in CH, p__Ascomycota (p < 0.05) in DH were observed, respectively (). The DESeq2 Volcano map exhibits that compared with AH abundance of Ascomycota (p < 0.05) and Mucoromycota (p < 0.05) were significantly lower in BH and Sebacinales (p < 0.05) was significantly lower in DH, respectively (). Regarding genus comparison in AH the abundance of Hydropisphaera (p < 0.001), Aphanoascus (p < 0.01), Lophotrichus (p < 0.05) and Dokmaia (p < 0.05) were significantly higher, while Occultifur (p < 0.001), Sarocladium (p < 0.01), Kazachstania (p < 0.01), Sebacina (p < 0.01), Nigrospora (p < 0.01), Inocybe (p < 0.01), Meyerozyma (p < 0.05), Filobasidium (p < 0.05), Pecoramyces (p < 0.05) and Gibberella were remarkably lower. The abundance of Lophotrichus (p < 0.0001), Aphanoascus (p < 0.0001), Chrysosporium (p < 0.001), Plenodomus (p < 0.01), Scopulariopsis (p < 0.01), Acrocalymma (p < 0.01), Dokmaia (p < 0.01), Aplosporella (p < 0.01), Mucor (p < 0.05) and Scytalidium were meaningfully higher in CH, while Occultifur (p < 0.0001), Stephanonectria (p < 0.0001), Coprinellus (p < 0.001), Stachybotrys (p < 0.01), Fusariella (p < 0.01), Symmetrospora (p < 0.01), Filobasidium (p < 0.01), Pleospora (p < 0.01), Candida (p < 0.01), Trichothecium (p < 0.01), Sarocladium (p < 0.01), Acrostalagmus (p < 0.01), Gibberella (p < 0.01), Inocyb (p < 0.05), Aureobasidium (p < 0.05), Kazachstania (p < 0.05) and Sporisorium were significantly lower. The abundance of Xeromyces (p < 0.0001), Dokmaia (p < 0.05), Lophotrichus (p < 0.05), Thermomyces (p < 0.05) and Aphanoascus (p < 0.05) were expressively higher in DH, while Inocybe (p < 0.01), Wickerhamomyces (p < 0.01), Sebacina (p < 0.01), Pichia (p < 0.05), Trichothecium (p < 0.05), Coprinellus (p < 0.05), Acrostalagmus (p < 0.05), Stephanonectria (p < 0.05), Occultifur (p < 0.05), Sarocladium (p < 0.05), Periconia (p < 0.05), Stachybotrys (p < 0.05), Candida (p < 0.05), Fusariella (p < 0.05) and Microascus (p < 0.05) were significantly lower. The abundance of Aphanoascus (p < 0.001), Cystofilobasidium (p < 0.001), Xeromyces (p < 0.001), Phoma (p < 0.01), Rhodotorul (p < 0.01), Wickerhamomyces (p < 0.01), Penicillium (p < 0.05), Ascochyta (p < 0.05), Trichoderma (p < 0.05) and Pichia (p < 0.05) were expressively higher in EH. While Stephanonectria (p < 0.001), Coprinellus (p < 0.001), Sarocladium (p < 0.01), Periconia (p < 0.01), Trichothecium (p < 0.01), Papiliotrema (p < 0.01), Occultifur (p < 0.01), Pleospora (p < 0.05) and Simplicillium (p < 0.05) were significantly lower (). Network analysis shows that phyla of Ascomycota and Mucoromycota were negatively related to the fungal microbiota of SCT sheep while, Basidiomycota, Blastocladiomycota, Chytridiomycota, Glomeromycota, Mortierellomycota, Zoopagomycota, Cnidaria and Anthophyta were positively related (). Genus of Scheffersomyces, Caecomyces, Pecoramyces, Naganishia and Phlebiopsis were negatively related to the microbiota, while Piromyces, Orpinomyces, Wickerhamomyces, Neocallimastix, Penicillium, Aspergillus, Sebacina, Inocybe, Wallemia, Mortierella, Cladosporium, Chaetomium, Talaromyces, Xeromyces, Hydropisphaera, Genea, Alternaria, Russula, Thermomyces and Aphanoascus were positively related ().
Figure 6. Beta diversity analysis of SCT sheep. a: NMDS, b: PCA, c: PCoA, d: Qiime2β, e: Group significance plots.
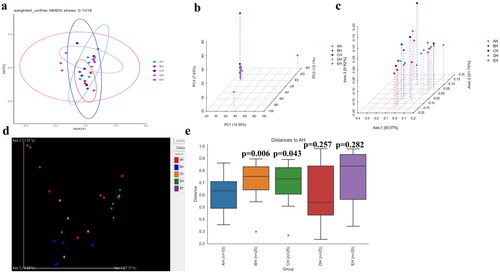
Figure 7. Exploring the significant different fungi species among SCT sheep via LEfSe. a: Phylum, b: Genera.
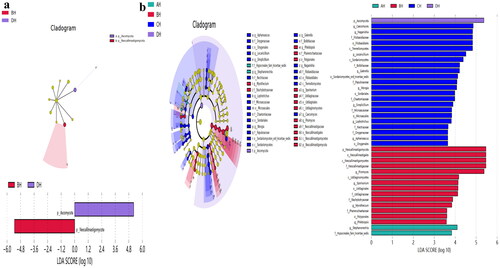
Figure 8. Discovering valid fungi species among SCT ruminants via DESeq2 volcano map a: Phylum, b: Genera.
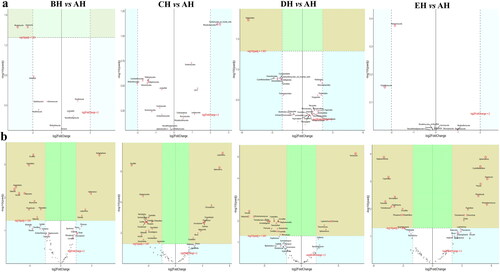
Figure 9. Network analysis of fungi microbiome of SCT sheep. a: Phylum, b: Genera.
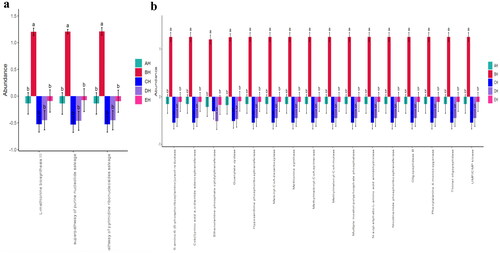
Function prediction analysis of fungi microbiota in SCT sheep
Fungi microbiota function prediction shows that L-methionine biosynthesis III, super pathway of purine nucleotide salvage and super pathway of pyrimidine ribonucleosides salvage were significantly different among the SCT sheep groups (). A total of 16 different enzymes were found significantly different among the five SCT sheep groups ().
Figure 10. Comparing fungi microbiota function of metaCys pathways (a) and enzyme abundance (b) in SCT sheep.
Discussion
Currently, researchers are paying attention to fungi microbiota due to their vital functions related to the health of their hosts.Citation26 Microbiome analysis is being widely used in forensic investigations,Citation27 diseases predictionCitation28 and epidemiological studies.Citation29
In this study we performed fungi microbiome analysis of SCT sheep with different feeds, and a total of 3,171,271 and 2,719,649 raw and filtered sequences were achieved in SCT sheep. The 111 shared ASVs were found in sheep and it was found that EH (268) had more shared ASVs compared to AH while in comparison with the same DH had less ASVs (171). There was no significant different alpha diversity index in present SCT sheep, which was in line with sheep in different feeding methods;Citation30 however, the results of the current study are not in agreement with dairy cattle fed with different dose of cellulose.Citation31 The primary phylum found in different sheep groups was Ascomycota in AH (44.64%), DH (54.29%) and EH (52.20%), and Neocallimastigomycota in BH (49.01%) and CH (35.81), respectively. These results are contradictory to the findings in citizen suffering inflammatory bowel disease and paediatric inhabitants,Citation32,Citation33 in which Ascomycota was found dominant phylum. Different results were found at genera level, Scheffersomyces was found main genera in AH (29.09%), CH (15.21%), DH (38.76%) and EH (18.37%), while Piromyces (31.97%) was the dominated genera in BH. Besides shifts of fungi structure in different taxa difference of feed also changed the composition of fungi microbiota in SCT sheep which was confirmed by grouping and clustering heat map along with evolutionary tree of species with heat map. The changed fungi microbiota affected functions of metaCys pathways and enzymes in SCT sheep.
Distinguished fungi species in SCT sheep were explored through LEfSe and it was discovered that p__Neocallimastigomycota and p__Ascomycota were higher in BH and DH respectively. Two genera in AH, 14 in BH, 22 in CH and 1 in DH were significantly higher, which proved altering of microbiota in SCT animals. For identifying different marker fungi species in sheep of the current study DESeq2 volcano map analysis was used which detected 3 phyla and 47 genera. Among those genus Sarocladium was discovered to be a pathogenic fungus.Citation34 The lower abundance of those genera in BH, CH and EH indicates that concentrated feed decreases fungal infection in SCT sheep. Kazachstania is a pathogen commonly reported in veterinary practice,Citation35 lower abundance of this genus in BH and DH shows that concentrated feed II can be used limit the growth of the aforementioned pathogen in sheep. Higher abundance of Gibberella was reported in Crohn’s disease.Citation36 The lower abundance of Gibberella in BH and CH suggests that concentrated feed I decreased the said hazardous fungi in SCT sheep. Lower abundance of Scytalidium was reported in ulcerative colitis,Citation37 more abundant of this genus in CH indicates that concentrated feed I promotes the colonization of Scytalidium in sheep. Previously, it has been reported that Coprinellus was found to be dominating in Cryptosporidium parvum infected yaks.Citation38 Lower abundance of this genera in CH, DH and EH demonstrates that concentrated feeds relieve the diarrhea in ruminants. The higher abundance of Symmetrospora, Filobasidium and Aureobasidium was reported in C. parvum infected yaks,Citation38 UC patientsCitation39 and newborns with bronchopulmonary dysplasia damage.Citation40 This confirms that concentrated feed I decrease pathogenic fungi genera in sheep. Species of Candida and Wickerhamomyces are to be opportunistic pathogensCitation37,Citation41 lower abundance of Candida in CH and DH along with Wickerhamomyces in DH and EH revealed that concentrated feed limited the pathogenic fungal growth in gut of sheep. Previous studies found higher abundance of Pichia in people with alcohol related liver damages,Citation42 lower abundance of this genus in DH demonstrates that concentrated feed II recover liver injury. It is reported that lower abundance of Penicillium in mice treated with fluoride,Citation43 more abundant of this genus in EH shows that concentrated feed# 3 promotes the these fungal in SCT sheep. Aphanoascus, Dokmaia, Occultifur, Sebacina, Stachybotrys and Stephanonectria are environmental genera,Citation44–49 which may have little relationship with the fungi microbiota in SCT sheep.
Conclusion
Concentrate feeding regulates the gut fungal biome of SCT sheep by enhancing the abundance of positive genera like Scytalidium and decreasing negative pathogenic fungal strains which include Sarocladium, Kazachstania, Gibberella, Scytalidium, Candida and Wickerhamomyce. These findings can be critical related to efficient feeding for Suffolk cross to Tibetan sheep on cold plateaus. Therefore, further studies need to conduct to related the results of this study for efficient feeding of Suffolk cross to Tibetan sheep on the plateaus.
Author contributions
YR and KL: research idea and methodology. YR and RCCW: reagents, materials, and analysis tools. KL: writing – original draft and preparation. KM, YR and KL: writing – review and editing. YR and KL: visualization and supervision. All authors known and approved the final manuscript.
Ethics approval and consent to participate
All the experiment procedures were performed under the ethics committee of Nanjing Agricultural University (NJAU.No20220520108).
Acknowledgement
No.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability
All raw sequences data were deposited in the NCBI Sequence Read Archive database under the following accession number: PRJNA1013103.
Additional information
Funding
References
- Sittipo P, Lobionda S, Lee YK, Maynard CL. Intestinal microbiota and the immune system in metabolic diseases. J Microbiol. 2018;56(3):1–13.
- Grice EA, Segre JA. The human microbiome: our second genome. Annu Rev Genomics Hum Genet. 2012;13(1):151–170.
- Hertli S, Zimmermann P. Molecular interactions between the intestinal microbiota and the host. Mol Microbiol. 2022;117(6):1297–1307.
- Lapiere A, Richard ML. Bacterial-fungal metabolic interactions within the microbiota and their potential relevance in human health and disease: a short review. Gut Microbes. 2022;14(1):2105610.
- Stewart D, Romo JA, Lamendella R, Kumamoto CA. The role of fungi in c. Difficile infection: an underappreciated transkingdom interaction. Fungal Genet Biol. 2019;129:1–6.
- Li F, Gao Y, Cheng W, Su X, Yang R. Gut fungal mycobiome: a significant factor of tumor occurrence and development. Cancer Lett. 2023;569:216302.
- Atarashi K, Suda W, Luo C, et al. Ectopic colonization of oral bacteria in the intestine drives th1 cell induction and inflammation. Science. 2017;358(6361):359–365.
- Wang ZK, Yang YS, Stefka AT, Sun G, Peng LH. Review article: fungal microbiota and digestive diseases. Aliment Pharmacol Ther. 2014;39(8):751–766.
- Hatoum R, Labrie S, Fliss I. Antimicrobial and probiotic properties of yeasts: from fundamental to novel applications. Front Microbiol. 2012;3:421.
- Ding X, Jiang H, Zhang R, et al. Comparative analysis of nasal microbial community between Tibetan sheep with different ages. Pak Vet J. 2023;43(4):723–731.
- Landy N, Kavyani A. Effects of using a multi‐strain probiotic on performance, immune responses and cecal microflora composition in broiler chickens reared under cyclic heat stress condition. Iran J Appl Anim Sci. 2013;3(4):703–708.
- You N, Xu J, Wang L, et al. Fecal fungi dysbiosis in nonalcoholic fatty liver disease. Obesity (Silver Spring). 2021;29(2):350–358.
- Mukhopadhya I, Hansen R, Meharg C, et al. The fungal microbiota of de-novo paediatric inflammatory bowel disease. Microbes Infect. 2015;17(4):304–310.
- Li K, Mehmood K, Zhang H, et al. Characterization of fungus microbial diversity in healthy and diarrheal yaks in gannan region of tibet autonomous prefecture. Acta Trop. 2018;182:14–26.
- Zmora N, Suez J, Elinav E. You are what you eat: diet, health and the gut microbiota. Nat Rev Gastroenterol Hepatol. 2019;16(1):35–56.
- Nel Van Zyl K, Whitelaw AC, Hesseling AC, Seddon JA, Demers A, Newton-Foot M. Fungal diversity in the gut microbiome of young south african children. BMC Microbiol. 2022;22(1):201.
- Harbuzov Z, Farberova V, Tom M, et al. Amplicon sequence variant-based meiofaunal community composition revealed by dada2 tool is compatible with species composition. Mar Genomics. 2022;65:100980.
- Rognes T, Flouri T, Nichols B, Quince C, Mahé F. Vsearch: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584.
- Rai SN, Qian C, Pan J, et al. Microbiome data analysis with applications to pre-clinical studies using qiime2: statistical considerations. Genes Dis. 2021;8(2):215–223.
- Wang Y, Sun F, Lin W, Zhang S. Ac-pcoa: adjustment for confounding factors using principal coordinate analysis. PLoS Comput Biol. 2022;18(7):e1010184.
- Ren Y, Zhaxi Y, Liu M, Idrees A, Li K. Revealing the fungi microbiome difference of suffolk cross with tibetan sheep on plateau. Pak Vet J. 2023;43(4):748–756.
- Wang H, Meng L, Mi L. Effects of Leymus chinensis hay and alfalfa hay on growth performance, rumen microbiota, and untargeted metabolomics of meat in lambs. Front Vet Sci. 2023;10:1256903.
- Yu X, Jiang W, Kosik RO, et al. Gut microbiota changes and its potential relations with thyroid carcinoma. J Adv Res. 2022;35:61–70.
- Shanmugam G, Lee SH, Jeon J. Ezmap: easy microbiome analysis platform. BMC Bioinformatics. 2021;22(1):179.
- Chen Y, Tian W, Shao Y, et al. Miscanthus cultivation shapes rhizosphere microbial community structure and function as assessed by illumina miseq sequencing combined with picrust and funguild analyses. Arch Microbiol. 2020;202(5):1157–1171.
- Huffnagle GB, Noverr MC. The emerging world of the fungal microbiome. Trends Microbiol. 2013;21(7):334–341.
- Kumari P, Prakash P, Yadav S, Saran V. Microbiome analysis: an emerging forensic investigative tool. Forensic Sci Int. 2022;340:111462.
- Butcher MC, Short B, Veena CLR, et al. Meta‐analysis of caries microbiome studies can improve upon disease prediction outcomes. APMIS. 2022;130(12):763–777.
- Hanson BM, Weinstock GM. The importance of the microbiome in epidemiologic research. Ann Epidemiol. 2016;26(5):301–305.
- Jize Z, Zhuoga D, Xiaoqing Z, et al. Different feeding strategies can affect growth performance and rumen functions in gangba sheep as revealed by integrated transcriptome and microbiome analyses. Front Microbiol. 2022;13:908326.
- Gharechahi J, Vahidi MF, Ding X, Han J, Salekdeh GH. Temporal changes in microbial communities attached to forages with different lignocellulosic compositions in cattle rumen. FEMS Microbiol Ecol. 2020;96(6):fiaa069.
- Sokol H, Leducq V, Aschard H, et al. Fungal microbiota dysbiosis in ibd. Gut. 2017;66(6):1039–1048.
- Chehoud C, Albenberg LG, Judge C, et al. Fungal signature in the gut microbiota of pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2015;21(8):1948–1956.
- Zhao Y, Ren X, Wu H, et al. Diversity and functional prediction of fungal communities in different segments of mongolian horse gastrointestinal tracts. BMC Microbiol. 2023;23(1):253.
- Vecherskii MV, Kuznetsova TA, Khayrullin DR, et al. Anthropogenic neighborhood impact on bacterial and fungal communities in polar bear feces. Animals (Basel). 2023;13(13):2067.
- Li Q, Wang C, Tang C, He Q, Li N, Li J. Dysbiosis of gut fungal microbiota is associated with mucosal inflammation in crohn’s disease. J Clin Gastroenterol. 2014;48(6):513–523.
- Jun X, Ning C, Yang S, et al. Alteration of fungal microbiota after 5-asa treatment in uc patients. Inflamm Bowel Dis. 2020;26(3):380–390.
- Lu S, Zou W, Chen X, et al. Effects of cryptosporidium parvum infection on intestinal fungal microbiota in yaks (bos grunniens). Microb Pathog. 2023;183:106322.
- van Thiel IAM, Rahman S, Hakvoort TBM, et al. Fecal filobasidium is associated with clinical remission and endoscopic response following fecal microbiota transplantation in mild-to-moderate ulcerative colitis. Microorganisms. 2022;10(4):737.
- Willis KA, Silverberg M, Martin I, et al. The fungal intestinal microbiota predict the development of bronchopulmonary dysplasia in very low birthweight newborns. medRxiv. 2023;
- Lim SJ, Mohamad Ali MS, Sabri S, Muhd Noor ND, Salleh AB, Oslan SN. Opportunistic yeast pathogencandida spp.: Secreted and membrane-bound virulence factors. Med Mycol. 2021;59(12):1127–1144.
- Hartmann P, Lang S, Zeng S, et al. Dynamic changes of the fungal microbiome in alcohol use disorder. Front Physiol. 2021;12:699253.
- Cao Q, Li R, Fu R, et al. Intestinal fungal dysbiosis in mice induced by fluoride. Chemosphere. 2020;245:125617.
- Li X, Li Y, Zhao X, et al. Restructured fungal community diversity and biological interactions promote metolachlor biodegradation in soil microbial fuel cells. Chemosphere. 2019;221:735–749.
- Lei W, Sijia L, Wen Z, et al. The effects of Cryptosporidium infection on gut fungi and enzyme abundance in Sus domesticus. Asian J. Agric. Biol. 2023;2023(4):2023114.
- Liang J, Zou R, Huang Y, et al. Structure and diversity of mycorrhizal fungi communities of different part of bulbophyllum tianguii in three terrestrial environments. Front Plant Sci. 2022;13:992184.
- Chafai W, EL Gabardi S, Douira A, Khalid A. Diversity and mycorrhizal potential of arbuscular mycorrhizal fungi in two natural soils in the eastern region of Morocco. Asian J. Agric. Biol. 2022;2022(2):202102101.
- Jabeen Z, Bukhari SA, Malik SA, Hussain G, Kamal S. Improved gut microbiota escalates muscle function rehabilitation and ameliorates oxidative stress following mechanically induced peripheral nerve injury in mice. Pak Vet J. 2023;43(4):707–713.
- Undugoda L, Kannangara S. Nature and activities of microfungi associated with the decomposition of rice straw in Sri Lanka. Asian J. Agric. Biol. 2022;2022(2):202103115.